

Abstract
Background
Conotruncal defects due to developmental abnormalities of the outflow tract (OFT) are an important cause of cyanotic congenital heart disease. Dysregulation of transcriptional programs tuned by NKX2–5, GATA6, and TBX1 have been implicated in abnormal OFT morphogenesis. However, there remains no consensus on how these transcriptional programs function in a unified gene regulatory network (GRN) within the OFT.
Methods
We generated mice harboring a 226-nucleotide deletion of a highly conserved cardiac enhancer containing two GATA-binding sites located approximately 9.4kb upstream of the transcriptional start site of Nkx2–5 (Nkx2–5∆enh) using CRISPR-Cas9 gene editing and assessed phenotypes. Cardiac defects in Nkx2–5∆enh/∆enh mice were structurally characterized using histology and scanning electron micrography, and physiologically assessed using electrocardiography, echocardiography, and optical mapping. Transcriptome analyses were performed using RNA sequencing (RNA-seq) and single cell RNA sequencing (scRNA-seq) datasets. Endogenous GATA6 interaction with and activity on the NKX2–5 enhancer was studied using chromatin immunoprecipitation sequencing and transposase accessible chromatin sequencing in human-induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs).
Results
Nkx2–5∆enh/∆enh mice recapitulated cyanotic conotruncal defects seen in patients with NKX2–5, GATA6, and TBX1 mutations. Nkx2–5∆enh/∆enh mice also exhibited defects in right Purkinje fiber network formation, resulting in right bundle branch block. Enhancer deletion reduced embryonic Nkx2–5 expression selectively in the right ventricle and OFT of mutant hearts, indicating that enhancer activity is localized to the anterior second heart field (A-SHF). Transcriptional profiling of the mutant OFT revealed downregulation of important genes involved in OFT rotation and septation, such as Tbx1, Pitx2, and Sema3c. Endogenous GATA6 interacted with the highly conserved enhancer in hiPSC-CMs and in wildtype mouse hearts. We found critical dose-dependency of cardiac enhancer accessibility on GATA6 gene dosage in hiPSC-CMs.
Conclusion
Our results using human and mouse models reveals an essential GRN of the OFT that requires an A-SHF enhancer to link GATA6 with NKX2–5-dependent rotation and septation gene programs.
Keywords: Congenital heart disease, conotruncal defects, right bundle branch block, gene regulatory network, non-coding DNA, anterior second heart field, enhancer, transcription factor
Introduction
Congenital heart disease (CHD) is a leading cause of birth defects with an estimated global prevalence of 1.8 cases per 100 live births.1 In subcategories of CHD, conotruncal defects caused by impaired development of the outflow tract (OFT) are reported to make up 6.7–12% of CHD cases.2–4 Cyanotic conotruncal defects, which typically manifest within the first week of life, confer extremely high mortality if uncorrected.5 Included in the category of cyanotic conotruncal defects are persistent truncus arteriosus (PTA), double outlet right ventricle (DORV), dextro-transposition of great arteries (d-TGA), and tetralogy of Fallot (TOF).6 While there have been major advances in diagnostic modalities and corrective surgical techniques for cyanotic conotruncal defects, our understanding of the molecular and genetic underpinnings of OFT morphogenesis remains rudimentary.
A genetic etiology for CHD has been identified in a third of cases, with approximately 23% linked to aneuploidies and copy number variations and the remaining 8% attributable to coding de novo variants (DNVs).7–9 More recently, the contribution of disruptive non-coding DNVs, which include cardiac enhancer regions, to CHD has been reported to be at a level similar to that observed for damaging coding DNVs.10 These data indicate that coding and non-coding DNA elements act cooperatively during heart development to regulate complex morphogenetic processes, such as cardiac looping, rotation, and septation. Therefore, a more comprehensive understanding of cardiac enhancers and how they regulate temporo-spatial expression of morphogenetic gene programs is not only critical to understanding normal heart development but also why congenital heart defects occur.
The OFT represents a particularly vulnerable site for CHD as it requires precise coordination of specification, migration, and maturation of multiple cell types, which include myocardial cells, cardiac neural crest cells (NCCs), endocardial cells, and vascular smooth muscle cells (VSMCs), to properly align and septate the pulmonic and systemic circulatory systems. Through classical genetic approaches and candidate gene analysis, mutations in lineage-determining transcription factors have been identified in patients with conotruncal defects and have provided important mechanistic insights into how transcriptional programs orchestrate OFT development. TBX1/22q11del is the most common human copy number variation, which causes DiGeorge Syndrome, a condition associated with DORV, PTA, and TOF.11–13 Pathologic coding variants in NKX2–5 and GATA6 have been identified in patients with conotruncal defects, with NKX2–5 mutations associated with TOF, PTA, and DORV, and GATA6 mutations associated with PTA and pulmonary stenosis14–17. Correspondingly, mouse models of Tbx1, Nkx2–5, and Gata6 deficiency recapitulate the spectrum of OFT abnormalities. Tbx1 hypomorphic mice exhibit PTA.18 Selective deletion of Nkx2–5 in second heart field myocardial precursors driven by Isl1-Ires-Cre (Nkx2–5flox/flox;Isl1-Ires-Cre) produces PTA and DORV.19 Deletion of Gata6 in cardiomyocytes and VSMCs using a SM22-cre driver causes PTA and DORV.20
The phenotypic redundancy of NKX2–5, TBX1, and GATA6 mutations indicates their shared role in morphogenetic programming of the OFT and suggests these transcription factors function in a ‘cardiac regulatory kernel’, essential for lineage determination and/or maturation of the OFT myocardium.21 The OFT, right ventricular myocardium, right bundle branch, and the majority of the right Purkinje fiber network (PFN) derive from anterior second heart field (A-SHF) precursor cells that originate from the splanchnic mesoderm and are added to the linear heart tube at the arterial pole.22, 23 The A-SHF-derived OFT myocardium represents a distinct regulatory domain that expresses important effector genes involved in rotation and septation, such as the homeobox transcription factor Pitx2 and the secreted chemotactic factor Sema3c, respectively. Pitx2 is essential for proper rotation and septation of the proximal or conal OFT myocardium to generate the right and left ventricular outflow tracts (RVOT and LVOT, respectively).24, 25 Sema3c is secreted by OFT cardiomyocytes to recruit cardiac NCCs into the conotruncal cushions of the distal or truncal OFT where they mature into VSMCs essential for aorticopulmonary septation.26 Together, Pitx2 and Sema3c ensure proper alignment of the RVOT with the pulmonary artery (PA) anteriorly and the LVOT with the aorta posteriorly. What remains unknown is the underlying genetic mechanism that links NKX2–5, TBX1, and GATA6 into a single regulatory network with Pitx2 and Sema3c in the OFT myocardium.
To address this important knowledge gap, we comprehensively studied a putative NKX2–5 enhancer that is highly conserved, is GATA-binding site dependent, and has restricted activity in A-SHF-derived structures to determine its importance in OFT and right ventricular development.27, 28 In order to functionally validate this enhancer region in vivo, we engineered transgenic mice harboring a 226-bp deletion within the cardiac enhancer region that removes two conserved GATA-binding sites. Enhancer site deletion selectively reduced Nkx2–5 expression in the right ventricle (RV) and OFT and produced an array of conotruncal defects as well as hypoplasia of the right PFN. Using this mouse model and human induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs), we define the developmental basis of OFT defects and identify a common genetic pathway that links GATA6 with NKX2–5-dependent morphogenetic programs through a single A-SHF enhancer.
Materials and Methods
Detailed materials and methods are described in Supplemental Material online. All animal experiments were approved under Institutional Animal Care and Use Committee of New York University Grossman School of Medicine protocol #PROTO201900150.
Data Availability
The RNA sequencing (RNA-seq) data are deposited in Gene Expression Omnibus (GSE227361). The chromatin immunoprecipitation sequencing (ChIP-seq) for GATA6 and assay of transposase accessible chromatin sequencing (ATAC-seq) data in hiPSC-CMs, and single cell RNA-seq (scRNA-seq) data from control and Pitx2hd−/− mouse hearts were obtained from previously published datasets.29, 30
Statistical analysis
GraphPad Prism v9.2.0 (GraphPad Software, MA, USA) was used for statistical comparisons except for sequencing data. Quantitative data were first evaluated by Shapiro-Wilk test for normal distribution, then two-tailed Student’s t-test was performed for comparison between 2 groups except where otherwise mentioned in the figure legend. The p values < 0.05 were considered statistically significant. Values are presented as mean ± SD. For survival analysis for 3 groups, logrank test was performed, and multiple comparison correction was applied as p value < 0.0167 to be significant. Additional details for sequencing data analyses are described in Supplemental Material.
Results
Generation of Nkx2–5 cardiac enhancer mutant mice
With the goal of defining essential genomic regulatory elements that govern OFT development, we focused on a highly conserved, cardiac enhancer located within the locus of chromosome 5: 173,245,843 to 173,245,543, positioned −10,637 to −10,337 bp upstream of the transcription start site of NKX2–5 in the human hg38 assembly (Figure 1A). The cardiac enhancer contains two GATA-binding sites within the most conserved region based on PhyloP basewise conservation analysis (Figure 1B). The corresponding location within the mouse genome is –9,435 to –8,922 bp upstream of the TSS of Nkx2–5 in the mouse mm10 assembly. Previous description of this enhancer region using transgenic LacZ reporter mice showed localized activity in the OFT and RV that was dependent on the 5’ GATA-binding site.27 These features made this enhancer a compelling candidate for targeted deletion in vivo.
Figure 1. Generation of Nkx2–5∆enh/∆enh mice engineered with a 226-bp deletion in a highly conserved enhancer region.

(A) Conservation state of the upstream NKX2–5 cardiac enhancer by the UCSC Genome Browser. NKX2–5 coding sequence (labeled in black) and an upstream minimal cardiac enhancer region (labeled in red) on the human hg38 assembly. Conservation score using phyloP (blue bars) and phastCons (green bars) reveals that sequences corresponding to the cardiac enhancer are highly conserved among vertebrates. Conserved elements identified by phastCons are also displayed in brown. Representative vertebrate genomes of selected species are displayed in gray. TSS, transcription start site. (B) A 301-bp region (labeled in red) on human hg38 assembly with 90% sequence similarity to the previously reported cardiac enhancer region on the mouse mm10 assembly with conserved GATA-binding sites (green arrows)(upper panel). A gap with double horizontal lines in the cardiac enhancer shows dissimilarity in sequence between human and mouse genomes. Further restriction of the conservation peaks above 50% threshold yielded a 241-bp region containing the two GATA-binding sites (lower panel) (C) Schematic of CRISPR-CAS9 deletion of a 226-bp fragment containing two conserved GATA-binding sites ≈9.4 kb upstream of the Nkx2–5 transcription start site in a mouse model with mm10 assembly. (D) PCR of genomic DNA from wildtype, heterozygous, and homozygous cardiac enhancer deletion (Nkx2–5∆enh) mice. (E) Sequencing of genomic DNA from wildtype Nkx2–5+/+ and homozygous Nkx2–5∆enh/∆enh mice showing deletion of the 226-bp fragment. (F) RT-qPCR from regional heart tissues at embryonic day E12.5 demonstrating reduced Nkx2–5 expression exclusively in the right ventricle (RV) and outflow tract (OFT). LV: left ventricle. Groups (n = 6 each) were compared by Student’s t-test. **p < 0.01. Data are presented as mean ± SD. (G) Genotype of viable offspring at each developmental stage from Nkx2–5∆enh/+ mating pairs. Number of viable Nkx2–5∆enh/∆enh at postnatal day (P) 10 is significantly lower than the expected Mendelian distribution.
BLAST-Like Alignment Tool (BLAT) in the UCSC Genome Browser identified a 301-bp region on the hg38 assembly with 90% sequence similarity to the cardiac enhancer region on the mm10 assembly. Further restriction of the conservation peaks above 50% threshold yielded a 241-bp region that contained the two GATA-binding sites (Figure 1B). To functionally validate this minimal cardiac enhancer in vivo, we knocked out a 226-bp fragment of the enhancer region containing the two GATA-binding sites (Nkx2–5∆enh) using CRISPR-Cas9 gene editing technology and confirmed the deletion (Figure 1C–E).31 RT-qPCR analysis of embryonic day (E) 12.5 hearts sectioned into atria, RV, left ventricle (LV), and OFT regions showed reduced transcript levels of Nkx2–5 exclusively in the RV and OFT with normal levels in the LV and atria in Nkx2–5∆enh/∆enh mice compared to Nkx2–5+/+ wildtype (WT) littermate controls (Figure 1F). These data demonstrate that the minimal enhancer functions as a cis-regulatory element of Nkx2–5 in the A-SHF.
Mice heterozygous for the minimal enhancer deletion (Nkx2–5∆enh/+) were grossly indistinguishable from WT littermates and displayed normal cardiac function assessed by electrocardiography (ECG) and transthoracic echocardiography (TTE) (data not shown). Upon intercrossing Nkx2–5∆enh/+ mice, embryos and offspring matched the expected Mendelian ratio until postnatal day (P) 1, after which most Nkx2–5∆enh/∆enh mice did not survive (Figure 1G). Nkx2–5∆enh/∆enh mice were cyanotic at birth and displayed high neonatal mortality rate (Figure 2A). Out of 72 offspring born from 8 consecutive litters examined for 10 days after birth, the homozygous mutant pups showed high postnatal mortality with most animals unable to survive beyond P2 (Figure 2B) (Logrank test for survival analysis among 3 groups, p < 0.0001; between Nkx2–5+/+ and Nkx2–5∆enh/+, p = 0.9336; between Nkx2–5+/+ and Nkx2–5∆enh/∆enh, p < 0.0001; between Nkx2–5∆enh/+ and Nkx2–5∆enh/∆enh, p < 0.0001).
Figure 2. Survival and pathological analysis of Nkx2–5∆enh/∆enh mice at postnatal (P) day 1.
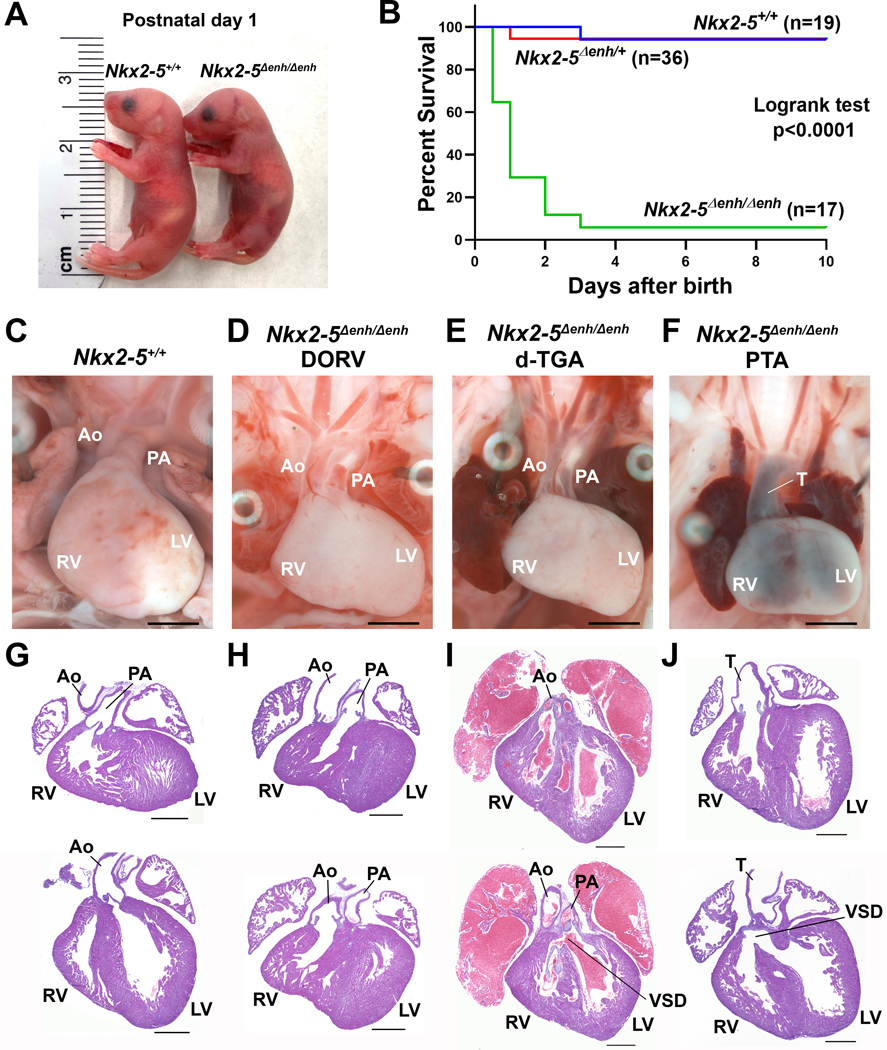
(A) Gross morphology of P1 Nkx2–5∆enh/∆enh and wildtype (Nkx2–5+/+) littermates. (B) Neonatal mouse survival curves in Nkx2–5+/+, Nkx2–5∆enh/+, and Nkx2–5∆enh/∆enh mice. P<0.0001 by logrank test for all three genotypes. (C, G) Representative wholemount and histological assessment of Nkx2–5+/+ hearts with normal anatomy. (D−F and H−J) Representative wholemount and histological assessment of Nkx2–5∆enh/∆enh hearts showing double-outlet right ventricle (DORV) (D, H), dextro-transposition of great arteries (d-TGA) with ventricular septal defect (VSD) (E, I), and persistent truncus-arteriosus (PTA) with VSD (F, J). PA, pulmonary artery; Ao, aorta; T, truncus arteriosus; LV, left ventricle; RV, right ventricle. Scale bars for C−F: 2 mm. Scale bars for G−J: 500 µm.
Nkx2–5∆enh/∆enh mice display cyanotic conotruncal defects
To determine the cause of neonatal cyanosis and lethality in Nkx2–5∆enh/∆enh mice, we examined mutant and WT littermates for cardiovascular defects in P1 pups using whole-mount and histological assessment. The most notable anatomical abnormalities in Nkx2–5∆enh/∆enh hearts were OFT abnormalities and ventricular septal defects (VSD). WT hearts displayed normal positioning of the aorta in connection with the LVOT posteriorly and PA in connection with the RVOT anteriorly (Figures 2C, G). Nkx2–5∆enh/∆enh mice displayed a spectrum of cyanotic conotruncal defects, including DORV (Figure 2D, H), d-TGA (Figure 2E, I), and PTA (Figure 2F, J), with PTA being the most prevalent condition (Table S1). Mutant hearts displayed DORV (17 %), where both the aorta and PA originate from the RV anteriorly in parallel configuration (Figure 2D, H), d-TGA (28 %), where the aorta connects to the RVOT anteriorly and the PA connects to the LVOT posteriorly (Figure 2E, I), and most commonly, PTA (44 %), which combines rotational defects (seen in DORV and d-TGA) with an aorticopulmonary septation defect resulting in a single truncus (Figure 2F, J).32–34 Therefore, Nkx2–5∆enh/∆enh mice recapitulate the full spectrum of conotruncal defects seen in patients and mouse models with GATA6, NKX2–5, and TBX1 mutations.18–20
Nkx2–5∆enh/∆enh embryonic hearts show defects in axial orientation of the conotruncal endocardial cushions
To explore the origins of the conotruncal defects in Nkx2–5∆enh/∆enh hearts, we performed histological analyses at E11.5 and scanning electron microscopy (SEM) at E12.5. Serial sections of mutant E11.5 embryos revealed defects in axial orientation of the conotruncal endocardial cushions (Figure 3). During normal OFT development (Figure 3B–E), the superior septal cushion (SSC) and inferior septal cushion (ISC) of the conotruncal cushions fuse medially to form the aortiopulmonary septum (Figure 3C) in a process that is dependent on migration of cardiac NCCs.35 The SSC forms the left cusps and ISC forms the right cusps of the aortic valve (AoV) and pulmonic valve (PV).35, 36 The anterior pulmonic intercalated cushion (APIC) and posterior aortic intercalated cushion (PAIC), which are positioned in perpendicular orientation to the SSC and ISC, form the anterior cusp of the PV and the posterior (non-coronary) cusp of the AoV, respectively (Figure 3B–D, red arrowhead pointing to the noncoronary cusp of the AoV). Axial alignment of the SSC and ISC cushions along the axis of the spinal cord (S) positions the PAIC (non-coronary cusp of the AoV) directly at 6 o’clock in the OFT (Figure 3C, D), which is essential for proper alignment of the AoV (Figure 3C) over the LVOT (Figure 3E). More distally (cranially) in the OFT, the conotruncal cushion axis is rotated clockwise, situating the PV (blue arrowhead) at approximately 1:30 on the clockface in the distal portion of RVOT (Figure 3B). Proper axial orientation of the conotruncal cushions (SSC, ISC, APIC, PAIC) ensures that the PA is connected to the RVOT anteriorly and the aorta is connected to the LVOT posteriorly as shown in the SEM of a normal E12.5 heart (Figure 3N).
Figure 3. Histological and electron micrographic assessment of Nkx2–5∆enh/∆enh embryonic hearts.

(A) Schematic of an embryonic day E11.5 heart with corresponding sections through the outflow tract (OFT) of Nkx2–5+/+ (B−E) and Nkx2–5∆enh/∆enh (F−I and J−M) hearts at select levels: distal level (green hashed lines)(B, F, J), aortic valve (AoV) level (blue hashed lines)(C, G, K), sub-AoV level (red hashed lines)(D, H, L), and proximal level (black hashed lines)(E, I, M). Magnified view of the OFT for each section is shown with a clockface demarcating the spinal cord (S) axis at 6 o’clock. Schematic diagrams show labeled anatomical structures of the OFT to the right of each magnified section. In the Nkx2–5+/+ heart (B−E), the superior and inferior septal cushions (SSC and ISC) are oriented along the axis of the spinal cord so that the posterior aortic intercalated cushion (PAIC)(panel D), which becomes the non-coronary cusp of the AoV (panel C, red arrowhead), is situated at 6 o’clock in the OFT. This allows for the AoV to connect with the left ventricular outflow tract (LVOT) below (panel E). More distally, the aorta (Ao) remains in the 6 o’ clock position with the right ventricular outflow tract (RVOT) looping around the aorta clockwise, ending with pulmonary valve (PV) (panel B, blue arrowhead) positioned at 1:30 on the clockface. Two Nkx2–5∆enh/∆enh mutant hearts are shown with different severities of rotation defects. The first mutant (F−I) shows insufficient counterclockwise rotation so that axis of the SSC, ISC, and PAIC (non-coronary cusp) are shifted rightward by 45 degrees to 7:30 on the clockface (panel G, red arrowhead), displacing the AoV off the LVOT and onto the proximal RVOT (pRVOT)(panel I). The anterior pulmonary intercalated cushion (APIC), which becomes the anterior valve (AV) of the PV, is also shifted clockwise but remains attached to the distal RVOT (dRVOT)(panel F, blue arrowhead). This results in double-outlet right ventricle (DORV) phenotype where the Ao is laterally displaced to the right and lies anteriorly with the pulmonary artery (PA) (panel F). The second mutant (J−M) shows more severe insufficient counterclockwise rotation so that SSC, ISC, and PAIC (non-coronary cusp) are shifted rightward by 105 degrees to 9:30 on the clockface (panel K, red arrowhead), allowing for connection of the AoV to the RVOT. The APIC and PV now sit at the 6 o’clock position (panel J and K, blue arrowhead), connecting the PV to the LVOT. This results in dextro-transposition of great arteries (d-TGA) phenotype where the Ao is anterolaterally displaced relative to the PA (panel J). Scale bars: 500 µm. RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; Ven, ventral; Dor, dorsal. (N–P) Scanned electron microscopic images of embryonic hearts at E12.5 in Nkx2–5∆enh/∆enh and wildtype littermates. Nkx2–5∆enh/∆enh hearts display rotational defects of the OFT. Scale bars: 200 µm.
In Nkx2–5∆enh/∆enh hearts (Figure 3F–I and 3J–M), the conotruncal cushion axis is insufficiently counterclockwise rotated so that the PAIC (non-coronary cusp of the AoV, red arrowhead) fails to reach the 6 o’clock position and is malpositioned anywhere between 7 o’clock to 10 o’clock. As shown in the first Nkx2–5∆enh/∆enh heart (Figure 3F–I), displacement of the PAIC to 7:30 on the clockface (Figure 3H) moves the non-coronary cusp of the AoV (Figure 3G, red arrowhead) away from its usual connection point with the LVOT (Figure 3I). This clockwise displacement of the AoV results in inappropriate connection of the AoV and aorta with the proximal RVOT (pRVOT) (Figure 3I). Correspondingly, the distal RVOT (dRVOT) and PV (blue arrowhead) are displaced to 2:30 on the clockface (Figure 3F, G). This places the aorta and PA in an equivalent anterior plane with both vessels connected to the RV, resulting in DORV phenotype (Figure 3F–I). SEM of a mutant E12.5 heart shows rightward displacement of the aorta, placing it in the same anterior plane as the PA (Figure 3O). If aorticopulmonary septation occurs, then DORV phenotype is the expected result. As shown in the second Nkx2–5∆enh/∆enh heart (Figure 3J–M), displacement of the PAIC to 9:30 on the clockface (Figure 3L) will inappropriately connect the AoV (red arrowhead pointing to the non-coronary cusp) with the RVOT anteriorly while displacement of the PV (blue arrowhead) to 6 o’ clock will allow for inappropriate connection of the PV and PA with the LVOT (Figure 3J–L), resulting in d-TGA phenotype. SEM of an E12.5 heart with d-TGA shows anterior displacement of the aorta and the PA positioned posteriorly (Figure 3P). PTA represents a combination of rotational abnormalities with aorticopulmonary septation defect.
To examine if the increased propensity for aorticopulmonary septation defects in Nkx2–5Δenh/Δenh hearts is secondary to deficiency in conotruncal cushion formation, we measured reconstructed three-dimensional SSC and ISC volumes from rendered histological sections (Figure S1). Comparisons of the conotruncal cushion volumes between WT and Nkx2–5Δenh/Δenh hearts showed no significant differences, suggesting that invasion of mesenchymal cells and secretion of cardiac jelly to form the conotruncal cushions are unperturbed in Nkx2–5 mutant hearts. Notably, the SSC and ISC axis again demonstrates under-rotation in Nkx2–5Δenh/Δenh hearts compared to WT littermate controls.
Nkx2–5∆enh/∆enh mice exhibit right ventricular conduction defects
Right-sided conduction system defects, in the form of incomplete and complete right bundle branch blocks, have been described in patients with d-TGA (33%), DORV (52%), and PTA (47%) prior to surgical correction.33, 37, 38 To assess whether Nkx2–5∆enh/∆enh mice display right-sided conduction abnormalities, we performed functional assessment using ECG and TTE. Surface ECG was recorded from P1 neonates. Nkx2–5∆enh/∆enh mice showed prolonged QRS interval duration compared to WT littermates (14.05 ± 1.00 ms vs 12.40 ± 0.84 ms, p = 0.0013), indicating interventricular conduction delay (Figure 4A, B). Other parameters including heart rate, P wave duration, and PR interval were comparable between mutant and WT mice (Figure 4B). TTE was performed at P1 to assess biventricular function and quantify mechanical dyssynchrony using speckle tracking technology. Schematic of two-dimensional strain analysis of short-axis view is shown in Figure 4C where a red dotted semi-circle represents the RV free wall endocardial border and a blue dotted ellipse represents the LV endocardial border used for speckle tracking. Representative circumferential strain activation analysis during a single cardiac cycle showed marked ventricular dyssynchrony between RV and LV in Nkx2–5∆enh/∆enh hearts where dotted lines represent time from onset of systole to peak systole (Figure 4D). Strain analysis of mutant hearts showed a significant activation delay in the RV by 10 % of cycle length when referenced to peak systolic strain in the LV compared to WT littermates (Figure 4E). There was no significant difference in biventricular function between WT and Nkx2–5∆enh/∆enh mice measured by circumferential strain (Figure S2A). Optical mapping of P1 WT hearts in sinus rhythm showed early activation from the RV and LV epicardial surface at the sites of right and left bundle branch exit, respectively. In contrast, Nkx2–5∆enh/∆enh hearts from P1 littermates displayed delayed RV epicardial activation relative to LV activation, indicative of right bundle branch block (Figure 4F, Figure S2B).
Figure 4. Physiological assessment of Nkx2–5∆enh/∆enh vs wildtype mice.

(A) Representative surface electrocardiogram traces in Nkx2–5∆enh/∆enh and wildtype littermates at birth (P1). (B) Comparisons of heart rate, P-wave duration, PR interval, and QRS interval between Nkx2–5∆enh/∆enh and wildtype mice. QRS interval is significantly prolonged in Nkx2–5∆enh/∆enh mice. **p < 0.01 by Student’s t-test. n = 9−10 per group. Data are presented as mean ± SD. (C) Representative B-mode snapshot of both ventricles in short-axis view. Endocardial segments used for circumferential strain are shown with dotted lines in red for the right ventricle (RV) and in blue for the left ventricle (LV). (D) Representative strain tracings of both ventricles in Nkx2–5∆enh/∆enh and wildtype mice with dotted vertical line representing peak systolic contraction. Nkx2–5∆enh/∆enh showed delayed relative RV activation with respect to LV. (E) Relative RV activation showing variability and delays in Nkx2–5∆enh/∆enh mice. *p < 0.05 by Student’s t-test. n = 6 per group. Data are presented as mean ± SD. (F) Representative activation maps of wildtype (left) and Nkx2–5∆enh/∆enh (right) hearts acquired by optical mapping at P1. Electrical activation of RV is delayed in Nkx2–5∆enh/∆enh compared to wildtype. RA, right atrium; RV, right ventricle; LA, left atrium; LV, left ventricle. Scale bars: 1 mm. (G) Representative whole-mount fluorescence images of the dissected RV at P1 to visualize Purkinje cells expressing Contactin2-eGFP. Nkx2–5∆enh/∆enh hearts show variable degrees of Purkinje fiber network hypoplasia, with a spectrum ranging from absent to near normal levels in the RV. n = 3 for wildtype and n = 8 for Nkx2–5∆enh/∆enh were examined. Scale bars: 2 mm. AVN, atrioventricular node; HB, His bundle; PF, Purkinje Fibers. (H) qPCR for Gja5 (encoding Cx40) and Gja1 (encoding Cx43) in the RV and LV at embryonic day E12.5. Gja5 expression was significantly reduced in the RV but not Gja1. **p < 0.01 by Student’s t-test. Expression of Gja5 in LV and Gja1 in RV were compared by Mann-Whitney U test. n = 6 per group. Data are presented as mean ± SD. (I) Immunofluorescence staining for troponin I (green) and Cx40 (red) in embryonic hearts at E12.5. Nkx2–5∆enh/∆enh hearts show less Cx40 expression in the trabecular myocardium in the RV compared to wildtype. n = 3 per group were examined. Scale bars: 200 µm.
Nkx2–5∆enh/∆enh mice exhibit variable degrees of hypoplasia of the right Purkinje fiber network
To elucidate the mechanism of right ventricular conduction delay in Nkx2–5∆enh/∆enh mice, we backcrossed mutant mice into a cardiac conduction system reporter (Contactin 2-eGFP) background.39 Whole-mount fluorescent images demonstrated a spectrum of defects of the right PFN in Nkx2–5∆enh/∆enh hearts (Figure 4G). Defects ranged from complete absence of the right PFN to variable degrees of PFN hypoplasia. To assess whether right PFN hypoplasia is an acquired defect due to hemodynamic abnormalities from conotruncal defects or a developmental programming abnormality, we evaluated Nkx2–5∆enh/∆enh and WT littermate hearts at E12.5 for differential expression of the Purkinje marker, Connexin 40 (Cx40, encoded by the gene Gja5). At this developmental timepoint, the interventricular septum is not completely formed and the hemodynamic load is shared equally between the LV and RV. We assessed for changes in Gja5 expression separately in the right and left ventricular chambers. Gja5 expression was significantly reduced in the RV but not in the LV in E12.5 Nkx2–5∆enh/∆enh mice compared to WT littermates (Figure 4H). In contrast, Connexin 43 (encoded by Gja1), which is expressed throughout the chamber myocardium, was unchanged in the RV and LV in mutant hearts (Figure 4H). We also observed reduced Cx40 expression in the RV but not the LV of E12.5 Nkx2–5∆enh/∆enh hearts by immunofluorescence staining (Figure 4I). These findings are consistent with other reports indicating that Purkinje cell recruitment from trabecular myocytes requires optimal dosing of Nkx2–5.40
The Nkx2–5 enhancer regulates a critical OFT transcriptome
To identify the Nkx2–5 enhancer-dependent transcriptome that regulates conotruncal development, we performed RNA-seq analysis from OFT tissue collected from E12.5 Nkx2–5∆enh/∆enh and WT littermates. Analysis of differentially expressed genes (DEGs) using an adjusted p value-cutoff (FDR < 0.1) showed reduced expression of Nkx2–5 as well as important OFT regulatory genes, Pitx2 and Sema3c, in mutant hearts (Figure 5A). These findings indicate that major rotation and septation gene programs are under the regulatory landscape of the Nkx2–5 enhancer. Using this threshold criterion, we identified 59 DEGs between Nkx2–5Δenh/Δenh versus WT littermate controls. A heatmap of the DEGs shows excellent genotype-dependent segregation including 20 upregulated and 39 downregulated transcripts in the Nkx2–5Δenh/Δenh OFT (Figure 5B). We performed integrative functional enrichment analysis using Enrichr Knowledge Graph (Enrichr-KG) to identify associations between DEGs and mammalian phenotypes and biological processes.41 Using this unbiased approach, we identified important gene-phenotype associations between Nkx2–5, Pitx2, Sema3c, Pcsk6/Pace4, Lama5 and the conotruncal defects, DORV and PTA. Mutant mouse models of each of these genes have been reported to exhibit DORV and/or PTA.19, 24, 26, 42–44 Notably, PCSK6 was identified as a candidate gene in a genome-wide linkage analysis using MOD score analysis in families with diverse CHD that included DORV, TGA, TOF, and VSD.45 Consistent with the conotruncal phenotype associations, we identified biological processes that were significantly downregulated in the Nkx2–5Δenh/Δenh OFT involved in NCC recruitment (Sema3c-dependent chemotaxis), OFT morphogenesis (cell morphogenesis involved in differentiation), and endocardial cushion formation (extracellular matrix organization, extracellular structure organization) (Figure 5C).
Figure 5. Transcriptome analysis of the embryonic outflow tract (OFT) in Nkx2–5∆enh/∆enh vs. wildtype mice.

(A) Volcano plot of all annotated genes in Nkx2–5∆enh/∆enh vs wildtype OFT. Significantly upregulated genes are highlighted in red and downregulated genes are in blue (adjusted p value < 0.1) with the verticle gray dashed line representing the boundary for identification of up- or down-regulated genes. The horizontal grey dashed line represents significance. Key genes (Nkx2–5, Pitx2 and Sema3c) in OFT development are colored in green. (B) Heatmap of differentially expressed genes (DEGs, adjusted p value < 0.1) between Nkx2–5∆enh/∆enh vs. wildtype OFT (n = 5 and 4, respectively). (C) Subnetwork analysis by Enrichr-KG. All DEGs from panel B were analyzed. The MGI Mammalian Phenotype and Gene Ontology Biological Process gene set libraries were selected for the analysis. Enrichment of phenotypes and descriptive biological processes are shown. (D) Validation qPCR of key genes in the OFT at E12.5. Genes are grouped based on assigned biological process. *p < 0.05, **p < 0.01, ****p < 0.0001 by Student’s t-test. Data are presented as mean ± SD. n = 6 per group. SHF, second heart field; VSMC, vascular smooth muscle cell; NCC, neural crest cell; ECM, extracellular matrix.
Due to the inherent variability of embryonic tissues, we also probed our RNAseq dataset using a threshold criterion of p value < 0.05 and base mean > 30, to expand our pool of candidate DEGs to be confirmed by RT-qPCR. A heatmap of the 1029 candidate DEGs again shows excellent genotype-dependent segregation including 469 upregulated and 560 downregulated transcripts in the Nkx2–5Δenh/Δenh OFT compared to WT littermates (Figure S3A). Functional enrichment analysis of the candidate DEGs using Enrichr revealed biological processes that were significantly downregulated in the Nkx2–5Δenh/Δenh OFT were implicated in OFT morphogenesis (cell morphogenesis involved in differentiation, outflow tract septum morphogenesis, heart development), endocardial cushion formation (extracellular matrix organization, epithelial cell development, regulation of BMP signaling pathway, regulation of Wnt signaling pathway, positive regulation of epithelial to mesenchymal transition), NCC recruitment and development (neural crest cell migration, neural crest cell development, positive regulation of cell migration), smooth muscle cell development (smooth muscle cell contraction), and determination of bilateral symmetry (Figure S3B and File S1). The biological processes that were significantly upregulated in the Nkx2–5Δenh/Δenh OFT included pathways involved in regulation of signal transduction cascades (negative regulation of ERBB signaling and MAPK cascade) and heart tube morphogenesis (embryonic heart tube morphogenesis, epithelial tube morphogenesis, and heart looping)(Figure S3B and File S2).
To validate the data obtained by RNA-seq, we performed RT-qPCR of key genes implicated in OFT morphogenesis, which include Nkx2–5, Sema3c, Pitx2, Pcsk6, Lama5, Tbx1, Isl1, Fgf10, and Tbx20. Expression levels by RT-qPCR of these genes were significantly up or downregulated in Nkx2–5Δenh/Δenh OFT in concordance with RNA-seq data (Figure 5D). Nkx2–5 is known to have an important role in mediating the transition of A-SHF progenitor cells to a ventricular myocyte lineage by down-regulating the progenitor gene program (Isl1, Fgf10, Tbx20, Bmp2).22, 46, 47 Consistent with these findings, our data shows that reduced Nkx2–5 expression in Nkx2–5Δenh/Δenh OFT myocardium is associated with a corresponding increase in Isl1, Fgf10, and Tbx20. Bmp2 expression was not significantly changed in the Nkx2–5Δenh/Δenh OFT (data not shown). The lack of change in Bmp2 was also noted in Nkx2–5flox/flox;Isl1-Ires-Cre mice,19 reflecting differences in Bmp2 regulation in global Nkx2–5 knockout (KO)46 versus selective Nkx2–5 deficiency in the A-SHF. In Nkx2–5flox/flox;Isl1-Ires-Cre mice, defects in A-SHF progenitor cell expansion into the OFT were attributable to down-regulation of R-spondin 3 (Rspo3), the secreted Wnt signaling agonist.19 In a similar fashion, the Nkx2–5Δenh/Δenh OFT showed downregulation of Wnt signaling on functional enrichment analysis and reduced expression of Rspo3 (Figure 5D).19
The persistence of the A-SHF progenitor program in mutant hearts is accompanied by a failure to upregulate critical pathways involved in OFT rotation and septation. Our transcriptomic analysis reveals that Tbx1, Pitx2, and Sema3c are downregulated in the Nkx2–5Δenh/Δenh OFT. Tbx1 is known to regulate Pitx2 expression in the OFT myocardium through a mechanism that likely involves Nkx2–5 as a cofactor.48 The combined reduction of Tbx1 and Pitx2 in the Nkx2–5Δenh/Δenh OFT is notable as compound heterozygous Tbx1+/−;Pitx2+/− mice manifest severe rotational abnormalities and conal septation defects, that include DORV, malpositioning of the aorta, and VSD.48
Our data also identify an important link between the Nkx2–5 enhancer and cardiac NCC migration and VSMC maturation pathways. Previously, it has been shown that Gata6 conditional KO mice exhibit PTA due to abnormal cardiac NCC migration/maturation as a result of reduced Sema3c production.14, 20 As Gata6 and Gata4 expression are unaffected in the Nkx2–5Δenh/Δenh OFT (Figure S4), the reduction in Sema3c can be attributed to reduced levels of Tbx1 and Nkx2–5, which have both been shown to regulate Sema3c expression in the OFT myocardium.19, 49, 50 Accompanied by the reduction in Sema3c, there was decreased expression in VSMC maturation markers, Acta2 and Myh11 (Figure 5D).51 These results suggest that defects in migration of cardiac NCCs and maturation of VSMCs contribute to the aorticopulmonary septation defects in the Nkx2–5Δenh/Δenh OFT.
To investigate whether Pitx2 influences Sema3c expression in the OFT myocardium, we analyzed a publicly available scRNA-seq dataset using a Pitx2hd−/− model, which knocks out the Pitx2 homeodomain region rendering it inactive.30 Within the dataset, the OFT cardiomyocyte cluster was identified (Figure 6A, Figure S5, File S3), and expression of key genes implicated in OFT morphogenesis and cell-cell communication was analyzed. Violin plots (Figure 6B) and DEG analysis (File S4) showed expected reduction in mutant Pitx2 transcript with no significant change in expression of Sema3c between Pitx2hd−/− and control OFT cardiomyocyte clusters. CellChat analysis further confirmed that interaction between OFT myocardium-derived Sema3c signal and its receptor complex Plxna2/Nrp1 expressed in VSMCs is unchanged between Pitx2hd−/− and control hearts (Figure 6C).50, 52 These results indicate that Sema3c is not transcriptionally regulated by Pitx2 in OFT myocardial cells.
Figure 6. Single cell RNA sequencing (scRNA-seq) of embryonic Pitx2 KO hearts indicates Pitx2 does not change Sema3c expression nor signaling pathway.

(A) UMAP plot of scRNA-seq from control and Pitx2hd−/− cardiac tissue at embryonic day 10.5.30 16 clusters are visualized in 2-dimensional embedding. (B) Violin plots of key genes associated with OFT septation and rotation in the OFT-CM cluster. (C) Circle plots showing cell-cell communication between different cell types for the Sema3c signaling pathway network in control and Pitx2hd−/− cardiac tissue. Control and Pitx2hd−/− plots show that most of the outgoing Sema3c signal patterns are unaltered. RV-CM, right ventricular cardiomyocytes; LA-CM, left atrial cardiomyocytes; LVS-CM, left ventricular septum cardiomyocytes; OFT-CM, outflow tract cardiomyocytes; EndoC, endocardial cells; Epi, epithelial cells; AVC-CM, atrioventricular canal cardiomyocytes; Mes, mesenchymal cells; EpiC, epicardial cells; RA-CM, right atrial cardiomyocytes; LV-CM, left ventricular cardiomyocytes; EndoV, endocardial valve cells; Immune, immune cells; SMC, smooth muscle cells; Mac, macrophages; Erythro, erythrocytes.
Chromatin accessibility of the A-SHF cardiac enhancer is GATA6 dependent in human cardiomyocytes
Observations from prior studies and our work support the hypothesis that GATA6 and NKX2–5 are within the same gene regulatory network (GRN) for OFT development. Both Gata6 conditional KO mice and Nkx2–5Δenh/Δenh mice exhibit high prevalence of PTA. GATA6 was previously shown to be a regulator of TBX1 and Sema3c expression.19, 20, 53 Analogously, Nkx2–5 enhancer deletion reduces Tbx1 and Sema3c expression in the OFT. However, Gata6 expression remains unchanged in the Nkx2–5Δenh/Δenh OFT, suggesting that GATA6 is upstream of NKX2–5 in the regulatory axis. We hypothesize that endogenous GATA6 interacts with and regulates the A-SHF enhancer in cardiomyocytes. To test this hypothesis, we utilized published ChIP-seq and ATAC-seq datasets generated from hiPSC-CMs to inform of GATA6 occupancy as well as GATA6-dependent chromatin accessibility of the conserved A-SHF NKX2–5 enhancer.29 Analysis of the GATA6 ChIP-seq dataset revealed over-representation of GATA binding motifs and occupancy around TSS’s (Figure S6). Endogenous GATA6 displayed enriched occupancy at the A-SHF NKX2–5 enhancer region corresponding with conserved GATA-binding sites (FDR-adjusted p value < 0.05 for differential peak-calling analysis) (Figure 7A). Analysis of ATAC-seq datasets in GATA6 isogenic WT, heterozygous, and homozygous KO hiPSC-CMs showed that chromatin accessibility of the enhancer region was dependent on GATA6 gene dosage (Figure S7, Figure 7A,B). There was a significant correlation between GATA6 genotype and normalized counts of ATAC-seq peaks in the cardiac enhancer region (Pearson correlation coefficient, r = – 0.9796075, p = 0.0006). Consistent with these findings, NKX2–5 expression levels were shown to be reduced in GATA6 homozygous KO hiPSC-CMs after day 4 of differentiation, and at day 12, NKX2–5 expression was reduced in a graded fashion based on GATA6 heterozygous and homozygous KO genotype.29 To verify interaction between endogenous Gata6, which is robustly expressed throughout the E12.5 WT heart (Figure S4),54 and the cardiac enhancer site during heart development, we isolated whole hearts from E12.5 WT mice and performed ChIP-qPCR. Endogenous Gata6 demonstrated occupancy of the enhancer region at both conserved GATA-binding sites (GS1 and GS2) in the developing hearts, similar to the human cardiomyocyte result (Figure 7C).
Figure 7. Endogenous GATA6 interacts with the NKX2–5 cardiac enhancer and determines enhancer accessibility in human cardiomyocytes.

(A) Visualization of GATA6 chromatin immunoprecipitation sequencing (ChIP-seq) and assay of transposase accessible chromatin sequencing (ATAC-seq) datasets in human-induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs). ChIP-seq for GATA6 and input control in day 4 hiPSC-CMs in blue. ATAC-seq in day 4 GATA6 isogenic wildtype (GATA6+/+) and mutants (GATA6+/− and GATA6−/−) in green. The conserved cardiac enhancer region shown in red containing two GATA-binding sites in black. A GATA6 ChIP-seq peak is identified in the cardiac enhancer region (adjusted p value < 0.05 by peak-calling analysis). Differential ATAC-seq peak analysis of the cardiac enhancer region based on GATA6 genotype. (B) Pearson correlation between normalized peak count in the cardiac enhancer region and experimental groups for ATAC-seq. GATA6 mutant allele correlates with decreased ATAC-seq peak counts. (C) ChIP-quantitative PCR (ChIP-qPCR) validation of GATA6 binding in embryonic day E12.5 mouse hearts. Conserved GATA-binding sites (red) in the cardiac enhancer region on the mouse mm10 assembly (left panel). Independent ChIP-qPCR for two GATA-binding sites, GS1 and GS2, in E12.5 mouse hearts showing GATA6 occupancy for both sites. **P < 0.01 by Student’s t-test. Data are presented as mean ± SD. n = 5 per group.
Discussion
Defining the GRN of the OFT is essential to understand how transcriptional programs orchestrate conotruncal morphogenesis. Here we define a key component of the OFT GRN that utilizes an A-SHF specific enhancer that is GATA6 dependent and regulates regional Nkx2–5 expression. Deletion of the minimal enhancer region recapitulates conotruncal defects seen in patients and mouse models of GATA6, NKX2–5, and TBX1 loss-of-function.14, 20, 55 Transcriptomic analysis of the mutant OFT reveals disruption of key transcriptional programs that coordinate rotation, septation, and maturation.
Through a single cardiac enhancer, we have linked major transcriptional programs that are associated with conotruncal defects. Our data shows that GATA6 occupies the NKX2–5 A-SHF enhancer and dictates its open probability in a dose dependent fashion in hiPSC-CMs. Nkx2–5 enrichment in the A-SHF, dictated by GATA6-dependent enhancer activation, simultaneously downregulates the Isl1-dependent progenitor cell program and upregulates OFT morphogenetic programs that include Tbx1, Pitx2, and Sema3c (Figure 8). Precise regulation of Isl1 is critical for appropriate expansion of the A-SHF progenitor pool. Isl1 is essential for specification and migration of cardiac progenitors in the splanchnic mesoderm to populate the A-SHF through activation of Fgf10 and Tbx20.22, 47, 56, 57 Consequently, mice deficient in Isl1 fail to form an OFT and RV.58 Nkx2–5 is an important repressor of Isl1, and mediates the transition to a myocardial lineage. Nkx2–5 overexpression in mouse ESC-derived cardiomyocytes causes downregulation of Isl1, Fgf10, and Tbx20.47 In reciprocal fashion, germline Nkx2–5 KO mice show upregulation of Isl1 and Fgf10, leading initially to progenitor cell overspecification followed by defective proliferation of A-SHF myocardial precursors.46 In agreement with these findings, Nkx2–5Δenh/Δenh mice display persistent upregulation of Isl1, Fgf10, and Tbx20 in the OFT. Nkx2–5Δenh/Δenh mice also show downregulation of Wnt signaling by functional enrichment analysis and reduced expression of Rspo3, similar to conditional Nkx2–5flox/flox;Isl1-Ires-Cre mice.19 The importance of Rspo3 in OFT development was shown using conditional Rspo3f/f;Isl1-Ires-Cre mice that recapitulate the DORV phenotype. In addition, transgenic Rspo3 overexpression in Nkx2–5flox/flox;Isl1-Ires-Cre mice was able to rescue the PTA phenotype.19 Our data demonstrate that precise temporo-spatial regulation of Nkx2–5 expression through activation of the A-SHF enhancer by GATA6 is essential to coordinate the shift from an Isl1-dependent progenitor cell program to an Nkx2–5-dependent, ventricular myocyte morphogenetic program.
Figure 8. Working model of the Gata6-dependent Nkx2–5 A-SHF enhancer gene regulatory network of the outflow tract (OFT).

GATA6 regulates OFT septation and rotation programs via direct regulation of the Nkx2–5 A-SHF enhancer. A-SHF, anterior second heart field; NCC, neural crest cell.
Transcriptomic analysis of the Nkx2–5∆enh/∆enh OFT supports a gene regulatory hierarchy where Nkx2–5 enrichment in the A-SHF turns on the Tbx1-dependent gene program, which includes Pitx2 and Sema3c (Figure 8). Our findings are consistent with prior work that has shown that Tbx1 selective deletion in Nkx2–5 expressing myocardial cells (Nkx2–5Cre/+;Tbx1flox/-) is sufficient to reproduce PTA.18 Our data extend these findings by placing Tbx1 downstream of Nkx2–5 and Gata6 in the regulatory axis in A-SHF myocardial cells. Tbx1 has been shown to regulate the expression of Pitx2 and Sema3c in the OFT myocardium.48, 49 To validate genetic interaction of Tbx1 and Pitx2 in vivo, Campione, Morrow, and colleagues generated double heterozygous mice (Tbx1+/−;Pitx2+/−), which displayed cyanotic conotruncal disease (DORV, malpositioning of the aorta, pulmonary trunk stenosis, and VSD) at a higher severity level and penetrance than in single heterozygous mice.48 Therefore, the combined reduction in Tbx1 and Pitx2 in Nkx2–5Δenh/Δenh mice may be sufficient to explain the high penetrance of cyanotic conotruncal disease. A putative enhancer for Pitx2, which contains a T-half site and a nearby Nkx2–5 binding motif, was shown to be synergistically responsive to Tbx1 and Nkx2–5 using heterologous expression systems.48
Our analysis of the Pitx2hd−/− scRNA-seq dataset30 showed that Pitx2 does not regulate Sema3c expression or Sema3c-dependent crosstalk from OFT myocardial cells. This finding is in agreement with previous work showing that cardiac NCC migration appears normal in Pitx2 KO mice.59 Our data indicates that Tbx1 likely represents the lowest transcriptional branchpoint in the GATA6-Nkx2–5-Tbx1 axis that can impact both Pitx2 and Sema3c expression, which explains why the main defect in Pitx2 KO mice is in OFT rotation rather than aorticopulmonary septation.24 Taken together, our data support a GATA6-NKX2–5-Tbx1-Pitx2c/Sema3c regulatory axis that is localized to the OFT myocardium through the Nkx2–5 A-SHF enhancer.
We also show that Pcsk6/Pace4 and Lama5 are also under the regulatory control of the A-SHF enhancer in the OFT. Pcsk6 is an endoprotease that regulates TGFβ-related signaling pathways, including BMP signaling, via proteolytic cleavage that converts inactive precursors to their active forms.42 Pcsk6 KO mice exhibit left/right patterning defects and severe cardiac malformations, including DORV, PTA, VSD, and dextrocardia. Although the mechanisms of conotruncal malformations were not explored, the authors hypothesize that Pcsk6 may regulate the autoinduction of nodal signaling, which has been shown to positively regulate Pitx2 expression. In addition, PCSK6 was identified as a candidate gene using genome-wide linkage analysis using MOD score analysis in families with mixed CHD phenotypes, including atrial septal defects, VSD, TOF, TGA, and DORV.45 Lama5 encodes Laminin α5, a major component of basement membranes, that has important roles in developmental processes, including neural tube closure, digit septation, and glomerulogenesis.60 Lama5 was identified in a mutational screen for CHD genes that also impact kidney development.43, 44 Lama5 mutants displayed DORV. Although the exact mechanisms by which Pcsk6 and Lama5 interact with Tbx1, Pitx2, and Sema3c in the OFT will need to be defined, our work now places all of these critical conotruncal modulatory genes under the regulatory landscape of the A-SHF enhancer.
The high penetrance of PTA and DORV phenotypes in Nkx2–5Δenh/Δenh and conditional Nkx2–5flox/flox;Isl1-Ires-Cre mouse models demonstrates that selective reduction of Nkx2–5 expression in the A-SHF markedly increases vulnerability to conotruncal defects.19 These observations highlight the importance of maintaining Nkx2–5 levels at an optimal balance between the A-SHF and the first heart field, which gives rise to the left ventricle and parts of the interventricular septum, to ensure normal OFT rotational alignment and aorticopulmonary septation. These findings are particularly important in light of our data that shows NKX2–5 A-SHF enhancer accessibility is highly dependent on GATA6 gene dosage, providing an important mechanistic link between GATA6 loss-of-function mutations and suboptimal NKX2–5 expression levels in the A-SHF that predispose to conotruncal defects.
Conduction defects and subsequent mechanical dyssynchrony in Nkx2–5Δenh/Δenh mice are due to hypoplasia of the right PFN. Normal PFN development occurs through several stages.40 The first stage involves early specification of conductive cells from cardiac progenitor cells that create a specialized conduction system scaffold.40 Subsequently, the conduction system expands through two phases of recruitment from the trabecular myocardium.40 The first wave of myocardial cell recruitment occurs during early fetal stages where bipotent trabecular myocytes exhibit progressive restriction toward a Purkinje lineage, and the second wave occurs in late fetal stages, when a high proportion of trabecular myocytes commit to a Purkinje lineage. The reduction of Gja5/Cx40 expression in embryonic trabecular myocytes in E12.5 Nkx2–5Δenh/Δenh hearts points to a defect in the first and likely second wave of recruitment. The absence of the right bundle branch in some mutant mice suggests that early specification of the right conduction system scaffold may be adversely affected in Nkx2–5Δenh/Δenh hearts.
The high penetrance of severe conotruncal abnormalities and right ventricular conduction system defects underscores the critical importance of this A-SHF enhancer for normal cardiac development. It is important to note that this enhancer site is located within a gene regulatory island that juxtaposes activating and repressing elements, which has been proposed to be essential for precise temporo-spatial control of Nkx2–5 expression during cardiogenesis.27 This hypothesis has recently been supported by the work of Zhang and colleagues, who knocked out a large 4.3 kb super-enhancer site, termed U1, which includes the A-SHF enhancer.61 Notably, U1 knockout mice displayed no phenotype and no change in Nkx2–5 expression. It is likely that the large-scale deletion of both activating and repressing regulatory elements resulted in a net neutral effect on Nkx2–5 expression. Furthermore, the authors identified another large 9.4 kb super-enhancer site, termed U2, that was frameshifted into closer proximity to the Nkx2–5 promoter through U1 deletion, possibly resulting in a compensatory effect. This was confirmed when U1 and U2 were both deleted, which caused reduced expression of Nkx2–5 in the E7.5 heart and embryonic lethality after E9.5 due to severe cardiac defects involving the atrioventricular canal, OFT, and RV, mimicking the Nkx2–5 germline null phenotype. In light of these results, our findings using the Nkx2–5∆enh model highlights the importance of applying a tailored approach to studying the role of individual enhancer sites on regional heart development.
In conclusion, our study delineates how a non-coding regulatory element acts cooperatively with coding transcriptional elements to coordinate OFT morphogenesis. The Nkx2–5Δenh/Δenh mouse model has functioned as an essential tool to decode the OFT GRN, unifying multiple transcriptional programs into a single GATA6-NKX2–5-Tbx1-Pitx2c/Sema3c regulatory axis that underlies cyanotic conotruncal defects. It will be of significant interest to determine if human variations within the minimal NKX2–5 A-SHF enhancer are associated with conotruncal defects. Our findings add to the growing body of evidence that implicates non-coding DNA elements as essential regulators of normal cardiac morphogenesis and underscores the need to validate non-coding DNA elements in vivo to fully decipher their role in governing major developmental events.
Supplementary Material
Clinical Perspective.
What Is New?
Loss of an Nkx2–5 anterior second heart field enhancer is sufficient to cause a broad spectrum of conotruncal defects and right bundle branch block in a murine model.
Our findings uncover a critical genomic mechanism that links multiple transcriptional programs implicated in conotruncal defects, such as NKX2–5, GATA6, and TBX1, with major rotation and septation gene programs into a single gene regulatory network.
What Are the Clinical Implications?
This study highlights the importance of defining the contribution of non-coding DNA elements to congenital heart disease (CHD).
Our findings support the expansion of genetic screening of CHD families in a research context to identify disease causing variants in select non-coding DNA elements that have impact on cardiac morphogenesis.
Comprehensive multimodality assessment of our mouse model provides important insights into the morphogenetic basis of conotruncal defects in CHD patients.
Acknowledgements
We are grateful to Mark Alu and Dani Hanks of the Experimental Pathology Research Laboratory at NYU Langone’s Laura and Isaac Perlmutter Cancer Center for their expert technical support, to the Microscopy Core at NYU Langone Health for scanning microscopy, to Arun Sharma, Steven Robert DePalma, and Lauren K Wasson for providing the GATA6 ChIP-seq and ATAC-seq datasets, and to Wilson Young for initial assistance in generation of Nkx2–5 cardiac enhancer mutant mice.
Sources of Funding
This work was supported by funding from the National Institutes of Health R01HL132073 and R01HL165130, a Fondation Leducq Transatlantic Network of Excellence Award, and generous support from the Ronald Altman family to Dr Park, American Heart Association grant 20POST35080180 to Dr Yamaguchi, National Institutes of Health grant T32-HL098129 to Dr Chang, Cancer Center Support Grant P30CA016087 (partially) to the Experimental Pathology Research Laboratory at NYU Langone, National Institutes of Health / National Center for Research Resources S10 RR023704-01A1 to the resource at the Microscopy Core at NYU Langone, and National Institutes of Health / National Cancer Institute 5P30CA016087 and National Institutes of Health / National Institute of Biomedical Imaging and Bioengineering Biomedical Technology Resource Center Grant P41-EB017183 to Preclinical Imaging Laboratory, a shared resource used for transthoracic echocardiography.
Non-standard Abbreviations and Acronyms
- AoV
aortic valve
- APIC
anterior pulmonary intercalated cushion
- A-SHF
anterior second heart field
- ATAC-seq
assay of transposase accessible chromatin sequencing
- CHD
congenital heart disease
- ChIP-seq
chromatin immunoprecipitation sequencing
- Cx40
connexin 40
- DNVs
de novo variants
- DORV
double outlet right ventricle
- d-TGA
dextro-transposition of great arteries
- ECG
electrocardiography
- ECM
extracellular matrix
- ERBB
erythroblastic leukemia viral oncogene homologue
- GRN
gene regulatory network
- hiPSC-CMs
human-induced pluripotent stem cell-derived cardiomyocytes
- ISC
inferior septal cushion
- KO
knockout
- LA
left atrium
- LV
left ventricle
- LVOT
left ventricular outflow tract
- MAPK
mitogen-activated protein kinase
- NCCs
neural crest cells
- OFT
outflow tract
- PAIC
posterior aortic intercalated cushion
- PFN
Purkinje fiber network
- PTA
persistent truncus arteriosus
- PV
pulmonary valve
- RA
right atrium
- RV
right ventricle
- RVOT
right ventricular outflow tract
- SHF
second heart field
- scRNA-seq
single cell RNA sequencing
- SSC
superior septal cushion
- TOF
tetralogy of Fallot
- TSS
transcription start site
- TTE
transthoracic echocardiography
- VSD
ventricular septal defect
- VSMCs
vascular smooth muscle cells
- WT
wildtype
Footnotes
Disclosures
None.
References
- 1.Collaborators GBDCHD. Global, regional, and national burden of congenital heart disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Child Adolesc Health. 2020;4:185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stavsky M, Robinson R, Sade MY, Krymko H, Zalstein E, Ioffe V, Novack V and Levitas A. Elevated birth prevalence of conotruncal heart defects in a population with high consanguinity rate. Cardiol Young. 2017;27:109–116. [DOI] [PubMed] [Google Scholar]
- 3.Egbe A, Uppu S, Stroustrup A, Lee S, Ho D and Srivastava S. Incidences and sociodemographics of specific congenital heart diseases in the United States of America: an evaluation of hospital discharge diagnoses. Pediatr Cardiol. 2014;35:975–82. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman JI and Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–900. [DOI] [PubMed] [Google Scholar]
- 5.Oster ME, Lee KA, Honein MA, Riehle-Colarusso T, Shin M and Correa A. Temporal trends in survival among infants with critical congenital heart defects. Pediatrics. 2013;131:e1502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laforest B and Zaffran S. Genetics of conotruncal anomalies. Surgery of Conotruncal Anomalies. 2016:607–621. [Google Scholar]
- 7.Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragan JD, Shin M and Correa A. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr Cardiol. 2011;32:1147–57. [DOI] [PubMed] [Google Scholar]
- 8.Kim DS, Kim JH, Burt AA, Crosslin DR, Burnham N, Kim CE, McDonald-McGinn DM, Zackai EH, Nicolson SC, Spray TL, Stanaway IB, Nickerson DA, Heagerty PJ, Hakonarson H, Gaynor JW and Jarvik GP. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J Thorac Cardiovasc Surg. 2016;151:1147–51 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung WC, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, Mital S, Russell MW, Gaynor JW, Deanfield J, Giardini A, Porter GA Jr., Srivastava D, Lo CW, Shen Y, Watkins WS, Yandell M, Yost HJ, Tristani-Firouzi M, Newburger JW, Roberts AE, Kim R, Zhao H, Kaltman JR, Goldmuntz E, Chung WK, Seidman JG, Gelb BD, Seidman CE, Lifton RP and Brueckner M. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, Zhou J, Qi H, Patel N, DePalma SR, Parfenov M, Homsy J, Gorham JM, Manheimer KB, Velinder M, Farrell A, Marth G, Schadt EE, Kaltman JR, Newburger JW, Giardini A, Goldmuntz E, Brueckner M, Kim R, Porter GA Jr., Bernstein D, Chung WK, Srivastava D, Tristani-Firouzi M, Troyanskaya OG, Dickel DE, Shen Y, Seidman JG, Seidman CE and Gelb BD. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat Genet. 2020;52:769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devriendt K, Eyskens B, Swillen A, Dumoulin M, Gewillig M and Fryns JP. The incidence of a deletion in chromosome 22Q11 in sporadic and familial conotruncal heart disease. Eur J Pediatr. 1996;155:721. [DOI] [PubMed] [Google Scholar]
- 12.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N and Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–73. [DOI] [PubMed] [Google Scholar]
- 13.Digilio M, Marino B, Capolino R and Dallapiccola B. Clinical manifestations of Deletion 22q11.2 syndrome (DiGeorge/Velo-Cardio-Facial syndrome). Images Paediatr Cardiol. 2005;7:23–34. [PMC free article] [PubMed] [Google Scholar]
- 14.Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, Takahashi T, Matsuoka R and Yamagishi H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci U S A. 2009;106:13933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Ji W, Wang J, Zhao P, Guo Y, Xu R, Chen S and Sun K. Identification of two novel GATA6 mutations in patients with nonsyndromic conotruncal heart defects. Mol Med Rep. 2014;10:743–8. [DOI] [PubMed] [Google Scholar]
- 16.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J and Kugler JD. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McElhinney DB, Geiger E, Blinder J, Benson DW and Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–5. [DOI] [PubMed] [Google Scholar]
- 18.Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA and Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–27. [DOI] [PubMed] [Google Scholar]
- 19.Cambier L, Plate M, Sucov HM and Pashmforoush M. Nkx2–5 regulates cardiac growth through modulation of Wnt signaling by R-spondin3. Development. 2014;141:2959–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lepore JJ, Mericko PA, Cheng L, Lu MM, Morrisey EE and Parmacek MS. GATA-6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. J Clin Invest. 2006;116:929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waardenberg AJ, Ramialison M, Bouveret R and Harvey RP. Genetic networks governing heart development. Cold Spring Harb Perspect Med. 2014;4:a013839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe Y, Zaffran S, Kuroiwa A, Higuchi H, Ogura T, Harvey RP, Kelly RG and Buckingham M. Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2–5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc Natl Acad Sci U S A. 2012;109:18273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang X, Wang G, Lin L, Lowe J, Zhang Q, Bu L, Chen Y, Chen J, Sun Y and Evans SM. HCN4 dynamically marks the first heart field and conduction system precursors. Circ Res. 2013;113:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bajolle F, Zaffran S, Kelly RG, Hadchouel J, Bonnet D, Brown NA and Buckingham ME. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ Res. 2006;98:421–8. [DOI] [PubMed] [Google Scholar]
- 25.Ma HY, Xu J, Eng D, Gross MK and Kioussi C. Pitx2-mediated cardiac outflow tract remodeling. Dev Dyn. 2013;242:456–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA and Raper JA. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128:3061–70. [DOI] [PubMed] [Google Scholar]
- 27.Lien CL, Wu C, Mercer B, Webb R, Richardson JA and Olson EN. Control of early cardiac-specific transcription of Nkx2–5 by a GATA-dependent enhancer. Development. 1999;126:75–84. [DOI] [PubMed] [Google Scholar]
- 28.Reecy JM, Li X, Yamada M, DeMayo FJ, Newman CS, Harvey RP and Schwartz RJ. Identification of upstream regulatory regions in the heart-expressed homeobox gene Nkx2–5. Development. 1999;126:839–49. [DOI] [PubMed] [Google Scholar]
- 29.Sharma A, Wasson LK, Willcox JA, Morton SU, Gorham JM, DeLaughter DM, Neyazi M, Schmid M, Agarwal R, Jang MY, Toepfer CN, Ward T, Kim Y, Pereira AC, DePalma SR, Tai A, Kim S, Conner D, Bernstein D, Gelb BD, Chung WK, Goldmuntz E, Porter G, Tristani-Firouzi M, Srivastava D, Seidman JG, Seidman CE and Pediatric Cardiac Genomics C. GATA6 mutations in hiPSCs inform mechanisms for maldevelopment of the heart, pancreas, and diaphragm. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill MC, Kadow ZA, Li L, Tran TT, Wythe JD and Martin JF. A cellular atlas of Pitx2-dependent cardiac development. Development. 2019;146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll KJ, Makarewich CA, McAnally J, Anderson DM, Zentilin L, Liu N, Giacca M, Bassel-Duby R and Olson EN. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc Natl Acad Sci U S A. 2016;113:338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neeb Z, Lajiness JD, Bolanis E and Conway SJ. Cardiac outflow tract anomalies. Wiley Interdiscip Rev Dev Biol. 2013;2:499–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calder L, Van Praagh R, Van Praagh S, Sears WP, Corwin R, Levy A, Keith JD and Paul MH. Truncus arteriosus communis. Clinical, angiocardiographic, and pathologic findings in 100 patients. Am Heart J. 1976;92:23–38. [DOI] [PubMed] [Google Scholar]
- 34.Gedikbasi A, Oztarhan K, Gul A, Sargin A and Ceylan Y. Diagnosis and prognosis in double-outlet right ventricle. Am J Perinatol. 2008;25:427–34. [DOI] [PubMed] [Google Scholar]
- 35.Martin PS, Kloesel B, Norris RA, Lindsay M, Milan D and Body SC. Embryonic Development of the Bicuspid Aortic Valve. J Cardiovasc Dev Dis. 2015;2:248–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savolainen SM, Foley JF and Elmore SA. Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol Pathol. 2009;37:395–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaher RM and Deuchar DC. The electrocardiogram in complete transposition of the great vessels. Br Heart J. 1966;28:265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krongrad E, Ritter DG, Weidman WH and DuShane JW. Hemodynamic and anatomic correlation of electrocardiogram in double-outlet right ventricle. Circulation. 1972;46:995–1004. [DOI] [PubMed] [Google Scholar]
- 39.Shekhar A, Lin X, Liu FY, Zhang J, Mo H, Bastarache L, Denny JC, Cox NJ, Delmar M, Roden DM, Fishman GI and Park DS. Transcription factor ETV1 is essential for rapid conduction in the heart. J Clin Invest. 2016;126:4444–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choquet C, Kelly RG and Miquerol L. Nkx2–5 defines distinct scaffold and recruitment phases during formation of the murine cardiac Purkinje fiber network. Nat Commun. 2020;11:5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evangelista JE, Xie Z, Marino GB, Nguyen N, Clarke DJB and Ma’ayan A. Enrichr-KG: bridging enrichment analysis across multiple libraries. Nucleic Acids Res. 2023;51:W168–W179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Constam DB and Robertson EJ. SPC4/PACE4 regulates a TGFbeta signaling network during axis formation. Genes Dev. 2000;14:1146–55. [PMC free article] [PubMed] [Google Scholar]
- 43.Gabriel GC, Pazour GJ and Lo CW. Congenital Heart Defects and Ciliopathies Associated With Renal Phenotypes. Front Pediatr. 2018;6:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X, Lo CW and Pazour GJ. Genetic link between renal birth defects and congenital heart disease. Nat Commun. 2016;7:11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flaquer A, Baumbach C, Pinero E, Garcia Algas F, de la Fuente Sanchez MA, Rosell J, Toquero J, Alonso-Pulpon L, Garcia-Pavia P, Strauch K and Heine-Suner D. Genome-wide linkage analysis of congenital heart defects using MOD score analysis identifies two novel loci. BMC Genet. 2013;14:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prall OW, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet H, Stennard FA, Wise N, Schaft D, Wolstein O, Furtado MB, Shiratori H, Chien KR, Hamada H, Black BL, Saga Y, Robertson EJ, Buckingham ME and Harvey RP. An Nkx2–5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorn T, Goedel A, Lam JT, Haas J, Tian Q, Herrmann F, Bundschu K, Dobreva G, Schiemann M, Dirschinger R, Guo Y, Kuhl SJ, Sinnecker D, Lipp P, Laugwitz KL, Kuhl M and Moretti A. Direct nkx2–5 transcriptional repression of isl1 controls cardiomyocyte subtype identity. Stem Cells. 2015;33:1113–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowotschin S, Liao J, Gage PJ, Epstein JA, Campione M and Morrow BE. Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development. 2006;133:1565–73. [DOI] [PubMed] [Google Scholar]
- 49.Theveniau-Ruissy M, Dandonneau M, Mesbah K, Ghez O, Mattei MG, Miquerol L and Kelly RG. The del22q11.2 candidate gene Tbx1 controls regional outflow tract identity and coronary artery patterning. Circ Res. 2008;103:142–8. [DOI] [PubMed] [Google Scholar]
- 50.Kodo K, Shibata S, Miyagawa-Tomita S, Ong SG, Takahashi H, Kume T, Okano H, Matsuoka R and Yamagishi H. Regulation of Sema3c and the Interaction between Cardiac Neural Crest and Second Heart Field during Outflow Tract Development. Sci Rep. 2017;7:6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Chen W, Li W, Li Y, Priest JR, Zhou B, Wang J and Zhou Z. Single-Cell RNA-Seq of the Developing Cardiac Outflow Tract Reveals Convergent Development of the Vascular Smooth Muscle Cells. Cell Rep. 2019;28:1346–1361 e4. [DOI] [PubMed] [Google Scholar]
- 52.Brown CB, Feiner L, Lu MM, Li J, Ma X, Webber AL, Jia L, Raper JA and Epstein JA. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001;128:3071–80. [DOI] [PubMed] [Google Scholar]
- 53.Jiang X, Li T, Liu S, Fu Q, Li F, Chen S, Sun K, Xu R and Xu Y. Variants in a cis-regulatory element of TBX1 in conotruncal heart defect patients impair GATA6-mediated transactivation. Orphanet J Rare Dis. 2021;16:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maitra M, Koenig SN, Srivastava D and Garg V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res. 2010;68:281–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang E, Hong N, Chen S, Fu Q, Li F, Yu Y and Sun K. Targeted sequencing identifies novel GATA6 variants in a large cohort of patients with conotruncal heart defects. Gene. 2018;641:341–348. [DOI] [PubMed] [Google Scholar]
- 56.Singh MK, Christoffels VM, Dias JM, Trowe MO, Petry M, Schuster-Gossler K, Burger A, Ericson J and Kispert A. Tbx20 is essential for cardiac chamber differentiation and repression of Tbx2. Development. 2005;132:2697–707. [DOI] [PubMed] [Google Scholar]
- 57.Golzio C, Havis E, Daubas P, Nuel G, Babarit C, Munnich A, Vekemans M, Zaffran S, Lyonnet S and Etchevers HC. ISL1 directly regulates FGF10 transcription during human cardiac outflow formation. PLoS One. 2012;7:e30677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cai C-L, Liang X, Shi Y, Chu P-H, Pfaff SL, Chen J and Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Developmental cell. 2003;5:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu C, Liu W, Palie J, Lu MF, Brown NA and Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129:5081–91. [DOI] [PubMed] [Google Scholar]
- 60.Miner JH and Li C. Defective glomerulogenesis in the absence of laminin alpha5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol. 2000;217:278–89. [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, Li CC, Li X, Liu Y, Wang Q, Zhang G, Xiong H, He A and Ai S. Novel enhancers conferring compensatory transcriptional regulation of Nkx2–5 in heart development. iScience. 2023;26:106509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing (RNA-seq) data are deposited in Gene Expression Omnibus (GSE227361). The chromatin immunoprecipitation sequencing (ChIP-seq) for GATA6 and assay of transposase accessible chromatin sequencing (ATAC-seq) data in hiPSC-CMs, and single cell RNA-seq (scRNA-seq) data from control and Pitx2hd−/− mouse hearts were obtained from previously published datasets.29, 30