

Abstract
High-quality genome assemblies and sophisticated algorithms have increased sensitivity for a wide range of variant types, and breakpoint accuracy for structural variants (SVs, ≥50 bp) has improved to near base pair precision. Despite these advances, many SV breakpoint locations are subject to systematic bias affecting variant representation. To understand why SV breakpoints are inconsistent across samples, we reanalyzed 64 phased haplotypes constructed from long-read assemblies released by the Human Genome Structural Variation Consortium (HGSVC). We identify 882 SV insertions and 180 SV deletions with variable breakpoints not anchored in tandem repeats (TRs) or segmental duplications (SDs). SVs called from aligned sequencing reads increase breakpoint disagreements by 2×–16×. Sequence accuracy had a minimal impact on breakpoints, but we observe a strong effect of ancestry. We confirm that SNP and indel polymorphisms are enriched at shifted breakpoints and are also absent from variant callsets. Breakpoint homology increases the likelihood of imprecise SV calls and the distance they are shifted, and tandem duplications are the most heavily affected SVs. Because graph genome methods normalize SV calls across samples, we investigated graphs generated by two different methods and find the resulting breakpoints are subject to other technical biases affecting breakpoint accuracy. The breakpoint inconsistencies we characterize affect ∼5% of the SVs called in a human genome and can impact variant interpretation and annotation. These limitations underscore a need for algorithm development to improve SV databases, mitigate the impact of ancestry on breakpoints, and increase the value of callsets for investigating breakpoint features.
The human reference genome (International Human Genome Sequencing Consortium 2001; Schneider et al. 2017) hosts annotations including genes (O'Leary et al. 2016; Frankish et al. 2021), regulatory regions (The ENCODE Project Consortium 2012; The ENCODE Project Consortium et al. 2020), and repeats (Benson 1999; Bailey et al. 2002; Smit 2013–2015), and it has become a universal coordinate system for describing genetic alterations across populations (International HapMap et al. 2007; Sudmant et al. 2015; The 1000 Genomes Project Consortium 2015; Audano et al. 2019; Abel et al. 2020; Collins et al. 2020; Karczewski et al. 2020; Beyter et al. 2021; Ebert et al. 2021) and diseases (Turner et al. 2017; ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020; Taliun et al. 2021). New high-quality references are emerging for humans, such as the T2T-CHM13v2.0 reference (Nurk et al. 2022), and a growing number of other species (Mouse Genome Sequencing Consortium 2002; Alonge et al. 2020; Jebb et al. 2020; Mao et al. 2021; Ferraj et al. 2023; Li et al. 2023), which play fundamental roles in modern genomics.
Variant discovery is largely based on aligning reads or assemblies to a reference genome to identify single-nucleotide variants (SNVs), small insertions and deletions (indels), and structural variants (SVs) including indels ≥50 bp, inversions, complex rearrangements, and chromosomal translocations. Imprecise SV breakpoints affect comparisons across samples, and although new methods are improving these comparisons (Ebert et al. 2021; English et al. 2022; Kirsche et al. 2023), error-free merging across many haplotypes has not yet been attained. Additionally, precise breakpoint features such as microhomology and nearby variants in cis are important signatures for predicting mechanisms of formation (Carvalho et al. 2011; Vogt et al. 2014; Beck et al. 2015; Carvalho and Lupski 2016), and the effect of breakpoint placement on these annotations is not well understood.
Recent advances in sequencing technology are now generating longer and more accurate reads capable of reaching into repetitive structures and spanning larger SVs. As a result, many new SV loci have been discovered, and SV yield per sample has increased from fewer than 10,000 SVs per genome to more than 25,000 (Chaisson et al. 2015; Audano et al. 2019; Ebert et al. 2021). Moreover, long reads routinely reveal the full sequence of SVs, which was not previously attainable. Long-read phased assemblies have now become a critical component for producing complete and accurate variant callsets spanning a range of variant types and sizes (Chaisson et al. 2019; Ebert et al. 2021; Garg et al. 2021; Liao et al. 2023). These advances enable more complete transposable element (TE) analysis, improve genotyping in short-read samples, and support new biological insights (Ebert et al. 2021; Ebler et al. 2022; Rozowsky et al. 2023).
Modern reference genomes are a single theoretical haplotype, and when reads containing nonreference alleles are aligned, it can create biases that are difficult to mitigate (Degner et al. 2009; Brandt et al. 2015; Eizenga et al. 2020). To support mapping and variant calling across diverse genomes, the Human Pangenome Reference Consortium (HPRC) is developing graph-based references encompassing many haplotypes simultaneously (Liao et al. 2023). Although in-graph haplotypes can be directly detected, variants absent from the graph reference still rely on calling differences between the sample and a graph path. Therefore, challenges with linear reference analyses will ultimately translate to graphs, especially for rare and somatic events often associated with disease (Vogt et al. 2014; Nattestad et al. 2018; Sakamoto et al. 2020; Wahlster et al. 2021; Rausch et al. 2023), and high-quality graphs for many nonhuman genomes are not yet available. In contrast, in-graph SVs are represented as a unique “bubble” in graph space with a common breakpoint across all variant haplotypes, and so, ambiguity merging across independent haplotypes may be eliminated.
Although contiguous high-accuracy assemblies are becoming routine, we find that SV breakpoints are still inconsistently placed across phased haplotypes (Fig. 1A), and many breakpoints do not represent the true site of rearrangements, which may impede downstream analyses. To quantify the effect on modern long-read variant discovery approaches, we reanalyze a callset derived from 64 phased haplotypes recently released by the HGSVC (Ebert et al. 2021). Because pangenomes may eliminate breakpoint ambiguity, we further assessed breakpoints in graphs using callsets recently released by the HPRC (Liao et al. 2023). We find discordance between approaches based on linear and graph references, and we identify systematic differences created by graph methods. Through this effort, we have revealed bias present in modern callsets and limitations that impede analysis on variant calls, which we can now target with improved methods and more informed analyses with current methods.
Figure 1.

Breakpoint differences and population structure. (A) A cartoon illustration of an insertion (top; blue) and deletion (bottom; red) called at different locations in different samples. The gray box represents the breakpoint difference for these SVs. (B) Maximum offset distances for variants in the merged callset, which is calculated as the distance between the left- and rightmost breakpoints across haplotypes for the same SV. (C) The number of unique breakpoints for each variant (vertical axis) does not scale with the number of haplotypes (horizontal axis). A blue line represents the x = y diagonal. Scatterplot points were jittered in each axis uniformly from −0.5 to 0.5 to show density. (D) For any pair of haplotypes, the proportion of offset SVs is stratified by same superpopulation (Same; green) or different superpopulation (Diff.; violet). The difference in means is significant for both insertions and deletions (Student's t-test of means), but a greater effect is seen for insertions. Notches indicate a 95% confidence interval around the median. (n.s.) Not significant, (*) 1 × 10−3 < P ≤ 1 × 10−2, (**) 1 × 10−4 < P ≤ 1 × 10−3, (***) P ≤ 1 × 10−4.
Results
Breakpoint offsets are prevalent in long-read SV callsets
We examined breakpoint placement for SVs across 64 phased haplotypes derived from 32 diverse samples released by the HGSVC (Ebert et al. 2021). In that study, variants were called independently on each assembled haplotype against the GRCh38 reference using minimap2 (Li 2018) and merged to a multihaplotype, nonredundant callset, which we use to identify variants with different breakpoint locations across the haplotype assemblies (Fig. 1A). In all our analyses, we exclude SVs in tandem repeats (TRs) in which alignment limitations and reference errors make accurate breakpoints difficult to analyze (Sulovari et al. 2019; Mikheenko et al. 2020). To quantify the effect of breakpoint differences per phased haplotype, we compared each of the 64 haplotypes with each other (2016 pairwise combinations of 64 haplotypes) and find on average 4.4% of insertions and 1.7% of deletions outside segmental duplications (SDs) have different breakpoints per haplotype pair, which increases to 9.8% of insertions and 8.8% of deletions for SVs anchored in SDs (Supplemental Table 1).
Although inconsistent breakpoints affect a small number of variants per haplotype pair, we find 5.9% of insertions and 3.1% of deletions disagree on breakpoint location in the callset merged across all 64 haplotypes excluding SDs, which increases to 17% of insertions and 21% of deletions in SDs (Table 1). We recreated the HGSVC callset using T2T-CHM13v2.0 (Nurk et al. 2022) as a reference (Methods) and find similar breakpoint disagreements (Table 1), indicating a systematic effect from existing methods that is not specific to one reference genome. Outside SDs, insertions vary by a median of 2.2 bp with 18% offset by ≥50 bp, and deletions by a median of 4.9 bp with 33% offset by ≥50 bp, resulting in nontrivial differences in SV representation (Fig. 1B; Table 1).
Table 1.
Summary of differential breakpoints in the merged callset
| Insertions |
Deletions |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reference | Repeat | No. of variants | Diff | Diff % | Med bp | No. of variants | Diff | Diff % | Med bp |
| GRCh38 | |||||||||
| No TR/SD | 14,961 | 882 | 5.9% | 2.2 | 5804 | 180 | 3.1% | 4.9 | |
| SD no TR | 2609 | 440 | 16.9% | 3.5 | 1922 | 402 | 20.9% | 5.0 | |
| All | 60,716 | 19,589 | 32.3% | 19.4 | 38,442 | 10,641 | 27.7% | 22.0 | |
| T2T-CHM13 | |||||||||
| No TR/SD | 15,180 | 888 | 5.8% | 3.4 | 11,191 | 411 | 3.7% | 3.7 | |
| SD no TR | 1752 | 419 | 23.9% | 4.5 | 2014 | 506 | 25.1% | 14.0 | |
| All | 46,862 | 12,833 | 27.4% | 19.9 | 40,420 | 11,625 | 28.8% | 24.6 | |
Breakpoint differences increase in both GRCh38 and T2T-CHM13v2.0. (No TR/SD) SV is not anchored in TRs or SDs, (SD no TR) SV is anchored in SDs but not TRs, (All) all variants including those TRs and SDs, (Diff) number of variants with different offsets in at least one haplotype, (Diff %) mean percentage of variants with different offsets in at least one haplotype, and (Med bp) median number of bases variants with different locations are shifted.
Finally, the number of distinct breakpoints for each variant does not scale linearly with the number of haplotypes harboring the SV (Fig. 1C), which further suggests that variant breakpoint differences are systematic and not random. We sought to identify the drivers of these breakpoint disagreements.
Diversity is a key driver of differential breakpoint placement
The HGSVC callset is a mix of two Pacific Biosciences (PacBio) technologies, continuous long read (CLR) with 8%–15% error rate and high-fidelity (HiFi) with a <1% error rate (Logsdon et al. 2020). To examine whether sequencing error in phased assemblies affects SV locations, we compared breakpoints in 21 CLR genomes with 11 HiFi genomes and find a marginally significant enrichment for differences in insertions (4.40% vs. 4.29%, P = 0.025, Student's t-test) but no enrichment for deletions (1.75% vs. 1.77%, P = 0.52, Student's t-test), which we confirmed with permutation tests (P = 0.012 insertions, P = 0.74 deletions, 100,000 permutations).
We next asked if polymorphisms in the human population might affect placement. The HGSVC callset was derived from samples spanning all five 1000 Genomes Project superpopulations composed of African, admixed American, East Asian, European, and South Asian populations. We observe that variant breakpoints differ more often when a pair of haplotypes was derived from different superpopulations for insertions (4.49% vs. 3.99%, p = 2.44 × 10−40, Welch's t-test, Cohen's d = 0.73) (Fig. 1D). Deletions also increased, but the effect did not reach significance (1.76% vs. 1.71%, P = 0.069, Welch's t-test) (Fig. 1D). Furthermore, there is a noticeable increase in offset distance when haplotypes are derived from different superpopulations (Supplemental Fig. 1); we confirmed these results with permutation tests (P < 1 × 10−5 insertions, P = 0.041 deletions, 10,000 permutations). Because polymorphic differences are expected to increase with evolutionary distance and diversity, these results point to allelic polymorphisms as a driver of breakpoint volatility.
Breakpoint offsets are more prevalent with TE-mediated SVs
TEs create tracts of homology throughout the genome resulting in TE-mediated rearrangements (TEMRs) (Sen et al. 2006; Han et al. 2008; Balachandran et al. 2022). TEs from the same family have highly similar sequences, and so there are many choices for breakpoint placement along TE copies (Fig. 2A). Although TEs may provide the homology necessary for duplications and deletions by nonallelic homologous recombination (NAHR), most show only short tracts of breakpoint homology and appear to be mediated by other repair processes (Balachandran et al. 2022). Therefore, accurately placing SV breakpoints within TEMRs is essential for understanding the mutational mechanisms underlying their formation (Morales et al. 2015).
Figure 2.

Breakpoints shift through homology and alter SV representation. (A) A nonreference sequence with chimeric TE copies (TE 1, light green; TE 2, dark green) at the breakpoints. (B) The maximum distance between differential breakpoints across haplotypes is greater in TEMR variants with a significant difference for insertions. (C) Maximum breakpoint offsets for non-TD (green) and TD (violet) SVs. Horns extending downward on TD deletions indicate that the 95% confidence interval for the median extends below the bottom quartile. (D) The sequence of a tandem duplication shifts with different alignment positions along the reference copy. The junction of the duplication copies (red dot) is located at the insertion breakpoint if the insertion is placed at the left or right end of the reference copy (top and bottom examples); otherwise, the junction is embedded within the insertion creating a chimera of duplication copies inside the SV insertion (middle examples). (E) Mapping a chimeric TD to the reference occurs in two pieces separated at the TD breakpoint (light and dark blue arrows), where each piece maps to one side of the reference insertion site (dashed line). Alignment programs often miss one or both fragments. (B,C) P-values are generated from t-tests of the mean. Notches indicate a 95% confidence interval around the median. Red arrows and numbers indicate the number of outlier points above the horizontal axis maximum. (n.s.) Not significant, (*) 1 × 10−3 < P ≤ 1 × 10−2, (**) 1 × 10−4 < P ≤ 1 × 10−3, (***) P ≤ 1 × 10−4.
Of the 1322 non-SD SVs with different breakpoints in the HGSVC callset, we find 112 SV insertions and 119 SV deletions are likely TEMRs (8.5% and 20.4% of differential variants outside SDs, respectively; Methods). We find TEMR insertions were significantly enriched for offset breakpoints (odds ratio [OR] = 4.18, P = 3.17 × 10−25, Fisher's exact test [FET]), as were TEMR deletions (OR = 3.20, P = 1.55 × 10−11, FET). TE homology is also associated with larger distances between breakpoints across haplotypes for insertions (15.17 vs. 2.50 bp, P = 1.45 × 10−8, Welch's t-test), but the breakpoint difference did not reach significance for deletions (46.71 vs. 10.93 bp, P = 0.065, Welch's t-test) (Fig. 2B). Because breakpoint distances are derived from absolute reference coordinates, which are not affected by homology, significant increases in these loci indicate their importance in mediating breakpoint differences.
Tandem duplications are heavily affected by differential breakpoints
Tandem duplications (TDs) are a common SV type in which a duplicate copy is inserted adjacent to its template. TDs may be driven by existing homology, including longer stretches of similar sequence leading to NAHR, or may occur in regions with little to no homology (Lee et al. 2007; Arlt et al. 2009; Menghi et al. 2016; Willis et al. 2017; Li et al. 2020). With short reads, TDs are detected by elevated read depth of the duplicated sequence combined with paired-end and split-read evidence at the duplication breakpoint, revealing the duplicated reference region (Alkan et al. 2011). However, long-read methods often identify TDs as SV insertions (Audano et al. 2019; Ebert et al. 2021). To quantify breakpoint precision in TDs, we identified 1843 SV insertions and 17 SV deletions as tandem events (Methods). We find that TD insertions are more likely to have differential breakpoints versus non-TD insertions (OR = 0.55, P = 1.45 × 10−9, FET), and the effect on TD deletions is small but significant (OR = 0.05, P = 3.14 × 10−7, FET). We observe greater average breakpoint distances in TD compared with non-TD SVs for insertions (9.37 bp vs. 2.20 bp, P = 1.07 × 10−13, Welch's t-test, Cohen's d = 0.45). A large increase in distance for deletions failed to reach significance (741.9 bp vs. 12.8 bp, P = 0.19, Welch's t-test, Cohen's d = 2.23) (Fig. 2C).
When sequences containing TDs are aligned, one copy is aligned to the reference and one copy appears inside the insertion. However, if the insertion is placed inside the duplicated reference locus instead of at one end, each duplication copy is split between the reference and the insertion creating a chimeric representation, and the duplication junction is embedded inside the insertion sequence (Fig. 2D). If the SV sequence is aligned to the reference to identify the duplicated locus, it maps in two fragments (Fig. 2E). The current alignment programs often miss one or both fragments, making TDs difficult to annotate from SV insertions. We find a chimeric representation for 14% (261) TD insertions and 47% (eight) TD deletions (Methods). Our TD annotation approach (Methods) specifically handles these fragmented alignments, which is now available as a pipeline (https://github.com/BeckLaboratory/dupmapper).
Whereas the chimeric structure of TDs may place breakpoints closer to true rearrangement sites, alignment artifacts can generate the same patterns. For example, a 13-kbp TD has one reference copy flanked by AluSx elements with an SV insertion representing one additional copy. The SV insertion breakpoint is placed between the Alu sites, splitting each copy between the reference and insertion sequences and producing a chimeric TD copy in the SV insertion (Supplemental Fig. 2). This SV may represent an Alu-mediated duplication with an incorrect breakpoint, or the reference may be the result of a TD deletion. In either case, polymorphisms that accumulate in both copies can produce similar alignments, and additional data are needed to discern these cases. TDs have the most variable breakpoints we have analyzed, leading to large differences in TD representation that may not accurately represent the SV.
Small polymorphisms surround offset SV breakpoints
As we have observed, variation in breakpoint placement increases when haplotypes are derived from different ancestral backgrounds. Therefore, we reasoned that small allelic polymorphisms near SVs might influence alignments. To identify these polymorphisms, we extracted the offset region around breakpoints from each haplotype assembly and compared them (Methods) (Fig. 3A). For SV insertions not anchored in SDs, we find on average 5.0 small variants on the left breakpoint versus 5.2 on the right breakpoint (P = 1.62 × 10−10, Welch's t-test) and a distinct peak 1 bp inside the rightmost breakpoint (Fig. 3B).
Figure 3.

Small variants accumulate at differential SV breakpoints. (A) SV insertions with different breakpoints (blue) in each haplotype pair were retrieved. Sequence around the left and right breakpoints was extracted (solid box) for both haplotypes including the differential locus (gray area between dashed lines) and 50 bp upstream and downstream. The red dashed line on the right side of each gray box marks the start and end position of the right-shifted variant. (B) Small variants accumulate at the upstream and downstream edges of the right-shifted variants where zero is the red line in A. Small polymorphisms occur most frequently 1 bp inside the right-shifted insertion sequence (red arrow).
To better understand the origin of these polymorphisms, we were able to confidently genotype 71 SNVs found inside the rightmost shifted breakpoints for non-TD SVs (Fig. 3B, red arrow) across all 64 haplotypes (Methods). For 59 SVs (83%), one SNV allele segregates exclusively with the rightmost SV breakpoint, and of these, 20 (34%) reached significance (P < 0.01, FET). These polymorphisms most likely arose by chance near SV breakpoints after the SV occurred. An additional 11 (15%) segregated with the rightmost breakpoint but were also identified in non-SV alleles, indicating either the SV or the SNV might be recurrent, and of these, two (18%) reached significance (P < 0.01, FET).
A model for breakpoint bias
Given these observations, we propose a model for the systematic bias driving breakpoint differences. Pairwise alignment algorithms make breakpoint choices based on a scoring system that increases with each matching aligned base and decreases with each mismatch, insertion, and deletion. When an SV has breakpoints in regions that are homologous, there are many choices for the breakpoint location with an equal score, but modern aligners such as minimap2 (Li 2018) “left-align” the breakpoint by placing it as far left as possible (Fig. 4A). However, when a small polymorphism is near an insertion SV, the score is penalized unless it can shift the SV through breakpoint homology and push the polymorphism inside the unaligned insertion sequence, thereby eliminating the effect of the SNV or indel on the alignment score (Fig. 4B).
Figure 4.
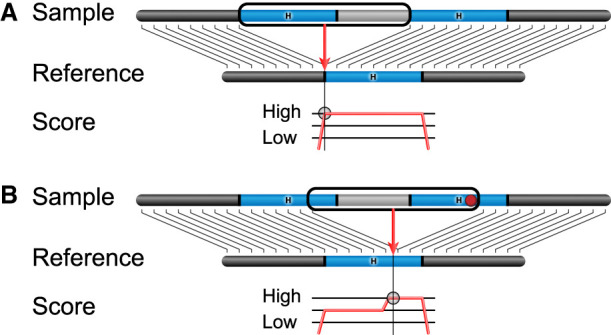
A model of breakpoint differences. SV insertion breakpoints in homologous sequence (“H,” blue) can be placed differently with little effect on the alignment score. (A) In the absence of differences between the homologous loci, aligners typically left-align the breakpoint consistently placing it on the left side of the homologous locus in the reference. (B) When a small polymorphism such as a SNP or indel is in the homologous region (red dot), the alignment score is penalized for the mismatching base. If the breakpoint is shifted so that the polymorphism falls inside the inserted sequence, the penalty is eliminated, and it appears to produce a better alignment. A different representation for this SV insertion is produced in samples with the SNP.
Breakpoint differences do not predict recurrence
Recurrent SVs driven by homology have been shown to arise in multiple independent genomes (Kolomietz et al. 2002; Porubsky et al. 2022) and sometimes lead to diseases (for review, see Carvalho and Lupski 2016). When we inspected 41 SV insertions of ≥1 kbp with breakpoint differences >10 bp against a phylogenetic tree (Methods), we find little evidence associating recurrence with breakpoint differences (Supplemental Fig. 3A,B). Of these, 27 (66%) were identified in the chimpanzee genomes (Supplemental Fig. 3C,D), indicating that they are likely ancestral deletions in which the deleted allele became the reference and, therefore, placing nondeleted sequences in multiple locations is not biologically meaningful. These breakpoint differences may not reflect biological origins and are not a strong indicator of recurrence.
Breakpoint homology annotations change with breakpoint placement
SVs are often mediated by tracts of homology. NAHR requires >100 bp of perfect homology, some double-strand break repair pathways can be mediated by short tracts of microhomology from 1 to 20 bp, nonhomologous end joining (NHEJ) requires no breakpoint homology, and alternative end-joining (alt-EJ) requires little or no microhomology (for review, see Carvalho and Lupski 2016; Iliakis et al. 2015). Mobile element insertions (MEIs) create homology in the form of target-site duplications, which are important for distinguishing true MEI polymorphisms from other SVs containing MEI sequences (Kazazian and Moran 1998; Zhou et al. 2020; Ebert et al. 2021). Accurate homology annotations are important for identifying SV mechanism and are a useful quality metric for SV callsets.
We used a recent update to our assembly-based variant caller, PAV, to estimate microhomology for all SV breakpoints (Methods). For each SV, we find that the number of different microhomology calls increases with the number of distinct breakpoints across haplotypes for insertions (ρ = 0.72, P < 1 × 10−100, Spearman's rank-order correlation [Spearman]) and deletions (ρ = 0.87, P < 1 × 10−100, Spearman), confirming that homology is altered with breakpoint placement (Supplemental Fig. 4). For insertions with consistent breakpoints (n = 6855), microhomology annotations varied by 2.16 bp on average, which rises to 21.91 bp on average with inconsistent breakpoints (n = 725) (P = 9.46 × 10−15, Welch's t-test, Cohen's d = 0.43). We see a similar effect on deletion microhomology, which varies by 0.01 bp across haplotypes with consistent breakpoints (n = 3399) and rises to 19.27 bp across haplotypes with inconsistent breakpoints (n = 172) (P = 1.01 × 10−16, Welch's t-test, Cohen's d = 1.77) (Supplemental Fig. 5).
As a result of imprecise breakpoints or polymorphisms within homologous loci, actual breakpoint homologies necessitate manual reconstruction, which is a tedious task and cannot easily scale with modern whole-genome analyses. Therefore, precise mechanisms are difficult to routinely annotate. For example, although SVs mediated by mobile elements with at least 85% identity are generally thought to be mediated by NAHR (Lam et al. 2010), a closer examination of breakpoints using modern long-read data shows that 20% have breakpoint features consistent with end-joining mechanisms, and few of the remaining 80% have homology tracts required for NAHR (Balachandran et al. 2022).
Read-based approaches have less consistent breakpoints
We examined breakpoints across all 32 HGSVC samples using three read-alignment callers, pbsv (https://github.com/PacificBiosciences/pbsv), Sniffles (Sedlazeck et al. 2018), and SVIM (Heller and Vingron 2019), as well as three assembly-based callers, PAV (Ebert et al. 2021), DipCall (Li et al. 2018), and SVIM-asm (Heller and Vingron 2021). Excluding SDs, we find read-based alignments place SVs at different locations across the samples for 15% to 36% of insertions and 15% to 52% of deletions (Supplemental Table 2). Assembly-based callers identified different breakpoints for the same SV for 3% to 6% of insertions and 2% to 3% of deletions (Supplemental Table 2). Consistent with our comparison of read-based callers, a recent study of TEMRs (Balachandran et al. 2022) finds that MANTA (Chen et al. 2016) places SVs more accurately than other short-read callers, and this may be attributable to breakpoint assemblies that MANTA performs. These results indicate that calling SVs from assemblies produces more consistent breakpoint representations for SVs.
Pangenome SV breakpoints disagree with linear reference alignments
Pangenome graphs are constructed from multiple haplotypes and can be used to negate differences in alignments. The PanGenome Graph Builder (PGGB) (Garrison et al. 2023) constructs graphs from multiple haplotypes simultaneously, and the Minigraph-Cactus (MC) approach iteratively adds haplotypes to a graph (Hickey et al. 2023). Both were featured in the recent pangenome drafts constructed from 94 phased assemblies derived from 47 diverse samples recently released by the HPRC (Liao et al. 2023).
Outside SDs, we identified all SVs that were present in more than one haplotype and matched an SV identified by MC (4851 insertions, 3240 deletions). We find that the MC breakpoint offset is greater than all the HGSVC offsets for 69% of SV insertions and 41% of SV deletions, with a majority of these resulting from not left-aligning in regions of breakpoint homology. For PGGB (4831 insertions, 3218 deletions), we find 13% of SV insertions and 15% of SV deletions have a greater offset. By manually inspecting SV differences, MC appears to place SV breakpoints irrespective of small variants and does not left-align against the reference path. For example, a 2.1-kbp insertion was identified in all HGSVC haplotypes (AF = 100%) with no breakpoint variation, but it is shifted by 43 bp in the MC graph (Fig. 5A). This variant inserted into a TE and had TE sequence at the breakpoint, creating a tract of imperfect microhomology, and MC aligned 43 bp from the insertion sequence to the reference. As a result, two false SNPs are found in all haplotypes with the SV and may mislead downstream analyses. For example, SNPs linked to SVs do suggest mechanisms of SV formation (Deem et al. 2011; Carvalho and Lupski 2016; Beck et al. 2019), although no point mutations were actually generated by this SV. This pattern was observed frequently in the MC callset.
Figure 5.

Pangenome graph breakpoints show systematic bias. (A) A variant with a different breakpoint in the MC graph versus GRCh38, with the red portion showing the imperfect homology between breakpoint placements, resulting in two SNPs called in the MC graph in all HPRC haplotypes (A > G and T > C). (Tig up) Contig sequence spanning from reference to inserted sequence, (Tig dn) contig sequence spanning from insertion sequence to the reference, and (Ref) reference sequence at the site of the insertion. (B) An SV insertion (blue) paired with deleted bases (red) yields a 101-bp net gain by minimap2 (mm2, HGSVC callset), PGGB, and MC. minimap2 calls three small deletions near the insertion breakpoint. PGGB calls one larger deletion but reinserts deleted bases (magenta boxes) into the insertion call, resulting in a larger SV insertion than minimap2. MC calls two insertions, two deletions, and a mismatch (black dot). (C) Alignments through breakpoints are shown for minimap2 and PGGB. Bases aligned to the reference are shown in red with matches in uppercase; inserted sequences are blue; and deleted bases are gray. The inserted sequence was not found in GRCh38 but was present in nonhuman primates and likely represents the deletion of ancestral sequence where the reference contains the derived (deleted) allele.
Many differences in the PGGB SVs are attributable to different breakpoint choices among largely equivalent representations. For example, a 162-bp imperfect VNTR expansion (27-bp motif) with one reference copy is inserted to the right of the reference copy rather than the left (Supplemental Fig. 6). More importantly, we find a distinct pattern of PGGB deleting and reinserting the same bases when calling variants in loci without clean breakpoints. In one example, minimap2 represents a 101-bp net gain as a 109-bp insertion with three deletions totaling 8 bp; PGGB calls a 118-bp insertion with a single 17-bp deletion; and MC calls a 105-bp insertion, a 5-bp insertion, two deletions totaling 9 bp, and a SNP (Fig. 5B). Further inspection of the breakpoints shows that 13 bases deleted by PGGB are reinserted as part of the SV insertion (Fig. 5C). This SV insertion sequence does not align to the human reference but is present in a chimpanzee (Pan troglodytes) assembly (Mao et al. 2021) on Chromosome 2 and is also in other primate genomes (Ebert et al. 2021; Mao et al. 2023). Therefore, the insertion is likely ancestral, and the deletion became the reference allele by chance. The minimap2 representation of this locus appears to be the most likely biological explanation for this event with small template switches within the replication fork, which is characteristic of double-strand break repair mechanisms mediated by microhomology (Hastings et al. 2009; Carvalho et al. 2013). The deletion and reinsertion of identical bases is less likely.
In addition to creating different representations of SVs, the area between breakpoints is often filled with small variants that are annotated differently across the haplotypes, which may impact the interpretation of variants. Coding sequences for 26 genes, on average, in MC and five genes, on average, in PGGB intersect variable breakpoints, with additional discrepancies in UTRs and ncRNAs (Supplemental Table 3). For example, we find a 180-bp insertion in ESYT3, in which minimap2 and PGGB place the breakpoint in an intron, but MC places it in an exon (Supplemental Fig. 7). These examples have now been corrected in MC (1.1), and when combined with left-aligned breakpoints, MC disagreements have been reduced to 7.3% for insertions and 9.4% for deletions. However, additional work is ongoing to refine SV breakpoints within graphs.
Discussion
Advances in long-read sequencing technology coupled with new phased assembly methods are producing more complete and more accurate variant callsets than was previously possible with shorter reads (Wenger et al. 2019; Ebert et al. 2021; Cheng et al. 2022; Liao et al. 2023; Rautiainen et al. 2023). This has facilitated a greater understanding of variation in the past 10 years, especially structural changes in human populations (Chaisson et al. 2015; Seo et al. 2016; Shi et al. 2016; Audano et al. 2019; Chaisson et al. 2019; Beyter et al. 2021; Ebert et al. 2021; Kim et al. 2022; Liao et al. 2023) and nonhuman species (Alonge et al. 2020; Rhie et al. 2021; Ferraj et al. 2023; Li et al. 2023). These advances continue to rival short-read technology by reducing costs, increasing availability, and improving read quality. In addition to detecting more SVs, long reads also capture the full SV sequence, which is important for detailed analyses of nonreference sequences and has already proven to be transformative in mobile element characterization (Ebert et al. 2021; Ferraj et al. 2023).
Although variant calling from assemblies has increased breakpoint accuracy, systematic errors still exist in the homologous regions responsible for mediating many SVs. Short reads are subject to reference biases, causing distant haplotypes to align less confidently to alternate reference alleles (Degner et al. 2009; Brandt et al. 2015). Although small polymorphisms are spanned by much longer flanking sequences with long reads and assemblies, this reference bias now manifests as different breakpoints for the same SV across samples and the loss of small polymorphisms near SVs. Recent developments in alignment methods have focused on better ways to seed and chain in complex regions (Jain et al. 2020; Prodanov and Bansal 2020; Li 2021; Sahlin et al. 2023), although little work has been performed to address the breakpoint biases we observe.
Breakpoint differences for the same SV affect variant interpretation, obscure annotations, hinder biological inference, and often embed the true breakpoint hundreds of bases inside the SV sequence. Because the polymorphisms causing breakpoint differences are systematically removed from the callset (Figs. 3B, 4B), there is a loss of the information required for mechanistic annotations (Deem et al. 2011; Carvalho and Lupski 2016; Beck et al. 2019) and signatures of selection around SV sites (Sabeti et al. 2002). We can alleviate some merging and variant comparison bias by tuning merging algorithms, and we are testing parameters in SV-Pop (Ebert et al. 2021) specifically targeting TDs; however, merged nonredundant callsets still contain anomalies as a result of breakpoint differences (Supplemental Fig. 8). By accounting for chimeric TDs, we can now produce better annotated SV insertions, and we have released a pipeline for this purpose (https://github.com/BeckLaboratory/dupmapper). Although alignment sensitivity may help to identify true breakpoint locations, our results suggest that this sensitivity also produces incorrect SV representations, such as chimeric TDs with breakpoints set well outside of the homologous loci that likely mediated them (Supplemental Fig. 2). These anomalies are largely technical artifacts driven by polymorphisms unrelated to the SV formation, are not signals of true SV biology, and are not reliable markers of recurrence. Producing more consistent SV representations in diverse samples will increase reproducibility and provide more predictable input for methods capable of identifying true breakpoints or recurrence using additional data such as polymorphisms and linkage information.
Although pangenome graphs normalize SV loci across samples, these methods are under active development to improve breakpoint precision and to address the issues outlined here. However, because rare and somatic variants cannot be captured by graph references, the same challenges must still be addressed.
Although this paper focuses on variants not anchored in complex repeats, some of the most impactful biology is emerging from complex loci that were once intractable (Ebert et al. 2021; Hallast et al. 2023; Liao et al. 2023; Logsdon et al. 2023; Mao et al. 2023). Complex rearrangements within these loci are driven by large and highly repetitive structures subject to the same breakpoint characteristics that we describe, except with kilobase-scale breakpoint homologies. As a result, we see breakpoint disagreements increase in SDs by 3×–4× for insertions and 7× for deletions using either GRCh38 or T2T-CHM13v2.0 as a reference. Although modern humans represent ∼260,000–350,000 years of evolution (Schlebusch et al. 2017), species of biological and medical import such as mice and nonhuman primates span 0.5 to 52 million years and show far greater sequence divergence and structural variation (Ferraj et al. 2023; Mao et al. 2023). As technology advances and more genomes are sequenced, our results suggest that breakpoint volatility will increase.
Perhaps most concerning is that a clear bias exists in human genome analyses, and populations with greater divergence from chosen references will be most impacted. Although pangenome references under development today may significantly improve diversity (Wang et al. 2022; Liao et al. 2023), they cannot reach all facets of human populations owing to technical, ethical, legal, and social constraints. Method development efforts aimed at eliminating this bias is one necessary step toward reclaiming information lost by current approaches and making modern genomics more equitable and accessible.
Methods
Resource table
A list of resources and software versions used for this work can be found in Supplemental Table 4.
Statistical analysis
Summary stats, such as mean and standard deviation, were performed with Python NumPy (v1.22.4) (Harris et al. 2020), and statistical tests including Student's t-test, Welch's t-test, F-test, and FET were performed with SciPy (1.9.3) (Virtanen et al. 2020). All tests were two-tailed. F-tests were used to determine if a Student's t-test was performed (F-test P-value ≥ 0.01) or a Welch's t-test (F-test P-value < 0.01).
P-values <1.0 × 10−100 are reported as P < 1.0 × 10−100. Extremely low P-values less than the smallest floating point value Python can represent (∼1 × 10−308 ± 1 × 10−15 on our system) are also reported as P < 1 × 10−100 in this paper.
Microhomology
The number of unique breakpoints was compared with the number of unique microhomology calls per merged variant. Neither the number of unique breakpoint locations or unique microhomology calls model a normal distribution (P < 1 × 10−100, scipy.stats.normaltest based on D'Agostino and Pearson's test), so we computed correlation based on Spearman's rank-order correlation coefficient.
Genome reference
We use the hg38-NoALT reference published with the HGSVC callset (Ebert et al. 2021; ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/technical/reference/20200513_hg38_NoALT/). This reference is the full primary assembly of the human genome build 38 (GRCh38/hg38), including unplaced and unlocalized contigs, but it does not include patches, alternates, or decoys.
Ebert callset
We acquired the version 2 (Freeze 4) merged callset from HGSVC (Ebert et al. 2021). We retained the same 32 population samples excluding the trio children used in the HGSVC publication. Frequencies and allele counts were adjusted to exclude child samples in the merged callset. We removed variants on unplaced and unlocalized contigs of the reference, including only variants on primary chromosome scaffolds. A merging bug in earlier versions of SV-Pop allowed for some long-range intersects, which we removed by requiring either (1) the maximum offset is less than or equal to the merged SV length or (2) the maximum offset difference was <400 bp (200 bp in either direction) and the maximum SV length difference was not >50% of the maximum SV length. These parameters mirror the expected results from the merging process without the long-range bug.
We obtained the Tandem Repeats Finder (TRF) (Benson 1999) and RepeatMasker (Smit 2013–2015) annotations from the UCSC Genome Browser (retrieved January 27, 2023; tracks “simpleRepeat” and “rmsk,” respectively) (Kent et al. 2002). From TRF, we used all loci. From RMSK, we used all loci annotated as “Low_complexity” or “Simple_repeat.” RMSK and TRF records within 200 bp were merged with BEDTools merge (v2.30.0) (Quinlan and Hall 2010), a 200-bp flank was added to all merged regions, and SVs were intersected with both TRF and RMSK. Insertions were marked as TRs if their insertion point was within a padded repeat region. Deletions were marked as TRs if either reference breakpoint was within a 200-bp padded repeat region. Intersects were performed with BEDTools intersect (v2.30.0) (Quinlan and Hall 2010).
SDs were annotated using the same process as TRs. The SD track was retrieved from the UCSC Genome Browser (January 28, 2023; track “genomicSuperDups”). Regions were merged within 200 bp, and a 200-bp flank was added, both operations with BEDTools. Insertion and deletion breakpoints were intersected with the merged and padded SD regions in the same way as simple repeats.
Distance between variant breakpoints is defined as before in SV-Pop (https://github.com/EichlerLab/svpop) as used by HGSVC for merging: min([start offset, end offset]), where “start offset” is the distance between the variant start positions, and “end offset” is the distance between the variant end positions, which may be different if the variant is a deletion (Ebert et al. 2021).
Pairwise comparisons were performed by selecting all combinations of 64 haplotypes among the 32 samples (2016 combinations of two haplotypes from a pool of 64). We obtained the original locations for each variant in all 64 haplotypes by tracing the merged call back through the sample to the original PAV call for each.
TEMR annotations
We labeled SVs as TEMRs if TE annotations at SV breakpoints were consistent with a rearrangement mediated by TE homology. For reference repeats, we obtained the RepeatMasker (RMSK) track (database: rmsk.txt.gz) for hg38 (retrieved January 3, 2023) from the UCSC Genome Browser. We retained only records with a repeat class of “LINE,” “SINE,” or “LTR” and with a minimum size of 100 bp. For deletions, we intersected the reference locations for each event independently (i.e., upstream breakpoint location and downstream breakpoint location) and annotated deletions as TEMRs if (1) both breakpoints intersected a TE annotation of the same type (e.g., Alu, ERV1, ERVK, L1, L2, etc.), and (2) each side of the breakpoint intersected a different TE (i.e., distinct TE events). For SV insertions, we intersected the reference breakpoint with the RMSK track. We additionally obtained RMSK annotations run on the merged callset by HGSVC (Ebert et al. 2021) and selected repeat annotations within 10 bp of each end of the insertion. We annotated insertions as TEs if (1) RMSK annotations at each end of the inserted sequence and at the reference breakpoint were all the same TE type, and (2) RMSK annotations at each end of the insertion sequence were not the same TE (i.e., distinct TE events). Breakpoint intersections with the RMSK track were performed with BEDTools intersect (v2.30.0).
TD identification
Insertion sequences in unique loci (excluding annotated SDs and TRs) were remapped to the reference with BLAST (v2.13.0) (Altschul et al. 1990) with parameters “-word_size 16 -perc_identity 95” against a BLAST database constructed from hg38-NoALT. We compiled a list of filter regions by including all TRs and RMSK annotations with a score of 50 or greater from the UCSC Genome Browser and merging records with BEDTools merge (v2.30.0). BLAST alignments were discarded if ≥50% of the alignment record intersected the TR and RMSK filter. We further filtered BLAST hits to include only records that mapped within 10% of the SV length from the insertion site or deletion breakpoints (e.g., 1-kbp INS, 100-bp window around the insertion site) with a minimum of 100 bp for small SVs. For deletions, we removed the deletion sequence alignment (i.e., remapping produces an alignment over the deletion). Alignments <30 bp were also excluded. Some redundant overlapping alignments remained and appeared to be driven by small TRs that were not in the reference, which were removed by keeping only the longest record if records overlapped by ≥80%. The same 80% overlap was performed in both reference space using aligned reference coordinates and in SV sequence space using coordinates from the SV sequence (i.e., the first base of the SV sequence is position zero). We selected SVs for which the total number of aligned bases on each side of the breakpoint was within 90% of the total SV size and ensured records with large gaps spanning more bases than were aligned did not contribute to the SV size calculation. We did not select records that had the expected alignment pattern (i.e., upstream SV sequence mapping downstream from the SV breakpoint and downstream SV sequence mapping upstream of the SV), although all the records left after the filtering process did show this pattern.
Small variants around breakpoints
Our goal was to identify small polymorphic differences between haplotypes that causes variant breakpoints to be placed differently. For each haplotype pair, we selected SV insertions and deletions with breakpoints placed at different sites and with breakpoints in unique loci (not TR or SD). We extracted the haplotype sequence from around the assembly, including a 50-bp flank on each side, and we extended one end appropriately to add flank, so that in the absence of other small variants, both sequences should start on the same base.
The sequences were aligned so that the SV with the rightmost breakpoint (in reference coordinates) was the reference and the SV with leftmost breakpoint variant was the query. Sequences were aligned with the “swalign” Python package (v0.3.7) using a global alignment and with match, mismatch, and gap scores from the minimap2 (short-gap scores from the default double-affine parameters):
aligner = swalign.LocalAlignment(
swalign.NucleotideScoringMatrix(2, −4),
gap_penalty = −4, gap_extension_penalty = −2,
globalalign=True)
Alignment align (“M” CIGAR operations) records were transformed to match/mismatch (“=” and “X” CIGAR operations), and using the known flanks added to each, we assigned variants to left flank, left breakpoint (intersecting the breakpoint), differential region, right breakpoint (intersecting the breakpoint), and right flank along with their relative position in each category.
Genotyping small breakpoint variants in all haplotypes
Small variants that accumulate at differential breakpoints are small polymorphisms, but because they are lost from the callset, we genotyped them into all 64 haplotypes to see which SVs contained the variants. We selected SNV polymorphisms within 2 bp of the shifted breakpoint inside the rightmost SV insertion, which according to our model, should be the polymorphisms contributing to breakpoint volatility. We extracted both SNV alleles with an 8-bp flank on each end and the variable base in the center (17-mer), and we removed any SNVs containing a ≥5-bp homopolymer run in the extracted sequence. For each SNV in each haplotype, we extracted assembly sequence around the SV site, including a 250-bp flank on each end and including the insertion sequence, if the haplotype contained the SV allele, and we matched both SNV alleles (including the 8-bp flank) to each extracted haplotype region.
We then counted the number of occurrences for each SNV allele in haplotypes not containing the SV while removing any records that genotyped both SNV alleles into the same haplotype. For each haplotype containing the SV, we counted the number of SNV alleles for the rightmost breakpoint separately from all other SV breakpoint positions together if there were more than one. This yields a table with counts for each SNV allele across three categories of haplotypes: non-SV haplotypes, haplotypes with the rightmost breakpoint, and haplotypes containing all other breakpoint locations. We removed SVs that did not yield useful genotypes, including ones where counts were zero for both SNV alleles in the rightmost SV breakpoint, other breakpoint haplotypes, or reference haplotypes. Any sites that genotype only one SNV allele in all three haplotype categories were also removed.
Microhomology
Microhomology is the span of perfectly matching bases at each end of a breakpoint, for example, the perfect homology at sites of ectopic recombination (i.e., NAHR), homology-directed repair, replication-based repair, or alt-EJ. We measured homology at breakpoints using an algorithm in PAV and previously validated as part of a TEMR project (Balachandran et al. 2022), where the region upstream of an SV sequence is matched with the downstream reference or contig and the region downstream from the SV sequence is matched with the upstream reference or contig. To compare haplotypes more consistently, we computed SV homology for insertions against the upstream and downstream contig where the SV was called and against the reference for deletions. We excluded all TD variants from homology because estimating breakpoint homology using this method counts whole TD copies as homologous.
Phylogeny
The chimpanzee genome was retrieved from a recent preprint (Mao et al. 2023) and run with PAV 2.3.4 on GRCh38 to generate a set of SNP calls, and we used haplotype 1 from the chimpanzee assembly as an outgroup. We obtained SNPs from the HGSVC callset (Ebert et al. 2021) for all 32 diverse samples.
RefSeq loci were obtained from the UCSC Genome Browser for hg38 (GRCh38). All exons within 5 kbp of SDs or centromeric loci were excluded. We further excluded exons within 5 kbp of copy number variable, complex, duplicated, or inverted loci identified in the high-coverage 1000 Genomes callset (Byrska-Bishop et al. 2022) and inversions identified in the HGSVC callset. We retained only SNPs intersecting the remaining exons.
We further excluded SNPs within 20 bp of TRs (UCSC Genome Browser track), homopolymers (≥4 bp) and dinucleotides (four or more dinucleotide repeats), where homopolymer error might generate false SNPs from alignment bias (we observed while validating events for Noyes et al. 2022), and SV insertions or deletions identified in the HGSVC callset or the high-coverage 1000 Genomes callset. This yielded 199,873 SNPs with 70,230 SNPs polymorphic among the human samples.
RAxML-NG (1.2.0) (Kozlov et al. 2019) was run on the set of SNPs with automatic bootstraping, 50 parsimony trees, and 50 random trees with the command-line “raxml-ng ‐‐all ‐‐model GTR + G ‐‐tree pars{50},rand{50} ‐‐seed = 102233 ‐‐msa snp_phy_hgsvc.phy.” We used the best tree for visualizing phylogeny and recurrence with the ETE3 (3.1.2) (Huerta-Cepas et al. 2016) Python package.
For visualization, we selected all SVs ≥1 kbp that were not annotated as intersecting reference TRs or SDs (annotations released with the HGSVC callset) with more than one breakpoint, with no more than three unique breakpoint sites (limiting colors for visualization), and with more than 10-bp maximum distance between breakpoints called for the same SV; that were identified in at least eight haplotypes; and that were callable in all 64 HGSVC and the chimpanzee assembly (by HGSVC annotations). This yielded 41 SVs to examine for signs of recurrence. To link SVs to chimp for visualization (gray bubbles in Supplemental Fig. 3), we used annotations released with the HGSVC callset for chimpanzee SV intersects.
Callset comparisons
We obtained PAV calls through the HGSVC callset (Ebert et al. 2021) that were subjected to an extensive high-throughput QC process. For this study, we additionally ran the latest available versions of pbsv 2.9.0 (https://github.com/PacificBiosciences/pbsv), Sniffles 2.0.7 (Sedlazeck et al. 2018), SVIM 2.0.0 (Heller and Vingron 2019), DipCall 0.3 (Li et al. 2018), and SVIM-asm 1.0.3 (Heller and Vingron 2021). We generated a merge of all samples (read-based methods) and haplotypes (assembly-based methods) with SV-Pop using the same parameters used to construct the HGSVC callest, and we retained only merged SVs matching an SV from the HGSVC callset to include only SVs passing QC in our comparisons. For each SV in the merge, we quantified the number of breakpoint locations across all samples or haplotypes.
Graph genome comparisons
Variants against GRCh38 for PGGB and MC graphs were obtained from the decomposed VCFs published by the HPRC (Supplemental Table 4; Liao et al. 2023). Variants were extracted for each sample using SV-Pop. Differences were manually investigated using the UCSC Genome Browser and custom browser tracks for HGSVC and HPRC variants.
Supplementary Material
Acknowledgments
P.A.A. and C.R.B. were supported by National Institutes of Health (NIH) National Institute of General Medical Sciences R35GM133600 and NIH National Cancer Institute P30CA034196. The Human Genome Structural Variation Consortium (HGSVC) provided published data, support, and feedback, and the HGSVC was supported by NIH National Human Genome Research Institute U24HG007497. We thank Parithi Balachandran for helping to check duplication detection tools and SV breakpoints. We thank Ardian Ferraj, Parithi Balachandran, and Kamari Weaver for manuscript proofreading. We thank Evan E. Eichler for providing feedback on graph genome comparisons. We thank Glenn Hickey and Benedict Paten for assistance with graph SV breakpoints and corresponding with us about future graph improvements.
Author contributions: P.A.A. and C.R.B. conceived the project and designed the study. P.A.A. executed analyses and prepared the manuscript. C.R.B. provided editing and obtained funding for the project.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278203.123.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no competing interests.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, Layer RM, Neale BM, Salerno WJ, Reeves C, et al. 2020. Mapping and characterization of structural variation in 17,795 human genomes. Nature 583: 83–89. 10.1038/s41586-020-2371-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Coe BP, Eichler EE. 2011. Genome structural variation discovery and genotyping. Nat Rev Genet 12: 363–376. 10.1038/nrg2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonge M, Wang X, Benoit M, Soyk S, Pereira L, Zhang L, Suresh H, Ramakrishnan S, Maumus F, Ciren D, et al. 2020. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182: 145–161.e23. 10.1016/j.cell.2020.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Arlt MF, Mulle JG, Schaibley VM, Ragland RL, Durkin SG, Warren ST, Glover TW. 2009. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet 84: 339–350. 10.1016/j.ajhg.2009.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. 2019. Characterizing the major structural variant alleles of the human genome. Cell 176: 663–675.e19. 10.1016/j.cell.2018.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007. 10.1126/science.1072047 [DOI] [PubMed] [Google Scholar]
- Balachandran P, Walawalkar IA, Flores JI, Dayton JN, Audano PA, Beck CR. 2022. Transposable element-mediated rearrangements are prevalent in human genomes. Nat Commun 13: 7115. 10.1038/s41467-022-34810-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Carvalho CM, Banser L, Gambin T, Stubbolo D, Yuan B, Sperle K, McCahan SM, Henneke M, Seeman P, et al. 2015. Complex genomic rearrangements at the PLP1 locus include triplication and quadruplication. PLoS Genet 11: e1005050. 10.1371/journal.pgen.1005050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Carvalho CMB, Akdemir ZC, Sedlazeck FJ, Song X, Meng Q, Hu J, Doddapaneni H, Chong Z, Chen ES, et al. 2019. Megabase length hypermutation accompanies human structural variation at 17p11.2. Cell 176: 1310–1324.e10. 10.1016/j.cell.2019.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyter D, Ingimundardottir H, Oddsson A, Eggertsson HP, Bjornsson E, Jonsson H, Atlason BA, Kristmundsdottir S, Mehringer S, Hardarson MT, et al. 2021. Long-read sequencing of 3622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet 53: 779–786. 10.1038/s41588-021-00865-4 [DOI] [PubMed] [Google Scholar]
- Brandt DY, Aguiar VR, Bitarello BD, Nunes K, Goudet J, Meyer D. 2015. Mapping bias overestimates reference allele frequencies at the HLA genes in the 1000 Genomes Project phase I data. G3 (Bethesda) 5: 931–941. 10.1534/g3.114.015784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. 2022. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185: 3426–3440.e19. 10.1016/j.cell.2022.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. 10.1038/nrg.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, McCall A, Pivnick EK, Hines-Dowell S, Seaver LH, et al. 2011. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet 43: 1074–1081. 10.1038/ng.944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Belmont JW, Hastings PJ, Lupski JR. 2013. Replicative mechanisms for CNV formation are error prone. Nat Genet 45: 1319–1326. 10.1038/ng.2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. 10.1038/nature13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10: 1784. 10.1038/s41467-018-08148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. 10.1093/bioinformatics/btv710 [DOI] [PubMed] [Google Scholar]
- Cheng H, Jarvis ED, Fedrigo O, Koepfli KP, Urban L, Gemmell NJ, Li H. 2022. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotechnol 40: 1332–1335. 10.1038/s41587-022-01261-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. 2020. A structural variation reference for medical and population genetics. Nature 581: 444–451. 10.1038/s41586-020-2287-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A. 2011. Break-induced replication is highly inaccurate. PLoS Biol 9: e1000594. 10.1371/journal.pbio.1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212. 10.1093/bioinformatics/btp579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert P, Audano PA, Zhu Q, Rodriguez-Martin B, Porubsky D, Bonder MJ, Sulovari A, Ebler J, Zhou W, Serra Mari R, et al. 2021. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372: eabf7117. 10.1126/science.abf7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebler J, Ebert P, Clarke WE, Rausch T, Audano PA, Houwaart T, Mao Y, Korbel JO, Eichler EE, Zody MC, et al. 2022. Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat Genet 54: 518–525. 10.1038/s41588-022-01043-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizenga JM, Novak AM, Sibbesen JA, Heumos S, Ghaffaari A, Hickey G, Chang X, Seaman JD, Rounthwaite R, Ebler J, et al. 2020. Pangenome graphs. Annu Rev Genomics Hum Genet 21: 139–162. 10.1146/annurev-genom-120219-080406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, et al. 2020. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583: 699–710. 10.1038/s41586-020-2493-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Menon VK, Gibbs RA, Metcalf GA, Sedlazeck FJ. 2022. Truvari: refined structural variant comparison preserves allelic diversity. Genome Biol 23: 271. 10.1186/s13059-022-02840-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraj A, Audano PA, Balachandran P, Czechanski A, Flores JI, Radecki AA, Mosur V, Gordon DS, Walawalkar IA, Eichler EE, et al. 2023. Resolution of structural variation in diverse mouse genomes reveals chromatin remodeling due to transposable elements. Cell Genomics 3: 100291. 10.1016/j.xgen.2023.100291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Jungreis I, Lagarde J, Loveland JE, Mudge JM, Sisu C, Wright JC, Armstrong J, Barnes I, et al. 2021. GENCODE 2021. Nucleic Acids Res 49: D916–D923. 10.1093/nar/gkaa1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Fungtammasan A, Carroll A, Chou M, Schmitt A, Zhou X, Mac S, Peluso P, Hatas E, Ghurye J, et al. 2021. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat Biotechnol 39: 309–312. 10.1038/s41587-020-0711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Guarracino A, Heumos S, Villani F, Bao Z, Tattini L, Hagmann J, Vorbrugg S, Marco-Sola S, Kubica C, et al. 2023. Building pangenome graphs. bioRxiv 10.1101/2023.04.05.535718 [DOI] [PubMed]
- Hallast P, Ebert P, Loftus M, Yilmaz F, Audano PA, Logsdon GA, Bonder MJ, Zhou W, Höps W, Kim K, et al. 2023. Assembly of 43 human Y chromosomes reveals extensive complexity and variation. Nature 621: 355–364. 10.1038/s41586-023-06425-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Lee J, Meyer TJ, Remedios P, Goodwin L, Batzer MA. 2008. L1 recombination-associated deletions generate human genomic variation. Proc Natl Acad Sci 105: 19366–19371. 10.1073/pnas.0807866105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, et al. 2020. Array programming with NumPy. Nature 585: 357–362. 10.1038/s41586-020-2649-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. 10.1371/journal.pgen.1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller D, Vingron M. 2019. SVIM: structural variant identification using mapped long reads. Bioinformatics 35: 2907–2915. 10.1093/bioinformatics/btz041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller D, Vingron M. 2021. SVIM-asm: structural variant detection from haploid and diploid genome assemblies. Bioinformatics 36: 5519–5521. 10.1093/bioinformatics/btaa1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey G, Monlong J, Ebler J, Novak AM, Eizenga JM, Gao Y, Human Pangenome Reference C, Marschall T, Li H, Paten B. 2023. Pangenome graph construction from genome alignments with Minigraph-Cactus. Nat Biotechnol. 10.1038/s41587-023-01793-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Serra F, Bork P. 2016. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol 33: 1635–1638. 10.1093/molbev/msw046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. 2020. Pan-cancer analysis of whole genomes. Nature 578: 82–93. 10.1038/s41586-020-1969-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliakis G, Murmann T, Soni A. 2015. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen 793: 166–175. 10.1016/j.mrgentox.2015.07.001 [DOI] [PubMed] [Google Scholar]
- International HapMap C, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, et al. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. 10.1038/nature06258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- Jain C, Rhie A, Zhang H, Chu C, Walenz BP, Koren S, Phillippy AM. 2020. Weighted minimizer sampling improves long read mapping. Bioinformatics 36: i111–i118. 10.1093/bioinformatics/btaa435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebb D, Huang Z, Pippel M, Hughes GM, Lavrichenko K, Devanna P, Winkler S, Jermiin LS, Skirmuntt EC, Katzourakis A, et al. 2020. Six reference-quality genomes reveal evolution of bat adaptations. Nature 583: 578–584. 10.1038/s41586-020-2486-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. 2020. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581: 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH Jr, Moran JV. 1998. The impact of L1 retrotransposons on the human genome. Nat Genet 19: 19–24. 10.1038/ng0598-19 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Jeon S, Kim Y, Kim C, Bhak J, Bhak J. 2022. KOREF_s1: phased, parental trio-binned Korean reference genome using long reads and Hi-C sequencing methods. GigaScience 11: giac022. 10.1093/gigascience/giac022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsche M, Prabhu G, Sherman R, Ni B, Battle A, Aganezov S, Schatz MC. 2023. Jasmine and Iris: population-scale structural variant comparison and analysis. Nat Methods 20: 408–417. 10.1038/s41592-022-01753-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E, Meyn MS, Pandita A, Squire JA. 2002. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 35: 97–112. 10.1002/gcc.10111 [DOI] [PubMed] [Google Scholar]
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35: 4453–4455. 10.1093/bioinformatics/btz305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HY, Mu XJ, Stütz AM, Tanzer A, Cayting PD, Snyder M, Kim PM, Korbel JO, Gerstein MB. 2010. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat Biotechnol 28: 47–55. 10.1038/nbt.1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247. 10.1016/j.cell.2007.11.037 [DOI] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2021. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37: 4572–4574. 10.1093/bioinformatics/btab705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Bloom JM, Farjoun Y, Fleharty M, Gauthier L, Neale B, MacArthur D. 2018. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat Methods 15: 595–597. 10.1038/s41592-018-0054-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, Khurana E, Waszak S, Korbel JO, Haber JE, et al. 2020. Patterns of somatic structural variation in human cancer genomes. Nature 578: 112–121. 10.1038/s41586-019-1913-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Gong M, Zhang X, Wang F, Liu Z, Zhang L, Yang Q, Xu Y, Xu M, Zhang H, et al. 2023. A sheep pangenome reveals the spectrum of structural variations and their effects on tail phenotypes. Genome Res 33: 463–477. 10.1101/gr.277372.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao WW, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, Lu S, Lucas JK, Monlong J, Abel HJ, et al. 2023. A draft human pangenome reference. Nature 617: 312–324. 10.1038/s41586-023-05896-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon GA, Vollger MR, Eichler EE. 2020. Long-read human genome sequencing and its applications. Nat Rev Genet 21: 597–614. 10.1038/s41576-020-0236-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon GA, Rozanski AN, Ryabov F, Potapova T, Shepelev VA, Mao Y, Rautiainen M, Koren S, Nurk S, Porubsky D, et al. 2023. The variation and evolution of complete human centromeres. bioRxiv 10.1101/2023.05.30.542849 [DOI] [PMC free article] [PubMed]
- Mao Y, Catacchio CR, Hillier LW, Porubsky D, Li R, Sulovari A, Fernandes JD, Montinaro F, Gordon DS, Storer JM, et al. 2021. A high-quality bonobo genome refines the analysis of hominid evolution. Nature 594: 77–81. 10.1038/s41586-021-03519-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Harvey WT, Porubsky D, Munson KM, Hoekzema K, Lewis AP, Audano PA, Rozanski A, Yang X, Zhang S, et al. 2023. Structurally divergent and recurrently mutated regions of primate genomes. bioRxiv 10.1101/2023.03.07.531415 [DOI] [PMC free article] [PubMed]
- Menghi F, Inaki K, Woo X, Kumar PA, Grzeda KR, Malhotra A, Yadav V, Kim H, Marquez EJ, Ucar D, et al. 2016. The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proc Natl Acad Sci 113: E2373–E2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko A, Bzikadze AV, Gurevich A, Miga KH, Pevzner PA. 2020. TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36: i75–i83. 10.1093/bioinformatics/btaa440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales ME, White TB, Streva VA, DeFreece CB, Hedges DJ, Deininger PL. 2015. The contribution of alu elements to mutagenic DNA double-strand break repair. PLoS Genet 11: e1005016. 10.1371/journal.pgen.1005016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. 10.1038/nature01262 [DOI] [PubMed] [Google Scholar]
- Nattestad M, Goodwin S, Ng K, Baslan T, Sedlazeck FJ, Rescheneder P, Garvin T, Fang H, Gurtowski J, Hutton E, et al. 2018. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res 28: 1126–1135. 10.1101/gr.231100.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MD, Harvey WT, Porubsky D, Sulovari A, Li R, Rose NR, Audano PA, Munson KM, Lewis AP, Hoekzema K, et al. 2022. Familial long-read sequencing increases yield of de novo mutations. Am J Hum Genet 109: 631–646. 10.1016/j.ajhg.2022.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. 2022. The complete sequence of a human genome. Science 376: 44–53. 10.1126/science.abj6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Höps W, Ashraf H, Hsieh P, Rodriguez-Martin B, Yilmaz F, Ebler J, Hallast P, Maria Maggiolini FA, Harvey WT, et al. 2022. Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell 185: 1986–2005.e26. 10.1016/j.cell.2022.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodanov T, Bansal V. 2020. Sensitive alignment using paralogous sequence variants improves long-read mapping and variant calling in segmental duplications. Nucleic Acids Res 48: e114. 10.1093/nar/gkaa829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Snajder R, Leger A, Simovic M, Giurgiu M, Villacorta L, Henssen AG, Fröhling S, Stegle O, Birney E, et al. 2023. Long-read sequencing of diagnosis and post-therapy medulloblastoma reveals complex rearrangement patterns and epigenetic signatures. Cell Genom 3: 100281. 10.1016/j.xgen.2023.100281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautiainen M, Nurk S, Walenz BP, Logsdon GA, Porubsky D, Rhie A, Eichler EE, Phillippy AM, Koren S. 2023. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat Biotechnol 41: 1474–1482. 10.1038/s41587-023-01662-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, et al. 2021. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592: 737–746. 10.1038/s41586-021-03451-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Gao J, Borsari B, Yang YT, Galeev T, Gürsoy G, Epstein CB, Xiong K, Xu J, Li T, et al. 2023. The EN-TEx resource of multi-tissue personal epigenomes & variant-impact models. Cell 186: 1493–1511.e40. 10.1016/j.cell.2023.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, et al. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. 10.1038/nature01140 [DOI] [PubMed] [Google Scholar]
- Sahlin K, Baudeau T, Cazaux B, Marchet C. 2023. A survey of mapping algorithms in the long-reads era. Genome Biol 24: 133. 10.1186/s13059-023-02972-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y, Xu L, Seki M, Yokoyama TT, Kasahara M, Kashima Y, Ohashi A, Shimada Y, Motoi N, Tsuchihara K, et al. 2020. Long-read sequencing for non-small-cell lung cancer genomes. Genome Res 30: 1243–1257. 10.1101/gr.261941.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebusch CM, Malmström H, Gunther T, Sjodin P, Coutinho A, Edlund H, Munters AR, Vicente M, Steyn M, Soodyall H, et al. 2017. Southern African ancient genomes estimate modern human divergence to 350,000 to 260,000 years ago. Science 358: 652–655. 10.1126/science.aao6266 [DOI] [PubMed] [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864. 10.1101/gr.213611.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, Schatz MC. 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15: 461–468. 10.1038/s41592-018-0001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen SK, Han K, Wang J, Lee J, Wang H, Callinan PA, Dyer M, Cordaux R, Liang P, Batzer MA. 2006. Human genomic deletions mediated by recombination between Alu elements. Am J Hum Genet 79: 41–53. 10.1086/504600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JS, Rhie A, Kim J, Lee S, Sohn MH, Kim CU, Hastie A, Cao H, Yun JY, Kim J, et al. 2016. De novo assembly and phasing of a Korean human genome. Nature 538: 243–247. 10.1038/nature20098 [DOI] [PubMed] [Google Scholar]
- Shi L, Guo Y, Dong C, Huddleston J, Yang H, Han X, Fu A, Li Q, Li N, Gong S, et al. 2016. Long-read sequencing and de novo assembly of a Chinese genome. Nat Commun 7: 12065. 10.1038/ncomms12065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green, P. 2013–2015. RepeatMasker Open-4.0. https://www.repeatmasker.org/.
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, et al. 2015. An integrated map of structural variation in 2504 human genomes. Nature 526: 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulovari A, Li R, Audano PA, Porubsky D, Vollger MR, Logsdon GA, Human Genome Structural Variation C, Warren WC, Pollen AA, Chaisson MJP, et al. 2019. Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc Natl Acad Sci 116: 23243–23253. 10.1073/pnas.1912175116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. 2021. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 590: 290–299. 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, Kronenberg ZN, Hormozdiari F, Raja A, Pennacchio LA, et al. 2017. Genomic patterns of de novo mutation in simplex autism. Cell 171: 710–722.e12. 10.1016/j.cell.2017.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. 2020. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17: 261–272. 10.1038/s41592-019-0686-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Bengesser K, Claes KB, Wimmer K, Mautner VF, van Minkelen R, Legius E, Brems H, Upadhyaya M, Högel J, et al. 2014. SVA retrotransposon insertion-associated deletion represents a novel mutational mechanism underlying large genomic copy number changes with non-recurrent breakpoints. Genome Biol 15: R80. 10.1186/gb-2014-15-6-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlster L, Verboon JM, Ludwig LS, Black SC, Luo W, Garg K, Voit RA, Collins RL, Garimella K, Costello M, et al. 2021. Familial thrombocytopenia due to a complex structural variant resulting in a WAC-ANKRD26 fusion transcript. J Exp Med 218: e20210444. 10.1084/jem.20210444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Antonacci-Fulton L, Howe K, Lawson HA, Lucas JK, Phillippy AM, Popejoy AB, Asri M, Carson C, Chaisson MJP, et al. 2022. The human pangenome project: a global resource to map genomic diversity. Nature 604: 437–446. 10.1038/s41586-022-04601-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Peluso P, Rowell WJ, Chang PC, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, et al. 2019. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37: 1155–1162. 10.1038/s41587-019-0217-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Frock RL, Menghi F, Duffey EE, Panday A, Camacho V, Hasty EP, Liu ET, Alt FW, Scully R. 2017. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 551: 590–595. 10.1038/nature24477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Emery SB, Flasch DA, Wang Y, Kwan KY, Kidd JM, Moran JV, Mills RE. 2020. Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Res 48: 1146–1163. 10.1093/nar/gkz1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.