

Abstract
Protein binding to negatively charged lipids is essential for maintaining numerous vital cellular processes where its dysfunction can lead to various diseases. One such protein that plays a crucial role in this process is lactadherin, which competes with coagulation factors for membrane binding sites to regulate blood clotting. Despite identifying key binding regions of these proteins through structural and biochemical studies, models incorporating membrane dynamics are still lacking. In this study, we report on the multimodal binding of lactadherin and use it to gain insight into the binding mechanisms of its C domain homologs, factor V and factor VIII. Molecular dynamics simulations enhanced with the highly mobile mimetic model enabled the determination of lactadherin’s multimodal binding on membranes that revealed critical interacting residues consistent with prior NMR and mutagenesis data. The binding occurred primarily via two dynamic structural ensembles: an inserted state and an unreported, highly conserved side-lying state driven by a cationic patch. We utilized these findings to analyze the membrane binding domains of coagulation factors V and VIII and identified their preferred membrane-bound conformations. Specifically, factor V’s C domains maintained an inserted state, while factor VIII preferred a tilted, side-lying state that permitted antibody binding. Insight into lactadherin’s atomistically-resolved membrane interactions from a multi-state perspective can guide new therapeutic opportunities in treating diseases related to blood coagulation.
Keywords: Molecular Dynamics, Lactadherin, MFG-E8, Coagulation Factor, C Domain, Membrane SPR, Ensemble, Acidic Lipids, Phosphatidylserine, POPS, Blood Clotting, Spike Insertion, Cationic Patch
Graphical Abstract
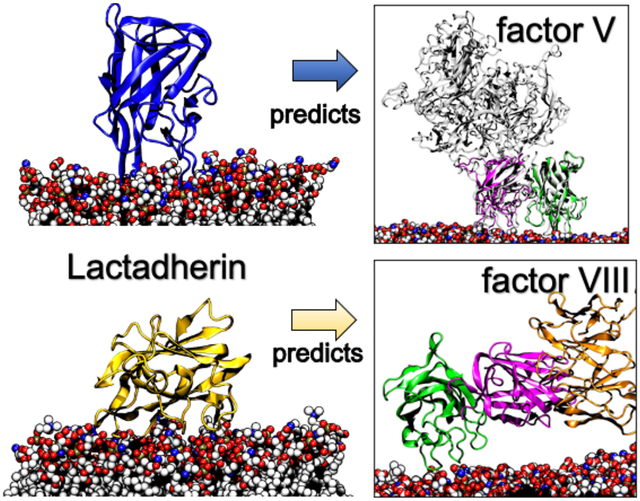
INTRODUCTION
Proteins that bind to acidic lipids are vital in regulating essential cellular processes like signal transduction, blood coagulation, and structural integrity.1,2 Errors occurring in this regulated binding process can lead to Alzheimer’s,3,4 cancer,5 and heart disease.6,7 Lactadherin (Lact), or MFG-E8, selectively binds to negatively charged phosphatidylserine (PS).8,9 Its capacity for specific PS binding culminates in numerous important biological roles such as phagocytosis of apoptotic cells, anti-inflammatory responses, tissue regeneration, homeostasis, angiogenesis, and blood clotting regulation.10 Despite the importance of Lact-PS binding in disease,11–13 detailed knowledge of its binding mechanism remains unclear.
Lact comprises of four functional domains: two epidermal growth factor (EGF)-like domains, EGF1 and EGF2, that bind to integrin receptors, and two discoidin C domains.10,14 Two crystal structures of Lact’s C2 membrane binding domain revealed that residues at the tips of its β-hairpin turns (or “spikes”) allowed Lact to bind to membranes,15,16 which were further validated by mutagenesis experiments.16 A solution nuclear magnetic resonance (NMR) study and structure of Lact reaffirmed the importance of the spike residues in recognizing PS lipids.17 The report also showed that mutating critical positively charged and hydrophobic residues, and caused dramatic decreases in PS binding. Although these experiments have identified key binding residues, while a dynamic binding model incorporating their combined action is still lacking.
Studies demonstrated Lact competes for phospholipid binding sites containing PS with blood coagulation factor V (fV), factor VIII (fVIII),18 and prothrombinase.19 Both fV and fVIII are integral components of the coagulation cascade. These factors undergo cleavage and subsequent activation to form critical complexes to enhance the coagulation response.20 Particularly, activated fV helps to form the prothrombinase complex with factor Xa, which can then cleave prothrombin into its activated state. Moreover, activated fVIII establishes a complex with factor IX, which in turn cleaves and activates factor X.21 Since hemostasis requires cells to redistribute PS lipids in its outer leaflet, Lact was hypothesized to regulate blood clotting through this competitive binding.18 The homology of Lact’s C2 binding domain with fV and fVIII offers one explanation for why Lact can compete with them for PS sites.22 It suggests a similar binding mechanism and effectively equivalent membrane interaction sites. A detailed understanding of Lact binding can therefore provide a framework for investigating coagulation factor function. Nonetheless, similarities of their binding mechanisms are still unknown at the molecular level.
Lact’s C2 discoidin domain also shares a comparable membrane binding capacity with Protein Kinase C’s (PKC) C2 domains.1,23 Despite being evolutionarily unrelated, both share structural resemblance and bind to PS-enriched membranes using three inter-strand loops. Interestingly, the PKC C2 domain also uses a secondary binding mode via a positively charged patch that is present on several C2 domain termed the cationic β-groove.23–26 This groove is conserved in synaptotagmin, a homologous protein with PKC, which studies demonstrated to be required for neurotransmitter release.27 This function suggests that the patch has crucial biological implications beyond membrane binding. However, despite Lact’s similarities with PKC, it remains unclear if Lact also participates in multimodal binding.
Herein, we report a two-state binding model for Lact and its key membrane-binding residues that enabled us to gain insight into the binding of coagulation factors fV and fVIII. To efficiently sample Lact membrane binding events, we utilized all-atom molecular dynamics (MD) simulations in conjunction with the highly mobile membrane mimetic model (HMMM)28,29 The extensive sampling captured converged membrane-bound trajectories that allowed us to determine how spike and non-spike residues interact with specific membrane regions and achieve an inserted state. Furthermore, we discovered a previously unreported side-lying state, which allowed Lact to engage in a secondary binding mode primarily driven by electrostatics. Using these insights, we investigated how the C domains of fV and fVIII can adopt either an inserted or side-lying state. Our proposed dynamic binding model provides an atomistic understanding of how coagulation factors bind to membranes and has implications for future therapeutic developments, such as anticoagulants based on Lact dynamic binding.
MATERIALS AND METHODS
Computational Methods
System Preparation: LactC2 Simulations
Ten replicates containing a single bovine Lactadherin C2 (LactC2) domain (PDB ID: 3BN6)16 were prepared and added approximately 20 Å above the membrane surface. The same initial starting configuration was used, with a starting orientation resembling an inserted state (θ tending to 0°, Figure S1). Membranes were first represented with the Highly Mobile Membrane Mimetic (HMMM) model using CHARMM-GUI.28,30,31 HMMM shortens the lipid tail and replaces it with hydrophobic organic solvent 1,1-dichloroethane (DCLE) (see Figure S10 for computational workflow). These HMMM membranes were used to capture and extensively sample LactC2-PS interactions since they increase lipid lateral diffusion in the membrane by 1–2 orders of magnitude while preserving an all-atom description and the accuracy of free energy of insertion into membrane interface.29 The increased lateral diffusion subsequently increases the sampling rate of lipid-lipid and lipid-ion interactions. This methodology was shown to sufficiently capture the peptide-lipid interactions while reproducing the effects of full-tail32–34. Moreover, using HMMM allowed LactC2 to sample a range of θ values before converging to either an inserted or side-lying state (Figure S1). Each replicate comprised a lipid bilayer of 120 lipids with a 70:30 palmitoyl-oleoyl phosphatidylserine (PS): palmitoyl-oleoyl phosphatidylcholine (PC) ratio and solvated with ~27,000 water molecules. CaCl2 was added to the system on both sides of the lipid bilayer in a concentration of 5 mM to mimic the lipid-ion ratio of the nanodiscs used in the SPR experiments (see Supplemental Methods).
We utilized a lipid bilayer with a high concentration of 70% PS to ensure robust capture and extensive sampling of the interactions between LactC2 and PS. It is necessary to use higher than normal amounts of PS when doing protein/membrane binding experiments with nanodiscs35,36. This is likely because such nanodiscs are mimicking a PS-rich nanodomains37. Importantly, the sizes of the membrane typically used in MD simulations, including this study, are similar to those of nanodiscs. Since this proportion of PS is beyond typical physiological ranges, we performed nanodisc surface plasmon resonance (SPR) experiments. We showed that Lact’s characteristic Ca2+ independent binding38 remained unaffected with the same lipid composition as our computational model. Specifically, we measured Lact’s binding with an affinity of 283 ± 67 nM in the presence of Ca2+ and 296 ± 83 nM in the absence of Ca2+ (Figure S3 and Supplemental Methods). Consequently, these experiments functioned as a verification and connection step for our simulations, affirming that Lact’s essential binding features can be observed even at elevated PS concentrations.
After 150 ns of HMMM production run simulations, we used the HMMM Builder module30 in the CHARMM-GUI to convert the short-tail lipids into a full-tailed representation and remove the DCLE solvent. These “full-tailed” systems were then subjected to an additional 100 ns of production molecular dynamics. All simulations were performed using the CHARMM36M force field.39. This workflow allowed us to take advantage of the increased lipid diffusion from HMMM to obtain LactC2-bound poses while ensuring it was stabilized in the traditional full-tailed membrane representation.
System Preparation: LactC1C2 & Factors V and VIII C Domain Constructs
We used AlphaFold2’s40,41 full structure of lactadherin (UniProt ID: Q95114) to generate a lactadherin C1-C2 (LactC1C2) construct. The EGF domains were removed since they do not participate in membrane binding.10,14. We aligned LactC1C2 to 3 LactC2 structures taken from the inserted ensemble (Figure 1B) and then used these aligned LactC1C2 structures as inputs for the CHARMM-GUI Membrane Builder. A 70:30 POPS:POPC composition was used to remain consistent with LactC2 simulations. A 111 Å XY box length was used with 50 mM of CaCl2 to neutralize the systems. LactC1C2 systems were solvated with ~37,000 water atoms. Three replicates were generated and equilibrated using the CHARMM-GUI equilibration protocol31 and then subjected to 200 ns of production MD. To obtain starting structures for LactC1C2 in a side-lying state, we extracted a snapshot from these simulations that started in an inserted state, where the linker between the C domains was in an extended conformation. This structure was then aligned to three structures from the LactC2 side-lying ensemble and used as inputs for CHARMM-GUI using identical system-building parameters. To generate coagulation factor V C1-C2 (fVC1C2) and factor VIII C1-C2 (fVIIIC1C2) constructs, we extracted the C1-C2 domains from their experimental structures (PDB ID 7KVE42 and PDB ID 7K66,43 respectively). We then aligned the membrane-binding C2 domain of the coagulation factor constructs to a conformation from LactC1C2 simulations where both of its C domains adopted an inserted state. These newly aligned structures of fVC1C2 and fVIIIC1C2 were then used as inputs to generate a membrane-protein system using CHARMM-GUI using the same modeling parameters for LactC1C2. Single replicates were created and then subjected to 200 ns of production MD (Table 1).
Figure 1.
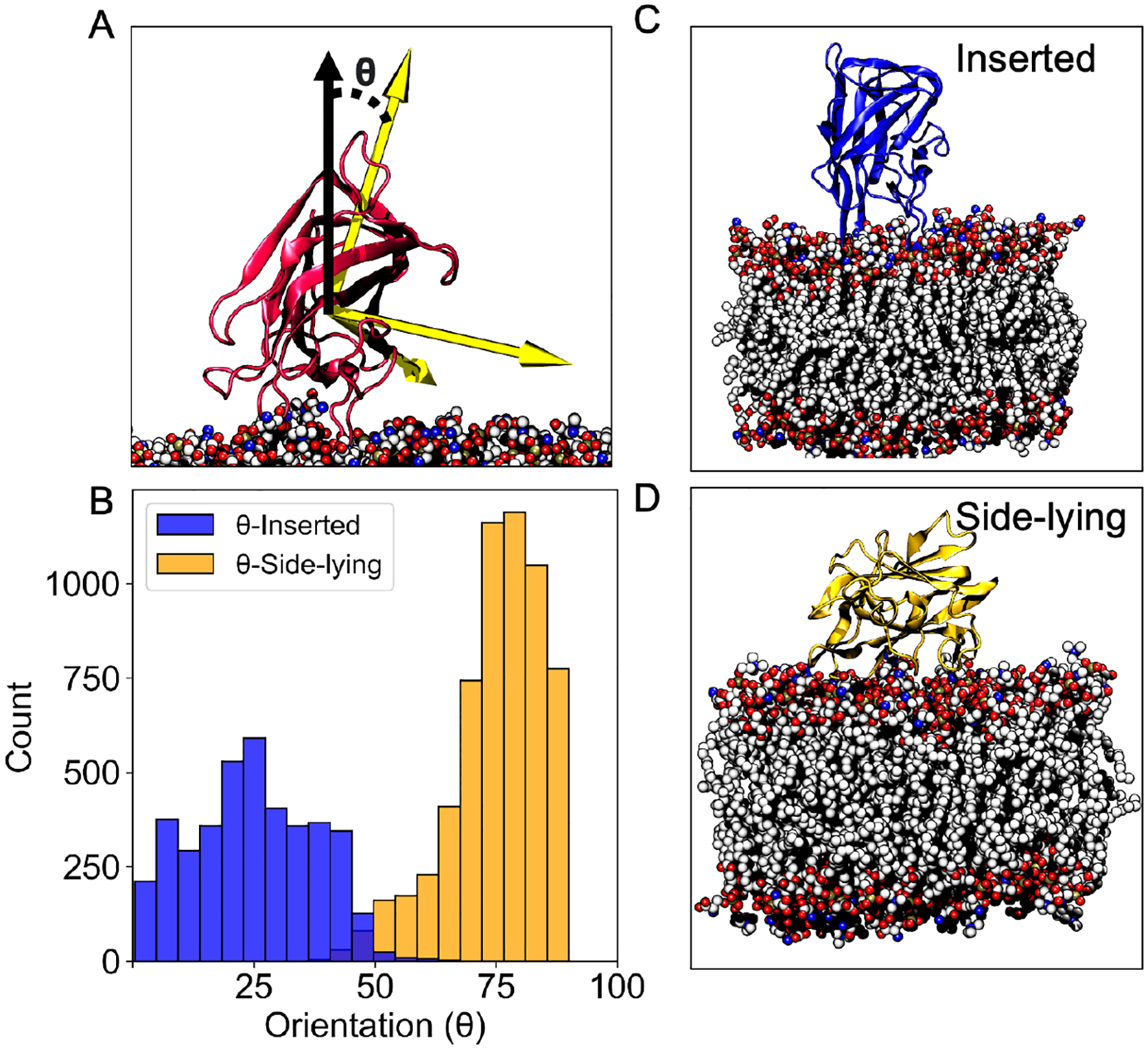
Tilt orientation relative to the membrane normal shows two distinct binding ensembles for LactC2. (A) The orientation (θ) was defined as the angle between the third principal axes (yellow arrow) and the membrane normal vector (solid black arrow). (B) Distribution of the angle θ observed from different ensembles (inserted and side-lying). Different colors distinguish between simulations of LactC2 converging to the inserted state (blue) and the side-lying state (yellow). Representative snapshots from simulations showing LactC2 in the inserted state (C) and the side-lying state (D).
Table 1.
Details summarizing simulations performed in this study.
| System | Membrane Representation | # Replicates | Time (ns) Per Replicate | Sample Size (N) per Replicate |
|---|---|---|---|---|
| LactC2 | HMMM | 10 | 150 | 1500 |
| LactC2 | Full-tail | 10 | 100 | 1000 |
| LactC1C2 (C2 Inserted) | Full-tail | 3 | 200 | 2000 |
| LactC1C2 (C2 Side-lying) | Full-tail | 3 | 200 | 2000 |
| fVC1C2 | Full-tail | 1 | 200 | 2000 |
| fvIIIc1c2 | Full-tail | 1 | 200 | 2000 |
Simulation Protocol
For all systems described in this work, we performed MD simulations using program NAMD 2.14.44 CHARMM36M force field39 parameters were used for the protein, lipids, and ions, while the CGenFF force field45 was used for DCLE molecules. Water molecules were represented by the TIP3P model.46 All simulations were performed at a constant temperature of 303 K and at a constant 1 atm of pressure. Constant pressure was maintained by the Langevin piston Nose-Hoover metho with a damping coefficient of 0.5 ps−1, a piston period of 100 fs, and a piston decay of 50 fs. Constant area was enforced to maintain membrane planarity. Constant temperature was maintained via Langevin dynamics with a damping coefficient of 0.5 ps−1. The non-bonded cutoff distance for short-range interactions was 12 Å with switching at 10 Å. The particle mesh Ewald (PME) method was used,48 with a 1 Å grid spacing, for long-range electrostatics. Hydrogen atom bond lengths were restrained with the SHAKE algorithim.49 The integration step was set to 2 fs. Frames were saved every 100 ps to be used for further analysis.
Analysis Software
All analysis was performed only on data from the full-tail representation. Statistical analysis of the MD trajectories was performed using the Python programming language and analysis libraries such as pytraj50,51 and MDAnalysis.52 All visualizations were performed using Visual Molecular Dynamics (VMD)53 unless otherwise stated. Electrostatic potential surfaces were calculated using the Adaptive Poisson–Boltzmann Solver (APBS) software.54 Clustal Omega55 was used to perform the multiple sequence alignments using UniProt accession IDs P00451, 2CP12259, 2CP21956, 2CP70490, 2CP79385, and 2CQ084. Sequence alignment was rendered using the Esprint webserver.56 The BLOSUM 80 matrix was used to color code each amino acid by similarity rendered in VMD. Orientation of Lactadherin was defined by first calculating principal axes. These axes of molecules are well-defined directions that often correspond to important symmetries. They were found by diagonalizing the moment of inertia tensor and calculated using VMD. Dynamic cross-correlation analysis was performed using the Bio3D package.57 All structures were first aligned to the linker region to understand the relative motions between the domains and remove correlative translational motions. Dynamic correlation analysis was performed on alpha-carbons only.
Experimental Materials and Methods
See Supplementary Information for details on nanodisc binding experiments.
RESULTS
Lactadherin’s C2 Domain Binds to The Membrane in Two Distinct Structural Ensembles
All results described herein were based on analysis of systems with full-tail lipids after 150 ns of converged sampling with HMMM lipids (see Methods for more details). To characterize LactC2’s orientation when binding to the membrane, we measured each replicate’s tilt angle θ between the third principal axis of the protein and the membrane normal (Figure 1A). For the first 150 ns of HMMM simulations, LactC2 orientation sampled wider angle distributions (Figure S1). These broader distributions were expected since HMMM is designed to increase the lateral lipid diffusion at least an order of magnitude and therefore enabling LactC2 to sample different membrane-bound poses with less computation. The consecutive full-tail lipid representation simulations converged to two tilt angles (Figure S1). A histogram of these angles revealed a bimodal distribution (Figure 1B), with one centered around 26°, which we will refer to as the “inserted state”, and the other at 77° that we call the “side-lying state”. In the inserted state LactC2 has at least one of its spikes inserted and interacting directly with the membrane (Figure 1C). In the side-lying state the side closest to spike 3 of LactC2 lays on the membrane surface. 4 out of 10 replicates converged to the inserted state, while the other 6 achieved the side-lying state (Figure S1). These findings suggests that LactC2 can bind to the membrane in two distinct binding modes.
Membrane Binding Contacts Correlate with Structural & NMR Experiments
To connect our computational model of LactC2 binding simulations to experiments, we first measured the insertion height of each spike residue (Figure 2A), defined as the z-component of the distance vector between each spike residue’s center-of-mass and phosphate-plane’s center-of-mass (Figure 2B). These calculations only contained data when LactC2 converged to the inserted state (replicates 2, 4, 6, and 8 (Figure S2)) since the others had minimal membrane-spike contacts. Data from the remaining replicates where LactC2 assumed a side-lying state were not considered in the analysis of this section. We found the hydrophobic residues that achieved the deepest insertion in our model were also observed in mutagenesis binding experiments16 (Figure 2C). These includes spike 1 residues: Trp26, Gly27, Leu28, and spike 3: residues Phe81, Gly82. Spike 1 residues were more likely to insert deeper into the membrane than spikes 2 and 3. Specifically, 7 out of 12 residues showed median insertion heights below the phosphate plane. On the other hand, the median insertion heights from other spike residues were above the phosphate plane (Figure 2C). The insertion height ranges also differed depending on the spike. Spike 1 residues showed a smaller range of insertion heights (except for Q43), while spike 2 and 3 residues exhibited a more significant variation. This range difference suggests that LactC2 can bind to membranes without simultaneous spike insertion.
Figure 2.
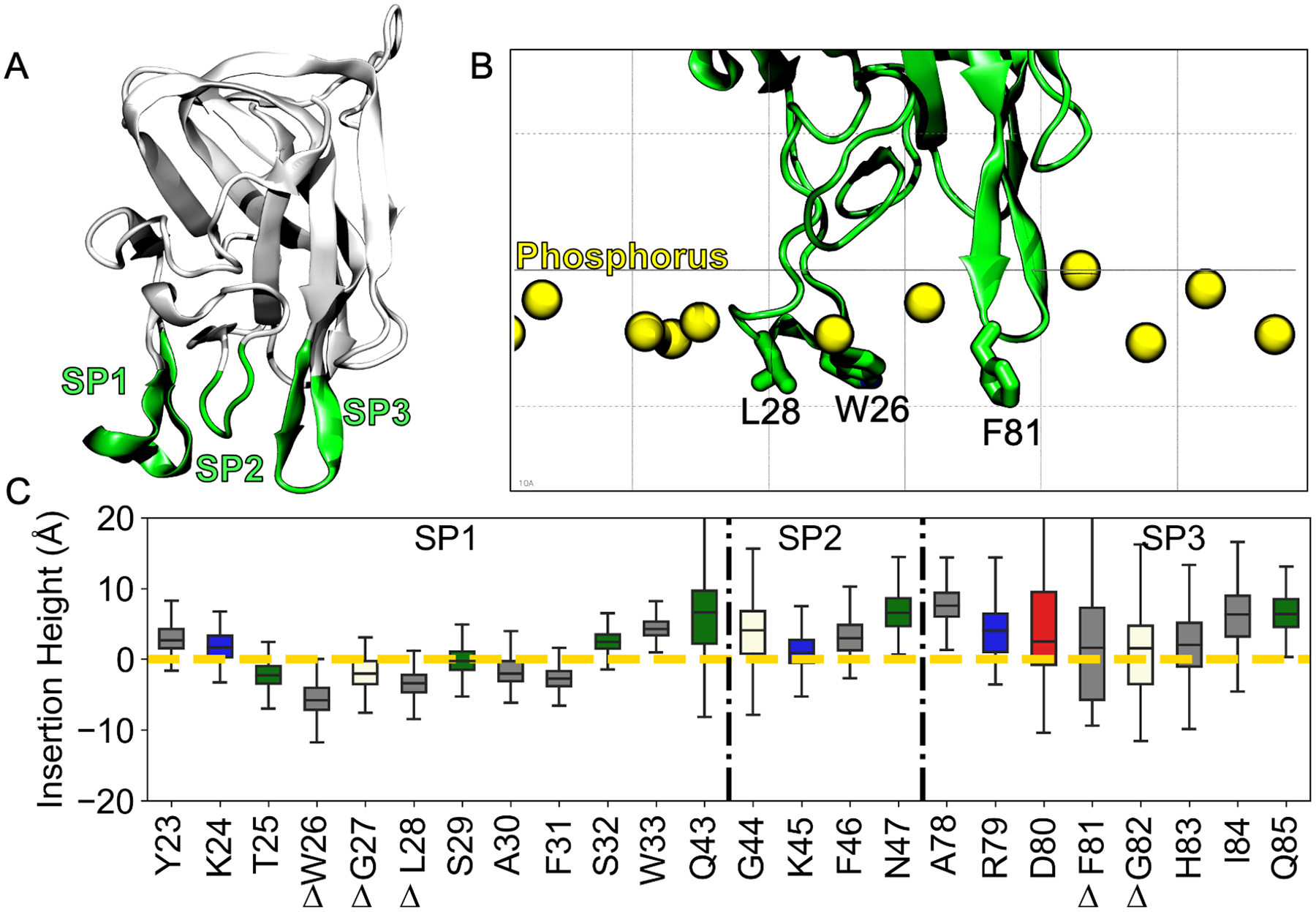
Insertion heights of each spike residue relative to the phosphate plane derived from LactC2 inserted state data. (A) Structure of LactC2 (PDB ID 3BN6)16 with its membrane binding spikes (green) labeled (B) Structural image showing how the insertion height was measured relative to the membrane phosphorus plane (yellow spheres). Hydrophobic residues that were experimentally shown to be important for binding are labeled.16 (C) Insertion height for each spike residue for replicates where all three spikes were fully inserted (colored by type: grey-hydrophobic, blue-basic, red-acidic, green-polar, vanilla-other). The yellow dashed line represents the phosphate plane. The black dashed-dotted line demarcates spike residues. Delta symbols represent critical membrane-binding residues from a prior study.16
To investigate how LactC2’s inserted state interacted with the membrane, we first determined the contact distribution between each residue and PS lipids (Figure 3A). A contact is defined if any atom on a particular residue approaches any component of PS within 2.5 Å (Figure 3B). Then, we decomposed this distribution into contacts into three lipid components: the headgroup α, phosphate β, and lipid tail γ (Figure 3C). We left out contact analysis with glycerol since the contacts were insignificant (Table 2). Spike residues were the most significant contributors to membrane contacts while in the inserted state with the spikes varying in their interactions with different lipid components. Specifically, spike 1 hardly contributed to the contacts of the serine headgroup α (Figure 3D) but majorly to lipid tail interactions γ (Figure 3F). Specifically, 3 residues from spike 1 contacted the lipid tail greater than 5000, indicating its crucial role in hydrophobic binding. On the other hand, spike 2 residues contributed minimally to the lipid tail with contacts less than 2000 and mainly towards the serine head and phosphate group. Spike 3 contained residues that contributed more than 2000 contacts with all lipid components, indicating its vital role in membrane binding. Most LactC2 interactions occurred at the negatively charged serine headgroup level (Table 2), followed by the lipid tail having the second most contacts. Overall, any residue contacts greater than 5000 occurred at either the serine headgroup (Figure 3D) or the lipid tail (Figure 3F), which suggests that both electrostatic and hydrophobic interactions equally drive Lact’s membrane binding and insertion.
Figure 3.
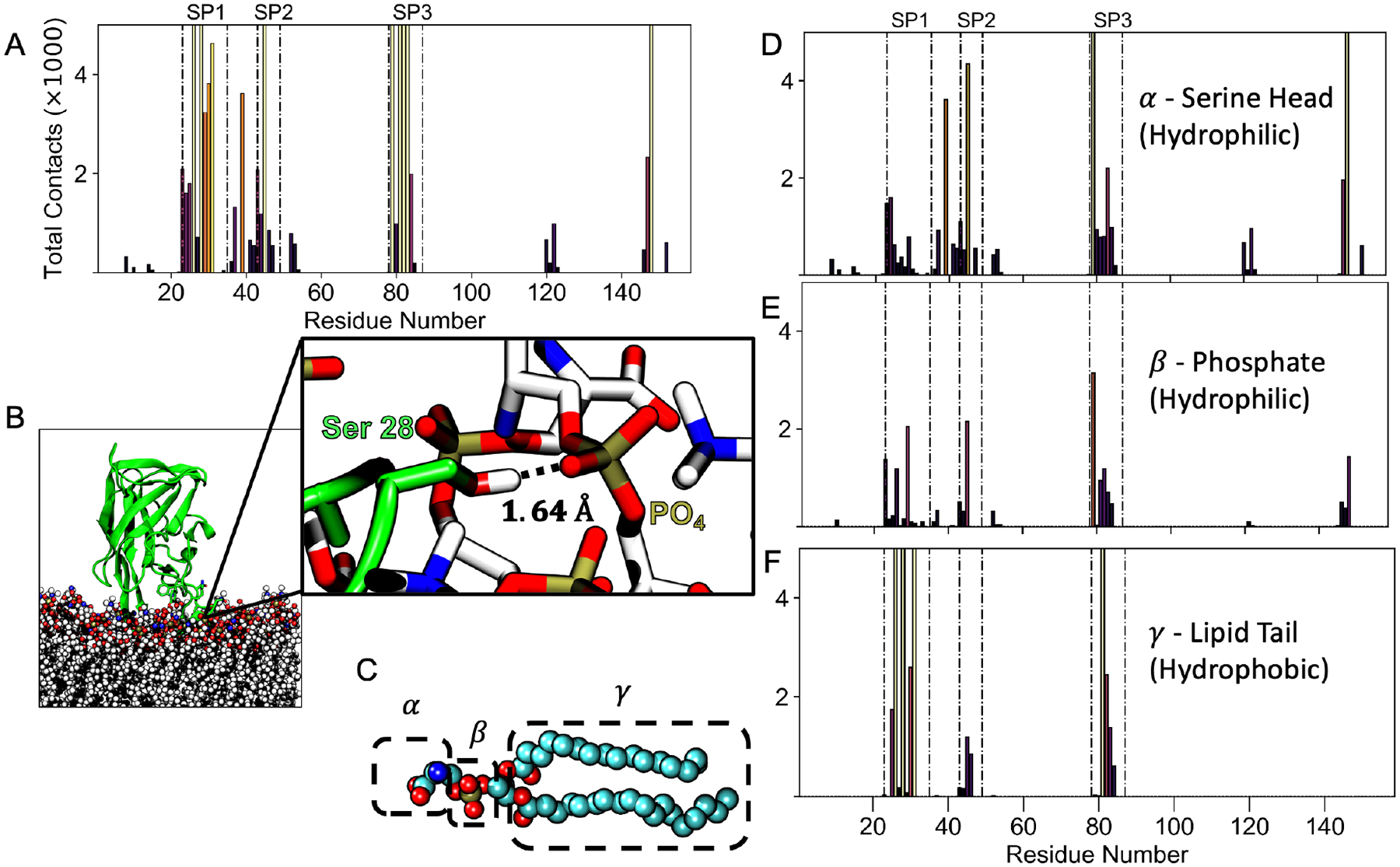
Contacts between LactC2 and the membrane for the inserted state. (A) Contact frequency distribution between LactC2 residues in the inserted ensemble and phosphatidylserines within 2.5Å. Each residue is colored by its peak intensity, with light colors corresponding to high intensity and dark colors corresponding to low. Total contacts above maximum are not shown for visual clarity. The dotted-dashed lines highlight the spike residues. (B) A representative simulation snapshot showing a contact between Ser 28 of spike 1 and a lipid phosphate group. The contact distribution was then decomposed to interactions with atoms belonging to different lipid components labeled as α, β, γ (C). They correspond to contact distributions from the serine headgroup (D), phosphate (E), and lipid tail (F).
Table 2.
Total contacts between LactC2 and each membrane component divided by the number of LactC2 residues.
| Lipid Component | Total Contacts per Residue |
|---|---|
| Choline (POPC) | 84.7 |
| Serine (POPS) | 327.8 |
| Phosphate | 115.9 |
| Glycerol | 25.2 |
| Lipid Tail | 253.1 |
Non-Spike Residues Like R148 Also Drive Lactadherin’s Membrane Binding
To reveal the role that non-spike residues played in membrane binding, we categorized contacts greater than 2000 based on spikes and non-spikes (Figure 4A). Arg 148 contacted the PS headgroup the most (contacts > 10,000), even if it is not considered a part of spikes 1–3. It is located on a β-hairpin opposite spike 2 (Figure 4B). Significant Arg 148 membrane interactions imply that non-spike residues aid Lact binding to membranes. We performed a sequence alignment of LactC2 from 4 other species to determine the evolutionary significance of this non-spike residue (Figure 4C). We found Arg 148 to be conserved across all organisms. Arg 39 also contacted the PS headgroup but minimally; an order of magnitude smaller than Arg 148. Although it belongs to the same loop as Spike 2, it is located further away from the spike residues and is thus we expected it to contribute less to membrane binding (Figure S4).
Figure 4.
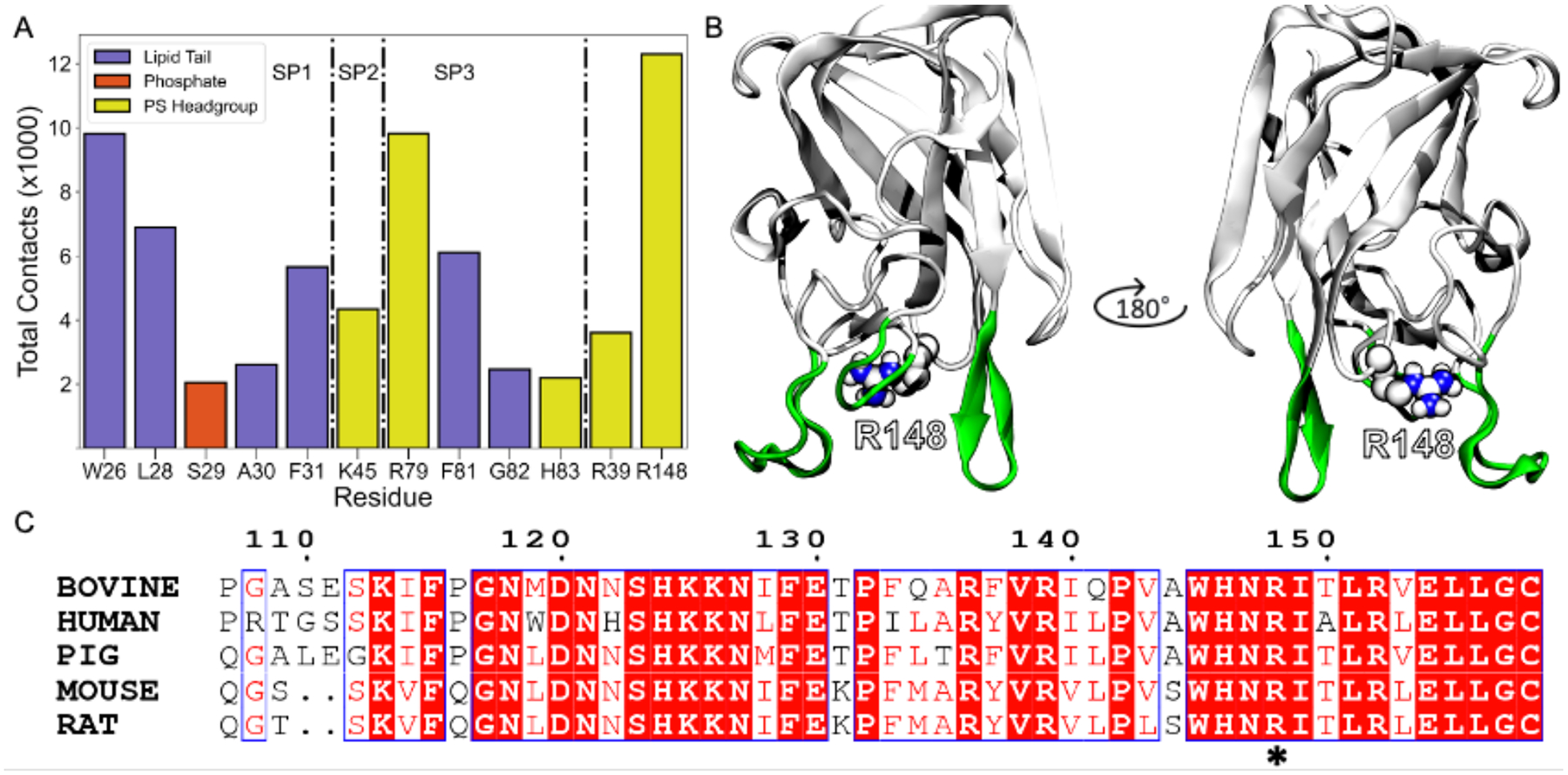
Non-spike residues (especially R148) contributions towards LactC2 binding and its evolutionary significance. (A) Total contacts for residues with at least 2000 contacts. Each residue is colored by the interacting lipid component. The dashed-dotted line highlights residues belonging to spikes 1, 2, 3, and non-spike residues (R39 and R148). (B) Location of R148 on LactC2 with spikes colored in green for positional reference. (C) Sequence alignment with four related species to bovine LactC2. The asterisk represents the position of R148 in the alignment.
Lactadherin Achieves the Side-Lying State via a Conserved Electrostatic Patch
To evaluate the evolutionary significance of the side-lying state (Figure 1B), we performed a multiple sequence alignment (MSA) between 5 related organisms (Figure 5A). We identified a highly conserved region in the MSA corresponding to a subset of membrane contacts in the side-lying state (SL) consisting of residues 119 to 130 (Figure 5B). Sequence alignment with homologs revealed 9 out of 11 residues were conserved (Figure 5C). Moreover, these contacts corresponding to the SL region were not present in the same analysis from the inserted ensemble (Figure 3A), which implies that Lact can utilize this cationic patch as a secondary binding mode.
Figure 5.
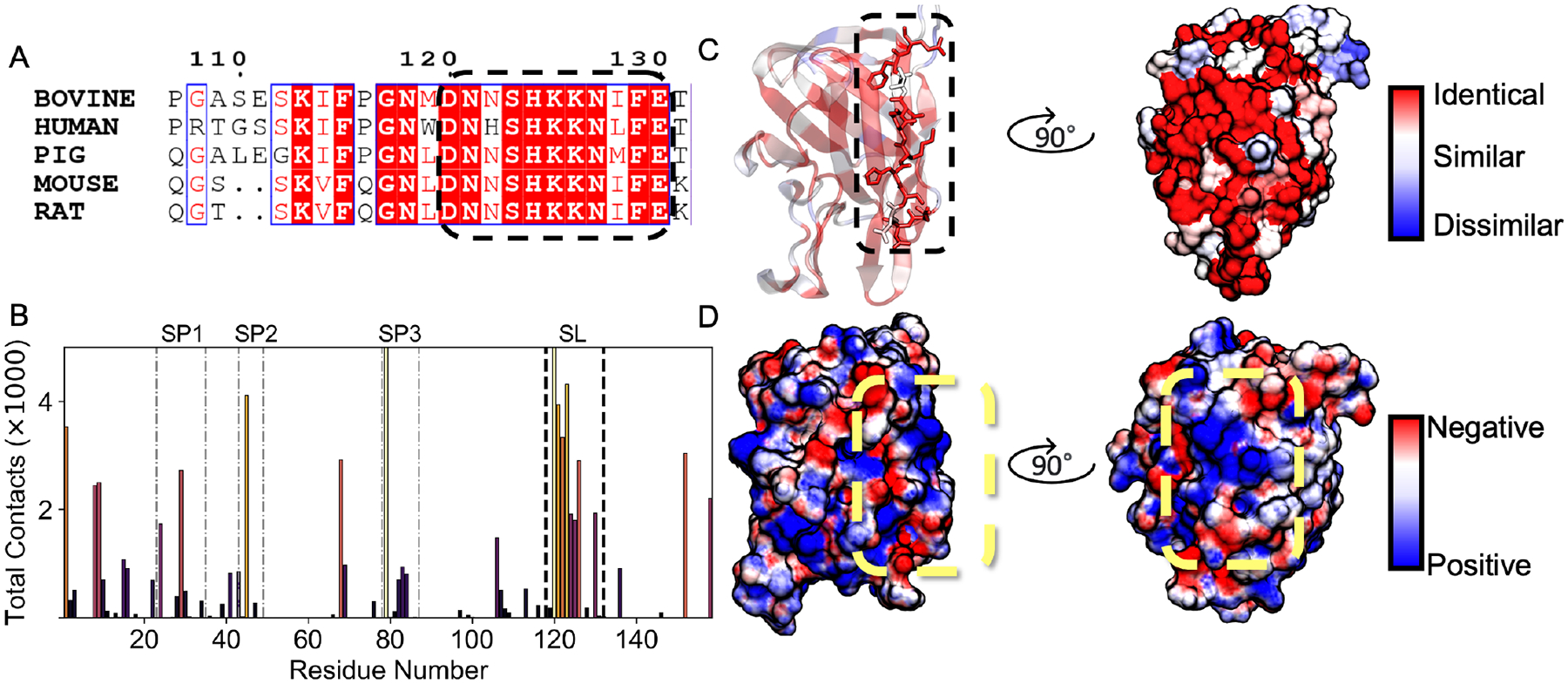
Side-lying conformation driven by conserved residues forming a positive electrostatic patch. (A) Sequence alignment with 4 homologs of bovine LactC2. The dashed oval represents the region where most contacts occur for LactC2 to achieve the Side-lying membrane-bound conformation. (B) Contact frequency distribution between LactC2 residues and phosphatidylserine within 2.5Å using data from the side-lying ensemble. Bar heights are colored by peak intensity, with light colors corresponding to a high contact number and dark colors corresponding to a low one. Faint dashed-dotted lines represent the 3 spikes, while dark black dashed lines represent side-lying (SL) positively charged patch contacts. (C) LactC2 is colored by sequence similarity ranging from identical residues (red) to similar (white) to dissimilar (blue). (D) Surface colored by electrostatic potential, from −3kBT/e(red) to +3kBT/e(blue). Yellow dashed oval identifies the positively charged patch.
To further understand the biophysical properties of these contacts, we calculated the electrostatic potential surface of LactC2. The surface corresponding to the highly conserved SL region formed a positively charged patch, indicating that electrostatic interactions play a critical role in achieving the side-lying conformation (Figure 5D). Our hypothesis that electrostatic interactions drive LactC2’s side-lying conformation was further supported when we decomposed the membrane contacts into different lipid moieties (Figure 6A). We found over 90% of SL contacts occur at the PS headgroup level (Figure 6B), which bears a net negative charge. Additionally, the SL residues consisted of predominantly polar and charged residues (Figure 6B). Asp 120 contributed the most contacts within the SL region through its interaction with the zwitterionic ammonium (NH3+) of the PS headgroup. Moreover, its evolutionary conservation further suggests its significance for stabilizing an SL conformation (Figure 5A). Little to no LactC2-membrane contacts occurred at the hydrophobic lipid tail level, which was consistent with our proposed insertion measurements showing that the spikes prefer to occupy space above the phosphate plane (Figure 6C). Despite the lack of spike residue insertion, some spikes still contributed toward the membrane contacts while in the side-lying state. LactC2’s 4 spike residues (S29, K45, K68, and R79) contributed to the top membrane contacts in contrast to the 10 inserted ensemble. Notably, Arg 79 from spike 3 contacted the membrane with an order of magnitude more than the other residues in the SL state (Figure 6B). These results demonstrate that charged residues primarily stabilize Lact’s side-lying state.
Figure 6.
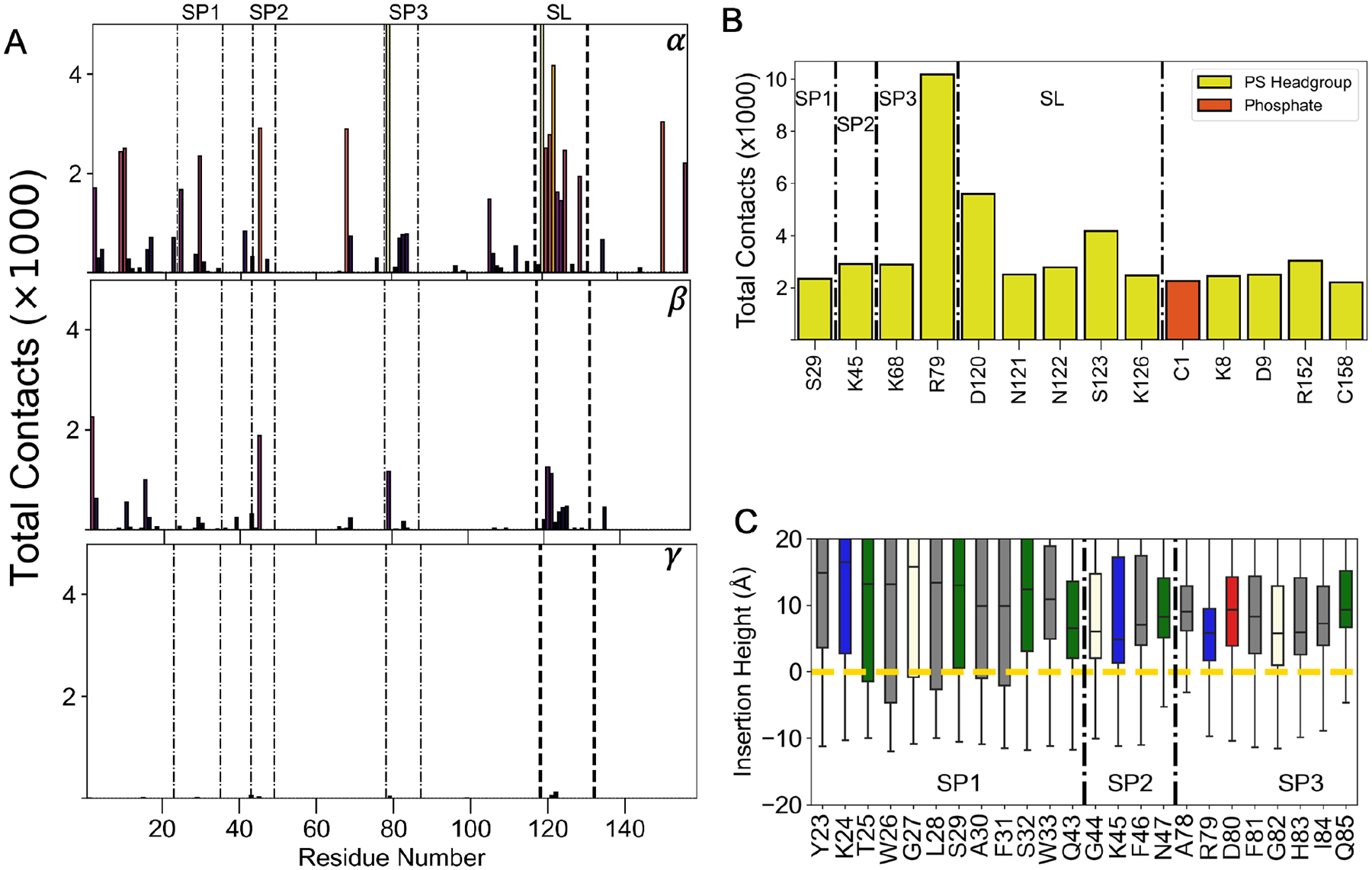
Characterizing LactC2’s side-lying interactions with the membrane. (A) Contact distribution between Lactadherin and atoms from different lipid moieties: α (serine headgroup), β (Phosphate), and γ (lipid tail). Contacts above the maximum are not shown for visual clarity. Spikes 1–3 are distinguished with dotted-dashed lines, and black dashed lines represent Side-lying (SL) conserved region contacts. (B) Contacts shown for residues with at least 2000 contacts and colored by their interacting lipid components. Residues belonging to spikes 1–3 are designated by letters SP. (C) Box plots of insertion height for each spike residues from Side-lying ensemble. Box plots are colored by residue type: grey-hydrophobic, blue-basic, red-acidic, green-polar, vanilla-other. Yellow dashed line represents the phosphate plane. Black dashed-dotted line demarcates spike residues 1–3.
Lactadherin Can Still Attain Both States in the Presence of its C1 Domain
To further test our hypothesis that LactC2 can achieve the inserted and side-lying state while membrane bound, we used AlphaFold40,41 to attach Lact’s C1 domain and build a Lactadherin C1-C2 construct (LactC1C2) (Figure 7A, 7B). We observed the RMSD of the C1 and C2 domains converge below 2.5Å with 200 ns of sampling across all replicates (Figure S5, S6). The low RMSD values indicate the stability of both domains as LactC1C2 binds to the membrane in either conformation. To evaluate the hypothesis that LactC1C2 can achieve a side-lying state (Figure 7B), we plotted the predicted alignment error generated by AlphaFold (Figure S7). AlphaFold predicted significant errors greater than 15 Å for inter-domain residue pairs between C1 (residues 1 – 159) and the C2 domain (residues 164–317). The high errors suggest Alphafold’s low confidence in the two domains’ relative position, indicating that a highly flexible linker connects them. This conclusion was consistent with our observations that the linker fully extended to attain the side-lying state.
Figure 7.
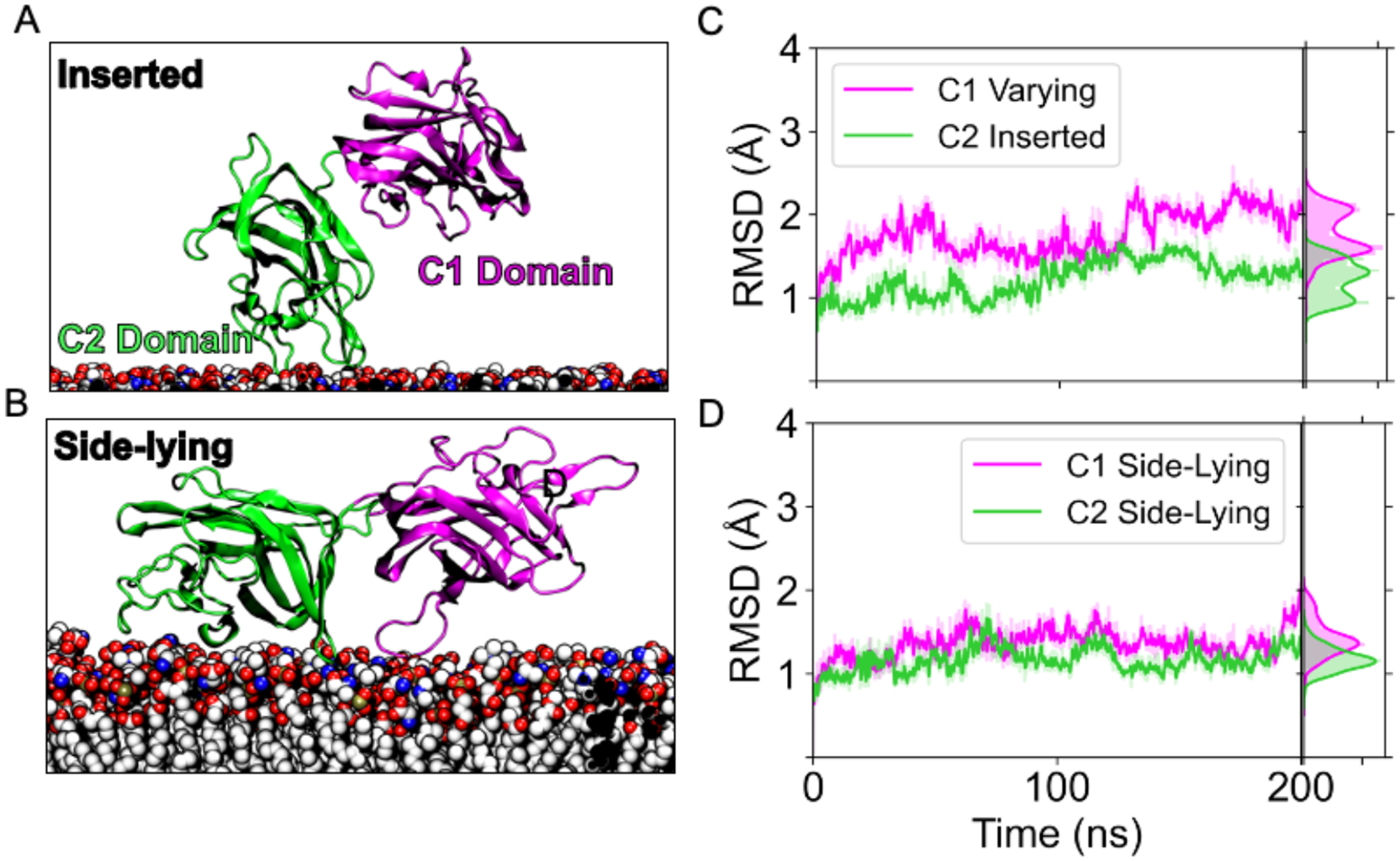
Stability of the C1 and C2 domains in two different membrane-bound states. (A) Snapshot from the simulations depicting the LactC1C2 construct when the C2 domain is in the inserted state. (B) Snapshot showing the same construct in the side-lying state. (C) RMSD trace of the C1 (magenta) in varying states and C2 domain (green) in the inserted state for a single replicate of 200 ns. (D) RMSD trace for the LactC1C2 where both domains maintain a side-lying state.
To investigate the membrane binding dynamics of LactC1C2’s C domains, we measured the tilt orientations of each one. In the inserted state (Figure 8A), the C2 orientation distribution centered at 24° where Lact’s spikes also interacted with the membrane. On the other hand, the C1 domain’s distribution was shifted toward 60°. The C1, therefore, takes on a more side-lying configuration as previously described (Figure 1D). In contrast, C1 sampled more orientations in the side-lying state (Figure 8B), indicating that this state allowed for more conformational heterogeneity. We then sought to understand the relative motions between these domains by performing dynamic cross-correlation analysis. Inter-domain residues showed little to no correlation regardless of taking on an inserted (Figure 8C) or the side-lying state (Figure 8D). Specifically, a few residue cross-correlations were −0.5 to −0.25, while most inter-domain residue pairs had no correlation. Therefore, C1 minimally influenced the motions of the C2 membrane binding domain and suggests that C2 acts as a stationary anchor tethered to C2 that allows it to explore more conformations.
Figure 8.
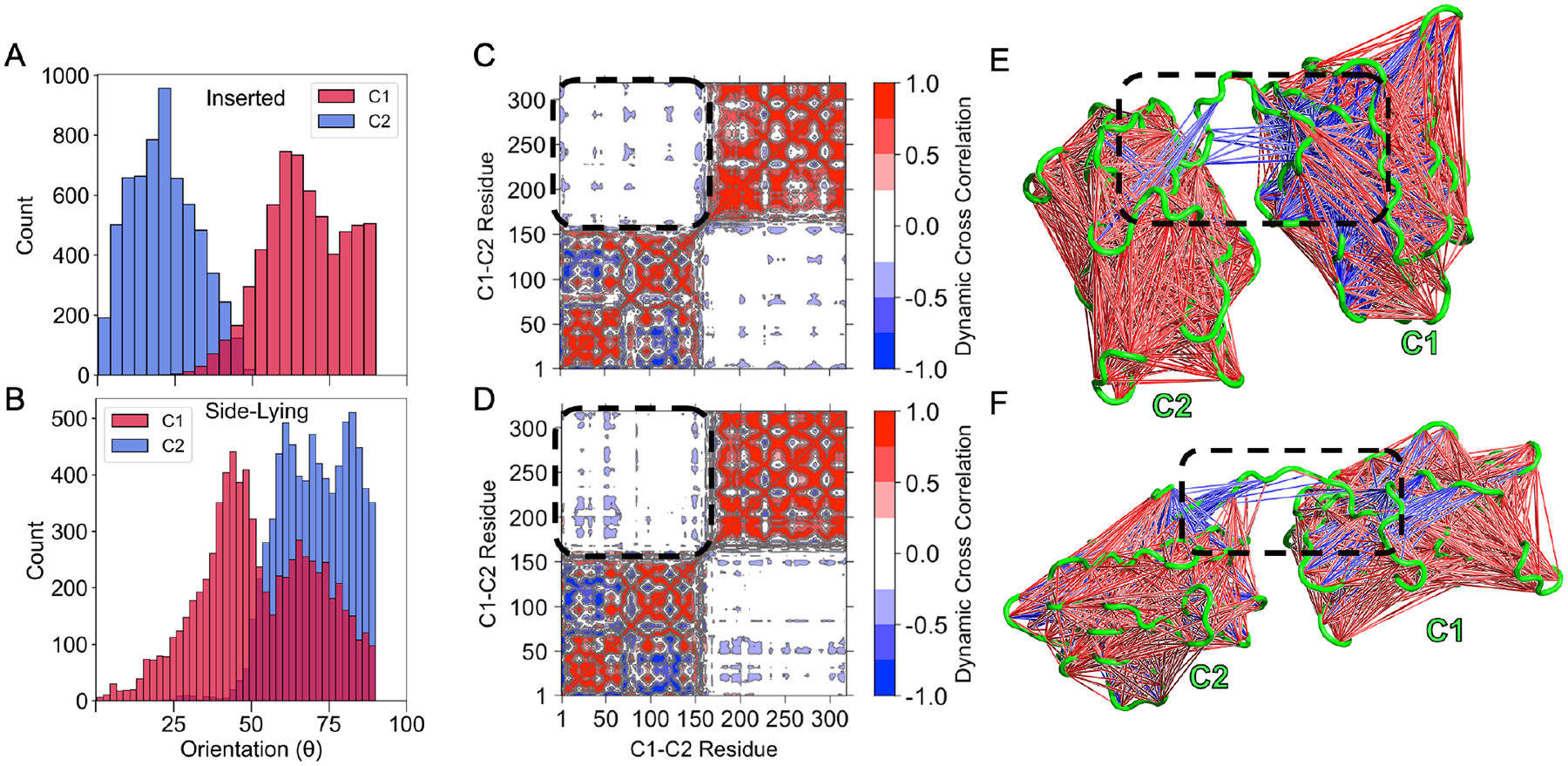
The C1 domain does not influence its neighboring membrane binding domain. (A) Tilt distribution of the C1 and C2 domain of lactadherin in the inserted state and (B) side-lying state relative to the membrane (defined in Figure 1B). Dynamic cross correlation matrix for the inserted state (C) and the side-lying state (D). Positive correlations are shown with red, and anticorrelations with blue. Regions corresponding interdomain correlations are encircled with a dashed line. Corresponding structural representations of these regions are shown for the inserted state (E) and the Side-lying state (F) to visualize the interdomain cross-correlations. Correlations of 0.6–0.8 (both positive and negative) are shown since larger ones were not present.
Lactadherin’s Membrane Bound State Enables New Investigations of Coagulation Factors
To apply our LactC1C2 membrane-bound models to probe clinically relevant homologs, we first simulated a coagulation factor V C1-C2 construct (fVC1C2) derived from a cryo-EM structure (PDB ID 7KVE)35. We assembled it by aligning 7KVE’s C2 to the same domain of LactC1C2 in the inserted state (Figure 9A). After converged sampling of 200 ns, the orientations of both domains were normally distributed where C1’s orientation peaked around 40°, while C2’s was around 17° (Figure 9B). These values suggest that the C1 domain takes on a more tilted orientation by almost 20°, while the C2 domain remains more upright with its spike regions interacting with the membrane. The distribution narrowness also indicates that fV’s C1 and C2 domains hardly fluctuate once bound to the membrane. Moreover, the structures of the domain were stable as the RMSD of the C1 domain converged to 3Å while the C2 domain converged to around 1.6 Å (Figure 9C). These convergences to low RMSD values demonstrate the structural stability of both domains while membrane bound. We also measured the contacts occurring with PS lipids. For the membrane-binding C2 domain, residue contacts greater than 2000 all consisted of spike residues (Figure 9D) (Table S1). For the C1 domain, the top residue contacts were R29, S38, K76, H77, Y78, and R145 (Table S1).
Figure 9.
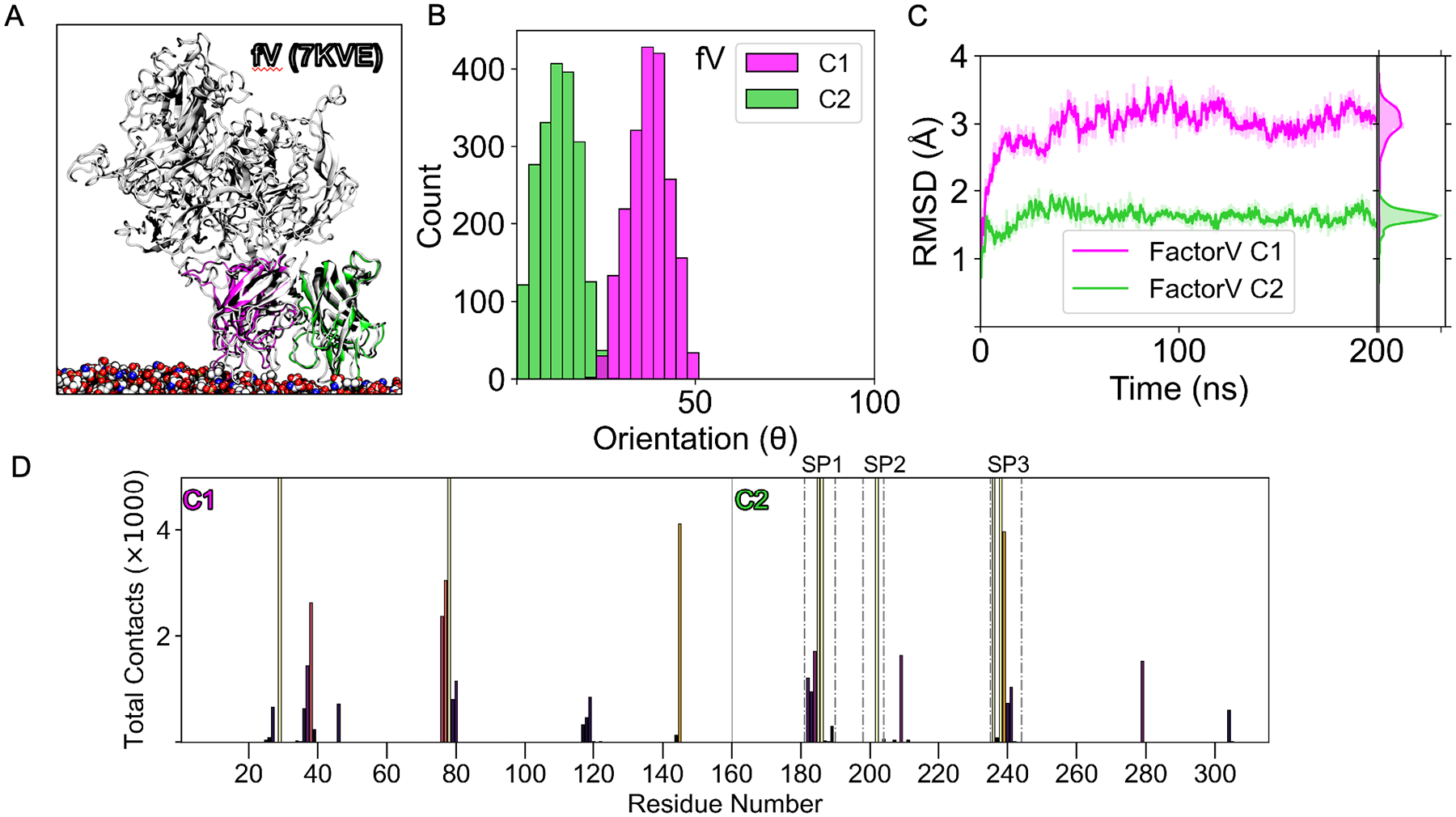
Factor V’s C-domains stabilizes in an inserted State. (A) An alignment of an equilibrated simulation snapshot of coagulation factor V (fV) C1 (magenta) and C2 (green) domain bound to the membrane with 3.3 Å cryo-EM structure (PDB ID: 7KVE).42 (B) Tilt distribution of the C1 and C2 domain of fV from the membrane (angle defined in Figure 1A). (C) RMSD trace of factor V’s C1 (magenta) and C2 domain (green) for 200 ns. (D) Contact frequency distribution between fVC1C2 residues and phosphatidylserine lipids within 2.5Å. The vertical line at residue 160 separates the distribution between the C1 and C2 domains. Each residue is colored by its peak intensity, with light colors corresponding to high intensity and dark colors corresponding to low. Total contacts above the maximum of 5000 are not shown for visual clarity. Spikes 1–3 of the C2 domain are distinguished with dotted-dashed lines. For this work fV residue numbering of C1 starts at 1, corresponding to 1879 in PDB ID 7KVE.42
Additionally, we simulated a coagulation factor VIII C1-C2 construct (fVIIIC1C2) based on the crystal structure (PDB ID: 7K66).43 We initiated simulations in the same fashion as the fV simulations by aligning the PDB structure’s C2 domain using LactC1C2 as a template. We measured the orientation distribution of both domains and found the C2 orientation to peak around 65° (Figure 10B), indicating that the domain favored a side-lying state. In contrast, the C1 domain did not show an apparent central tendency, thus, suggesting it can sample more orientations and attain both a side-lying and inserted state (Figure S8). Regarding membrane contacts, C1 contributed minimally (Figure S10), with most coming from the C2 domain (Table S2), which implies that the C1 domain did not play a role in membrane binding as it did previously with fVC1C2 (Figure 9D). In addition, the RMSD converged for the C1 to ~1.7 Å and C2 to ~1 Å, indicating fVIIIC2C2’s structural stability throughout the 200 ns (Figure 10D). To evaluate which conformation the antibody structure from 7K6643 could best align to, we compared alignments to fVIIC1C2 in both states. Interestingly, due to membrane clashing, antibody alignments did not fit when both domains were in the inserted state (Figure 10A). On the other hand, when both domains were side-lying, a fit was possible from the lack of steric clashes (Figure 10B).
Figure 10.
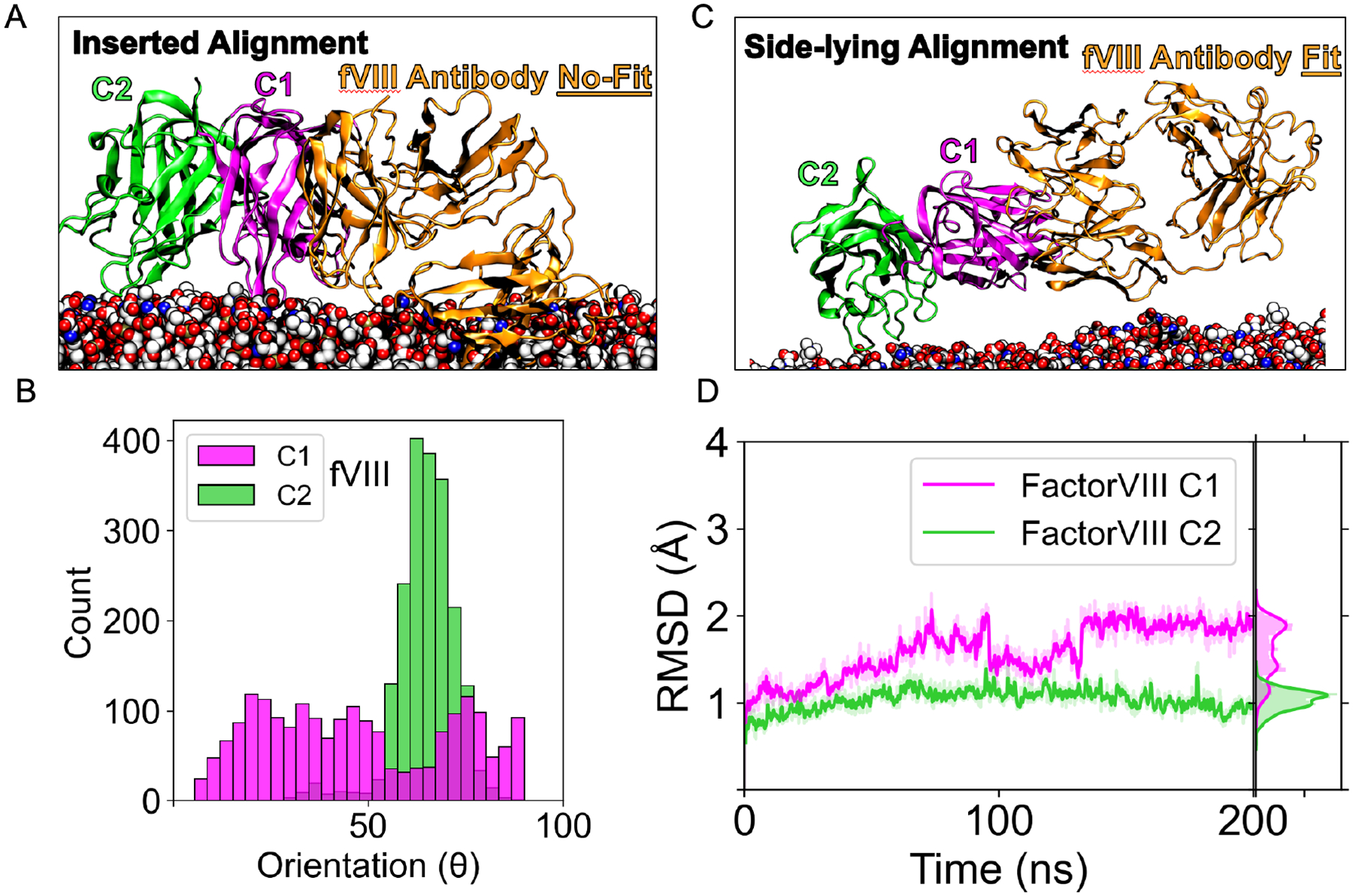
Factor VIII’s side-lying conformation permits antibody fitting. (A) An alignment of the factor VIII C1C2 (PDB ID 7K66)43 with its antibody inhibitor (orange) in with a simulation snapshot in the inserted state showing significant steric clashes with the membrane model. (B) Tilt orientation distribution of the C1 and C2 domain of coagulation factor VIII from the membrane (angle defined in Figure 1A). (C) Alignment of the factor VIII C1C2 (PDB ID 7K66)43 with its antibody inhibitor (orange) with a snapshot in the side-lying state. (D) RMSD trace of the C1 (magenta) and C2 domain (green) for factor VIII for 200 ns.
DISCUSSION
Here we describe a dynamic two-state binding model for Lact that provide insights on how coagulation factors bind to the membrane with atomistic resolution. Although previous studies of Lact have identified its spikes as crucial for binding, determining the possible conformations of its membrane-bound states is challenging to ascertain based on single mutations or structural analysis without the bilayer. Our investigation revealed that Lact interacts with distinct membrane regions by adopting either an inserted or a side-lying conformation. The side binding mode was driven by a conserved positively charged patch that maintained protein contact with the membrane surface. Our model also enabled us to characterize dynamics of the membrane binding domains of fV and fVIII at the amino acid level. We found that fV uses spikes from both C domains for membrane binding, while fVIII’s C domains prefer a tilted, side-lying conformation. The C1 domain showed more extensive conformational sampling without interacting with the membrane and permitted antibody-C1 binding.
Spike Residues in Lactadherin Play Distinct and Specific Roles in Membrane Binding
Lact achieved its inserted state (Figure 1C) by interacting with distinct membrane components and reaching different insertion heights. Hydrophobic residues, specifically those on spike 1 (W26, L28) and spike 3 (F81), were critical for Lact to insert deeply into the bilayer (Figure 2B). Interestingly, these identical residues were previously predicted to significantly reduce Lact binding activity.16 Despite the lacking evidence for PS headgroup interactions, we determined H83, R148, K45, R79, and G82 (Figure 4A) to contact the polar region of the membrane considerably and agree with previous docking results.16 Surprisingly, we also observed non-spike residues R36 and R148 as important membrane interaction contributors, with R148 contacting the membrane more than all spike residues (Figure 4A). We found this residue to be highly conserved among other organisms (Figure 4C), and an NMR study demonstrated it as a top interacting residue with PS.17 The agreement between our model and experimental data suggests that non-spike residues could also be vital for membrane binding. Thus, additional residues adjacent to the spikes should be considered for future investigations of PS binding for other related C2 domains.
Despite characterizing essential hydrophobic and hydrophilic interactions, it remained unclear how Lact’s spikes cooperated to achieve membrane binding. We, therefore, decomposed Lact-membrane interactions into different lipid components (Figure 3) and found that each spike preferred a specific membrane region. For instance, spike 1 predominantly favored hydrophobic tail interactions, while spike 2 preferred the PS headgroup. Spike 3 contained residues interacting with the headgroup and lipid tail (Figure 3D, E, F). Each spike’s interaction specificity toward a membrane component also explains why we observed a tilt distribution for the inserted state (Figure 1B). A spike preference for different membrane heights would incline Lact while bound and suggests that optimal binding occurs at an angle rather than the commonly idealized vertical fashion. Since another report58 demonstrated a similar tilted conformation in fVIII’s homologous C2 domain, the configuration could affect coagulation cascade regulation. Nonetheless, more evidence would be needed to further elucidate the functional advantage of a tilted conformation binding to PS-enriched membranes.
The Importance of the Cationic Patch for LactC2’s Multimodal Binding to PS Lipids
LactC2 achieved its side-lying state primarily through electrostatic headgroup contacts (Figure 5D, 6B) at the surface membrane level (Figure 6C). Despite LactC2’s contact distribution (Figure 3A) agreeing with chemical shift perturbation data,17 there were minimal perturbation signals accordant with the SL region that we identified (i.e., residues 120–130) (Figure 6A). One reason for this inconsistency could be from the PS titrations employed during the NMR experiments, which might have resulted in an insufficient binding surface for Lact to achieve a side-lying state. This could especially be the case since a previous study demonstrated the sensitivity of binding to PS concentration and micelle size.8
Although preceding studies have not explicitly mentioned a side-lying state for Lact, our findings are supported by studies of PKC C2 domain, which reported a remarkably similar binding model1 (Figure 1). Both proteins bind to PS lipids using three key inter-strand loops and share similar β-sandwich structures, despite lacking an evolutionary relationship. The majority of PKC C2 domains contain a cationic patch on the concave face of the C2 β-sandwich, known as the cationic β-groove.23 In one study, PKC used its lysine-rich patch as a secondary lipid binding site25 resembling the mechanism we identified (Figure 5). Fluorescence experiments also showed an increased C2 incline angle in the presence of PIP224 similar to the increased tilt angle of LactC2’s side-lying state (Figure 2B).
Additionally, the conservation of this patch (Figure 5A) suggests that it plays a crucial functional role in Lact binding that was previously unrecognized. The evidence of sequence conservation and similar binding models observed in other proteins support the notion that a secondary binding mode could be a critical and general feature for proteins that bind to PS lipids. The identification of a multimodal binding model for Lact could lead to a better understanding of its competitive binding with annexin V, where Lact demonstrated more efficient binding to PS expressed in apoptotic cells.19,59,60 By reducing the positive charge in the cationic patch region, similar to the study of synaptotagmin,61 one could investigate the relationship between our findings and Lact’s PS binding efficiency. Our model would predict that decreasing the positive charge in this region would result in a decrease in Lact’s binding efficiency, as it would reduce its ability to participate in multiple binding modes.
We investigated the impact of Lact’s tandem C1 domain on its binding properties, as previous studies primarily focused on the C2 domain. Our results indicated that the linker between the C1 and C2 domains is sufficiently flexible to allow LactC1C2 to adopt both the inserted and side-lying states (Figure 7). AlphaFold’s predictions supported this finding, by predicting large alignment errors for interdomain residue pairs (Figure S7). Interestingly, the C1 and C2 domains exhibited independent motions (Figure 8C, 8D), implying that the C2 domain primarily functions as a membrane anchor while the C1 domain moves freely. This novel insight into the motion of the C1 domain may provide a mechanistic basis for how Lact mediates binding between different PS membranes.9 Given that the EGF domain connects directly to C1,10 these new domains would likely restrict the extension of the side-lying conformation (Figure 8B). However, since they do not participate in membrane binding and are situated away from the bilayer, we would predict that the presence of these domains only partially prevents the side-lying conformation from being achieved. Since the EGF domains bind to integrin receptors,14 C1’s conformational flexibility could be essential for effective integrin receptor binding.
The Role of C Domains in Hemostasis with Insights from fV and fVIII Conformational Changes
Our fVC1C2 construct maintained an inserted conformation (Figure 9B) with spike residues from both the C1 and C2 domains contacting the membrane (Figure 9D). Reports of fV’s crystal and cryo-EM structure hypothesized that C1 and C2 participate in membrane binding based on their side-by-side organization.42,62 This hypothesis finds further support in an additional structural study,63 which features activated fV within the prothrombin and prothrombinase complex. Specifically, the cryo-EM structures reveal how the Gla domains and prothrombin align with the same plane of fVa’s C domains for membrane interaction. When fV is in complex form, its C1 and C2 domains were predicted to orient themselves perpendicular to the membrane, mirroring the ‘inserted’ conformation predicted by the fVC1C2 model (Figure 9B). Our computational predictions with the available structural data on fV—both in isolation and when complexed with coagulation cascade cofactors—suggest that the C domains of fV primarily adopt an inserted state rather than a side-lying state.
Moreover, these experimental structural models lacked an explicit bilayer and thus the exact residue contacts were difficult to obtain. Alanine scanning mutations of the C1 domain established that residues Y1956 and R2023 were critical for lipid binding,64 which we also determined to contact the membrane significantly (Table S1). Likewise, our predictions (Table S1) agreed with studies showing that the C2’s W2063 and W2064 participated in high-affinity PS binding.65 The structural, mutagenesis, and computational results suggest that both fV’s C domains could be necessary for membrane binding in an inserted state (Figure 9A), which was previously unclear due to the lack of dynamical data. If fV favors a single membrane-bound state, it could explain the underlying mechanism of why Lact efficiently competes with fV18 since Lact would participate in multimodal binding (Figure 1). Put differently, Lact’s access to an additional binding mode (i.e., the side-lying, SL, state) that its coagulation counterpart does not utilize could provide a mechanistic account for its more efficient binding. Considering that Lact and fV compete for the same PS sites, this model could offer a mechanism for downregulating PS-dependent physiological processes, such as blood coagulation.
In contrast, fVIIIC1C2’s C2 domain preferred a tilted, side-lying conformation, while the C1 (Figure 10B) showed more extensive conformational sampling without membrane interactions (Figure S9). Prior reports have reported similar changes in the C domains. For example, an electron microscopy model proposed the C2 domain inclined 60°, which we captured in our model (Figure 10B), while C1 preferred to be unbound away from the membrane.66 A cryo-EM study proposed a similar model with C2 only bound with an extended C1 state.67 We also determined that this side-lying conformation permitted antibody-C1 binding, where significant antibody-membrane steric clashes disappeared in this state (Figure 10C) compared to an upright, inserted state. Crystallographic studies similarly demonstrated how the C2 domain inclined after being complexed with the 2A9 antibody, suggesting that a conformational change resulted from its binding.43,58 Collectively, these results suggest that the side-lying state of fVIII could have implications in hemostasis that were before uncertain from structural analysis lacking a lipid bilayer. One function of a side-lying conformation could be that it allows the proper binding of other cofactors like factor IXa and its membrane-binding Gla domain.36,68 We corroborated this hypothesis by showing how fVIII’s additional A1-A3 domains could still align without membrane clashes in the side-lying state (Figure S8). Secondly, a side-lying conformation could be necessary to expose the C1 for both antibody and von Willebrand factor binding.43,69
CONCLUSION
Current Lact binding models primarily derive from static structures. Although they are a critical first stage in identifying key membrane interacting regions, determining the membrane bound ensemble remains difficult especially when considering an explicit bilayer. This study presents a new framework for understanding Lact’s multi-state binding and its possible therapeutic application to coagulation factor interactions with lipids. To develop more robust models that can lead to promising treatment opportunities, it will be essential to gain a more precise understanding of the structure-function relationship from the binding states identified in this study.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to Prof. Martin Gruebele for insightful discussions.
Funding Sources
Any funds used to support the research of the manuscript should be placed here (per journal style). The authors are grateful for the National Institutes of Health Transformative Research Award (R01 GM123455) and NHLBI grant R35 HL135823. K.J.C. acknowledges support from the NSF-GRFP and the Ford Foundation Predoctoral Fellowship. A.M.D. was supported by a postdoctoral fellowship from the National Center for Supercomputing Resource and Cyprus Institute. T.V.P. acknowledges support from the NIH (R01-GM141298), the Department of Chemistry, the School of Chemical Sciences, and the Office of the Vice Chancellor for Research (RSOCR Award 4703) at University of Illinois at Urbana-Champaign. The authors acknowledge the computational resources from the Texas Advanced Computing Center (TACC) at the University of Texas at Austin made available from the Extreme Science and Engineering Discovery Environment (XSEDE) Grant TG-MCB130112, which is supported by National Science Foundation Grant ACI-1053575.
ABBREVIATIONS
- Lact
lactadherin
- LactC1C2
lactadherin C1-C2 construct
- fV
factor V
- fVIII
factor VIII
- fVC1C2
factor V C1-C2 construct
- fVIIIC1C2
factor VIII C1-C2 construct
- HMMM
highly mobile mimetic model
- MD
molecular dynamics
- PS
phosphatidylserine
- EGF
epidermal growth factor
- NMR
solution nuclear magnetic resonance
- PKC
Protein Kinase C’s
- PS
palmitoyl-oleoyl phosphatidylserine
- PC
palmitoyl-oleoyl phosphatidylcholine
Footnotes
Supporting Information. A listing of the contents of each file supplied as Supporting Information should be included. For instructions on what should be included in the Supporting Information as well as how to prepare this material for publications, refer to the journal’s Instructions for Authors.
The following files are available free of charge.
Additional analysis figures (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lemmon MA Membrane Recognition by Phospholipid-Binding Domains. Nat. Rev. Mol. Cell Biol 2008, 9 (2), 99–111. 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- (2).Leventis PA; Grinstein S The Distribution and Function of Phosphatidylserine in Cellular Membranes. Annu. Rev. Biophys 2010, 39 (1), 407–427. 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- (3).Aziz M; Jacob A; Matsuda A; Wang P Review: Milk Fat Globule-EGF Factor 8 Expression, Function and Plausible Signal Transduction in Resolving Inflammation. Apoptosis 2011, 16 (11), 1077–1086. 10.1007/s10495-011-0630-0. [DOI] [PubMed] [Google Scholar]
- (4).Cheyuo C; Aziz M; Wang P Neurogenesis in Neurodegenerative Diseases: Role of MFG-E8. Front. Neurosci 2019, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Neutzner M; Lopez T; Feng X; Bergmann-Leitner ES; Leitner WW; Udey MC MFG-E8/Lactadherin Promotes Tumor Growth in an Angiogenesis-Dependent Transgenic Mouse Model of Multistage Carcinogenesis. Cancer Res. 2007, 67 (14), 6777–6785. 10.1158/0008-5472.CAN-07-0165. [DOI] [PubMed] [Google Scholar]
- (6).McLaughlin S; Murray D Plasma Membrane Phosphoinositide Organization by Protein Electrostatics. Nature 2005, 438 (7068), 605–611. 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- (7).Di Paolo G; De Camilli P Phosphoinositides in Cell Regulation and Membrane Dynamics. Nature 2006, 443 (7112), 651–657. 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- (8).Shi J; Heegaard CW; Rasmussen JT; Gilbert GE Lactadherin Binds Selectively to Membranes Containing Phosphatidyl-l-Serine and Increased Curvature. Biochim. Biophys. Acta BBA - Biomembr 2004, 1667 (1), 82–90. 10.1016/j.bbamem.2004.09.006. [DOI] [PubMed] [Google Scholar]
- (9).Andersen MH; Graversen H; Fedosov SN; Petersen TE; Rasmussen JT Functional Analyses of Two Cellular Binding Domains of Bovine Lactadherin. Biochemistry 2000, 39 (20), 6200–6206. 10.1021/bi992221r. [DOI] [PubMed] [Google Scholar]
- (10).Kamińska A; Enguita FJ; Stępień EŁ Lactadherin: An Unappreciated Haemostasis Regulator and Potential Therapeutic Agent. Vascul. Pharmacol 2018, 101, 21–28. 10.1016/j.vph.2017.11.006. [DOI] [PubMed] [Google Scholar]
- (11).Ait-Oufella H; Kinugawa K; Zoll J; Simon T; Boddaert J; Heeneman S; Blanc-Brude O; Barateau V; Potteaux S; Merval R; Esposito B; Teissier E; Daemen MJ; Lesèche G; Boulanger C; Tedgui A; Mallat Z Lactadherin Deficiency Leads to Apoptotic Cell Accumulation and Accelerated Atherosclerosis in Mice. Circulation 2007, 115 (16), 2168–2177. 10.1161/CIRCULATIONAHA.106.662080. [DOI] [PubMed] [Google Scholar]
- (12).Oshima K; Yasueda T; Nishio S; Matsuda T MFG-E8: Origin, Structure, Expression, Functions and Regulation. In MFG-E8 and Inflammation; Wang P, Ed.; Springer Netherlands: Dordrecht, 2014; pp 1–31. 10.1007/978-94-017-8765-9_1. [DOI] [Google Scholar]
- (13).Villoutreix BO; Miteva MA Discoidin Domains as Emerging Therapeutic Targets. Trends Pharmacol. Sci 2016, 37 (8), 641–659. 10.1016/j.tips.2016.06.003. [DOI] [PubMed] [Google Scholar]
- (14).Andersen MH; Berglund L; Rasmussen JT; Petersen TE Bovine PAS-6/7 Binds AVβ5 Integrin and Anionic Phospholipids through Two Domains. Biochemistry 1997, 36 (18), 5441–5446. 10.1021/bi963119m. [DOI] [PubMed] [Google Scholar]
- (15).Lin L; Huai Q; Huang M; Furie B; Furie BC Crystal Structure of the Bovine Lactadherin C2 Domain, a Membrane Binding Motif, Shows Similarity to the C2 Domains of Factor V and Factor VIII. J. Mol. Biol 2007, 371 (3), 717–724. 10.1016/j.jmb.2007.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shao C; Novakovic VA; Head JF; Seaton BA; Gilbert GE Crystal Structure of Lactadherin C2 Domain at 1.7Å Resolution with Mutational and Computational Analyses of Its Membrane-Binding Motif*. J. Biol. Chem 2008, 283 (11), 7230–7241. 10.1074/jbc.M705195200. [DOI] [PubMed] [Google Scholar]
- (17).Ye H; Li B; Subramanian V; Choi B-H; Liang Y; Harikishore A; Chakraborty G; Baek K; Yoon HS NMR Solution Structure of C2 Domain of MFG-E8 and Insights into Its Molecular Recognition with Phosphatidylserine. Biochim. Biophys. Acta BBA - Biomembr 2013, 1828 (3), 1083–1093. 10.1016/j.bbamem.2012.12.009. [DOI] [PubMed] [Google Scholar]
- (18).Shi J; Gilbert GE Lactadherin Inhibits Enzyme Complexes of Blood Coagulation by Competing for Phospholipid-Binding Sites. Blood 2003, 101 (7), 2628–2636. 10.1182/blood-2002-07-1951. [DOI] [PubMed] [Google Scholar]
- (19).Shi J; Pipe SW; Rasmussen JT; Heegaard CW; Gilbert GE Lactadherin Blocks Thrombosis and Hemostasis in Vivo: Correlation with Platelet Phosphatidylserine Exposure. J. Thromb. Haemost 2008, 6 (7), 1167–1174. 10.1111/j.1538-7836.2008.03010.x. [DOI] [PubMed] [Google Scholar]
- (20).Green D Coagulation Cascade. Hemodial. Int 2006, 10 (S2), S2–S4. 10.1111/j.1542-4758.2006.00119.x. [DOI] [PubMed] [Google Scholar]
- (21).Lenting PJ; van Mourik JA; Mertens K The Life Cycle of Coagulation Factor VIII in View of Its Structure and Function. Blood 1998, 92 (11), 3983–3996. 10.1182/blood.V92.11.3983. [DOI] [PubMed] [Google Scholar]
- (22).Baumgartner S; Hofmann K; Bucher P; Chiquet-Ehrismann R The Discoidin Domain Family Revisited: New Members from Prokaryotes and a Homology-Based Fold Prediction. Protein Sci. 1998, 7 (7), 1626–1631. 10.1002/pro.5560070717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cho W; Stahelin RV Membrane Binding and Subcellular Targeting of C2 Domains. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 2006, 1761 (8), 838–849. 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- (24).Kohout SC; Corbalán-García S; Gómez-Fernández JC; Falke JJ C2 Domain of Protein Kinase Cα: Elucidation of the Membrane Docking Surface by Site-Directed Fluorescence and Spin Labeling. Biochemistry 2003, 42 (5), 1254–1265. 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Corbalán-Garcıá S; Garcıá-Garcıá J; Rodríguez-Alfaro JA; Gómez-Fernández JC A New Phosphatidylinositol 4,5-Bisphosphate-Binding Site Located in the C2 Domain of Protein Kinase Cα*. J. Biol. Chem 2003, 278 (7), 4972–4980. 10.1074/jbc.M209385200. [DOI] [PubMed] [Google Scholar]
- (26).Corbalan-Garcia S; Gómez-Fernández JC Signaling through C2 Domains: More than One Lipid Target. Biochim. Biophys. Acta BBA - Biomembr 2014, 1838 (6), 1536–1547. 10.1016/j.bbamem.2014.01.008. [DOI] [PubMed] [Google Scholar]
- (27).Wu Z; Ma L; Courtney NA; Zhu J; Landajuela A; Zhang Y; Chapman ER; Karatekin E Polybasic Patches in Both C2 Domains of Synaptotagmin-1 Are Required for Evoked Neurotransmitter Release. J. Neurosci 2022, 42 (30), 5816–5829. 10.1523/JNEUROSCI.1385-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ohkubo YZ; Pogorelov TV; Arcario MJ; Christensen GA; Tajkhorshid E Accelerating Membrane Insertion of Peripheral Proteins with a Novel Membrane Mimetic Model. Biophys. J 2012, 102 (9), 2130–2139. 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pogorelov TV; Vermaas JV; Arcario MJ; Tajkhorshid E Partitioning of Amino Acids into a Model Membrane: Capturing the Interface. J. Phys. Chem. B 2014, 118 (6), 1481–1492. 10.1021/jp4089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Qi Y; Cheng X; Lee J; Vermaas JV; Pogorelov TV; Tajkhorshid E; Park S; Klauda JB; Im W CHARMM-GUI HMMM Builder for Membrane Simulations with the Highly Mobile Membrane-Mimetic Model. Biophys. J 2015, 109 (10), 2012–2022. 10.1016/j.bpj.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y; Jo S; Pande VS; Case DA; Brooks CLI; MacKerell AD Jr.; Klauda JB; Im W CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 2016, 12 (1), 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Vermaas JV; Baylon JL; Arcario MJ; Muller MP; Wu Z; Pogorelov TV; Tajkhorshid E Efficient Exploration of Membrane-Associated Phenomena at Atomic Resolution. J. Membr. Biol 2015, 248 (3), 563–582. 10.1007/s00232-015-9806-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Baylon JL; Vermaas JV; Muller MP; Arcario MJ; Pogorelov TV; Tajkhorshid E Atomic-Level Description of Protein–Lipid Interactions Using an Accelerated Membrane Model. Biochim. Biophys. Acta BBA - Biomembr 2016, 1858 (7, Part B), 1573–1583. 10.1016/j.bbamem.2016.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Muller MP; Jiang T; Sun C; Lihan M; Pant S; Mahinthichaichan P; Trifan A; Tajkhorshid E Characterization of Lipid–Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem. Rev 2019, 119 (9), 6086–6161. 10.1021/acs.chemrev.8b00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Denisov IG; Sligar SG Nanodiscs for Structural and Functional Studies of Membrane Proteins. Nat. Struct. Mol. Biol 2016, 23 (6), 481–486. 10.1038/nsmb.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tavoosi N; Davis-Harrison RL; Pogorelov TV; Ohkubo YZ; Arcario MJ; Clay MC; Rienstra CM; Tajkhorshid E; Morrissey JH Molecular Determinants of Phospholipid Synergy in Blood Clotting*. J. Biol. Chem 2011, 286 (26), 23247–23253. 10.1074/jbc.M111.251769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hallock MJ; Greenwood AI; Wang Y; Morrissey JH; Tajkhorshid E; Rienstra CM; Pogorelov TV Calcium-Induced Lipid Nanocluster Structures: Sculpturing of the Plasma Membrane. Biochemistry 2018, 57 (50), 6897–6905. 10.1021/acs.biochem.8b01069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Waehrens LN; Heegaard CW; Gilbert GE; Rasmussen JT Bovine Lactadherin as a Calcium-Independent Imaging Agent of Phosphatidylserine Expressed on the Surface of Apoptotic HeLa Cells. J. Histochem. Cytochem 2009, 57 (10), 907–914. 10.1369/jhc.2009.953729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14 (1), 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Varadi M; Anyango S; Deshpande M; Nair S; Natassia C; Yordanova G; Yuan D; Stroe O; Wood G; Laydon A; Žídek A; Green T; Tunyasuvunakool K; Petersen S; Jumper J; Clancy E; Green R; Vora A; Lutfi M; Figurnov M; Cowie A; Hobbs N; Kohli P; Kleywegt G; Birney E; Hassabis D; Velankar S AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50 (D1), D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ruben EA; Rau MJ; Fitzpatrick JAJ; Di Cera E Cryo-EM Structures of Human Coagulation Factors V and Va. Blood 2021, 137 (22), 3137–3144. 10.1182/blood.2021010684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gish JS; Jarvis L; Childers KC; Peters SC; Garrels CS; Smith IW; Spencer HT; Doering CB; Lollar P; Spiegel PC Jr. Structure of Blood Coagulation Factor VIII in Complex with an Anti–C1 Domain Pathogenic Antibody Inhibitor. Blood 2021, 137 (21), 2981–2986. 10.1182/blood.2020008940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem 2010, 31 (4), 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- (47).Feller SE; Zhang Y; Pastor RW; Brooks BR Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys 1995, 103 (11), 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- (48).Darden T; York D; Pedersen L Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- (49).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- (50).Nguyen H; Case DA; Rose AS NGLview–Interactive Molecular Graphics for Jupyter Notebooks. Bioinformatics 2018, 34 (7), 1241–1242. 10.1093/bioinformatics/btx789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Roe DR; Cheatham TEI PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9 (7), 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- (52).Michaud-Agrawal N; Denning EJ; Woolf TB; Beckstein O MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem 2011, 32 (10), 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graph 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- (54).Jurrus E; Engel D; Star K; Monson K; Brandi J; Felberg LE; Brookes DH; Wilson L; Chen J; Liles K; Chun M; Li P; Gohara DW; Dolinsky T; Konecny R; Koes DR; Nielsen JE; Head-Gordon T; Geng W; Krasny R; Wei G-W; Holst MJ; McCammon JA; Baker NA Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27 (1), 112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Sievers F; Higgins DG The Clustal Omega Multiple Alignment Package. In Multiple Sequence Alignment: Methods and Protocols; Katoh K, Ed.; Methods in Molecular Biology; Springer US: New York, NY, 2021; pp 3–16. 10.1007/978-1-0716-1036-7_1. [DOI] [PubMed] [Google Scholar]
- (56).Robert X; Gouet P Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42 (W1), W320–W324. 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Grant BJ; Skjærven L; Yao X-Q The Bio3D Packages for Structural Bioinformatics. Protein Sci. 2021, 30 (1), 20–30. 10.1002/pro.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Smith IW; d’Aquino AE; Coyle CW; Fedanov A; Parker ET; Denning G; Spencer HT; Lollar P; Doering CB; Spiegel PC Jr. The 3.2 Å Structure of a Bioengineered Variant of Blood Coagulation Factor VIII Indicates Two Conformations of the C2 Domain. J. Thromb. Haemost 2020, 18 (1), 57–69. 10.1111/jth.14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Dasgupta SK; Guchhait P; Thiagarajan P Lactadherin Binding and Phosphatidylserine Expression on Cell Surface-Comparison with Annexin A5. Transl. Res 2006, 148 (1), 19–25. 10.1016/j.lab.2006.03.006. [DOI] [PubMed] [Google Scholar]
- (60).Bouter A; Carmeille R; Gounou C; Bouvet F; Degrelle SA; Evain-Brion D; Brisson AR Review: Annexin-A5 and Cell Membrane Repair. Placenta 2015, 36, S43–S49. 10.1016/j.placenta.2015.01.193. [DOI] [PubMed] [Google Scholar]
- (61).Borden CR; Stevens CF; Sullivan JM; Zhu Y Synaptotagmin Mutants Y311N and K326/327A Alter the Calcium Dependence of Neurotransmission. Mol. Cell. Neurosci 2005, 29 (3), 462–470. 10.1016/j.mcn.2005.03.015. [DOI] [PubMed] [Google Scholar]
- (62).Adams TE; Hockin MF; Mann KG; Everse SJ The Crystal Structure of Activated Protein C-Inactivated Bovine Factor Va: Implications for Cofactor Function. Proc. Natl. Acad. Sci 2004, 101 (24), 8918–8923. 10.1073/pnas.0403072101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ruben EA; Summers B; Rau MJ; Fitzpatrick JAJ; Di Cera E Cryo-EM Structure of the Prothrombin-Prothrombinase Complex. Blood 2022, 139 (24), 3463–3473. 10.1182/blood.2022015807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Saleh M; Peng W; Quinn-Allen MA; Macedo-Ribeiro S; Fuentes-Prior P; Bode W; Kane WH The Factor V C1 Domain Is Involved in Membrane Binding: Identification of Functionally Important Amino Acid Residues within the C1 Domain of Factor V Using Alanine Scanning Mutagenesis. Thromb. Haemost 2004, 91 (1), 16–27. 10.1160/TH03-04-0222. [DOI] [PubMed] [Google Scholar]
- (65).Peng W; Quinn-Allen MA; Kim SW; Alexander KA; Kane WH Trp2063 and Trp2064 in the Factor Va C2 Domain Are Required for High-Affinity Binding to Phospholipid Membranes but Not for Assembly of the Prothrombinase Complex. Biochemistry 2004, 43 (14), 4385–4393. 10.1021/bi035763o. [DOI] [PubMed] [Google Scholar]
- (66).Stoilova-McPhie S; Villoutreix BO; Mertens K; Kemball-Cook G; Holzenburg A 3-Dimensional Structure of Membrane-Bound Coagulation Factor VIII: Modeling of the Factor VIII Heterodimer within a 3-Dimensional Density Map Derived by Electron Crystallography. Blood 2002, 99 (4), 1215–1223. 10.1182/blood.V99.4.1215. [DOI] [PubMed] [Google Scholar]
- (67).Stoilova-McPhie S; Lynch GC; Ludtke S; Pettitt BM Domain Organization of Membrane-Bound Factor VIII. Biopolymers 2013, 99 (7), 448–459. 10.1002/bip.22199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ngo JCK; Huang M; Roth DA; Furie BC; Furie B Crystal Structure of Human Factor VIII: Implications for the Formation of the Factor IXa-Factor VIIIa Complex. Structure 2008, 16 (4), 597–606. 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- (69).Chiu P-L; Bou-Assaf GM; Chhabra ES; Chambers MG; Peters RT; Kulman JD; Walz T Mapping the Interaction between Factor VIII and von Willebrand Factor by Electron Microscopy and Mass Spectrometry. Blood 2015, 126 (8), 935–938. 10.1182/blood-2015-04-641688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.