

Abstract
Introduction
Juvenile-onset Huntington’s disease (JOHD) is characterized by a unique motor phenotype relative to patients with adult-onset Huntington’s Disease (AOHD). This study characterized motor progression of JOHD to propose improved outcome measures for this group.
Methods:
We used linear mixed effect regression models to compare progression of the Unified Huntington’s Disease Rating Scale (UHDRS) Total Motor Score (TMS) and the chorea score between patients with JOHD and AOHD. We then evaluated all 31 subscales that make up the UHDRS over time within patients with JOHD to identify measures that may be used to track motor progression most reliably.
Results:
The JOHD cohort had faster TMS progression compared to AOHD (p=0.006) but no group difference in the rate of change of chorea. Patients with JOHD did not show significant change in any of the chorea subscales. The subscales that changed most reliably over time amongst patients with JOHD were dysarthria, upper extremity dystonia, tandem walking, gait, bilateral pronate/supinate, bilateral finger-tapping, and tongue protrusion. When these subscales were summed, they progressed at a faster rate (7.07%, 95% CI [5.96 – 8.18]) than the TMS (4.92%, 95% CI [3.95 – 5.89]).
Conclusion:
While the TMS changes at a significant rate in JOHD subjects, not all subscales that make up the TMS accurately represent the unique motor features of JOHD. A JOHD-specific scale performed better at tracking motor progression relative to the TMS. This scale may improve clinical care for patients with JOHD and allow for the development of more efficient clinical trials.
Keywords: Juvenile-Onset Huntington Disease, motor progression, Enroll-HD
INTRODUCTION
Huntington’s Disease (HD) is a neurodegenerative disease caused by a CAG repeat expansion in the huntingtin gene. Patients with HD experience psychiatric symptoms and progressive cognitive and motor decline.[1] Clinical diagnosis is based on the onset of motor abnormalities, which generally occurs between the ages of 40-50 years.[2] Longer CAG repeat expansions are associated with earlier disease onset and faster rate of progression.[2]
A small proportion of patients with HD have exceptionally long expansions leading to juvenile-onset HD (JOHD). Patients with JOHD generally present prior to the age of 21 years [3, 4] with hypokinetic symptoms (i.e., bradykinesia and rigidity) that persist throughout the disease.[5] In contrast, patients with adult-onset HD (AOHD) generally present with hyperkinetic movements, such as chorea, with hypokinetic features becoming more prominent in the late stages of the disease.[6]
Patients with JOHD are often excluded from clinical trials.[7] For example, the TETRA-HD[8] study that evaluated the use of tetrabenazine for chorea only included participants with a maximum CAG of 54, likely indirectly excluding patients with JOHD. The primary outcome measure for this study was the Unified Huntington’s Disease Rating Scale (UHDRS) [9] and the chorea subscale was a key secondary outcome of this study. These are reasonable outcome measures for patients with AOHD but may not apply to patients with JOHD. Despite the known differences in the presentation of motor symptoms between patients with AOHD and JOHD, there is not a JOHD-specific motor scale. This lack of a reliable outcome measure that addresses the unique phenotype of patients with JOHD has likely contributed to a lack of clinical trials in this population. The aim of this study was to leverage the Enroll-HD database to better characterize the components of the UHDRS that may perform best at tracking motor progression for patients with JOHD.
METHODS
Study Design
Enroll-HD is a longitudinal, observational research platform to facilitate research in HD.[10] Data sets are collected annually on participants in this multicenter longitudinal observational study. Data are monitored for quality and accuracy using a risk-based monitoring approach. Enroll-HD includes individuals with pre-manifest HD, motor-manifest HD, and controls. For the present analysis, we included data from subjects who had an HD gene mutation (≥ 36 CAG repeats) and a reported age of clinical diagnosis (ACD) of HD from the HD clinical characteristics form within Enroll-HD (variable ‘hddiagn’). This variable was chosen to limit recall bias introduced by other patient-reported measure of age of symptom onset. Furthermore, we excluded patient visits where the time since clinical diagnosis was >10 years. Of note, some participants entered the Enroll-HD study in the pre-manifest phase of the disease and received a clinical diagnosis during their time in the Enroll study. These incident participants would have a disease duration of 0 and beyond. Other participants entered the Enroll-HD study having already received a clinical diagnosis of HD (prevalent participants). These participants would have a baseline disease duration greater than zero. Specifically, not all participants have a baseline disease duration value of zero. Subjects <18 years old are excluded from the Enroll-HD study, unless they are symptomatic. Consequently, nearly all subjects in the JOHD group were prevalent users. There were only three incident JOHD users who received a diagnosis between the age of 18 and 21 years. Baseline visit refers to the first available visit where the subject has a known ACD. Given the longitudinal nature of the analyses performed, participants were excluded if they only had one eligible visit available. Participants were also excluded if they had missing or incomplete data regarding use of antipsychotic medications, tetrabenazine, or dopaminergic medications. Medications were defined by the WHO Anatomical Chemical Index Numbers. Specifically, antipsychotics were defined as those with index numbers starting with N05A. Tetrabenazine and deutetrabenazine were defined as those with an index number of N07XX, and dopaminergic medications were defined as those with the numbers N04BA, N04BB, N04BC, N04BD, or N04BX.
Participants meeting the above criteria were divided into two groups according to their reported ACD. Subjects with a reported ACD > 21 years and a CAG repeat length <55 were considered to have AOHD. Participants with a reported ACD of ≤ 21 years and a CAG ≥ 55 were considered to have JOHD. A CAG cutoff was implemented to decrease heterogeneity within the JOHD group, and was based on previous studies [3, 11]. We investigated the rate of motor progression during the first 5 years of participation in the Enroll-HD study.
As noted above, heterogeneity can exist within a cohort of JOHD patients. For example, a patient with a CAG of 80 may have a significantly different course of motor symptoms compared to a subject with a CAG repeat of 60. We performed additional analyses after dividing the JOHD group into those with pediatric-onset HD (clinical diagnosis received prior to the age of 18; pHD) and non-pediatric JOHD (clinical diagnosis received at the age of 18 or above; npJOHD).
We identified 31,421 observations from 9,614 individuals with HD. This included 9,524 patients with AOHD and 90 patients with JOHD (Supplemental Figure 1; Table 1). We first compared the rate of change of the UHDRS total motor score (TMS) between groups. Additionally, we calculated a chorea score (sum of UHDRS chorea subscales; maximum score of 28) and compared the annualized change of this score between groups.
Table 1:
Baseline Demographics
| AOHD | JOHD | p-value | |
|---|---|---|---|
| N | 9,524 | 90 | N/A |
| Age, mean ± S.D. | 51.8 ± 12.0 | 19.4 ± 4.8 | <0.001 |
| Males, N (%) | 4,645 (48.8) | 40 (44.4) | 0.477 |
| BMI, mean ± S.D. | 25.1 ± 5.0 | 22.5 ± 4.9 | <0.001 |
| CAG, mean ± S.D. | 43.6 ± 3.0 | 67.4 ± 10.7 | <0.001 |
| Disease Duration, mean ± S.D. | 2.1 ± 2.3 | 2.9 ± 2.8 | 0.001 |
| Age of Clinical Dx, mean ± S.D. | 49.7 ± 12.0 | 16.5 ± 4.3 | <0.001 |
| Years Followed, mean ± S.D. | 2.63 ± 1.59 | 2.63 ± 1.61 | 0.991 |
| Disease Burden, mean ± S.D. | 390.4 ± 86.7 | 582.9 ± 119.6 | <0.001 |
| TMS, mean ± S.D. | 29.7 ± 17.4 | 40.5 ± 20.5 | <0.001 |
| Antipsychotic Use, N (%) | 2680 (28.1) | 24 (26.7) | 0.848 |
| Tetrabenazine Use, N (%) | 1142 (12.0) | 9 (10.0) | 0.677 |
| Levodopa Use, N (%) | 363 (3.8) | 12 (13.3) | <0.001 |
AOHD, Adult-Onset HD; CAG, Cytosine-Adenine-Guanine; Dx, Diagnosis; HD, Huntington Disease; JOHD, Juvenile-Onset HD; TMS, Total Motor Score
Amongst the JOHD individuals, we evaluated the annualized change for all 31 subscales, individually, to better understand which scores had the largest rate of change with the lowest variability in the JOHD population, based on calculated t-values. Based on these results, we chose the 10 subscales with the highest t-values and summed them to create a new JOHD-specific score.
Statistical Analysis
We constructed linear mixed effect regression (LMER) models to investigate the trajectory of the TMS and Chorea scores between the AOHD and JOHD groups. The predictor variable of interest was the interaction between group and time in study. Baseline age, sex, and CAG repeat length were included as covariates in all models. We also controlled for use of VMAT-2 inhibitors, antipsychotics, and dopaminergic medications, given their potential to impact motor scores. Lastly, the baseline score of the dependent variable was included in each model. All models included a random slope and intercept per participant.
We used similar LMER models to evaluate each individual subscale from the UHDRS amongst patients with JOHD only. The models were like those described above. The primary difference was that the predictor variable of interest for these models was time in study given that there was only one group. Results were adjusted using the Bonferroni method to account for multiple comparisons and were considered significant if the adjusted p-value (q) was < 0.05. All analyses were performed in RStudio version 4.0.5.
Data Sharing
The Enroll-HD database is made available by CHDI to qualified researchers.
Ethical Compliance
All Enroll-HD sites were required to obtain and maintain local Ethics Committee approvals. Participants must have signed informed consent forms for their data to be included in the datasets. The authors confirm that the approval of the institutional review board at the University of Iowa was not required for this work, which is deemed a secondary data analysis of de-identified data.
RESULTS
There were expected differences between groups at baseline (Table 1). The JOHD group had a mean annualized increase in TMS that was 2.64 points per year faster (95% confidence interval [1.65 – 3.62], p<0.001) than the AOHD group (Supplemental Figure 2A). In contrast, there was not a significant difference in the mean annualized rate of change of the chorea score between groups (Mean Difference = 0.06, 95% CI [−0.36 – 0.48], p=0.772; Supplemental Figure 2B).
There were 38 (42.2%) subjects with pHD within the JOHD group (Supplemental Table 2). The annualized change of the TMS was faster in the pHD and npJOHD groups compared to the AOHD group, but the pHD and npJOHD groups did not significantly differ from one another (Supplemental Figure 3A). There were no significant differences in the rate of change of the chorea score between any of the groups (Supplemental Figure 3B).
Next, we investigated the annualized rate of change of all 31 subscales that make up the UHDRS amongst the JOHD subjects only. After correcting for multiple comparisons, none of the chorea subscales changed significantly over time amongst the JOHD subjects (Supplemental Table 2; Figure 1A). The ten variables that had the most significant change over time were dysarthria (t=8.38), dystonia: LUE (t=6.73), gait (t=6.55), tandem walking (t=6.46), pronate/supinate: R hand (t=6.38), dystonia: RUE (t=6.04), finger tapping: R hand (t=5.98), finger tapping: L hand (t=5.59), tongue protrusion (t=5.52), and pronate/supinate: L hand (t=5.19). These 10 variables were summed to create a new JOHD Scale (maximum possible score of 40). We then evaluated the annualized rate of change of this new JOHD Scale relative to the TMS. Additionally, we created a simplified motor variable that included all the TMS subscales, except for the chorea scores, given that none of these variables changed significantly over time amongst JOHD individuals. This variable is referred to as the Abbreviated Motor Scale and has a maximum possible score of 96 points.
Figure 1.
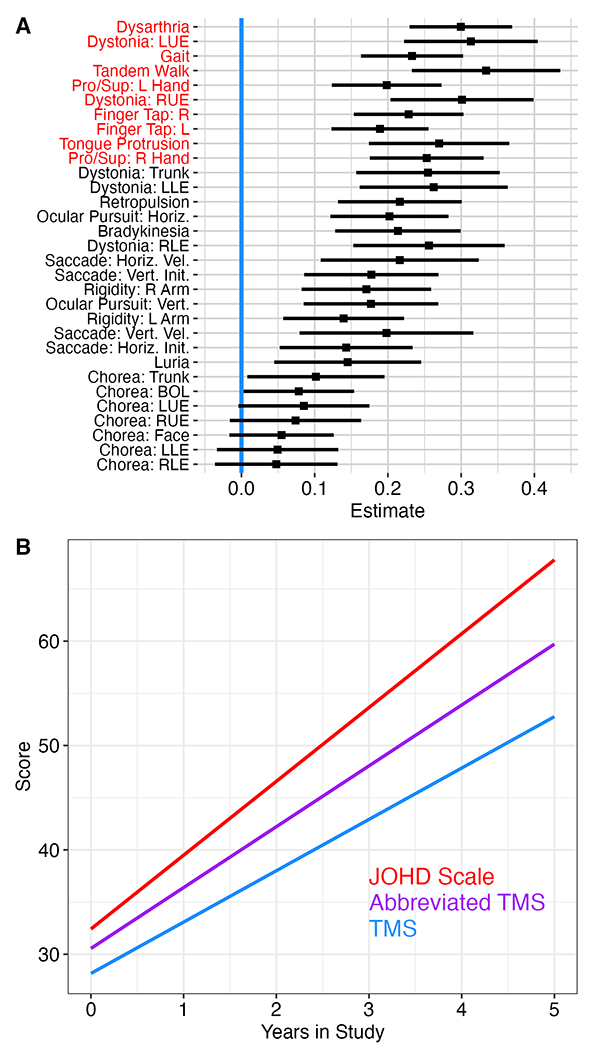
A) None of the chorea subscales showed significant change over time in JOHD participants after correcting for multiple comparisons. The variables highlighted in red represent the 10 variables with the largest t-values that were used to calculate the JOHD Scale.
B) The proposed JOHD Scale (red) changed at the fastest rate at more than 7% per year. The Abbreviated TMS Scale (purple) performed better than the TMS Scale (blue).
Given that the scale of each of these outcome variables was different, we standardized each of them to the percent of the maximum possible total score. For example, a participant with a TMS of 25 would have a standardized score of 19.5% ((25/128)*100); a score of 25 points on the JOHD scale, however, would equate to a standardized score of 62.5% ((25/40)*100). Based on these standardized scores, the mean annualized change in TMS score amongst JOHD participants was 4.92% (95% CI [3.95 – 5.89], p<0.0001). The mean annualized change in the abbreviated TMS was 5.83% (95% CI [4.77 – 6.90], p<0.0001). Lastly, the JOHD Scale changed at an annualized rate of 7.07% (95% CI [5.96 – 8.18], p<0.0001). These differences are outlined in Figure 1B.
We also assessed these three scales in the pHD and npJOHD groups. There was not a statistically significant difference in the trajectory of change between groups in the JOHD Scale (p=0.156), the Abbreviated TMS Scale (p=0.242), or the TMS (p=0.231).
We performed post-hoc sample-size analyses aimed to compare the number of participants required for a placebo-controlled clinical trial utilizing the TMS or the proposed JOHD Scale. Specifically, we propose a hypothetical, two-arm study measuring the average change of an outcome measure over one year. The goal would be to have 80% power to detect a 25% difference between groups using a simple, two-sample, two-sided, independent samples t-test with an accepted type 1 error rate of 5%. If the TMS scale was used as the primary outcome measure, the study would require 155 participants in each arm (total sample size of, at least, 310 participants). In contrast, this same study could be conducted with 99 participants per group (total sample size of, at least, 198 participants) if the proposed JOHD Scale was used.
DISCUSSION
The motor phenotype of JOHD is unique relative to patients with AOHD. We have replicated previous findings demonstrating that the TMS progresses at a significantly faster rate in patients with JOHD compared to patients with AOHD.[3] However, the rate of change of chorea scores are highly variable in patients with JOHD and do not predictably worsen in this rare group of patients. Consequently, we identified the ten UHDRS subscales that worsened most predictably over time in JOHD. Dystonia in the upper extremities, finger tapping, and pronation/supination of both hands, dysarthria, gait, and tandem walking measurements were the most predictive measures in JOHD. The sum of these 10 measures changed more than 7% per year in patients with JOHD compared to less than 5% per year for the TMS. In fact, the JOHD Scale changed at an annual rate that was nearly 44% faster relative to the annual rate of change of the TMS. Furthermore, there were no significant differences in the rate of progression of the JOHD Scale between patients with pHD and npJOHD. This suggests that this scale adequately tracks disease progression across the wide spectrum of phenotypes seen in patients with JOHD. Based on our findings, we simulated a hypothetical interventional study for patients with JOHD and found that use of the JOHD Scale as the primary outcome measure resulted in reduction in the total number of required participants by more than 33% compared to a similar study that used the TMS as the primary outcome measure. Consequently, this newly proposed JOHD Scale may represent a more clinically meaningful outcome measure that may be employed by both clinicians to monitor the progression of patients with JOHD over time as well as researchers who are planning clinical trials aimed as symptom management or disease modification in patients with JOHD.
There are limitations to this work. First, the possibility of recall bias exists. Subjects with manifest HD may be asked to recall their ACD. This may lead to inaccuracies in the data provided. Second, there is potential for a selection bias to occur. Diagnosis of JOHD can be difficult and prolonged [12]. Therefore, subjects in the Enroll-HD study who have JOHD may have received that diagnosis because of more significant or unique symptoms that precipitated a diagnosis. Despite these limitations, these results present new information for the HD community that may serve to better characterize motor progression in JOHD. The rarity of JOHD is a major challenge in the conduct of sufficiently powered clinical trials. As a result, there is a critical need to better characterize the motor phenotype of JOHD to develop outcome measures that most closely track with disease progression. The proposed JOHD Scale is a step towards this goal, but further validation is necessary.
Supplementary Material
ACKNOWLEDGEMENTS
Enroll-HD is a longitudinal observational study for Huntington’s disease families intended to accelerate progress towards therapeutics; it is sponsored by CHDI Foundation, a nonprofit biomedical research organization exclusively dedicated to developing therapeutics for HD. Enroll-HD would not be possible without the vital contribution of the research participants and their families.
This work was supported by the National Institutes of Health (NIH) (K23-NS117736 to JLS).
Funding Sources and Conflict of Interest:
This work was supported by the National Institute of Neurological Disorders and Stroke (NIH K23-NS117736 to JLS). The funding agency had no part in data analysis or the preparation of this manuscript.
Financial Disclosures for the previous 12 months:
Dr. Schultz receives salary support from the Michael J. Fox Foundation for Parkinson’s Disease Research.
Dr. Killoran serves as principal site investigator for studies sponsored by CHDI, NINDS, Sage Pharmaceuticals, and UCB Pharmaceuticals. She receives salary support for this work.
No other authors report financial disclosures for the previous 12 months.
Footnotes
Author Disclosures
The authors report no potential conflicts of interest related to this work.
REFERENCES
- 1.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell, 1993. 72(6): p. 971–83. [DOI] [PubMed] [Google Scholar]
- 2.Roos RA, Huntington’s disease: a clinical review. Orphanet J Rare Dis, 2010. 5: p. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fusilli C, et al. , Biological and clinical manifestations of juvenile Huntington’s disease: a retrospective analysis. Lancet Neurol, 2018. 17(11): p. 986–993. [DOI] [PubMed] [Google Scholar]
- 4.Schultz JL, et al. , Longitudinal Clinical and Biological Characteristics in Juvenile-Onset Huntington’s Disease. Mov Disord, 2023. 38(1): p. 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cronin T, Rosser A, and Massey T, Clinical Presentation and Features of Juvenile-Onset Huntington’s Disease: A Systematic Review. J Huntingtons Dis, 2019. 8(2): p. 171–179. [DOI] [PubMed] [Google Scholar]
- 6.Reilmann R, Parkinsonism in Huntington’s disease. Int Rev Neurobiol, 2019. 149: p. 299–306. [DOI] [PubMed] [Google Scholar]
- 7.Stout JC, Juvenile Huntington’s disease: left behind? Lancet Neurol, 2018. 17(11): p. 932–933. [DOI] [PubMed] [Google Scholar]
- 8.Huntington Study G, Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology, 2006. 66(3): p. 366–72. [DOI] [PubMed] [Google Scholar]
- 9.Unified Huntington’s Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord, 1996. 11(2): p. 136–42. [DOI] [PubMed] [Google Scholar]
- 10.Landwehrmeyer GB, et al. , Data Analytics from Enroll-HD, a Global Clinical Research Platform for Huntington’s Disease. Mov Disord Clin Pract, 2017. 4(2): p. 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tereshchenko A, et al. , Brain structure in juvenile-onset Huntington disease. Neurology, 2019. 92(17): p. e1939–e1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ribai P, et al. , Psychiatric and cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients. Arch Neurol, 2007. 64(6): p. 813–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.