

Abstract
One of the strongest drivers in evolution is the struggle to survive a host–pathogen battle. This pressure selects for diversity among the factors directly involved in this battle, including virulence factors deployed by pathogens, their corresponding host targets, and host immune factors. A logical outcome of this diversification is that over time, the sequence of many immune factors will not be evolutionarily conserved across a broad range of species. Thus, while universal sequence conservation is often hailed as the hallmark of the importance of a particular gene, the immune system does not necessarily play by these rules when defending against co-evolving pathogens. This loss of sequence conservation is in contrast to many signaling pathways in development and basic cell biology that are not targeted by pathogens. In addition to diversification, another consequence of host–pathogen battles can be an amplification in gene number, thus leading to large gene families that have sequence relatively specific to a particular strain, species, or clade. Here we highlight this general theme across a variety of pathogen virulence factors and host immune factors. We summarize the wide range and number across species of these expanded, lineage-specific host–pathogen factors including ubiquitin ligases, nucleotide-binding leucine-rich repeat receptors, GTPases, and proteins without obvious biochemical function but that nonetheless play key roles in immunity.
Keywords: gene families, host–pathogen, immunity, pathogenesis, species-specific
Introduction
Molecular interactions between pathogenic microbes and host organisms are key drivers of genome evolution [1]. This competition between hosts and pathogens creates pressure to diversify the sequence of proteins directly involved in this battle, as each side struggles to survive and pass on their genes. After generations of co-evolution, the weapons used in host/pathogen battles can diversify enough to appear specific to certain species and lineages, and thus no longer appear to be evolutionarily conserved across a broad range of species.
The loss of conservation in host immune factors is in contrast to the extensive conservation of factors involved in many signaling pathways not targeted by co-evolving pathogens. For example, Homeobox transcription factors specify a head/tail body axis in the nematode Caenorhabditis elegans, in the fruit fly Drosophila melanogaster, and in vertebrates [2,3]. However, it is difficult to find examples of immune pathways conserved across this range of phylogeny. Indeed, even within immune signaling pathways that are conserved between species, there can be key differences. For example, the Toll receptor that provides immunity in D. melanogaster is activated by endogenous ligands, while Toll-like receptors in mammals are activated by exogenous, pathogen-derived ligands [4]. Furthermore, many ‘critical’ immune regulators have been lost in certain clades, such as the transcription factor NFκB, which is present in vertebrates and D. melanogaster but has been lost in C. elegans [5], and the RIG-I double-stranded RNA receptor, which is present in vertebrates and C. elegans, but likely lost in D. melanogaster [6]. Thus, it is difficult to determine a core set of immune regulators across species, because of the diversification and loss that occurs during host–pathogen conflicts. While factors not necessarily involved in immunity also can undergo diversification due to their role in detecting a changing environment (such as chemosensory receptors [7]), increased gene diversification across species is often the hallmark of a factor involved in a host–pathogen battle. If immune factors are conserved, it may be an indication of their importance in defense, but it is also possible they were less important for defense against co-evolving pathogens, because they were not targeted by these pathogens in the recent evolutionary past [8].
In addition to sequence diversification across species, a common theme for genes involved in host–pathogen conflicts is a large variation in their number across different species or clades. Gene duplication is thought to be the ‘fuel for evolution’, and these duplication events appear to go into overdrive during host–pathogen conflicts, which can create dramatically expanded gene families [9]. Diversification and expansion of immune genes are at the heart of the vertebrate adaptive immune system, with this process happening within a lifetime, instead of across generations. Diversification can occur through immune locus rear-rangement and/or hypermutation, followed by expansion, which occurs because specific cell types that carry these genes will proliferate, to enable a learned response to previous pathogen exposure. Such adaptive immune genes include the immunoglobulin B-cell and T-cell receptors in jawed vertebrates, as well as the variable lymphocyte receptors found in jawless vertebrates [10,11]. These are factors that diversify and expand in number in somatic tissues. Here, we discuss expansion and diversification of innate immune genes in germline tissues, which are thus genes passed onto the next generation. We also focus on pathogen factors subject to the same phenomena. While excellent reviews have described diversification and expansion of individual gene classes [12,13], here we cover several related examples, to illustrate the commonality of the evolution of species-specific, giant gene families from host–pathogen battles (Fig. 1). We also aim to highlight the value in studying these species-specific gene families, as their function may be conserved, even if their sequences are not.
Fig. 1.
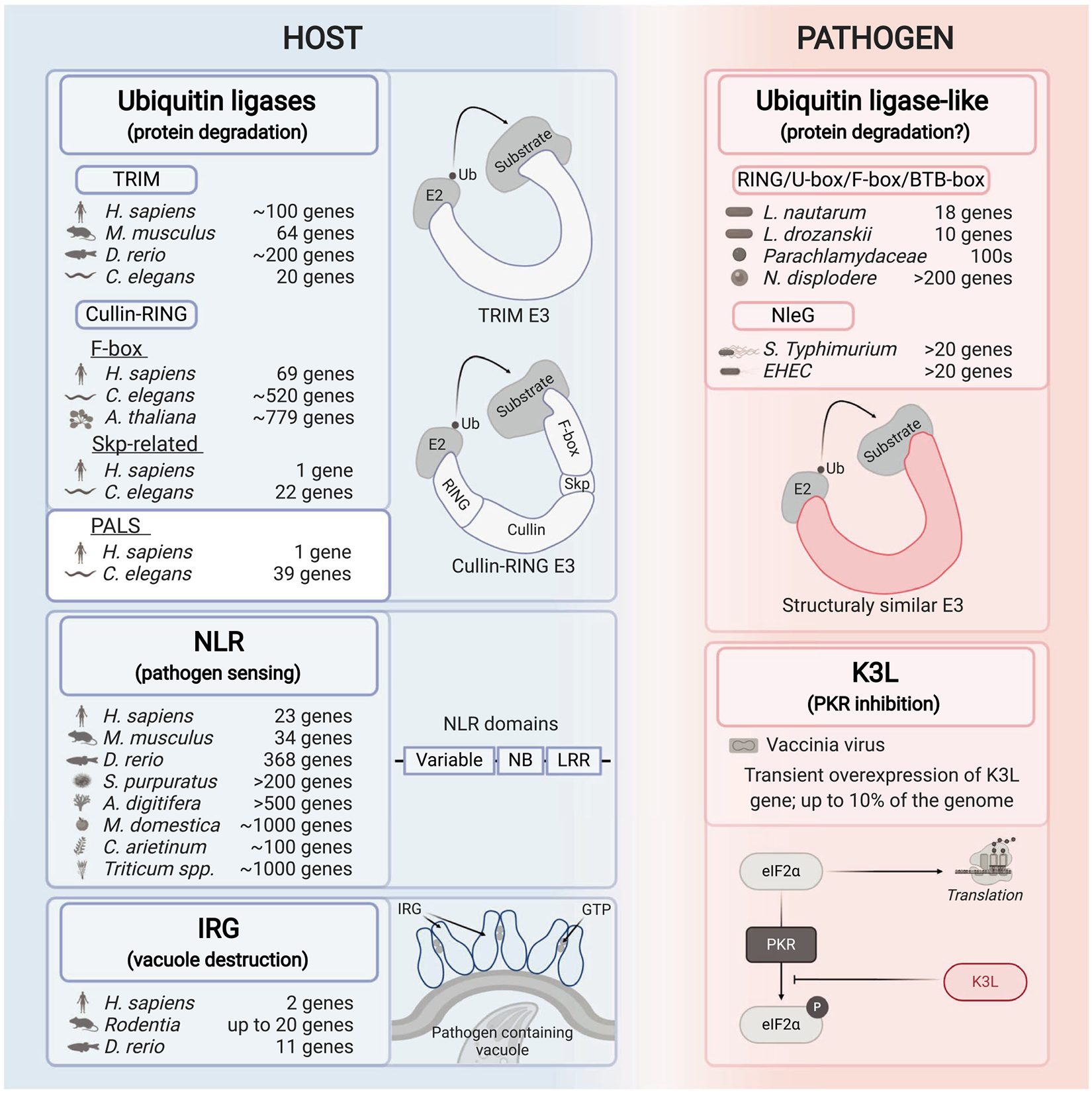
Model illustrating the mechanism and number of host–pathogen genes that have expanded in certain lineages. Selected host (left) and pathogen (right) expanded gene families involved in the host–pathogen battles are shown in the figure. The known or predicted functions for each described family/factor are given in the parentheses. The Latin species names and the number of genes per species are listed for every presented expanded gene family. Graphical illustrations of the mechanisms of function or functional domains are depicted in each panel.
Expansion and diversification of ubiquitin ligases on both sides of host–pathogen battles
Protein degradation is a critical battleground between hosts and pathogens that have resulted in expanded and diverse gene families. In particular, the ubiquitin–proteasome system, which is a crucial degradation system in eukaryotic cells, has seen considerable diversification. In brief, the ubiquitin–proteasome system involves sequential reactions with E1, E2, and then E3 ubiquitin ligases that ultimately catalyze the transfer of a 7 kD ubiquitin protein tag onto target proteins, which can flag them for degradation by the proteasome [14]. E3 ligases come in two major classes, HECT-type and RING/U-box, and these can be found in single-subunit ubiquitin ligases, as well as multi-subunit enzymes, such as the cullin-RING ligases (CRLs). All of these classes of E3 ubiquitin ligases have evidence of being entangled in host–pathogen conflicts, as described below.
The tripartite motif-containing (TRIM) ubiquitin ligases [15,16] are a notable family of host single-subunit E3 ubiquitin ligases that include factors which serve to restrict viral infection. Studies indicate that there are ~ 100 TRIM genes in humans (Homo sapiens), with 11 specific to humans and African apes, and 7 specific just to humans, and certain individuals that harbor extensive copy number variation [17]. There are fewer TRIM genes in other organisms, with ~ 64 in the mouse Mus musculus, ~ 20 in C. elegans, and < 10 in D. melanogaster. Strikingly, it appears that the zebrafish Danio rerio genome has about 200 TRIM genes [18], some of which appear to be undergoing positive (diversifying) selection and to be induced by viral infection, suggesting that these genes may be involved in a co-evolutionary battle against fish pathogens. One of the best-studied TRIM proteins is the TRIM5α ubiquitin ligase, which was identified in the primate lineage as a viral restriction factor for human immunodeficiency virus (HIV) and other retroviruses. TRIM5α from rhesus (Old World monkeys) can bind the capsid of HIV, trigger ubiquitylation and innate immune signaling to block viral replication, whereas the human version of TRIM5α has only weak restriction ability [19,20]. Importantly, human TRIM5α can target proteins in other viruses for ubiquitylation and proteasomal degradation [21,22], indicating that it has maintained function, but has different specificity than rhesus TRIM5α. Recent work has shown that human TRIM5α can also target human endogenous retroelements [23], indicating there is greater substrate range for this ligase than previously thought.
The most numerous ubiquitin ligases are the multi-subunit CRLs, which are modular and can form a wide range of different enzyme complexes [24]. Within this group, a subset are the Skp-Cullin-F-box ligases, which have four subunits: a RING domain protein that recruits an E2 ligase, a Cullin, a Skp protein, and an F-box adaptor protein that binds to the substrate to be ubiquitylated. Given the large number of F-box proteins in different species, theoretically these ligases would provide an enormous potential for targeting pathogen proteins for destruction. While humans have 69 F-box proteins, there have been expansions in this class of proteins in the mustard plant Arabidopsis thaliana, which has ~ 779 family members [25], as well as in the model nematode C. elegans, which has ~ 520 family members [26] (Fig. 1). While there are hints that CRLs regulate susceptibility to viral and microsporidia infection in C. elegans [27,28], robust functional roles have not yet been assigned. It is intriguing that both A. thaliana and C. elegans appear to lack professional immune cells, so speculatively it is possible that the expansion of F-box proteins evolved to increase the detection capability of nonprofessional immune cells in both of these lineages.
Interestingly, another expanded gene family called the pals gene family in C. elegans is involved in immunity against intracellular pathogens [29]. Although their biochemical function is unknown, pals genes in C. elegans have very distant sequence homology to F-box genes, and are found in clusters in the genome sometimes near F-box genes. Mouse and human genomes have one pals gene of unknown function, while C. elegans has expanded to 39 genes, and related species Caenorhabditis briggsae only has eight genes [30]. Two of the C. elegans pals genes, pals-22 and pals-25, act as antagonistic paralogs and comprise an ‘ON/OFF switch’ for the intracellular pathogen response, a recently described transcriptional response that promotes immunity against viruses and microsporidia [29].
On the pathogen side, bacteria have evolved a wide range of ubiquitin ligases with diverse enzymatic capabilities and sequences. Given that bacteria lack eukaryotic-like ubiquitylation machinery, it is thought that all ubiquitylation factors from bacteria function solely in host cells, where they can promote survival of bacteria through altering degradation of various protein targets [31]. For example, the facultative intracellular pathogen Legionella pneumophila, which infects amoeba and humans, uses SidE family effectors to catalyze a ubiquitin conjugation event that is independent of E1 and E2 enzymes to target host GTPases. These SidE effectors define a ‘new type’ of E3 ligase. Interestingly, there are other bacterial ubiquitin ligases that have structural and functional similarity to canonical E3 ligases but have no primary sequence similarity with these enzymes. These ubiquitin ligases include the Salmonella enterica serovar Typhimurium effector SopA, which targets host factors to regulate inflammation and has structural similarity to HECT-type E3 ligases. In addition, there is a family of at least 20 effectors in the NleG family in S. Typhimurium and Enterohemorrhagic Escherichia coli (EHEC) that has structural similarity but no primary sequence similarity to RING type ubiquitin ligases [32]. NleG effectors in EHEC have been shown to catalyze degradation of host targets in both the nucleus and the cytoplasm of mammalian cells [33]. The lack of primary sequence similarity to canonical E3 ligases in these bacterial effectors may represent convergent evolution. Alternatively, these ligases may have been acquired from a eukaryote through horizontal gene transfer, and then underwent such extreme sequence divergence that is no longer possible to find a common ancestor.
Several distinct types of pathogens produce F-box adaptor proteins, which can then act together with host core components in multi-subunit cullin-RING ligases. Such F-box effectors are used by viruses [34,35] as well as by bacterial pathogens such as the plant pathogen Agrobacterium tumefaciens [36], and by L. pneumophila [37]. A recent metagenomics study of 80 Legionella genomes spanning 58 species found that two thirds of the species had either F-box or U-box containing genes [38]. Most genomes only had one to three such genes, but Legionella nautarum and Legionella drozanskii contained expanded numbers of 18 and 10, respectively. Other studies have indicated that F-box containing genes in Legionella spp. are undergoing more rapid diversification than other eukaryotic-like domains or housekeeping genes [39]. In addition, extensive expansion of gene families containing F-box and BTB-box (another ubiquitin ligase adaptor) and RING/U-box domains have been seen in obligate intracellular pathogens from the Parachlamy-diaceae family [40]. Here there are hundreds of genes containing these ubiquitylation domains across a variety of gene families and they appear to be undergoing rapid birth/death. They also have disparity in copy number among closely related organisms, perhaps reflecting the impact of a host–pathogen battle.
The theme of diversification and expansion in factors implicated in virulence is an especially notable characteristic for microsporidia, which are fungal-related pathogens that include over 1400 highly diverged species [41]. As a phylum, microsporidia appear to have a wide host range, with most animals likely susceptible to at least one microsporidian species. Like viruses, all microsporidia are obligate intracellular pathogens, and many species replicate in direct contact with host cytoplasm. Microsporidian genomes are marked by extreme contraction, having lost many metabolic and other signaling pathways. Somewhat paradoxically however, they tend to have extremely large, clade-specific gene families. For example, Nematocida displodere, which infects the nematode C. elegans, has only 2278 predicted genes, but 10% of these genes are contained in a single giant gene family that is specific to the Nematocida genus (Fig. 1), with only 1–3 members found in Nematocida species other than N. displodere [42]. One clue to the function of these proteins is a RING domain, which is a domain found in ubiquitin ligases. Importantly, localized proteomics studies have demonstrated the microsporidian ‘host-exposed’ proteins are enriched for those that belong to these giant gene families, further supporting the model that they are directly engaged in host–pathogen battles [43]. Because of a lack of genetic tools in microsporidia, the function of these expanded, species-specific gene families is unknown, but they appear to be a common feature of microsporidia genomes, which otherwise are the smallest known eukaryotic genomes.
In summary, there is a vast range of genes in both pathogens and hosts that share little or no direct sequence similarity with each other, but encode ubiquitin ligases or have similarity to ubiquitin ligases, and belong to gene families that have undergone dramatic expansion in various lineages.
Expansion and diversification of host NLR receptors in plants and animals
Perhaps the best-characterized examples of diversification and expansion of innate immune genes are the R genes in plants [13,44]. Many of these R genes encode nucleotide-binding domain and leucine-rich repeat (NLR) proteins, which are intracellular immune receptors. In general, plant NLRs are activated by sensing the effects of virulence factors, either through detecting modification of a separate protein (a ‘guardee’, as part of the guard hypothesis), or through sensing virulence factor-mediated modification of the NLR protein itself [13]. Once activated, NLR proteins trigger the hyper-sensitive response, which leads to local cell death as well as a systemic immune response.
Plant NLRs are characterized by three broad domains, including leucine-rich repeats (LRRs) at the C terminus, a nucleotide-binding domain in the middle, and one of a variety of domains at the N terminus. The N-terminal domain determines downstream signal transduction [45] and enables classification of NLRs based on whether this N-terminal domain is a Toll-like/interleukin 1 domain or not (TIR-type vs. non-TIR-type NLRs). Interestingly, these two classes of NLRs have evolved differently in different plant species, and some plants have expanded one or the other class of NLRs. For example, genes encoding TIR-type NLRs are more numerous in Arabidopsis spp. and Brassicaceae, non-TIR-type family members are more numerous in potato (Solanum sp.) and grape-vine (Vitis sp.), whereas apple (Malus domestica) has similar gene numbers of both classes [45].
Impressively, there is 100-fold variation in the number of NLR genes across plant genomes, with only a few dozen NLR genes found in crop fruits like papaya, to several thousand in wheat [13]. This variation in number is not just due to genome size; chick-pea (Cicer arietinum) and apple genomes are of similar size, but contain ~ 100 and ~ 1000 NLR family members, respectively [45] (Fig. 1). In addition, rapid expansions and contractions appear to have occurred in closely related strains and species. Given that many NLR proteins sense pathogen effectors and promote resistance, it seems likely that the expansions occurred because they improved pathogen resistance. Conversely, the drive for contraction may be due to the collateral damage from increased gene number, as several NLR alleles have been linked to autoimmunity in Arabidopsis species [13].
Animals also use NLR proteins as intracellular pathogen receptors. Despite an intriguing similarity in domain structure, recent analyses suggest that animal and plant NLRs are not derived from a common ancestor [46], although the nucleotide-binding domain may have evolved from distinct lineages of a common prokaryotic ancestral ATPase [44,47]. Animal NLRs can trigger immune responses either through detecting pathogen components directly, or through sensing the effects of pathogen virulence factors. From this perspective, plant NLRs share a common mode of activation with animal NLRs, as both are also activated by nucleotide-dependent oligomerization, and higher-order assembly. For animal NLRs, the higher-order assembly provides a platform for oligomerization of signaling components, and here the proximal downstream signaling mechanisms are better understood than in plants, and include oligomerization and activation of enzymes like caspase-1, which triggers cell death and cytokine release.
Similar to NLRs in plants, there are many examples of NLR gene number variation across animal species. Most analyses of NLR function have been in mammals, with humans and mice having 23 and 34 NLR family members, respectively. However, a fish-specific subgroup of NLR genes expanded and diversified substantially, with the number of NLR members varying significantly among different fish species and clades [12]. For example, the zebrafish genome contains 368 coding and 37 pseudo NLR-C genes (similar to the common carp Cyprinus carpio), northern pike (Esox lucius) has 50, whereas spotted gar (Lepisosteus oculatus) has 10 NLR-C genes. NLR expansions are not only characteristic for fish, but also for some invertebrate animals, especially those living in aquatic environments. In particular, coral Acropora digitifera has an extraordinarily expanded repertoire of NLR genes, with over 500 family members [48]. Besides interactions with pathogens, a candidate driver of NLR diversification in corals is their obligatory symbiotic relationship with dinoflagellates [48]. Another species of aquatic invertebrates with NLR gene expansion is the purple sea urchin, whose genome contains over 200 family members [49,50] (Fig. 1). Exactly how plants and animals both evolved to use NLRs in defense is unclear. It is clear, however, that the combination of domains found in NLR proteins is critical host defense factors against co-evolving pathogens across a wide range of phylogeny, which has led to their diversification and expansion.
Expansion and diversification of vertebrate interferon-inducible GTPases and pathogen rhoptry kinases
As described above, NLRs in both animals and plants detect pathogens to trigger downstream signaling cascades leading to cell death and systemic immune responses. In contrast, interferon-inducible GTPases are intracellular receptors that directly target intracellular pathogen proteins or the intracellular pathogens themselves for destruction [51,52]. There are four classes of interferon-inducible GTPases, including the immunity-related GTPases (IRGs), which we describe here, because they have undergone expansion in gene number in some species, and there are functional insights about their roles in immunity.
The IRG genes arose in the chordate lineage, and their number varies across vertebrate genomes, with humans having two genes, rodents having as many as 20 genes, zebrafish 11 genes, and birds having lost IRG genes altogether. The battles between IRG proteins and intracellular pathogens have been well characterized for the mouse IRG proteins that are engaged in an arms race with rhoptry kinases from the protozoan parasite Toxoplasma gondii. IRG proteins are activated by binding to GTP, and they directly localize to intracellular vacuoles containing T. gondii, as well as to vacuoles containing other intracellular pathogens such as the bacterium Chlamydia trachomatis and the microsporidium Encephalitozoon cuniculi [53,54]. The targeting of IRG proteins to these vacuoles leads to their lysis, which then releases parasites into the cytosol, resulting in parasite death. While the exact mechanisms of parasite death as well as vacuole lysis are still being defined, GTP hydrolysis by IRG proteins is required, and there are several E3 ubiquitin ligases and ubiquitin that are also recruited to the vacuole [55,56]. As mentioned above, IRG genes have expanded in rodent genomes, and they are highly polymorphic among wild strains of mice [57], while on the pathogen side, rhoptry kinases have expanded and diversified in T. gondii genomes. Certain versions of these rhoptry kinases can inactivate certain versions of the IRG proteins, illustrating that the outcome of a host–pathogen battle depends on exactly which strain of host and pathogen species are interacting.
Real-time observation of transient expansion of viral virulence factor K3L
Key insights about the diversification and expansion of gene families have come from evolutionary analyses of viruses. Their fast replication times allow them to sample sequence space much more rapidly than their hosts, and many viruses have a high mutation rate, which further accelerates the diversification of their genomes [58]. Positive selection has been described for viral factors, as well as host factors, involved in the vertebrate protein kinase R (PKR) antiviral response to halt mRNA translation [59]. Here, phosphorylation of the eukaryotic translation initiation factor eIF2a by host PKR inhibits eIF2a activity. As access to translation machinery is critical for viral replication, poxviruses use eIF2-mimics like the viral protein K3L to inhibit PKR [60–63], and thus prevent translation from being inhibited. Evolutionary analysis reveals that primate hosts have diversified PKR amino acid residues targeted by K3L, and K3L in turn has mutated amino acids, likely in order to keep pace with changes in host PKR. Of note, it is not known exactly which viral factor was challenging primate PKR in the evolutionary past, although it may have been a K3L-like protein from an ancient poxvirus.
Knowledge of PKR/K3L interactions provided the basis for directed evolution studies with the model poxvirus vaccinia, which illustrated in real time how an expansion in gene number can increase viral success. Dramatically, upon selecting for viruses better adapted to infect host cells, the vaccinia genome acquired multiple duplications of the anti-host factor K3L, which caused up to 7–10% increase in genome size [64]. Overexpression of K3L gene product was necessary and sufficient for the improved success of the virus. Notably, this gene amplification was transient, and ultimately the gene number contracted down with selection for better efficacy of one of the mutated K3L copies. This ‘accordion model’ for genome evolution of DNA viruses provides an example of micro-evolution of gene families undergoing expansion and contraction in host–pathogen conflicts.
Concluding thoughts about shared evolutionary themes in host–pathogen battles
How do gene families evolve over time? As beautifully illustrated by the directed evolution studies with poxviruses, the large size of a gene family can be a transient phenomenon [64]. Perhaps many of the giant gene families described above will contract in the future, either due to genetic drift, or because of the detrimental effects of these large families. For example, it is important to remember that hosts may be battling multiple types of pathogens, and in some cases resistance to one pathogen can cause susceptibility to another pathogen [29,65–67]. And while viruses have extreme pressures to reduce copy number after there has been expansion and mutation to generate a more beneficial gene variant, other organisms have genomes that can be more tolerant of additional copy number in their genomes.
Which situations create the strongest drive to amplify and diversify gene families? Situations of direct contact between host and pathogen proteins, and in particular, specific domains of these proteins, will create some of the strongest pressure for diversification and expansion. Viruses are among the most exposed pathogens, performing all their replication without their own membrane structure, although they are known to shield themselves using membranes derived from hosts [68]. Next most exposed include obligate intracellular pathogens that replicate within their own membranes, but without a separate host membrane compartment surrounding them, which includes many microsporidia species like those in the Nematocida genus [69,70]. This intense evolutionary pressure on microsporidia may have led to the large, lineage-specific gene families commonly observed in their genomes [43]. Pathogens such as Toxoplasma sp., Plasmodium sp., and Chlamydia sp. replicate inside separate, host membrane-bound compartments, but they are also obligate intracellular pathogens and thus dependent on navigating the changing environment of a host cell for replication. Here too these organisms appear able to secrete hundreds of proteins to interface with host cells and have demonstrated diversification and expansion in gene families [71].
In terms of protein class, there may be varying structural constraints in terms of how many different three-dimensional structures a given protein scaffold can adopt. For proteins that have intrinsic disordered regions perhaps there is less need to amplify gene number, if the intrinsically disordered region can adopt several different conformations. For example, the Mx interferon-inducible GTPases, which arose in the chordate lineage like interferon-inducible IRG GTPases, have pathogen-interacting domains that are predicted to be intrinsically disordered, and these proteins appear able to restrict a wide range of viruses, likely through interacting with diverse viral proteins [72]. Interestingly, they do not have greatly varying gene number in vertebrates, which could be due to chance, or perhaps due to the ability to adopt multiple conformations without mutation. A recent mutational analysis of the TRIM5α restriction factor indicated that remarkably, most random mutations in the v1 loop of TRIM5α that binds viral capsid led to an increase in restriction capability [73]. This finding is in contrast to mutational analyses of highly conserved proteins where random mutations are more likely to decrease function. Perhaps such mutational resilience in TRIM5α has facilitated an increase in the diversity and number of TRIM genes.
Another issue related to protein structure that may be in common among immune gene families that increase in number is that they tend to form higher-order structures. For example, NLRs, IRGs and TRIM proteins have all been shown to engage in higher-order assembly upon activation [44,51,74]. Domains that participate in protein–protein interactions may be more tolerant of ‘new members’ in an oligomer that could adopt new specificities, either for targeting pathogens or for modifying downstream signaling reactions. An interesting example of how a duplicated gene can acquire new function and thus be maintained in the genome was demonstrated for nonsense-mediated RNA decay (NMD) factor UPF3A, which was acquired in the vertebrate lineage. Here, UPF3A is part of the multi-subunit NMD complex, and serves as a rheostat to control this process [75]. Perhaps members of some large gene families involved in immunity also act to diminish outputs, and thus tune responses based on the risk/reward ratio of collateral damage vs. defense. Indeed, immune regulators like the PALS-22/PALS-25 antagonistic paralogs in C. elegans and many NLR antagonistic paralogs like RRS-1/RPS4 in Arabidopsis appear to have arisen from gene duplication events and encode negative and positive regulators respectively, which physically associate and regulate immune activation [28,29,76,77].
In summary, large, species-specific gene families are a common outcome of host–pathogen battles. And while it is commonly thought that large gene families have redundancy, genomes are not tolerant of redundancy for long, and duplicated genes either need to be ‘fixed’ due to a novel function, or they will be lost to genetic drift. Of note, forward genetic approaches have been successful in identifying function for many individual genes from large gene families, such as the many specific variants of NLR genes that play roles in plant defense against specific pathogen variants, and the pals genes that regulate C. elegans defense against natural pathogens [13,29,78,79]. Further exploration of such gene families represents a potentially rich source of biochemical information. As illustrated by the L. pneumophila SidE effectors that function as ubiquitin ligases despite a lack of primary sequence similarity with canonical ligases, there can be functional and conceptual conservation in such highly diverged factors, despite a lack of obvious sequence similarity.
Acknowledgements
We thank Gira Bhabha, Matthew Daugherty, Spencer Gang, Bretta McCall, Eillen Tecle, David Wang and Elina Zuniga and the anonymous reviewers for helpful comments on the manuscript. Figure was created with BioRender.com. This study was supported by NIH R01 GM114139 to ERT and American Heart Association postdoctoral fellowship to VL.
Abbreviations
- A. thaliana
Arabidopsis thaliana
- ATPase
adenylpyrophosphatase
- BTB
broad-complex, Tramtrack, and Bric-à-brac
- C terminus
carboxyl terminus
- C. elegans
Caenorhabditis elegans
- CRL
cullin-RING E3 Ligase
- D. melanogaster
Drosophila melanogaster
- DNA
deoxyribonucleic acid
- E1
ubiquitin-activating enzyme
- E2
ubiquitin-conjugating enzyme
- E3
ubiquitin ligase
- EHEC
enterohemorrhagic Escherichia coli
- eIF2a
eukaryotic translation initiation factor 2A
- GTP
guanosine triphosphate
- GTPase
guanosine triphosphate hydrolase
- HECT
homologous to the E6-AP carboxyl terminus
- HIV
human immunodeficiency virus
- IRG
immunity-related GTPases
- L. pneumophila
Legionella pneumophila
- LRR
leucine-rich repeat
- mRNA
messenger ribonucleic acid
- Mx
myxovirus resistance
- N terminus
amino terminus
- N. displodere
Nematocida displodere
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NleG
non-LEE-encoded effector G
- NLR
nucleotide-binding domain and leucine-rich repeat
- NMD
nonsense-mediated RNA Decay
- pals
protein containing ALS2cr12 (ALS2CR12) signature
- PKR
protein kinase R
- R genes
resistance genes
- RIG-I
retinoic acid-inducible gene I
- RING
really interesting new gene
- RNA
ribonucleic acid
- S. Typhimurium
Salmonella enterica serovar Typhimurium
- SidE
substrate of Icm/Dot transporter E
- SIV
Simian immunodeficiency virus
- Skp
S-phase kinase-associated protein
- SopA
Salmonella outer protein A
- sp.
species (singular)
- spp.
species (plural)
- T. gondii
Toxoplasma gondii
- TIR
toll-like/interleukin 1 domain
- TRIM
tripartite motif-containing
- UPF3A
up-frameshift suppressor 3A
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Daugherty MD & Malik HS (2012) Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 46, 677–700. [DOI] [PubMed] [Google Scholar]
- 2.Pearson JC, Lemons D & McGinnis W (2005) Modulating Hox gene functions during animal body patterning. Nat Rev Genet 6, 893–904. [DOI] [PubMed] [Google Scholar]
- 3.Gaunt SJ (2018) Hox cluster genes and collinearities throughout the tree of animal life. Int J Dev Biol 62, 673–683. [DOI] [PubMed] [Google Scholar]
- 4.Nie L, Cai SY, Shao JZ & Chen J (2018) Toll-like receptors, associated biological roles, and signaling networks in non-mammals. Front Immunol 9, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irazoqui JE, Urbach JM & Ausubel FM (2010) Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev 10, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sowa JN, Jiang H, Somasundaram L, Tecle E, Xu G, Wang D & Troemel ER (2020) The Caenorhabditis elegans RIG-I homolog DRH-1 mediates the intracellular pathogen response upon viral infection. J Virol 94, e01173–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benton R (2015) Multigene family evolution: perspectives from insect chemoreceptors. Trends Ecol Evol 30, 590–600. [DOI] [PubMed] [Google Scholar]
- 8.Maltez VI & Miao EA (2016) Reassessing the evolutionary importance of inflammasomes. J Immunol 196, 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lespinet O, Wolf YI, Koonin EV & Aravind L (2002) The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res 12, 1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flajnik MF (2018) A cold-blooded view of adaptive immunity. Nat Rev 18, 438–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alder MN, Herrin BR, Sadlonova A, Stockard CR, Grizzle WE, Gartland LA, Gartland GL, Boydston JA, Turnbough CL Jr & Cooper MD (2008) Antibody responses of variable lymphocyte receptors in the lamprey. Nat Immunol 9, 319–327. [DOI] [PubMed] [Google Scholar]
- 12.Chang MX, Xiong F, Wu XM & Hu YW (2020) The expanding and function of NLRC3 or NLRC3-like in teleost fish: recent advances and novel insights. Dev Comp Immunol 114, 103859. [DOI] [PubMed] [Google Scholar]
- 13.Tamborski J & Krasileva KV (2020) Evolution of plant NLRs: from natural history to precise modifications. Annu Rev Plant Biol 71, 355–378. [DOI] [PubMed] [Google Scholar]
- 14.Zheng N & Shabek N (2017) Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem 86, 129–157. [DOI] [PubMed] [Google Scholar]
- 15.van Gent M, Sparrer KMJ & Gack MU (2018) TRIM proteins and their roles in antiviral host defenses. Annu Rev Virol 5, 385–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatakeyama S (2017) TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci 42, 297–311. [DOI] [PubMed] [Google Scholar]
- 17.Han K, Lou DI & Sawyer SL (2011) Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet 7, e1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langevin C, Levraud JP & Boudinot P (2019) Fish antiviral tripartite motif (TRIM) proteins. Fish Shellfish Immunol 86, 724–733. [DOI] [PubMed] [Google Scholar]
- 19.Lukic Z & Campbell EM (2012) The cell biology of TRIM5alpha. Curr HIV/AIDS Rep 9, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P & Sodroski J (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853. [DOI] [PubMed] [Google Scholar]
- 21.Chiramel AI, Meyerson NR, McNally KL, Broeckel RM, Montoya VR, Mendez-Solis O, Robertson SJ, Sturdevant GL, Lubick KJ, Nair V et al. (2019) TRIM5alpha restricts flavivirus replication by targeting the viral protease for proteasomal degradation. Cell Rep 27, 3269–3283, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatziioannou T, Perez-Caballero D, Yang A, Cowan S & Bieniasz PD (2004) Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc Natl Acad Sci USA 101, 10774–10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volkmann B, Wittmann S, Lagisquet J, Deutschmann J, Eissmann K, Ross JJ, Biesinger B & Gramberg T (2020) Human TRIM5alpha senses and restricts LINE-1 elements. Proc Natl Acad Sci USA 117, 17965–17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K, Deshaies RJ & Liu X (2020) Assembly and regulation of CRL ubiquitin ligases. Adv Exp Med Biol 1217, 33–46. [DOI] [PubMed] [Google Scholar]
- 25.Xu G, Ma H, Nei M & Kong H (2009) Evolution of F-box genes in plants: different modes of sequence divergence and their relationships with functional diversification. Proc Natl Acad Sci USA 106, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas JH (2006) Adaptive evolution in two large families of ubiquitin-ligase adapters in nematodes and plants. Genome Res 16, 1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bakowski MA, Desjardins CA, Smelkinson MG, Dunbar TA, Lopez-Moyado IF, Rifkin SA, Cuomo CA & Troemel ER (2014) Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLoS Pathog 10, e1004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panek J, Gang SS, Reddy KC, Luallen RJ, Fulzele A, Bennett EJ & Troemel ER (2020) A cullin-RING ubiquitin ligase promotes thermotolerance as part of the intracellular pathogen response in Caenorhabditis elegans. Proc Natl Acad Sci USA 117, 7950–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddy KC, Dror T, Underwood RS, Osman GA, Elder CR, Desjardins CA, Cuomo CA, Barkoulas M & Troemel ER (2019) Antagonistic paralogs control a switch between growth and pathogen resistance in C. elegans. PLoS Pathog 15, e1007528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leyva-Diaz E, Stefankakis N, Carerra I, Glenwinkel L, Wang G, Driscoll M & Hobert O (2017) pals-22, a member of an expanded C. elegans gene family, controls silencing of repetitive DNA. bioRxiv [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashida H & Sasakawa C (2017) Bacterial E3 ligase effectors exploit host ubiquitin systems. Curr Opin Microbiol 35, 16–22. [DOI] [PubMed] [Google Scholar]
- 32.Wu B, Skarina T, Yee A, Jobin MC, Dileo R, Semesi A, Fares C, Lemak A, Coombes BK, Arrowsmith CH et al. (2010) NleG type 3 effectors from enterohaemorrhagic Escherichia coli are U-Box E3 ubiquitin ligases. PLoS Pathog 6, e1000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valleau D, Little DJ, Borek D, Skarina T, Quaile AT, Di Leo R, Houliston S, Lemak A, Arrowsmith CH, Coombes BK et al. (2018) Functional diversification of the NleG effector family in enterohemorrhagic Escherichia coli. Proc Natl Acad Sci USA 115, 10004–10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Correa RL, Bruckner FP, de Souza Cascardo R & Alfenas-Zerbini P (2013) The role of F-Box proteins during viral infection. Int J Mol Sci 14, 4030–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y & Tan X (2020) Viral manipulations of the cullin-RING ubiquitin ligases. Adv Exp Med Biol 1217, 99–110. [DOI] [PubMed] [Google Scholar]
- 36.Magori S & Citovsky V (2011) Hijacking of the host SCF ubiquitin ligase machinery by plant pathogens. Front Plant Sci 2, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lomma M, Dervins-Ravault D, Rolando M, Nora T, Newton HJ, Sansom FM, Sahr T, Gomez-Valero L, Jules M, Hartland EL et al. (2010) The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell Microbiol 12, 1272–1291. [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Valero L, Rusniok C, Carson D, Mondino S, Perez-Cobas AE, Rolando M, Pasricha S, Reuter S, Demirtas J, Crumbach J et al. (2019) More than 18,000 effectors in the Legionella genus genome provide multiple, independent combinations for replication in human cells. Proc Natl Acad Sci USA 116, 2265–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kenzaka T, Yasui M, Baba T, Nasu M & Tani K (2018) Positive selection in F-box domain (lpp0233) encoded in Legionella pneumophila strains. Biocontrol Sci 23, 53–59. [DOI] [PubMed] [Google Scholar]
- 40.Domman D, Collingro A, Lagkouvardos I, Gehre L, Weinmaier T, Rattei T, Subtil A & Horn M (2014) Massive expansion of ubiquitination-related gene families within the Chlamydiae. Mol Biol Evol 31, 2890–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han B & Weiss LM (2017) Microsporidia: obligate intracellular pathogens within the fungal kingdom. Microbiol Spectr 5. 10.1128/microbiolspec.FUNK-0018-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luallen RJ, Reinke AW, Tong L, Botts MR, Felix MA & Troemel ER (2016) Discovery of a natural microsporidian pathogen with a broad tissue tropism in Caenorhabditis elegans. PLoS Pathog 12, e1005724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reinke AW, Balla KM, Bennett EJ & Troemel ER (2017) Identification of microsporidia host-exposed proteins reveals a repertoire of rapidly evolving proteins. Nat Commun 8, 14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J & Chai J (2020) Molecular actions of NLR immune receptors in plants and animals. Sci China Life Sci 63, 1–14. [DOI] [PubMed] [Google Scholar]
- 45.Borrelli GM, Mazzucotelli E, Marone D, Crosatti C, Michelotti V, Vale G & Mastrangelo AM (2018) Regulation and evolution of NLR genes: a close interconnection for plant immunity. Int J Mol Sci 19, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urbach JM & Ausubel FM (2017) The NBS-LRR architectures of plant R-proteins and metazoan NLRs evolved in independent events. Proc Natl Acad Sci USA 114, 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leipe DD, Koonin EV & Aravind L (2004) STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J Mol Biol 343, 1–28. [DOI] [PubMed] [Google Scholar]
- 48.Hamada M, Shoguchi E, Shinzato C, Kawashima T, Miller DJ & Satoh N (2013) The complex NOD-like receptor repertoire of the coral Acropora digitifera includes novel domain combinations. Mol Biol Evol 30, 167–176. [DOI] [PubMed] [Google Scholar]
- 49.Rast JP, Smith LC, Loza-Coll M, Hibino T & Litman GW (2006) Genomic insights into the immune system of the sea urchin. Science 314, 952–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hibino T, Loza-Coll M, Messier C, Majeske AJ, Cohen AH, Terwilliger DP, Buckley KM, Brockton V, Nair SV, Berney K et al. (2006) The immune gene repertoire encoded in the purple sea urchin genome. Dev Biol 300, 349–365. [DOI] [PubMed] [Google Scholar]
- 51.Pilla-Moffett D, Barber MF, Taylor GA & Coers J (2016) Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol 428, 3495–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haller O, Staeheli P, Schwemmle M & Kochs G (2015) Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol 23, 154–163. [DOI] [PubMed] [Google Scholar]
- 53.Al-Zeer MA, Al-Younes HM, Braun PR, Zerrahn J & Meyer TF (2009) IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One 4, e4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferreira-da-Silva Mda F, Springer-Frauenhoff HM, Bohne W & Howard JC (2014) Identification of the microsporidian Encephalitozoon cuniculi as a new target of the IFNgamma-inducible IRG resistance system. PLoS Pathog 10, e1004449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coers J & Haldar AK (2015) Ubiquitination of pathogen-containing vacuoles promotes host defense to Chlamydia trachomatis and Toxoplasma gondii. Commun Integr Biol 8, e1115163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haldar AK, Foltz C, Finethy R, Piro AS, Feeley EM, Pilla-Moffett DM, Komatsu M, Frickel EM & Coers J (2015) Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc Natl Acad Sci USA 112, E5628–E5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lilue J, Muller UB, Steinfeldt T & Howard JC (2013) Reciprocal virulence and resistance polymorphism in the relationship between Toxoplasma gondii and the house mouse. Elife 2, e01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drake JW (1999) The distribution of rates of spontaneous mutation over viruses, prokaryotes, and eukaryotes. Ann N Y Acad Sci 870, 100–107. [DOI] [PubMed] [Google Scholar]
- 59.Elde NC, Child SJ, Geballe AP & Malik HS (2009) Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457, 485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gale M Jr, Tan SL, Wambach M & Katze MG (1996) Interaction of the interferon-induced PKR protein kinase with inhibitory proteins P58IPK and vaccinia virus K3L is mediated by unique domains: implications for kinase regulation. Mol Cell Biol 16, 4172–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davies MV, Elroy-Stein O, Jagus R, Moss B & Kaufman RJ (1992) The vaccinia virus K3L gene product potentiates translation by inhibiting double-stranded-RNA-activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J Virol 66, 1943–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beattie E, Tartaglia J & Paoletti E (1991) Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology 183, 419–422. [DOI] [PubMed] [Google Scholar]
- 63.Carroll K, Elroy-Stein O, Moss B & Jagus R (1993) Recombinant vaccinia virus K3L gene product prevents activation of double-stranded RNA-dependent, initiation factor 2 alpha-specific protein kinase. J Biol Chem 268, 12837–12842. [PubMed] [Google Scholar]
- 64.Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP & Malik HS (2012) Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150, 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hodgkin J, Felix MA, Clark LC, Stroud D & Gravato-Nobre MJ (2013) Two Leucobacter strains exert complementary virulence on Caenorhabditis including death by worm-star formation. Curr Biol 23, 2157–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boxx GM & Cheng G (2016) The roles of type I interferon in bacterial infection. Cell Host Microbe 19, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colon-Thillet R, Hsieh E, Graf L, McLaughlin RN Jr, Young JM, Kochs G, Emerman M & Malik HS (2019) Combinatorial mutagenesis of rapidly evolving residues yields super-restrictor antiviral proteins. PLoS Biol 17, e3000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T et al. (2010) Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Troemel ER, Felix MA, Whiteman NK, Barriere A & Ausubel FM (2008) Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol 6, 2736–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szumowski SC, Botts MR, Popovich JJ, Smelkinson MG & Troemel ER (2014) The small GTPase RAB-11 directs polarized exocytosis of the intracellular pathogen N. parisii for fecal-oral transmission from C. elegans. Proc Natl Acad Sci USA 111, 8215–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Ooij C, Tamez P, Bhattacharjee S, Hiller NL, Harrison T, Liolios K, Kooij T, Ramesar J, Balu B, Adams J et al. (2008) The malaria secretome: from algorithms to essential function in blood stage infection. PLoS Pathog 4, e1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mitchell PS, Emerman M & Malik HS (2013) An evolutionary perspective on the broad antiviral specificity of MxA. Curr Opin Microbiol 16, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tenthorey JL, Young C, Sodeinde A, Emerman M & Malik HS (2020) Mutational resilience of antiviral restriction favors primate TRIM5alpha in host-virus evolutionary arms races. Elife 9, e59988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li X & Sodroski J (2008) The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol 82, 11495–11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shum EY, Jones SH, Shao A, Dumdie J, Krause MD, Chan WK, Lou CH, Espinoza JL, Song HW, Phan MH et al. (2016) The antagonistic gene paralogs Upf3a and Upf3b govern nonsense-mediated RNA decay. Cell 165, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le Roux C, Huet G, Jauneau A, Camborde L, Tremousaygue D, Kraut A, Zhou B, Levaillant M, Adachi H, Yoshioka H et al. (2015) A receptor pair with an integrated decoy converts pathogen disabling of transcription factors to immunity. Cell 161, 1074–1088. [DOI] [PubMed] [Google Scholar]
- 77.Sarris PF, Duxbury Z, Huh SU, Ma Y, Segonzac C, Sklenar J, Derbyshire P, Cevik V, Rallapalli G, Saucet SB et al. (2015) A plant immune receptor detects pathogen effectors that target WRKY transcription factors. Cell 161, 1089–1100. [DOI] [PubMed] [Google Scholar]
- 78.Reddy KC, Dror T, Panek J, Chen K, Lim ES, Wang D & Troemel ER (2017) Intracellular pathogen response pathway promotes proteostasis in C. elegans. bioRxiv [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reddy KC, Dror T, Sowa JN, Panek J, Chen K, Lim ES, Wang D & Troemel ER (2017) An intracellular pathogen response pathway promotes proteostasis in C. elegans. Curr Biol 27, 3544–3553 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]