

Abstract

Globally, the pharmaceutical industry has been facing challenges from nitroso drug substance-related impurities (NDSRIs). In the current study, we synthesized and developed a rapid new UPLC-MS/MS method for the trace-level quantification of ciprofloxacin NDSRIs and a couple of N-nitroso impurities simultaneously. (Q)-SAR methodology was employed to assess and categorize the genotoxicity of all ciprofloxacin N-nitroso impurities. The projected results were positive, and the cohort of concern (CoC) for all three N-nitroso impurities indicates potential genotoxicity. AQbD-driven I-optimal mixture design was used to optimize the mixture of solvents in the method. The chromatographic resolution was accomplished using an Agilent Poroshell 120 Aq-C18 column (150 mm × 4.6 mm, 2.7 μm) in isocratic elution mode with 0.1% formic acid in a mixture of water, acetonitrile, and methanol in the ratio of 475:500:25 v/v/v at a flow rate of 0.5 mL/min. Quantification was carried out using triple quadrupole mass detection with electrospray ionization (ESI) in a multiple reaction monitoring technique. The finalized method was validated successfully, affording ICH guidelines. All N-nitroso impurities revealed excellent linearity over the concentration range of 0.00125–0.0250 ppm. The Pearson correlation coefficient of each N-nitroso impurity was >0.999. The method accuracy recoveries ranged from 93.98 to 108.08% for the aforementioned N-nitrosamine impurities. Furthermore, the method was effectively applied to quantify N-nitrosamine impurities simultaneously in commercially available formulated samples, with its efficiency recurring at trace levels. Thus, the current method is capable of determining the trace levels of three N-nitroso ciprofloxacin impurities simultaneously from the marketed tablet dosage forms for commercial release and stability testing.
1. Introduction
Ciprofloxacin (COX) is chemically 1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid and the molecular formula and molecular weight are C17H18FN3O3 and 331.34 Da, respectively. It is an antibiotic agent and belongs to the class of fluoroquinolones. It is slightly soluble in ethanol and methanol but insoluble in ether, acetone, and chloroform. It is used to treat the urinary tract and pneumonia bacterial infections.1 In addition to treating bacterial infections, it can be used to treat joint, bone, skin, sexually transmitted infections, typhoid fever, lower respiratory tract infections, plague, salmonella, and anthrax. COX is an appropriate treatment for patients with predisposing factors for Gram-negative infections and is also used for chronic bacterial prostatitis.2,3 FDA-approved COX ophthalmic solution is used for treating corneal ulcers and conjunctivitis caused by susceptible strains.
COX pharmacokinetics exhibited interethnic variability, with Asians exhibiting an increased bioavailability compared to Mexicans and Caucasians. It is the most extensively utilized fluoroquinolone antibiotic in treating Pseudomonas aeruginosa.4 However, in spite of the significant amount of research designed to develop COX powder for inhalation, no formulations are commercially available.5−7 An antibacterial effect of COX-coated poly(lactic acid)-based 3D discs was demonstrated against Escherichia coli. It is, therefore, possible in the future to evaluate the antibacterial activity of polylactic acid-based COX-containing 3D products.8,9
In recent years, pharmaceutical companies and regulatory agencies have placed increasing emphasis on controlling genotoxic and cohort-of-concern impurities in drug substances and products. During the synthetic process, certain potential genotoxic impurities (PGIs) can emerge from byproducts, reagents, and intermediates of the drug substance and drug products.10 Developing a highly sensitive analytical method for routine analysis is relatively stimulating since acceptable intake limits for these impurities are much lower. Furthermore, there are sensitivity concerns related to GTI assessment in the low-ppm range and reactivity or stability issues, which cause additional problems.11,12
N-Nitrosamines, which can refer to any compound bearing an N-nitroso functional group, have been labeled as a cohort of concern owing to their potential to cause cancer in humans when consumed in a significant amount over an extended period of time. A number of factors and conditions can lead to the production of nitrosamine impurities in active pharmaceutical ingredients (APIs).13 Some common conditions include using nitrosating agents in the presence of secondary or tertiary amines during the same manufacturing step or different steps, using starting materials and intermediates contaminated by processes or using raw materials containing residual nitrosamines. In-depth studies have shown that there are many more factors at stake in the formation of nitrosamine impurities besides the presence of nitrites and amines during manufacturing.14−16
Several APIs and impurities are accountable for being nitrosated owing to the variety of potential routes for forming nitrosamines, either during the synthetic route of the API, during drug product manufacturing, or during finished formulation packaging and storage. Recently, the United States Food and Drug Administration (USFDA) disclosed that some drug products contain nitrosamine impurities, such as API-derived complex nitrosamines, also known as nitrosamine drug substance-related impurities (NDSRIs),17 which are a sort of nitrosamines with structural similarities to the API and can be stimulated during the drug product’s storage or through the manufacturing of the drug substances. It has also been demonstrated through research that nitrite impurities present in excipients at parts per million levels can lead to the occurrence of NDSRIs. The presence of nitrite impurities in a number of commonly used excipients, including water, has been found to contribute to the occurrence of NDSRIs in specific drug products to a greater extent.18−20
A product recall is the result of careful pharmacovigilance, which is integral to drug regulation. It is one of the most serious reasons for product recalls to detect unacceptable levels of carcinogenic impurities.21 Some batch tablets of Chantix (varenicline) were voluntarily recalled by Pfizer in August 2021, owing to the existence of N-nitroso-varenicline detected at the above-established acceptable daily intake (ADI).22 Similarly, some batches of irbesartan tablets and irbesartan combination hydrochlorothiazide tablets were recalled by Lupin Pharmaceuticals Inc. in October 2021 due to the presence of N-nitroso-irbesartan higher than the specification limit.23 In March 2022, Pfizer recalled five lots of quinapril tablets with the Accupril brand due to the elevated levels of the N-nitroso-quinapril.24 Also, in March 20, 2022, Sandoz, Inc. recalled 13 lots of orphenadrine citrate 100 mg strength extended-release tablets due to the presence of the N-nitroso-orphenadrine impurity25 and Pfizer Canada recalled the Inderal-LA (propranolol hydrochloride) different strengths of extended capsules due to the presence of N-nitroso propranolol.26 Based on their higher potency, the animal carcinogenicity established for this structural class, such as N-nitrosamine impurities, is classified as a “cohort of concern (CoC)″. According to the International Agency for Research on Cancer (IARC), these impurities are categorized into probable or possible carcinogens. Considering a lack of compound-specific limits and the futile process for establishing acceptable intake limits (AIs) over NDSRIs,27 health regulatory bodies such as the USFDA, the International Conference on Harmonization (ICH), and the European Medicines Agency (EMA) have proposed a limit of 18 ng/day as a precautionary measure based on robust rodent carcinogenicity data from structurally related surrogates.28−30
The chemical reactions that the nitrosamine commences into a biological transformation are stimulated by the N-nitroso functional group. The nitrosamine is metabolically triggered by cytochrome P450 enzymes and forms alkyl diazonium ions through the hydroxylation of the α-carbon atom adjacent to the N-nitroso group. The alkyl diazonium ion can interact with DNA.31 COX N-nitrosamine piperizine (secondary amine) is the accountable moiety to form the N-nitrosamine and forms an alkyl diazonium ion. The possible pathway of the N-nitroso group to form an alkyl diazonium ion and its interaction with DNA is shown in Figure 1.
Figure 1.

Possible pathway of the N-nitroso group interaction with DNA via the cyclic alkyl diazonium ion.
Modifying the synthetic pathway, comprehending the entire process of manufacturing, and removing nitrosamine formation sources are all recommended for achieving regulatory compliance. The presence of nitrosamines in pharmaceutical products has emerged as a significant concern for regulatory authorities aiming to mitigate the potential carcinogenic and mutagenic impacts on patients.32 The three-step mitigation initiatives were proposed by regulatory agencies. The initial approach is derived from scientific research that demonstrates the preventive effects of commonly employed antioxidants on the production of nitrosamines. Another technique is based on the observation that the occurrence of nitrosamines can be reliably anticipated in acidic environments. As a result, under basic or neutral circumstances, the reaction kinetics are significantly reduced, and the final step is that the presence of nitrosamines is minimized.33,34
Global Substance Registration System (GSRS) is a system for registering ingredients in medicines. The system was developed through collaboration between the National Center for Advancing Translational Sciences (NCATS), the EMA, and the FDA health informatics division. The United States Pharmacopeia (USP) is collaborating with the FDA and the NCATS to use GSRS’s expertise to address cutting-edge and emerging informatics needs. A total of 41.4% of APIs and 30.2% of API impurities are predicted to be nitrosamine precursors based on data from the GSRS database.35 After removing tertiary amines as nitrosamine precursors, it is, however, acknowledged that 14.7% of APIs and 12.8% of API impurities can serve as precursors for nitrosamine.
In recent years, quality by design (QbD)-centered design of experiments (DoEs) have gained extreme reputation in the optimization, screening, and robustness of analytical method parameters. A traditional method development strategy is either a one-variable-at-a-time (OVAT) or trial-and-error (TAE) approach. There is, however, no information disclosed about the interaction results of two or more variables in either of these approaches. To understand the multiple variable interaction effects and to identify probable risks and failures, a statistical quality-by-design approach is useful.36
A simple centroid mixture design (SCMD) is a type of experimental design often used in analytical method development to optimize the conditions for a particular analytical method. SCMD is useful in optimizing the method conditions from a mixture of components to identify a suitable ratio for the intended use. A set of factors or variables that can affect the capability of an analytical method are identified, including the type of reagents and concentrations, pH, temperature, and sample preparation conditions.37 In centroid design, the experimental conditions are chosen based on a simplex-centroid design, which is a type of design that involves selecting a central point (the centroid) within a simplex, which is a geometric shape defined by the range of each factor being studied.38 The design involves creating a simplex, which is a triangle in a multidimensional space with each vertex representing a pure component. The centroid of the simplex represents the center of the design space, which is typically the average of the pure components.
A detailed literature survey indicates that as of now, no research articles have been reported on the synthesis, separation, and trace-level quantification of N-nitroso COX impurities. In the current study, we aimed to synthesize the COX-NDSRI and a couple of COX N-nitroso impurities, assess the genotoxicity using (Q)-SAR models, and develop a simple isocratic UPLC-MS/MS method and optimize using a quality by design-based design of experiments for quantifying trace-level N-nitroso COX impurities. The chemical structures of COX, COX-NDSRI, and COX-N-nitroso impurity-1 and COX-N-nitroso impurity-2 are shown in Figure 2.
Figure 2.
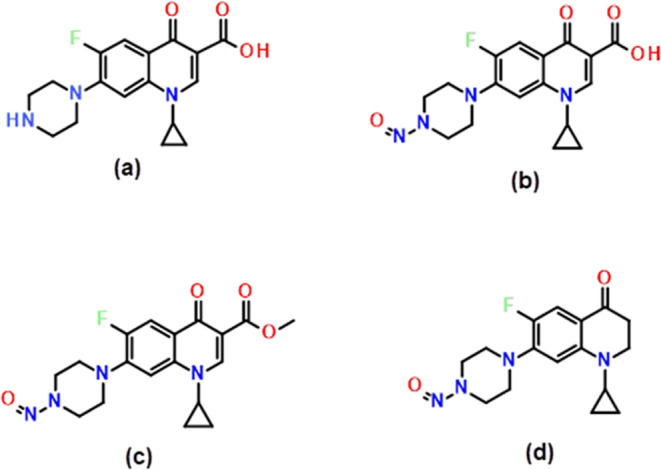
Chemical Structures of (a) COX API, (b) COX NDSRI, (c) COX N-nitroso impurity-1, and (d) COX N-nitroso impurity-2.
2. Materials and Methods
2.1. Software
The chemical structures of COX and COX-N-nitroso impurities were depicted using ChemDraw Professional 15.0. Both Derek version: Derek Nexus: 6.2.0, Knowledge: Derek KB 2022 1.0, Knowledge version: 1.0 with Knowledge Date: 06 January 6, 2022, and Sarah version: Sarah Nexus: 3.2.0, Nexus version: Nexus: 1.9, Model: Sarah Model 2022.1, were the two main (Q) SAR methods used. Bacteria are the species, and in vitro, mutagenicity is the end point. MassLynx software version 4.2 was used to control all UPLC and MS acquisition and processing parameters. Design Expert software version 13 (Stat-Ease Inc., Minneapolis) was utilized for the design of experiments and optimization data for the I-optimal centroid design. All acquisition and process parameters of LC and MS were measured using Masslynx software version 4.2 (Waters Corporation, Milford, Massachusetts).
2.2. Reagents
The reference standard for COX (purity = 99.63%) was procured from M/S Synpure laboratories (Hyderabad, India). The COX-NDSRI (purity = 99.56%), N-nitroso impurity-1 (purity = 99.58%), and N-nitroso impurity-2 (purity = 99.68%) were synthesized. COX, COX- NDSRI, N-nitroso impurity-1, and N-nitroso impurity-2 were characterized by NMR and MS. Formic acid was procured from Fisher Scientific (Waltham, MA). LC-MS-grade acetonitrile and methanol were acquired from Merck Life Sciences (Mumbai, India). Millipore Millex-GV hydrophilic PVDF 0.22 μm filters were procured from Millipore (Burlington, Massachusetts). Throughout the analysis, Millipore Milli-Q high-purity water from the purification system was used (Bedford, MA). An analytical balance from Mettler-Toledo (Model: XPE205, Columbus, Ohio) was utilized for weighing the impurity standard. The centrifuge was obtained from Eppendorf (Model:5810R, Hemburg, Germany).
2.3. Mobile Phase Preparation
To a 1000 mL dried and clean flask were added 475 mL of Milli-Q water, 500 mL of acetonitrile, and 25 mL of methanol to add 1 mL of formic acid and mix thoroughly and ultrasonicate for degassing.
2.4. Diluent
To a 1000 mL dried and clean flask were added 200 mL of Milli-Q water and 800 mL of acetonitrile to add 1 mL of formic acid and mix thoroughly and ultrasonicate for degassing. 0.1% formic acid in water and acetonitrile composition in the ratio of 20:80 v/v was used as a diluent for the preparation of standards and sample solutions.
2.5. Standard Impurity Diluted Stock Solution Preparation
The suitable amounts of each impurity were dissolved in a diluent solution to prepare a stock mixture of COX N-nitroso impurities (1000 ng/mL). The stock solution was further diluted to prepare a 10 ng/mL diluted stock solution.
2.6. Sample Preparation
A sample solution of 2 mg/mL was prepared by accurately weighing about 800 mg of COX into a 10 mL volumetric flask, dissolving in 5 mL of diluent, and sonicating for about 15 min to dissolve completely. The flask was equilibrated to room temperature and made up to the mark with the diluent. The resultant sample solution was filtered through 0.22 μm Millex-GV hydrophilic PVDF syringe filters.
2.7. Formulation Tablet Sample Preparation
10 tablets of three commercially available branded tablets of COX were ground separately using a mortar and pestle to a fine powder. An equivalent of 800 mg of COX powder was weighed into a 10 mL volumetric flask with addition of 5 mL of diluent and sonicated for 30 min to dissolve. It was made up with the diluent to the mark after bringing the flask to room temperature and centrifuged for 10 min at 4500 rpm. The resultant supernatant sample solution was filtered through 0.22 μm Millipore Millex-GV hydrophilic PVDF syringe filters.
2.8. UPLC-MS/MS Operating Conditions
The chromatographic investigation was accomplished using a Waters Acquity H-Class UHPLC system equipped with a quaternary solvent manager (QSM), sample manager- Flow Through Needle (FTN), column manager with eCord, and TUV/PDA detector. MRM analysis was conducted employing a Waters Xevo TQ-XS MS system with an ESI ion source.
An Agilent Poroshell 120 Aq-C18 column (150 mm × 4.6 mm, 2.7 μm) was used to achieve the chromatographic separations. With an acquisition duration of 8 min and a flow rate of 0.5 mL/min with isocratic mode, the mobile phase consisted of 0.1% formic acid in water, acetonitrile, and methanol in the composition of 475:500:25 (v/v/v). While the autosampler temperature was controlled at 15 °C, the column temperature was kept at 35 °C with 10 μL injection volume.
The highly sensitive and specific analytical technique multiple reaction monitoring (MRM) has become increasingly important in clinical research, drug development, and metabolomics research. MRM is highly selective for targeted analytes. By the selection of precursor and product ions specific to the analyte of interest, MRM can discriminate the targeted analyte from other molecules. The MRM technique is capable of detecting and quantifying the impurities accurately at very low concentrations.39 It helps us to ensure that the final product meets regulatory requirements.
In the current study, the MRM technique with electrospray ionization positive mode was utilized for the detection and quantification of all three N-nitroso impurities. The m/z values of 361.16/331.12 for N-nitroso COX, 375.16/345.12 for N-nitroso impurity-1, and 319.10/289.12 for N-nitroso impurity-2 were selected for the detection and quantification in MRM mode. The ESI source parameters, specifically capillary voltage (kV), desolvation gas flow, and desolvation temperature, were maintained at 3.5 kV, 900 L/h, and 450 °C, respectively. The cone voltage (kV) was set as 32 for both N-nitroso COX and N-nitroso impurity-1 and 25 for N-nitroso impurity-2. A collision energy of 20 eV was used for both N-nitroso COX and N-nitroso impurity-1 with 18 eV used for N-nitroso impurity-2. The MRM and ESI source parameters are listed in Table 1. The first 3 min of the sample solution was bypassed from the mass detector; since the COX peak elutes about 3 min to avoid its interference on trace-level nitrosamine impurity quantification and mass detector contamination.
Table 1. Optimized MRM Mass Spectrometry Conditions for N-Nitroso COX Impurities in Electron Spray Positive Ion Mode.
| compound | precursor ion (m/z) | product ion (m/z) | spectral window (min) | collision energy (eV) | cone voltage (V) | capillary voltage (kV) |
|---|---|---|---|---|---|---|
| N-nitroso COX impurity-1 | 375.16 | 345.12 | 3.40–3.65 | 20 | 32 | 3.50 |
| COX-NDSRI | 361.16 | 331.12 | 4.25–4.60 | 20 | 32 | 3.50 |
| N-nitroso COX impurity-2 | 319.10 | 289.12 | 7.10–7.50 | 18 | 25 | 3.50 |
3. Results and Discussion
The reference standards for the small and potent nitrosamines such as N-nitroso-diisopropylamine (NDIPA), N-nitroso-dimethylamine (NDMA), N-nitroso-N-methyl-4-aminobutyric acid (NMBA), N-ethyl-N-nitroso-2-propanamine (NEIPA), N-nitroso-methyl phenylamine (NMPA), N-nitroso-di-n-propylamine (NDPA), N-nitroso-di-n-butylamine (NDBA), and N-nitroso-diethylamine (NDEA) are available in compendial databases, including USP and Ph. Eur./EDQM. In the case of NDSRI, the situation is quite different from the aforementioned small nitrosamines. So far, NDSRI compendial standards are not available with any pharmacopoeias. Due to the increase in business opportunities and demand for commercial standards, suppliers have been covering more commercial standards over the past few months. However, the list of NDSRIs is quite large; therefore, it requires a lot more time to cover all of them.40 Hence, we synthesized the COX-NDSRI- and COX-related N-nitrosamine impurities from the COX drug substance.
3.1. Synthesis of COX Impurities and Corresponding N-Nitroso Impurities
3.1.1. Synthesis of Methyl 1-Cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (Compound 2)
To a stirred solution of 1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (compound 1) (1 g, 0.003 mol, 1.0 equiv) in 15 mL of methanol cooled to 0 °C was added a catalytic amount of H2SO4 (0.1 mL); then, the temperature was increased to 70 °C and maintained for 16 h. The reaction mixture was diluted with 150 mL of ethyl acetate and washed with a saturated NaHCO3 solution. The organic layer was dried over Na2SO4 and concentrated. The obtained crude was purified by silica gel (100–200 mesh) column chromatography with gradient elution mode, and compound 2 was eluted in EtOAc:hexane (30:70). The obtained yield was 85% (0.88 g). [M + H] 346.12.1H NMR (500 MHz, DMSO-d6): δ(ppm) 8.44(s, 1H), 7.76 (d, J = 14.0 Hz, 1H), 7.42 (d, J = 7.5 Hz, 1H), 3.73 (s, 3H), 3.67 (m, 1H), 3.16 (t, J = 4.5 Hz, 4H), 2.90 (t, J = 4.5 Hz, 4H), 1.25 (m, 2H), 1.11 (m, 2H).
3.1.2. Synthesis of 1-Cyclopropyl-6-fluoro-7-(piperazin-1-yl)-2,3-dihydroquinolin-4(1H)-one (Compound 3)
To a stirred solution of 1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid-compound 1 (1g, 0.003 mol, 1.0 equiv) in t-BuOH (10 vol) cooled to 0 °C was added H2SO4 (0.29 g, 0.003 mol, 1.0 equiv), and then, the temperature was raised to 90 °C and maintained for 12 h. The reaction mixture was quenched with saturated NaHCO3 solution and extracted with EtOAc (2 × 100 mL); the organic layer was washed with brine solution and dried over Na2SO4 and concentrated. The obtained crude was purified by silica gel (100–200 mesh) column chromatography with gradient elution mode, and compound 3 was eluted in EtOAc:hexane(30:70). The obtained yield was 76% (0.66 g). [M + H] 290.16.1H NMR (500 MHz, DMSO-d6): δ(ppm) 7.28 (d, 1H), 6.72 (d, J = 13.5 Hz, 1H), 3.42 (t, J = 7.0 Hz, 2H), 3.14 (t, J = 4.5 Hz, 4H), 2.90 (t, J = 4.5 Hz, 4H), 2.45 (t, J = 3.0 Hz, 2H), 2.35 (m, 1H), 0.87 (m, 2H), 0.62 (m, 2H).
3.1.3. Synthesis of N-Nitroso COX (Compound 1a), Methyl 1-Cyclopropyl-6-fluoro-7-(4-nitroso piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (Compound 2a), and 1-Cyclopropyl-6-fluoro-7-(4-nitroso piperazin-1-yl)-2,3-dihydroquinolin-4(1H)-one (Compound 3a)
To a stirred solution of amine compounds 1–3 (1.0 equiv) in water (10 mL) cooled to 0 °C were added NaNO2 (2.0 equiv) portion-wise slowly and acetic acid (1.0 equiv) and stirred overnight at RT (room temperature). Ice-cold water was added to the reaction mass and extracted with EtOAc (2 × 100 mL). The organic layer was dried in Na2SO4 and concentrated.
The obtained crude compound1awas purified by silica gel (100–200 mesh) column chromatography with gradient elution mode, and compound1awas eluted in EtOAc:hexane (70:30). The obtained yield was 68% (0.74 g). [M + H] 361.16. 1H NMR (500 MHz, DMSO-d6): δ(ppm) 15.17 (s, 1H),8.68 (s, 1H), 7.98 (d, J = 13.0 Hz,1H), 7.63 (d, J = 7.5 Hz,1H), 4.48 (t, J = 5.0 Hz, 2H), 3.98 (t, J = 5.0 Hz, 4H), 3.84 (m, 1H), 3.64 (t, J = 5.0 Hz, 2H), 3.42 (t, J = 5.0 Hz, 2H), 1.34 (m, 2H), 1.21 (m, 2H).
The obtained crude compound2awas purified by silica gel (100–200 mesh) column chromatography with gradient elution mode, and compound2awas eluted in EtOAc:hexane (35:65). The obtained yield was 65% (0.42 g). [M + H] 375.04. 1H NMR (500 MHz, DMSO-d6): δ(ppm) 8.46 (s, 1H), 7.82 (d, J = 13.0 Hz, 1H), 7.50 (d, J = 7.5 Hz, 1H), 4.46 (t, J = 5.0 Hz, 2H), 3.97 (t, J = 5.0 Hz, 4H), 3.74 (m, 1H), 3.64 (t, J = 5.0 Hz, 2H),3.62 (s, 3H), 3.28 (t, J = 5.0 Hz, 2H), 1.28 (m, 2H), 1.25 (m, 2H).
The obtained crude compound3awas purified by silica gel (100–200 mesh) column chromatography with gradient elution mode, and compound3awas eluted in EtOAc:hexane (20:80). The obtained yield was 70% (0.38 g). [M + H] 319.07. 1H NMR (500 MHz, CDCl3): δ(ppm) 7.59 (d, J = 13.0 Hz,1H), 6.76 (d, J = 7.0 Hz, 1H), 4.47 (t, J = 5.0 Hz, 2H), 4.04 (t, J = 5.5 Hz, 2H), 3.51 (t, J = 6.5 Hz, 2H), 3.46 (t, J = 5.5 Hz, 2H), 3.21 (t, J = 5.5 Hz, 2H), 2.63 (t, 6.5 Hz, 2H), 2.32 (m, 1H), 0.92 (m, 2H), 0.89 (m, 2H). Synthetic schemes of impurities and corresponding N-nitroso impurities are shown in Figure 3.
Figure 3.

Synthetic scheme of (A) COX impurities and corresponding N-nitroso impurities and (B) reaction mechanism of the formation N-N = O functional group.
The NMR data clearly showed that in the formation of the N-nitroso group from cyclic secondary amine-(−NH), the splitting patterns of the piperizine ring protons were changed significantly, indicating the formation of N-nitroso (−N-N=O). The corresponding NMR and mass spectra are shown in the Supporting Information file (Figures S1–S3).
3.2. (Q)-SAR Prediction Data
Quantitative structure–activity relationship ((Q)-SAR) predictions are an important part of hazard characterization as per ICH M7(R1) classification. In this case, genotoxicity might be evaluated in accordance with the warning structure(s) of substances used in toxicological research. (Q)-SAR methods with a high throughput capability can generate predictions for hundreds of impurity compounds in a very short amount of time. Two reciprocal (Q)-SAR methodologies are often used to determine the bacterial reverse mutation test results: expert rule-based and statistics-based.41−43 COX, N-nitroso COX, and N-nitroso impurity structures were placed in Nexus 2.5.0. The ICH classification model was utilized for the classification as per ICH M7 guidelines. The obtained results are given in Table 2. According to the established results of three N-nitroso impurities, Derek is plausible, while Sarah is positive and CoC.
Table 2. (Q)-SAR Prediction Summary for Genotoxicity of Three N-Nitroso Impurities.

TD50 (threshold dose or median toxic dose) values of N-nitrosamines “Lhasa” are reimplementations of the original “Gold” TD50 values mentioned in the carcinogenic potency database. As compared to simple N-methyl, N-ethyl -N-nitrosamines (NDEA-TD50 = 0.0265 mg/kg/day or NDMA-TD50 = 0.096 mg/kg/day), the carcinogenicity and mutagenicity of nitrosamines with larger alkyl sides or cycloalkyl rings do not show the highest carcinogenic potency. Piperazine is the accountable moiety for N-nitroso COX formation and other N-nitroso COX-related impurities. N-Nitroso piperazine had TD50 = 8.78 mg/kg/day (Gold) and 6.04 mg/kg/day (Lhasa).44
As noted in ICH M7, the “CoC” group cannot be regulated by the TTC limit due to its higher potency than most carcinogenic compounds. The maximum daily dose (MDD) for COX was 1500 mg/day.45 Consequently, the acceptable intake of the COX-NDSRI content is 0.012 ppm ((18 ng/day)/(1500 mg/day)) as per ICH M7 guidelines. Therefore, 0.012 ppm can be considered a specification limit, and the LOQ level is defined as 10% of the specification limit, i.e., 0.0125 ppm.
3.3. Chromatographic Method Development
We aimed to develop an accurate, precise, and selective LC-MS/MS method to separate and simultaneously quantify three N-nitroso impurities in COX and its formulations. For initial method development, preparing the mixture solution of all compounds (COX, COX-NDSRI, COX N-nitroso impurity-1, and COX N-nitroso impurity-2) equivalent to 1 mg/mL solution was used for the study of chromatographic separation, and the LC-MS method event flow state was set to waste in order to prevent the MS contamination. A few chromatographic columns were evaluated to obtain a good peak symmetry and separation of the components. However, the poor peak shapes and coelution of N-nitroso COX impurity-1 and N-nitroso COX were observed while utilizing the ACQUITY BEH C18 (100 mm × 2.1 mm, 1.7 μm) column. Similarly, while using Symmetry C18 (100 mm × 2.1 mm, 1.7 μm) column, both N-nitroso COX impurity-1 and N-nitroso COX were coeluted due to having similar structural features and retention times, thus potentially causing inaccurate quantification of N-nitroso COX impurity-1 and N-nitroso COX. An Agilent Poroshell Aq-C18, 150 mm × 3.0 mm, 2.7 μm, column was found to be suitable with good applicability in terms of acceptable resolution, good peak shape, and the response of the analytes. The COX is eluting prior to all components from the sample; therefore, it is simpler to exclude the introduction of COX to the mass detector. The flow state in the MS method acquisition parameters was set to divert the eluent of 0.0–3.1 min to waste and the rest of the eluent into the mass spectrometer to detect the peaks of interest. The composed mobile phase applied was 0.1% formic acid aqueous solution, 0.01 mol/L ammonium acetate, and 0.1% trifluoroacetic acid aqueous solution and was used for initial screening, and it was found that the 0.1% formic acid in aqueous solution was suitable for the analysis. The chromatography efficiency was evaluated with different diluents. However, based on the solubility of all N-nitroso impurities chosen, the diluent was 0.1% formic acid in water and acetonitrile (20:80) v/v. Different sonication times were evaluated and finalized at 15 min for sample preparation, bearing in mind the solubility and extraction efficiency. The filter study was also evaluated against the unfiltered centrifuged solution of COX, and the three N-nitrosamine peak areas are comparable. COX and three N-nitrosamine impurities are not retained by the Millex GV PVDF filters. Therefore, the filter is appropriate for this study without retaining COX and its genotoxic impurities. The initial method development trials to separate COX-NDSRI from COX and other N-nitroso impurities with 0.1% formic acid in water and acetonitrile found inadequate separation with the longer chromatographic run time. Hence, a small amount of methanol, along with water and acetonitrile, is introduced into the mobile phase. The isocratic elution mode was used with a mobile phase composition of 0.1% formic acid in the mixture of water, acetonitrile, and methanol in the ratio of 500:450:50 v/v/v and found better peak shape, resolution, and response. The pump flow rate was set at 0.5 mL/min, and the column temperature was maintained at 35 °C. Since the isocratic mobile phase with three major components, such as water, acetonitrile, and methanol, is used in the mobile phase, using a mixture design to optimize the mobile phase compositions using a systematic QbD approach is appropriate.
3.4. Method Optimization Using Mixture Design
The I-Optimal mixture design tool was utilized for the optimization of water, acetonitrile, and methanol components (mL/L) in the mobile-phase composition in order to achieve better resolution between COX and N-nitroso COX impurities. The three-component mixture design is depicted by a symmetrical triangle in a two-dimensional space. The design of experiments (DoEs) was mainly employed to discriminate and enhance the critical method parameters (CMPs). To understand the critical significance of the analytical method, a mixture design was employed to find CMPs and each of their unique interaction effects. The designated CMPs were identified based on the initial experiments and the adherences, the percentage of water in the mobile phase (CMP-A), the percentage of acetonitrile in the mobile phase (CMP-B), and the percentage of methanol in the mobile phase (CMP-C). The critical quality attributes (CQAs) are identified as the resolution between COX and N-nitroso COX impurity-1 (R1), the resolution between N-Nitroso COX impurity-1 and N-nitroso COX (R2), and the retention time of the late-eluting N-nitroso COX impurity-2 (R3). CQAs are measurable as numerical variables and are quantitative.
According to the USP guideline <621>,46 chromatographic parameters are altered from lower to higher. The I-Optimal mixture design with three components, two replicates, and three lack-of-fit points, a total of 11 experiments, was chosen. The DoE study employed the standard solution that contained 2 mg/mL COX and each N-nitroso COX impurity. The experimental designs with variables (CMPs) and responses (CQAs) are shown in Table 3.
Table 3. Experimental Designs with Variables (CMPs) and Replies (CQAs).
| component 1 | component 2 | component 3 | response 1 | response 2 | response 3 | |
|---|---|---|---|---|---|---|
| run | A: water (mL/L) | B: acetonitrile (mL/L) | C: methanol (mL/L) | R1 | R2 | R3 (min) |
| 1 | 512 | 463 | 25 | 14.63 | 10.19 | 9.87 |
| 2 | 525 | 438 | 37 | 17.18 | 11.51 | 11.59 |
| 3 | 512 | 425 | 63 | 16.06 | 11.14 | 11.09 |
| 4 | 550 | 425 | 25 | 19.2 | 12.4 | 12.66 |
| 5 | 512 | 425 | 63 | 16.32 | 11.31 | 11.31 |
| 6 | 475 | 462 | 63 | 12.99 | 9.1 | 8.92 |
| 7 | 487 | 475 | 38 | 13.65 | 9.38 | 9.13 |
| 8 | 512 | 463 | 25 | 14.84 | 10.31 | 9.65 |
| 9 | 475 | 500 | 25 | 11.95 | 7.24 | 7.08 |
| 10 | 487 | 437 | 76 | 13.2 | 9.96 | 9.81 |
| 11 | 475 | 425 | 100 | 12.79 | 9.67 | 9.64 |
The chosen model was analyzed, and the ANOVA test demonstrated that the model p-values are significant with <0.05 for all responses. Similarly, the lack-of-fit P values are not significant, with >0.05 for all responses. The model and lack-of-fit P values demonstrate that the model is suitable for the current study.
The design model was examined and navigated using the adjusted R2, predicted R2, regression coefficient (R2), and adequate precision. The data seemed to be excellent, as evidenced by the closeness of the values between the predicted R2 (0.9292, 0.8326, and 0.9414 for responses R1, R2, and R3, respectively) and adjusted R2 values (0.9463, 0.9830, and 0.9627 for responses R1, R2, and R3, respectively), which was obtained to be <0.2. A ratio of more than 4 is enviable when measuring the signal-to-noise ratio with allowable precision. The perceived ratio (26.93, 37.62, and 34.52 for responses R1, R2, and R3) shows that an ample signal and design can be used to traverse the design space. Data from the ANOVA, including P values, significance level, model R2, predicted R2, and modified R2, are shown in Table 4.
Table 4. Summary of ANOVA Results.
| response |
P values |
significance
level |
||||||
|---|---|---|---|---|---|---|---|---|
| model | lack of fit | model | lack of fit | R2 | adjusted R2 | predicted R2 | adeq precision | |
| R1 | <0.0001 | 0.0786 | significant | not Significant | 0.9570 | 0.9463 | 0.9292 | 26.9320 |
| R2 | <0.0001 | 0.1882 | significant | not Significant | 0.9915 | 0.9830 | 0.8326 | 37.6232 |
| R3 | <0.0001 | 0.1955 | significant | not Significant | 0.9702 | 0.9627 | 0.9414 | 34.5227 |
To distinguish the interactions between the variables and the effect of factors, trace (Piepel) plots and numerical plots were examined for each response. The model graphs revealed that response R1 increases with the increase of percentage water composition (CMP-A) and decreases with the increase in the percentage of acetonitrile (CMP-B) and the percentage of methanol (CMP-C) in mobile-phase composition. R2 increases with the increase of the water composition (CMP-A), decreases with the increase in the percentage of acetonitrile (CMP-B), and decreases with the percentage of methanol (CMP-C) in the mobile-phase composition. Similarly, R3 increases with the increase in water composition (CMP-A) and decreases with the increase in the percentage of acetonitrile (CMP-B). R3 remains unchanged with an increase in the percentage of methanol (CMP-C) in the mobile-phase composition. Normal plots, predicted vs actual plots, trace plots, and triangle 2D contour graphs are shown in Figure 4.
Figure 4.

Normal plots, predicted vs actual plots, trace plots, and triangle 2D contour graphs R1: resolution between COX and N-nitroso COX impurity-1, R2: resolution between N-nitroso COX impurity-1, and N-nitroso COX, and R3: retention time of N-nitroso COX impurity-2.
To optimize the responses R1, R2, and R3, the best optimum chromatographic conditions have been found using the numerical and graphical optimization approaches, as shown in Figures 5 and 6.
Figure 5.

Numerical optimization plots.
Figure 6.

Surface plots and overlay plot.
The water (CMP-A), the acetonitrile (CMP-B), and the methanol (CMP-C) compositions in the mobile phase were determined as 475, 500, and 25 mL/L, respectively, and the corresponding responses were 11.84, 7.32, and 7.36 min. The retention times of COX, N-nitroso impurity-1, COX-NDSRI, and N-nitroso impurity-2 were observed at 2.16, 3.42, 4.20, and 6.86 min, respectively, and the corresponding typical chromatogram is shown in Figure 7. The diverted valve switching technique was used to avoid the introduction of highly concentrated COX (80 mg/mL) to the mass detector to protect the mass spectrometer.
Figure 7.

Typical chromatogram of separation of COX-N-nitroso impurities.
3.5. Method Validation
The current optimized LC-MS/MS method was successfully validated in compliance with ICH guidelines,47 USP <1225>,48 and published journals49,50 and guidelines. The method validation was verified in terms of specificity, linearity, limit of detection, limit of quantitation, method and intermediate precision, accuracy, robustness, and solution stability.
3.5.1. Specificity
In the current method, the specificity of the analytical method was demonstrated by the ability of the LC-MS chromatographic system to distinguish between the diluent and individual impurities. The specificity was assessed by observing the retention times of COX, COX-NDSRI, N-nitroso impurity-1, and N-nitroso impurity-2, along with diluent solutions subjected to LC/MS analysis. The results exposed that all peaks were well separated, and no coeluting peaks were detected at the retention times of COX NDSRI, N-nitroso COX impurity-1, and N-nitroso impurity-2, thus enabling the precise and accurate quantification of the above-mentioned N-nitroso impurities in the COX drug substance as well as in drug products. The typical MRM chromatograms indicating method specificity can be found in Figure 8a–e.
Figure 8.

MRM chromatograms of (a) diluent blank, (b) placebo, (c) COX N-nitroso impurity-1, (d) COX-NDSRI, and (e) COX N-nitroso impurity-2.
3.5.2. Determination of the Limit of Detection (LOD) and Limit of Quantitation (LOQ)
The values of the LOD and LOQ of COX NDSRI, COX N-nitroso impurity-1, and N-nitroso impurity-2 were estimated by using the signal-to-noise ratio method (S/N) of 3 and 10, respectively. The LOD and LOQ were determined by preparing the known concentrations of standard solutions, which were injected into LC-MS spectrometry. The repeatability (precision) of LOQ was also performed by six replicate injections of these N-nitroso impurities and the % RSD value. The measured value of LOD and LOQ for all N-nitroso impurities was 0.03 and 0.1 ng/mL, respectively. The calculated results are shown in Table 5, and the typical LOQ MRM chromatograms are shown in Figure 9, respectively.
Table 5. Summary of Method Validation Results.
| results |
|||
|---|---|---|---|
| method validation parameter | COX-NDSRI | COX-N-Nitroso imp-1 | COX-N-Nitroso imp-2 |
| specificity | |||
| should be no interference from the diluent | no interference | no interference | no interference |
| precision | |||
| % content (n = 6, % RSD < 10.0) | 0.81 | 0.76 | 0.69 |
| intermediate precision | |||
| % content (n = 6, % RSD < 10.0) | |||
| analyst-1 | 0.95 | 1.20 | 1.45 |
| analyst-2 | 1.05 | 1.10 | 1.30 |
| limit of detection (LOD) | |||
| LOD (ng/mL) | 0.03 | 0.03 | 0.03 |
| S/N value (≥3) | 6.16 | 3.79 | 4.89 |
| limit of quantitation (LOQ) | |||
| LOQ (ng/mL) | 0.10 | 0.10 | 0.10 |
| S/N value (≥10) | 33.67 | 17.19 | 21.19 |
| LOQ precision | |||
| % content (n = 6, % RSD < 10.0) | 1.68 | 1.72 | 1.98 |
| linearity | |||
| range, (μg/mL) | 0.0012–0.025 | 0.0012–0.025 | 0.0012–0.025 |
| slope | 0.0027 | 0.0018 | 0.0099 |
| intercept | –0.0722 | 0.2774 | –0.4012 |
| Pearson coefficient (>0.999) | 0.9996 | 0.9994 | 0.9992 |
| accuracy in pure COX (%) (n = 3, average percentage) | |||
| the level at 0.00125 ppm mean ± SD | 94.80 ± 0.65 | 95.10 ± 0.65 | 96.90 ± 0.65 |
| the level at 0.0125 ppm mean ± SD | 98.24 ± 0.27 | 99.14 ± 0.49 | 98.94 ± 0.89 |
| the level at 0.025 ppm mean ± SD | 105.78 ± 0.14 | 107.18 ± 0.34 | 107.08 ± 0.36 |
| accuracy in formulated COX (%) (n = 3, average percentage) | |||
| the level at 0.00125 ppm mean ± SD | 93.98 ± 0.49 | 95.70 ± 0.65 | 96.30 ± 0.65 |
| the level at 0.0125 ppm mean ± SD | 98.94 ± 0.32 | 97.54 ± 0.27 | 98.14 ± 0.17 |
| the level at 0.025 ppm mean ± SD | 100.58 ± 0.14 | 107.18 ± 0.14 | 108.08 ± 0.14 |
| solution stability | |||
| peak area (0–48 h, % difference with initial < 10.0) | 1.48 | 1.63 | 1.12 |
Figure 9.

Typical LOQ MRM chromatograms of (a) COX N-nitroso impurity-1, (b) COX-NDSRI, and (c) COX N-nitroso impurity-2.
3.5.3. Linearity
The linearity of the method was investigated over the concentration range between 0.1 and 2.0 ng/mL, translating from 0.00125 ppm (1.2 ppb) to 0.025 ppm (25 ppb) with respect to 80 mg/mL sample concentration for all three N-nitroso impurities. In the construction of a set of calibration standards, a diluted stock solution (10 ng/mL) was prepared as described in Section 2.5. Furthermore, it was diluted to produce the following final concentrations: 0.1, 0.5, 0.8, 1.0, 1.2, 1.5, and 2.0 ng/mL (0.0012, 0.006, 0.01, 0.012, 0.015, 0.019, and 0.025 ppm (μg/g), correspondingly). The method linearity for all N-nitroso impurities was examined at seven distinct concentrations varying from 0.012 to 0.025 ppm (μg/g). The slope (a), intercept (b), and Pearson correlation coefficient (r) were determined by using the linear regression equation with least squares. Calculation of the linear regression equation was performed between peak area and analyte concentration. These results denote a good correlation between the peak areas and the concentrations of all three N-nitroso impurities with data summarized in Table 5.
3.5.4. Precision
In the current method, precision was evaluated by determining the % RSD of the contents of all three N-nitroso impurities from six replicate injections at a concentration of the specification-level spiked solution. Furthermore, intermediate precision is also measured on a different day by a different analyst by determining the % RSD of the contents of all three N-nitroso impurities from a total of 12 replicate injections of spiked solution at the specification level. The results are revealed in Table 5.
3.5.5. Accuracy and Recovery Study
The accuracy of the method was estimated by the triplicate preparation at three levels, 0.0012, 0.012, and 0.025 ppm, to the pure and formulated samples of COX by using the standard addition method. One preparation for each concentration was injected in triplicate. The acceptance criterion for recovery was 80–120%. The obtained recoveries were between 93.98 and 108.08% (Table 5).
3.5.6. Robustness Study
The robustness of the method states “the capability of an analytical method to remain unaltered by even small changes in method conditions”. During routine use, it provides an indication of its consistency. The flow rate (mL/min), column temperature (°C), collision gas flow (L/Hr), and desolvation temperature (°C) were changed in order to assess the robustness of the current method. The spiked sample solution with all three N-nitroso impurities at the specification level (0.0125 ppm) to the COX drug substance at the concentration of 80 mg/mL was injected into LCMS for the method robustness evaluation. The optimized flow rate was altered by ±0.01 mL/min from the actual flow rate of 0.5 mL/min, the column temperature (°C) was altered by ±1.0 °C from the actual 35 °C, the collision gas flow was altered by ±50 L/Hr from the actual 900 L/Hr, and the desolvation gas temperature was altered by ±50 °C from the actual 450 °C. The optimized UPLC-MS/MS method was designed to be consistent; as a result, the absolute percentage difference in content with respect to nominal conditions for all three N-nitroso impurities was not greater than 3.12 under any altered conditions. The robustness results are shown in Table 6.
Table 6. Summarized Results of the Robustness Study.
| content
(%) |
% absolute
difference with respect to the nominal condition |
||||||
|---|---|---|---|---|---|---|---|
| altered condition | change | COX-NDSRI | COX-N-Nitroso impurity-1 | COX-N-Nitroso impurity-2 | COX-NDSRI | COX-N-Nitroso impurity-1 | COX-N-Nitroso impurity-2 |
| flow rate (mL/min) | 0.45 | 0.0123 | 0.0127 | 0.0124 | 1.60 | 1.60 | 3.12 |
| 0.50 | 0.0125 | 0.0129 | 0.0128 | ||||
| 0.55 | 0.0124 | 0.0126 | 0.0126 | 0.80 | 2.32 | 1.56 | |
| column temp (°C) | 33.0 | 0.0124 | 0.0125 | 0.0125 | 0.80 | 3.12 | 2.34 |
| 35.0 | 0.0125 | 0.0129 | 0.0128 | ||||
| 37.0 | 0.0123 | 0.0127 | 0.0124 | 1.60 | 1.56 | 3.12 | |
| collision gas flow (L/h) | 850 | 0.0127 | 0.0127 | 0.0126 | 1.60 | 1.56 | 1.56 |
| 900 | 0.0125 | 0.0129 | 0.0128 | ||||
| 950 | 0.0124 | 0.0128 | 0.0125 | 0.80 | 0.77 | 2.34 | |
| desolvation temp (°C) | 400 | 0.0124 | 0.0128 | 0.0124 | 0.80 | 0.77 | 3.12 |
| 450 | 0.0125 | 0.0129 | 0.0128 | ||||
| 500 | 0.0126 | 0.0127 | 0.0125 | 0.80 | 1.56 | 2.34 | |
3.5.7. Solution Stability
COX and three N-nitroso COX impurities were tested for solution stability by leaving spiked and unspiked samples in capped LC vials for 48 h at 25 °C in an autosampler. We determined the concentration of each impurity against a freshly prepared standard solution, and none of the N-nitroso impurities showed any significant changes. Hence, we concluded that the impurities in the sample solution remained stable at ambient temperature (25 °C) for at least 48 h. The % absolute difference was calculated with respect to the peak areas of the freshly prepared COX and all three N-nitroso impurities.
3.6. Developed Optimized Method and Their Pharmaceutical Application
Five different commercially available formulation samples were investigated using our validated UPLC-MS/MS method to quantify the above-mentioned N-nitrosamine impurities accurately. The estimated amount of COX-NDSRI was in the range of 0.015–0.045 ppm. The test concentration of COX and the COX formulation was 80 mg/mL in triplicate determinations. Both COX N-nitroso impurity-1 and N-nitroso impurity-2 were not detected in all five batch formulation samples.
4. Conclusions
The current study synthesized the ciprofloxacin nitroso drug substance-related impurity (NDSRI) and a couple of COX-related N-nitroso impurities and identified them by using NMR and mass spectroscopic data. Two complementary (Q)-SAR methodologies were employed to assess and categorize the N-nitroso COX-related impurities, which were found to be Class 3 category with a cohort of concern, indicating potential genotoxicity. Based on the available literature and regulatory guidelines, COX-NDSRI acceptable intake was considered to be 18 ng/day; hence, it is a need to control and quantify the impurities at low levels. A QbD-based isocratic new simple, accurate, and rapid UPLC-MS/MS method was optimized for the simultaneous trace-level quantification of N-Nitroso COX impurities. This method offered precise and highly sensitive quantification of N-Nitrosamine impurities simultaneously. The method was successfully validated and presented good linearity, accuracy, repeatability, and robustness. Furthermore, the method was successfully applied to N-Nitrosamine impurity quantification in ciprofloxacin drug substance and formulated samples, recurring its high efficiency at low levels. Consequently, the current method can serve as a precise and accurate method to quantify three N-nitroso COX impurities simultaneously from the marketed tablet dosage forms for commercial release and stability sample testing.
Acknowledgments
The authors sincerely thank the Gitam University management.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c05170.
1H-NMR spectrum of COX-API and COX-NDSRI, mass spectrum of COX-NDSRI, 1H-NMR spectrum of COX-impurity-1 and N-nitroso COX-impurity-1, mass spectrum of N-nitroso COX-Impurity-1, 1H-NMR spectrum of COX-impurity-2 and N-nitroso COX-impurity-2, mass spectrum of N-nitroso COX-impurity-2, 1H-NMR spectrum of COX-API and COX-NDSRI, mass spectrum of COX-NDSRI, 1H-NMR spectrum of COX-impurity-1 and N-nitroso COX-impurity-1, mass spectrum of N-nitroso COX-impurity-1, 1H-NMR spectrum of COX-impurity-2 and N-nitroso COX-impurity-2, and mass spectrum of N-nitroso COX-impurity-2 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- 2023https://pubchem.ncbi.nlm.nih.gov/compound/ciprofloxacin#section=NCI-Thesaurus-Code (accessed May 25).
- Azizli E.; Muluk N. B.; Sezer C. V.; Kutlu; H M.; Cingi C. Evaluation of ciprofloxacin used as an intranasal antibiotic. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 82–91. 10.26355/eurrev_202212_30489. [DOI] [PubMed] [Google Scholar]
- Zang W.; Li D.; Gao L.; Gao S.; Hao P.; Bian H. The Antibacterial Potential of Ciprofloxacin Hybrids against Staphylococcus aureus. Curr. Top. Med. Chem. 2022, 22, 1020–1034. 10.2174/1568026622666220317162132. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Miao X.; Zheng Y. D.; Liu J.; He Q. Y.; Ge R.; Sun X. Ciprofloxacin-Resistant Staphylococcus aureus Displays Enhanced Resistance and Virulence in Iron-Restricted Conditions. J. Proteome. Res. 2021, 20, 2839–2850. 10.1021/acs.jproteome.1c00077. [DOI] [PubMed] [Google Scholar]
- Shariati A.; Arshadi M.; Khosrojerdi M. A.; Abedinzadeh M.; Ganjalishahi M.; Maleki A.; Heidary M.; Khoshnood S. The resistance mechanisms of bacteria against ciprofloxacin and new approaches for enhancing the efficacy of this antibiotic. Front. Public. Health. 2022, 10, 1025633 10.3389/fpubh.2022.1025633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhajj N.; O’Reilly N. J.; Cathcart H. Developing ciprofloxacin dry powder for inhalation: A story of challenges and rational design in the treatment of cystic fibrosis lung infection. Int. J. Pharm. 2022, 613, 121388 10.1016/j.ijpharm.2021.121388. [DOI] [PubMed] [Google Scholar]
- Wilson R.; Welte T.; Polverino E.; De Soyza A.; Greville H.; O’Donnell A.; Alder J.; Reimnitz P.; Hampel B. Ciprofloxacin dry powder for inhalation in non-cystic fibrosis bronchiectasis: a phase II randomised study. Eur. Respir. J. 2013, 41, 1107–15. 10.1183/09031936.00071312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruth E.; Mammadov E. Antibacterial activity of ciprofloxacin-impregnated 3D-printed polylactic acid discs: an in vitro study. J. Infect. Dev. Ctries. 2022, 16, 484–490. 10.3855/jidc.15267. [DOI] [PubMed] [Google Scholar]
- Alzahrani A.; Youssef A. A. A.; Nyavanandi D.; Tripathi S.; Bandari S.; Majumdar S.; Repka M. A.Design and optimization of ciprofloxacin hydrochloride biodegradable 3D printed ocular inserts: Full factorial design and in-vitro and ex-vivo evaluations: Part II. Int. J. Pharm. 2023, 631, 122533 10.1016/j.ijpharm.2022.122533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guideline on control of impurities of pharmacopeial substances: Compliance with the European pharmacopeia general monograph “Substances for pharmaceutical use” and general chapter “control of impurities in substances for pharmaceutical use”
- Control of impurities of pharmacopoeial substances - Scientific guideline | European Medicines Agency (europa.eu) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, S2 (R1) 2011https://database.ich.org/sites/default/files/S2_R1_Guideline.pdf.
- National Pharmacopoeia Commission . Guidelines for the Control of Genotoxic Impurities (First draft) 2019https://www.chp.org.cn/ydw/upload/userfiles/20190123/641548209919095.pdf.
- Holzgrabe U. Nitrosated Active Pharmaceutical Ingredients – Lessons Learned?. J. Pharm. Sci. 2023, 5, 1210–1215. 10.1016/j.xphs.2023.01.021. [DOI] [PubMed] [Google Scholar]
- Tuesuwan B.; Vongsutilers V. Nitrosamine Contamination in Pharmaceuticals: Threat, Impact, and Control. J. Pharm. Sci. 2021, 11, 3118–3128. 10.1016/j.xphs.2021.04.021. [DOI] [PubMed] [Google Scholar]
- Rodríguez R. L.; James A.; McManus; Murphy N. S.; Ott A. M.; Burns M. J. Pathways for N-Nitroso Compound Formation: Secondary Amines and Beyond. Org. Process. Res. Dev. 2020, 24, 1558–1585. 10.1021/acs.oprd.0c00323. [DOI] [Google Scholar]
- Wang Y.; You S.; Ruan M.; Wang F.; Ma C.; Lu C.; Yang G.; Chen Z.; Gao M. The Use of Potassium/Sodium Nitrite as a Nitrosating Agent in the Electrooxidative N -Nitrosation of Secondary Amines. Eur. J. Org. Chem. 2021, 22, 3289–3293. 10.1002/ejoc.202100363. [DOI] [Google Scholar]
- Andrew T.Mutagenic Impurities: Strategies for Identification and Control: N-Nitrosamines; John Wiley & Sons, Inc, 2022; pp 269–320 10.1002/9781119551249.ch10. [DOI] [Google Scholar]
- Ruth B.; Joerg S.; Sebastian H.; Christian K.; Grace K.; Bert L.; Giorgio B.; Mark H.; Marc F.; Leonardo A.; Yongmei W.; Youssi B. A Nitrite Excipient Database: A Useful Tool to Support N-Nitrosamine Risk Assessments for Drug Products. J. Pharm. Sci. 2023, 112, 1615–1624. 10.1016/j.xphs.2022.04.016. [DOI] [PubMed] [Google Scholar]
- Horne S.; Vera M. D.; Nagavelli L. R.; Sayeed V. A.; Heckman L.; Johnson D.; Berger D.; Yip Y. Y.; Krahn C. L.; Sizukusa L. O.; Rocha N. F. M.; Bream R. N.; Ludwig J.; Keire D. A.; Condran G. Regulatory Experiences with Root Causes and Risk Factors for Nitrosamine Impurities in Pharmaceuticals. J. Pharm. Sci. 2023, 5, 1166–1182. 10.1016/j.xphs.2022.12.022. [DOI] [PubMed] [Google Scholar]
- Ashworth I. W.; Dirat O.; Teasdale A.; Whiting M. Potential for the Formation of N-Nitrosamines during the Manufacture of Active Pharmaceutical Ingredients: An Assessment of the Risk Posed by Trace Nitrite in Water. Org. Process. Res. Dev. 2020, 9, 1629–1646. 10.1021/acs.oprd.0c00224. [DOI] [Google Scholar]
- Bharate S. S. Critical Analysis of Drug Product Recalls due to Nitrosamine Impurities. J. Med. Chem. 2021, 64, 2923–2936. 10.1021/acs.jmedchem.0c02120. [DOI] [PubMed] [Google Scholar]
- FDA 2021 . Pfizer expands voluntary nationwide recall to include all lots of CHANTIX (Varenicline) tablets due to N-nitroso varenicline content 2023https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/pfizer-expands-voluntary-nationwide-recall-include-all-lots-chantixr-varenicline-tablets-due-n (accessed May 13).
- https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/lupin-pharmaceuticals-inc-issues-voluntarily-nationwide-recall-all-irbesartan-tablets-and-irbesartan.
- FDA 2022 . Pfizer voluntary nationwide recall of lots of ACCUPRIL (Quinapril HCl) due to N-nitroso-quinapril content 2022https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/pfizer-voluntary-nationwide-recall-lots-accuprilr-quinapril-hcl-due-n-nitroso-quinapril-content (accessed May 23).
- FDA 2022 . Sandoz, Inc. Issues nationwide recall of 13 lots of orphenadrine citrate 100 mg extended-release tablets due to presence of a nitrosamine impurity 2023https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/sandoz-inc-issuesnationwide-recall-13-lots-orphenadrine-citrate-100-mg-extended-release-tablets-due (accessed May 13).
- Health Canada 2022 . Pfizer recalls Inderal-LA (propranolol hydrochloride) capsules due to a nitrosamine impurity 2023https://recalls-rappels.canada.ca/en/alert-recall/pfizer-recalls-inderal-propranolol-hydrochloride-capsules-due-nitrosamine-impurity (accessed May 13).
- Justin M.; Jörg S.; Christoph S. N-Nitrosamines Impurities in Pharmaceuticals. The Abrupt Challenges that Resulted, the Evolving Science, and the Regulatory Framework. J. Pharm. Sci. 2023, 5, 1161–1162. 10.1016/j.xphs.2023.01.016. [DOI] [PubMed] [Google Scholar]
- Ponting D. J.; Dobo K. L.; Kenyon M. O.; Kalgutkar A. S. Strategies for Assessing Acceptable Intakes for Novel N-Nitrosamines Derived from Active Pharmaceutical Ingredients. J. Med. Chem. 2022, 23, 15584–15607. 10.1021/acs.jmedchem.2c01498. [DOI] [PubMed] [Google Scholar]
- Dobo K. L.; Kenyon M. O.; Dirat O.; Engel M.; Fleetwood A.; Martin M.; Mattano S.; Musso A.; McWilliams J. C.; Papanikolaou A.; Parris P.; Whritenour J.; Yu S.; Kalgutkar A. S.Practical and Science-Based Strategy for Establishing Acceptable Intakes for Drug Product N-Nitrosamine Impurities. Chem. Res. Toxicol. 2022, 35, 475–489. 10.1021/acs.chemrestox.1c00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidance for Industry U.S. Department of Health and Human Services Food and Drug Administration . Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs), Center for Drug Evaluation and Research (CDER), Pharmacology/Toxicology (accessed August 30, 2023).
- Schlingemann J.; Burns M. J.; Ponting D. J.; Martins Avila C.; Romero N. E.; Jaywant M. A.; Smith G. F.; Ashworth I. W.; Simon S.; Saal C.; Wilk A. The Landscape of Potential Small and Drug Substance Related Nitrosamines in Pharmaceuticals. J. Pharm. Sci. 2023, 5, 1287–1304. 10.1016/j.xphs.2022.11.013. [DOI] [PubMed] [Google Scholar]
- ICH M7(R1) . Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk, Current Step 4 version dated 31 March, 2017https://database.ich.org/sites/default/files/M7_R1_Guideline.pdf.
- 2023https://www.fda.gov/drugs/mitigation-strategies-nitrosamine-drug-substance-related-impurities-quality-and-bioequivalence (accessed May 13).
- 2023https://www.fda.gov/drugs/drug-safety-and-availability/updates-possible-mitigation-strategies-reduce-risk-nitrosamine-drug-substance-related-impuritiesaccessed May 13.
- Peryea T.; Southall N.; Miller M.; Katzel D.; Anderson N.; Neyra J.; Stemann S.; Nguyễn Đ.-T.; Amugoda D.; Newatia A.; Ghazzaoui R.; Johanson E.; Diederik H.; Callahan L.; Switzer F. Global Substance Registration System: consistent scientific descriptions for substances related to health. Nucleic. Acids. Res. 2021, 49, D1179–D1185. 10.1093/nar/gkaa962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tome T.; Zigart N.; Casar Z.; Obreza A. Development and optimization of liquid chromatography analytical methods by using AQbD principles: overview and recent advances. Org. Process. Res. Dev. 2019, 23, 1784–1802. 10.1021/acs.oprd.9b00238. [DOI] [Google Scholar]
- Presenza L.; Fabrício L. F.; de F.; Galvão J. A.; Vieira T. M. F. Simplex-centroid mixture design as a tool to evaluate the effect of added flours for optimizing the formulation of native Brazilian freshwater fish burger. LWT 2022, 156, 113008 10.1016/j.lwt.2021.113008. [DOI] [Google Scholar]
- BahramParvar M.; Tehrani M. M.; Razavi S. M. A.; Koocheki A. Application of simplex-centroid mixture design to optimize stabilizer combinations for ice cream manufacture. J. Food. Sci. Technol. 2015, 52, 1480–1488. 10.1007/s13197-013-1133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Freue G. V.; Borchers C. H. Multiple reaction monitoring (MRM): principles and application to coronary artery disease. Circ. Cardiovasc. Genet. 2012, 5, 378. 10.1161/CIRCGENETICS.111.959528. [DOI] [PubMed] [Google Scholar]
- Ashworth I. W.; Blanazs A.; Byrne J. J.; Dirat O.; Fennel J. W.; Kuhl N.; Wells S. L.; Whiting M. P.. Approaches and Considerations for the Investigation and Synthesis of N-Nitrosamine Drug Substance-Related Impurities (NDSRIs) Org. Process. Res. Dev. 2023 10.1021/acs.oprd.3c00084. [DOI]
- Ridings J. E.; Barratt M. D.; Cary R.; Earnshaw C. G.; Eggington C. E.; Ellis M. K.; Judson P. N.; Langowski J. J.; Marchant C. A.; Payne M. P.; Watson W. P.; Yih T. D. Computer prediction of possible toxic action from chemical structure: an update on the DEREK system. Toxicology 1996, 1–3, 267–279. 10.1016/0300-483x(95)03190-q. [DOI] [PubMed] [Google Scholar]
- Cross K. P.; Ponting D. J. Developing structure-activity relationships for N-nitrosamine activity. Comput. Toxicol. 2021, 20, 100186 10.1016/j.comtox.2021.100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R.; Thresher A.; Ponting D. J. Utilisation of parametric methods to improve percentile-based estimates for the carcinogenic potency of nitrosamines. Regul. Toxicol. Pharmacol. 2021, 121, 104875 10.1016/j.yrtph.2021.104875. [DOI] [PubMed] [Google Scholar]
- Ponting D. J.; Dobo K. L.; Kenyon M. P.; Kalgutkar A. S. Strategies for Assessing Acceptable Intakes for Novel N-Nitrosamines Derived from Active Pharmaceutical Ingredients. J. Med. Chem. 2022, 23, 15584–15607. 10.1021/acs.jmedchem.2c01498. [DOI] [PubMed] [Google Scholar]
- 2023https://www.drugs.com/dosage/cipro.html (accessed May 13).
- United states Pharmacopeial Convention, Rockville, Maryland, USA . USP General Chapter Chromatography, USP-43-NF38, 6853. https://doi.org.10.31003/uspnf_M99380_07_01.
- ICH Q2(2) International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Topic Q2(R2): validation of analytical procedures: Text and methodology, (2005) 2023https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1_Guideline.pdf (accessed May 13).
- United states Pharmacopeial Convention, Rockville, Maryland, USA . USP General Chapter Validation of compendial procedures, USP-43-NF38, 8166 2023https://doi.org.10.31003/uspnf_M99945_04_01 (accessed May 13).
- Muchakayala S. K.; Katari N. K.; Saripella K. K.; Schaaf H.; Marisetti V. M.; Kowtharapu L. P.; Jonnalagadda S. B. AQbD based green UPLC method to determine mycophenolate mofetil impurities and Identification of degradation products by QToF LCMS. Sci. Rep. 2022, 12, 19138 10.1038/s41598-022-22998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakka S.; Katari N. K.; Muchakayala S. K.; Jonnalagadda S. B.; Babu M. S. S.. Isolation, identification, structural elucidation, and toxicity prediction using (Q)-SAR models of two degradants: AQbD-driven LC method to determine the Roxadustat impurities. Talanta Open, 2023, 7, 100221. 10.1016/j.talo.2023.100221. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.