

SUMMARY
Pathobionts employ unique metabolic adaptation mechanisms to maximize their growth in disease conditions. Adherent–invasive Escherichia coli (AIEC), a pathobiont enriched in the gut mucosa of patients with inflammatory bowel disease (IBD), utilizes diet-derived L-serine to adapt to the inflamed gut. Therefore, the restriction of dietary L-serine starves AIEC and limits its fitness advantage. Here, we find that AIEC can overcome this nutrient limitation by switching the nutrient source from the diet to the host cells in the presence of mucolytic bacteria. During diet-derived L-serine restriction, the mucolytic symbiont Akkermansia muciniphila promotes the encroachment of AIEC to the epithelial niche by degrading the mucus layer. In the epithelial niche, AIEC acquires L-serine from the colonic epithelium and thus proliferates. Our work suggests that the indirect metabolic network between pathobionts and commensal symbionts enables pathobionts to overcome nutritional restriction and thrive in the gut.
Keywords: inflammatory bowel disease, L-serine, adherent-invasive Escherichia coli, Akkermansia muciniphila, intestinal mucus barrier
INTRODUCTION
Microbial metabolism plays a critical role in cooperation and competition within the microbial community (Passalacqua et al., 2016). Microbial metabolism rapidly responds to environmental stimulation, such as host immune activation, dietary modification, and gut inflammation, to adapt to the surrounding microenvironment (Becattini et al., 2021; Desai et al., 2016; Kitamoto et al., 2020; Sonnenburg et al., 2005). For example, commensal symbionts reprogram the transcription of their metabolic genes in response to host immune activation and alter their metabolic functions within several hours (Becattini et al., 2021). Likewise, gut inflammation alters the luminal microenvironment, including nutrient availability and oxygen levels, which, in turn, contributes to the alteration of the gut microbial composition and function (Rigottier-Gois, 2013; Stecher, 2015). Metatranscriptome studies have shown that gut inflammation upregulates stress response pathways and downregulates polysaccharide utilization and fermentation in a murine model of colitis (Ilott et al., 2016; Schwab et al., 2014). In addition, chronic intestinal inflammation upregulates stress response genes, including small heat shock proteins, which protect commensal Escherichia coli from oxidative stress (Patwa et al., 2011). These disease-specific microbial transcriptional signatures have also been observed in patients with inflammatory bowel disease (IBD) (Schirmer et al., 2018). However, the impact of the transcriptional adaptation of microbes on host–microbe interaction and disease course is largely unknown.
The gut microbiota plays a fundamental role in the pathogenesis of IBD (Ananthakrishnan, 2015; Sugihara and Kamada, 2021). Potentially pathogenic members of the commensal bacteria, termed pathobionts, have been identified in IBD patients and observed to trigger or exacerbate inflammation in the gut. Adherent-invasive Escherichia coli (AIEC) is the most studied pathobiont associated with IBD. The prevalence of AIEC increases in the ileal and colonic mucosae of IBD patients compared to non-IBD control subjects (Nadalian et al., 2021). AIEC strains harbor several virulence genes related to the ability to adhere and invade the intestinal epithelial cells (IECs) and thus are associated with the exacerbation of intestinal inflammation and fibrosis (Carvalho et al., 2009; Imai et al., 2019). In IBD, AIEC may exploit unique strategies to gain a growth advantage over competing, nonpathogenic, symbiont E. coli strains. We have reported that AIEC reprograms metabolic gene transcription in the inflamed gut to adapt to the inflammatory microenvironment (Kitamoto et al., 2020). In particular, AIEC upregulates L-serine metabolism pathways that are crucial in acquiring a growth advantage over symbiont E. coli strains. Interestingly, as luminal L-serine is supplied by diet, the modulation of dietary L-serine can regulate intraspecific competition between AIEC and commensal E. coli (Kitamoto et al., 2020). Thus, dietary modification can be an effective strategy to treat pathobiont-driven diseases, such as IBD.
L-serine is a nonessential amino acid that supports several metabolic processes crucial for the growth and survival of mammalian and bacterial cells, especially under disease conditions (Newman and Maddocks, 2017). For example, L-serine metabolism is markedly upregulated in cancer cells and immune cells and plays a central role in their survival and growth (Ma et al., 2017; Maddocks et al., 2013; Rodriguez et al., 2019). Moreover, consistent with gut bacteria, L-serine used in the proliferation of cancer cells and immune cells is also supplied by the diet, and therefore a lack of dietary L-serine can inhibit the proliferation of these cells (Ma et al., 2017; Maddocks et al., 2017).
Here, we report the impact of dietary L-serine on the host–microbe interaction during gut inflammation. As the deprivation of diet-derived L-serine limits the fitness advantage of AIEC over commensal E. coli, we anticipated that dietary L-serine restriction would improve gut inflammation in mice colonized with conventional microbiota (specific pathogen–free [SPF] mice). However, to our surprise, diet-derived L-serine restriction exacerbated dextran sodium sulfate (DSS)–induced colitis in SPF mice. In our quest to explain this unexpected phenotype, we discovered that Akkermansia muciniphila, a commensal symbiont capable of degrading mucin, is expanded in colitic mice under dietary L-serine restriction. The expansion of A. muciniphila results in a massive erosion of the colonic mucus layer, thereby allowing AIEC to relocate close to the host epithelial cells. In the epithelial niche, AIEC can acquire L-serine from the host epithelial cells, whereby it can overcome dietary L-serine restriction and proliferate. Thus, the mucolytic bacteria, such as A. muciniphila, can serve as an indirect metabolic supporter for AIEC by licensing the acquisition of host-derived nutrients.
RESULTS
L-serine metabolism is disturbed in the gut microbiota of IBD patients
Our previous study showed that IBD-associated AIEC uses amino acid metabolism, particularly L-serine catabolism, to adapt to the inflamed gut. Consistent with this notion, the IBD-associated AIEC strain LF82 rapidly consumed L-serine, more than other amino acids in the cultured media (Figure S1), indicating that AIEC prefers L-serine as a nutrient source. However, it remains unclear whether L-serine plays a central metabolic role in the more complex microbiota of IBD patients. To assess the microbial L-serine metabolism, we first analyzed data available in the Inflammatory Bowel Disease Multi’omics Database (IBDMBD) of the Integrative Human Microbiome Project (iHMP), which integrates metagenomic, metatranscriptomic, metaproteomic, and metabolic data on the microbiome of IBD (Figure 1A). As previous studies have reported (Lloyd-Price et al., 2019; Morgan XC, 2012; Vich Vila A, 2018), the abundance of Enterobacteriaceae, including E. coli, is significantly higher in patients with ulcerative colitis (UC) and Crohn’s disease (CD), the two most common forms of IBD, than in non-IBD controls (Figure 1B). L-serine is biosynthesized from intermediates of the glycolysis pathway or from L-glycine, and it converts to pyruvate, which is a necessary substrate for gluconeogenesis and the tricarboxylic acid cycle (Figure 1C, right). Metagenomic analysis showed that the abundance of phosphoglycerate dehydrogenase (PHGDH), the rate-limiting enzyme for serine biosynthesis from the glycolysis pathway, was significantly reduced in both UC and CD patients compared to non-IBD controls (Figure 1C, left). Conversely, the abundance of serine dehydratase (SDH), the enzyme that catalyzes the conversion of L-serine to pyruvate, was significantly higher in the gut microbiota of UC and CD patients compared to non-IBD controls (Figure 1C, left). Although metatranscriptomic profiles were more varied between individuals than metagenomic profiles, these genes were also transcriptionally changed in IBD patients (Figure 1D). These results suggested that bacteria that utilize L-serine, such as AIEC, are enriched in the microbiota of IBD patients. In addition to microbial metabolism, we also confirmed the amino acid levels in the feces of patients with IBD using the iHMP metabolome data (Figure 1E). Compared with non-IBD controls, essential amino acids, particularly valine and histidine, were significantly higher in patients with IBD. Importantly, nonessential amino acids, including serine, glutamate, and arginine, were significantly lower in IBD patients than in non-IBD controls. Thus, it is likely that the gut microbiota of IBD patients consumes more L-serine than the gut microbiota of non-IBD controls.
Figure 1. L-serine metabolism is disturbed in the gut microbiota of IBD patients.
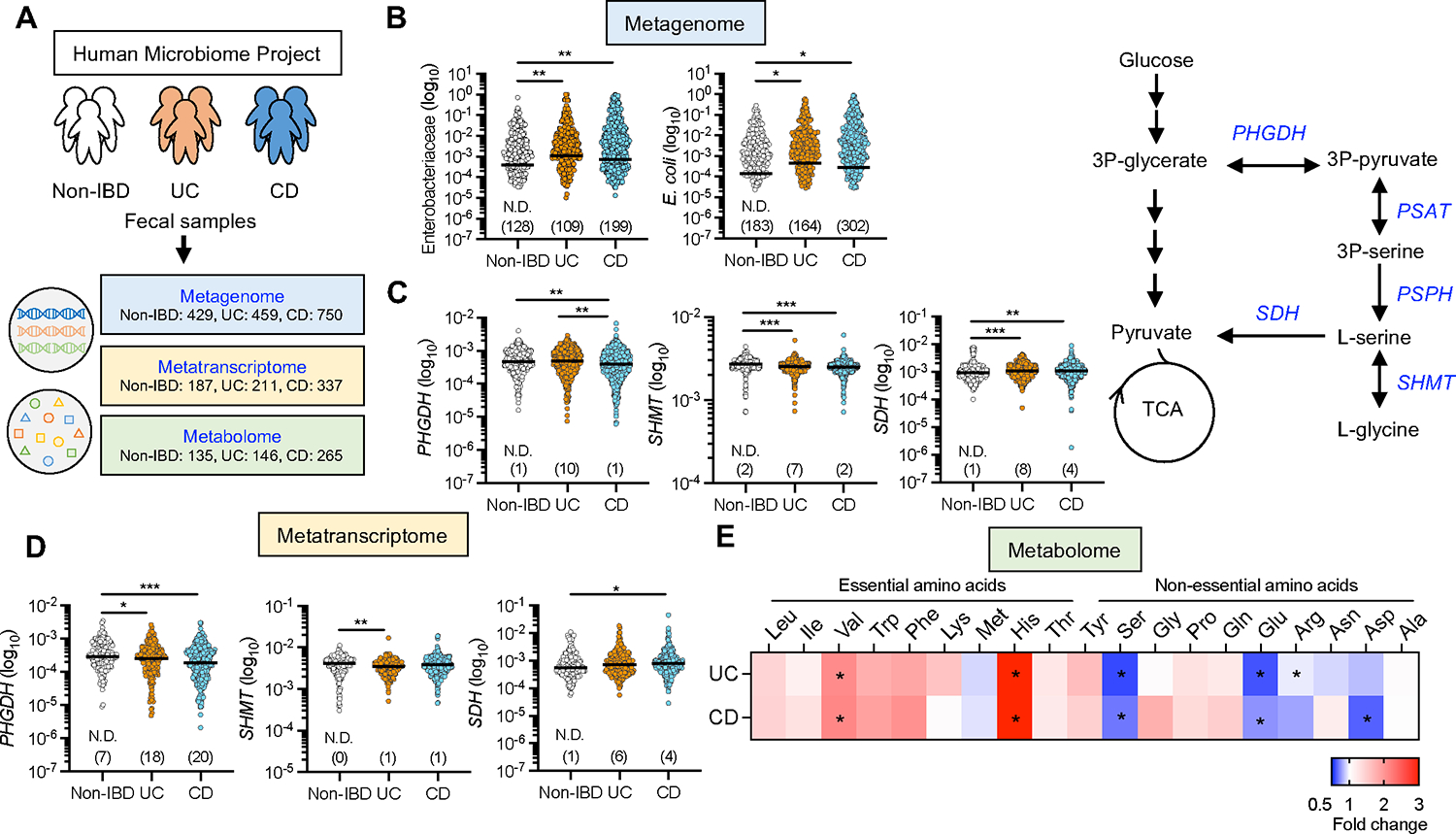
(A) Metagenomics, metatranscriptomics, and metabolomics data were downloaded from the public resource, the second phase of the Integrative Human Microbiome Project (HMP2 or iHMP) – the Inflammatory Bowel Disease Multi’omics Database.
(B) Abundance of Enterobacteriaceae and E. coli in the metagenomics database.
(C) Abundance of PHGDH, SHMT, and SDH in the metagenomics database (left). Schematic of L-serine metabolism (right).
(D) Abundance of PHGDH, SHMT, and SDH in the metatranscriptomics database.
(E) Abundance of fecal amino acids in the metabolomics database. The heatmap indicates the fold change (UC or CD/non-IBD).
Dots indicate individual people, with median (Metagenome: N = 429 Non-IBD, 459 UC, 750 CD. Metatranscriptome: N = 187 Non-IBD, 211 UC, 337 CD. Metabolome: N = 135 Non-IBD, 146 UC, 265 CD). The numbers in parentheses indicate the number of null values. *p < 0.05, **p < 0.01, ***p < 0.001 by Kruskal–Wallis test with Dunn test for multiple comparisons. PHGDH, phosphoglycerate dehydrogenase; SHMT, serine hydroxymethyltransferase; SDH, serine dehydratase. See also Figure S1.
The deprivation of dietary L-serine exacerbates DSS-induced colitis
Given that the gut microbiota of IBD patients appeared to be enriched with L-serine utilizers, including AIEC, we hypothesized that limiting L-serine availability may suppress the growth of potential pathobionts, thereby reducing the susceptibility to colitis. As luminal L-serine levels are mainly regulated by diet-derived L-serine (Kitamoto et al., 2020), we next examined the impact of dietary L-serine deprivation on intestinal inflammation. To this end, specific pathogen–free (SPF) mice were fed either a defined amino acid control diet (Ctrl) or an L-serine deficient (ΔSer) diet, as previously defined (Kitamoto et al., 2020; Maddocks et al., 2013). L-glycine was removed from the ΔSer diet as L-serine and L-glycine may be interconverted (Pizer LI, 1964). Mice were treated with 1.5% DSS for 5 days to induce colitis, followed by conventional water for 2 days (Figure 2A). Unexpectedly, the ΔSer diet–fed mice lost significantly more body weight and had a higher disease activity index (DAI) than the Ctrl diet–fed mice (Figures 2B and 2C). Likewise, mice fed the ΔSer diet had a greater degree of inflammation in the colon than the mice fed the Ctrl diet (Figures 2D–2F). Notably, the ΔSer diet did not affect body weight, colon length, and histology in the DSS-untreated mice (Figures 2B–2F). To uncover the mechanism by which the restriction of dietary L-serine exacerbates colitis, we focused on the role of the gut microbiota. As shown in Figures 2G–2J, the ΔSer diet did not worsen colitis in germ-free (GF) mice. We confirmed the same phenotype in SPF mice by depleting the gut microbiota with a cocktail of broad-spectrum antibiotics (Figure S2). These results suggest that the gut microbiota is required for the exacerbation of colitis caused by the restriction of dietary L-serine.
Figure 2. Deprivation of dietary L-serine exacerbates gut inflammation in DSS-induced colitis through the gut microbiota.

(A) SPF C57BL/6 mice were fed the control diet (Ctrl) or the ΔSer diet for 3 days, then given 1.5% DSS for 5 days, followed by conventional water for 2 days. On day 7 post-DSS, all mice were euthanized.
(B and C) Body weight and DAI were monitored during the 5-day DSS treatment.
(D–F) Colon length, representative histological images of colon sections (scale bar, 200 μm), and histology scores were evaluated.
(G) GF Swiss Webster mice were fed a Ctrl diet or a ΔSer diet for 3 days and then treated with 1.5% DSS for 5 days. On day 5 post-DSS, all mice were euthanized.
(H–J) Colon length, representative histological images of colon sections (Scale bar, 200 μm), and histology scores were assessed.
(B–D and H–J) Data pooled from two independent experiments (N = 7–10). (E, F) Data are representative of two independent experiments (N = 5). Dots indicate individual mice, with mean ± SEM. N.S., not significant, *p < 0.05, **p < 0.01, ***p < 0.001 by 1-way ANOVA or 2-way ANOVA with Tukey post hoc test. See also Figure S2.
Dietary L-serine starvation leads to the blooms of pathotype E. coli in the inflamed gut
We next analyzed the gut microbiota isolated from Ctrl diet– and ΔSer diet–fed mice to identify the bacterial taxa that may be associated with the severe inflammation observed in ΔSer diet–fed mice. We found that the restriction of dietary L-serine affected the microbial composition during inflammation, while in the steady state it had little influence (Figure 3A). Linear discriminant analysis effect size (LEfSe) further identified the bacterial families over- and under-represented after the dietary change. LEfSe analysis showed that Verrucomicrobiaceae and Enterobacteriaceae families were over-represented in ΔSer diet–fed mice, whereas Sutterellaceae and Porphyromonadaceae families were under-represented (Figure 3B). Interestingly, the abundance of E. coli, which belongs to the Enterobacteriaceae family, was significantly higher in the colitic mice fed the ΔSer diet rather than the Ctrl diet (Figure 3C and Figure S3). To further characterize E. coli accumulated in ΔSer diet–fed mice, the presence of genes associated with pathogenic features of E. coli was examined. In this context, the abundance of E. coli harboring genes associated with adhesion and invasion to host epithelial cells (vat, fimH, fliC, ompA, ompC, and ibeA) and metabolic adaptation (pduC, chuA, fyuA) was quantified (Cieza et al., 2015; Dogan et al., 2014; Dreux et al., 2013; Ellermann et al., 2019; Gibold et al., 2016; Rolhion et al., 2010; Rolhion et al., 2007; Sevrin et al., 2020; Viladomiu et al., 2021). Notably, the abundance of those genes was significantly enriched in ΔSer diet–fed mice than in Ctrl diet–fed mice during colitis (Figure 3D). This result suggests that E. coli strains accumulated in ΔSer diet–fed mice may be pathotype E. coli, such as AIEC. Similarly, the abundance of Akkermansia muciniphila, a major bacterial species in the Verrucomicrobiaceae family, was significantly increased in the L-serine deficient condition (Figure 3C). These data indicate that the restriction of dietary L-serine leads to the unexpected blooms of E. coli harboring AIEC pathotypes, together with other commensal symbionts, such as A. muciniphila, in the inflamed gut.
Figure 3. Deprivation of dietary L-serine fosters blooms of pathotype E. coli and A. muciniphila in the inflamed gut.

(A) Feces were collected from Ctrl diet– and ΔSer diet–fed mice with and without DSS treatment, and DNA was isolated. Gut microbiota was analyzed by 16S rRNA sequencing.
(B) Significantly enriched bacterial taxa in Ctrl diet–fed mice (blue bars) and ΔSer diet–fed mice (red bars) were identified by LEfSe analysis.
(C) The relative abundance of A. muciniphila and E. coli was each quantified by qPCR.
(D) The heat map shows the abundance of E. coli virulence genes in ΔSer diet–fed colitis mice compared to Ctrl diet–fed colitis mice.
Data are representative of two independent experiments (N = 4–5). Dots indicate individual mice, with mean ± SEM. N.S., not significant. ***p < 0.001 by 1-way ANOVA with Tukey post hoc test.
A. muciniphila enables AIEC to relocate to the epithelial niche by degrading the mucus layer
As AIEC requires L-serine for its fitness in the inflamed gut (Kitamoto et al., 2020), we did not expect dietary L-serine restriction to induce an AIEC bloom during inflammation. The suppression of AIEC growth by dietary L-serine restriction in the setting of intraspecific competition (Kitamoto et al., 2020) suggests that other bacterial species in SPF mice may act as metabolic supporters for AIEC to overcome the nutrient limitation. In this regard, we focused on A. muciniphila as a metabolic supporter for AIEC. We first assessed the growth kinetics of A. muciniphila and E. coli during colitis. As colitis progressed, the abundance of A. muciniphila gradually decreased in Ctrl diet–fed mice (Figure 4A). In contrast, when dietary L-serine was restricted, A. muciniphila was markedly increased on day 1 after DSS treatment and it maintained a higher abundance until day 7 compared (Figure 4A). As A. muciniphila does not require L-serine for its growth (Derrien et al., 2004), it may have a growth advantage over other commensal gut bacteria under the nutrient-restricted condition. Of note, the abundance of E. coli (likely enriched with AIEC) was unchanged in the early stage of colitis but dramatically increased at 7 days after DSS treatment with L-serine starvation (Figure 4A). These results suggest that under these conditions, the expansion of A. muciniphila may trigger the subsequent proliferation of AIEC.
Figure 4. Disruption of colonic mucus barrier under L-serine starvation enhances the encroachment of AIEC to the epithelial niche.

(A) Relative abundance of A. muciniphila and E. coli during DSS treatment were each assessed by qPCR.
(B and C) Colonic sections were stained with AB/PAS, and the thickness of the inner mucus layer was measured (scale bar, 100 μm).
(D) Intestinal permeability was assessed with FITC–dextran.
(E) Immunostaining (MUC2, green; DAPI, blue) and FISH (EUB338 probe, red,) of Carnoy’s solution–fixed colonic sections (scale bar, 100 μm).
(F) SPF C57BL/6 mice were fed the Ctrl diet or the ΔSer diet for 3 days, then given 1.5% DSS for 5 days, followed by conventional water for 2 days. Mice were infected with each strain of E. coli (1 × 109 CFU/mouse) on days 5 and 6. On day 7 post-DSS, all mice were euthanized.
(G) Homogenates of colon tissues were cultured on LB agar plates supplemented with ampicillin or streptomycin. The number of viable bacteria was estimated by counting the CFUs and calculating the fold change (ΔSer diet/Ctrl diet).
Data are representative of two independent experiments (N = 4–5). Dots indicate individual mice, with mean ± SEM or geographic mean ± SD. N.S., not significant, *p < 0.05, **p < 0.01, ***p < 0.001 by 1-way ANOVA with Tukey post hoc test or unpaired t test.
A. muciniphila is a mucolytic bacterium capable of degrading mucus by several glycoside hydrolases that target the host mucus glycans (Derrien et al., 2004). Thus, the expansion of A. muciniphila may cause mucus barrier dysfunction. Consistent with this notion, we observed that the thickness of the inner mucus layer was significantly reduced in the ΔSer diet–fed mice after DSS treatment, with the expansion of A. muciniphila (Figures 4B and 4C). Notably, in the steady state (i.e., without expanding A. muciniphila), the ΔSer diet had no apparent effect on the mucus barrier (Figures 4B and 4C). As intestinal mucus acts as a physical barrier that keeps luminal antigens, including resident microbiota, distant from the host epithelial cells (Johansson et al., 2008; Van der Sluis et al., 2006), a defective intestinal mucus barrier may result in the penetration of luminal antigens and increase the risk of colitis (Johansson et al., 2014; Van der Sluis et al., 2006). Consistent with this notion, dietary L-serine deprivation increased intestinal permeability after DSS treatment (Figure 4D). Also, degradation of the mucus layer by dietary L-serine restriction brought the luminal bacteria closer to the IECs (Figure 4E). Notably, dietary L-serine deprivation significantly facilitated the encroachment of Enterobacteriaceae in colon tissues of colitis mice (Figure S3). Thus, the A. muciniphila expansion may promote the encroachment of luminal bacteria, including AIEC, close to the colonic epithelium, and it may contribute to the increased susceptibility to colitis. To validate whether disruption of the mucus barrier under L-serine restriction facilitates the localization of AIEC to the epithelial niche, SPF mice were fed the Ctrl diet or the ΔSer diet to induce the A. muciniphila expansion and subsequent mucus barrier disruption in colitic mice (Figure 4F). After disrupting the mucus layer by the feeding of the ΔSer diet, followed by DSS treatment, mice were challenged exogenously with AIEC strains (LF82 and CUMT8) or commensal E. coli strains (HS and MG1655) (Figure 4F). As shown in Figure 4G, the number of colonic mucosa-associated AIEC strains, but not commensal E. coli strains, was significantly higher in the ΔSer diet–fed mice (disrupted mucus layer) than in the Ctrl diet–fed mice (intact mucus layer).
To further examine the direct interaction between A. muciniphila and AIEC in the gut, we cocultured A. muciniphila and AIEC in the presence of a human-derived colonoid monolayer (HCM). As A. muciniphila is an obligate anaerobe (Derrien et al., 2004), we used a two-chamber system that enables the coculture of anaerobic bacteria in the upper anaerobic chamber and the HCM supplemented with oxygen in the lower aerobic chamber (Figure 5A) (Lauder et al., 2020). In this culture system, the HCM secreted mucin and formed a thick mucus layer on its surface (Figure 5B). After coculture with A. muciniphila, the mucus layer was dramatically reduced (Figure 5B). In contrast, exposure to AIEC strain LF82 did not affect the mucus layer on the HCM (Figure 5B). Notably, the presence of A. muciniphila facilitated the encroachment of AIEC LF82 to the HCM (Figure 5B). This consequence was further validated by enumeration of mucosa-associated AIEC. Consistent with the observations of immunofluorescence staining, the association of AIEC LF82 with the HCM was significantly increased in the presence of A. muciniphila (Figure 5C). Conversely, the number of nonadherent AIEC LF82 (i.e., floating in media) was even reduced when cocultured with A. muciniphila (Figure 5C). These findings suggest that A. muciniphila–mediated mucus degradation facilitates the encroachment of AIEC to the epithelial niche.
Figure 5. A. muciniphila-mediated mucus disruption facilitates adhesion of AIEC to IECs.
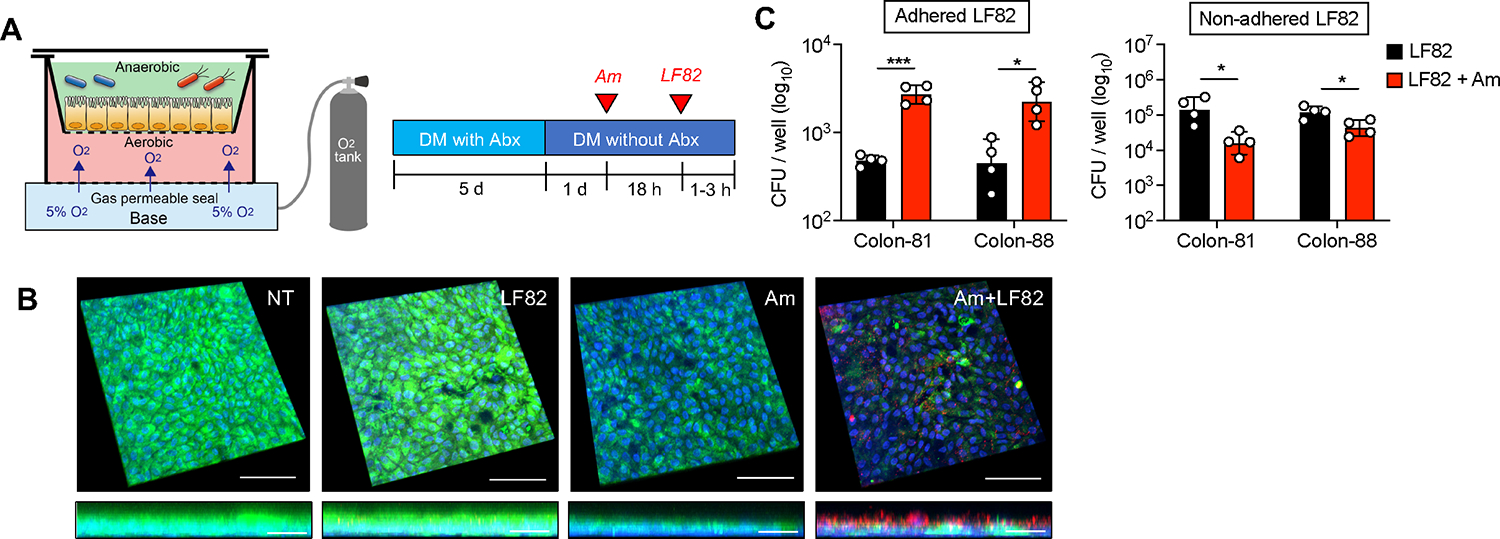
(A) Assembly of anaerobic coculture system. The human-derived colonoid monolayer (HCM) from each donor (colon-81 and colon-88) was differentiated for 6 days by differentiation media (DM) with or without antibiotics (Abx). A. muciniphila was infected for 18 h, and then AIEC LF82 was infected for 1–3 h.
(B) Immunofluorescence staining of MUC2 (green), E. coli (red), and DAPI (blue). Scale bar, 100 μm (XYZ axis) and 20 μm (XZ axis).
(C) Cell-associated AIEC LF82 was cultured on LB agar plates supplemented with ampicillin. The number of viable bacteria was estimated by counting the CFUs.
Data are representative of two independent experiments (N = 4). Dots indicate individual mice, with geographic mean ± SD. N.S., not significant. *p < 0.05, ***p < 0.001 by unpaired t test.
AIEC and A. muciniphila cooperate to promote gut inflammation under dietary L-serine restriction
Thus far, we determined that A. muciniphila, which can proliferate independent of L-serine, expands when dietary L-serine is restricted and facilitates the epithelial localization of AIEC strains in the gut by degrading the mucus layer. However, the link between AIEC–A. muciniphila interaction and the increased susceptibility to colitis remained unclear. Hence, we next examined the involvement of this bacterial cooperation in the exacerbation of colitis under dietary L-serine restricted conditions. To this end, we generated gnotobiotic mice colonized by AIEC and A. muciniphila. To assess the importance of the mucus-degrading capacity of A. muciniphila, we used a consortium of nonmucolytic bacterial strains as the base bacterial community. We modified a known synthetic human gut microbiota (SM) model (Desai et al., 2016). The original SM consortium is composed of 14 fully sequenced human commensal gut bacteria representing the five dominant phyla, which collectively possess essential core metabolic capabilities (Desai et al., 2016). We removed 4 species of mucolytic bacteria (Bacteroides caccae, B. thetaiotaomicron, Barnesiella intestinihominis, and A. muciniphila) and commensal E. coli from the original consortium. We defined this new base consortium composed of 9 species of nonmucolytic commensal symbionts as the “nonmucolytic synthetic human gut microbiota” (NmSM) (Figure 6A). To evaluate the interaction between AIEC and A. muciniphila, we added AIEC LF82 with or without A. muciniphila to the base NmSM community (NmSM+LH82 and NmSM+LF82+Am) (Figure 6A). Likewise, for the control groups, the commensal E. coli strain HS replaced AIEC LF82 (NmSM+HS and NmSM+ HS+Am) (Figure 6A). All mice were fed with the ΔSer diet, and colitis was induced by DSS treatment (Figure 6A). In the absence of A. muciniphila, and under dietary L-serine restriction, the colonization of AIEC LF82 did not exacerbate colitis compared to the colonization of HS (Figures 6B–6F). The presence of A. muciniphila did not alter the susceptibility to colitis in the commensal E. coli HS–colonized mice, although colitis led to a marked expansion of A. muciniphila (Figures 6B–6F). These results indicated that A. muciniphila can gain a growth advantage over other commensal symbionts in the gut when dietary L-serine intake is limited; however, A. muciniphila per se is not colitogenic. Notably, unlike commensal E. coli, cocolonization of AIEC LF82 and A. muciniphila significantly exacerbated colitis (Figures 6B–6F). These results suggest that the colonization of AIEC alone is not sufficient to exacerbate colitis, whereas A. muciniphila may promote the colitogenic capability of AIEC. Consistent with the severity of colitis, the presence of A. muciniphila promoted the expansion of AIEC LF82, but not commensal E. coli HS, in both fecal and mucosal compartments (Figures 6G–6I). These results demonstrated a causal role of the interaction between AIEC and A. muciniphila in the exacerbation of colitis. It is noteworthy that this pathogenic interaction is only observed in the absence of dietary L-serine. In the gnotobiotic mice fed the Ctrl diet (i.e., L-serine–sufficient), the cocolonization of AIEC LF82 and A. muciniphila did not increase the susceptibility to colitis (Figure S4). The maintenance of the intact mucus layer may explain this phenotype (Figures S4B and S4C). Additionally, we evaluated transcripts of glycoside hydrolase (GH) enzyme associated with the degradation of mucus glycan (Desai et al., 2016) in A. muciniphila. As expected, dietary L-serine restriction significantly up-regulated the transcripts of GH enzymes, including GH20, 33, 84, and 109 (Figure S4D), suggesting that A. muciniphila enhanced mucus degradation under dietary L-serine deprivation.
Figure 6. AIEC and A. muciniphila cooperatively exacerbate colitis under L-serine restriction.
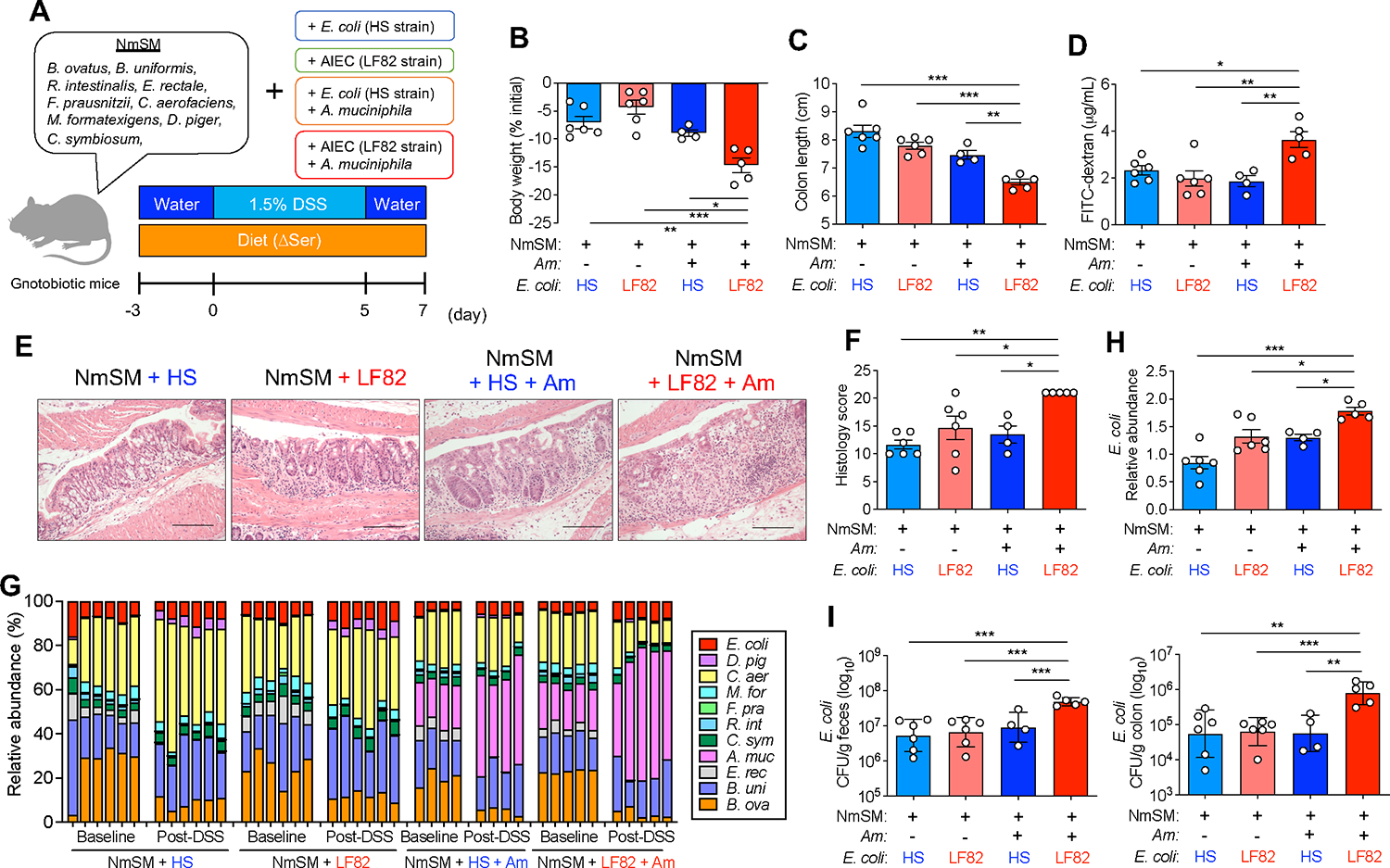
(A) Experimental protocol and the composition of the nonmucolytic synthetic human gut microbiota (NmSM) for the gnotobiotic mouse experiments.
(B and C) Body weight and colon length were measured 7 days post DSS treatment.
(D) Intestinal permeability was assessed with FITC–dextran.
(E and F) Representative histological images of colonic sections stained with HE (scale bar, 200 μm) and the histology scores.
(G and H) The relative abundance of each bacterial strain at baseline and post-DSS treatment was assessed by qPCR. Fold change of E. coli abundance (post-DSS/baseline) was calculated.
(I) Homogenates of feces or colon tissues were cultured on LB agar plates. The number of viable bacteria was estimated by counting the CFUs.
Data are representative of two independent experiments (N = 5–6). Dots indicate individual mice, with mean ± SEM or geographic mean ± SD. N.S., not significant. *p < 0.05, **p < 0.01, ***p < 0.001 by 1-way ANOVA with Tukey post hoc test. See also Figure S3.
AIEC exploits host-derived L-serine to counteract dietary L-serine deprivation
We demonstrated that mucolytic bacteria promote the relocation of AIEC to the epithelial niche, where it can evade nutrient (i.e., L-serine) restriction, proliferate, and facilitate intestinal inflammation. However, the mechanism by which AIEC proliferates in the epithelial niche under dietary L-serine restriction remained unclear. We hypothesized that AIEC exploits host-derived nutrients in the epithelial niche, as some pathogens can liberate host-derived nutrients for their growth (Eisenreich et al., 2010). To determine if AIEC uses host-derived nutrients for its growth, we first compared the growth of LF82 cultured with or without IECs. As shown in Figure 7A, the coculture of AIEC LF82 and IECs significantly enhanced the growth of AIEC LF82 compared with the host cell–free condition. This bacterial growth enhancement by IECs was not observed in the commensal E. coli strain HS, nor in the LF82 ΔfimH mutant strain that lacks genes involved in adhesion to IECs (Figure 7B), which suggests that bacterial adhesion to IECs is required for the utilization of host-derived nutrients. To identify the host-derived nutrients used by AIEC in the epithelial niche, we analyzed the transcriptomic changes of AIEC LF82 induced by the association with the HCM (Figure S5A). RNA-seq analysis demonstrated that epithelial association significantly altered the transcriptional profiles of AIEC LF82 (Figure S5B). The Gene Ontology (GO) enrichment analysis showed that the coculture of AIEC LF82 and HCM upregulated the pathways involved in AIEC growth, including ribosome biosynthesis and protein folding, response to stress, and sugar transport (Figure S5C). In contrast, the pathways related to chemotaxis and amino acid biosynthesis were downregulated (Figure S5C). In contrast, the presence of AIEC had a minor impact on the transcriptomic profiles of the HCM (Figures S5D and S5E). Notably, AIEC LF82 upregulated genes related to L-serine transport (sdaC and tdcC) and metabolism (sdaA) and downregulated genes related to L-serine biosynthesis (serA and serC) when associated with the HCM (Figure 7C), indicating that AIEC acquires L-serine from the host epithelium and uses it for its growth. In fact, IEC presence did not promote the growth of the ΔtdcΔsda (ΔTS) mutant AIEC LF82 strain, which lacks two major L-serine utilization gene operons and is incapable of utilizing L-serine (Kitamoto et al., 2020), unlike its effect on LF82 WT (Figure 7D). Consistent with this finding, the association with AIEC LF82 significantly reduced the concentration of free intracellular L-serine in the IECs, whereas the association with LF82 ΔTS had no effect (Figure 7E), indicating that AIEC LF82 consumes L-serine in the infected host cells. Further, we confirmed that although host-derived L-serine is not required for the initial AIEC adhesion and invasion, it is vital for AIEC growth after it associates with the host cells. For example, early phase (1 h) adherence and invasion of AIEC did not differ between the WT and the ΔTS mutant AIEC LF82 strains (Figures 7F and 7G). However, in the later phase (3 h), proliferation of AIEC after adherence and invasion was significantly impaired in the ΔTS mutant strain compared to the WT strain (Figures 7F and 7G). Interestingly, the deprivation of L-serine from the media facilitated the adhesion and invasion of AIEC LF82 to IECs (Figure 7H). This evidence suggests that AIEC enhances its growth after adhering to host cells by utilizing host-derived L-serine.
Figure 7. L-serine utilization by AIEC is a partial requirement for the exacerbation of colitis under L-serine deprivation.
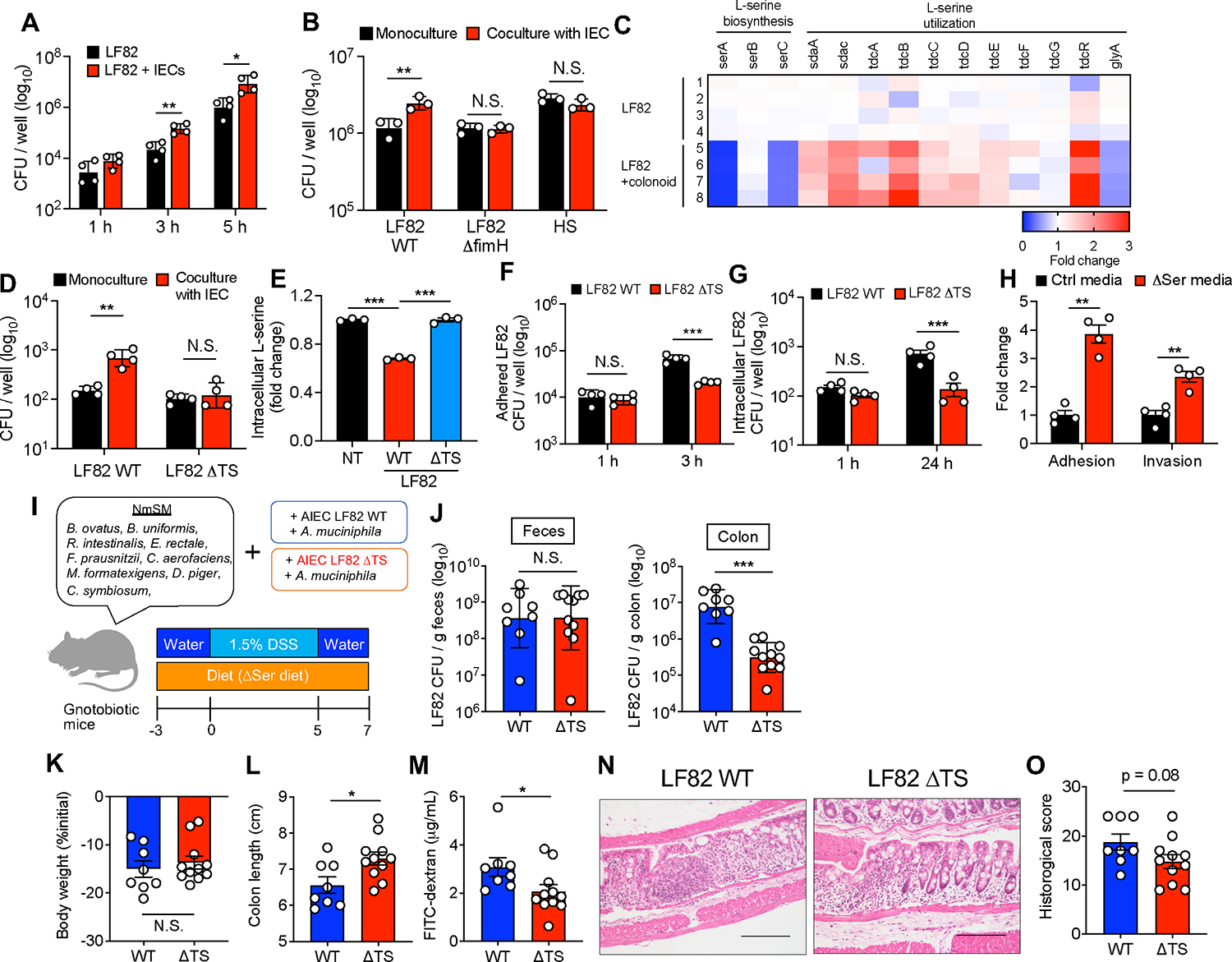
(A and B) E. coli strains were cultured with and without T84 cells. After 1–5 h infection, the CFUs of total bacteria, including adhered and nonadhered bacteria, were counted.
(C) AIEC LF82 was monocultured or cocultured with the human-derived colonoid monolayer (HCM) for 3 h, and the transcriptomic profiles were evaluated by RNA-seq. The heat map shows fold changes of L-serine metabolism genes (LF82 + HCM/LF82).
(D) LF82 WT or ΔTS mutant strains were infected in T84 cells for 5 h, and then the CFUs of total bacteria were counted.
(E) Fold changes of intracellular L-serine after infection of T84 cells with AIEC strain LF82.
(F) T84 cells were infected with LF82 WT or ΔTS mutant strains. After 3 h, adhesion bacteria were counted.
(G) T84 cells were infected with LF82 WT or ΔTS mutant strains. After 1 h, the cells were cultured with gentamicin (100 μg/mL) for 24 h. Intracellular bacteria were plated on LB agar plates and counted.
(H) LF82 and T84 cells were cocultured in the Ctrl media or ΔSer media. After a 3 h infection, adhesion and invasion bacteria were plated on LB agar plates and counted.
(I) Experimental design. GF mice were colonized by nonmucolytic synthetic human gut microbiota (NmSM) and A. muciniphila with LF82 WT or ΔTS mutant strains.
(J) On day 7 post-DSS, all mice were euthanized, and the LF82 burden in the colon and feces was assessed.
(K and L) Body weight and colon length.
(M) Intestinal permeability was evaluated by FITC–dextran assay.
(N and O) Representative histological images (scale bar, 200 μm) and histology scores were evaluated.
(A–H) Data are representative of two or three independent experiments (N = 3–4). (J–O) Data pooled from two independent experiments (N = 4–7). Dots indicate individual mice, with mean ± SEM or geographic mean ± SD. N.S., not significant. *p < 0.05, **p < 0.01, ***p < 0.001 by 1-way ANOVA with Tukey post hoc test or unpaired t test. See also Figure S4.
Lastly, we assessed the extent to which the utilization of host-derived L-serine by AIEC is linked to the susceptibility to colitis. To this end, NmSM-colonized gnotobiotic mice were colonized with either AIEC LF82 WT or ΔTS mutant AIEC LF82 together with A. muciniphila (Figure 7I). Both groups of mice were fed the ΔSer diet, followed by a DSS challenge. The colonization of LF82 WT or the ΔTS mutant was comparable in feces, whereas the number of AIEC associated with the colonic mucosa was significantly lower in the ΔTS mutant strain than in the LF82 WT strain (Figure 7J), suggesting that AIEC LF82 may exploit host-derived L-serine in the epithelial niche. Furthermore, consistent with the impaired proliferation of AIEC in the epithelial niche, the mice colonized with the LF82 ΔTS mutant strain displayed an attenuated degree of colitis compared to the mice colonized with the LF82 WT strain (Figures 7K–7O).
DISCUSSION
In this study, we show that the biosynthesis and utilization of L-serine are disturbed in the gut microbiota of patients with IBD, which is consistent with our recent research showing that the IBD-associated pathobiont AIEC upregulates L-serine catabolism in the inflamed gut (Kitamoto et al., 2020). We had expected that the deprivation of dietary L-serine would attenuate inflammation by suppressing the expansion of pathobionts, such as AIEC. However, we observed that dietary L-serine restriction leads to the expansion of AIEC and subsequent exacerbation of colitis. This unexpected and adverse effect of dietary L-serine deprivation is context dependent. Dietary L-serine restriction promotes the abnormal expansion of AIEC only when it coexists with mucolytic bacteria, such as A. muciniphila. The fact that L-serine is a crucial nutrient for the growth of various gut bacteria, including AIEC, but not A. muciniphila, gives A. muciniphila a growth advantage over other commensal microbes under L-serine restriction. Notably, the blooms of A. muciniphila per se are not detrimental. However, amassed A. muciniphila facilitates the encroachment of AIEC to the epithelial niche by degrading the mucus barrier. As a result, AIEC can reside in the epithelial niche and counteract dietary L-serine restriction by extracting host-derived nutrients. Notably, AIEC can acquire L-serine pooled in the host colonic epithelium. This insight advances the understanding of the complex interplay among pathobionts, symbionts, and host cells in the context of gastrointestinal diseases, such as IBD.
Dietary amino acids are vital nutrients for maintaining intestinal homeostasis and the gut microbiota (Sugihara et al., 2018). L-serine is thought to be a conditionally essential amino acid as it plays a critical role in the cellular metabolisms of both mammalian and bacterial cells only under certain conditions (Kitamoto et al., 2020; Ma et al., 2017; Maddocks et al., 2017; Rodriguez et al., 2019). Several biosynthetic and signaling pathways require L-serine, including the synthesis of other amino acids, the production of phospholipids, and the provision of one-carbon units to the folate cycle, which are used for the de novo synthesis of nucleotides (Yang and Vousden, 2016). L-serine also contributes to the production of glutathione, which is essential for reducing toxic oxidants and metabolic byproducts. Thus, L-serine supports several metabolic processes that are crucial for growth and adaptation to the microenvironment. It has been reported that some E. coli strains rapidly consume L-serine, as compared to other amino acids (Prüss et al., 1994). Notably, E. coli promotes the consumption of L-serine under heat stress (Matthews and Neidhardt, 1989). Consistently, our work has demonstrated that bacteria belonging to the Enterobacteriaceae family, including E. coli, use L-serine to adapt to the inflamed gut (Kitamoto et al., 2020). Thus, some bacteria, most likely pathogens, utilize L-serine metabolism to counteract environmental stress. The multi’omics database shows that bacteria that catabolize L-serine (i.e., bacteria-harboring L-serine dehydratase [SDH]) are significantly enriched in patients with IBD compared to non-IBD controls. In contrast, bacteria that synthesize L-serine or convert L-glycin to L-serine (i.e., bacteria-harboring phosphoglycerate dehydrogenase [PHGDH] or serine hydroxymethyltransferase [SHMT], respectively) are decreased in IBD, particularly in CD, compared to IBD controls. These results suggest that L-serine consumers are enriched in IBD. Although we need to clarify the mechanism by which gut bacteria consume L-serine in IBD, certain bacteria may use L-serine to adapt to the inflammatory microenvironment. How the concentration of luminal L-serine is regulated remains unclear. Diet is the primary source of luminal L-serine, as dietary deprivation significantly reduces its concentration in the gut lumen (Kitamoto et al., 2020). Regarding the consumption of L-serine, both host cells and bacteria are L-serine utilizers in the gut (Caballero-Flores et al., 2020; Ma et al., 2017; Nagao-Kitamoto et al., 2016). Generally, L-serine is contained in protein-rich foods, such as meat, fish, eggs, and soybeans. A recent systematic review has shown that fiber and calcium intakes are insufficient in IBD, whereas protein intake meets or exceeds the recommended amount (Lambert et al., 2021). Therefore, the reduced concentration of L-serine in the gut lumen of individuals with IBD may be caused by the excessive consumption of L-serine by the gut microbiota or the host cells rather than by the insufficient intake of protein. As mentioned, L-serine plays a pivotal role in the fitness of certain bacteria, including pathobionts such as AIEC, particularly when exposed to environmental stress. Consequently, pathobionts may evolve backup systems to evade the shortage of such a vital nutrient. This seems to be a strategy that pathobionts use to maintain a fitness advantage over commensal competitors in disease conditions. In the present study, we found that AIEC can switch the source of L-serine from the diet to the host cells when dietary L-serine is limited. To acquire host-derived L-serine, AIEC relocates its colonizing niche from the gut lumen to the mucosa. Given the evidence that the availability of L-serine in the gut lumen is decreased in patients with IBD, the enrichment of mucosa-associated AIEC in IBD may be triggered by the perturbed amino acid homeostasis in the gut lumen.
As L-serine metabolism plays a vital role in the survival of some gut microbes, particularly pathobionts, this metabolic pathway can be a therapeutic target for pathobiont-driven inflammatory diseases, such as IBD. Our previous work showed that the dietary deprivation of L-serine suppresses AIEC expansion (Kitamoto et al., 2020). In the current study, dietary L-serine deprivation resulted in the unexpected expansion of AIEC. Thus, the therapeutic effects of the dietary intervention are context dependent. In other words, the same dietary therapy could be either beneficial or detrimental. The factor that dictates the efficacy of dietary interventions may be the basal microbiota composition of individuals. For example, dietary L-serine restriction may not significantly impact patients who are not colonized by AIEC or colonized by AIEC without coexistent mucolytic bacteria. In individuals colonized by AIEC and metabolic competitors for AIEC (e.g., certain commensal E. coli strains) but lacking mucolytic bacteria, dietary L-serine restriction can facilitate the competitive elimination of AIEC by the commensal E. coli strains. Indeed, gnotobiotic mice colonized by the microbiota from a patient with CD displayed a reduction of Enterobacteriaceae and the attenuation of colitis when L-serine was removed from the diet (Kitamoto et al., 2020). This result implies that this individual had a sufficient number of metabolic competitors for AIEC and the restriction of dietary L-serine prompted the competitive elimination of AIEC. On the other hand, as shown in the current study, in the presence of mucolytic bacteria, AIEC can overcome nutritional restriction and exacerbate colitis. Notably, A. muciniphila has a protective role in some individuals with metabolic diseases, such as obesity and diabetes mellitus (Dao et al., 2016; Everard et al., 2013), and therefore, it has been proposed as a probiotic bacterium (Cani and de Vos, 2017). Consistently, in the present study, A. muciniphila colonization per se did not exacerbate colitis, even under dietary L-serine restriction (with a noticeable A. muciniphila expansion). However, A. muciniphila can serve as a metabolic supporter for AIEC, and hence, indirectly contribute to the pathogenesis of colitis. Thus, the balance of pathobionts and their metabolic competitors and supporters may determine the outcomes of dietary interventions.
Nutritional competition is one of the main strategies used by commensal symbionts to prevent the colonization and proliferation of commensal pathobionts and exogenous pathogens (Cameron and Sperandio, 2015; Guo et al., 2020). To overcome this symbiont-mediated colonization resistance, pathogens have evolved various strategies (Perez-Lopez et al., 2016). For example, some pathogens use unique nutrients, such as ethanolamine, that commensal symbionts cannot use (Thiennimitr et al., 2011). Likewise, relocation of the living niche provides an escape from the nutritional competition. For example, Citrobacter rodentium, a mouse pathogen used to model human infections with enteropathogenic E. coli (EPEC) and enterohemorrhagic E. coli (EHEC), resides on the intestinal epithelial surface by expressing the locus of enterocyte effacement (LEE) virulence factor. In this new niche, C. rodentium can acquire specific nutrients not accessible to commensal symbionts residing in the luminal niche (Kamada et al., 2012). However, unlike C. rodentium or other enteropathogens, commensal pathobionts, such as AIEC, lack the LEE virulence factors, and therefore the niche relocation is rarely executed in the steady-state (i.e., intact gut microbiota and mucus barrier). These pathobionts are, therefore, not classified as obligate pathogens, but rather as opportunistic pathogens. In other words, pathobionts have a poor ability to proliferate and establish infection in the healthy hosts. Instead, pathobionts bloom only in conditions that compromise the host’s defenses, including the colonization resistance by the healthy gut microbiota. In this context, we found that mucolytic bacteria, such as A. muciniphila, can serve as metabolic supporters for AIEC in the gut. Although A. muciniphila may not directly promote the growth of AIEC, A. muciniphila licenses AIEC to acquire alternative nutrients by facilitating its niche relocation.
Some pathogens, like AIEC, which can reside in the epithelial niche, can obtain nutrients from infected host cells for replication. For example, EPEC exploits nutrients from infected host cells by using injectisome components, which enable the pathogen to thrive in competitive niches (Pal et al., 2019). As AIEC strains lack injectisome components, it is plausible that AIEC exploits a distinct mechanism to extract host-derived nutrients when associated with the epithelial niche. In this regard, AIEC may invade the colonic epithelium to acquire nutrients from the host cells rather than extracting nutrients from the cell surface, as cellular invasion is a unique pathogenic feature of AIEC strains compared to other pathogenic E. coli strains (Glasser et al., 2001; Lapaquette et al., 2010). Further research is required to identify the mechanisms by which AIEC exploits host-derived nutrients. Our present study shows that AIEC uses L-serine pooled in the host epithelium. This notion was supported by the upregulation of L-serine utilization genes in AIEC when associated with the IECs, along with the reduced free L-serine concentration in the IECs. Moreover, the growth promotion of AIEC due to the AIEC–IEC interaction does not occur if the AIEC strain is incapable of utilizing L-serine (i.e., ΔTS mutant AIEC LF82). Along with the aforementioned critical roles of L-serine that enable AIEC to adapt to the inflammatory environment, our results confirm that L-serine is a major nutrient extracted from the host cells by AIEC in this setting. However, AIEC may extract and use other host-derived nutrients in addition to L-serine. In this context, several pathogens exploit host glycan metabolites as nutrient sources. For example, Salmonella enterica serovar Typhimurium and Clostridioides difficile use sialic acid liberated from host mucus glycans by Bacteroides thetaiotaomicron (Ng et al., 2013). EHEC uses fucose, also liberated from host glycans by B. thetaiotaomicron, for the regulation of virulence factor expression and colonization in the gut (Pacheco et al., 2012). AIEC strains can also use fucose as a nutrient source through propanediol dehydratase (Viladomiu et al., 2017; Viladomiu et al., 2021). Consistent with this notion, AIEC upregulated genes related to the metabolism of other possible host-derived nutrients (e.g., mannose, N-acetylglucosamine) when associated with IECs. These other metabolites may compensate for the growth of AIEC, at least to some extent, in the absence of L-serine. Nevertheless, the studies by us and others as described suggest that L-serine plays a major role among diet- and host-derived nutrients accessible to AIEC in regulating its fitness.
Considered in their entirety, our results demonstrate that the pathogenic capacity of commensal pathobionts, such as AIEC, is context dependent. In the steady-state gut, pathobionts may behave as nonpathogenic commensals. Pathobionts may be detrimental only when metabolic supporters are present. Notably, the metabolic supporters, such as mucolytic bacteria, per se are not detrimental. Also, the balance of luminal nutrients, regulated by diet, is essential to elicit the interaction between pathobionts and metabolic supporters. Therefore, the complex pathobiont–symbiont interactions dictate the success of dietary interventions. Hence, a personalized dietary intervention adapted to the composition of an individual’s gut microbiota is required to treat IBD effectively.
LIMITATIONS OF THE STUDY
Although our study provides a comprehensive understanding of the diet–microbe interactions in the inflamed gut, several limitations deserve mention. First, we need to clarify how dietary L-serine restriction expands A. muciniphila in the inflamed gut. As A. muciniphila is decreased in the mice-fed a control diet, A. mucniphila may exploit unique strategies to overcome the inflammatory microenvironment under L-serine deprivation. Regarding clinical relevance, we also need to investigate the interaction between AIEC and other mucolytic bacteria, such as Ruminococcus gnavus, which is reported to be a pathobiont involved in the pathogenesis of IBD. As some clinical studies have shown the reduced abundance of A. muciniphila in IBD patients, it is possible that other mucolytic bacteria also license AIEC to colonize the epithelial niche in human IBD. Second, we show that AIEC exploits L-serine provided by host IECs; however, the mechanism by which L-serine is required for the colonization and replication of AIEC in IECs remains unclear. AIEC preferentially consumes L-serine in the gut, and moreover, L-serine is required to adapt to the inflammatory microenvironment. Therefore, it is conceivable that AIEC uses L-serine for the stress response and the energy source for bacterial growth. Elucidation of these limitations will advance the understanding of diet–microbe interactions during gut inflammation.
STAR METHOD
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and fulfilled by the Lead Contact, Nobuhiko Kamada (nkamada@umich.edu).
Material Availability
Bacterial strains used in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Bacterial 16S rRNA sequencing data and bacterial RNA sequencing data reported in this paper have been deposited and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 goat anti-rabbit IgG | Invitrogen | A11008 |
| Alexa Fluor 555 goat anti-mouse IgG | Invitrogen | A32727 |
| Alexa Fluor 555-conjugated EUB338 probe | Invitrogen | N/A |
| Anti-E. coli LPS antibody | Abcam | ab35654 |
| Mucin 2 antibody (H-300) | Santa Cruz Biotechnology | sc-15334 |
| Bacterial and Virus Strains | ||
| Adherent-invasive Escherichia coli: LF82 strain | Darfeuille-Michaud et al. Gastroenterology. 1998 | N/A |
| Adherent-invasive Escherichia coli: LF82 ΔTS mutant strain | Kitamoto et al. Nat Microbiol. 2020 | N/A |
| Adherent-invasive Escherichia coli: LF82 ΔfimH mutant strain | Imai et al. Mucosal Immunol. 2019 | N/A |
| Adherent-invasive Escherichia coli: CUMT8 strain | Kitamoto et al. Nat Microbiol. 2020 | N/A |
| Akkermansia muciniphila: DSM 22959, type strain | DMSZ | DMS 22959 |
| Bacteroides ovatus: DSM 1896, type strain | DMSZ | DSM 1896 |
| Bacteroides uniformis: ATCC 8492, type strain | ATCC | ATCC 8492 |
| Clostridium symbiosum: DSM 934, type strain | DMSZ | DSM 934 |
| Collinsella aerofaciens: DSM 3979, type strain | DSMZ | DSM 3979 |
| Desulfovibrio piger: ATC 29098, type strain | ATCC | ATCC 29098 |
| Escherichia coli: HS strain | ATCC | N/A |
| Escherichia coli: MG1655 strain | Miranda et al. Infect Immun. 2004 | N/A |
| Eubacterium rectale: DSM 17629, A1-86 | DSMZ | DSM 17629 |
| Faecalibacterium prausnitzii: DSM 17677, A2-165 | DSMZ | DSM 17677 |
| Marvinbryantia formatexigens: DSM 14469, type strain, l-52 | DSMZ | DSM 14469 |
| Roseburia intestinalis: DSM 14610 type strain, L1-82 | DSMZ | DSM 14610 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetic acid | Thermo Fisher Scientific | A38S |
| Alcian blue | Sigma-Aldrich | A5268 |
| Ampicillin | Sandoz Inc | 0781-3408 |
| BHI agar | BD Difco | BD241830 |
| Bovine Serum Albumins | Thermo Fisher Scientific | BP1600 |
| Chloroform | Thermo Fisher Scientific | 423550250 |
| DAPI | Sigma-Aldrich | D9542 |
| Dextran Sodium Sulfate | MP Biomedical | 0216011090 |
| DMEM | Gibco | 11320033 |
| Ethanol | Thermo Fisher Scientific | BP28184 |
| FITC-dextran | Sigma-Aldrich | FD4 |
| HBSS | Thermo Fisher Scientific | 14170112 |
| Isopropanol | Thermo Fisher Scientific | BP26324 |
| Kanamycin | Thermo Fisher Scientific | BP906-5 |
| LB agar | Thermo Fisher Scientific | BP1425 |
| Lithium chloride solution | Invitrogen | AM9480 |
| Metronidazole | Fagron | 804380 |
| Methanol, extra dry | Thermo Fisher Scientific | 326950025 |
| Neomycin | AG Scientific | N-1053 |
| Paraformaldehyde 4% | Thermo Fisher Scientific | AAJ19943K2 |
| PAS staining kit | Sigma-Aldrich | 95B-1KT |
| PBS | Gibco | 10010023 |
| Phenol:Chloroform:Isoamyl Alcohol (ph: 8) | Thermo Fisher Scientific | 15593031 |
| Phenol:Chloroform:Isoamyl Alcohol (ph: 4.5) | Thermo Fisher Scientific | AM9722 |
| Permount™ Mounting Medium | Thermo Fisher Scientific | SP15 |
| RNAprotect | QIAGEN | 76506 |
| RNAlater Stabilization Solution | Invitrogen | AM7020 |
| Sheep blood | Hemostat Laboratories | DSB250 |
| SDS | Thermo Fisher Scientific | BP166 |
| Sodium Acetate (3 M), pH 5.5 | Invitrogen | AM9740 |
| Sodium Chloride | Thermo Fisher Scientific | BP358 |
| Spermine | Sigma-Aldrich | S3256 |
| Tris-HCl, 1M | Thermo Fisher Scientific | BP1758-100 |
| Vancomycin | APP Pharmaceuticals | 63323028420 |
| Critical Commercial Assays | ||
| BioMasher® II Micro Tissue Homogenizers | DWK Life Sciences | 749625-0030 |
| E.Z.N.A.Total RNA Kit I | OMEGA | R6834-01 |
| E.Z.N.A. Bacterial RNA Kit | OMEGA | R6950-01 |
| DL-Serine Assay Kit | Sigma-Aldrich | MAK352 |
| DNA Removal Kit | Invitrogen | AM1906 |
| DNeasy Blood & Tissue Kit | QIAGEN | 69506 |
| High-Capacity RNA-to-cDNA Kit | Thermo Fisher Scientific | 4387406 |
| Protein Concentrators PES, 10K MWCO | Thermo Fisher Scientific | 88513 |
| Radiant™ SYBR Green Lo-ROX qPCR Kits | Alkali Scientific | QS1020 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| TrueVIEW® Autofluorescence Quenching Kit | Vector Laboratories | SP-8400-15 |
| Deposited Data | ||
| 16S rRNA and RNA sequencing data | This paper | PRJNA763203 |
| Experimental Models: Cell Lines | ||
| T84 cell line | ATCC | CCL-248 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J: WT | The Jackson Laboratory | JAX 000664 |
| Mouse: Germ-free Swiss Webster | Germ-free Facility at University N/A of Michigan | N/A |
| Software and Algorithms | ||
| Mothur v1.39.0 | Schloss et al. Appl Environ Microbiol. 2009 | N/A |
| Prism 9.2 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | Schneider et al. Nat Methods. 2012 | http://imagej.net/Welcome |
| Other | ||
| Amino acid-based control diet | Envigo | TD.130595 |
| Serine and glycine deficient diet | Envigo | TD.140546 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human data
Metagenomics, metatranscriptomics, and metabolomics data were downloaded from the public resource, the second phase of the Integrative Human Microbiome Project (HMP2 or iHMP) – the Inflammatory Bowel Disease Multi’omics Database (https://ibdmdb.org/). The description and collection of samples, and the data preprocessing are explained in a previous study (Lloyd-Price et al., 2019). The samples included in the current analysis are described in Figure 1A.
Animal
SPF C57BL/6 mice were housed by the Unit for Laboratory Animal Medicine at the University of Michigan. GF Swiss Webster mice were obtained from the Germ-Free Mouse Facility at the University of Michigan. GF mice were housed in flexible film isolators, and their germ-free status was confirmed weekly by aerobic and anaerobic culture. Female and male mice, age 6 – 12 wk, were used in all experiments. Mice were fed either a control amino acid–based diet (Ctrl., TD.130595) or an L-serine deficient diet (ΔSer, TD.140546), which had been used previously (Kitamoto et al., 2020; Maddocks et al., 2013). The custom diets were manufactured by Envigo (Madison, WI) and sterilized by gamma irradiation. All animal studies were performed in accordance with protocols reviewed and approved by the Institutional Animal Care and Use Committee at the University of Michigan.
Cell culture
T84 cells derived from a human colorectal carcinoma cell line were purchased from ATCC (Gaithersburg, MD) and cultured in Ham’s F-12 nutrient mixture + DMEM (1:1) supplemented with 10% FBS and an antibiotic solution (penicillin-streptomycin). The human stem cell–derived colonoid line was cultured as described in the protocol from the Translational Tissue Modeling Laboratory at the University of Michigan (https://www.umichttml.org). Histologically normal colon tissue from subjects (donors 81, 84, and 88) in our previous studies was used for the colonoid culture (Dame et al., 2018; Tsai et al., 2018). The collection and use of human colonic tissue for the colonoids were approved by the Institutional Review Boards (IRBMED) at the University of Michigan. The experiment was conducted according to the principles stated in the Declaration of Helsinki.
METHOD DETAILS
Histology
The colon tissues were quickly removed and immediately preserved overnight in 4% paraformaldehyde for regular histology assessment or in Carnoy’s fixative (60% dry methanol, 30% chloroform, 10% glacial acetic acid) for mucus barrier evaluation. The preserved colon tissues were then incubated in 70% ethanol or dry methanol, respectively, and processed into paraffin-embedded tissue sections (4–5 μm) and stained with HE for histological assessment. Histological inflammation was scored at the In-Vivo Animal Core in the Unit for Laboratory Animal Medicine at the University of Michigan. A veterinary pathologist performed a blind evaluation of the histological scores. Inflammation and epithelial loss were assessed for severity based on the most severe lesion in each section (i.e., 0, none; 1, mild; 2, moderate; 3, severe; 4, marked). Lesion extent was assessed as the percent of the section affected (0, 0%; 1, 1%–25%; 2, 26%–50%; 3, 51%–75%; 4, 76%–100%). The extent and severity scores for inflammation and epithelial cell loss were multiplied to give a total score for each parameter (range 0–16). The total scores for each parameter were summed to give a total colitis score (range 0–32).
DNA extraction, qPCR, and 16S rRNA sequencing
Fecal DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD), according to a procedure used in a previous study (Nagao-Kitamoto et al., 2016). In the gnotobiotic experiments, fecal DNA was isolated by phenol:chloroform:isoamyl alcohol, as previously described (Steimle et al., 2021). Briefly, 500 μL buffer A (200 mM NaCl, 200 mM Tris, 20 mM EDTA), 210 μL 20% SDS (filter sterilized), and 500 μL phenol:chloroform:isoamyl alcohol (125:24:1, pH 8.0, Thermo Fischer Scientific, Waltham, MA) were added to the cecal content. The mixture was subjected to bead beating for 3 min at 4°C and centrifuged at 4°C (17,000g for 3 min). The aqueous phase was recovered, and 500 μL phenol:chloroform:isoamyl alcohol (pH 8.0) was added. After mixing with a vortex mixer, the mixture was centrifuged again at 4°C (17,000g for 3 min). The aqueous phase was recovered and 500 μL of chloroform was added and mixed by inversion. After centrifuging (17,000g) for 3 min, the aqueous phase was transferred to new tubes, and 60 μL 3 M sodium acetate (pH 5.2) and 600 μL isopropanol were added. After incubation for 60 min at −20°C, the mixture was centrifuged at 4°C (13,000 rpm for 20 min). The pellet was washed with 70% ethanol, and resuspended in nuclease-free water. DNA was further cleaned using the DNeasy Blood and Tissue Kit (Qiagen). qPCR was performed using a Radiant SYBR Green Lo-ROX qPCR Kit (Alkali Scientific, Fort Lauderdale, FL). To eliminate the inhibitory effect of dextran sodium sulfate (DSS) on qPCR, spermine was added to the PCR (Krych et al., 2018). The relative expression of the target bacteria was calculated using universal 16S primers as a reference. In the gnotobiotic experiments, the relative abundance of bacteria was calculated using the standard curves obtained from the monoculture of each strain (Steimle et al., 2021). The primer sets used for amplification are listed in Table S1. For the 16S rRNA sequencing, PCR and library preparation were performed at the Microbiome Core at the University of Michigan. The V4 region of the 16S rRNA-encoding gene was amplified from extracted DNA using the barcoded dual-index primers, as reported previously (Kozich et al., 2013). Samples were amplified, normalized, and sequenced on the MiSeq system. Raw sequences were analyzed using mothur (v1.33.3)(Schloss et al., 2009). Operational taxonomic units (OTUs) (>97% identity) were curated and converted to relative abundance using mothur. We performed LEfSe to identify significant differentially abundant OTUs.
Measurement of the thickness of the colonic mucus layer
To measure the thickness of the colonic inner mucus layer, the colonic sections were stained with alcian blue/periodic acid–Schiff (AB/PAS) according to the following protocol: 1) deparaffinize and hydrate in distilled water, 2) 3% acetic acid for 3 min, 3) AB solution for 15 min, 4) wash in running tap water for 2 min, 4) periodic acid solution for 5 min, 5) rinse with distilled water, 6) Schiff reagent for 15 min, 7) wash in running tap water for 5 min, 8) Gill hematoxylin solution for 90 sec, 9) wash in running tap water for 5 min, 10) dehydrate and clear in xylene, 11) cover with a coverslip. We used the images captured of all the available fecal masses of each mouse for quantification, although this number was variable, and generally, the colitic mice had fewer colonic fecal masses. The thickness of the mucus layer in the colonic sections was measured using ImageJ (Schneider et al., 2012).
FISH and immunofluorescence staining
FISH and MUC2 immunofluorescence staining were performed according to a previous study, with slight modifications (Johansson and Hansson, 2012). Briefly, paraffin-embedded colon sections were deparaffinized and hydrated. Sections were then incubated with 2 μg Alexa555-conjugated EUB338 (5′-GCTGCCTCCCGTAGGAGT-3′) in 200 μL hybridization buffer (20 mM Tris-HCl, 0.9 M NaCl, 0.1% (w/v) SDS) at 50°C. After overnight incubation, sections were rinsed in wash buffer (20 mM Tris-HCl, 0.9 M NaCl) at 50°C for 15 min. Sections were blocked with 1% BSA in PBS at room temperature for 1 h and then incubated with anti-MUC2 antibody (H-300; Santa Cruz Biotechnology, Dallas, TX) at 4°C for 6 h. After washing with PBS, sections were incubated with Alexa 488–conjugated rabbit polyclonal antibody (Invitrogen, Thermo Fisher Scientific, Waltham, MA) and DAPI at room temperature for 1 h. To reduce autofluorescence, the sections were treated with an autofluorescence quenching kit (Vector Laboratories, Burlingame, CA), according to the manufacturer’s instruction. The slides were stored overnight, in the dark, at room temperature, and then visualized using the Nikon Eclipse TE2000-S inverted microscope (Nikon USA, Melville, NY). For the in vitro experiment, immunofluorescence staining of MUC2 and E. coli was performed as previously described, with slight modifications (Liu et al., 2020). Briefly, the human-derived colonoid monolayer (HCM) was fixed with Carnoy’s solution (90% dry methanol and 10% glacial acetic acid), washed 2 times with PBS, permeabilized with 0.1% Triton-X for 10 min, and blocked with 1% BSA/PBS for 30 min. Cells were then incubated with MUC2 antibody (H-300; Santa Cruz Biotechnology) and anti–E. coli LPS antibody (2D7/1; Abcam, Waltham, MA) in 1% BSA/PBS overnight at 4°C. The cells were then washed with PBS twice for 5 min followed by incubation with Alexa Fluor 488–conjugated goat anti-rabbit antibody (Invitrogen), Alexa Fluor 555–conjugated goat anti-mouse antibody (Invitrogen), and DAPI for 1 h at room temperature. Stained cells were analyzed using a Nikon A1 confocal microscope.
Intestinal permeability assay
Intestinal permeability assay was performed using FITC–dextran, as described previously (Chassaing et al., 2014). Briefly, mice were deprived of food for 4 h and then gavaged with 0.6 mg/g body weight 4 kDa FITC–dextran (FD4, Sigma-Aldrich St. Louis, MO). Blood was collected after 4 h, and fluorescence intensity was measured (excitation, 485 nm; emission, 520 nm). FITC–dextran concentrations were determined using a standard curve generated by the serial dilution of FITC–dextran.
Gnotobiotic experiments
GF Swiss Webster mice were colonized by a consortium of human synthetic microbiota, as reported previously (Desai et al., 2016), with a few modifications. The composition of the bacteria consortium is shown in Figure 6A and Figure S4A. Bacteria were anaerobically (i.e., 85% N2, 10% H2, 5% CO2) cultured in their respective media at 37°C with final absorbance (600 nm) readings ranging from about 1.0. Bacteroides ovatus (DSMZ 1896), B. uniformis (ATCC 8492), Clostridium symbiosum (DSMZ 934), Collinsella aerofaciens (DSMZ 3979), and E. coli (ATCC HS and CD patient–derived LF82) were cultured in TYG medium (Moore, 1977). Modified chopped meat medium (Hehemann et al., 2012) was used to culture A. muciniphila (DSMZ 22959) and Eubacterium rectale (DSMZ 17629). Roseburia intestinalis (DSMZ 14610), Faecalibacterium prausnitzii (DSMZ 17677), and Marvinbryantia formatexigens (DSMZ 14469) were cultured in YCFA medium (Ze et al., 2012). Desulfovibrio piger (ATCC 29098) was cultured in ATCC 1249 medium. Bacterial cultures were mixed in equal volumes, and the mice were orally gavaged with 0.2 mL of this mixture. After a 14-day reconstitution, the mice were fed a sterilized custom diet and treated with DSS.
E. coli infection in vivo and in vitro
For E. coli infection in vivo, mice were infected with each E. coli strain (1 × 109 colony forming units (CFU)/mouse) (Darfeuille-Michaud et al., 1998; Kitamoto et al., 2020; Miranda et al., 2004). To assess E. coli colonization, homogenates of feces and colon tissues were cultured on LB agar plates with ampicillin or streptomycin. The number of viable bacteria was estimated by plate counting the number of CFUs. In the in vitro experiments, T84 cells or the HCM were infected with E. coli at an MOI = 1–10 (2 × 105–2 × 106 CFU/well) as described in the respective Figures 5, 7, and S4. After infection, the cells were centrifuged at 1,000g for 10 min at 24°C and maintained at 37°C. In the bacterial adhesion assay, cells were washed three times with PBS and then lysed with 0.1% Triton X-100 (Sigma-Aldrich) in deionized water. In the bacterial invasion assay, cells were infected with E. coli for 1–3 h and then cultured with gentamycin (100 μg/mL) to kill extracellular E. coli. After incubation, the cells were washed 3 times with PBS and then lysed with 0.1% Triton X-100 in deionized water. Lysed cells were diluted and plated on LB agar plates to determine the number of CFUs corresponding to the total number of cell-associated bacteria.
Coculture of anaerobic bacteria with primary human colon monolayers
The human-derived colonoids were provided by the Translational Tissue Modeling Laboratory at the University of Michigan. To generate the monolayers, the three-dimensional human colonoids were dissociated into a single-cell suspension and plated on collagen IV (Sigma-Aldrich)–coated transwell inserts (0.4 μm pore size, 0.33 cm2, polyester [PET], Costar, Corning, Tewksbury, MA) in a 24-well plate. After 24 h, the growth medium was replaced with differentiation medium (Watson et al., 2014). Differentiation was completed in a 5% O2, 5% CO2, balanced N2 environment. The medium was refreshed every 48 h for 6 days. Transepithelial electrical resistance (TEER) was determined to be present in an intact monolayer (>400 Ω/cm2), which is suitable for an anaerobic coculture system. TEER was recorded using an electrical volt/ohm meter (World Precision Instruments, Sarasota, FL). The transwell plates were set in the apical chambers (Coy Laboratory Products, Grass Lake, MI) of an anaerobic chamber (5% CO2, balance N2 environment), and cultured in an anaerobic coculture system as shown in Figure 6A (Lauder et al., 2020). Briefly, setting up the apical chamber, a 24-well gas-permeable plate was placed on the base and sealed in place using double-sided adhesive tape. The entire apparatus was placed in an anaerobic chamber (90% N2, 5% H2, 5% CO2) to allow the growth of anaerobic bacteria in the apical wells. 5.0% O2 was pumped from an external tank through the base of the plate to supply oxygen to the basolateral side of the monolayer. The basal compartment was maintained in a 5% O2, 5% CO2, balance N2 environment, whereas the apical chamber was maintained in a 5% CO2, balance N2 environment. The colonoid monolayer was cultured with differentiation medium in the basolateral wells and minimal coculture medium in the apical wells. To evaluate the impact of A. muciniphila on the integrity of the mucus layer and the adhesion of AIEC, the apical well was infected with A. muciniphila (4 × 104 CFU/well) for 18 h, and then with LF82 (4 × 103 CFU/well) for 3 h. LF82 adhesion was assessed as described.
Bacterial RNA extraction
Bacterial RNA was extracted from cecal content by a standard phenol-chloroform method as described earlier with few modifications. Briefly, 500 μl of Buffer A (200 mM NaCl, 200 mM Tris, 20 mM EDTA), 210 μl of 20% SDS (filter sterilized) and 500 μl of phenol:chloroform:isoamyl alcohol (125:24:1, pH 4.5; Fischer Scientific, USA) were added to cecal content. The mixture was applied bead beating for 3 min at 4 °C and centrifuged at 4 °C (13000 rpm for 3 min). The aqueous phase was recovered and then 500 μl of phenol:chloroform:isoamyl alcohol solution was added. After mixing with vortex, the mixture was centrifuged again at 4 °C (13,000 rpm for 3 min) and the aqueous phase was recovered. 1 volume of pre-chilled ethanol and 0.1 volume of a 3M sodium acetate (pH: 5.2) were added to the aqueous phase. The resulting solution was then mixed by gentle inversion and incubated for 30 min at −20 °C. Afterward, the mixture was centrifuged at 4 °C (13,000 rpm for 20 min). The pellet was washed with 70% ethanol, and then resuspended in nuclease-free water. For RNA-seq, bacterial RNA was isolated with a bacterial RNA isolation kit (Omega Bio-tek, Norcross, GA), according to the manufacturer’s protocol. Isolated RNA was treated with DNase (Thermo Fisher Scientific) and then cleaned with an RNeasy Mini Kit (QIAGEN). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The qPCR was performed using SYBR Green Supermix (Radiant molecular tolls).
Bacterial RNA-sequencing
Library preparation and sequencing of the RNA-seq libraries were performed in the Advanced Genomics Core at the University of Michigan. Briefly, RNA was assessed for quality using the Agilent TapeStation system (Agilent, Santa Clara, CA). Samples were prepared using the New England BioLabs (Ipswitch, MA) NEBNext Ultra II Directional RNA Library Prep Kit for Illumina, the NEBNext rRNA Depletion Kit (Bacteria), the NEBNext rRNA Depletion Kit (Human/Mouse/Rat), and the NEBNext Multiplex Oligos for Illumina (Unique Dual Index for Primer Pairs), where 145 ng total RNA was ribosomal depleted using the human/mouse/rat and bacteria rRNA depletion modules. The rRNA-depleted RNA was then fragmented for 7–10 min determined by the RIN (RNA Integrity Number) of input RNA as per protocol and copied into first-strand cDNA using reverse transcriptase and dUTP mix. The samples underwent end repair and dA-tailing, followed by ligation of the NEBNext adapters. The products were purified and enriched by PCR to create the final cDNA libraries, which were checked for quality and quantity by the Agilent TapeStation system and Qubit (Thermo Fisher Scientific). The samples were pooled and sequenced on the Illumina NovaSeq 6000 S4 paired-end 150 bp, according to the manufacturer’s recommended protocols (Illumina, San Diego, CA. bcl2fastq2 Conversion Software (Illumina) was used to generate de-multiplexed Fastq files. Paired-end reads were mapped on LF82 genomic sequence CU651637 by bowtie2 and reads of individual LFL82 genes were counted by HTSeq as described (Kitamoto et al., 2020). Human transcripts were mapped on human cDNA sequences GRCg38 and counted by Salmon (https://pubmed.ncbi.nlm.nih.gov/28263959/). For functional GO enrichment analysis, over-represented and under-represented bacterial genes were identified by LEfSe and then analyzed using the Gene Ontology Resource (http://geneontology.org)
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 9.3.0 (GraphPad Software San Diego, CA). The numbers of animals used for individual experiments, details of the statistical tests used, and pooled values for several biological replicates are indicated in the respective figure legends. Differences between the two groups were evaluated using the two-tailed Student t test or the Mann–Whitney U test. 1-way ANOVA or the Kruskal–Wallis test followed by the Tukey correction or the Dunn test was performed for the comparison of more than 3 groups. Differences of p < 0.05 were considered significant. Statistically significant differences are shown with asterisks as follows: *p < 0.05, **p < 0.01, ***p < 0.001, whereas N.S. indicates comparisons that are not significant.
Supplementary Material
Acknowledgements
The authors wish to thank the University of Michigan Center for Gastrointestinal Research (NIH 5P30DK034933), and the Host Microbiome Initiative, the Germ-Free Mouse Facility, the In-Vivo Animal Core, the Advanced Genomics Core, the School of Dentistry Histology Core, the Rogel Cancer Center Tissue & Molecular Pathology Shared Resource, the Microscopy Core, the Metabolomics Core, and the Translational Tissue Modeling Laboratory, all at the University of Michigan. We also thank Peter Kuffa, Yadong Mao, and Ingrid L. Bergin for experimental assistance and Eric C Martens for providing bacterial strains of SM. This work was supported by National Institutes of Health grants DK110146, DK108901, DK125087, DK119219, and AI142047 (to N.K.), the Kenneth Rainin Foundation Innovator Award and Synergy Award (to N.K.), a JSPS Postdoctoral Fellowship for Research Abroad (to K.S., S.K., and H.N.-K.), the Uehara Memorial Foundation Postdoctoral Fellowship Award (to K.S., and S.K.), and the Crohn’s and Colitis Foundation (to K.S., H.N.-K., and N.K.), and the Office of the Assistant Secretary of Defense for Health Affairs endorsed by the Department of Defense through the Peer-Reviewed Cancer Research Program under Award No. W81XWH2010547 (to S.K.).
Footnotes
Disclosures:
The authors declare no competing interests.
Reference
- Ananthakrishnan AN (2015). Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol 12, 205–217. 10.1038/nrgastro.2015.34. [DOI] [PubMed] [Google Scholar]
- Becattini S, Sorbara MT, Kim SG, Littmann EL, Dong Q, Walsh G, Wright R, Amoretti L, Fontana E, Hohl TM, and Pamer EG (2021). Rapid transcriptional and metabolic adaptation of intestinal microbes to host immune activation. Cell Host Microbe. 10.1016/j.chom.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero-Flores G, Pickard JM, Fukuda S, Inohara N, and Nunez G (2020). An Enteric Pathogen Subverts Colonization Resistance by Evading Competition for Amino Acids in the Gut. Cell Host Microbe 28, 526–533 e525. 10.1016/j.chom.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron EA, and Sperandio V (2015). Frenemies: Signaling and Nutritional Integration in Pathogen-Microbiota-Host Interactions. Cell Host Microbe 18, 275–284. 10.1016/j.chom.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, and de Vos WM (2017). Next-Generation Beneficial Microbes: The Case of Akkermansia muciniphila. Front Microbiol 8, 1765. 10.3389/fmicb.2017.01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, and Darfeuille-Michaud A (2009). Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med 206, 2179–2189. 10.1084/jem.20090741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Malleshappa M, and Vijay-Kumar M (2014). Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol 104, 15 25 11–15 25 14. 10.1002/0471142735.im1525s104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieza RJ, Hu J, Ross BN, Sbrana E, and Torres AG (2015). The IbeA invasin of adherent-invasive Escherichia coli mediates interaction with intestinal epithelia and macrophages. Infect Immun 83, 1904–1918. 10.1128/IAI.03003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dame MK, Attili D, McClintock SD, Dedhia PH, Ouillette P, Hardt O, Chin AM, Xue X, Laliberte J, Katz EL, et al. (2018). Identification, isolation and characterization of human LGR5-positive colon adenoma cells. Development 145. 10.1242/dev.153049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, Kayser BD, Levenez F, Chilloux J, Hoyles L, et al. (2016). Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65, 426–436. 10.1136/gutjnl-2014-308778. [DOI] [PubMed] [Google Scholar]
- Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, and Colombel JF (1998). Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 115, 1405–1413. 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- Derrien M, Vaughan EE, Plugge CM, and de Vos WM (2004). Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol 54, 1469–1476. 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353 e1321. 10.1016/j.cell.2016.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al. (2014). Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis 20, 1919–1932. 10.1097/MIB.0000000000000183. [DOI] [PubMed] [Google Scholar]
- Dreux N, Denizot J, Martinez-Medina M, Mellmann A, Billig M, Kisiela D, Chattopadhyay S, Sokurenko E, Neut C, Gower-Rousseau C, et al. (2013). Point mutations in FimH adhesin of Crohn’s disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog 9, e1003141. 10.1371/journal.ppat.1003141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenreich W, Dandekar T, Heesemann J, and Goebel W (2010). Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol 8, 401–412. 10.1038/nrmicro2351. [DOI] [PubMed] [Google Scholar]
- Ellermann M, Gharaibeh RZ, Fulbright L, Dogan B, Moore LN, Broberg CA, Lopez LR, Rothemich AM, Herzog JW, Rogala A, et al. (2019). Yersiniabactin-Producing Adherent/Invasive Escherichia coli Promotes Inflammation-Associated Fibrosis in Gnotobiotic Il10−/− Mice. Infect Immun 18, e00587–00519. 10.1128/IAI.00587-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, et al. (2013). Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A 110, 9066–9071. 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibold L, Garenaux E, Dalmasso G, Gallucci C, Cia D, Mottet-Auselo B, Fais T, Darfeuille-Michaud A, Nguyen HT, Barnich N, et al. (2016). The Vat-AIEC protease promotes crossing of the intestinal mucus layer by Crohn’s disease-associated Escherichia coli. Cell Microbiol 18, 617–631. 10.1111/cmi.12539. [DOI] [PubMed] [Google Scholar]
- Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, and Darfeuille-Michaud A (2001). Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect Immun 69, 5529–5537. 10.1128/iai.69.9.5529-5537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Kitamoto S, and Kamada N (2020). Microbial adaptation to the healthy and inflamed gut environments. Gut Microbes 12, 1857505. 10.1080/19490976.2020.1857505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehemann JH, Kelly AG, Pudlo NA, Martens EC, and Boraston AB (2012). Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc Natl Acad Sci U S A 109, 19786–19791. 10.1073/pnas.1211002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilott NE, Bollrath J, Danne C, Schiering C, Shale M, Adelmann K, Krausgruber T, Heger A, Sims D, and Powrie F (2016). Defining the microbial transcriptional response to colitis through integrated host and microbiome profiling. ISME J 10, 2389–2404. 10.1038/ismej.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai J, Kitamoto S, Sugihara K, Nagao-Kitamoto H, Hayashi A, Morhardt TL, Kuffa P, Higgins PDR, Barnich N, and Kamada N (2019). Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol 12, 632–643. 10.1038/s41385-019-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, Ghishan FK, Carvalho FA, Gewirtz AT, Sjovall H, and Hansson GC (2014). Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 63, 281–291. 10.1136/gutjnl-2012-303207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, and Hansson GC (2012). Preservation of mucus in histological sections, immunostaining of mucins in fixed tissue, and localization of bacteria with FISH. Methods Mol Biol 842, 229–235. 10.1007/978-1-61779-513-8_13. [DOI] [PubMed] [Google Scholar]
- Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, and Hansson GC (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 105, 15064–15069. 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, and Nunez G (2012). Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336, 1325–1329. 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto S, Alteri CJ, Rodrigues M, Nagao-Kitamoto H, Sugihara K, Himpsl SD, Bazzi M, Miyoshi M, Nishioka T, Hayashi A, et al. (2020). Dietary L-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat Microbiol 5, 116–125. 10.1038/s41564-019-0591-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, and Schloss PD (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79, 5112–5120. 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krych L, Kot W, Bendtsen KMB, Hansen AK, Vogensen FK, and Nielsen DS (2018). Have you tried spermine? A rapid and cost-effective method to eliminate dextran sodium sulfate inhibition of PCR and RT-PCR. J Microbiol Methods 144, 1–7. 10.1016/j.mimet.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Lambert K, Pappas D, Miglioretto C, Javadpour A, Reveley H, Frank L, Grimm MC, Samocha-Bonet D, and Hold GL (2021). Systematic review with meta-analysis: dietary intake in adults with inflammatory bowel disease. Aliment Pharmacol Ther 54, 742–754. 10.1111/apt.16549. [DOI] [PubMed] [Google Scholar]
- Lapaquette P, Glasser AL, Huett A, Xavier RJ, and Darfeuille-Michaud A (2010). Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol 12, 99–113. 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauder E, Kim K, Schmidt TM, and Golob JL (2020). Organoid-derived adult human colonic epithelium responds to co-culture with a probiotic strain of Bifidobacterium longum. BioRxiv. 10.1101/2020.07.16.207852. [DOI] [Google Scholar]
- Liu L, Saitz-Rojas W, Smith R, Gonyar L, In JG, Kovbasnjuk O, Zachos NC, Donowitz M, Nataro JP, and Ruiz-Perez F (2020). Mucus layer modeling of human colonoids during infection with enteroaggragative E. coli. Sci Rep 10, 10533. 10.1038/s41598-020-67104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, et al. (2017). Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 25, 345–357. 10.1016/j.cmet.2016.12.011. [DOI] [PubMed] [Google Scholar]
- Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, and Vousden KH (2013). Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546. 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. 10.1038/nature22056. [DOI] [PubMed] [Google Scholar]
- Matthews R, and Neidhardt F (1989). Elevated serine catabolism is associated with the heat shock response in Escherichia coli. J Bacteriol 171, 2619–2625. 10.1128/jb.171.5.2619-2625.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda RL, Conway T, Leatham MP, Chang DE, Norris WE, Allen JH, Stevenson SJ, Laux DC, and Cohen PS (2004). Glycolytic and gluconeogenic growth of Escherichia coli O157:H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect Immun 72, 1666–1676. 10.1128/IAI.72.3.1666-1676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore ECLHW (1977). Anaerobe laboratory manual, 4th edition Edition (Blacksburg; ). [Google Scholar]
- Morgan XC, T.T., Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 16 (13), R79. 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadalian B, Yadegar A, Houri H, Olfatifar M, Shahrokh S, Asadzadeh Aghdaei H, Suzuki H, and Zali MR (2021). Prevalence of the pathobiont adherent-invasive Escherichia coli and inflammatory bowel disease: a systematic review and meta-analysis. J Gastroenterol Hepatol 36, 852–863. 10.1111/jgh.15260. [DOI] [PubMed] [Google Scholar]
- Nagao-Kitamoto H, Shreiner AB, Gillilland MG 3rd, Kitamoto S, Ishii C, Hirayama A, Kuffa P, El-Zaatari M, Grasberger H, Seekatz AM, et al. (2016). Functional Characterization of Inflammatory Bowel Disease-Associated Gut Dysbiosis in Gnotobiotic Mice. Cell Mol Gastroenterol Hepatol 2, 468–481. 10.1016/j.jcmgh.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AC, and Maddocks ODK (2017). Serine and Functional Metabolites in Cancer. Trends Cell Biol 27, 645–657. 10.1016/j.tcb.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, and Sonnenburg JL (2013). Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99. 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, and Sperandio V (2012). Fucose sensing regulates bacterial intestinal colonization. Nature 492, 113–117. 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal RR, Baidya AK, Mamou G, Bhattacharya S, Socol Y, Kobi S, Katsowich N, Ben-Yehuda S, and Rosenshine I (2019). Pathogenic E. coli Extracts Nutrients from Infected Host Cells Utilizing Injectisome Components. Cell 177, 683–696 e618. 10.1016/j.cell.2019.02.022. [DOI] [PubMed] [Google Scholar]
- Passalacqua KD, Charbonneau ME, and O’Riordan MXD (2016). Bacterial Metabolism Shapes the Host-Pathogen Interface. Microbiol Spectr 4. 10.1128/microbiolspec.VMBF-0027-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwa LG, Fan TJ, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, and Hansen JJ (2011). Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology 141, 1842–1851 e1841–1810. 10.1053/j.gastro.2011.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Lopez A, Behnsen J, Nuccio SP, and Raffatellu M (2016). Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol 16, 135–148. 10.1038/nri.2015.17. [DOI] [PubMed] [Google Scholar]
- Pizer LI PM (1964). Nutritional and regulatory aspects of serine metabolism in Escherichia coli. J Bacteriol 88, 611–619. 10.1128/JB.88.3.611-619.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüss BM, Nelms JM, Park C, and Wolfe AJ (1994). Mutations in NADH-ubiquinone oxidoreductase of Escherichia coli affect growth on mixed amino acids. J Bacteriol 176, 2143–2150. 10.1128/jb.176.8.2143-2150.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigottier-Gois L (2013). Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J 7, 1256–1261. 10.1038/ismej.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V, Friedman A, Manfredi MG, Weinberg SE, Rabinowitz JD, and Chandel NS (2019). Serine Metabolism Supports Macrophage IL-1beta Production. Cell Metab 29, 1003–1011 e1004. 10.1016/j.cmet.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolhion N, Barnich N, Bringer MA, Glasser AL, Ranc J, Hebuterne X, Hofman P, and Darfeuille-Michaud A (2010). Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut 59, 1355–1362. 10.1136/gut.2010.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolhion N, Carvalho FA, and Darfeuille-Michaud A (2007). OmpC and the sigma(E) regulatory pathway are involved in adhesion and invasion of the Crohn’s disease-associated Escherichia coli strain LF82. Mol Microbiol 63, 1684–1700. 10.1111/j.1365-2958.2007.05638.x. [DOI] [PubMed] [Google Scholar]
- Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW, Ananthakrishnan AN, Andrews E, Barron G, Lake K, et al. (2018). Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol 3, 337–346. 10.1038/s41564-017-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541. 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Berry D, Rauch I, Rennisch I, Ramesmayer J, Hainzl E, Heider S, Decker T, Kenner L, Muller M, et al. (2014). Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J 8, 1101–1114. 10.1038/ismej.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrin G, Massier S, Chassaing B, Agus A, Delmas J, Denizot J, Billard E, and Barnich N (2020). Adaptation of adherent-invasive E. coli to gut environment: Impact on flagellum expression and bacterial colonization ability. Gut Microbes 11, 364–380. 10.1080/19490976.2017.1421886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg J, Xu J, Leip D, Chen C, Westover B, Weatherford J, Buhler J, and Gordon J (2005). Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307, 1955–1959. 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- Stecher B (2015). The Roles of Inflammation, Nutrient Availability and the Commensal Microbiota in Enteric Pathogen Infection. Microbiol Spectr 3. 10.1128/microbiolspec.MBP-0008-2014. [DOI] [PubMed] [Google Scholar]
- Steimle A, De Sciscio A, Neumann M, Grant ET, Pereira GV, Ohno H, Martens EC, and Desai MS (2021). Constructing a gnotobiotic mouse model with a synthetic human gut microbiome to study host-microbe cross talk. STAR Protoc 2, 100607. 10.1016/j.xpro.2021.100607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara K, and Kamada N (2021). Diet-Microbiota Interactions in Inflammatory Bowel Disease. Nutrients 13. 10.3390/nu13051533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara K, Morhardt TL, and Kamada N (2018). The Role of Dietary Nutrients in Inflammatory Bowel Disease. Front Immunol 9, 3183. 10.3389/fimmu.2018.03183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, and Baumler AJ (2011). Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A 108, 17480–17485. 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YH, Czerwinski M, Wu A, Dame MK, Attili D, Hill E, Colacino JA, Nowacki LM, Shroyer NF, Higgins PDR, et al. (2018). A Method for Cryogenic Preservation of Human Biopsy Specimens and Subsequent Organoid Culture. Cell Mol Gastroenterol Hepatol 6, 218–222 e217. 10.1016/j.jcmgh.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Buller HA, Dekker J, Van Seuningen I, Renes IB, and Einerhand AW (2006). Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129. 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Vich Vila A IF, Collij V, Jankipersadsing SA, Gurry T, Mujagic Z, Kurilshikov A, Bonder MJ, Jiang X, Tigchelaar EF, Dekens J, Peters V, Voskuil MD, Visschedijk MC, van Dullemen HM, Keszthelyi D, Swertz MA, Franke L, Alberts R, Festen EAM, Dijkstra G, Masclee AAM, Hofker MH, Xavier RJ, Alm EJ, Fu J, Wijmenga C, Jonkers DMAE, Zhernakova A, Weersma RK (2018). Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci Transl Med 19 (10), eaap8914. 10.1126/scitranslmed.aap8914. [DOI] [PubMed] [Google Scholar]
- Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos J, Woo V, Teng F, Tran N, Sczesnak A, et al. (2017). IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote T H 17-dependent inflammation. Sci Transl Med 9, eaaf9655. 10.1126/scitranslmed.aaf9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladomiu M, Metz ML, Lima SF, Jin WB, Chou L, Bank JRILC, Guo CJ, Diehl GE, Simpson KW, Scherl EJ, and Longman RS (2021). Adherent-invasive E. coli metabolism of propanediol in Crohn’s disease regulates phagocytes to drive intestinal inflammation. Cell Host Microbe 29, 607–619 e608. 10.1016/j.chom.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CL, Mahe MM, Munera J, Howell JC, Sundaram N, Poling HM, Schweitzer JI, Vallance JE, Mayhew CN, Sun Y, et al. (2014). An in vivo model of human small intestine using pluripotent stem cells. Nat Med 20, 1310–1314. 10.1038/nm.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, and Vousden KH (2016). Serine and one-carbon metabolism in cancer. Nat Rev Cancer 16, 650–662. 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- Ze X, Duncan SH, Louis P, and Flint HJ (2012). Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J 6, 1535–1543. 10.1038/ismej.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bacterial 16S rRNA sequencing data and bacterial RNA sequencing data reported in this paper have been deposited and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 goat anti-rabbit IgG | Invitrogen | A11008 |
| Alexa Fluor 555 goat anti-mouse IgG | Invitrogen | A32727 |
| Alexa Fluor 555-conjugated EUB338 probe | Invitrogen | N/A |
| Anti-E. coli LPS antibody | Abcam | ab35654 |
| Mucin 2 antibody (H-300) | Santa Cruz Biotechnology | sc-15334 |
| Bacterial and Virus Strains | ||
| Adherent-invasive Escherichia coli: LF82 strain | Darfeuille-Michaud et al. Gastroenterology. 1998 | N/A |
| Adherent-invasive Escherichia coli: LF82 ΔTS mutant strain | Kitamoto et al. Nat Microbiol. 2020 | N/A |
| Adherent-invasive Escherichia coli: LF82 ΔfimH mutant strain | Imai et al. Mucosal Immunol. 2019 | N/A |
| Adherent-invasive Escherichia coli: CUMT8 strain | Kitamoto et al. Nat Microbiol. 2020 | N/A |
| Akkermansia muciniphila: DSM 22959, type strain | DMSZ | DMS 22959 |
| Bacteroides ovatus: DSM 1896, type strain | DMSZ | DSM 1896 |
| Bacteroides uniformis: ATCC 8492, type strain | ATCC | ATCC 8492 |
| Clostridium symbiosum: DSM 934, type strain | DMSZ | DSM 934 |
| Collinsella aerofaciens: DSM 3979, type strain | DSMZ | DSM 3979 |
| Desulfovibrio piger: ATC 29098, type strain | ATCC | ATCC 29098 |
| Escherichia coli: HS strain | ATCC | N/A |
| Escherichia coli: MG1655 strain | Miranda et al. Infect Immun. 2004 | N/A |
| Eubacterium rectale: DSM 17629, A1-86 | DSMZ | DSM 17629 |
| Faecalibacterium prausnitzii: DSM 17677, A2-165 | DSMZ | DSM 17677 |
| Marvinbryantia formatexigens: DSM 14469, type strain, l-52 | DSMZ | DSM 14469 |
| Roseburia intestinalis: DSM 14610 type strain, L1-82 | DSMZ | DSM 14610 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetic acid | Thermo Fisher Scientific | A38S |
| Alcian blue | Sigma-Aldrich | A5268 |
| Ampicillin | Sandoz Inc | 0781-3408 |
| BHI agar | BD Difco | BD241830 |
| Bovine Serum Albumins | Thermo Fisher Scientific | BP1600 |
| Chloroform | Thermo Fisher Scientific | 423550250 |
| DAPI | Sigma-Aldrich | D9542 |
| Dextran Sodium Sulfate | MP Biomedical | 0216011090 |
| DMEM | Gibco | 11320033 |
| Ethanol | Thermo Fisher Scientific | BP28184 |
| FITC-dextran | Sigma-Aldrich | FD4 |
| HBSS | Thermo Fisher Scientific | 14170112 |
| Isopropanol | Thermo Fisher Scientific | BP26324 |
| Kanamycin | Thermo Fisher Scientific | BP906-5 |
| LB agar | Thermo Fisher Scientific | BP1425 |
| Lithium chloride solution | Invitrogen | AM9480 |
| Metronidazole | Fagron | 804380 |
| Methanol, extra dry | Thermo Fisher Scientific | 326950025 |
| Neomycin | AG Scientific | N-1053 |
| Paraformaldehyde 4% | Thermo Fisher Scientific | AAJ19943K2 |
| PAS staining kit | Sigma-Aldrich | 95B-1KT |
| PBS | Gibco | 10010023 |
| Phenol:Chloroform:Isoamyl Alcohol (ph: 8) | Thermo Fisher Scientific | 15593031 |
| Phenol:Chloroform:Isoamyl Alcohol (ph: 4.5) | Thermo Fisher Scientific | AM9722 |
| Permount™ Mounting Medium | Thermo Fisher Scientific | SP15 |
| RNAprotect | QIAGEN | 76506 |
| RNAlater Stabilization Solution | Invitrogen | AM7020 |
| Sheep blood | Hemostat Laboratories | DSB250 |
| SDS | Thermo Fisher Scientific | BP166 |
| Sodium Acetate (3 M), pH 5.5 | Invitrogen | AM9740 |
| Sodium Chloride | Thermo Fisher Scientific | BP358 |
| Spermine | Sigma-Aldrich | S3256 |
| Tris-HCl, 1M | Thermo Fisher Scientific | BP1758-100 |
| Vancomycin | APP Pharmaceuticals | 63323028420 |
| Critical Commercial Assays | ||
| BioMasher® II Micro Tissue Homogenizers | DWK Life Sciences | 749625-0030 |
| E.Z.N.A.Total RNA Kit I | OMEGA | R6834-01 |
| E.Z.N.A. Bacterial RNA Kit | OMEGA | R6950-01 |
| DL-Serine Assay Kit | Sigma-Aldrich | MAK352 |
| DNA Removal Kit | Invitrogen | AM1906 |
| DNeasy Blood & Tissue Kit | QIAGEN | 69506 |
| High-Capacity RNA-to-cDNA Kit | Thermo Fisher Scientific | 4387406 |
| Protein Concentrators PES, 10K MWCO | Thermo Fisher Scientific | 88513 |
| Radiant™ SYBR Green Lo-ROX qPCR Kits | Alkali Scientific | QS1020 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| TrueVIEW® Autofluorescence Quenching Kit | Vector Laboratories | SP-8400-15 |
| Deposited Data | ||
| 16S rRNA and RNA sequencing data | This paper | PRJNA763203 |
| Experimental Models: Cell Lines | ||
| T84 cell line | ATCC | CCL-248 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J: WT | The Jackson Laboratory | JAX 000664 |
| Mouse: Germ-free Swiss Webster | Germ-free Facility at University N/A of Michigan | N/A |
| Software and Algorithms | ||
| Mothur v1.39.0 | Schloss et al. Appl Environ Microbiol. 2009 | N/A |
| Prism 9.2 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | Schneider et al. Nat Methods. 2012 | http://imagej.net/Welcome |
| Other | ||
| Amino acid-based control diet | Envigo | TD.130595 |
| Serine and glycine deficient diet | Envigo | TD.140546 |