

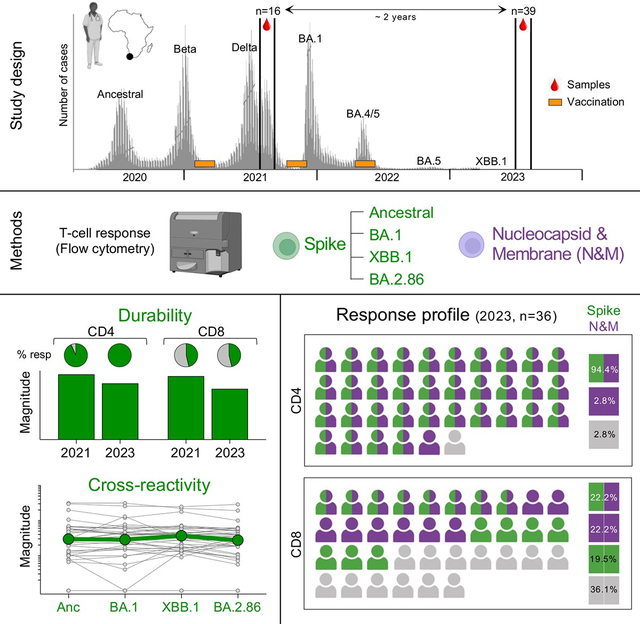
SUMMARY
Ongoing SARS-CoV-2 evolution has given rise to recombinant Omicron lineages that dominate globally (XBB.1), as well as the emergence of hypermutated variants (BA.2.86). In this context, durable and cross-reactive T-cell immune memory is critical for continued protection against severe COVID-19. We examined T-cell responses to SARS-CoV-2 approximately 1.5 years since Omicron first emerged. We describe sustained CD4+ and CD8+ spike-specific T-cell memory responses in healthcare workers in South Africa (n=39), who were vaccinated and experienced at least one SARS-CoV-2 infection. Spike-specific T cells were highly cross-reactive with all Omicron variants tested, including BA.2.86. Abundant nucleocapsid and membrane-specific T cells were detectable in most participants. The bulk of SARS-CoV-2-specific T-cell responses had an early-differentiated phenotype, explaining their persistent nature. Overall, hybrid immunity leads to the accumulation of spike and non-spike T cells evident 3.5 years after the start of the pandemic, with preserved recognition of highly mutated SARS-CoV-2 variants.
Graphical Abstract
INTRODUCTION
Sustained and cross-reactive immunological memory in the post-pandemic period is essential for continued protection from severe outcomes of COVID-19. Viral evolution has led to the emergence and dominance of the XBB recombinant sub-lineages of Omicron1. Recently, the novel Omicron subvariant BA.2.86 was described, with up to 60 amino acid changes compared to ancestral SARS-CoV-2 in the spike protein, and over 30 changes in spike compared to the BA.2 and XBB.1.5 variants. Due to its hyper-mutated nature, BA.2.86 has been classified as a “variant under monitoring” by WHO, and as of November 01, 2023, it has been identified in 6,819 sequences from 47 countries. This is likely an underestimate of BA.2.86 prevalence, given the current limited SARS-CoV-2 surveillance effort. Recent studies have evaluated the neutralization sensitivity of BA.2.862–5. As anticipated, BA.2.86 shows extensive immune evasion relative to ancestral SARS-CoV-2 in sera collected prior to the Omicron wave2; although in BA.1-infected individuals, the degree of neutralization of BA.2.86 was similar to that of XBB lineages currently dominating globally. However, the ability of spike-specific T cells to cross-recognize BA.2.86 spike has not yet been investigated. While it has been demonstrated that spike T-cell responses generated upon natural infection and vaccination against the ancestral SARS-CoV-2 spike are highly cross-reactive against Omicron BA.16–9, it is important to determine whether the extensive mutations in BA.2.86 spike could hinder its recognition by spike memory T-cell responses in individuals who have been infected and/or vaccinated during the course of the COVID-19 pandemic.
It is now clearly established that the SARS-CoV-2-specific antibody response (generated upon infection or vaccination) wanes relatively quickly10 and shows reduced neutralization activity against each new variant of concern (VOC) that dominates circulation, resulting in sub-optimal protection against SARS-CoV-2 infection. In contrast, memory T-cell responses to SARS-CoV-2 can persist for up to a year following exposure to the SARS-CoV-2 spike protein and maintain robust cross-reactivity against VOCs11–14. As we find ourselves three and a half years into the COVID-19 pandemic, where infection waves are smaller and booster vaccination is limited in most parts of the world due to restricted eligibility or availability, it is critical to monitor long-term immunity to SARS-CoV-2. Immune mechanisms of protection are complex and multifactorial, including neutralizing and Fc-mediated functions of antibodies, memory B cells, memory T cell function, T cell specificity and mucosal responses15.
In this study, we focused on investigating the durability of T cell responses. We included 39 healthcare workers, with a documented history of SARS-CoV-2 infection and vaccination, to determine whether their prevailing spike-specific memory T-cell responses in mid-late 2023, could cross-recognize the BA.2.86 sub-lineage. T-cell cross-reactivity was assessed in both Omicron-infected and -uninfected participants. In parallel, paired samples obtained two years apart were used to explore SARS-CoV-2-specific T-cell durability, including assessing the contribution of non-spike T cell responses to overall T-cell immunity.
RESULTS
Cross-reactivity of spike-specific T-cell response to Omicron variants
We measured T-cell responses to spike in blood samples (n=39) collected between July and September 2023. At this timepoint (T2 in Table 1 and Fig. S1), 28.2% (11/39) of the participants had received one dose of Ad26.COV2.S vaccine, 56.4% (22/39) two vaccine doses and 15.4% (6/39) three vaccine doses. The median time since last vaccination was ~21 months (IQR: 20.2–24.4). Twenty-two participants (56.4%) had a documented SARS-CoV-2 infection prior to the onset of the Omicron wave, and all experienced a breakthrough infection during the Omicron wave, at a median of 19.4 months (IQR: 17.8–19.9) before sample collection. We measured cytokine production (IFN-γ, IL-2 and TNF-α) in response to peptide pools covering the full ancestral, BA.1, XBB.1 or BA.2.86 spike protein (Fig. 1A). Fig. 1B shows the frequency of memory CD4+ T-cells to each SARS-CoV-2 spike tested. Notably, most participants (94.9%) still exhibited a robust ancestral spike-specific CD4+ T-cell response (median: 0.031%, IQR: 0.018–0.059) one and a half years after their last known SARS-CoV-2 infection. When comparing ancestral, BA.1, XBB.1 and BA.2.86 spike, we observed no significant difference in the frequency of spike-specific CD4+ T cells between the variants (Fig. 1B). For each Omicron sub-lineage, the fold change in the frequency of spike-specific CD4+ T-cell responses, relative to the ancestral spike, was calculated (Fig. 1C). Overall, the spike-specific CD4+ T-cell response was highly preserved (≥90%) against all Omicron variants tested, including the hyper-mutated BA.2.86. We also assessed the cross-reactivity of spike-specific CD8+ T-cell responses. In contrast to the CD4 compartment, the proportion of CD8 responders to ancestral spike was strikingly lower (~40%), and this was consistent amongst all three Omicron sub-lineages tested. While the median magnitude of spike-specific CD8+ T-cell responses was similar across all variants (Fig. 1D), the fold change within individual participants was variable. A fraction of participants who did not have a detectable CD8 response to ancestral spike (5/24, 20.8%) gained a CD8 response to one or more Omicron sub-lineages, likely reflecting de novo generation of a CD8 response to their Omicron breakthrough infection. In participants who had a detectable CD8+ T-cell response to ancestral spike, at least 50% of the CD8+ T-cell response was preserved against Omicron sub-lineages in most participants (10/15 for BA.1 and BA.2.86 and 11/15 for XBB.1), while a small fraction of individuals exhibited a reduction (>50%) or loss in T-cell reactivity to Omicron spike (5/15 for BA.1 and BA.2.86 and 4/15 for XBB.1) (Fig. 1E).
Table 1:
Clinical characteristics of study participants.
| T1 samples (pre-BA.1 wave) | T2 samples (post-BA.1 wave) | |
|---|---|---|
|
| ||
| n | n = 16 | n = 39 |
| Age at enrolment (median, IQR) | 51 [38–54] | 47 [31–54] |
| Gender (% female) | 87.5% | 84.2% |
| Sampling dates | July - Sept 2021 | July - Sept 2023 |
|
| ||
| Vaccination history | ||
| 1 vaccine dose (%, n) | 100% | 28.2% (n=11) |
| 2 vaccine doses (%, n) | 0% | 56.4% (n=22) |
| 3 vaccine doses (%, n) | 0% | 15.4% (n=6) |
| Months since last vaccination (median, IQR) | 5.2 [5–6] | 20.7 [20.2–24.4] |
|
| ||
| Infection history | ||
| Prior recorded infectiona (%, n) | 56.2% (n=9) | 56.4% (n=22) |
| Omicron BTI (%, n) | na | 100% (n=39) |
| Months since last recorded infection (median, IQR) | 8.4 [7–13]b | 19.4 [17.8–19.9]c |
|
| ||
| Paired samples | n = 15 | |
| Months between T2 and T1 samples (median, IQR) | 23.9 [23.3–24.1] | |
T1 samples were collected approximately 4–6 months prior to the Omicron BA.1 wave and T2 samples were collected 2 years later, approximately 1.5 years after the BA.1 wave (see Figure S1). The majority of participants (89.7%) were vaccinated with Ad26.COV2.S. Three participants received a heterologous vaccination regimen (Ad26.COV2.S and BNT162b2) and one participant received 3 doses of the BNT162b2 vaccine. Prior infection and breakthrough infection were determined by PCR (‘recorded infection’) or by Nucleocapsid seroconversion or a two-fold increase in Nucleocapsid-specific IgG.
IQR: Interquartile range; BTI: breakthrough infection;
: Ancestral SARS-CoV-2 or Beta variant infection; na: Not applicable;
: PCR data available for 5/9 participants with documented infection;
: PCR data available for 15/39 participants.
Figure 1. CD4+ and CD8+ T-cell responses to SARS-CoV-2 ancestral, BA.1, XBB.1 or BA.2.86 spike.
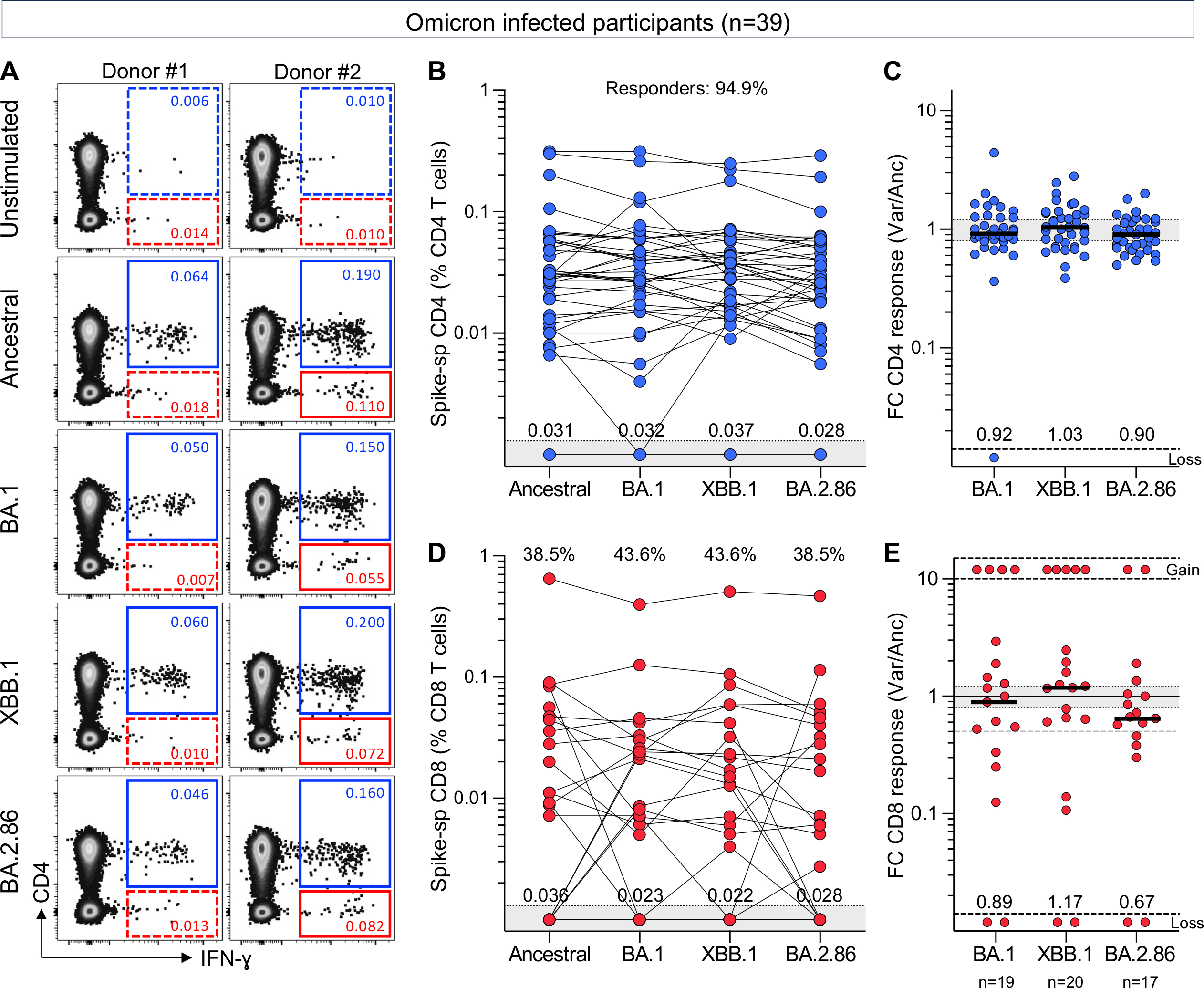
(A) Representative examples of IFN-γ production in response to ancestral, BA.1, XBB.1 or BA.2.86 spike in two individuals. The frequency of IFN-γ+ cells is expressed as a percentage of total CD4+ (blue) or CD8+ T cells (red).
(B and D) Frequency of spike-specific CD4+ T cells (B) and CD8+ T cells (D) producing any cytokine (IFN-γ, IL-2 or TNF-α) in 39 participants with confirmed Omicron infection. The proportion of responders is indicated at the top and median frequencies of spike-specific T cells in responders are indicated at the bottom of the graph.
(C and E) Fold change in frequency of spike-specific CD4+ T cells (C) and CD8+ T cells (E) between ancestral and SARS-CoV-2 variants in participants with confirmed Omicron infection. Medians are indicated. Gained responses are depicted on top and lost responses at the bottom. No significant differences were observed between variants using Friedman test with Dunn’s multiple comparisons post-test.
Since all participants had experienced an Omicron breakthrough infection, potentially prompting the development of de novo T-cell responses targeting mutated epitopes of spike16, we also assessed T-cell cross-reactivity at an earlier timepoint, obtained before Omicron emergence (see T1 in Table 1 and Fig. S1). Comparable results to post-Omicron-infected participants were found, demonstrating that spike-specific T-cell responses were highly cross-reactive with BA.1, XBB.1 and BA.2.86 (Fig. S2) and Omicron lineage cross-reactivity was not dependent on having been Omicron-infected.
Nucleocapsid and membrane-specific T-cell response significantly contribute to the memory SARS-CoV-2 adaptative immune response.
To gain a more comprehensive understanding of the profile of SARS-CoV-2-specific memory T-cell responses, we defined the extent to which non-spike proteins contribute to SARS-CoV-2 immunological memory. Specifically, our focus was on the T-cell response to SARS-CoV-2 nucleocapsid and membrane proteins, as these two proteins, in addition to spike, have demonstrated the highest immunogenicity in symptomatic patients17–21. T-cell responses for both spike and nucleocapsid and membrane (N&M) were available for 36 participants (Fig. 2A). CD4+ T-cell responses to N&M were detectable in 97.2% (35/36) of participants, with magnitudes comparable to those elicited toward spike (Fig. 2A, left panel). In fact, there was an association between the frequency of spike- and N&M-specific CD4+ T cells (r=0.64, p=2.3×10−5). In the CD8 compartment, similar proportions of responders (44.4%) were observed to spike and N&M (Fig. 2A, right panel). However, no association was found between spike and N&M responses (r=0.28, p=0.09), as previously reported22. The overall profile of SARS-CoV-2 CD4+ and CD8+ T-cell responses targeting spike and N&M was diverse amongst participants (Fig. 2B). It is noteworthy that most participants (34/36, 94.4%) had CD4 responses targeting both spike and N&M. In contrast, CD8 responders (23/36, 63.9%) were evenly divided amongst those who targeted both spike and N&M (9/23, 39.1%), those targeting spike exclusively (7/23, 30.4%) and those targeting N&M exclusively (7/23, 30.4%). Thus, quantifying non-spike responses increased the ability to detect CD8 responses to SARS-CoV-2 in those who were persistently spike-hyporesponsive despite multiple vaccinations. The contribution of N&M-specific T cells to SARS-CoV-2-specific memory responses is further illustrated in Fig. 2C, showing the profile of SARS-CoV-2 T-cell responses and clinical characteristics for each participant.
Figure 2. Profile of ancestral SARS-CoV-2 spike- and nucleocapsid and membrane-specific T-cell response ~3.5 years after the start of the COVID-19 pandemic.
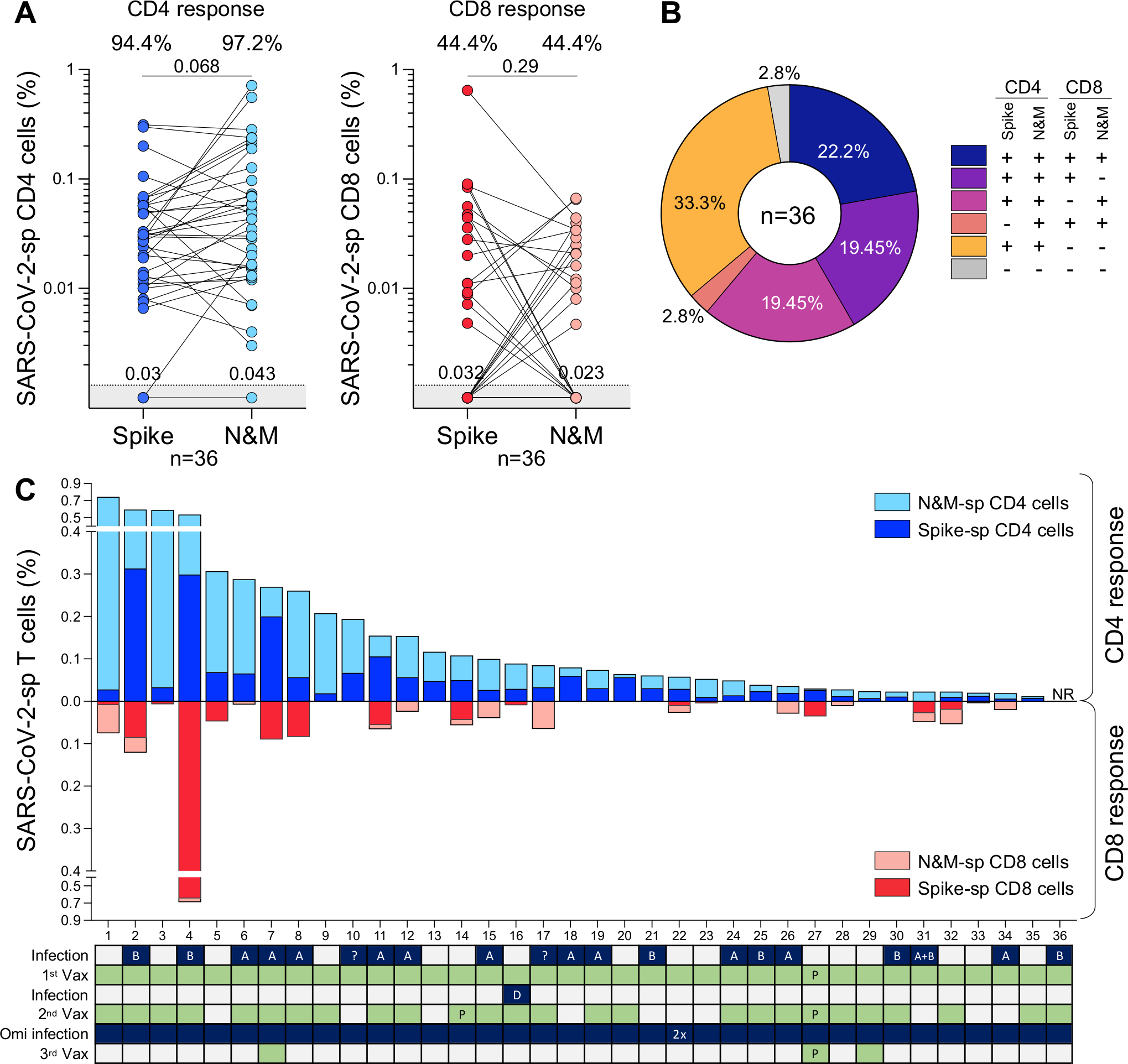
(A) Frequency of spike- and nucleocapsid and membrane (N&M)-specific CD4+ (left) and CD8+ T cells (right) in 36 participants sampled between July and September 2023. Proportion of responders is indicated on top and median responses at the bottom of the graph. Statistical comparisons were assessed using Wilcoxon matched-pairs signed rank test.
(B) Distribution of spike- and N&M-specific CD4+ and CD8+ T-cell responses. Each slice of pie represents a response pattern, as indicated.
(C) Total magnitude of spike- and N&M-specific CD4+ and CD8+ T-cell responses. The SARS-CoV-2 infection and vaccination histories of each participant are indicated. A: ancestral SARS-CoV-2 infection, B: Beta variant, D: Delta variant, ?: unknown variant infection. All vaccinations were Ad26.COV2.S, unless indicated with “P” for Pfizer/BNT162b2.
Durability of SARS-CoV-2 T-cell responses
Although a specific T cell-based measure of protection has yet to be defined, accumulating evidence suggests that T cells contribute to the control of SARS-CoV-2, indicated by their associations with COVID-19 symptoms and outcomes23,24. Thus, to measure T-cell maintenance, we compared the frequencies of T cells specific for spike and N&M in 15 paired samples. The samples were taken at two timepoints, 2 years apart: T1 (~4–6 months prior to the Omicron BA.1 wave) and T2 (~1.5 years after the BA.1 wave) (Table 1). Between the two timepoints, 60% (9/15) of the participants received a booster vaccination (median: 20.5 months before T2 sampling) and all experienced an Omicron breakthrough infection (median 19.4 months before T2 sampling). Fig. 3A shows the frequency of CD4+ T-cell responses to ancestral spike and N&M in these participants. No significant change was observed in the frequency of spike-specific CD4+ T cells between T1 and T2 (median: 0.036% and 0.031%, respectively), with a median fold-change variation of 0.91 (Fig. 3B), demonstrating a preservation of spike CD4+ T cell responses over time. Similar sustained levels of CD4+ T-cell responses were observed against BA.1, XBB.1 and BA.2.86 (Fig. S3A and S3B). All participants with an undetectable N&M-specific CD4+ T-cell response (n=4) had mounted a response by T2, after breakthrough infection (Fig. 3A). In the remaining 10 participants assessed, the preservation of N&M-specific CD4+ T cells varied, with a median fold change of 0.57 (ranging from 0.028 to 1.54) (Fig. 3B). Within the CD8 compartment, only a small number of the paired participants exhibited detectable spike- or N&M-specific CD8+ responses (Fig. 3C). The evolution of CD8+ T-cell responses from T1 to T2 was highly variable amongst participants, showing newly acquired, sustained, or lost responses (Fig. 3D). Similar patterns were observed for CD8+ T-cell responses against BA.1, XBB.1 or BA.2.86 (Fig. S3C and S3D).
Figure 3. Longitudinal assessment of the maintenance and memory profile of ancestral SARS-CoV-2-specific T-cell responses over 2 years.
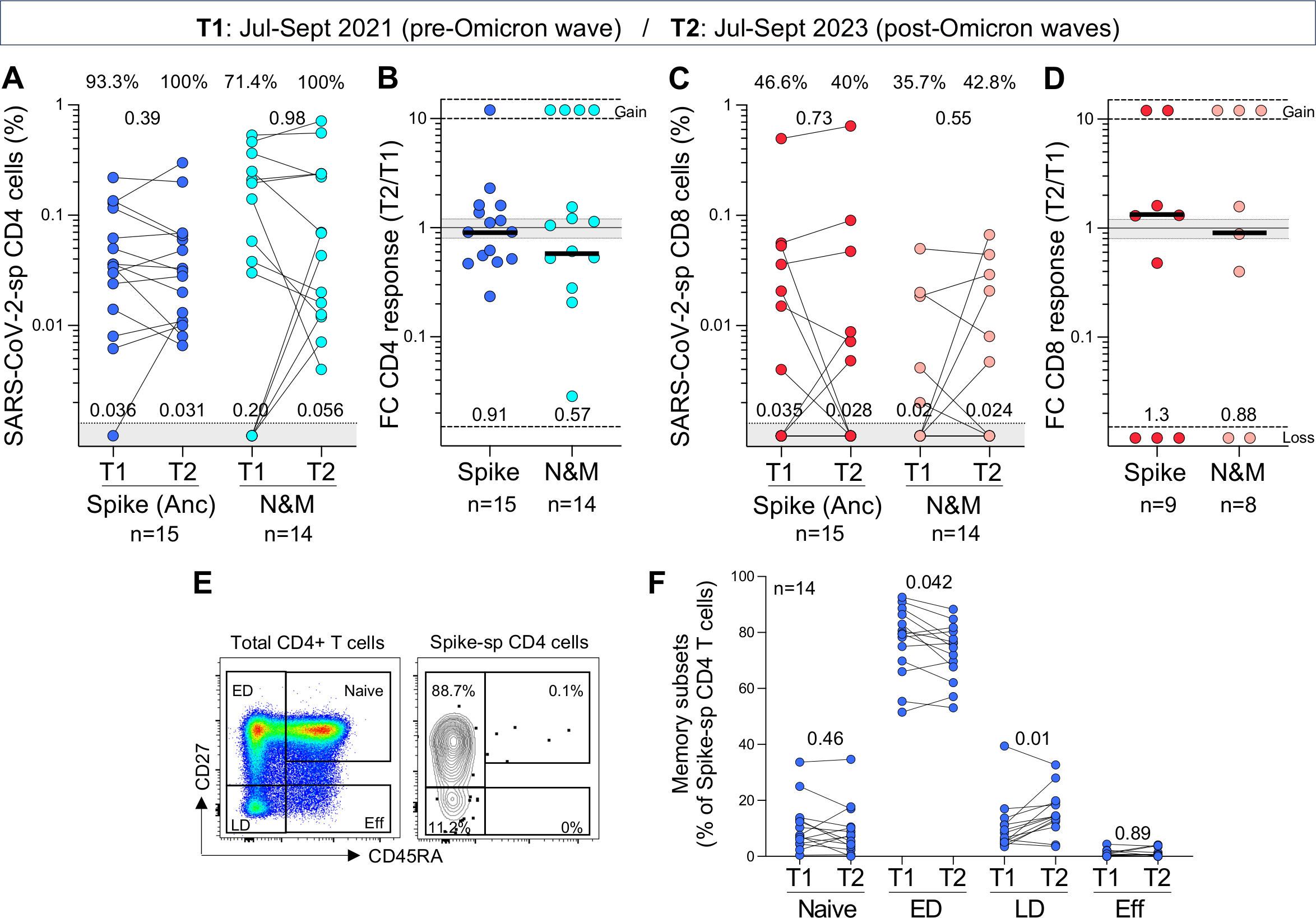
(A and C) Frequency of ancestral spike- and nucleocapsid and membrane (N&M)-specific CD4+ (A) and CD8+ T cells (C) in paired samples (n=15 for Spike and n=14 for N&M). T1 and T2 samples were collected between July and September 2021 and July and September 2023, respectively. Medians frequencies of spike- and N&M-specific T cells in responders are indicated at the bottom of the graphs. Statistical comparisons were assessed using Wilcoxon matched-pairs signed rank test.
(B and D) Fold change in frequency of SARS-CoV-2-specific CD4+ (B) and CD8+ T cells (D) between T2 and T1 in responders. Bars represent medians, and median fold change is indicated at the bottom of each graph. Gained responses are depicted on top and lost responses at the bottom.
(E) Representative plots of the memory differentiation profile of total CD4+ T cells and ancestral spike-specific CD4+ T cells. ED: early differentiated cells, LD: late differentiated cells and Eff: Effector cells.
(F) Comparison of the memory profile of spike-specific CD4+ T cells (n=14) at T1 and T2. Statistical comparisons were assessed using Wilcoxon matched-pairs signed rank test.
Lastly, we defined the memory profile of spike-specific T cells. Using differentiation markers CD45RA and CD27, we identified four memory subsets, namely naïve (CD45RA+CD27+), early differentiated (CD45RA−CD27+), late differentiated (CD45RA−CD27−) and effector (CD45RA+CD27−) (Fig. 3E). Ancestral spike-specific CD4+ T cells exhibited primarily an ED memory profile at T1 (Fig. 3F). At T2, there was a marginal decrease in the proportion of ED spike-specific CD4+ T cells (median 75.4% vs 79.5% at T1, p=0.042), counterbalanced by an increase in cells exhibiting a late differentiated profile (p=0.01). Of note, the memory profile of CD4+ T cells recognising BA.1, XBB.1 and BA.2.86 spike was similar to ancestral spike, characterized by a predominance of ED memory cells (Fig. S3E). Due to the limited number of paired samples with a spike-specific CD8+ T-cell response, we could not reliably compare the memory phenotype at both timepoints. However, the memory profile of SARS-CoV-2-specific CD8+T cells was defined at T2 (Fig. S3F). Unlike the CD4 response, spike-specific CD8+ T cells exhibited a more diverse memory profile within each participant, consisting of a median of ~40% of ED cells, ~20% of LD cells and ~20% of effector cells.
DISCUSSION
In this study, we observed robust circulating memory T cells persisting in healthcare workers in mid-late 2023, >1.5 years after the global Omicron wave, with most individuals receiving their last vaccination prior to Omicron breakthrough infection. All participants experienced mild primary or breakthrough infections. Immunological memory has previously been observed for over eight months after vaccination or infection, regardless of the severity of disease25 and to our knowledge these data represent the most recent T-cell response measurements reported from the post-pandemic period. Most parts of the world now experience ongoing viral circulation1. Thus, maintenance could be related to recurrent exposure to SARS-CoV-2, expanding the T-cell memory pool, or highly durable memory responses persisting from prior vaccination and infection. Phenotypic analysis revealed that SARS-CoV-2-specific T cells exhibit predominantly an early differentiated memory phenotype, consistent with several studies26,27. It is noteworthy that SARS-CoV-1 responses were detectable up to 17 years after infection28,29. Together, these data suggest a high capacity for the T-cell response to persist long term and provide recall responses upon SARS-CoV-2 re-exposure, even in the absence of booster vaccination or viral exposure.
We demonstrate that T-cell responses can effectively cross-recognize SARS-CoV-2 variants, including XBB.1 whose sub-lineages currently dominate globally1. Moreover, cross-reactivity was preserved to the highly mutated BA.2.86. This retention of T-cell reactivity across variants is consistent with many published studies6–9,30, emphasizing the potency of T-cell responses against a backdrop of diminishing neutralizing antibody responses with successively more highly evolved variants2–5. Importantly, we observed that Omicron sub-lineage cross-reactivity was readily detectable even before Omicron infection, suggesting that the T-cell response to conserved spike epitopes, included in the first-generation vaccines, may provide adequate cross-recognition. This is reassuring, given that the availability of updated booster vaccines based on XBB.1.5 in late 202331 is largely restricted to high income countries. The stable preservation of CD4 T-cell responses across variants suggests that most targeted epitopes are located in non-variable parts of spike or that mutations do not affect epitope recognition32. In contrast, as previously reported6,9, the preservation of CD8+ T-cell responses to variants is more heterogenous. To note, Koutsakos and colleagues demonstrated that spike epitope-specific CD8+ T cells correlated with accelerated viral clearance after breakthrough infection16, highlighting the importance of CD8 T-cell cross-protection. Our results emphasize that while variant mutations may lead to the occasional loss of epitope cross-recognition, they could also result in the creation of new immunogenic epitopes after breakthrough infection. Detailed follow-up studies are underway to map de novo epitope specificities and loss of epitope recognition after breakthrough infection. Together, these data demonstrate that highly resilient and adaptable T-cell responses are present in most individuals in the post-pandemic period.
An important consideration of hybrid immunity is that infection delivers additional T-cell targets from the SARS-CoV-2 proteome. We showed that non-spike T-cell responses constituted a sizable portion of the overall SARS-CoV-2 response, expanding the breadth of the response from vaccination. This was particularly striking for CD8 responses, with a third of responders targeting nucleocapsid and membrane in the absence of a CD8 spike response, consistent with epitope repertoires highly dependent on the HLA background of the individual25,32. Unlike the antibody neutralization response, T cells targeting spike or non-spike antigens have the potential to clear infected cells and limit viral replication. Since non-spike proteins are not under relentless selective pressure from neutralizing antibodies, accumulation of mutations is limited, ensuring a high degree of T-cell cross-reactivity to emerging variants33. For these reasons, conserved non-spike Sarbecovirus epitopes are being included in pan-coronavirus vaccines in development34.
Overall, we report durable, broad and highly cross-reactive post-pandemic T-cell responses in healthcare workers who were vaccinated and infected with SARS-CoV-2. The results of this study demonstrate that long-term immunological T-cell memory persists in the background of heterogenous exposure history, and withstands continued and extensive viral variation, providing immune resources for protection from severe outcomes of SARS-CoV-2 infection. However, it must be noted that COVID-19 exposure histories globally are now highly complex and divergent, making it challenging to draw generalizations and simple interpretations of the immune responses observed in different populations.
Limitations of the study
We may have underestimated the number of additional infections in subsequent smaller waves that have followed the initial Omicron BA.1 wave. Testing is no longer free or easily accessible, SARS-CoV-2 has ceased to be a notifiable disease and asymptomatic infections are more likely, given substantial population immunity35. Memory responses after recorded Omicron breakthrough infections may thus have been boosted with further exposures, influencing durability, magnitude and cross-reactivity. Furthermore, while structural proteins are the dominant T-cell targets36, we did not measure responses to non-structural components of the viral proteome 17 and thus may have underestimated the total T-cell response. In particular, SARS-CoV-2 non-structural and accessory proteins elicit robust T cell responses in asymptomatic or abortive infection37,38. Indeed, the contribution of different specificities of T cells among the larger breadth of memory T cell responses to preventing severe disease is unknown. In addition, we limited our analysis to Th1 cytokine production, but significantly higher and sustained IL-10-producing SARS-CoV-2-specific T cells have been observed in vaccinated persons with prior infection compared to those vaccinated alone39, an interesting finding that warrants further investigation. Lastly, we were restricted to measuring T cells in circulation, but infection can lead to sequestration of memory T cells to the respiratory tract that may not reflect the magnitude or specificity of responses measured in blood, and that may offer protection at the site of infection40,41. Further studies are needed to examine the durability of these tissue resident memory T cells.
STAR★METHODS:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Wendy Burgers (wendy.burgers@uct.ac.za).
Materials availability
Materials will be made available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
Participants included in this study (n=40) were selected from a longitudinal study of healthcare workers (HCW) enrolled from Groote Schuur Hospital (Cape Town, Western Cape, South Africa) 42,43. Participants were selected based on the availability of peripheral blood mononuclear cells (PBMC) in our biorepository. We used samples collected at two timepoints: 1) Timepoint 1 samples (T1, n=16) were collected between July and September 2021 (4–6 months prior to the Omicron BA.1 wave). At this timepoint, 9 out of 16 (56.25%) had a recorded SARS-CoV-2 infection and all participants received one dose of the Ad26.COV2.S vaccine (Johnson and Johnson/Janssen) 5.2 months [IQR: 5–6] prior to blood collection; 2) Timepoint 2 samples (T2, n=39) were collected between July and September 2023, approximately 1.5 years after the BA.1 wave. At this timepoint, 28.2% (11/39) of the participants had received one Ad26.COV2.S vaccine dose, 56.4% (22/39) two vaccine doses and 15.4% (6/39) three vaccine doses. The majority of participants (89.7%) were vaccinated with Ad26.COV2.S. Three participants received a heterologous vaccination regimen (Ad26.COV2.S and BNT162b2) and one participant received 3 doses of the BNT162b2 vaccine. The median time since last vaccination was ~21 months (IQR: 20.2–24.4). Twenty-two participants (56.4%) had a documented SARS-CoV-2 infection, prior to the onset of the Omicron wave; and all experienced a breakthrough infection during the Omicron wave, at a median time of 19.4 month (IQR: 17.8–19.9) before sample collection. The specific Omicron sub-lineage was not sequenced, but it is likely that most participants were BA.1-infected due to the dominance of this sub-variant and the timing of infections. Prior infection or breakthrough infection (BTI) were determined by a positive PCR or by Nucleocapsid (N) seroconversion or a two-fold increase in nucleocapsid IgG. The demographic and clinical characteristics of participants, at each timepoint, are summarized in Table 1. The landscape of SARS-CoV-2 waves and vaccination timeline with time of sample collection is depicted in Fig. S1. The study was approved by the University of Cape Town Human Research Ethics Committee (HREC 190/2020 and 291/2020), and written informed consent was obtained from all participants
METHOD DETAILS
Isolation of peripheral blood mononuclear cells (PBMC)
Blood was collected in heparin tubes and processed within 4 hours of collection. PBMC were isolated by density gradient sedimentation using Ficoll-Paque (Amersham Biosciences, Little Chalfont, UK) as per the manufacturer’s instructions. The PBMC were then cryopreserved in freezing media consisting of heat-inactivated fetal bovine serum (FBS, Thermofisher Scientific, Waltham, MA, USA) containing 10% dimethyl sulfoxide (DMSO) and stored in liquid nitrogen until use.
SARS-CoV-2 antigens
To measure SARS-CoV-2 spike-specific T-cell responses, we used custom mega pools of peptides. These peptides (15-mer overlapping by 10 amino acids) spanned the entire spike protein corresponding to the ancestral Wuhan sequence (GenBank: MN908947), Omicron B.1.1.529 (BA.1), XBB.1 and BA.2.86. The list of mutations for the Omicron sub-lineage compared to the ancestral Wuhan sequence is provided in Table S1. To measure SARS-CoV-2 nucleocapsid and membrane protein T-cell responses, we used commercial synthetic SARS-CoV-2 Pep-Tivator peptides (Miltenyi Biotec, Woking, UK), consisting of 15-mer sequences with 11 amino acids overlap, covering the complete sequence of the nucleocapsid (N, GenBank MN908947.3, Protein QHD43423.2) and membrane protein (M, GenBank MN908947.3, Protein QHD43419.1).
Cell stimulation and flow cytometry staining
Cryopreserved PBMC were thawed, washed, and rested for 4 hours in RPMI 1640 (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% heat inactivated FBS. After resting, cells were seeded in a 96-well V-bottom plate at ~2 ×106 cells/well. Cells were stimulated with SARS-CoV-2 mega pools spanning the entire Spike (S) protein of the ancestral, Omicron BA.1, XBB.1 and BA.2.86 variants (1 μg/mL), as well the ancestral Membrane (M) and Nucleocapsid (N) proteins. All stimulations were performed in the presence of Brefeldin A (10 μg/mL, Sigma-Aldrich) and co-stimulatory antibodies against CD28 (clone 28.2) and CD49 (clone L25) (1 μg/mL each; BD Biosciences, San Jose, CA, USA). As a background control, PBMC were incubated with co-stimulatory antibodies, Brefeldin A and an equimolar amount of DMSO. After 16 hours of stimulation, cells were washed, stained with LIVE/DEAD™ Fixable Near-IR Stain (Invitrogen, Carlsbad, CA, USA) and subsequently surface stained with the following antibodies: CD14 APC-Cy7 (HCD14, Biolegend, San Diego, CA, USA), CD19 APC-Cy7 (HIB19, Biolegend), CD4 PE-Cy7 (L200, BD Biosciences), CD8 BV510 (RPA-8, Biolegend), CD27 PE-Cy5 (1A4, Beckman Coulter, Brea, CA, USA) and CD45RA BV605 (HI100, Biolegend). Cells were then fixed and permeabilized using Cytofix/Cytoperm buffer (BD Biosciences) and stained with CD3 BV785 (OKT3), TNF-α FITC (Mab11) and IL-2 PE/Dazzle™ 594 (MQ1–17H12) from Biolegend and IFN-γ Alexa 700 (B27, BD Biosciences). After intracellular cytokine staining, cells were washed and fixed in 1% Paraformaldehyde (ThermoFisher Scientific). Samples were acquired on a BD Fortessa flow cytometer using FACSDiva software and analysed using FlowJo (v10, FlowJo LLC, Ashland, OR, USA). Cells were gated on singlets, CD14−CD19−, live CD3+ T cells. Results are expressed as the frequency of total CD4+ or CD8+ T cells expressing IFN-γ, TNF-α or IL-2. The gating strategy is presented is Fig. S4. Due to high TNF-α backgrounds, cells producing TNF-α alone were excluded from the analysis. All data are presented after background subtraction (from the frequency of cytokine produced in unstimulated cells). To define the memory phenotype of SARS-CoV-2-specific T cells, a cut-off of 30 events was used.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using Prism (v10.3.1; GraphPad Software Inc, San Diego, CA, USA). Non-parametric tests were used for all comparisons. The Friedman test with Dunn’s correction was used for multiple groups comparison, the Spearman rank test for correlations, and the Wilcoxon matched pairs test for paired samples. A P value <0.05 was considered statistically significant. Details of statistical analyses performed for each experiment are described in the figure legends.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the study participants, the clinical staff at Groote Schuur Hospital for their dedication. This study was supported by Wellcome Trust (226137/Z/22/Z), South African Medical Research Council (SA-MRC) with funds received from South African Department of Science and Innovation (DSI), Bill and Melinda Gates Foundation (INV-046743), Poliomyelitis Research Foundation (21/65) and Wellcome Centre for Infectious Diseases Research in Africa supported by core funding from Wellcome Trust (203135/Z/16/Z and 222754). W.A.B. and C.R. are supported by EDCTP2 program of European Union’s Horizon 2020 program (TMA2016SF-1535-CaTCH-22 to W.A.B and TMA2017SF-1951-TB-SPEC to C.R.). R.N. is supported by Harry Crossley Foundation. N.A.B.N. acknowledges funding from SA-MRC, MRC UK, NRF, and Lily and Ernst Hausmann Trust. M.A.H. is supported by the Fogarty International Center of the National Institutes of Health and the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) under award number D43 TW010559. This project has been funded in part with Federal funds from National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93021C00016 to A.G. and Contract No. 75N93019C00065 to A.S. For the purposes of open access, the authors have applied a CC-BY public copyright license to any author-accepted version arising from this submission.
Footnotes
DECLARATION OF INTERESTS
A.S. is a consultant for AstraZeneca Pharmaceuticals, Calyptus Pharmaceuticals, Inc, Darwin Health, EmerVax, EUROIMMUN, F. Hoffman-La Roche Ltd, Fortress Biotech, Gilead Sciences, Granite bio., Gritstone Oncology, Guggenheim Securities, Moderna, Pfizer, RiverVest Venture Partners, and Turnstone Biologics. A.G. is a consultant for Pfizer. LJI has filed for patent protection for various aspects of T cell epitope and vaccine design work. All other authors declare no competing interests.
REFERENCES
- 1.Roemer C, Sheward DJ, Hisner R, Gueli F, Sakaguchi H, Frohberg N, Schoenmakers J, Sato K, O’Toole A, Rambaut A, et al. (2023). SARS-CoV-2 evolution in the Omicron era. Nat Microbiol. 10.1038/s41564-023-01504-w. [DOI] [PubMed] [Google Scholar]
- 2.Khan K, Lustig G, Reedoy K, Jule Z, Römer C, Karim F, Ganga Y, Bernstein M, Baig Z, Mahlangu B, et al. (2023). Evolution and neutralization escape of the SARS-CoV-2 BA.2.86 subvariant. Preprint in MedRxiv. 10.1101/2023.09.08.23295250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheward DJ, Yang Y, Westerberg M, Oling S, Muschiol S, Sato K, Peacock TP, Hedestam GBK, Albert J, and Murrell B (2023). Sensitivity of the SARS-CoV-2 BA.2.86 variant to prevailing neutralising antibody responses. Lancet Infect Dis. 10.1016/S1473-3099(23)00588-1. [DOI] [PubMed] [Google Scholar]
- 4.Yang S, Yu Y, Jian F, Song W, Yisimayi A, Chen X, Xu Y, Wang P, Wang J, Yu L, et al. (2023). Antigenicity and infectivity characterisation of SARS-CoV-2 BA.2.86. Lancet Infect Dis. 10.1016/S1473-3099(23)00573-X. [DOI] [PubMed] [Google Scholar]
- 5.Lasrado N, Collier AY, Hachmann NP, Miller J, Rowe M, Schonberg ED, Rodrigues SL, LaPiana A, Patio RC, Anand T, et al. (2023). Neutralization escape by SARS-CoV-2 Omicron subvariant BA.2.86. Vaccine. 10.1016/j.vaccine.2023.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keeton R, Tincho MB, Ngomti A, Baguma R, Benede N, Suzuki A, Khan K, Cele S, Bernstein M, Karim F, et al. (2022). T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 603, 488–492. 10.1038/s41586-022-04460-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarke A, Coelho CH, Zhang Z, Dan JM, Yu ED, Methot N, Bloom NI, Goodwin B, Phillips E, Mallal S, et al. (2022). SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell 185, 847–859 e811. 10.1016/j.cell.2022.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y, Cai C, Grifoni A, Muller TR, Niessl J, Olofsson A, Humbert M, Hansson L, Osterborg A, Bergman P, et al. (2022). Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat Med 28, 472–476. 10.1038/s41591-022-01700-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naranbhai V, Nathan A, Kaseke C, Berrios C, Khatri A, Choi S, Getz MA, Tano-Menka R, Ofoman O, Gayton A, et al. (2022). T cell reactivity to the SARS-CoV-2 Omicron variant is preserved in most but not all individuals. Cell 185, 1041–1051 e1046. 10.1016/j.cell.2022.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levin EG, Lustig Y, Cohen C, Fluss R, Indenbaum V, Amit S, Doolman R, Asraf K, Mendelson E, Ziv A, et al. (2021). Waning Immune Humoral Response to BNT162b2 Covid-19 Vaccine over 6 Months. N Engl J Med 385, e84. 10.1056/NEJMoa2114583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo L, Wang G, Wang Y, Zhang Q, Ren L, Gu X, Huang T, Zhong J, Wang Y, Wang X, et al. (2022). SARS-CoV-2-specific antibody and T-cell responses 1 year after infection in people recovered from COVID-19: a longitudinal cohort study. Lancet Microbe 3, e348–e356. 10.1016/S2666-5247(22)00036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng C, Shi J, Fan Q, Wang Y, Huang H, Chen F, Tang G, Li Y, Li P, Li J, et al. (2021). Protective humoral and cellular immune responses to SARS-CoV-2 persist up to 1 year after recovery. Nat Commun 12, 4984. 10.1038/s41467-021-25312-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung JH, Rha MS, Sa M, Choi HK, Jeon JH, Seok H, Park DW, Park SH, Jeong HW, Choi WS, and Shin EC (2021). SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat Commun 12, 4043. 10.1038/s41467-021-24377-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, Grifoni A, Ramirez SI, Haupt S, Frazier A, et al. (2021). Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 371. 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldblatt D, Alter G, Crotty S, and Plotkin SA (2022). Correlates of protection against SARS-CoV-2 infection and COVID-19 disease. Immunol Rev 310, 6–26. 10.1111/imr.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koutsakos M, Reynaldi A, Lee WS, Nguyen J, Amarasena T, Taiaroa G, Kinsella P, Liew KC, Tran T, Kent HE, et al. (2023). SARS-CoV-2 breakthrough infection induces rapid memory and de novo T cell responses. Immunity 56, 879–892 e874. 10.1016/j.immuni.2023.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarke A, Sidney J, Kidd CK, Dan JM, Ramirez SI, Yu ED, Mateus J, da Silva Antunes R, Moore E, Rubiro P, et al. (2021). Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep Med 2, 100204. 10.1016/j.xcrm.2021.100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelde A, Bilich T, Heitmann JS, Maringer Y, Salih HR, Roerden M, Lubke M, Bauer J, Rieth J, Wacker M, et al. (2021). SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat Immunol 22, 74–85. 10.1038/s41590-020-00808-x. [DOI] [PubMed] [Google Scholar]
- 19.Saini SK, Hersby DS, Tamhane T, Povlsen HR, Amaya Hernandez SP, Nielsen M, Gang AO, and Hadrup SR (2021). SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8(+) T cell activation in COVID-19 patients. Sci Immunol 6. 10.1126/sciimmunol.abf7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loyal L, Braun J, Henze L, Kruse B, Dingeldey M, Reimer U, Kern F, Schwarz T, Mangold M, Unger C, et al. (2021). Cross-reactive CD4(+) T cells enhance SARS-CoV-2 immune responses upon infection and vaccination. Science 374, eabh1823. 10.1126/science.abh1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dang TTT, Anzurez A, Nakayama-Hosoya K, Miki S, Yamashita K, de Souza M, Matano T, and Kawana-Tachikawa A (2023). Breadth and Durability of SARS-CoV-2-Specific T Cell Responses following Long-Term Recovery from COVID-19. Microbiol Spectr 11, e0214323. 10.1128/spectrum.02143-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thieme CJ, Anft M, Paniskaki K, Blazquez-Navarro A, Doevelaar A, Seibert FS, Hoelzer B, Konik MJ, Berger MM, Brenner T, et al. (2020). Robust T Cell Response Toward Spike, Membrane, and Nucleocapsid SARS-CoV-2 Proteins Is Not Associated with Recovery in Critical COVID-19 Patients. Cell Rep Med 1, 100092. 10.1016/j.xcrm.2020.100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, Belanger S, Abbott RK, Kim C, Choi J, et al. (2020). Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 183, 996–1012 e1019. 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Bert N, Clapham HE, Tan AT, Chia WN, Tham CYL, Lim JM, Kunasegaran K, Tan LWL, Dutertre CA, Shankar N, et al. (2021). Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J Exp Med 218. 10.1084/jem.20202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sette A, Sidney J, and Crotty S (2023). T Cell Responses to SARS-CoV-2. Annu Rev Immunol 41, 343–373. 10.1146/annurev-immunol-101721-061120. [DOI] [PubMed] [Google Scholar]
- 26.Goel RR, Painter MM, Apostolidis SA, Mathew D, Meng W, Rosenfeld AM, Lundgreen KA, Reynaldi A, Khoury DS, Pattekar A, et al. (2021). mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science 374, abm0829. 10.1126/science.abm0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Painter MM, Johnston TS, Lundgreen KA, Santos JJS, Qin JS, Goel RR, Apostolidis SA, Mathew D, Fulmer B, Williams JC, et al. (2023). Prior vaccination promotes early activation of memory T cells and enhances immune responses during SARS-CoV-2 breakthrough infection. Nat Immunol 24, 1711–1724. 10.1038/s41590-023-01613-y. [DOI] [PubMed] [Google Scholar]
- 28.Ng OW, Chia A, Tan AT, Jadi RS, Leong HN, Bertoletti A, and Tan YJ (2016). Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine 34, 2008–2014. 10.1016/j.vaccine.2016.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, Chng MHY, Lin M, Tan N, Linster M, et al. (2020). SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 584, 457–462. 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- 30.Riou C, Keeton R, Moyo-Gwete T, Hermanus T, Kgagudi P, Baguma R, Valley-Omar Z, Smith M, Tegally H, Doolabh D, et al. (2022). Escape from recognition of SARS-CoV-2 variant spike epitopes but overall preservation of T cell immunity. Sci Transl Med 14, eabj6824. 10.1126/scitranslmed.abj6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krammer F, and Ellebedy AH (2023). Variant-adapted COVID-19 booster vaccines. Science 382, 157–159. 10.1126/science.adh2712. [DOI] [PubMed] [Google Scholar]
- 32.Karsten H, Cords L, Westphal T, Knapp M, Brehm TT, Hermanussen L, Omansen TF, Schmiedel S, Woost R, Ditt V, et al. (2022). High-resolution analysis of individual spike peptide-specific CD4(+) T-cell responses in vaccine recipients and COVID-19 patients. Clin Transl Immunology 11, e1410. 10.1002/cti2.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer S, Blaas I, Bollineni RC, Delic-Sarac M, Tran TT, Knetter C, Dai KZ, Madssen TS, Vaage JT, Gustavsen A, et al. (2023). Prevalent and immunodominant CD8 T cell epitopes are conserved in SARS-CoV-2 variants. Cell Rep 42, 111995. 10.1016/j.celrep.2023.111995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang CY, Peng WJ, Kuo BS, Ho YH, Wang MS, Yang YT, Chang PY, Shen YH, and Hwang KP (2023). Toward a pan-SARS-CoV-2 vaccine targeting conserved epitopes on spike and non-spike proteins for potent, broad and durable immune responses. PLoS Pathog 19, e1010870. 10.1371/journal.ppat.1010870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madhi SA, Kwatra G, Myers JE, Jassat W, Dhar N, Mukendi CK, Blumberg L, Welch R, Izu A, and Mutevedzi PC (2023). Sustained Low Incidence of Severe and Fatal COVID-19 Following Widespread Infection Induced Immunity after the Omicron (BA.1) Dominant in Gauteng, South Africa: An Observational Study. Viruses 15. 10.3390/v15030597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, Rawlings SA, Sutherland A, Premkumar L, Jadi RS, et al. (2020). Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 181, 1489–1501 e1415. 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swadling L, Diniz MO, Schmidt NM, Amin OE, Chandran A, Shaw E, Pade C, Gibbons JM, Le Bert N, Tan AT, et al. (2022). Pre-existing polymerase-specific T cells expand in abortive seronegative SARS-CoV-2. Nature 601, 110–117. 10.1038/s41586-021-04186-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samandari T, Ongalo JB, McCarthy KD, Biegon RK, Madiega PA, Mithika A, Orinda J, Mboya GM, Mwaura P, Anzala O, et al. (2023). Prevalence and functional profile of SARS-CoV-2 T cells in asymptomatic Kenyan adults. J Clin Invest 133. 10.1172/JCI170011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodda LB, Morawski PA, Pruner KB, Fahning ML, Howard CA, Franko N, Logue J, Eggenberger J, Stokes C, Golez I, et al. (2022). Imprinted SARS-CoV-2-specific memory lymphocytes define hybrid immunity. Cell 185, 1588–1601 e1514. 10.1016/j.cell.2022.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grau-Exposito J, Sanchez-Gaona N, Massana N, Suppi M, Astorga-Gamaza A, Perea D, Rosado J, Falco A, Kirkegaard C, Torrella A, et al. (2021). Peripheral and lung resident memory T cell responses against SARS-CoV-2. Nat Commun 12, 3010. 10.1038/s41467-021-23333-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roukens AHE, Pothast CR, Konig M, Huisman W, Dalebout T, Tak T, Azimi S, Kruize Y, Hagedoorn RS, Zlei M, et al. (2022). Prolonged activation of nasal immune cell populations and development of tissue-resident SARS-CoV-2-specific CD8(+) T cell responses following COVID-19. Nat Immunol 23, 23–32. 10.1038/s41590-021-01095-w. [DOI] [PubMed] [Google Scholar]
- 42.Keeton R, Tincho MB, Suzuki A, Benede N, Ngomti A, Baguma R, Chauke MV, Mennen M, Skelem S, Adriaanse M, et al. (2023). Impact of SARS-CoV-2 exposure history on the T cell and IgG response. Cell Rep Med 4, 100898. 10.1016/j.xcrm.2022.100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keeton R, Richardson SI, Moyo-Gwete T, Hermanus T, Tincho MB, Benede N, Manamela NP, Baguma R, Makhado Z, Ngomti A, et al. (2021). Prior infection with SARS-CoV-2 boosts and broadens Ad26.COV2.S immunogenicity in a variant-dependent manner. Cell Host Microbe 29, 1611–1619 e1615. 10.1016/j.chom.2021.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.