

Abstract
Introduction:
Exposures to particulate matter (PM) from combustion sources can exacerbate pre-existing asthma. However, the cellular and molecular mechanisms by which PM promotes the exacerbation of asthma remain elusive. We used a house dust mite (HDM)-induced mouse model of asthma to test the hypothesis that inhaled DCB230, which are PM containing environmentally persistent free radicals (EPFRs), will aggravate asthmatic responses.
Methods:
Groups of 8–10-week-old C57BL/6 male mice were exposed to either air or DCB230 aerosols at a concentration of 1.5 mg/m3 4 h/day for 10 days with or without prior HDM-induction of asthma.
Results:
Aerosolized DCB230 particles formed small aggregates (30–150 nm). Mice exposed to DCB230 alone showed significantly reduced lung tidal volume, overexpression of the Muc5ac gene, and dysregulation of 4 inflammation related genes, Ccl11, Ccl24, Il-10, and Tpsb2. This suggests DCB230 particles interacted with the lung epithelium inducing mucous hypersecretion and restricting lung volume. In addition to reduced lung tidal volume, compared to respective controls, the HDM+DCB230-exposed group exhibited significantly increased lung tissue damping and up-regulated expression of Muc5ac, indicating that in this model, mucous hypersecretion may be central to pulmonary dysfunction. This group also showed augmented lung eosinophilic inflammation accompanied by an up-regulation of 36 asthma related genes. Twelve of these genes are part of IL-17 signaling, suggesting that this pathway is critical for DCB230 induced toxicity and adjuvant effects in lungs previously exposed to HDM.
Conclusion:
Our data indicate that inhaled DCB230 can act as an adjuvant, exacerbating asthma through IL-17-mediated responses in a HDM mouse model.
Keywords: Environmentally persistent free radicals (EPFRs), particulate matter, inhalation, air pollution, asthma, oxidative stress
INTRODUCTION
According to the American Lung Association, in 2022, more than 18.8 million Americans lived in an area where the concentration of particulate matter with an aerodynamic diameter smaller than 2.5 µm (PM2.5) exceeded annual exposure limit of 12 µg/m3 (American Lung Association, 2022). Exposures to PM2.5 may contribute to the premature deaths of 200,000 Americans (Collins et al. 2022). Aggravation of respiratory diseases by environmental exposures is a leading cause of morbidity and mortality worldwide (Jiang et al. 2016; Chen et al. 2020). As the portal of entry to inhaled pollutants, the lungs are equipped with robust defense and repair mechanisms. Inhaled ultrafine particles (aerodynamic diameter smaller than 100 nm), however, have been shown capable of escaping macrophage phagocytosis, thus avoiding clearance mechanisms in the deep lungs, leading to increased particle-lung epithelial interactions (Hahn et al. 1977; Ferrin et al. 1992; Daigle et al. 2003; Stoeger et al. 2006). This is in contrast with inhaled PM2.5, which are mainly cleared by alveolar macrophage phagocytosis (Mack et al. 2000). Epidemiological data have shown that acute exposures to high levels of PM or sub-chronic/chronic exposures to lower levels of PM are associated with decreased lung function across the lifespan (Pope, 1989; Pope, 2000; Atkinson et al. 2001; Schwartz et al. 2004). The mechanism(s), however, responsible for inhaled PM-induced respiratory dysfunction are still poorly understood. Further, exposures to PM from combustion can exacerbate pre-existing respiratory diseases, including asthma (Li et al. 2009; Li et al. 2010; Saravia et al. 2014; Castaneda et al. 2017; Rice et al. 2018; Xu et al. 2020; Keirsbulck et al. 2023). Asthma is an inflammatory lung disease characterized by airflow obstruction and pronounced eosinophilic or neutrophilic influx in severe cases (Gurczynski and Moore, 2018; Xu et al. 2020). In the United States, asthma affects 25 million people, and worldwide it affects more than 334 million individuals (Gurczynski and Moore, 2018; Xu et al. 2020). Thus, the worldwide prevalence of asthma is high, making it a widespread chronic respiratory disease.
Adjuvant effects of PM on asthmatic responses have been described (Diaz-Sanchez et al. 1997; Gilliland et al. 2004; Steerenberg et al. 2004; Matsumoto et al. 2006; Kleinman et al. 2007; Stevens et al. 2008; Li et al. 2009). An adjuvant can be defined in immunology as a substance, molecule or particle that acts as an activator of innate immunity. In other words, PM is a substance that can amplify immune responses when present together with a specific antigen (Kuroda et al. 2013). Adjuvant effects of PM evidenced by the potentiating effect of an allergen (ovalbumin) were worse for PM collected in Europe in winter compared to the other seasons (Steerenberg et al. 2004). While oxidative stress often is regarded as the culprit in PM-elicited pulmonary dysfunction, health effects associated with environmental PM exposures are not fully explained by PM elemental constituents (Wang et al. 2014; Franklin et al. 2015). Another important factor of PM affecting respiratory health is particle size, with fine and ultrafine particles (50 to 200 nm) producing a stronger adjuvant effect than do micron-sized counterparts, with sizes between 2,000 and 20,000 nm (Coban et al. 2010a and 2010b). Thus, based on PM sizes, differing immune-pulmonary responses have been observed in animal models (Xu et al. 2020). Other PM constituents, including the oxidant potential of PM, have been investigated (Balakrishna et al. 2009; Li et al. 2009; Balakrishna et al. 2011; Saravia et al. 2014; Harding et al. 2021). The oxidant potential of particles has also been shown to impact particle-induced immune-inflammatory responses (Balakrishna et al. 2009; Li et al. 2009; Balakrishna et al. 2011; Kuroda et al. 2013; Saravia et al. 2014). In a mouse model with ovalbumin sensitization, aggravation of respiratory effects was induced by redox chemicals present on the surface of PM (Li et al. 2009). Thus far, the adjuvant effects of PM have been shown to depend on particle size, chemical composition, including oxidant potential, and source or geographical origin of the PM (Steerenberg et al. 2004). The mechanisms, however, by which the radical content of PM leads to adjuvant effects during ongoing allergen sensitization remain unclear.
Environmentally persistent free radicals (EPFRs) are emerging pollutants studied by our research groups and others worldwide (Dellinger et al. 2000; Dellinger et al. 2001; Valavanidis et al., 2006; Lomnicki et al. 2008; Khachatryan et al. 2011; Yang et al. 2015; Jia et al. 2017; Vejerano et al. 2018) for over two decades. EPFRs are organic pollutants chemically bound to particles through a transition metal, resulting in complex pollutant-particle systems with a surface-stabilized radical. These pollutant-particle systems are also known as semiquinone radicals (Valavanidis et al., 2006; Yang et al. 2015; Jia et al. 2017). Most importantly, our research group has demonstrated that EPFRs form and persist on airborne PM near industrial sites (Gehling et al. 2015). EPFRs are an understudied type of airborne pollutant that have previously been shown to affect asthma pathogenesis (Saravia et al. 2014; Harding et al. 2021). Recently we reported that mice inhaling ultrafine EPFRs for 10 days exhibited significant endothelial dysfunction and reduced lung function, without any signs of pulmonary or systemic inflammation (Harmon et al. 2021). Previously we demonstrated that EPFRs affect P450 function, which plays a key role in the induction of oxidative stress responses through the generation of reactive oxygen species (ROS) (Balakrishna et al. 2009; Balakrishna et al. 2011; Harmon et al. 2018). In addition, our group showed that short-term inhalation exposures to EPFR-aerosols initiates inflammatory responses in the lungs of rodents (Balakrishna et al. 2009; Balakrishna et al. 2011). These lung responses were characterized by an influx of inflammatory cells in the bronchoalveolar lavage fluid (BALF), increased smooth muscle mass in the peribronchial regions, and increased bronchial hyperreactivity in response to methacholine challenge (Balakrishna et al. 2009; Balakrishna et al. 2011). All of these outcomes suggest that EPFR exposure may be partially responsible for the observed worldwide increase in cases of asthma and asthma exacerbations.
Currently, the cellular and molecular mechanisms by which PM promotes the onset or exacerbation of asthma remain elusive. The majority of previous studies investigating the adjuvant effects of PM on allergen sensitization (mainly ovalbumin) used the intranasal or intratracheal instillation routes to administer both the PM as well as the allergens. Thus, very few studies (Li et al. 2010; Saravia et al. 2014) used the inhalation route to expose rodents to PM, as would occur in real-life exposure scenarios. Whether EPFR contained on PM are a trigger for aggravation of asthma, delay the resolution of asthmatic responses or increase the risk and/or sensitivity for asthma onset is currently unknown. Nonetheless, the EPFR component of PM may disrupt the redox homeostasis in the lungs and lead to increased susceptibility to an allergen exposure. Our current model was designed to test whether exposures to EPFR containing PM during the resolution phases of asthmatic responses would promote a microenvironment facilitating exacerbation of pulmonary inflammation. We used a house-dust mite (HDM)-induced mouse model of asthma to test the hypothesis that inhaled ultrafine PM containing EPFRs will cause lung damage, leading to reduced lung function and aggravation of asthmatic responses. Our study is doubly innovative. First, we used the inhalation route to expose mice to PM containing EPFRs, and second, we used HDM as an allergen, and both the exposure route and the allergen that we used are relevant to human real-world exposure scenarios, thus allowing for a better understanding of the role of inhaled EPFR on asthmatic responses.
METHODS
General experimental study design
We used 8- to 10-week-old C57BL/6 male mice purchased from Jackson Laboratory (Bar Harbor, ME). For these first sets of studies, we elected to use only male mice since recent reports revealed male dominated sex-based differences regarding exposures to PM2.5 and subsequent cardiovascular responses (Kuzma et al. 2020; Xia et al. 2021). We exposed the mice to PM containing EPFRs, which are PM consisting of 1, 2-dichlorobenzene chemisorbed at 230 °C to copper oxide (CuO)/silica particles, with a primary particle size of 20 – 40 nm, commonly named DCB230. Two groups of randomly assigned C57BL/6 male mice (n = 7 – 14 per group) were whole-body exposed to DCB230 aerosols at a targeted total particulate matter (TPM) concentration of 1.5 mg/m3, 4 h/day for 10 days with or without previous HDM sensitization. Similarly, two additional groups were exposed to filtered air with or without HDM (n = 7 – 14 per group) (Figure 1). The targeted concentration of 1.5 mg/m3 was chosen to represent high PM2.5 exposure levels measured in China, with levels reaching a maximum of 994 µg/m3, which was recorded in 2012 in Beijing, China (San Martini et al. 2015). Thus, while high, this exposure concentration is environmentally relevant since similar levels are observed in very populated cities throughout Asia (San Martini et al. 2015). As we reported previously (Harmon et al. 2021), the mice were exposed to EPFR content in the range of 1.8e13 – 5.3e13 EPFR/m3. These EPFR levels are also environmentally relevant as an EPFR ambient air concentration range of 1×1017 – 4×1019 spins/g, equivalent to 3.9e12 – 1.3e15 EPFR/m3, was observed in PM from Memphis, TN (Harmon et al. 2021). We previously reported the exposure data as well as the cardiopulmonary responses of the mice exposed for 10 days to either solely filtered air or DCB230 aerosols alone, in a separate publication (Harmon et al. 2021). In the current study, we are presenting additional and complementary data regarding the mice that were exposed to HDM for 3 weeks followed by exposures to either filtered air or DCB230 aerosols for 10 consecutive days (Figure 1). The HDM was purchased from Stallergenes Greer (Lenoir, NC; D. pteronyssinus, catalogue number: XPB70DA2.5). HDM powder was suspended in saline. Each HDM treated mouse received 50 µg of HDM per instillation. The mice were anesthetized with isoflurane and 50 µg of HDM was administered via intranasal instillation of 10 μL per nostril of the HDM solution. This HDM treatment was administered once a week for three consecutive weeks, as previously described (Noël et al. 2021). Mice were humanely euthanized on day 10 immediately after the last 4-hour exposure to either air or DCB230. All mice were housed in an AAALAC-approved animal care facility at the Louisiana State University School of Veterinary Medicine under a 12-hour light/dark cycle (from 6:00 am to 6:00 pm). The mice had access to water and food ad libitum, except during the 4-hour exposure periods. Mice were housed and handled in accord with the NIH Guide for the Care and Use of Laboratory Animals. All procedures and protocols were approved by the Louisiana State University Institutional Animal Care and Use Committee.
Figure 1.
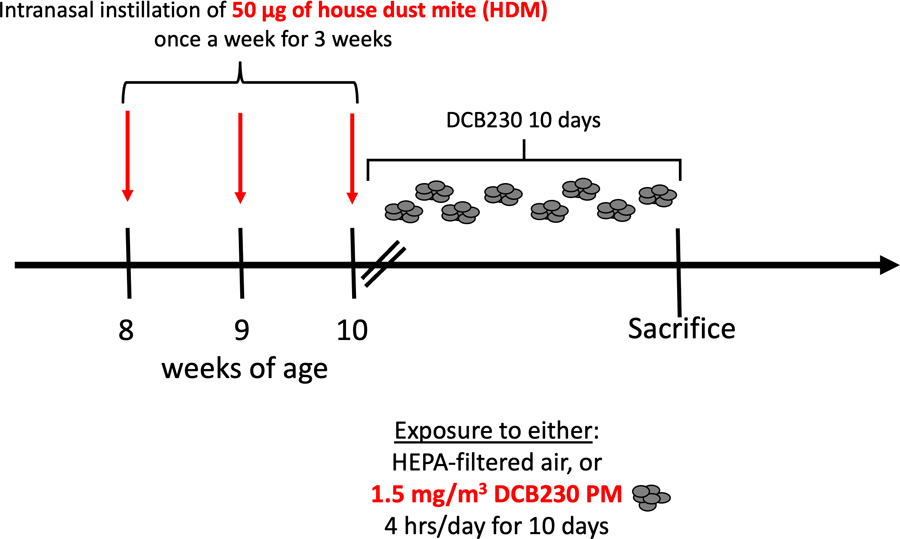
Schematic of the overall experimental study design.
Animal exposures to DCB230 aerosols
We used an integrated aerosol whole-body inhalation exposure system to closely mimic, in mice, human exposures to EPFRs, and to evaluate aggravation of asthmatic responses and pulmonary dysfunction induced by sub-acute (10 days) exposures to DCB230 aerosols. Mice were exposed in an 18 L inhalation chamber where a maximum of 16 mice can be exposed at a time. The mice are housed in individual compartments of a stainless-steel mesh cage placed inside the chamber, enabling exposures to 1.5 mg/m3 of DCB230 particles for 4 hours a day for 10 consecutive days. Control mice were exposed concurrently to filtered air in an identical 18 L chamber. These exposure set-ups were previously described (Harmon et al. 2021; Aryal et al. 2023). In brief, we placed 130 mg of DCB230 particles in a small glass container connected to a Venturi via a Tygon tube. Thus, aerosols composed of DCB230 particles were generated by a Venturi-based dry-particle dispersion technique, dispersing the aerosol at the top of the exposure chamber. Dilution air with average flow rates of ~ 5 L/min was added to create a mixing effect inside the exposure chamber. The aerosol from the chamber was exhausted at flow rates ranging between 2 and 4 L/min. The TPM mass concentration inside the exposure chamber was assessed off-line and in real-time, using gravimetric measurements and a DustTrak II (Model 8530, TSI Inc., Shoreview, MN), respectively. The gravimetric concentration was determined by sampling and collecting DCB230 aerosols throughout the 4-hour exposure on 25 mm diameter glass fiber filters (Catalog #AP4002500, Millipore Sigma, Burlington, MA). The filters were weighed before and after each daily exposure on a Sartorius MC5 microbalance. We also sampled and collected aerosolized DCB230 particles on transmission electron microscopy (TEM) precoated carbon Formvar copper grids (Catalog #01810, Ted Pella, Inc., Redding, CA), which were lightly glued onto a glass fiber filter placed inside a cassette. TEM images acquired using a JEOL JEM-1010 microscope were used to evaluate the morphology and the structure of the DCB230 particle agglomerates. Chamber temperature and relative humidity were monitored continuously during the daily 4-hour exposures. A summary of the exposure characterization data is presented in Figure 2.
Figure 2. Characterization of DCB230 particulate matter (PM).
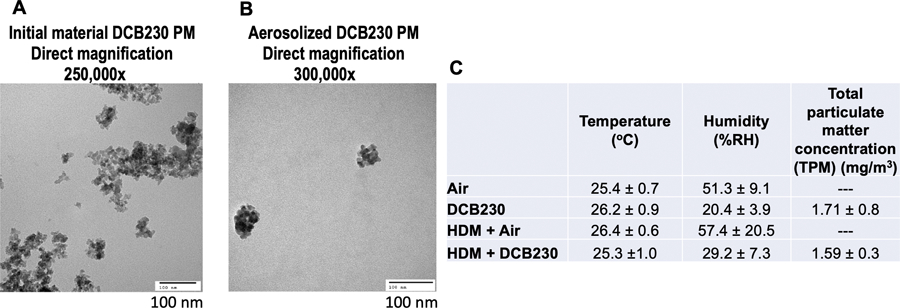
A) Initial DCB230 PM prior to aerosolization. B) Aerosolized DCB230 form small aggregates (< 150 nm). Images obtained by transmission electron microscopy (TEM) at 250,000x and 300,000x magnification. Scale bar = 100 nm. C) DCB230 PM exposure characterization during animal studies. Average values ± standard deviation (SD).
Lung function testing
After the end of the last 4-hour exposure on day 10, we conducted lung function testing using two distinct approaches. First, we evaluated lung function via plethysmography in mice from all four groups (n = 7 – 8 mice per group). A whole-body plethysmograph from Buxco (Troy, NY) was used to record lung function parameters every minute for 5 minutes. These measurements were then used to assess lung tidal volume, as described (Harmon et al. 2021). Second, we performed invasive lung function testing using the flexiVent system (SCIREQ, Montreal, QC, Canada) to accurately determine lung parameters in both HDM-treated groups, as described (Noël et al. 2021). Airway function was measured in ketamine and xylazine anesthetized mice (n = 6 per group). Each anesthetized mouse was connected to a ventilator for approximately 30 min. Lung function was assessed using the single-compartment and the constant-phase models to determine lung resistance as well as lung tissue damping. These parameters were evaluated through a dose-response curve based on challenge with the bronchoconstrictor methacholine, which was delivered in incremental concentrations (0, 12.5, 25, and 50 mg/mL of methacholine dissolved in saline). After the invasive lung function procedure, the mice were humanely euthanized with an intraperitoneal injection of Beuthanasia-D (Schering-Plough, NJ) (0.1 mL/10 g of body weight). No mice that were used to assess lung function with the flexiVent were used for any subsequent tissue collection and analyses.
Broncho-alveolar lavage fluid (BALF) collection and cytology analyses
Immediately following the last 4-hour exposure to either air or DCB230 on day 10, the mice that were not assessed with the flexiVent were humanely euthanized by an intraperitoneal injection of Beuthanasia-D (Schering-Plough, NJ) (0.1 mL/10 g of body weight). Following euthanasia, a small incision was made in the trachea of each mouse, a cannula was inserted, and we lavaged the lungs twice with 0.5 mL of saline. The BALF was pooled and collected on ice prior to being processed for cytology analysis. BALF slides were prepared using a cytospin and cells were stained with a HEMA-TEK automated slide stainer (modified Wright’s stain) (Catalog #23-036222, Fisher Scientific, Waltham, MA). A board-certified clinical pathologist counted 300 cells on each slide to determine the BALF differential cell counts.
Lung histopathology assessment
Following BALF collection, the right lung was inflated with 0.02 M periodate, 0.1 M lysine, 0.25% paraformaldehyde (PLP) fixative in phosphate buffer (pH 7.4). Subsequently, using standard histology processes, the lung tissues were sectioned and stained using hematoxylin and eosin (H&E). A board-certified veterinary pathologist conducted the histopathology assessment of the lung tissues for both HDM treated groups. The lung tissue slides were evaluated based on the following criteria: overall inflammation, bronchial eosinophils, bronchial neutrophils, bronchial lymphocytes, alveolar inflammatory cells, bronchial damage, and mucous hyperplasia. Each of the criteria were assessed based on an arbitrary scale of 0 to 4. 0 = normal; 1= minimal; 2 = mild; 3 = moderate; 4 = severe.
Lung RNA extraction and gene expression through PCR array
After euthanasia and BALF collection, a small piece of the left lung was placed in RNA Later (Catalog #76106, Qiagen, Germantown, MD) and stored at −80°C for subsequent RNA extraction. We isolated the RNA from the lungs of mice using the Qiagen miRNeasy Mini Kit (catalog #7217004, Qiagen, Germantown, MD) with Trizol/chloroform extraction, following the manufacturer’s recommendations. We used a NanoDrop ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE) to determine the RNA concentration and purity.
As described in Harmon et al. (2021), we evaluated lung gene expression using a RT² Profiler™ PCR Array from Qiagen. We followed the manufacturer’s recommended procedures to prepare cDNA and for the preparation of the Master mix, using the RT2 First Strand Kit (catalog #330401, Qiagen, Germantown, MD) and the RT2 SYBR Green qPCR Master mix (catalog #330503, Qiagen, Germantown, MD), respectively. We used the RT² Profiler™ PCR Array for Mouse Allergy & Asthma (Catalog #330231 PAMM-067Z; Qiagen, Germantown, MD) to evaluate the expression of 84 genes related to asthmatic responses. We followed the manufacturer’s recommendation to run the samples on an Applied Biosystems 7300 Real Time PCR System. The resulting threshold cycle (Ct) values were analyzed using the Qiagen GeneGlobe data analysis software, which expressed the results for each gene analyzed using the ΔΔCt method. Results are presented as fold-changes of the treatment groups (DCB230, HDM + Air, and HDM + DCB230) compared to the Air group.
Ingenuity pathway analysis
Using the lung gene expression results we obtained through the PCR array, we analyzed genes that were differentially expressed (fold-change > |1.5|) in the HDM + DCB230 group using Qiagen’s Ingenuity Pathway Analysis (IPA) software. This enabled the identification of networks and canonical pathways associated with the dysregulated genes.
Protein extraction and Western blot
After euthanasia and BALF collection, a small piece of the left lung was flash-frozen on dry ice and stored at −80°C for subsequent protein extraction. As previously described in Noël et al. (2016), to extract the proteins, the lung tissue samples were mechanically broken by lightly hammering the samples that were placed in aluminum foil and snap-frozen in liquid nitrogen. The samples were then rapidly transferred into test tubes containing 300 μL of RIPA lysis buffer (Santa Cruz Biotechnology, Dallas, TX, USA), and three 2.5 mm zirconia/silica beads (Biospec Products Inc.). Subsequently, we used a TissueLyser II (Qiagen, USA) at a frequency of 25 MHz for 2 min to completely lyse the tissue samples. Lysed samples were centrifuged (13,000 g for 5 min at 4 °C) and the protein concentration was measured in the supernatant using a BCA protein assay kits (Thermo Scientific, Waltham, MA, USA). Remaining supernatant was stored at −80 °C.
Fifteen micrograms of extracted proteins from each sample were analyzed by Western blotting, as previously described in Noël et al. (2016). Briefly, extracted proteins were incubated in Laemmli sample buffer (Bio Rad Laboratories Inc., Hercules, CA, USA) containing 2-mercaptoethanol at 95 °C for five minutes. Proteins were then resolved by SDS-PAGE on an Any kD™ Mini-PROTEAN® TGX™ Gel (Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 100 volts, then resolved proteins were transferred onto PVDF membranes (Immobilon-P, pore size of 0.45 μm, Millipore Inc., USA) using the Trans-Blot Turbo™ Transfer System (Bio-Rad Laboratories), before detection with antibodies (ACTB as control, and IL-17; both from Cell Signaling Technology Inc., Danvers, MA, USA). The images of blots were captured with ChemiDoc™ Touch imaging system (Bio-Rad Laboratories), and captured images were analyzed with Image Lab 5.2 (BioRAD Laboratories).
In situ hybridization to label IL-17A transcripts using RNAscope
The lung tissues collected for histopathology assessment (as described above) were embedded in paraffin. RNA chromogenic in situ hybridization for the labeling of IL-17A transcripts was performed in 5 µm paraffin-embedded lung section slides according to manufacturer’s instructions, utilizing an RNAScope mouse probe for IL-17A in C1 (green) (#319571) and RNAscope 2.5HD duplex reagent kit (#322430) (ACDBio, Newark, CA). Slides were mounted using VectaMount permanent mounting medium (H-5000, Vector Labs, Burlington, Canada) and images were captured using a NanoZoomer 2.0-HT slide scanner (Hamamatsu Photonics, Japan).
Statistical procedures
We used GraphPad Prism software to perform the statistical analyses. The data for lung function, BALF cytology, gene and protein expression are expressed as mean ± standard error of the mean (SEM). When analyzing the results between the two HDM treated groups, we used a Student t-test, while a one-way analysis of variance (ANOVA) followed by a Tukey’s post-hoc test, was used when the results from the 4 groups (Air, DCB230, HDM + Air, HDM + DCB230) were compared. The histopathology results are expressed as median ± interquartile range, and we used the Mann-Whitney U test to examine statistical differences between the two HDM treated groups. For all assays, results were considered statistically significant with a p-value < 0.05 or a fold-change > | 1.5 | for the PCR array gene expression data.
RESULTS
Inhaled ultrafine PM containing EPFRs results in a decline in lung function and aggravates HDM-induced lung tissue damping
TEM characterization of the initial DCB230 particles (starting material) showed that these PM had a primary particle size between 20 – 40 nm and agglomerated into open structures, reaching sizes up to 200 nm (Figure 2A). TEM images of the aerosolized DCB230 particles sampled inside the inhalation chamber showed that these particles formed small aggregates mainly in the ultrafine size range (30 up to 150 nm) (Figure 2B), suggesting that the aerosolization process fragments these agglomerates of particles into smaller structures. We found that the inhalation of these DCB230 particles for 4 hours per day for 10 consecutive days, significantly reduced the tidal volume of mice, as previously reported in Harmon et al. (2021) (Figure 3A). Further, this decrease in tidal volume was maintained in the mice exposed for 3 weeks to the HDM allergen prior to the inhalation of DCB230 particles for 10 days (Figure 3A). Thus, there was no additive or synergetic effect on the lung tidal volume associated with the HDM sensitization plus the inhalation of DCB230 particles. With respect to invasive lung function measures in the HDM treated groups, although the respiratory system resistance increased with incremental doses of methacholine, there was no significant differences between the two HDM groups (Figure 3B). We found, however, that the additional exposure to DCB230 particles significantly increased the values of lung tissue damping at the methacholine doses of 25 and 50 mg/mL (Figure 3C). Thus, while we did not observe significant differences in the overall constriction level (i.e., resistance) of the lungs, we did observe a significant increase in tissue damping, a lung parameter associated with lung tissue resistance, that mirrors the energy dissipated in the deep lungs (Darrah et al. 2016; Dylag et al. 2020). Together, these data show that inhaled ultrafine PM containing EPFRs triggered an aggravated lung physiological response when pre-exposed to HDM allergens.
Figure 3. DCB230 PM decreased lung function in male mice.
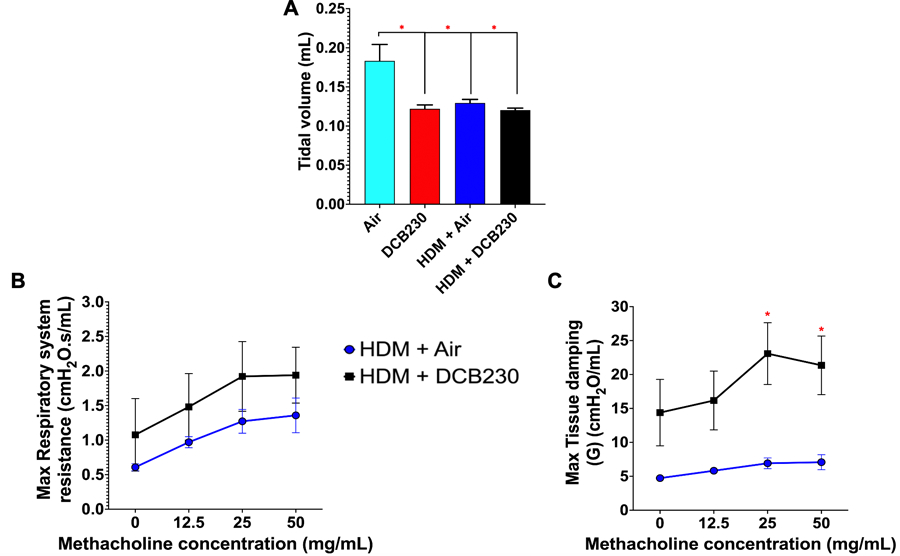
A) DCB230 PM significantly reduces tidal volume. Lung function obtained via whole-body plethysmography. N = 7 – 8 mice per group. ANOVA followed by a Tukey post-hoc test. * p < 0.05. B) DCB230 PM does not significantly affect the respiratory system resistance in an HDM-induced mouse model of asthma. C) HDM + DCB230 PM significantly increases lung tissue damping in an HDM-induced mouse model of asthma. Lung function data obtained via the flexiVent. N = 6 per group. Student t-test. * p < 0.05. All data are expressed as mean ± standard error of the mean (SEM).
Inhaled ultrafine PM containing EPFRs exacerbates HDM-induced eosinophilic inflammation
As reported in Harmon et al. (2021), we showed that whole-body inhalation of DCB230 for 10 days does not induce any significant influx of inflammatory cells in the lungs (Figure 4). We used a HDM treatment paradigm known to induce an asthmatic phenotype (Noël et al. 2021) to sensitize mice to the HDM allergen prior to the exposure to the DCB230 particles. We found that HDM sensitization for three weeks followed by exposure to air for 10 days resulted in a not significant increase of the percentage of leukocytes in the BALF (lymphocytes 1.1% ± 0.5; neutrophils 1.0% ± 0.5, and eosinophils 3% ± 2) (Figure 4), suggesting that the HDM-induction of asthma was in the resolution phase at the time of assessment. However, when this HDM sensitization was followed by the inhalation of DCB230 particles we observed a significantly increased percentage of eosinophils in the BALF (39% ± 12) (Figure 4). Thus, it is possible that the residual HDM sensitization in conjunction with the PM-induced inflammation triggered the adjuvant effect. Overall, these results suggest that inhaled ultrafine PM containing EPFRs induced an eosinophilic inflammatory reaction and acted as an adjuvant, i.e., enhanced the inflammatory responses to a prior allergen sensitization, which exacerbated the asthma and/or delayed its resolution.
Figure 4. HDM + DCB230 PM aggravated eosinophilic pulmonary inflammation in male mice.
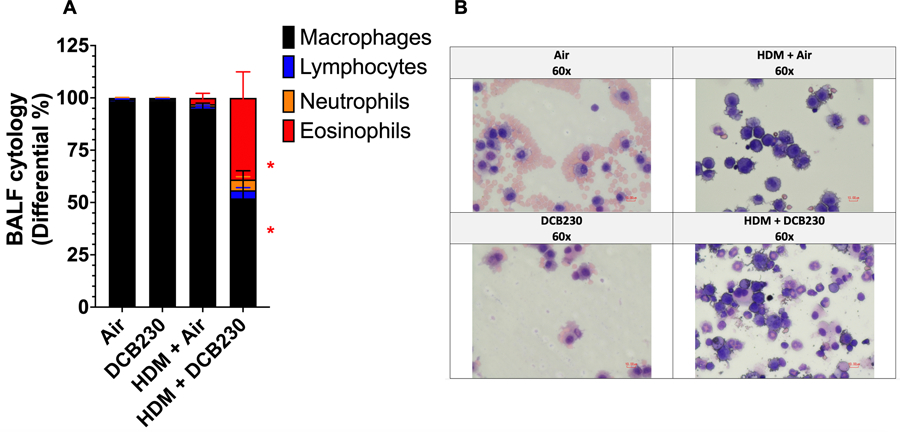
A) HDM+DCB230 PM significantly increases the percentage of eosinophils in the broncho-alveolar lavage fluid (BALF) of exposed mice. B) Representative images of BALF slides. Images obtained by light microscopy at 60x magnification. N = 7 – 8 mice per group. ANOVA followed by a Tukey post-hoc test. * p < 0.05. All data are expressed as mean ± standard error of the mean (SEM).
Inhaled ultrafine PM containing EPFRs worsens HDM-induced lung damage
We previously showed normal lung architecture in mice following exposures to DCB230 particles for 10 days (Harmon et al. 2021). In the allergen model described here, with HDM sensitization followed by 10 days of exposure to air, we observed minimal changes of the lung structure compared to controls, in terms of inflammatory cell recruitment, as evidenced by a histopathology score of 2 for overall inflammation and bronchial eosinophils (Figure 5). For the mice sensitized to HDM and subsequently exposed to DCB230, we had a significantly increased histopathology score of 3 for overall inflammation, as well as high histopathology scores for bronchial eosinophils (score of 3), bronchial lymphocytes (score of 2.5), alveolar inflammatory cells (score of 2) and mucous hyperplasia (score of 1), when compared to the HDM + Air group (Figure 5). Overall, the lung tissue data (Figure 5) support the inflammatory responses observed in the BALF (Figure 4). These data suggest that inhaled ultrafine PM containing EPFRs exacerbated inflammatory cell infiltration in the lung parenchyma of mice previously sensitized to the HDM allergen.
Figure 5. DCB230 PM aggravated HDM-induced lung damage in male mice.
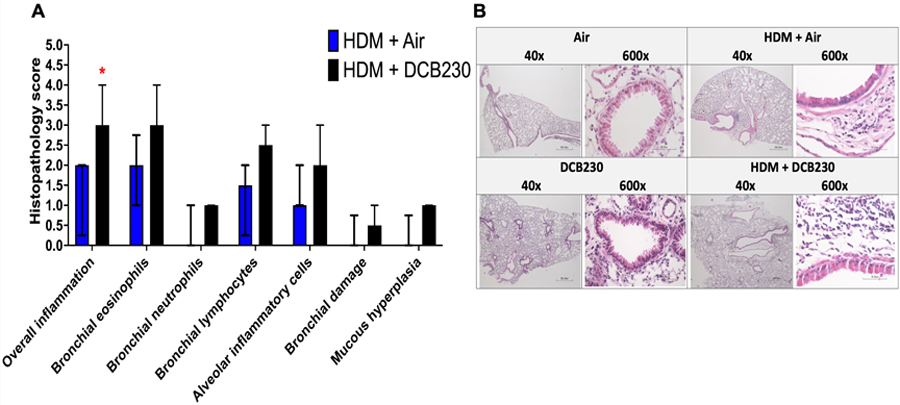
A) Histopathology score scale : 0 = normal; 1= minimal; 2 = mild; 3 = moderate; 4 = severe. B) Representative images of lung tissue slides stained with H&E. Images obtained by light microscopy at 40x and 600x magnification. N = 8 mice per group. Mann Whitney U test performed between two HDM treated groups. * p < 0.05. Data are expressed as median ± interquartile range.
Following HDM sensitization, inhaled ultrafine PM containing EPFRs up-regulate asthma-related genes focused on the IL-17 pathway
We previously showed that daily inhalation of DCB230 particles for 10 days up-regulated Cyp1b1 lung gene expression by ~2-fold (Harmon et al. 2021). Cyp1b1 is a gene involved in the biotransformation of xenobiotics and whose transcription is activated by AhR, a signaling pathway central to oxidative stress responses (Jacob et al. 2011; Kim et al. 2020). This suggests that the up-regulation of the Cyp1b1 lung gene can be considered a biomarker of exposure to inhaled DCB230 particles, which are ultrafine PM that contain free radicals. In addition, using a PCR array evaluating the expression of 84 genes related to allergy and asthma, compared to the air group, we found that 10 days of exposure to DCB230 alone up-regulated the lung gene expression of Ccl11 (1.6-fold) and Ccl24 (1.6-fold; (Figure 6A), which are two protein-coding genes for eosinophil chemoattractants (Isgro et al. 2013; Wu et al. 2014). Further, DCB230 exposure also increased the lung gene expression of tryptase beta 2 (Tpsb2) (2.9-fold) (Figure 6A), a gene coding for a mast cell serine protease that plays a role in stimulating bronchoconstriction (Choy et al. 2018). This was accompanied by the up-regulation of Il-10 (1.6-fold) (Figure 6A), a cytokine that has both pro and anti-inflammatory properties and is implicated in airway hyperresponsiveness (Makela et al. 2000), as well as Muc5ac (2.3-fold) (Figure 6A), a gene coding for secretory mucins of the tracheo-bronchial region, indicative of mucus hypersecretion (Wang et al. 2012). Taken together, these molecular effects suggest that inhaled DCB230 particles alone can affect lung function, as evidenced physiologically by decreased lung tidal volume (Figure 3A) and up-regulation of Tpsb2, Il-10, and Muc5ac (Figure 6A). The observed up-regulation of Ccl11, Ccl24 and Il-10 (Figure 6A) furthermore suggest an increased susceptibility for aggravated eosinophilic asthmatic responses (Figure 4).
Figure 6. HDM + DCB230 PM up-regulated multiple asthma-related genes in male mice.
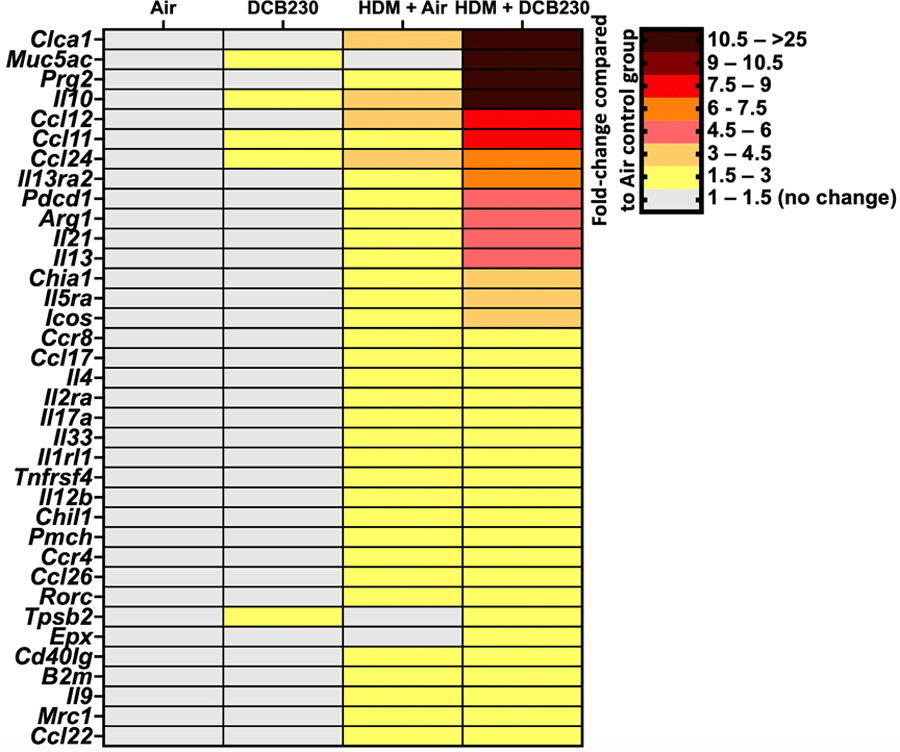
Heatmaps of significantly dysregulated genes assessed by a PCR array specific for 84 genes related to allergy & asthma. N = 4 mice per group. Results are presented as fold-changes of the treatment groups (DCB230, HDM + Air, and HDM + DCB230) compared to the Air group. Results were considered statistically significant with a fold-change > | 1.5 |.
Regarding the HDM treated groups, we found that 3 weeks of sensitization to the HDM allergen followed by 10 days of exposure to air up-regulated the expression of 33 genes related to allergy and asthma in the lungs, with fold-changes ranging from 1.5 to 3.7 compared to the air group (Figure 6A). HDM sensitization followed by 10 days of exposure to DCB230 particles up-regulated the expression of 36 genes in the lungs, with fold-changes extending from 1.5- to greater than 25-fold compared to the air group (Figure 6A). Although the up-regulated expression of 33 genes related to allergy and asthma were common to both HDM groups (Figure 6A), importantly, for 29 of those genes (87.9%), the magnitude of up-regulation was greater in the mice exposed to HDM followed by the inhalation of DCB230 particles (Figure 6A). Further, when comparing the HDM + Air and HDM + DCB230 groups, 3 genes were uniquely up-regulated in the HDM + DCB230 group, namely Muc5ac, Tpsb2, and eosinophil peroxidase (Epx) (Figure 6A). Muc5ac and Tpsb2 carried over from the baseline lung molecular effects of inhaling DCB230 particles alone for 10 days (Figure 6A), while Epx is a protein-coding gene expressed in eosinophils (Percopo et al. 2019). Thus, the molecular signature induced by HDM sensitization followed by inhalation of DCB230 particles (Figure 6A) supports the lung function (Figure 3), BALF cytology (Figure 4), and lung tissue damage (Figure 5) results we observed when comparing both HDM treated groups. Overall, these data suggest that inhaled ultrafine PM containing EPFRs enhanced the up-regulation of asthma-related genes in the lungs of mice that were pre-sensitized to HDM.
To better understand these genotypic changes, we conducted IPA analyses to elucidate the molecular mechanisms underlying the aggravated asthmatic phenotype we observed when mice were first sensitized to HDM and then exposed by inhalation to DCB230 particles (Figure 3 to 6). We evaluated the networks associated with the 36 up-regulated genes in the HDM + DCB230 group (Figure 7). Based on the IPA results, the main canonical pathways that were up-regulated in the HDM + DCB230 group included the IL-17 pathway (12 up-regulated genes), the T helper cell differentiation pathway (10 up-regulated genes), the airway inflammation in asthma pathway (10 up-regulated genes), and the Th2 pathway (12 up-regulated genes) (Figure 7). While IL-17 pathways have been associated with severe neutrophilic asthma (Nakajima and Hirose, 2010), activation of Th2 pathways are related to eosinophilic asthma (Georg and Brightling, 2016). More recently, however, it was demonstrated in a mouse model of asthma that dysregulated genes associated with both Th17 (IL-17) and Th2 pathways led to eosinophilic inflammation (Choy et al. 2015), thus supporting our results (Figures 4 to 6) at the lung tissue, cellular and molecular levels.
Figure 7. HDM + DCB230 PM exposure affected lung molecular networks associated with Th2 and Th17 responses in male mice.

The significantly dysregulated genes assessed by PCR array were used to assess the canonical pathways involved in the HDM + DCB230 PM pulmonary responses using Ingenuity Pathway Analysis (IPA). Red denotes up-regulation, i.e., the expression of this gene was up-regulated compared to the respective air control group.
Since the HDM + DCB230-exposed mice had 5.1% ± 1.3 of neutrophils in the BALF (Figure 4) and a 2.5-fold increase in the gene expression of Il17a (Figure 6), added to the fact that IL-17 is associated with increased severity of asthmatic responses, we further investigated the protein expression of IL-17 in the lungs (Figure 8A–C), as well as the pulmonary localization of mRNA transcript for Il-17a using in situ hybridization (RNAscope) (Figure 8D). This enabled us to better characterize the role of inhaled DCB230 particles in the severity of the aggravated asthmatic response we observed following sensitization to the HDM allergen. We found that the protein expression of IL-17 in the lungs of the mice exposed to HDM + DCB230 was 1.8-fold higher than in the lungs of mice exposed to HDM + Air (Figure 8C). Also, since each green punctum of the in situ hybridized lung tissue represents one transcript of Il-17a, it appears that the main source of IL-17a in our model are the bronchial epithelial cells (Figure 8D). Overall, these results suggest that following HDM sensitization, inhaled ultrafine PM containing EPFRs led to asthmatic responses, which seem to be associated with the activation of IL-17 molecular pathways (Figures 6 – 8).
Figure 8. HDM + DCB230 PM exposure increased the protein expression of IL-17 compared to HDM + Air.
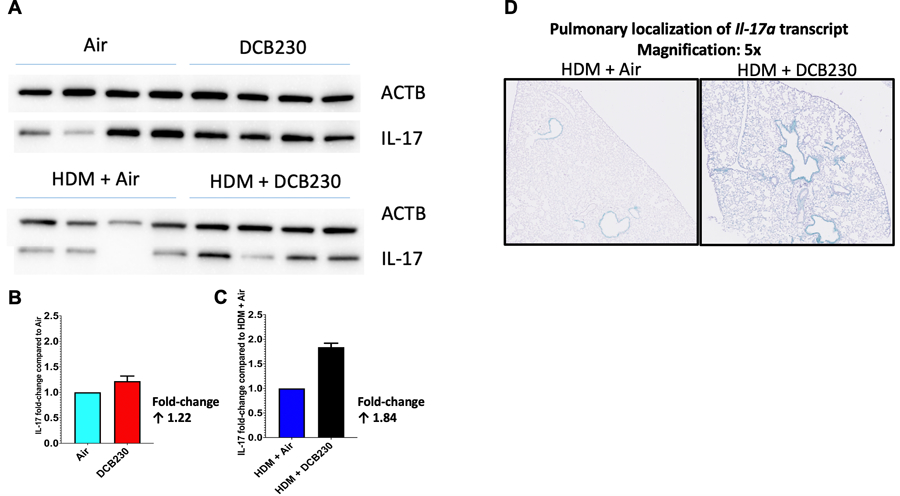
A) Protein expression of IL-17 assessed by western blot. N = 4 mice per group. Results are presented as fold-changes of the treatment group compared to the respective air control group, e.g., DCB230 vs. Air and HDM + DCB230 vs. HDM + Air. Results were normalized to ACTB. B) Quantification of the protein expression of IL-17 expressed as fold-change of DCB230 over Air. The Air control group was normalized to 1. C) Quantification of the protein expression of IL-17 expressed as fold-change of HDM + DCB230 over HDM + Air. The HDM + Air control group was normalized to 1. D) Representative microscopy images of RNA chromogenic in situ hybridization for labeling of IL-17A transcripts on 5 µm paraffin embedded lungs section slides. Each green dot represents the localization for one transcript of Il-17a. Magnification: 5x.
DISCUSSION
The mechanisms by which PM, as a risk factor, influences the severity of asthma pathogenesis are not fully elucidated. The majority of experimental studies aiming to investigate the adjuvant effects of PM have used ovalbumin models to simulate asthmatic responses (Steerenberg et al. 2003; Stoeger et al. 2006; Li et al. 2009; Li et al. 2010; Castaneda et al. 2017). Few studies, including from our research group (Saravia et al. 2014), used HDM as an allergen. HDM has an advantage over ovalbumin as HDM is a natural allergen to which humans are ubiquitously exposed. Hence, the use of HDM in an allergy model enhances the human exposure relevance, as well as the asthmatic phenotypic responses, and thus, the translational impact of the animal study. In the current study, we showed that mice exposed to DCB230 alone overexpressed the Muc5ac gene (2.3-fold) and dysregulated the expression of 4 inflammation related genes, Ccl11, Ccl24, Il-10 and Tpsb2 (Figure 6). Also, in addition to reduced lung tidal volume (Figure 3), compared to their respective controls, the HDM+DCB230-exposed group exhibited significantly increased lung tissue damping (Figure 3) and a vastly up-regulated (24-fold) gene expression of Muc5ac (Figure 6), indicating that in this model, mucous hypersecretion could play a central role in restricting lung function (Ritzmann et al. 2022). This study also showed augmented eosinophilic inflammation in BALF and lung tissue (Figures 4 & 5), accompanied by an up-regulation of 36 asthma related genes, mainly Th2 mediators (Figure 6). Twelve of these genes, including Ccl2, Ccl11, Ccl17, Ccl22, Il-17a, Il-21, are part of the IL-17 canonical pathway (Figure 7), suggesting that this pathway is critical for DCB230 to induce toxicity and adjuvant effects in the lungs following HDM treatment. Taken together, our data indicate that inhaled PM containing EPFRs act as an adjuvant to exacerbate asthma through IL-17-mediated responses in an HDM-induced mouse model of asthma (Figures 3 – 8).
An increasing body of epidemiological and experimental evidence suggests that the adjuvant effects of PM, including diesel exhaust particles, may be due to their ability to increase oxidative stress responses (Diaz-Sachez et al. 1997; Steerenberg et al. 2003; Gilliland et al. 2004; Kleinman et al. 2007; Stevens et al. 2008; Li et al. 2009). It was previously demonstrated that, compared to fine and ultrafine particles without oxidants, mice intranasally exposed to PM that had high oxidant potential exhibited worse pulmonary inflammation following ovalbumin challenge via enhanced Th2 polarization (Li et al. 2009). This study highlighted the association between the oxidant potential of PM and their adjuvant effect following a secondary exposure to an allergen. It is well-known that oxidative stress is central to PM-mediated pulmonary injury and that PM containing EPFR are redox-active particles (Kelley et al. 2013). The mechanisms underlying these effects include the fact that EPFR can increase the generation of ROS, creating oxidative lung damage resulting in increased epithelial permeability, which leads to the production of cytokines and chemokines, key players in the establishment of the inflammatory responses via, among others, Nfkb signaling pathway, and airway remodeling (Balakrishna et al. 2009; Balakrishna et al. 2011; Saravia et al. 2014; Xu et al. 2020; Harding et al. 2021). Also, the physico-chemical characteristics of inhaled aerosols will influence the pulmonary deposition and clearance of the particles in the respiratory tract (Mack et al. 2020). In this study, the DCB230 aerosols were composed of fine and ultrafine particles (Figure 2), allowing for the PM-containing EPFRs to reach the deep lungs, and induce oxidative stress responses in the bronchial and alveolar regions. In addition, the pulmonary clearance mechanisms through the mucociliary escalator and alveolar macrophages, involve redox-sensitive signaling that is mediated through oxidative stress transcription factors, including Nrf2, thus also contributing to oxidative stress responses (Castaneda et al. 2017). In the present study, no inflammation was detected in the BALF of DCB230-exposed mice (Figure 4). Based on the size of the inhaled DCB230 particles (< 150 nm) (Figure 2), it is possible that these particles may not be detected by alveolar macrophages (Kreyling et al. 2002; Yang et al. 2008), whose optimal size for detection occurs at approximatively 500 to 3,000 nm (Kreyling et al. 2002; Yang et al. 2008). This suggests that aerosolized DCB230 did not induce an inflammatory reaction through macrophage phagocytosis (Morrissette et al. 1999; Kreyling et al. 2002; Yang et al. 2008; Mack et al. 2020). Inhaled DCB230 particles would therefore reside longer in the alveolar region and have the ability to interact with the pulmonary epithelium. Our data are consistent with DCB230 particles interacting with the lung epithelium, inducing mucous hypersecretion (Figure 5), which subsequently contributes to restricting lung volume (Figure 3) (Ritzmann et al. 2022). In addition, the lung molecular signatures induced by the DCB230 exposure alone, with the up-regulation of Muc5ac, Ccl11, Ccl24, Il-10 and Tpsb2 (Figure 6A), indicate that this exposure may prime pulmonary responses for mucus hypersecretion (Muc5ac) (Wang et al. 2012), airway hyperresponsiveness (Il-10 and Tpsb2) (Makela et al. 2000; Choy et al. 2018), and eosinophilic inflammation (Ccl11, Ccl24, Il-10) (Isgro et al. 2013; Wu et al. 2014). These all are hallmark effects of asthma (Xu et al. 2020) and therefore highlight the mechanisms by which EPFR exposures alone can predispose the lung to exacerbation of ongoing pulmonary inflammation. Thus, this baseline effect of DCB230 exposure alone can lead to pulmonary oxidative stress and irritation, which can increase the sensitivity of the lungs and contribute to the exacerbation of inflammatory responses when the lungs were previously sensitized with an allergen. This further suggests that inhaled EPFR particles can create a microenvironment that primes the lungs for enhanced immune and inflammatory responses. Thus far, the mechanisms of toxicity related to the pulmonary effects of EPFRs can be summarized as related to oxidative stress and immune dysregulation (Saravia et al. 2014; Harmon et al. 2018; Harding et al. 2021), pathways that are both central to lung injury and chronic diseases, including asthma.
Multiple studies have highlighted an association between exposure to PM2.5 and increased incidence and exacerbation of asthma (Matsumoto et al. 2006; Kleinman et al. 2007; Stevens et al. 2008; Li et al. 2009; Saravia et al. 2014; Xu et al. 2020). The pathophysiological effects of asthma include airway inflammation, mainly driven by either eosinophils or neutrophils, airway hyperreactivity, increased mucus production and airflow obstruction, as well as airway remodeling (Gurczynski and Moore, 2018; Xu et al. 2020). In mice, it was previously demonstrated that exposure to PM alone without a subsequent allergen exposure, induced Th2 pulmonary inflammation (Huang et al. 2017). Thus, results of the present study support the idea that PM exposure alone can contribute to the onset of asthma via Th2-mediated pathways (Huang et al. 2017). Inhalation exposures of mice to HDM and DCB230 particles, have recapitulated phenotypic asthmatic responses. More specifically, we showed that DCB230 particles triggered pulmonary inflammation when a prior sensitization to HDM had occurred (Figures 3 – 6). This was characterized by the increased presence mainly of eosinophils in the BALF (39%; p < 0.05), also in addition to neutrophils (5.1%) and lymphocytes (3.9%) (Figure 4). The results were further supported at the tissue level with histopathology indicating bronchial eosinophils and lymphocytes, as well as inflammatory cells in the alveolar region (Figure 5). Further, at the molecular level, we found that key genes involved in Th2-mediated inflammation were significantly up-regulated in the HDM + DCB230 group (Figure 6). Il-4, a key cytokine involved in inducing Th2 responses (Kuroda et al. 2013), was up-regulated by 1.5-fold in the HDM + Air group, whereas its up-regulation was 2.8-fold in the HDM + DCB230 group (Figure 6). Il-5 is another protein-coding gene that plays a crucial role in eosinophil diapedesis (Huang et al. 2017), and gene expression for the receptor of this protein (Il-5ra) was up-regulated by 2.2-fold in the HDM + Air-exposed mice, while in the HDM + DCB230-exposed mice, the up-regulation reached 3.8-fold (Figure 6). Moreover, gene expression for Il-13, a protein-coding gene that can activate both macrophages and eosinophils (Huang et al. 2017), was up-regulated by 2.3-fold in the HDM + Air-exposed group compared to 4.6-fold in the HDM + DCB230-exposed group (Figure 6). Further, Il-13 is implicated in airway remodeling through mucus secretion, which correlates with the gene expression for Muc5ac in the HDM + DCB230-exposed mice (0-fold increase in the HDM + Air-exposed group, versus 24.7-fold upregulation in the HDM + DCB230-exposed group) (Figure 6). Il-13 is also involved in airway hyperreactivity, affecting the constriction level of the respiratory tract, leading to difficulty breathing (Huang et al. 2017; Castaneda et al. 2017). As mentioned above, the EPFR component of PM may cause irritation in the airways, which can trigger bronchial hyperreactivity. This is supported by our lung function data in the HDM + DCB230-exposed mice (Figure 3). When comparing both HDM treated groups, we observed significantly increased tissue damping in the HDM + DCB230-exposed mice compared to the HDM + Air-exposed group (Figure 3C). Since no significant difference was observed for the respiratory system resistance between the two HDM-treated groups (Figure 3B), the resistance of the large airways in those mice was not changed, but HDM + DCB230 co-exposure had an effect on the peripheral lung compartment, as evidenced by the increased tissue damping (Figure 3C) (Darrah et al. 2016). Further, an increase in tissue damping translates into a requirement of more work or energy to expand the lungs (Darrah et al. 2016; Dylag et al. 2020). Overall, we showed that exposure to HDM + DCB230 results in altered tissue mechanics of the lung parenchyma compared to HDM + Air treated mice. These results suggest that HDM + DCB230 exposure mainly had an impact on the mechanics of the lung tissue rather than on the mechanics of the airways, demonstrating that exposures to EPFR can lead to pulmonary dysfunction. Overall, since increased eosinophils in the lungs are a hallmark effect of asthmatic responses and lead to epithelial injury, mucus production and airway hyperresponsiveness (Huang et al. 2017), taken together, our data show that EPFRs potentiate HDM-induced pulmonary inflammation and asthma (Figures 3 – 6). Thus, these results clearly indicate that the EPFR component of PM can act as an adjuvant when present with an allergen.
We previously reported the role of the aryl hydrocarbon receptor (AhR), a key transcription factor that plays a central role in the metabolism of xenobiotics, and of its downstream targets related to biotransformation, Cyp1a1 and Cyp1b1, in EPFR-induced pulmonary injury (Harding et al. 2021; Harmon et al. 2018; Harmon et al. 2021; Aryal et al. 2023). Also, AhR has been shown to be implicated in PM-related asthma onset and exacerbation, since it was previously demonstrated that PM can activate Th17 signaling pathway via the AhR receptor (Huang et al. 2015; Huang et al. 2017; Xu et al. 2020; Harding et al. 2021). In addition, IL-17A is a pleiotropic protein associated with severe asthma as well as increased risk of exacerbation (Castaneda et al. 2017; Huang et al. 2017; Gurczynski and Moore, 2018; Ritzmann et al. 2022). In our model, at the gene (2.5-fold up-regulation) and protein (1.8-fold increase) levels, IL-17A was significantly increased in the HDM + DCB230-exposed mice compared to their respective HDM + Air controls (Figures 6 and 8). These results are supported by other research groups who showed, using the inhalation route, that rodents exposed to PM collected from Los Angeles, CA acted as an adjuvant and amplified immune responses by enhancing pulmonary Th2 inflammation, which was characterized by an increase in Il-17a gene expression (Li et al. 2010). Thus, this study highlighted that PM in the presence of an allergen could activate both Th2 and Th17 immunity pathways. Further, it was recently demonstrated that asthmatic responses can be characterized by the activation of two major molecular pathways, including 1) Th2 signaling pathway and its related genes, dominated by IL-13 and resulting in eosinophilic inflammation, and 2) Th17 signaling pathway and its related genes, dominated by IL-17A and resulting in neutrophilic inflammation (Choy et al. 2015). Also, an asthmatic response can be characterized by a mix of Th2 and Th17 signaling pathways (Choy et al. 2015). In addition, the synergistic effect of IL-13 and IL-17A on asthmatic pulmonary inflammation leading to exacerbation was previously demonstrated (Lajoie et al. 2010; Hall et al. 2017; Kim et al. 2019). Thus, the molecular signatures of asthma pathogenesis imply multipart shared communication pathways between Th2 and Th17 responses, and it was previously demonstrated that increased eosinophilic inflammation was associated with increased expression of both Th2- and Th17-related genes (Choy et al. 2015). This is similar to what we observed in our current study, where mice exposed to HDM + DCB230 exhibited eosinophilic inflammation in the BALF and lung tissue (Figures 4 & 5) plus activation of molecular pathways involving genes related to Th2 (Ccr4, Ccr8, Icos, Il-1rl1, Il-2ra, Il-4, Il-9, Il-10, Il-13, Il-12b, Il-33, Tnfrsf4) and Th17 (Ccl2, Ccl11, Ccl17, Ccl22, Cd40lg, Il-4, Il-12b, Il-13, Il-17a, Il-21, Il-33, Muc5ac) signaling (Figures 6 & 7). Moreover, as a key protein involved in the mucosal barrier, Il-17 plays a crucial role in lung epithelial cell homeostasis (Gurczynski and Moore, 2018). IL-17A can stimulate epithelial cells to up-regulate the expression of the Muc5ac gene, and by this mean facilitate goblet cell hyperplasia, an indicator of airway remodeling in asthma (Chen et al. 2003; Hashimoto et al. 2004; Xia et al. 2014; Gurczynski and Moore, 2018; Ritzmann et al. 2022). Thus, our molecular results with the increased gene expression for Muc5ac (Figure 6) also correlate with our lung function data for both DCB230 exposures, with and without the presence of HDM (Figure 3), since mucus hypersecretion can narrow the airways and lead to decreased lung function (Ritzmann et al. 2022). Taken together, in our study, the increased expression of IL-17A at both the gene and protein expression levels (Figures 6 and 8) suggest that the asthmatic response following EPFR exposure is amplified due to the development of a more severe phenotype of eosinophilic asthma (Figures 4 & 5), decline in lung function (Figure 3), as well as airway remodeling (Figure 5), which are most probably driven by the up-regulation of IL-17A produced mainly by bronchial epithelial cells (Figures 6 and 8) (Castaneda et al. 2017; Huang et al. 2017; Gurczynski and Moore, 2018; Ritzmann et al. 2022). Also, since Il-13 was up-regulated in the HDM + DCB230 group, the enhanced asthmatic response observed in that group may also be due to the synergetic effect of Il-13 (4.6-fold) and Il-17a (2.5-fold) up-regulation (Figure 6) (Lajoie et al. 2010; Hall et al. 2017; Kim et al. 2019). Overall, these molecular, cellular and physiological responses indicate that EPFR exposures can lead to adjuvant effects and aggravate asthma through molecular mechanisms associated not only with Th2 signaling, but also with Th17 responses in an HDM-induced mouse model of asthma.
Nearly a decade ago, our research group reported that neonatal mice exposed to DCB230 particles via nebulization at a concentration of 200 µg/m3 from post-natal day 3 to post-natal day 17, and receiving HDM treatment, exhibited immunosuppression of the classical asthmatic reaction (decreased pulmonary eosinophilia, airway hyper responsiveness, and immunoglobulin responses), compared to HDM only treated neonates (Saravia et al. 2014). This immunosuppressive response at an early age may dampen the response of the immune system to subsequent viral or bacterial pathogens, and thus negatively impact the respiratory health of neonates both early and later in life (Saravia et al. 2014). The long-term effects of this early life exposure to EPFR and HDM also were evaluated. Mice that were previously exposed to EPFR and HDM in early life, when re-challenged with HDM as adults, exhibited an aggravated pulmonary inflammation response compared to respective controls (Saravia et al. 2014). This increased response in adulthood was characterized by increased numbers of Th2, Th17 and Treg cells in the lungs, as well as increased respiratory system resistance (Saravia et al. 2014). It is important to note here that the immune system of an infant or a neonatal mouse is underdeveloped in terms of maturity compared to the immune system of an adult; therefore, the responses in a neonate versus an adult mouse model are expected to be different. Overall, this study laid the groundwork for experimental evidence supporting the fact that inhalation of EPFR from PM may alter the immune responses of the lungs and mediate the inflammatory responses following a secondary or tertiary exposure to an allergen (Saravia et al. 2014). Thus, our current results (Figures 3–7) are supportive of our previous report in adult mice (Saravia et al. 2014; Harding et al. 2021) showing that EPFR exposures are associated with asthma exacerbation through Th2- and Th17-mediated pathways.
There are numerous differences between the exposure conditions we used in this study and those from other similar studies (Li et al. 2009; Li et al. 2010; Saravia et al. 2014; Castaneda et al. 2017; Xu et al. 2020; Harding et al. 2021), in terms of experimental factors, including the use of different route of exposures, concentrations of PM, composition of PM, experimental timing of the allergen exposure, type of allergen used, mouse strain, mouse age, etc. Nonetheless, the results of previous studies (Li et al. 2009; Li et al. 2010; Saravia et al. 2014; Harding et al. 2021) and our current study, all point in the same direction and demonstrate that the EPFR content of PM is associated with aggravation of asthmatic responses, regardless of whether the exposure to the aeroallergen occurs before or after the PM exposure. Thus, our results, obtained by whole-body inhalation, are supported by previous studies that used the intranasal and intratracheal instillation routes to expose rodents to PM and allergens, and show that PM containing EPFRs can act as an adjuvant and exacerbate pulmonary inflammation following the exposure to an allergen. Our study also demonstrated that even low exposure levels of inhaled EPFR (~1.5 mg/m3) can result in an adjuvant effect. Whether there is a threshold concentration of inhaled EPFR that would trigger this increased immune response to an allergen is currently unknown. However, epidemiological studies have repeatedly shown that small increases in ambient PM concentrations, by as low as 10 µg/m3, lead to significantly increased morbidity and mortality (Levy et al. 2000; Kloog et al. 2013; Chen et al. 2021). Thus, the mounting evidence accumulated thus far from our research groups (Figure 3–8; Saravia et al. 2014; Harding et al. 2021) as well as others (Li et al. 2009) highly suggests that EPFR may be the PM component that is the missing link between air pollution exposure and asthma exacerbation.
This study has many strengths, including its innovative features of using of the inhalation route and using an environmentally relevant EPFR exposure concentration, which helped further our understanding of the mechanisms whereby the EPFR content of PM act as an adjuvant in previously HDM-sensitized lungs. As previously mentioned, due to the higher prevalence of cardiovascular diseases in males, for this first set of experiments, only male mice were used. We acknowledge that this is a limitation to our study since sex-based differences, in terms of biological outcomes, could be conceivable for female counterparts. Asthma prevalence is higher in adolescent and adult females compared to males (Zein et al. 2015). Thus, our results collected from adult male mice may differ in a female mouse asthma model. We (Cahill et al. 2022) and others (Weiss et al. 2021; Mostafa et al. 2022) previously reported that pulmonary inflammatory responses following HDM treatment were different in terms of the major types of leukocytes present in the BALF of exposed mice. While HDM exposure seems to induce pulmonary neutrophilia/eosinophilia in male mice, in female mice, more severe asthmatic phenotypes were observed, with predominance of eosinophils and lymphocytes in BALF (Weiss et al. 2021; Cahill et al. 2022; Mostafa et al. 2022). Thus, the data reported here in male mice may not be entirely generalizable to the female population. We are currently conducting additional studies exposing both male and female mice to various concentrations of EPFR on PM2.5. These studies utilize multiple PM types with different radical contents, e.g., EPFR 2.1e−16 radical/g vs. EPFR 5.5e−17 radical/g, as recently published by our group (Aryal et al. 2023). This will enable the establishment of a comprehensive comparative approach with multiple PM-related EPFR content to elucidate the mechanism of toxicity of EPFRs. Also, while we acknowledge that we did not measure the total number of inflammatory cells present in the BALF, which may be a more accurate measure of lung inflammation, based on the clear increase in the percentage of inflammatory cells (neutrophils and eosinophils) present in the BALF of the HDM + DCB230-treated mice (Figure 4), we are confident that the reported percentages are reflective of the lung inflammatory response, as supported by the histopathology evaluation of the lung tissues (Figure 5). Other limitations of our study include the fact that we did not perform invasive lung function testing and in situ hybridization of IL-17 in the two baseline groups (Air and DCB230 alone), since the majority of the biological outcomes assessed for these groups were collected and analyzed as part of our previously published report (Harmon et al. 2021). Nonetheless, the results presented here add to the cumulative evidence showing that the EPFR content of PM could contribute to the exacerbation of asthmatic responses in a mouse model.
In summary, EPFR is a relevant chemical entity which directly leads to oxidative stress-mediated effects in tissues (Figures 3–8; Balakrishna et al. 2009; Balakrishna et al. 2011; Saravia et al. 2014; Harmon et al. 2018; Harding et al. 2021; Harmon et al. 2021; Aryal et al. 2023). Pre-exposures, or sensitization, to allergens can make the airways more sensitive to inhaled environmental pollutants, including PM. In the present study, following HDM sensitization, which alters the lung epithelial barrier, and thus increases the sensitivity of the lungs to a subsequent environmental pollutant, the inhalation of PM containing EPFRs led to pulmonary inflammation on an already primed lung. Thus, the immuno-toxicological effects of EPFRs may be associated with its pro-inflammatory and pro-allergic properties, and by these associations, EPFRs can modulate the lung immuno-inflammatory responses in the context of a prior sensitization to an allergen. Hence, EPFR act as an adjuvant by priming the innate immune responses of the lungs to enhance inflammation during an ongoing allergic response. Further, we showed the association between the EPFR content of PM and the adjuvant effect following a previous exposure to HDM, an aeroallergen. Overall, our data suggest that PM adjuvant toxicity may be associated with the EPFR content, and that, in addition to Th2 signaling pathways, the EPFR adjuvant mechanism involves Th17 signaling driven by increased levels of IL-17A. Nonetheless, more research on EPFRs is urgently needed since EPFRs are emerging contaminants with currently no epidemiological studies addressing their risk on human health. While experimental evidence accumulated thus far make a strong case for monitoring airborne levels of EPFR, translation of scientific evidence into strategies to better protect public health is imperative.
FUNDING
Funding for this research was provided by the Louisiana State University Biomedical Collaborative Research Program (A. Noël, A. L. Penn, K. J. Varner and T. R. Dugas), by the Louisiana Governor’s Biotechnology Initiative Grant GBI-BOR#013 (A. L. Penn), and the National Institute of Environmental Health Sciences Grants R21ES03006202 (K. J. Varner and T. R. Dugas) and P42ES01364808A1 (K. J. Varner, T. R. Dugas and A. Noël).
Footnotes
DISCLOSURES
No conflicts of interest are declared by the authors.
DATA AVAILABILITY STATEMENT
The data-sets analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- American Lung Association. State of the Air 2023. https://www.lung.org/research/sota/key-findings/year-round-particle-pollution
- Aryal A, Noël A, Khachatryan L, Cormier SA, Chowdhury PH, Penn A, Dugas TR, Harmon AC. Environmentally persistent free radicals: Methods for combustion generation, whole-body inhalation and assessing cardiopulmonary consequences. Environ Pollut 2023. Jul 11:122183. doi: 10.1016/j.envpol.2023.122183. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson RW, Anderson HR, Sunyer J, Ayres J, Baccini M, Vonk JM, Boumghar A, Forastiere F, Forsberg B, Touloumi G, Schwartz J, Katsouyanni K. Acute effects of particulate air pollution on respiratory admissions: results from APHEA 2 project. Air Pollution and Health: a European Approach. Am J Respir Crit Care Med 2001. Nov 15;164(10 Pt 1):1860–6. doi: 10.1164/ajrccm.164.10.2010138. [DOI] [PubMed] [Google Scholar]
- Balakrishna S, Lomnicki S, McAvey KM, Cole RB, Dellinger B, Cormier SA. Environmentally persistent free radicals amplify ultrafine particle mediated cellular oxidative stress and cytotoxicity. Particle and Fibre Toxicology 2009;6(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishna S, Saravia J, Thevenot P, Ahlert T, Lominiki S, Dellinger B, Cormier SA. Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs. Part Fibre Toxicol 2011. Mar 9;8:11. doi: 10.1186/1743-8977-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill KM, Johnson TK, Perveen Z, Schexnayder M, Xiao R, Heffernan LM, Langohr IM, Paulsen DB, Penn AL, Noël A. In utero exposures to mint-flavored JUUL aerosol impair lung development and aggravate house dust mite-induced asthma in adult offspring mice. Toxicology 2022. Jul;477:153272. doi: 10.1016/j.tox.2022.153272. Epub 2022 Jul 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda AR, Bein KJ, Smiley-Jewell S, Pinkerton KE. Fine particulate matter (PM2.5) enhances allergic sensitization in BALB/c mice. J Toxicol Environ Health A 2017;80(4):197–207. doi: 10.1080/15287394.2016.1222920. Epub 2017 May 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem 2003. May 9;278(19):17036–43. doi: 10.1074/jbc.M210429200. Epub 2003 Mar 6. [DOI] [PubMed] [Google Scholar]
- Chen Y, Jiao Z, Chen P, Fan L, Zhou X, Pu Y, Du W, Yin L. Short-term effect of fine particulate matter and ozone on non-accidental mortality and respiratory mortality in Lishui district, China. BMC Public Health 2021. Sep 13;21(1):1661. doi: 10.1186/s12889-021-11713-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Li SW, Lin CD, Huang MZ, Lin HJ, Chin CY, Lai YR, Chiu CH, Yang CY, Lai CH. Fine Particulate Matter Exposure Alters Pulmonary Microbiota Composition and Aggravates Pneumococcus-Induced Lung Pathogenesis. Front Cell Dev Biol 2020;8:570484. Epub 20201026. doi: 10.3389/fcell.2020.570484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy David, Trivedi Neil, Dressen Amy, Babina Magda, Ahuja Rahul, Yi Tangsheng, Jackman Janet, Ji Guiquan, Hackney Jason, Orozco-Guerra Luz, Chang Diana, Caughey George, Yaspan Brian. Tryptase loss-of-function mutations reduce tryptase expression and predict asthmatic response to anti-IgE therapy. European Respiratory Journal Sep 2018, 52 (suppl 62) OA1652; DOI: 10.1183/13993003.congress-2018.OA1652 [DOI] [Google Scholar]
- Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, Jia G, Ohri CM, Doran E, Vannella KM, Butler CA, Hargadon B, Sciurba JC, Gieseck RL, Thompson RW, White S, Abbas AR, Jackman J, Wu LC, Egen JG, Heaney LG, Ramalingam TR, Arron JR, Wynn TA, Bradding P. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 2015. Aug 19;7(301):301ra129. doi: 10.1126/scitranslmed.aab3142. [DOI] [PubMed] [Google Scholar]
- Coban C, Igari Y, Yagi M, Reimer T, Koyama S, Aoshi T, Ohata K, Tsukui T, Takeshita F, Sakurai K, Ikegami T, Nakagawa A, Horii T, Nuñez G, Ishii KJ, Akira S. Immunogenicity of whole-parasite vaccines against Plasmodium falciparum involves malarial hemozoin and host TLR9. Cell Host Microbe 2010a. Jan 21;7(1):50–61. doi: 10.1016/j.chom.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Coban C, Yagi M, Ohata K, Igari Y, Tsukui T, Horii T, Ishii KJ, Akira S. The malarial metabolite hemozoin and its potential use as a vaccine adjuvant. Allergol Int 2010b. Jun;59(2):115–24. doi: 10.2332/allergolint.10-RAI-0194. Epub 2010 Apr 24. [DOI] [PubMed] [Google Scholar]
- Collins TW, Grineski SE, Shaker Y, Mullen CJ. Communities of color are disproportionately exposed to long-term and short-term PM(2.5) in metropolitan America. Environ Res 2022;214(Pt 4):114038. Epub 20220810. doi: 10.1016/j.envres.2022.114038. [DOI] [PubMed] [Google Scholar]
- Darrah RJ, Mitchell AL, Campanaro CK, Barbato ES, Litman P, Sattar A, Hodges CA, Drumm ML, Jacono FJ. Early pulmonary disease manifestations in cystic fibrosis mice. J Cyst Fibros 2016. Nov;15(6):736–744. doi: 10.1016/j.jcf.2016.05.002. Epub 2016 May 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle CC, Chalupa DC, Gibb FR, Morrow PE, Oberdörster G, Utell MJ, Frampton MW. Ultrafine particle deposition in humans during rest and exercise. Inhal Toxicol 2003. May;15(6):539–52. doi: 10.1080/08958370304468. [DOI] [PubMed] [Google Scholar]
- Diaz-Sanchez D, Garcia MP, Wang M, Jyrala M, Saxon A. Nasal challenge with diesel exhaust particles can induce sensitization to a neoallergen in the human mucosa. J Allergy Clin Immunol 1999. Dec;104(6):1183–8. doi: 10.1016/s0091-6749(99)70011-4. [DOI] [PubMed] [Google Scholar]
- Dellinger B, Pryor WA, Cueto B, Squadrito GL, Deutsch WA. The role of combustion-generated radicals in the toxicity of PM2. 5. Proceedings of the Combustion Institute 2000;28(2):2675–2681. [Google Scholar]
- Dellinger B, Pryor WA, Cueto R, Squadrito GL, Hegde V, Deutsch WA. Role of free radicals in the toxicity of airborne fine particulate matter. Chemical Research in Toxicology 2001;14(10):1371–1377. [DOI] [PubMed] [Google Scholar]
- Dylag AM, Haak J, Yee M, O’Reilly MA. Pulmonary mechanics and structural lung development after neonatal hyperoxia in mice. Pediatr Res 2020. Jun;87(7):1201–1210. doi: 10.1038/s41390-019-0723-y. Epub 2019 Dec 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferin J, Oberdörster G, Penney DP. Pulmonary retention of ultrafine and fine particles in rats. Am J Respir Cell Mol Biol 1992. May;6(5):535–42. doi: 10.1165/ajrcmb/6.5.535. [DOI] [PubMed] [Google Scholar]
- Franklin BA, Brook R, Pope CA III. Air pollution and cardiovascular disease. Current Problems in Cardiology 2015;40(5):207–238. [DOI] [PubMed] [Google Scholar]
- Gilliland FD, Li YF, Saxon A, Diaz-Sanchez D. Effect of glutathione-S-transferase M1 and P1 genotypes on xenobiotic enhancement of allergic responses: randomised, placebo-controlled crossover study. Lancet 2004. Jan 10;363(9403):119–25. doi: 10.1016/S0140-6736(03)15262-2. [DOI] [PubMed] [Google Scholar]
- Gehling W, Dellinger B. Environmentally persistent free radicals and their lifetimes in PM2. 5. Environmental Science and Technology 2013;47(15):8172–8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis 2016. Jan;7(1):34–51. doi: 10.1177/2040622315609251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurczynski SJ, Moore BB. IL-17 in the lung: the good, the bad, and the ugly. Am J Physiol Lung Cell Mol Physiol 2018. Jan 1;314(1):L6–L16. doi: 10.1152/ajplung.00344.2017. Epub 2017 Aug 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn FF, Newton GJ, Bryant PL. 1977. In vitro phagocytosis of respirable-sized monodisperse particles by alveolar macrophages. In: Pulmonary Macrophages and Epithelial Cells: Proceedings of the Sixteenth Annual Hanford Biology Symposium, September 1976, Richland, WA: (Sanders C, Schneider R, Dagle G, Ragan H, eds). ERDA Symposium Series 43. Oak Ridge, TN:Energy Research and Development Administration, 424–436. [Google Scholar]
- Hall SL, Baker T, Lajoie S, Richgels PK, Yang Y, McAlees JW, van Lier A, Wills-Karp M, Sivaprasad U, Acciani TH, LeCras TD, Myers JB, Kovacic MB, Lewkowich IP. IL-17A enhances IL-13 activity by enhancing IL-13-induced signal transducer and activator of transcription 6 activation. J Allergy Clin Immunol 2017. Feb;139(2):462–471.e14. doi: 10.1016/j.jaci.2016.04.037. Epub 2016 Jun 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding JN, Gross M, Patel V, Potter S, Cormier SA. Association between particulate matter containing EPFRs and neutrophilic asthma through AhR and Th17. Respir Res 2021. Oct 26;22(1):275. doi: 10.1186/s12931-021-01867-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon AC, Hebert VY, Cormier SA, Subramanian B, Reed JR, Backes WL, Dugas TR. Particulate matter containing environmentally persistent free radicals induces AhR-dependent cytokine and reactive oxygen species production in human bronchial epithelial cells. PLoS One 2018. Oct 11;13(10):e0205412. doi: 10.1371/journal.pone.0205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon AC, Noël A, Subramanian B, Perveen Z, Jennings MH, Chen YF, Penn AL, Legendre K, Paulsen DB, Varner KJ, Dugas TR. Inhalation of particulate matter containing free radicals leads to decreased vascular responsiveness associated with an altered pulmonary function. Am J Physiol Heart Circ Physiol 2021. Oct 1;321(4):H667–H683. doi: 10.1152/ajpheart.00725.2020. Epub 2021 Aug 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Graham BS, Ho SB, Adler KB, Collins RD, Olson SJ, Zhou W, Suzutani T, Jones PW, Goleniewska K, O’Neal JF, Peebles RS Jr. Respiratory syncytial virus in allergic lung inflammation increases Muc5ac and gob-5. Am J Respir Crit Care Med 2004. Aug 1;170(3):306–12. doi: 10.1164/rccm.200301-030OC. Epub 2004 May 6. [DOI] [PubMed] [Google Scholar]
- Huang SK, Zhang Q, Qiu Z, Chung KF. Mechanistic impact of outdoor air pollution on asthma and allergic diseases. J Thorac Dis 2015. Jan;7(1):23–33. doi: 10.3978/j.issn.2072-1439.2014.12.13. Erratum in: J Thorac Dis. 2015 Oct;7(10):E521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KL, Liu SY, Chou CC, Lee YH, Cheng TJ. The effect of size-segregated ambient particulate matter on Th1/Th2-like immune responses in mice. PLoS One 2017. Feb 28;12(2):e0173158. doi: 10.1371/journal.pone.0173158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isgrò M, Bianchetti L, Marini MA, Bellini A, Schmidt M, Mattoli S. The C-C motif chemokine ligands CCL5, CCL11, and CCL24 induce the migration of circulating fibrocytes from patients with severe asthma. Mucosal Immunol 2013. Jul;6(4):718–27. doi: 10.1038/mi.2012.109. Epub 2012 Nov 14. [DOI] [PubMed] [Google Scholar]
- Jia H, Zhao S, Nulaji G, et al. Environmentally persistent free radicals in soils of past coking sites: distribution and stabilization. Environmental Science and Technology 2017;51(11):6000–6008. [DOI] [PubMed] [Google Scholar]
- Jiang XQ, Mei XD, Feng D. Air pollution and chronic airway diseases: what should people know and do? Journal of Thoracic Disease 2016;8(1):E31–E40. doi: 10.3978/j.issn.2072-1439.2015.11.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Hartz AM, Potin S, Coumoul X, Yousif S, Scherrmann JM, Bauer B, Declèves X. Aryl hydrocarbon receptor-dependent upregulation of Cyp1b1 by TCDD and diesel exhaust particles in rat brain microvessels. Fluids Barriers CNS 2011. Aug 25;8:23. doi: 10.1186/2045-8118-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MA, Hebert VY, Thibeaux TM, Orchard MA, Hasan F, Cormier SA, Thevenot PT, Lomnicki SM, Varner KJ, Dellinger B, Latimer BM, Dugas TR. Model combustion-generated particulate matter containing persistent free radicals redox cycle to produce reactive oxygen species. Chem Res Toxicol 2013. Dec 16;26(12):1862–71. doi: 10.1021/tx400227s. Epub 2013 Nov 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachatryan L, Dellinger B. Environmentally persistent free radicals (EPFRs)-2. Are free hydroxyl radicals generated in aqueous solutions? Environmental Science and Technology 2011;45(21):9232–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Kim KW, Lee SM, Lee DH, Park S, Son BS, Park MK. Overexpression of the Aryl Hydrocarbon Receptor (Ahr) Mediates an Oxidative Stress Response following Injection of Fine Particulate Matter in the Temporal Cortex. Oxid Med Cell Longev 2020. Dec 28;2020:6879738. doi: 10.1155/2020/6879738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman MT, Sioutas C, Froines JR, Fanning E, Hamade A, Mendez L, Meacher D, Oldham M. Inhalation of concentrated ambient particulate matter near a heavily trafficked road stimulates antigen-induced airway responses in mice. Inhal Toxicol 2007;19 Suppl 1:117–26. doi: 10.1080/08958370701495345. [DOI] [PubMed] [Google Scholar]
- Keirsbulck M, Savouré M, Lequy E, Chen J, de Hoogh K, Vienneau D, Goldberg M, Zins M, Roche N, Nadif R, Jacquemin B. Long-term exposure to ambient air pollution and asthma symptom score in the CONSTANCES cohort. Thorax 2023. Jan;78(1):9–15. doi: 10.1136/thoraxjnl-2021-218344. Epub 2022 Mar 2. [DOI] [PubMed] [Google Scholar]
- Kim D, McAlees JW, Bischoff LJ, Kaur D, Houshel LK, Gray J, Hargis J, Davis X, Dudas PL, Deshmukh H, Lewkowich IP. Combined administration of anti-IL-13 and anti-IL-17A at individually sub-therapeutic doses limits asthma-like symptoms in a mouse model of Th2/Th17 high asthma. Clin Exp Allergy 2019. Mar;49(3):317–330. doi: 10.1111/cea.13301. Epub 2018 Nov 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloog I, Ridgway B, Koutrakis P, Coull BA, Schwartz JD. Long- and short-term exposure to PM2.5 and mortality: using novel exposure models. Epidemiology 2013. Jul;24(4):555–61. doi: 10.1097/EDE.0b013e318294beaa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, Oberdörster G, Ziesenis A. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A 2002. Oct 25;65(20):1513–30. doi: 10.1080/00984100290071649. [DOI] [PubMed] [Google Scholar]
- Kuźma Ł, Struniawski K, Pogorzelski S, Bachórzewska-Gajewska H, Dobrzycki S. Gender Differences in Association between Air Pollution and Daily Mortality in the Capital of the Green Lungs of Poland-Population-Based Study with 2,953,000 Person-Years of Follow-Up. J Clin Med 2020. Jul 23;9(8):2351. doi: 10.3390/jcm9082351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie S, Lewkowich IP, Suzuki Y, Clark JR, Sproles AA, Dienger K, Budelsky AL, Wills-Karp M. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol 2010. Oct;11(10):928–35. doi: 10.1038/ni.1926. Epub 2010 Aug 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JI, Hammitt JK, Spengler JD. Estimating the mortality impacts of particulate matter: what can be learned from between-study variability? Environ Health Perspect 2000. Feb;108(2):109–17. doi: 10.1289/ehp.00108109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang M, Bramble LA, Schmitz DA, Schauer JJ, Sioutas C, Harkema JR, Nel AE. The adjuvant effect of ambient particulate matter is closely reflected by the particulate oxidant potential. Environ Health Perspect 2009. Jul;117(7):1116–23. doi: 10.1289/ehp.0800319. Epub 2009 Mar 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Harkema JR, Lewandowski RP, Wang M, Bramble LA, Gookin GR, Ning Z, Kleinman MT, Sioutas C, Nel AE. Ambient ultrafine particles provide a strong adjuvant effect in the secondary immune response: implication for traffic-related asthma flares. Am J Physiol Lung Cell Mol Physiol 2010. Sep;299(3):L374–83. doi: 10.1152/ajplung.00115.2010. Epub 2010 Jun 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomnicki S, Truong H, Vejerano E, Dellinger B. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter. Environmental Science and Technology 2008;42(13):4982–4988. [DOI] [PubMed] [Google Scholar]
- Mack SM, Madl AK, Pinkerton KE. Respiratory Health Effects of Exposure to Ambient Particulate Matter and Bioaerosols. Compr Physiol 2019. Dec 18;10(1):1–20. doi: 10.1002/cphy.c180040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä MJ, Kanehiro A, Borish L, Dakhama A, Loader J, Joetham A, Xing Z, Jordana M, Larsen GL, Gelfand EW. IL-10 is necessary for the expression of airway hyperresponsiveness but not pulmonary inflammation after allergic sensitization. Proc Natl Acad Sci U S A 2000. May 23;97(11):6007–12. doi: 10.1073/pnas.100118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Hiramatsu K, Li Y, Azuma A, Kudoh S, Takizawa H, Sugawara I. Repeated exposure to low-dose diesel exhaust after allergen challenge exaggerates asthmatic responses in mice. Clin Immunol 2006. Nov;121(2):227–35. doi: 10.1016/j.clim.2006.08.003. Epub 2006 Sep 18. [DOI] [PubMed] [Google Scholar]
- Morrissette N, Gold E, Aderem A. The macrophage--a cell for all seasons. Trends Cell Biol 1999. May;9(5):199–201. doi: 10.1016/s0962-8924(99)01540-8. [DOI] [PubMed] [Google Scholar]
- Mostafa DHD, Hemshekhar M, Piyadasa H, Altieri A, Halayko AJ, Pascoe CD, Mookherjee N. Characterization of sex-related differences in allergen house dust mite-challenged airway inflammation, in two different strains of mice. Sci Rep 2022. Dec 2;12(1):20837. doi: 10.1038/s41598-022-25327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Hirose K. Role of IL-23 and Th17 Cells in Airway Inflammation in Asthma. Immune Netw 2010. Feb;10(1):1–4. doi: 10.4110/in.2010.10.1.1. Epub 2010 Feb 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noël A, Xiao R, Perveen Z, Zaman HM, Rouse RL, Paulsen DB, Penn AL. Incomplete lung recovery following sub-acute inhalation of combustion-derived ultrafine particles in mice. Part Fibre Toxicol 2016. Feb 24;13:10. doi: 10.1186/s12989-016-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noël A, Perveen Z, Xiao R, Hammond H, Le Donne V, Legendre K, Gartia MR, Sahu S, Paulsen DB, Penn AL. Mmp12 Is Upregulated by in utero Second-Hand Smoke Exposures and Is a Key Factor Contributing to Aggravated Lung Responses in Adult Emphysema, Asthma, and Lung Cancer Mouse Models. Front Physiol 2021. Nov 29;12:704401. doi: 10.3389/fphys.2021.704401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percopo CM, Krumholz JO, Fischer ER, Kraemer LS, Ma M, Laky K, Rosenberg HF. Impact of eosinophil-peroxidase (EPX) deficiency on eosinophil structure and function in mouse airways. J Leukoc Biol 2019. Jan;105(1):151–161. doi: 10.1002/JLB.3AB0318-090RR. Epub 2018 Oct 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CA III. Respiratory disease associated with community air pollution and a steel mill, Utah Valley. American Journal of Public Health 1989;79(5):623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CA III. What do epidemiologic findings tell us about health effects of environmental aerosols? Journal of Aerosol Medicine 2000;13(4):335–354. [DOI] [PubMed] [Google Scholar]
- Porter M, Karp M, Killedar S, Bauer SM, Guo J, Williams D, Breysse P, Georas SN, Williams MA. Diesel-enriched particulate matter functionally activates human dendritic cells. Am J Respir Cell Mol Biol 2007. Dec;37(6):706–19. doi: 10.1165/rcmb.2007-0199OC. Epub 2007 Jul 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice MB, Rifas-Shiman SL, Litonjua AA, Gillman MW, Liebman N, Kloog I, Luttmann-Gibson H, Coull BA, Schwartz J, Koutrakis P, Oken E, Mittleman MA, Gold DR. Lifetime air pollution exposure and asthma in a pediatric birth cohort. J Allergy Clin Immunol 2018. May;141(5):1932–1934.e7. doi: 10.1016/j.jaci.2017.11.062. Epub 2018 Feb 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzmann F, Lunding LP, Bals R, Wegmann M, Beisswenger C. IL-17 Cytokines and Chronic Lung Diseases. Cells 2022. Jul 6;11(14):2132. doi: 10.3390/cells11142132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martini Federico M., Hasenkopf Christa A., and Roberts David C. “Statistical analysis of PM2. 5 observations from diplomatic facilities in China.” Atmospheric Environment 110 (2015): 174–185. [Google Scholar]
- Saravia J, You D, Thevenot P, Lee GI, Shrestha B, Lomnicki S, Cormier SA. Early-life exposure to combustion-derived particulate matter causes pulmonary immunosuppression. Mucosal Immunol 2014. May;7(3):694–704. doi: 10.1038/mi.2013.88. Epub 2013 Oct 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J Air pollution and children’s health. Pediatrics 2004;113(Supplement 3):1037–1043. [PubMed] [Google Scholar]
- Steerenberg PA, Withagen CE, Dormans JA, van Dalen WJ, van Loveren H, Casee FR. Adjuvant activity of various diesel exhaust and ambient particles in two allergic models. J Toxicol Environ Health A 2003. Aug 8;66(15):1421–39. doi: 10.1080/15287390306415. [DOI] [PubMed] [Google Scholar]
- Steerenberg PA, Withagen CE, van Dalen WJ, Dormans JA, Cassee FR, Heisterkamp SH, van Loveren H. Adjuvant activity of ambient particulate matter of different sites, sizes, and seasons in a respiratory allergy mouse model. Toxicol Appl Pharmacol 2004. Nov 1;200(3):186–200. doi: 10.1016/j.taap.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Stevens T, Krantz QT, Linak WP, Hester S, Gilmour MI. Increased transcription of immune and metabolic pathways in naive and allergic mice exposed to diesel exhaust. Toxicol Sci 2008. Apr;102(2):359–70. doi: 10.1093/toxsci/kfn006. Epub 2008 Jan 11. Erratum in: Toxicol Sci. 2008 Oct;105(2):435. [DOI] [PubMed] [Google Scholar]
- Stoeger T, Reinhard C, Takenaka S, Schroeppel A, Karg E, Ritter B, Heyder J, Schulz H. Instillation of six different ultrafine carbon particles indicates a surface area threshold dose for acute lung inflammation in mice. Environ Health Perspect 2006. Mar;114(3):328–33. doi: 10.1289/ehp.8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valavanidis A, Fiotakis K, Vlahogianni T, et al. Characterization of atmospheric particulates, particle-bound transition metals and polycyclic aromatic hydrocarbons of urban air in the centre of Athens (Greece). Chemosphere 2006;65(5):760–768. [DOI] [PubMed] [Google Scholar]
- Vejerano EP, Rao G, Khachatryan L, Cormier SA, Lomnicki S. Environmentally Persistent Free Radicals: Insights on a New Class of Pollutants. Environl Science and Technology 2018;52(5):2468–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Xu Z, Wang R, Al-Hijji M, Salit J, Strulovici-Barel Y, Tilley AE, Mezey JG, Crystal RG. Genes associated with MUC5AC expression in small airway epithelium of human smokers and non-smokers. BMC Med Genomics 2012. Jun 7;5:21. doi: 10.1186/1755-8794-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Beelen R, Stafoggia M, et al. Long-term exposure to elemental constituents of particulate matter and cardiovascular mortality in 19 European cohorts: results from the ESCAPE and TRANSPHORM projects. Environment International 2014;66:97–106. [DOI] [PubMed] [Google Scholar]
- Weiss K, Wanner N, Queisser K, Frimel M, Nunn T, Myshrall T, Sangwan N, Erzurum S, Asosingh K. Barrier Housing and Gender Effects on Allergic Airway Disease in a Murine House Dust Mite Model. Immunohorizons 2021. Jan 21;5(1):33–47. doi: 10.4049/immunohorizons.2000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Zhou J, Bi H, Li L, Gao W, Huang M, Adcock IM, Barnes PJ, Yao X. CCL11 as a potential diagnostic marker for asthma? J Asthma 2014. Oct;51(8):847–54. doi: 10.3109/02770903.2014.917659. Epub 2014 May 13. [DOI] [PubMed] [Google Scholar]
- Xia W, Bai J, Wu X, Wei Y, Feng S, Li L, Zhang J, Xiong G, Fan Y, Shi J, Li H. Interleukin-17A promotes MUC5AC expression and goblet cell hyperplasia in nasal polyps via the Act1-mediated pathway. PLoS One 2014. Jun 3;9(6):e98915. doi: 10.1371/journal.pone.0098915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Fang F, Montgomery S et al. Sex differences in associations of fine particulate matter with non-accidental deaths: an ecological time-series study. Air Qual Atmos Health 14, 863–872 (2021). 10.1007/s11869-021-00985-0 [DOI] [Google Scholar]
- Xu X, Zhang J, Yang X, Zhang Y, Chen Z. The Role and Potential Pathogenic Mechanism of Particulate Matter in Childhood Asthma: A Review and Perspective. J Immunol Res 2020. Jan 17;2020:8254909. doi: 10.1155/2020/8254909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Peters JI, Williams RO 3rd. Inhaled nanoparticles--a current review. Int J Pharm 2008. May 22;356(1–2):239–47. doi: 10.1016/j.ijpharm.2008.02.011. Epub 2008 Feb 16. [DOI] [PubMed] [Google Scholar]
- Yang J, Pan B, Li H, et al. Degradation of p-nitrophenol on biochars: role of persistent free radicals. Environmental Science and Technology 2015;50(2):694–700. [DOI] [PubMed] [Google Scholar]
- Zein JG, Erzurum SC. Asthma is Different in Women. Curr Allergy Asthma Rep 2015. Jun;15(6):28. doi: 10.1007/s11882-015-0528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data-sets analyzed during the current study are available from the corresponding author on reasonable request.