

Abstract
Enzymes can be engineered at the level of their amino acid sequences to optimize key properties such as expression, stability, substrate range, and catalytic efficiency—or even to unlock new catalytic activities not found in nature. Because the search space of possible proteins is vast, enzyme engineering usually involves discovering an enzyme starting point that has some level of the desired activity followed by directed evolution to improve its “fitness” for a desired application. Recently, machine learning (ML) has emerged as a powerful tool to complement this empirical process. ML models can contribute to (1) starting point discovery by functional annotation of known protein sequences or generating novel protein sequences with desired functions and (2) navigating protein fitness landscapes for fitness optimization by learning mappings between protein sequences and their associated fitness values. In this Outlook, we explain how ML complements enzyme engineering and discuss its future potential to unlock improved engineering outcomes.
Short abstract
Machine learning can complement enzyme engineering by helping to discover enzymes with desired function and to accelerate the optimization of enzyme fitness.
1. Introduction: The Current Approach to Enzyme Engineering
Engineered proteins are important for medicine, chemical manufacturing, biotechnology, energy, agriculture, consumer products, and more. Antibodies, for example, can be engineered to enhance their binding and specificity as therapeutics, whereas the stabilities and activities of enzymes can be improved under process conditions to obtain greener and more efficient chemical syntheses.1−3 At its core, protein engineering is a design problem: the goal is to generate and/or alter a protein’s amino acid sequence to encode a desired function. “Fitness” is a numerical quantification of that desired function, which may include multiple features that contribute to overall performance. Altering fitness is equivalent to traversing the protein’s fitness landscape, which is a surface in high-dimensional space that maps sequence to fitness. Protein engineering is challenging because accurate biophysical prediction methods for determining protein fitness are rare or nonexistent, and the search space of possible proteins is beyond-astronomically large.4 To make matters worse, functional proteins are scarce in the space of all protein sequences, and finding an optimal sequence on this protein fitness landscape is NP-hard, as there is no known polynomial-time solution.5
In this Outlook we focus on engineering enzymes, which have applications in areas ranging from chemical synthesis and plastic degradation to diagnostics, protein therapeutics, and gene editing.2,3 Enzyme engineering poses some unique challenges: catalysis is more complex than binding and may involve multiple substrates, cofactors, and elementary steps. Furthermore, typical experimental screening methods for measuring enzymatic fitness are lower throughput than binding assays, for which powerful positive and negative selections can usually be devised. Enzymes are often engineered to enhance their native functions, or alternatively to target “promiscuous” activities, such as reactivity on non-native substrates or even non-native reactivities (Figure 1A).6 Due to the challenges of modeling catalysis and the limited throughput of meaningful assays, enzyme engineers often use directed evolution (DE) to optimize these features.7,8
Figure 1.

The enzyme engineering workflow. Enzyme engineering begins with a discovery phase to identify an enzyme with initial activity (desired function). If fitness is not sufficient, the enzyme is then optimized using DE. (A) Enzyme discovery involves screening for desired activities, which could include native activity or promiscuous activities. (B) Enzyme starting points can be found in known proteins or by (C) diversification of enzymes using various computational methods to generate starting sequences that are more stable and evolvable. (D, E) In its simplest form, optimization using DE involves generating a pool of protein variants, identifying one with improved fitness, and using this variant as the starting point for the next generation of mutation and screening. DE can be thought of as a greedy hill climb on a protein fitness landscape. The natural ordering of sequences in the DE fitness landscape is that all sequences are surrounded by their single mutant neighbors.25
At a high level, engineering an enzyme involves discovering an enzyme with some initial level of activity (satisfying some but not all desired properties), followed by fitness improvement using DE (Figure 1).9 Thus, the first step of an enzyme engineering workflow involves identifying (or designing) an enzyme with some measurable fitness. Consider engineering an enzyme to catalyze a new chemical reaction. To find a new activity that is related to a known activity, one might screen previously engineered enzymes for “promiscuous” activity for the desired function (Figure 1A).10,11 If none is detected, it may be necessary to explore other known enzymes or proteins in annotated databases (Figure 1B).12 Those with active sites amenable to accommodating a particular substrate, evolvable folds, cofactors relevant to a desired activity, or similar mechanisms may be valid starting points. Unfortunately, these approaches rely too much on experimental intuition and luck, and such an Edisonian search through existing proteins is inefficient and often ineffective. Even if activity is found, the enzyme might need to be stabilized so that it has suitable behavior for screening or can undergo further evolution, and it must express well in the host organism, such as bacteria or yeast. Computationally assisted methods such as chimeragenesis and ancestral sequence reconstruction have emerged to propose diverse protein starting points (sometimes having higher stability, evolvability, different substrate scopes) (Figure 1C).13−15 Methods aided by software suites such as Rosetta have been successful in redesigning enzymes and enhancing their stabilities,16−21 but de novo enzyme design is still nascent and works well only for relatively simple reactions.22−24 Because enzyme activity is influenced by a complex mix of poorly understood factors, most de novo designed enzymes must be further optimized.
Once a suitable enzyme with measurable function is identified, fitness can be improved by DE and related techniques.7,8 DE sidesteps the need to understand protein sequence-fitness relationships and optimizes protein fitness by performing greedy hill climbing on the protein fitness landscape (Figure 1D).1,4,25 In its simplest form, DE involves accumulating beneficial mutations by making mutations to the protein (mutagenesis) and screening for variant(s) with higher performance on target properties (Figure 1E). The targeted properties can change during optimization by changing the screening criteria, and informative screens can investigate multiple properties simultaneously. Recombination is often used to shuffle beneficial mutations so that screening can identify mutation combinations that further increase fitness.26,27 DE takes advantage of the fact that functional sequences are clustered in sequence space, i.e., functional sequences are surrounded by many other functional sequences, and smooth uphill paths exist in the landscape.25 However, DE is limited because screening can only explore a limited, local region within the sequence search space. Additionally, because DE largely follows a smooth path taking one mutation step at a time, so it can become stuck at a local fitness optimum.
Recently, machine learning (ML) has emerged as a useful tool for enzyme engineering, both for the discovery of functional enzymes, which is the focus of the first section of this Outlook, and for navigating protein fitness landscapes for fitness optimization, which is the focus of the second section. We encourage readers to read other reviews summarizing recent advancements in these areas.28−37 ML is particularly well suited for the challenges of enzyme engineering, as generative models can take advantage of patterns in known protein sequences and supervised models can learn from labels of protein properties such as various measures of fitness. In this Outlook, we explain existing methods where ML is used to assist enzyme engineering, and we propose ML-related research efforts that can have the most beneficial impact for engineering outcomes. Ultimately, we believe that the steps of ML-assisted enzyme engineering can be integrated toward fully automated engineering of many desired properties.
2. Discovery of Functional Enzymes with Machine Learning
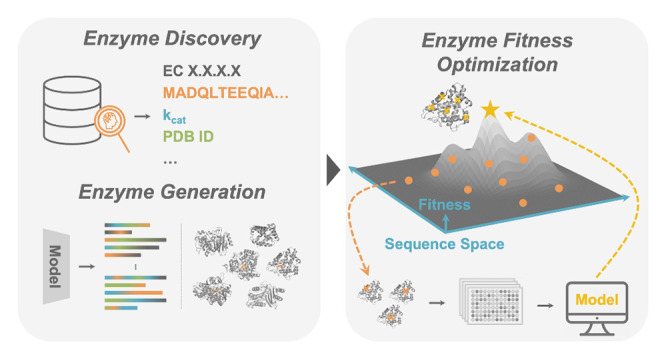
A starting point for enzyme engineering is usually identified either from a search of existing sequences or by generating new candidates. ML methods have emerged to help with both approaches (Figure 2). Classification methods can annotate protein sequence/structure databases and uncover previously unannotated proteins with a desired function, while generative models using deep learning can design novel proteins with desired functions.
Figure 2.

Opportunities for the discovery of functional enzymes using machine learning. Identifying functional enzymes as starting points for optimization of their properties is a key challenge in enzyme engineering. Many useful enzymes could be discovered amidst already known, but unannotated, protein sequences. (A) ML models can classify sequences based on their EC numbers. (B) Generalized LLMs could annotate proteins in databases and scientific literature, and (C) AI could act as a structural biologist and organic chemist to discern if certain reactions might work based on catalytic/structural motifs. Alternatively, emerging deep learning methods can look beyond the sequences explored by natural evolution and design novel functional enzymes. This problem can be treated as (D) pure sequence generation or (E) generation toward a target structure. Future work should focus on identifying promiscuous and evolvable enzymes.
2.1. Annotation of Enzyme Activity among Known Proteins
Approximately 250 million protein sequences are catalogued in the UniProt database, but less than 0.3% are annotated with function.38 Thus, hundreds of millions of known proteins have not been explored as starting points for enzyme engineering. If these proteins could be accurately annotated, protein engineers would have access to a wealth of diverse candidates for engineering. While enzyme engineers have long been using multiple sequence alignments (MSAs) and homology to predict the functions of unannotated protein sequences,39 ML classification models extend these approaches and draw from more complete features describing protein sequences and structures to predict more specific functions, such as type of reactivity and kcat.34,40−48 Focusing on known sequences without annotations, many of these methods aim to classify enzyme sequences based on their enzyme commission (EC) numbers, which is a hierarchical classification scheme that divides enzymes into general classes and then further subclasses, based on their catalytic activities (Figure 2A).
In particular, contrastive learning-enabled enzyme annotation (CLEAN) has demonstrated state-of-the-art success at accurately classifying enzyme sequences based on their EC numbers.40 Upon wet-lab validation, CLEAN accurately characterized all four EC hierarchical numbers of understudied halogenase enzymes with 87% accuracy, which is significantly better than the next-best method at 40% accuracy. Impressively, CLEAN also correctly identified an enzyme with three different EC numbers, corresponding to promiscuous activities, where promiscuity prediction was framed as multitask classification.49 Promiscuous activities, which can include similar reactivity on new substrates or entirely different reactivity (Figure 1A), are often the starting points for evolving enzymes for non-natural activity. Thus, enzyme functional annotation efforts should include efforts to annotate these sorts of promiscuous activities for use in future enzyme discovery pipelines.11,40 Many promiscuous activities are difficult to detect or simply have not been tested; it will be critical to perform experimental assays to update enzyme function databases. Text mining of literature using large language models (LLMs) based on generative pretrained transformer (GPT) architectures could also help identify missing labels and update existing databases by extracting knowledge from scientific literature which has not been included in existing databases (Figure 2B).
We suggest a few other strategies to improve functional annotation efforts. EC numbers do not capture a quantitative notion of similarity between reactions, so enzyme activity prediction would benefit from a learned continuous representation of the similarity between activities, where reactions, substrates, and products are numerically encoded. This could resemble current efforts to encode chemical structures and predict the outcomes of reactions in synthetic organic chemistry.50−53 Databases will be useful for the curation and standardization of enzyme reaction data.54,55 Overall, there is also still room to develop better benchmarks for enzyme discovery, to measure the effectiveness of various models and representations.56
Recently, there has been an explosion in protein structure data from ML-enabled protein structure prediction tools such as AlphaFold2 and others57−62 and databases of unannotated protein structures. Clustering similar structures is one way to annotate for function.63 Alternatively, many enzymes have common “modules,” or recurring residue arrangements, which perform similar reactions.64 The structures of active sites in unlabeled protein structures could be compared to existing structures to identify new, diverse sets of proteins with given function, using models trained on sequence and structure.65 Structures could also be physically modeled to predict their interactions with different substrates. In principle, an ML model could be trained to combine multimodal information such as spatial descriptors of protein structures with an LLM trained on information about chemical reactions.66,67 This artificial intelligence (AI) model would act as protein structural biologist and organic chemist. By synthesizing these two forms of knowledge, the model could perform the laborious work of sifting through and identifying viable protein structures for desired reactivity (Figure 2C).68,69 Finally, it is also possible to go beyond known protein sequences and expand the search for functional enzymes to microbial dark matter: metagenomic analysis has only scratched the surface of these genomes.70
2.2. Generating New Proteins with Deep Learning
While many functional enzymes could be discovered through annotation of known protein sequences, generating entirely new sequences not explored by evolution could also be useful, as these could unlock unseen combinations of properties and, potentially, non-natural activities. Chimeragenesis, an approach to generating energetically favorable proteins based on recombining functional homologous proteins,14,26 has inspired development of deep learning approaches to assemble compatible structural domains in enzymes.71 Similarly, sets of mutations that are calculated to be energetically favorable using physics-based simulations (FuncLib) can be introduced in or near protein active sites to construct diversified proteins with high stability; by virtue of their sequence changes, they also exhibit promiscuous activities.17,18,72−74 Efforts to combine structure design methods75−77 and ancestral sequence reconstruction15,75,78−80 with data-driven models could help identify improved enzyme variants with diversified substrate scope and enhanced stability/evolvability as starting points for enzyme engineering. However, generating proteins with non-native activities will be more challenging.
While the above methods can generate diverse sequences, these sequences are still quite similar to naturally occurring sequences, which means that vast regions of protein sequence space remain underexplored. Recently, significant efforts have focused on using deep learning to design enzymes with low similarity to known sequences or structures. These efforts are reviewed elsewhere in great detail.24,35,81−85 In general, these methods fall into one of two main categories: (1) pure sequence generation and (2) structure design (finding a sequence that folds to a target structure or scaffold).
In pure sequence generation, protein language models (PLMs) can be conditioned by a known enzyme family to generate novel sequences with that function, without direct consideration of structure (Figure 2D).86−98 Models with transformer architectures have generated enzymes such as lysozymes, malate dehydrogenases, and chorismate mutases: for the best models, up to 80% of wet-lab validated sequences expressed and functioned.88,90 Some of these generated sequences have low sequence identity (<40%) to known proteins and may be quite different from those explored by evolution, thus potentially unlocking combinations of properties not found in nature. Variational autoencoders (VAEs) have been used to generate phenylalanine hydroxylases and luciferases, with wet-lab validation achieving 30–80% success rates.86,87,96 Generative adversarial networks (GANs) were also applied to the generation of malate dehydrogenases, with 24% success rate.95 Alternatively, a diffusion model such as EvoDiff could achieve better coverage of protein functional and structural space during generation.99 Despite these successes, for many methods, only a small fraction of proposed sequences are functional in the wet lab, and those that do function are often quite similar to known sequences. Simulating the structures of generated proteins, filtering them based on evolutionary likelihood, and doing other quality checks significantly increased the hit rate of functional enzymes from generative models,100 but there is still much room for improvement. So far, these models have been demonstrated on large enzyme families; achieving the same success on smaller enzyme families poses a challenge.
It is also possible to design desired enzyme scaffolds/structures (Figure 2E).101−113 One approach is hallucination, where a search algorithm uses a structure predictor to find a sequence that folds to the right structure.103,110,35 Luciferases with high luminescence and selectivity were engineered using deep-learning-assisted protein design, by combining hallucination with Rosetta sequence design.107 One of the wet-lab-validated designs demonstrated catalytic activity comparable to natural luciferases, with much higher substrate selectivity: the active site and the enzyme scaffold were both entirely different from naturally occurring luciferases. More recently, methods such as ProteinMPNN and RFdiffusion have achieved particular success for designing a broad range of proteins with targeted structures,104,108 where design success was validated by measuring the similarity between the target structure and the designed structure as predicted by AlphaFold2. ProteinMPNN is an inverse folding model, which is a class of models where the input to the model is a structure, and the output is a sequence. RFdiffusion is a diffusion model, where the input is a condition based on desired structure or symmetry (along with random coordinates), and the output is the coordinates of the generated structure. Still, additional wet-lab studies are needed to determine if designed enzymes can express, fold, and function.
Enzyme design still has a lot of room for growth. Designs could provide diverse starting points for further engineering of desired activities, including activities that fall outside known EC numbers. While most current success involves generating protein scaffolds or activities that are already known, it will be exciting to see more efforts that focus on generating enzymes that do not resemble those in nature and/or exhibit non-natural activities. In protein engineering, certain protein folds are more evolvable for certain reasons, including elevated stability114,115 that is imparted by residues outside the active site,116,117 balanced with flexibility to change conformation and accommodate new substrates and reactions.118 Proteins that express well in a host organism for evolution are also preferred. Generative models have the potential to address this need for enzymes that are better starting points than natural enzymes: for example, ProteinMPNN was able to design wet-lab validated enzymes with higher expression and thermostability.119 With proper labels about enzyme activity on different substrates, generative design models could be conditioned to generate enzymes with several of these desirable attributes. Future research that could address this need would be highly impactful for enzyme engineering.
3. Navigating Protein Fitness Landscapes Using Machine Learning
Most enzyme starting points identified during the discovery stage need to be further optimized to achieve desired performance levels. DE and related techniques have demonstrated success in navigating protein fitness landscapes to optimize various properties. However, DE screens or selections can sample only a small fraction of sequences in a protein fitness landscape. DE can additionally be inefficient because focusing on single mutants ignores the nonadditive effects of accumulating multiple mutations (epistasis),120,121 which is commonly observed when residues interact, such as in an enzyme active site or through a cofactor or substrate. Thus, a DE campaign can get stuck at a local optimum, even when high fitness sequences are nearby (Figure 3A). To address this limitation, protein fitness prediction methods using supervised ML models have emerged to learn a mapping between protein sequences and their associated fitness values to approximate protein fitness landscapes.122−124 These models can then predict the fitnesses of previously unseen protein variants, increasing screening efficiency by evaluating proteins in silico and expanding exploration to a greater scope of sequences compared to conventional DE approaches.125,126 At the same time, zero-shot (ZS) predictors—such as implicit fitness constraints learned from naturally occurring protein sequences (evolutionary conservation)—can also guide the prediction of protein fitness.127−129
Figure 3.

Opportunities for machine learning models to help navigate protein fitness landscapes. (A) ML models can allow for bigger jumps in sequence space by proposing combinations of mutations that would not be achieved by traditional DE. The role of nonadditivity between mutation effects, or epistasis, should be explored further to understand when ML offers an advantage. (B) The role of ZS scores to predict protein fitness without any labeled assay data needs to be better understood for different protein families and functions. Finally, ML-assisted protein fitness optimization could benefit from (C) multimodal representations that capture physically relevant descriptors of proteins to predict multiple relevant properties and (D) active learning with deep learning models tailored toward proteins and uncertainty quantification.
For a protein of length N, there are ∼20N possible sequences in the search space. ML models trained on the order of 102–103 labeled sequences (typical for an informative enzyme screen) would be unable to accurately extrapolate on such a large search space. As a result, current ML-assisted protein engineering approaches operate on constrained design spaces. Chimeragenesis has been explored as one way to constrain the search space, and various ML efforts have demonstrated success and utility on these landscapes.122,130−133 This approach can only introduce naturally occurring protein motifs, which can generate diverse proteins with native function while improving properties like stability. However, chimeragenesis is less likely to improve other properties, such as novel reactivity, because it retains conserved residues such as those important for native activity. More promising protein fitness prediction efforts focus on variants with one or several point mutations from a parent protein, by building training libraries using random mutagenesis134 or combinatorial site saturation mutagenesis. Still, artificially constraining the search space in these ways neglects certain important considerations. Using random mutagenesis to create a training library captures very limited epistasis,135 whereas building a meaningful combinatorial mutagenesis library requires choosing a few sites relevant to increasing fitness while still introducing epistasis, and these choices are often not obvious.
There remain many open questions about when ML-assisted protein fitness prediction is useful and how to improve it for better protein engineering outcomes, which we have summarized into the following guiding questions: (1) How should ML be used to determine the best combinations of multiple mutations on epistatic and nonepistatic protein fitness landscapes? (2) Which ZS predictors are useful in the context of native and non-native function? (3) How can ML approaches be improved to identify protein variants with high fitness more efficiently? The considerations are highlighted in Figure 3. Answering these questions is critical for advancing ML-assisted protein fitness optimization and will require new ML methods as well as new sequence-fitness data sets.
3.1. Combining Mutations on Epistatic and Nonepistatic Protein Fitness Landscapes
ML-assisted directed evolution (MLDE) is a specific implementation which uses supervised ML to predict the fitnesses of protein variants with multiple mutations. MLDE was demonstrated on the GB1 data set—this data set is from a combinatorial library in which four residues (with high degrees of epistasis136,137) were mutated simultaneously to all possible amino acids and fitness was measured by binding to an immunoglobulin protein. On this particular protein fitness landscape, MLDE was more effective than traditional protein engineering methods: it outperformed baselines such as DE using a single-step greedy walk.138 MLDE allowed for bigger jumps in sequence space to avoid getting stuck at local optima, which are more prevalent on highly epistatic (rugged) landscapes (Figure 3A).129 ML methods may be particularly beneficial where few samples are measured by assays and used for training (the low N regime).133,139 In a wet-lab validation, MLDE was used to identify a combination of mutations that resulted in an enzyme that could perform enantioselective carbon–silicon bond formation with high yield.138
Still, methods are needed to evaluate the prevalence of epistasis in a chosen design space to predict the utility of using MLDE over traditional approaches. As the number of simultaneously mutated residues increases, so will the epistatic complexity of the fitness landscape, and thus MLDE should be evaluated on combinatorial libraries with differing numbers of mutated sites. It is important to understand where epistatic interactions confound optimization by simple hill climbing (DE). Interacting residues near the active site of enzymes are likely to have more epistatic combinations of mutations, and the effects of mutations at these sites may be harder to predict.140 Similarly, studies should also explore how fitness landscapes are similar or different between different types of proteins, i.e., binding proteins, enzymes, and synthetic landscapes developed using evolutionary priors.141 Ultimately, combinatorial mutagenesis data sets on additional protein families are necessary for understanding when MLDE is useful. In addition to developing high-throughput assays to map protein sequences to fitnesses,142−146 it will be important to develop general and realistic mathematical models to describe protein fitness landscapes (Figure 3A).141,147−150
Alternatively, if a design space is believed to have minimal epistasis, it may be effective to assume that single mutation effects are largely additive and use recombination of beneficial mutations to find improvements. In current DE workflows, beneficial mutations found in experimental screens are mixed using methods such as DNA shuffling or StEP recombination.7,27 Experimental screens usually measure only a fraction of all possible single mutants, unless all sites are subjected to saturation mutagenesis, which can be time- and cost-prohibitive. Several promising studies have shown that supervised ML models can generally extrapolate well from a subset of single mutants to all possible single mutants of a protein on deep mutational scanning (DMS) landscapes, looking at natural function.127,151 These studies should be extended to understand how effective ML is for predicting recombination outcomes or choosing sites for further exploration.
3.2. Developing a Better Understanding of Zero-Shot Predictors for Different Protein Families and Functions
ZS predictors can help guide engineering toward higher protein fitness without any labeled data from experimental screens. In focused-training MLDE (ftMLDE), sampling training libraries with variants having favorable ZS scores yielded ML models with better performance than random sampling.129 Single mutant fitness prediction is also improved by combining sequence encodings with ZS scores,127 and proteins can possibly be engineered toward higher fitness using evolutionary ZS scores alone.152 For example, antibodies were engineered toward higher binding affinity using PLM likelihoods128 and higher virus neutralization using inverse folding models153 despite only screening 20–30 variants per round. Luciferase and chorismate mutase enzyme variants with higher stability and activity have also been identified using evolutionary ZS scores.154−157 The potential to improve protein engineering outcomes using ZS scores has warranted significant attention (reviewed here158), as calculating ZS scores does not require collecting fitness labels through expensive experimental assays. However, a method based purely on evolutionary conservation may have limitations.
Many ZS predictors have only been extensively evaluated on data sets measuring native function or activity, such as the ProteinGym DMS data sets.151 For example, ZS scores based on MSAs can predict protein variants that are more likely based on evolutionary conservation and coevolution.151,159−162 Likelihoods derived from PLMs trained on known protein sequences88,94,151,163−171 and inverse folding models108,172,173 are also able to learn these implicit evolutionary and biochemical constraints (Figure 3B). There are additional efforts to improve the accuracy of ZS predictors by using structure and reducing bias toward variants with many mutations.174,175 However, none of these models capture function that is not found in nature, and most studies have focused on well-studied protein families. Thus, ZS predictors need to be evaluated on proteins from different families for native and non-native functions.
Engineering enzymes for non-native activity can be challenging because many mutations that are beneficial to activity are also destabilizing.115,176,177 Proteins can tolerate such destabilizing mutations only up to a threshold, beyond which the protein will be unfolded.114 Thus, computed stability (ΔΔGmut-wt) as a ZS score will be more correlated with fitness if the protein is marginally stable,178 as destabilization is more likely to cause loss of function in these proteins, such as on GB1.129,179 A highly stable protein, on the other hand, can tolerate multiple destabilizing mutations before it loses function; stability effects will likely not be correlated with activity for such a protein. In short, the predictive power of various ZS scores should be evaluated on existing and future data sets, to understand whether protein function, family, or other biochemical insights can be used to decide which ZS scores will be useful for a particular engineering goal.
3.3. Expanding the Power of ML Methods to Optimize Protein Fitness
There is also a critical need to improve supervised ML approaches to better capture patterns in data to more efficiently identify variants with high fitness. Developing higher throughput screens to obtain more data is one way to achieve improved model performance, but that of course will also improve the performance of the laboratory approach alone. In this Outlook, we focus on computational approaches that can lead to better predictions from ML models.
There is significant potential for developing more effective representations of proteins, and alongside them, evaluation metrics for these representations.180,181 The most simplified encodings used in ML models linking sequence to fitness include one-hot encodings of amino acid types and Georgiev parameters capturing fixed amino acid descriptors.182 As an alternative, learned embeddings can be extracted from PLMs, such as those mentioned above. While these representations can offer performance boosts for certain tasks,183 they have not yet offered significant performance boosts compared to simple sequence encodings for supervised fitness prediction in MLDE129 or relevant protein engineering benchmarks such as predicting multimutant fitness from the fitness effects of single mutations.165,181 Fine-tuning and semisupervised learning are other strategies to augment model performance when only a small amount of labeled data is available; this has shown initial promise but should be explored further.184 Additional benchmarks are needed to evaluate whether learned embeddings are more effective for ML-assisted protein fitness prediction.
As an alternative to PLMs, there are efforts to improve representations of proteins using multimodal data (Figure 3C). It is generally agreed that for many proteins, sequence determines structure, and structure strongly influences function. Thus, there have been efforts to enrich protein representations by incorporating structural information using voxels, contact maps, or graph neural networks.185−192 However, these have not led to significant performance improvements, likely because variant structures vary in subtle yet impactful ways which are challenging to model and extremely difficult to observe experimentally, despite an explosion in protein structure prediction tools. Many available protein structures may be quite noisy or inaccurate. In addition, proteins do not carry out their functions as static structures, which means that features such as dynamics and conformational changes, which could be generated using physics-based simulations or measured with experimental spectroscopic methods, could be useful.193−198 Because many protein fitness tasks involve variants with very few mutations from a parent protein, future efforts should explore whether representations can be learned locally on protein variants199 as opposed to global databases. Potentially these representations could then be fine-tuned for fitness prediction.
There has also been limited work exploring active-site focused representations,199−201 as the shape and electronics of an enzyme active site can strongly influence its reactivity.202 A related approach is taken by MutCompute, which trains a model to classify wild-type amino acids, based on their neighboring structural microenvironments.75,76 MutCompute was successfully used in wet-lab experiments to enhance the activity of hydrolases for PET depolymerization (plastic degradation).77 Joint protein–substrate representations have been studied to predict enzymatic activity for various substrate transformations, but these joint models did not perform better than independent models.203,204 Additionally, there exist deep learning methods that can dock substrates with proteins to predict their joint structures.205,206 A future generalized enzyme fitness prediction model would be able to incorporate multimodal information about both protein and substrate and simultaneously predict important properties such as expression, stability, and activity for various reactions (Figure 3C).207 Such models would be highly practical and impactful.
Protein fitness optimization is well suited for active learning on an expanded search space, and this area of research has significant room for growth (Figure 3D).31,132,208,209 Broadly, active learning is an iterative cycle that alternates between wet-lab experiments to synthesize/screen enzymes and computational modeling to propose the next set of enzymes to test, typically guided by uncertainty quantification. The goal of finding a protein variant with maximum (or at least greatly improved) fitness, is particularly aligned with Bayesian optimization (BO), which is a form of active learning. Several studies have used Gaussian process models with BO to optimize chimeric proteins.122,130,131,133 In an early wet-lab example, P450 enzyme thermostability was improved efficiently using an iterative BO approach.122 However, to engineer new enzymatic activities, protein variants with point mutations may be more interesting and promising to explore.210−214 BO approaches with adaptive sampling have been tested on existing data sets,215−218 and meta learning has been explored as way to utilize clean and noisy data for antibody engineering.219 An active-learning approach would more efficiently find solutions in larger design spaces, thus allowing protein engineers to expand their search to sequences with increased numbers of mutations at increased numbers of sites simultaneously mutated. An added advantage over DE is that BO allows for optimization of multiple properties simultaneously in a mathematically principled way.220
At the same time, new classes of ML models should be developed for protein fitness prediction to take advantage of uncertainty and introduce helpful inductive biases for the domain.221,222 There exist methods that take advantage of inductive biases and prior information about proteins, such as the assumption that most mutation effects are additive or incorporation of biophysical knowledge into models as priors.223−229 Another method biases the search toward variants with fewer mutations, which are more likely to be stable and functional.230 Domain-specific self-supervision has been explored by training models on codons rather than amino acid sequences.90,231,232 There are also efforts to utilize calibrated uncertainty about predicted fitnesses of proteins that lie out of the domain of previously screened proteins from the training set, but there is a need to expand and further test these methods in real settings.208,233 It is still an open question whether supervised models can extrapolate beyond their training data to predict novel proteins.234,235 More expressive deep learning methods, such as deep kernels,236,237 could be explored as an alternative to Gaussian processes for uncertainty quantification in BO. Overall, there is significant potential to improve ML-based protein fitness prediction to help guide the search toward proteins with ideal fitness.
4. Conclusion: Toward General, Self-Driven Protein Engineering
ML can complement many steps in existing enzyme engineering workflows, and it will play an increasingly important role in the future. Before beginning an enzyme fitness improvement campaign, classification models and generative ML models have the potential to unlock new enzymes with diverse functions, evolvabilities, and folds. Afterward, supervised ML offers a unique opportunity to accelerate protein fitness optimization by more efficiently choosing which protein variants to synthesize and screen, and it can suggest protein variants that would not normally be considered by the limited scope of DE.
On the computational side, there remain many open questions about how to use ML for enzyme engineering, and which ML-assisted methods would have the most real-world impact if successful. In this Outlook, we have suggested that discovery and generation should focus on identifying promiscuous and evolvable enzymes with new activities and folds. A wealth of diverse protein starting points remain to be discovered, and ML is well suited to identify patterns and efficiently sift through the haystack of existing proteins. ML has also demonstrated utility for navigating protein fitness landscapes, but we believe that a greater understanding of epistasis and the role of various ZS predictors is needed. Furthermore, ML models mapping sequence to fitness would benefit from improved representations of protein variants, utilization of uncertainty in predictions, and tailored models with inductive biases relevant to proteins. Here, ML allows for bigger jumps in protein sequence space than would be possible with DE. Perhaps in the future, the optimization step may not even be necessary if protein fitness information can be incorporated into generative models as part of the discovery step.
Protein fitness improvement is poised to become a fully automated process, with implications across many industries. There is already work on developing automated evolution systems and integrating these into active learning workflows where data generated from automated experiments can train and refine ML models to suggest beneficial variants to explore further.132,238,239 These “design-build-test-learn” cycles would enable continuous optimization of enzymes and other proteins (Figure 4), as they can for small molecules.240 LLMs could power these automated systems, with AI flexibly adapting to perform new types of syntheses and screens with robotic scripts written on the fly.241−244 At the same time, multiple desirable properties and activity for multiple reactions could be optimized simultaneously during protein engineering campaigns, powered by generalized ML models that can utilize multimodal representations of proteins. With ever increasing amounts of data on protein structures and sequence-fitness pairs, and new tools to conduct experiments245−248 and make ML methods for proteins more accessible to the broader community,249 the future of ML-assisted protein engineering is bright.
Figure 4.
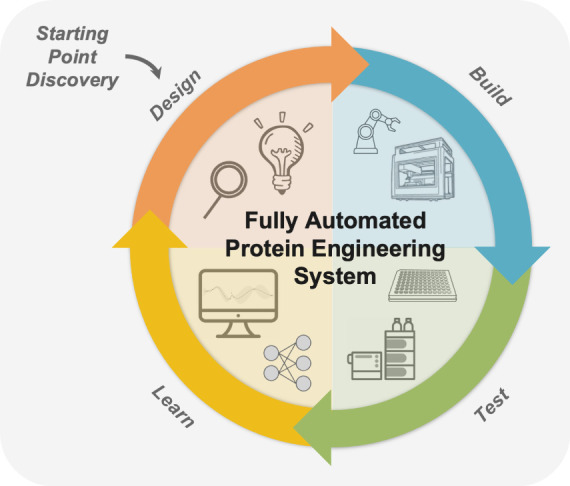
A fully self-driven protein engineering system as an active learning “design-build-test-learn” cycle assisted by machine learning. Emerging ML-assisted methods will provide an increased diversity of protein starting points that possess desired function and are highly evolvable. Automated robotic systems will synthesize protein variants and test them for various properties using experimental assays. Supervised ML models will then be trained to learn a mapping between protein features and their properties. Finally, design algorithms will propose new variants to test in the next iteration and update robotic scripts on the fly. This protein engineering system will perform automated end-to-end discovery and optimization of proteins for desired functions.
Acknowledgments
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0022218. This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. This work was also supported by an Amgen Chem-Bio-Engineering Award (CBEA) and by the NSF Division of Chemical, Bioengineering, Environmental and Transport Systems (CBET 1937902). J.Y. and F.Z.L are partially supported by National Science Foundation Graduate Research Fellowships. The authors thank Kadina Johnston and Sabine Brinkmann-Chen for helpful discussions and critical reading of the manuscript.
Glossary
Abbreviations
- DE
Directed Evolution
- ML
Machine Learning
- MSA
Multiple Sequence Alignment
- EC
Enzyme Commission
- LLM
Large Language Model
- GPT
Generative Pretrained Transformer
- AI
Artificial Intelligence
- PLM
Protein Language Model
- VAE
Variational Autoencoder
- GAN
Generative Adversarial Network
- ZS
Zero-shot
- MLDE
Machine Learning-Assisted Directed Evolution
- DMS
Deep Mutational Scanning
- BO
Bayesian Optimization
The authors declare no competing financial interest.
References
- Arnold F. H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem., Int. Ed. 2018, 57 (16), 4143–4148. 10.1002/anie.201708408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E. L.; Finnigan W.; France S. P.; Green A. P.; Hayes M. A.; Hepworth L. J.; Lovelock S. L.; Niikura H.; Osuna S.; Romero E.; Ryan K. S.; Turner N. J.; Flitsch S. L. Biocatalysis. Nat. Rev. Methods Primer 2021, 1 (1), 46. 10.1038/s43586-021-00044-z. [DOI] [Google Scholar]
- Buller R.; Lutz S.; Kazlauskas R. J.; Snajdrova R.; Moore J. C.; Bornscheuer U. T. From Nature to Industry: Harnessing Enzymes for Biocatalysis. Science 2023, 382 (6673), eadh8615 10.1126/science.adh8615. [DOI] [PubMed] [Google Scholar]
- Romero P. A.; Arnold F. H. Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Bio 2009, 10, 866–876. 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce N. A.; Winfree E. Protein Design Is NP-Hard. Protein Eng. Des. Sel. 2002, 15 (10), 779–782. 10.1093/protein/15.10.779. [DOI] [PubMed] [Google Scholar]
- Chen K.; Arnold F. H. Engineering New Catalytic Activities in Enzymes. Nat. Catal. 2020, 3 (3), 203–213. 10.1038/s41929-019-0385-5. [DOI] [Google Scholar]
- Packer M. S.; Liu D. R. Methods for the Directed Evolution of Proteins. Nat. Rev. Genet. 2015, 16 (7), 379–394. 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xue P.; Cao M.; Yu T.; Lane S. T.; Zhao H. Directed Evolution: Methodologies and Applications. Chem. Rev. 2021, 121 (20), 12384–12444. 10.1021/acs.chemrev.1c00260. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T.; Huisman G. W.; Kazlauskas R. J.; Lutz S.; Moore J. C.; Robins K. Engineering the Third Wave of Biocatalysis. Nature 2012, 485 (7397), 185–194. 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- Miller D. C.; Athavale S. V.; Arnold F. H. Combining Chemistry and Protein Engineering for New-to-Nature Biocatalysis. Nat. Synth. 2022, 1 (1), 18–23. 10.1038/s44160-021-00008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveson-Gower R. B.; Mayer C.; Roelfes G. The Importance of Catalytic Promiscuity for Enzyme Design and Evolution. Nat. Rev. Chem. 2019, 3 (12), 687–705. 10.1038/s41570-019-0143-x. [DOI] [Google Scholar]
- Knight A. M.; Kan S. B. J.; Lewis R. D.; Brandenberg O. F.; Chen K.; Arnold F. H. Diverse Engineered Heme Proteins Enable Stereodivergent Cyclopropanation of Unactivated Alkenes. ACS Cent. Sci. 2018, 4 (3), 372–377. 10.1021/acscentsci.7b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedbrook C. N.; Rice A. J.; Yang K. K.; Ding X.; Chen S.; LeProust E. M.; Gradinaru V.; Arnold F. H. Structure-Guided SCHEMA Recombination Generates Diverse Chimeric Channelrhodopsins. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (13), E2624–E2633. 10.1073/pnas.1700269114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt C. A.; Martinez C.; Wang Z.-G.; Mayo S. L.; Arnold F. H. Protein Building Blocks Preserved by Recombination. Nat. Struct. Biol. 2002, 9 (7), 553–558. 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- Merkl R.; Sterner R. Ancestral Protein Reconstruction: Techniques and Applications. Biol. Chem. 2016, 397 (1), 1–21. 10.1515/hsz-2015-0158. [DOI] [PubMed] [Google Scholar]
- Alford R. F.; Leaver-Fay A.; Jeliazkov J. R.; O’Meara M. J.; DiMaio F. P.; Park H.; Shapovalov M. V.; Renfrew P. D.; Mulligan V. K.; Kappel K.; Labonte J. W.; Pacella M. S.; Bonneau R.; Bradley P.; Dunbrack R. L.; Das R.; Baker D.; Kuhlman B.; Kortemme T.; Gray J. J. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13 (6), 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenzweig A.; Goldsmith M.; Hill S. E.; Gertman O.; Laurino P.; Ashani Y.; Dym O.; Unger T.; Albeck S.; Prilusky J.; Lieberman R. L.; Aharoni A.; Silman I.; Sussman J. L.; Tawfik D. S.; Fleishman S. J. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63 (2), 337–346. 10.1016/j.molcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Lipsh R.; Avizemer Z.; Ashani Y.; Goldsmith M.; Leader H.; Dym O.; Rogotner S.; Trudeau D. L.; Prilusky J.; Amengual-Rigo P.; Guallar V.; Tawfik D. S.; Fleishman S. J. Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72 (1), 178–186. 10.1016/j.molcel.2018.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J. J.; Goldenzweig A.; Hoch S.; Fleishman S. J. PROSS 2: A New Server for the Design of Stable and Highly Expressed Protein Variants. Bioinformatics 2021, 37 (1), 123–125. 10.1093/bioinformatics/btaa1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman J. K.; Weitzner B. D.; Lewis S. M.; Adolf-Bryfogle J.; Alam N.; Alford R. F.; Aprahamian M.; Baker D.; Barlow K. A.; Barth P.; Basanta B.; Bender B. J.; Blacklock K.; Bonet J.; Boyken S. E.; Bradley P.; Bystroff C.; Conway P.; Cooper S.; Correia B. E.; Coventry B.; Das R.; De Jong R. M.; DiMaio F.; Dsilva L.; Dunbrack R.; Ford A. S.; Frenz B.; Fu D. Y.; Geniesse C.; Goldschmidt L.; Gowthaman R.; Gray J. J.; Gront D.; Guffy S.; Horowitz S.; Huang P.-S.; Huber T.; Jacobs T. M.; Jeliazkov J. R.; Johnson D. K.; Kappel K.; Karanicolas J.; Khakzad H.; Khar K. R.; Khare S. D.; Khatib F.; Khramushin A.; King I. C.; Kleffner R.; Koepnick B.; Kortemme T.; Kuenze G.; Kuhlman B.; Kuroda D.; Labonte J. W.; Lai J. K.; Lapidoth G.; Leaver-Fay A.; Lindert S.; Linsky T.; London N.; Lubin J. H.; Lyskov S.; Maguire J.; Malmström L.; Marcos E.; Marcu O.; Marze N. A.; Meiler J.; Moretti R.; Mulligan V. K.; Nerli S.; Norn C.; Ó’Conchúir S.; Ollikainen N.; Ovchinnikov S.; Pacella M. S.; Pan X.; Park H.; Pavlovicz R. E.; Pethe M.; Pierce B. G.; Pilla K. B.; Raveh B.; Renfrew P. D.; Burman S. S. R.; Rubenstein A.; Sauer M. F.; Scheck A.; Schief W.; Schueler-Furman O.; Sedan Y.; Sevy A. M.; Sgourakis N. G.; Shi L.; Siegel J. B.; Silva D.-A.; Smith S.; Song Y.; Stein A.; Szegedy M.; Teets F. D.; Thyme S. B.; Wang R. Y.-R.; Watkins A.; Zimmerman L.; Bonneau R. Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Methods 2020, 17 (7), 665–680. 10.1038/s41592-020-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J.; Borg J.; Stricher F.; Nys R.; Rousseau F.; Serrano L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33, W382–W388. 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J. B.; Zanghellini A.; Lovick H. M.; Kiss G.; Lambert A. R.; St Clair J. L.; Gallaher J. L.; Hilvert D.; Gelb M. H.; Stoddard B. L.; Houk K. N.; Michael F. E.; Baker D. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329 (5989), 309–313. 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvet I.; Ortmayer M.; Zhao J.; Crawshaw R.; Ennist N. M.; Levy C.; Roy A.; Green A. P.; Baker D. Design of Heme Enzymes with a Tunable Substrate Binding Pocket Adjacent to an Open Metal Coordination Site. J. Am. Chem. Soc. 2023, 145 (26), 14307–14315. 10.1021/jacs.3c02742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.-S.; Boyken S. E.; Baker D. The Coming of Age of de Novo Protein Design. Nature 2016, 537 (7620), 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- Smith J. M. Natural Selection and the Concept of a Protein Space. Nature 1970, 225, 563–564. 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- Drummond D. A.; Silberg J. J.; Meyer M. M.; Wilke C. O.; Arnold F. H. On the Conservative Nature of Intragenic Recombination. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (15), 5380–5385. 10.1073/pnas.0500729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Giver L.; Shao Z.; Affholter J. A.; Arnold F. H. Molecular Evolution by Staggered Extension Process (StEP) in Vitro Recombination. Nat. Biotechnol. 1998, 16 (3), 258–261. 10.1038/nbt0398-258. [DOI] [PubMed] [Google Scholar]
- Yang K. K.; Wu Z.; Arnold F. H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16 (8), 687–694. 10.1038/s41592-019-0496-6. [DOI] [PubMed] [Google Scholar]
- Wittmann B. J.; Johnston K. E.; Wu Z.; Arnold F. H. Advances in Machine Learning for Directed Evolution. Curr. Opin. Struct. Biol. 2021, 69, 11–18. 10.1016/j.sbi.2021.01.008. [DOI] [PubMed] [Google Scholar]
- Freschlin C. R.; Fahlberg S. A.; Romero P. A. Machine Learning to Navigate Fitness Landscapes for Protein Engineering. Curr. Opin. Biotechnol. 2022, 75, 102713 10.1016/j.copbio.2022.102713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hie B. L.; Yang K. K. Adaptive Machine Learning for Protein Engineering. Curr. Opin. Struct. Biol. 2022, 72, 145–152. 10.1016/j.sbi.2021.11.002. [DOI] [PubMed] [Google Scholar]
- Ferguson A. L.; Ranganathan R. 100th Anniversary of Macromolecular Science Viewpoint: Data-Driven Protein Design. ACS Macro Lett. 2021, 10 (3), 327–340. 10.1021/acsmacrolett.0c00885. [DOI] [PubMed] [Google Scholar]
- Mardikoraem M.; Woldring D.. Machine Learning-Driven Protein Library Design: A Path Toward Smarter Libraries. In Yeast Surface Display; Traxlmayr M. W., Ed.; Springer U.S.: New York, NY, 2022; pp 87–104 10.1007/978-1-0716-2285-8_5. [DOI] [PubMed] [Google Scholar]
- Yu T.; Boob A. G.; Volk M. J.; Liu X.; Cui H.; Zhao H. Machine Learning-Enabled Retrobiosynthesis of Molecules. Nat. Catal. 2023, 6 (2), 137–151. 10.1038/s41929-022-00909-w. [DOI] [Google Scholar]
- Strokach A.; Kim P. M. Deep Generative Modeling for Protein Design. Curr. Opin. Struct. Biol. 2022, 72, 226–236. 10.1016/j.sbi.2021.11.008. [DOI] [PubMed] [Google Scholar]
- Johnston K. E.; Fannjiang C.; Wittmann B. J.; Hie B. L.; Yang K. K.; Wu Z.. Machine Learning for Protein Engineering; 2023.
- Kouba P.; Kohout P.; Haddadi F.; Bushuiev A.; Samusevich R.; Sedlar J.; Damborsky J.; Pluskal T.; Sivic J.; Mazurenko S. Machine Learning-Guided Protein Engineering. ACS Catal. 2023, 13 (21), 13863–13895. 10.1021/acscatal.3c02743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium; Bateman A.; Martin M.-J.; Orchard S.; Magrane M.; Ahmad S.; Alpi E.; Bowler-Barnett E. H.; Britto R.; Bye-A-Jee H.; Cukura A.; Denny P.; Dogan T.; Ebenezer T.; Fan J.; Garmiri P.; Da Costa Gonzales L. J.; Hatton-Ellis E.; Hussein A.; Ignatchenko A.; Insana G.; Ishtiaq R.; Joshi V.; Jyothi D.; Kandasaamy S.; Lock A.; Luciani A.; Lugaric M.; Luo J.; Lussi Y.; MacDougall A.; Madeira F.; Mahmoudy M.; Mishra A.; Moulang K.; Nightingale A.; Pundir S.; Qi G.; Raj S.; Raposo P.; Rice D. L.; Saidi R.; Santos R.; Speretta E.; Stephenson J.; Totoo P.; Turner E.; Tyagi N.; Vasudev P.; Warner K.; Watkins X.; Zaru R.; Zellner H.; Bridge A. J.; Aimo L.; Argoud-Puy G.; Auchincloss A. H.; Axelsen K. B.; Bansal P.; Baratin D.; Batista Neto T. M.; Blatter M.-C.; Bolleman J. T.; Boutet E.; Breuza L.; Gil B. C.; Casals-Casas C.; Echioukh K. C.; Coudert E.; Cuche B.; De Castro E.; Estreicher A.; Famiglietti M. L.; Feuermann M.; Gasteiger E.; Gaudet P.; Gehant S.; Gerritsen V.; Gos A.; Gruaz N.; Hulo C.; Hyka-Nouspikel N.; Jungo F.; Kerhornou A.; Le Mercier P.; Lieberherr D.; Masson P.; Morgat A.; Muthukrishnan V.; Paesano S.; Pedruzzi I.; Pilbout S.; Pourcel L.; Poux S.; Pozzato M.; Pruess M.; Redaschi N.; Rivoire C.; Sigrist C. J. A.; Sonesson K.; Sundaram S.; Wu C. H.; Arighi C. N.; Arminski L.; Chen C.; Chen Y.; Huang H.; Laiho K.; McGarvey P.; Natale D. A.; Ross K.; Vinayaka C. R.; Wang Q.; Wang Y.; Zhang J. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51 (D1), D523–D531. 10.1093/nar/gkac1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak W. S.; Tran S.; Marcheschi R.; Bertolani S.; Thompson J.; Baker D.; Liao J. C.; Siegel J. B. Integrative Genomic Mining for Enzyme Function to Enable Engineering of a Non-Natural Biosynthetic Pathway. Nat. Commun. 2015, 6 (1), 10005 10.1038/ncomms10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T.; Cui H.; Li J. C.; Luo Y.; Jiang G.; Zhao H. Enzyme Function Prediction Using Contrastive Learning. Science 2023, 379 (6639), 1358–1363. 10.1126/science.adf2465. [DOI] [PubMed] [Google Scholar]
- Zheng L.; Shi S.; Fang P.; Zhang H.; Pan Z.; Huang S.; Xia W.; Li H.; Zeng Z.; Zhang S.; Chen Y.; Lu M.; Li Z.; Zhu F.. AnnoPRO: An Innovative Strategy for Protein Function Annotation Based on Image-like Protein Representation and Multimodal Deep Learning. bioRxiv; preprint, 2023 10.1101/2023.05.13.540619. [DOI] [Google Scholar]
- Bileschi M. L.; Belanger D.; Bryant D. H.; Sanderson T.; Carter B.; Sculley D.; Bateman A.; DePristo M. A.; Colwell L. J. Using Deep Learning to Annotate the Protein Universe. Nat. Biotechnol. 2022, 40 (6), 932–937. 10.1038/s41587-021-01179-w. [DOI] [PubMed] [Google Scholar]
- Feehan R.; Franklin M. W.; Slusky J. S. G. Machine Learning Differentiates Enzymatic and Non-Enzymatic Metals in Proteins. Nat. Commun. 2021, 12 (1), 3712. 10.1038/s41467-021-24070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson A. M.; Mofrad M. R. K.. Fine-Tuning Protein Embeddings for Generalizable Annotation Propagation. bioRxiv; preprint, 2023 10.1101/2023.06.22.546084. [DOI] [Google Scholar]
- Li F.; Yuan L.; Lu H.; Li G.; Chen Y.; Engqvist M. K. M.; Kerkhoven E. J.; Nielsen J. Deep Learning-Based Kcat Prediction Enables Improved Enzyme-Constrained Model Reconstruction. Nat. Catal. 2022, 5 (8), 662–672. 10.1038/s41929-022-00798-z. [DOI] [Google Scholar]
- Thurimella K.; Mohamed A. M. T.; Graham D. B.; Owens R. M.; La Rosa S. L.; Plichta D. R.; Bacallado S.; Xavier R. J.. Protein Language Models Uncover Carbohydrate-Active Enzyme Function in Metagenomics. bioRxiv; preprint, 2023 10.1101/2023.10.23.563620. [DOI] [Google Scholar]
- Derry A.; Altman R. B. Explainable Protein Function Annotation Using Local Structure Embeddings. bioRxiv; preprint, 2023 10.1101/2023.10.13.562298. [DOI] [Google Scholar]
- Buton N.; Coste F.; Le Cunff Y. Predicting Enzymatic Function of Protein Sequences with Attention. Bioinformatics 2023, 39 (10), btad620 10.1093/bioinformatics/btad620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visani G. M.; Hughes M. C.; Hassoun S. Enzyme Promiscuity Prediction Using Hierarchy-Informed Multi-Label Classification. Bioinformatics 2021, 37 (14), 2017–2024. 10.1093/bioinformatics/btab054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller P.; Laino T.; Gaudin T.; Bolgar P.; Hunter C. A.; Bekas C.; Lee A. A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5 (9), 1572–1583. 10.1021/acscentsci.9b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.; Swanson K.; Jin W.; Coley C.; Eiden P.; Gao H.; Guzman-Perez A.; Hopper T.; Kelley B.; Mathea M.; Palmer A.; Settels V.; Jaakkola T.; Jensen K.; Barzilay R. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59 (8), 3370–3388. 10.1021/acs.jcim.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutter D.; Schwaller P.; Reymond J.-L. Predicting Enzymatic Reactions with a Molecular Transformer. Chem. Sci. 2021, 12 (25), 8648–8659. 10.1039/D1SC02362D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4 (2), 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid E.; Probst D.; Green W. H.; Madsen G. K. H. EnzymeMap: Curation, Validation and Data-Driven Prediction of Enzymatic Reactions. bioRxiv; preprint, 2023 10.26434/chemrxiv-2023-jzw9w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach S.; Dienhart H.; Range J.; Malzacher S.; Spöring J.-D.; Rother D.; Pinto M. F.; Martins P.; Lagerman C. E.; Bommarius A. S.; Høst A. V.; Woodley J. M.; Ngubane S.; Kudanga T.; Bergmann F. T.; Rohwer J. M.; Iglezakis D.; Weidemann A.; Wittig U.; Kettner C.; Swainston N.; Schnell S.; Pleiss J. EnzymeML: Seamless Data Flow and Modeling of Enzymatic Data. Nat. Methods 2023, 20 (3), 400–402. 10.1038/s41592-022-01763-1. [DOI] [PubMed] [Google Scholar]
- Groth P. M.; Michael R.; Salomon J.; Tian P.; Boomsma W.. FLOP: Tasks for Fitness Landscapes Of Protein Wildtypes. bioRxiv; preprint, 2023 10.1101/2023.06.21.545880. [DOI] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M.; DiMaio F.; Anishchenko I.; Dauparas J.; Ovchinnikov S.; Lee G. R.; Wang J.; Cong Q.; Kinch L. N.; Schaeffer R. D.; Millán C.; Park H.; Adams C.; Glassman C. R.; DeGiovanni A.; Pereira J. H.; Rodrigues A. V.; van Dijk A. A.; Ebrecht A. C.; Opperman D. J.; Sagmeister T.; Buhlheller C.; Pavkov-Keller T.; Rathinaswamy M. K.; Dalwadi U.; Yip C. K.; Burke J. E.; Garcia K. C.; Grishin N. V.; Adams P. D.; Read R. J.; Baker D. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373 (6557), 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z.; Akin H.; Rao R.; Hie B.; Zhu Z.; Lu W.; Smetanin N.; Verkuil R.; Kabeli O.; Shmueli Y.; dos Santos Costa A.; Fazel-Zarandi M.; Sercu T.; Candido S.; Rives A. Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model. Science 2023, 379 (6637), 1123–1130. 10.1126/science.ade2574. [DOI] [PubMed] [Google Scholar]
- Wu R.; Ding F.; Wang R.; Shen R.; Zhang X.; Luo S.; Su C.; Wu Z.; Xie Q.; Berger B.; Ma J.; Peng J.. High-Resolution de Novo Structure Prediction from Primary Sequence. bioRxiv; preprint, 2022 10.1101/2022.07.21.500999. [DOI] [Google Scholar]
- Krishna R.; Wang J.; Ahern W.; Sturmfels P.; Venkatesh P.; Kalvet I.; Lee G. R.; Morey-Burrows F. S.; Anishchenko I.; Humphreys I. R.; McHugh R.; Vafeados D.; Li X.; Sutherland G. A.; Hitchcock A.; Hunter C. N.; Baek M.; DiMaio F.; Baker D.. Generalized Biomolecular Modeling and Design with RoseTTAFold All-Atom. bioRxiv; preprint, 2023 10.1101/2023.10.09.561603. [DOI] [PubMed] [Google Scholar]
- Chowdhury R.; Bouatta N.; Biswas S.; Floristean C.; Kharkar A.; Roy K.; Rochereau C.; Ahdritz G.; Zhang J.; Church G. M.; Sorger P. K.; AlQuraishi M. Single-Sequence Protein Structure Prediction Using a Language Model and Deep Learning. Nat. Biotechnol. 2022, 40 (11), 1617–1623. 10.1038/s41587-022-01432-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio-Hernandez I.; Yeo J.; Jänes J.; Mirdita M.; Gilchrist C. L. M.; Wein T.; Varadi M.; Velankar S.; Beltrao P.; Steinegger M. Clustering-Predicted Structures at the Scale of the Known Protein Universe. Nature 2023, 622, 637. 10.1038/s41586-023-06510-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riziotis I. G.; Ribeiro A. J. M.; Borkakoti N.; Thornton J. M. The 3D Modules of Enzyme Catalysis: Deconstructing Active Sites into Distinct Functional Entities. J. Mol. Biol. 2023, 435 (20), 168254 10.1016/j.jmb.2023.168254. [DOI] [PubMed] [Google Scholar]
- Hu B.; Tan C.; Xia J.; Zheng J.; Huang Y.; Wu L.; Liu Y.; Xu Y.; Li S. Z.. Learning Complete Protein Representation by Deep Coupling of Sequence and Structure. bioRxiv; preprint, 2023 10.1101/2023.07.05.547769. [DOI] [Google Scholar]
- Ock J.; Guntuboina C.; Barati Farimani A. Catalyst Energy Prediction with CatBERTa: Unveiling Feature Exploration Strategies through Large Language Models. ACS Catal. 2023, 13 (24), 16032–16044. 10.1021/acscatal.3c04956. [DOI] [Google Scholar]
- Luo R.; Sun L.; Xia Y.; Qin T.; Zhang S.; Poon H.; Liu T.-Y. BioGPT: Generative Pre-Trained Transformer for Biomedical Text Generation and Mining. Brief. Bioinform. 2022, 23 (6), bbac409 10.1093/bib/bbac409. [DOI] [PubMed] [Google Scholar]
- Gligorijević V.; Renfrew P. D.; Kosciolek T.; Leman J. K.; Berenberg D.; Vatanen T.; Chandler C.; Taylor B. C.; Fisk I. M.; Vlamakis H.; Xavier R. J.; Knight R.; Cho K.; Bonneau R. Structure-Based Protein Function Prediction Using Graph Convolutional Networks. Nat. Commun. 2021, 12 (1), 1–14. 10.1038/s41467-021-23303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Rué Casamajo A.; Finnigan W.; Schnepel C.; Barker R.; Morrill C.; Heath R. S.; De Maria L.; Turner N. J.; Scrutton N. S. Structure-Based Design of Small Imine Reductase Panels for Target Substrates. ACS Catal. 2023, 13, 12310–12321. 10.1021/acscatal.3c02278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlopoulos G. A.; Baltoumas F. A.; Liu S.; Selvitopi O.; Camargo A. P.; Nayfach S.; Azad A.; Roux S.; Call L.; Ivanova N. N.; Chen I. M.; Paez-Espino D.; Karatzas E.; Acinas S. G.; Ahlgren N.; Attwood G.; Baldrian P.; Berry T.; Bhatnagar J. M.; Bhaya D.; Bidle K. D.; Blanchard J. L.; Boyd E. S.; Bowen J. L.; Bowman J.; Brawley S. H.; Brodie E. L.; Brune A.; Bryant D. A.; Buchan A.; Cadillo-Quiroz H.; Campbell B. J.; Cavicchioli R.; Chuckran P. F.; Coleman M.; Crowe S.; Colman D. R.; Currie C. R.; Dangl J.; Delherbe N.; Denef V. J.; Dijkstra P.; Distel D. D.; Eloe-Fadrosh E.; Fisher K.; Francis C.; Garoutte A.; Gaudin A.; Gerwick L.; Godoy-Vitorino F.; Guerra P.; Guo J.; Habteselassie M. Y.; Hallam S. J.; Hatzenpichler R.; Hentschel U.; Hess M.; Hirsch A. M.; Hug L. A.; Hultman J.; Hunt D. E.; Huntemann M.; Inskeep W. P.; James T. Y.; Jansson J.; Johnston E. R.; Kalyuzhnaya M.; Kelly C. N.; Kelly R. M.; Klassen J. L.; Nusslein K.; Kostka J. E.; Lindow S.; Lilleskov E.; Lynes M.; Mackelprang R.; Martin F. M.; Mason O. U.; McKay R. M.; McMahon K.; Mead D. A.; Medina M.; Meredith L. K.; Mock T.; Mohn W. W.; Moran M. A.; Murray A.; Neufeld J. D.; Neumann R.; Norton J. M.; Partida-Martinez L. P.; Pietrasiak N.; Pelletier D.; Reddy T. B. K.; Reese B. K.; Reichart N. J.; Reiss R.; Saito M. A.; Schachtman D. P.; Seshadri R.; Shade A.; Sherman D.; Simister R.; Simon H.; Stegen J.; Stepanauskas R.; Sullivan M.; Sumner D. Y.; Teeling H.; Thamatrakoln K.; Treseder K.; Tringe S.; Vaishampayan P.; Valentine D. L.; Waldo N. B.; Waldrop M. P.; Walsh D. A.; Ward D. M.; Wilkins M.; Whitman T.; Woolet J.; Woyke T.; Iliopoulos I.; Konstantinidis K.; Tiedje J. M.; Pett-Ridge J.; Baker D.; Visel A.; Ouzounis C. A.; Ovchinnikov S.; Buluc A.; Kyrpides N. C. Unraveling the Functional Dark Matter through Global Metagenomics. Nature 2023, 622 (7983), 594–602. 10.1038/s41586-023-06583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsh-Sokolik R.; Khersonsky O.; Schroder S. P.; de Boer C.; Hoch S.-Y.; Davies G. J.; Overkleeft H. S.; Fleishman S. J. Combinatorial Assembly and Design of Enzymes. Science 2023, 379 (6628), 195–201. 10.1126/science.ade9434. [DOI] [PubMed] [Google Scholar]
- Weinstein J. Y.; Martí-Gómez C.; Lipsh-Sokolik R.; Hoch S. Y.; Liebermann D.; Nevo R.; Weissman H.; Petrovich-Kopitman E.; Margulies D.; Ivankov D.; McCandlish D. M.; Fleishman S. J. Designed Active-Site Library Reveals Thousands of Functional GFP Variants. Nat. Commun. 2023, 14 (1), 2890. 10.1038/s41467-023-38099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber-Zucker S.; Mateljak I.; Goldsmith M.; Kupervaser M.; Alcalde M.; Fleishman S. J. Designed High-Redox Potential Laccases Exhibit High Functional Diversity. ACS Catal. 2022, 12 (21), 13164–13173. 10.1021/acscatal.2c03006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez De Santos P.; Mateljak I.; Hoang M. D.; Fleishman S. J.; Hollmann F.; Alcalde M. Repertoire of Computationally Designed Peroxygenases for Enantiodivergent C–H Oxyfunctionalization Reactions. J. Am. Chem. Soc. 2023, 145 (6), 3443–3453. 10.1021/jacs.2c11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff R.; Cole A. W.; Diaz D. J.; Morrow B. R.; Donnell I.; Annapareddy A.; Gollihar J.; Ellington A. D.; Thyer R. Discovery of Novel Gain-of-Function Mutations Guided by Structure-Based Deep Learning. ACS Synth. Biol. 2020, 9 (11), 2927–2935. 10.1021/acssynbio.0c00345. [DOI] [PubMed] [Google Scholar]
- Kulikova A. V.; Diaz D. J.; Loy J. M.; Ellington A. D.; Wilke C. O. Learning the Local Landscape of Protein Structures with Convolutional Neural Networks. J. Biol. Phys. 2021, 47 (4), 435–454. 10.1007/s10867-021-09593-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Diaz D. J.; Czarnecki N. J.; Zhu C.; Kim W.; Shroff R.; Acosta D. J.; Alexander B. R.; Cole H. O.; Zhang Y.; Lynd N. A.; Ellington A. D.; Alper H. S. Machine Learning-Aided Engineering of Hydrolases for PET Depolymerization. Nature 2022, 604 (7907), 662–667. 10.1038/s41586-022-04599-z. [DOI] [PubMed] [Google Scholar]
- Foley G.; Mora A.; Ross C. M.; Bottoms S.; Sützl L.; Lamprecht M. L.; Zaugg J.; Essebier A.; Balderson B.; Newell R.; Thomson R. E. S.; Kobe B.; Barnard R. T.; Guddat L.; Schenk G.; Carsten J.; Gumulya Y.; Rost B.; Haltrich D.; Sieber V.; Gillam E. M. J.; Bodén M. Engineering Indel and Substitution Variants of Diverse and Ancient Enzymes Using Graphical Representation of Ancestral Sequence Predictions (GRASP). PLOS Comput. Biol. 2022, 18 (10), e1010633 10.1371/journal.pcbi.1010633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livada J.; Vargas A. M.; Martinez C. A.; Lewis R. D. Ancestral Sequence Reconstruction Enhances Gene Mining Efforts for Industrial Ene Reductases by Expanding Enzyme Panels with Thermostable Catalysts. ACS Catal. 2023, 13 (4), 2576–2585. 10.1021/acscatal.2c03859. [DOI] [Google Scholar]
- Joy J. B.; Liang R. H.; McCloskey R. M.; Nguyen T.; Poon A. F. Y. Ancestral Reconstruction. PLOS Comput. Biol. 2016, 12 (7), e1004763 10.1371/journal.pcbi.1004763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferruz N.; Heinzinger M.; Akdel M.; Goncearenco A.; Naef L.; Dallago C. From Sequence to Function through Structure: Deep Learning for Protein Design. Comput. Struct. Biotechnol. J. 2023, 21, 238–250. 10.1016/j.csbj.2022.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S.; Huang P.-S. Structure-Based Protein Design with Deep Learning. Curr. Opin. Chem. Biol. 2021, 65, 136–144. 10.1016/j.cbpa.2021.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferruz N.; Höcker B. Controllable Protein Design with Language Models. Nat. Mach. Intell. 2022, 4 (6), 521–532. 10.1038/s42256-022-00499-z. [DOI] [Google Scholar]
- Winnifrith A.; Outeiral C.; Hie B.. Generative Artificial Intelligence for de Novo Protein Design. arXiv 2023. https://arxiv.org/abs/2310.09685. [DOI] [PubMed]
- Wu Z.; Johnston K. E.; Arnold F. H.; Yang K. K. Protein Sequence Design with Deep Generative Models. Curr. Opin. Chem. Biol. 2021, 65, 18–27. 10.1016/j.cbpa.2021.04.004. [DOI] [PubMed] [Google Scholar]
- Sevgen E.; M?ller J.; Lange A.; Parker J.; Quigley S.; Mayer J.; Srivastava P.; Gayatri S.; Hosfield D.; Korshunova M.; Livne M.; Gill M.; Ranganathan R.; Costa A. B.; Ferguson A. L.. ProT-VAE: Protein Transformer Variational AutoEncoder for Functional Protein Design. bioRxiv; preprint, 2023 10.1101/2023.01.23.525232. [DOI] [Google Scholar]
- Praljak N.; Lian X.; Ranganathan R.; Ferguson A. L. ProtWave-VAE: Integrating Autoregressive Sampling with Latent-Based Inference for Data-Driven Protein Design. ACS Synth. Biol. 2023, 12 (12), 3544–3561. 10.1021/acssynbio.3c00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani A.; Krause B.; Greene E. R.; Subramanian S.; Mohr B. P.; Holton J. M.; Olmos J. L.; Xiong C.; Sun Z. Z.; Socher R.; Fraser J. S.; Naik N. Large Language Models Generate Functional Protein Sequences across Diverse Families. Nat. Biotechnol. 2023, 41, 1099. 10.1038/s41587-022-01618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durairaj J.; Waterhouse A. M.; Mets T.; Brodiazhenko T.; Abdullah M.; Studer G.; Akdel M.; Andreeva A.; Bateman A.; Tenson T.; Hauryliuk V.; Schwede T.; Pereira J.. What Is Hidden in the Darkness? Deep-Learning Assisted Large-Scale Protein Family Curation Uncovers Novel Protein Families and Folds. bioRxiv; preprint, 2023 10.1101/2023.03.14.532539. [DOI] [Google Scholar]
- Zvyagin M.; Brace A.; Hippe K.; Deng Y.; Zhang B.; Bohorquez C. O.; Clyde A.; Kale B.; Perez-Rivera D.; Ma H.; Mann C. M.; Irvin M.; Gregory Pauloski J.; Ward L.; Hayot-Sasson V.; Emani M.; Foreman S.; Xie Z.; Lin D.; Shukla M.; Nie W.; Romero J.; Dallago C.; Vahdat A.; Xiao C.; Gibbs T.; Foster I.; Davis J. J.; Papka M. E.; Brettin T.; Stevens R.; Anandkumar A.; Vishwanath V.; Ramanathan A.. GenSLMs: Genome-Scale Language Models Reveal SARS-CoV-2 Evolutionary Dynamics. bioRxiv; preprint, 2022 10.1101/2022.10.10.511571. [DOI] [Google Scholar]
- Verkuil R.; Kabeli O.; Du Y.; Wicky B. I.; Milles L. F.; Dauparas J.; Baker D.; Ovchinnikov S.; Sercu T.; Rives A. Language Models Generalize beyond Natural Proteins 2022, 10.1101/2022.12.21.521521. [DOI] [Google Scholar]
- Sgarbossa D.; Lupo U.; Bitbol A.-F.. Generative Power of a Protein Language Model Trained on Multiple Sequence Alignments. bioRxiv; preprint, 2022 10.1101/2022.04.14.488405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijkamp E.; Ruffolo J. A.; Weinstein E. N.; Naik N.; Madani A. ProGen2: Exploring the Boundaries of Protein Language Models. Cell Syst. 2023, 14 (11), 968–978. 10.1016/j.cels.2023.10.002. [DOI] [PubMed] [Google Scholar]
- Shin J.-E.; Riesselman A. J.; Kollasch A. W.; McMahon C.; Simon E.; Sander C.; Manglik A.; Kruse A. C.; Marks D. S. Protein Design and Variant Prediction Using Autoregressive Generative Models. Nat. Commun. 2021, 12 (1), 2403. 10.1038/s41467-021-22732-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repecka D.; Jauniskis V.; Karpus L.; Rembeza E.; Rokaitis I.; Zrimec J.; Poviloniene S.; Laurynenas A.; Viknander S.; Abuajwa W.; Savolainen O.; Meskys R.; Engqvist M. K. M.; Zelezniak A. Expanding Functional Protein Sequence Spaces Using Generative Adversarial Networks. Nat. Mach. Intell. 2021, 3 (4), 324–333. 10.1038/s42256-021-00310-5. [DOI] [Google Scholar]
- Hawkins-Hooker A.; Depardieu F.; Baur S.; Couairon G.; Chen A.; Bikard D. Generating Functional Protein Variants with Variational Autoencoders. PLOS Comput. Biol. 2021, 17 (2), e1008736 10.1371/journal.pcbi.1008736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Cheng X.; Geng Y.; Li S.; Zeng X.; Wang B.; Gong J.; Liu C.; Zeng A.; Dong Y.; Tang J.; Song L.. xTrimoPGLM: Unified 100B-Scale Pre-Trained Transformer for Deciphering the Language of Protein. bioRxiv; preprint, 2023 10.1101/2023.07.05.547496. [DOI] [Google Scholar]
- Ferruz N.; Schmidt S.; Höcker B. ProtGPT2 Is a Deep Unsupervised Language Model for Protein Design. Nat. Commun. 2022, 13 (1), 4348. 10.1038/s41467-022-32007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamdari S.; Thakkar N.; Van Den Berg R.; Lu A. X.; Fusi N.; Amini A. P.; Yang K. K.. Protein Generation with Evolutionary Diffusion: Sequence Is All You Need. bioRxiv; preprint, 2023 10.1101/2023.09.11.556673. [DOI] [Google Scholar]
- Johnson S. R.; Fu X.; Viknander S.; Goldin C.; Monaco S.; Zelezniak A.; Yang K. K.. Computational Scoring and Experimental Evaluation of Enzymes Generated by Neural Networks. bioRxiv; preprint, 2023 10.1101/2023.03.04.531015. [DOI] [PMC free article] [PubMed]
- Ni B.; Kaplan D. L.; Buehler M. J. Generative Design of de Novo Proteins Based on Secondary-Structure Constraints Using an Attention-Based Diffusion Model. Chem. 2023, 9 (7), 1828–1849. 10.1016/j.chempr.2023.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K. E.; Yang K. K.; Berg R.; van den Zou J. Y.; Lu A. X.; Amini A. P.. Protein Structure Generation via Folding Diffusion. arXiv November 23, 2022. http://arxiv.org/abs/2209.15611. [DOI] [PMC free article] [PubMed]
- Wicky B. I. M.; Milles L. F.; Courbet A.; Ragotte R. J.; Dauparas J.; Kinfu E.; Tipps S.; Kibler R. D.; Baek M.; DiMaio F.; Li X.; Carter L.; Kang A.; Nguyen H.; Bera A. K.; Baker D. Hallucinating Symmetric Protein Assemblies. Science 2022, 378 (6615), 56–61. 10.1126/science.add1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. L.; Juergens D.; Bennett N. R.; Trippe B. L.; Yim J.; Eisenach H. E.; Ahern W.; Borst A. J.; Ragotte R. J.; Milles L. F.; Wicky B. I. M.; Hanikel N.; Pellock S. J.; Courbet A.; Sheffler W.; Wang J.; Venkatesh P.; Sappington I.; Torres S. V.; Lauko A.; De Bortoli V.; Mathieu E.; Ovchinnikov S.; Barzilay R.; Jaakkola T. S.; DiMaio F.; Baek M.; Baker D. De Novo Design of Protein Structure and Function with RFdiffusion. Nature 2023, 620 (7976), 1089–1100. 10.1038/s41586-023-06415-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trippe B. L.; Yim J.; Tischer D.; Baker D.; Broderick T.; Barzilay R.; Jaakkola T.. Diffusion Probabilistic Modeling of Protein Backbones in 3D for the Motif-Scaffolding Problem. arXiv June 8, 2022. http://arxiv.org/abs/2206.04119.
- Hie B.; Candido S.; Lin Z.; Kabeli O.; Rao R.; Smetanin N.; Sercu T.; Rives A.. A High-Level Programming Language for Generative Protein Design. bioRxiv; preprint, 2022 10.1101/2022.12.21.521526. [DOI] [Google Scholar]
- Yeh A. H.-W.; Norn C.; Kipnis Y.; Tischer D.; Pellock S. J.; Evans D.; Ma P.; Lee G. R.; Zhang J. Z.; Anishchenko I.; Coventry B.; Cao L.; Dauparas J.; Halabiya S.; DeWitt M.; Carter L.; Houk K. N.; Baker D. De Novo Design of Luciferases Using Deep Learning. Nature 2023, 614 (7949), 774–780. 10.1038/s41586-023-05696-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauparas J.; Anishchenko I.; Bennett N.; Bai H.; Ragotte R. J.; Milles L. F.; Wicky B. I. M.; Courbet A.; de Haas R. J.; Bethel N.; Leung P. J. Y.; Huddy T. F.; Pellock S.; Tischer D.; Chan F.; Koepnick B.; Nguyen H.; Kang A.; Sankaran B.; Bera A. K.; King N. P.; Baker D. Robust Deep Learning-Based Protein Sequence Design Using ProteinMPNN. Science 2022, 378 (6615), 49–56. 10.1126/science.add2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anishchenko I.; Pellock S. J.; Chidyausiku T. M.; Ramelot T. A.; Ovchinnikov S.; Hao J.; Bafna K.; Norn C.; Kang A.; Bera A. K.; DiMaio F.; Carter L.; Chow C. M.; Montelione G. T.; Baker D. De Novo Protein Design by Deep Network Hallucination. Nature 2021, 600, 547–552. 10.1038/s41586-021-04184-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Lisanza S.; Juergens D.; Tischer D.; Watson J. L.; Castro K. M.; Ragotte R.; Saragovi A.; Milles L. F.; Baek M.; Anishchenko I.; Yang W.; Hicks D. R.; Expòsit M.; Schlichthaerle T.; Chun J.-H.; Dauparas J.; Bennett N.; Wicky B. I. M.; Muenks A.; DiMaio F.; Correia B.; Ovchinnikov S.; Baker D. Scaffolding Protein Functional Sites Using Deep Learning. Science 2022, 377 (6604), 387–394. 10.1126/science.abn2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norn C.; Wicky B. I. M.; Juergens D.; Liu S.; Kim D.; Tischer D.; Koepnick B.; Anishchenko I.; Baker D.; Ovchinnikov S.; Coral A.; Bubar A. J.; Boykov A.; Valle Perez A. U.; MacMillan A.; Lubow A.; Mussini A.; Cai A.; Ardill A. J.; Seal A.; Kalantarian A.; Failer B.; Lackersteen B.; Chagot B.; Haight B. R.; Tastan B.; Uitham B.; Roy B. G.; de Melo Cruz B. R.; Echols B.; Lorenz B. E.; Blair B.; Kestemont B.; Eastlake C. D.; Bragdon C. J.; Vardeman C.; Salerno C.; Comisky C.; Hayman C. L.; Landers C. R; Zimov C.; Coleman C. D.; Painter C. R.; Ince C.; Lynagh C.; Malaniia D.; Wheeler D. C.; Robertson D.; Simon V.; Chisari E.; Kai E. L. J.; Rezae F.; Lengyel F.; Tabotta F.; Padelletti F.; Bostrom F.; Gross G. O.; McIlvaine G.; Beecher G.; Hansen G. T.; de Jong G.; Feldmann H.; Borman J. L.; Quinn J.; Norrgard J.; Truong J.; Diderich J. A.; Canfield J. M.; Photakis J.; Slone J. D.; Madzio J.; Mitchell J.; Stomieroski J. C.; Mitch J. H.; Altenbeck J. R.; Schinkler J.; Weinberg J. B.; Burbach J. D.; Sequeira da Costa J. C.; Bada Juarez J. F.; Gunnarsson J. P.; Harper K. D.; Joo K.; Clayton K. T.; DeFord K. E.; Scully K. F.; Gildea K. M.; Abbey K. J.; Kohli K. L.; Stenner K.; Takacs K.; Poussaint L. L.; Manalo L. C.; Withers L. C.; Carlson L.; Wei L.; Fisher L. R.; Carpenter L.; Ji-hwan M.; Ricci M.; Belcastro M. A.; Leniec M.; Hohmann M.; Thompson M.; Thayer M. A.; Gaebel M.; Cassidy M. D.; Fagiola M.; Lewis M.; Pfutzenreuter M.; Simon M.; Elmassry M. M.; Benevides N.; Kerr N. K.; Verma N.; Shannon O.; Yin O.; Wolfteich P.; Gummersall P.; Tłuscik P.ł; Gajar P.; Triggiani P. J.; Guha R.; Mathew Innes R. B.; Buchanan R.; Gamble R.; Leduc R.; Spearing R.; dos Santos Gomes R. L. C.; Estep R. D.; DeWitt R.; Moore R.; Shnider S. G.; Zaccanelli S. J.; Kuznetsov S.; Burillo-Sanz S.; Mooney S.; Vasiliy S.; Butkovich S. S.; Hudson S. B.; Pote S. L.; Denne S. P.; Schwegmann S. A.; Ratna S.; Kleinfelter S. C.; Bausewein T.; George T. J.; de Almeida T. S.; Yeginer U.; Barmettler W.; Pulley W. R.; Wright W. S.; Willyanto; Lansford W.; Hochart X.; Gaiji Y. A. S.; Lagodich Y.; Christian V. Protein Sequence Design by Conformational Landscape Optimization. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (11), e2017228118 10.1073/pnas.2017228118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; AlQuraishi M. Generating Novel, Designable, and Diverse Protein Structures by Equivariantly Diffusing Oriented Residue Clouds. arXiv June 6, 2023. http://arxiv.org/abs/2301.12485.
- Subramanian A. M.; Thomson M. Unexplored Regions of the Protein Sequence-Structure Map Revealed at Scale by a Library of Foldtuned Language Models. bioRxiv; preprint, 2023 10.1101/2023.12.22.573145. [DOI] [Google Scholar]
- Bloom J. D.; Labthavikul S. T.; Otey C. R.; Arnold F. H. Protein Stability Promotes Evolvability. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (15), 5869–5874. 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. Stability Effects of Mutations and Protein Evolvability. Curr. Opin. Struct. Biol. 2009, 19 (5), 596–604. 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Kipnis Y.; Chaib A. O.; Vorobieva A. A.; Cai G.; Reggiano G.; Basanta B.; Kumar E.; Mittl P. R. E.; Hilvert D.; Baker D. Design and Optimization of Enzymatic Activity in a de Novo B-barrel Scaffold. Protein Sci. 2022, 31 (11), e4405 10.1002/pro.4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A. E.; Fernandez D.; Liu J.; Eguchi R. R.; Huang P.-S. De Novo Design of a Highly Stable Ovoid TIM Barrel: Unlocking Pocket Shape towards Functional Design. BioDesign Res. 2022, 2022, 1–13. 10.34133/2022/9842315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. Protein Dynamism and Evolvability. Science 2009, 324 (5924), 203–207. 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- Sumida K. H.; Núñez-Franco R.; Kalvet I.; Pellock S. J.; Wicky B. I. M.; Milles L. F.; Dauparas J.; Wang J.; Kipnis Y.; Jameson N.; Kang A.; De La Cruz J.; Sankaran B.; Bera A. K.; Jiménez-Osés G.; Baker D.. Improving Protein Expression, Stability, and Function with ProteinMPNN. bioRxiv; preprint, 2023 10.1101/2023.10.03.560713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miton C. M.; Buda K.; Tokuriki N. Epistasis and Intramolecular Networks in Protein Evolution. Curr. Opin. Struct. Biol. 2021, 69, 160–168. 10.1016/j.sbi.2021.04.007. [DOI] [PubMed] [Google Scholar]
- Starr T. N.; Thornton J. W. Epistasis in Protein Evolution. Protein Sci. 2016, 25 (7), 1204–1218. 10.1002/pro.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P. A.; Krause A.; Arnold F. H. Navigating the Protein Fitness Landscape with Gaussian Processes. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (3), E193–E201. 10.1073/pnas.1215251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardikoraem M.; Woldring D. Protein Fitness Prediction Is Impacted by the Interplay of Language Models, Ensemble Learning, and Sampling Methods. Pharmaceutics 2023, 15 (5), 1337. 10.3390/pharmaceutics15051337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Verma D.; Sheridan R. P.; Liaw A.; Ma J.; Marshall N. M.; McIntosh J.; Sherer E. C.; Svetnik V.; Johnston J. M. Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model. 2020, 60 (6), 2773–2790. 10.1021/acs.jcim.0c00073. [DOI] [PubMed] [Google Scholar]
- Bryant D. H.; Bashir A.; Sinai S.; Jain N. K.; Ogden P. J.; Riley P. F.; Church G. M.; Colwell L. J.; Kelsic E. D. Deep Diversification of an AAV Capsid Protein by Machine Learning. Nat. Biotechnol. 2021, 39 (6), 691–696. 10.1038/s41587-020-00793-4. [DOI] [PubMed] [Google Scholar]
- Saito Y.; Oikawa M.; Nakazawa H.; Niide T.; Kameda T.; Tsuda K.; Umetsu M. Machine-Learning-Guided Mutagenesis for Directed Evolution of Fluorescent Proteins. ACS Synth. Biol. 2018, 7 (9), 2014–2022. 10.1021/acssynbio.8b00155. [DOI] [PubMed] [Google Scholar]
- Hsu C.; Nisonoff H.; Fannjiang C.; Listgarten J. Learning Protein Fitness Models from Evolutionary and Assay-Labeled Data. Nat. Biotechnol. 2022, 40 (7), 1114–1122. 10.1038/s41587-021-01146-5. [DOI] [PubMed] [Google Scholar]
- Hie B. L.; Shanker V. R.; Xu D.; Bruun T. U. J.; Weidenbacher P. A.; Tang S.; Wu W.; Pak J. E.; Kim P. S.. Efficient Evolution of Human Antibodies from General Protein Language Models. Nat. Biotechnol. 2023 10.1038/s41587-023-01763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann B. J.; Yue Y.; Arnold F. H. Informed Training Set Design Enables Efficient Machine Learning-Assisted Directed Protein Evolution. Cell Syst. 2021, 12 (11), 1026–1045. 10.1016/j.cels.2021.07.008. [DOI] [PubMed] [Google Scholar]
- Bedbrook C. N.; Yang K. K.; Robinson J. E.; Mackey E. D.; Gradinaru V.; Arnold F. H. Machine Learning-Guided Channelrhodopsin Engineering Enables Minimally Invasive Optogenetics. Nat. Methods 2019, 16 (11), 1176–1184. 10.1038/s41592-019-0583-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh J. C.; Fahlberg S. A.; Pfleger B. F.; Romero P. A. Machine Learning-Guided Acyl-ACP Reductase Engineering for Improved in Vivo Fatty Alcohol Production. Nat. Commun. 2021, 12 (1), 5825. 10.1038/s41467-021-25831-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp J. T.; Bremer B. J.; Romero P. A. Self-Driving Laboratories to Autonomously Navigate the Protein Fitness Landscape. bioRxiv; preprint, 2023 10.1101/2023.05.20.541582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedbrook C. N.; Yang K. K.; Rice A. J.; Gradinaru V.; Arnold F. H. Machine Learning to Design Integral Membrane Channelrhodopsins for Efficient Eukaryotic Expression and Plasma Membrane Localization. PLOS Comput. Biol. 2017, 13 (10), e1005786 10.1371/journal.pcbi.1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond D. A.; Iverson B. L.; Georgiou G.; Arnold F. H. Why High-Error-Rate Random Mutagenesis Libraries Are Enriched in Functional and Improved Proteins. J. Mol. Biol. 2005, 350 (4), 806–816. 10.1016/j.jmb.2005.05.023. [DOI] [PubMed] [Google Scholar]
- Park Y.; Metzger B. P. H.; Thornton J. W.. The Simplicity of Protein Sequence-Function Relationships. bioRxiv; preprint, 2023 10.1101/2023.09.02.556057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson C. A.; Wu N. C.; Sun R. A Comprehensive Biophysical Description of Pairwise Epistasis throughout an Entire Protein Domain. Curr. Biol. 2014, 24 (22), 2643–2651. 10.1016/j.cub.2014.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N. C.; Dai L.; Olson C. A.; Lloyd-Smith J. O.; Sun R. Adaptation in Protein Fitness Landscapes Is Facilitated by Indirect Paths. eLife 2016, 5, e16965 10.7554/eLife.16965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Kan S. B. J.; Lewis R. D.; Wittmann B. J.; Arnold F. H. Machine Learning-Assisted Directed Protein Evolution with Combinatorial Libraries. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (18), 8852–8858. 10.1073/pnas.1901979116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Khimulya G.; Alley E. C.; Esvelt K. M.; Church G. M. Low-N Protein Engineering with Data-Efficient Deep Learning. Nat. Methods 2021, 18 (4), 389–396. 10.1038/s41592-021-01100-y. [DOI] [PubMed] [Google Scholar]
- Van Der Flier F. J.; Estell D.; Pricelius S.; Dankmeyer L.; Van Stigt Thans S.; Mulder H.; Otsuka R.; Goedegebuur F.; Lammerts L.; Staphorst D.; Van Dijk A. D. J.; De Ridder D.; Redestig H. What Makes the Effect of Protein Mutations Difficult to Predict? bioRxiv; preprint, 2023 10.1101/2023.09.25.559319. [DOI] [Google Scholar]
- Thomas N.; Agarwala A.; Belanger D.; Song Y. S.; Colwell L.. Tuned Fitness Landscapes for Benchmarking Model-Guided Protein Design. bioRxiv; preprint, 2022 10.1101/2022.10.28.514293. [DOI] [Google Scholar]
- Wrenbeck E. E.; Azouz L. R.; Whitehead T. A. Single-Mutation Fitness Landscapes for an Enzyme on Multiple Substrates Reveal Specificity Is Globally Encoded. Nat. Commun. 2017, 8 (1), 15695. 10.1038/ncomms15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markin C. J.; Mokhtari D. A.; Sunden F.; Appel M. J.; Akiva E.; Longwell S. A.; Sabatti C.; Herschlag D.; Fordyce P. M. Revealing Enzyme Functional Architecture via High-Throughput Microfluidic Enzyme Kinetics. Science 2021, 373 (6553), eabf8761 10.1126/science.abf8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markin C. J.; Mokhtari D. A.; Du S.; Doukov T.; Sunden F.; Cook J. A.; Fordyce P. M.; Herschlag D. Decoupling of Catalysis and Transition State Analog Binding from Mutations throughout a Phosphatase Revealed by High-Throughput Enzymology. Proc. Natl. Acad. Sci. U. S. A. 2023, 120 (29), e2219074120 10.1073/pnas.2219074120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure A. J.; Domingo J.; Schmiedel J. M.; Hidalgo-Carcedo C.; Diss G.; Lehner B. Mapping the Energetic and Allosteric Landscapes of Protein Binding Domains. Nature 2022, 604 (7904), 175–183. 10.1038/s41586-022-04586-4. [DOI] [PubMed] [Google Scholar]
- Faure A. J.; Martí-Aranda A.; Hidalgo-Carcedo C.; Schmiedel J. M.; Lehner B.. The Genetic Architecture of Protein Stability. bioRxiv; preprint, 2023 10.1101/2023.10.27.564339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilborg D.; Alenicheva A.; Grisoni F. Exposing the Limitations of Molecular Machine Learning with Activity Cliffs. J. Chem. Inf. Model. 2022, 62, 5938–5951. 10.1021/acs.jcim.2c01073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Costa S.; Hinds E. C.; Freschlin C. R.; Song H.; Romero P. A. Inferring Protein Fitness Landscapes from Laboratory Evolution Experiments. PLOS Comput. Biol. 2023, 19 (3), e1010956 10.1371/journal.pcbi.1010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman S. A.; Weinberger E. D. The NK Model of Rugged Fitness Landscapes and Its Application to Maturation of the Immune Response. J. Theor. Biol. 1989, 141 (2), 211–245. 10.1016/S0022-5193(89)80019-0. [DOI] [PubMed] [Google Scholar]
- Papkou A.; Garcia-Pastor L.; Escudero J. A.; Wagner A. A Rugged yet Easily Navigable Fitness Landscape. Science 2023, 382 (6673), eadh3860 10.1126/science.adh3860. [DOI] [PubMed] [Google Scholar]
- Notin P.; Dias M.; Frazer J.; Marchena-Hurtado J.; Gomez A.; Marks D. S.; Gal Y.. Tranception: Protein Fitness Prediction with Autoregressive Transformers and Inference-Time Retrieval. arXiv May 27, 2022. http://arxiv.org/abs/2205.13760.
- Hie B. L.; Yang K. K.; Kim P. S. Evolutionary Velocity with Protein Language Models Predicts Evolutionary Dynamics of Diverse Proteins. Cell Syst. 2022, 13 (4), 274–285. 10.1016/j.cels.2022.01.003. [DOI] [PubMed] [Google Scholar]
- Shanker V. R.; Bruun T. U. J.; Hie B. L.; Kim P. S.. Inverse Folding of Protein Complexes with a Structure-Informed Language Model Enables Unsupervised Antibody Evolution. bioRxiv; preprint, 2023 10.1101/2023.12.19.572475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W. J.; Liu D.; Wang X.; Zhang A.; Wei Q.; Nandi A.; Dong S.; Warshel A. Enhancing Luciferase Activity and Stability through Generative Modeling of Natural Enzyme Sequences. Proc. Natl. Acad. Sci. U. S. A. 2023, 120 (48), e2312848120 10.1073/pnas.2312848120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ W. P.; Figliuzzi M.; Stocker C.; Barrat-Charlaix P.; Socolich M.; Kast P.; Hilvert D.; Monasson R.; Cocco S.; Weigt M.; Ranganathan R. An Evolution-Based Model for Designing Chorismate Mutase Enzymes. Science 2020, 369 (6502), 440–445. 10.1126/science.aba3304. [DOI] [PubMed] [Google Scholar]
- Xie W. J.; Asadi M.; Warshel A. Enhancing Computational Enzyme Design by a Maximum Entropy Strategy. Proc. Natl. Acad. Sci. U. S. A. 2022, 119 (7), e2122355119 10.1073/pnas.2122355119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W.; Cheng P.; Mao C.; Tang J.; Yang S.; Gu Q.; Han W.; Chen Y.; Zhou J.; Li W.; Pan A.; Zhao S.; Huang X.; Zhang J.; Zhu S.; Wang S.-Q.. Zero-Shot Prediction of Mutation Effects on Protein Function with Multimodal Deep Representation Learning. In Review; preprint, 2023 10.21203/rs.3.rs-3358917/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W. J.; Warshel A.. Harnessing Generative AI to Decode Enzyme Catalysis and Evolution for Enhanced Engineering. bioRxiv; preprint, 2023 10.1101/2023.10.10.561808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf T. A.; Ingraham J. B.; Poelwijk F. J.; Schärfe C. P. I.; Springer M.; Sander C.; Marks D. S. Mutation Effects Predicted from Sequence Co-Variation. Nat. Biotechnol. 2017, 35 (2), 128–135. 10.1038/nbt.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesselman A. J.; Ingraham J. B.; Marks D. S. Deep Generative Models of Genetic Variation Capture the Effects of Mutations. Nat. Methods 2018, 15 (10), 816–822. 10.1038/s41592-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer J.; Notin P.; Dias M.; Gomez A.; Min J. K.; Brock K.; Gal Y.; Marks D. S. Disease Variant Prediction with Deep Generative Models of Evolutionary Data. Nature 2021, 599 (7883), 91–95. 10.1038/s41586-021-04043-8. [DOI] [PubMed] [Google Scholar]
- Rao R. M.; Liu J.; Verkuil R.; Meier J.; Canny J.; Abbeel P.; Sercu T.; Rives A.. MSA Transformer. In Proceedings of the 38th International Conference on Machine Learning; Meila M., Zhang T., Eds.; Proceedings of Machine Learning Research; PMLR, 2021; Vol. 139, pp 8844–8856.
- Meier J.; Rao R.; Verkuil R.; Liu J.; Sercu T.; Rives A.. Language Models Enable Zero-Shot Prediction of the Effects of Mutations on Protein Function. In Advances in Neural Information Processing Systems; Ranzato M., Beygelzimer A., Dauphin Y., Liang P. S., Vaughan J. W., Eds.; Curran Associates, Inc., 2021; Vol. 34, pp 29287–29303. [Google Scholar]
- Alley E. C.; Khimulya G.; Biswas S.; AlQuraishi M.; Church G. M. Unified Rational Protein Engineering with Sequence-Based Deep Representation Learning. Nat. Methods 2019, 16 (12), 1315–1322. 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R.; Bhattacharya N.; Thomas N.; Duan Y.; Chen X.; Canny J.; Abbeel P.; Song Y. S.. Evaluating Protein Transfer Learning with TAPE. 2019. [PMC free article] [PubMed]
- Rives A.; Meier J.; Sercu T.; Goyal S.; Lin Z.; Liu J.; Guo D.; Ott M.; Zitnick C. L.; Ma J.; Fergus R. Biological Structure and Function Emerge from Scaling Unsupervised Learning to 250 Million Protein Sequences. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (15), e2016239118 10.1073/pnas.2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. K.; Fusi N.; Lu A. X.. Convolutions Are Competitive with Transformers for Protein Sequence Pretraining. bioRxiv; preprint, 2022 10.1101/2022.05.19.492714. [DOI] [PubMed] [Google Scholar]
- Brandes N.; Ofer D.; Peleg Y.; Rappoport N.; Linial M. ProteinBERT: A Universal Deep-Learning Model of Protein Sequence and Function. Bioinformatics 2022, 38 (8), 2102–2110. 10.1093/bioinformatics/btac020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnaggar A.; Heinzinger M.; Dallago C.; Rehawi G.; Wang Y.; Jones L.; Gibbs T.; Feher T.; Angerer C.; Steinegger M.; Bhowmik D.; Rost B. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44 (10), 7112–7127. 10.1109/TPAMI.2021.3095381. [DOI] [PubMed] [Google Scholar]
- Hesslow D.; Zanichelli N.; Notin P.; Poli I.; Marks D.. RITA: A Study on Scaling Up Generative Protein Sequence Models. arXiv May 11, 2022. http://arxiv.org/abs/2205.05789.
- Dunham A. S.; Beltrao P.; AlQuraishi M. High-Throughput Deep Learning Variant Effect Prediction with Sequence UNET. Genome Biol. 2023, 24 (1), 110. 10.1186/s13059-023-02948-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. K.; Yeh H.; Zanichelli N.. Masked Inverse Folding with Sequence Transfer for Protein Representation Learning. bioRxiv; preprint, 2022 10.1101/2022.05.25.493516. [DOI] [PubMed] [Google Scholar]
- Hsu C.; Verkuil R.; Liu J.; Lin Z.; Hie B.; Sercu T.; Lerer A.; Rives A.. Learning Inverse Folding from Millions of Predicted Structures. In Proceedings of the 39th International Conference on Machine Learning; Chaudhuri K., Jegelka S., Song L., Szepesvari C., Niu G., Sabato S., Eds.; Proceedings of Machine Learning Research; PMLR, 2022; Vol. 162, pp 8946–8970.
- Cheng J.; Novati G.; Pan J.; Bycroft C.; Žemgulytė A.; Applebaum T.; Pritzel A.; Wong L. H.; Zielinski M.; Sargeant T.; Schneider R. G.; Senior A. W.; Jumper J.; Hassabis D.; Kohli P.; Avsec Ž. Accurate Proteome-Wide Missense Variant Effect Prediction with AlphaMissense. Science 2023, 381 (6664), eadg7492 10.1126/science.adg7492. [DOI] [PubMed] [Google Scholar]
- Shaw A.; Spinner H.; Shin J.; Gurev S.; Rollins N.; Marks D.. Removing Bias in Sequence Models of Protein Fitness. bioRxiv; preprint, 2023 10.1101/2023.09.28.560044. [DOI] [Google Scholar]
- Tokuriki N.; Stricher F.; Serrano L.; Tawfik D. S. How Protein Stability and New Functions Trade Off. PLoS Comput. Biol. 2008, 4 (2), e1000002 10.1371/journal.pcbi.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboyama K.; Dauparas J.; Chen J.; Laine E.; Mohseni Behbahani Y.; Weinstein J. J.; Mangan N. M.; Ovchinnikov S.; Rocklin G. J. Mega-Scale Experimental Analysis of Protein Folding Stability in Biology and Design. Nature 2023, 620 (7973), 434–444. 10.1038/s41586-023-06328-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna D. M.; Goldstein R. A. Why Are Proteins Marginally Stable?. Proteins Struct. Funct. Genet. 2002, 46 (1), 105–109. 10.1002/prot.10016. [DOI] [PubMed] [Google Scholar]
- Nisthal A.; Wang C. Y.; Ary M. L.; Mayo S. L. Protein Stability Engineering Insights Revealed by Domain-Wide Comprehensive Mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (33), 16367–16377. 10.1073/pnas.1903888116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detlefsen N. S.; Hauberg S.; Boomsma W.. What Is a Meaningful Representation of Protein Sequences? ArXiv201202679 Cs Q-Bio 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallago C.; Mou J.; Johnston K. E.; Wittmann B. J.; Bhattacharya N.; Goldman S.; Madani A.; Yang K. K.. FLIP: Benchmark Tasks in Fitness Landscape Inference for Proteins. bioRxiv; preprint, 2021 10.1101/2021.11.09.467890. [DOI] [Google Scholar]
- Georgiev A. G. Interpretable Numerical Descriptors of Amino Acid Space. J. Comput. Biol. 2009, 16 (5), 703–723. 10.1089/cmb.2008.0173. [DOI] [PubMed] [Google Scholar]
- Michael R.; Kæstel-Hansen J.; Groth P. M.; Bartels S.; Salomon J.; Tian P.; Hatzakis N. S.; Boomsma W.. Assessing the Performance of Protein Regression Models. bioRxiv; preprint, 2023 10.1101/2023.06.18.545472. [DOI] [Google Scholar]
- Schmirler R.; Heinzinger M.; Rost B.. Fine-Tuning Protein Language Models Boosts Predictions across Diverse Tasks. bioRxiv; preprint, 2023 10.1101/2023.12.13.571462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T.; Berger B.. Learning Protein Sequence Embeddings Using Information from Structure. 2019.
- Gelman S.; Fahlberg S. A.; Heinzelman P.; Romero P. A.; Gitter A. Neural Networks to Learn Protein Sequence–Function Relationships from Deep Mutational Scanning Data. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (48), e2104878118 10.1073/pnas.2104878118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X.; Wang Z.; Yang K. K.; Su J.; Du H.; Zheng Q.; Guo G.; Yang M.; Yang F.; Yuan F.. Sequence vs. Structure: Delving Deep into Data-Driven Protein Function Prediction. bioRxiv; preprint, 2023 10.1101/2023.04.02.534383. [DOI] [Google Scholar]
- Qabel A.; Ennadir S.; Nikolentzos G.; Lutzeyer J. F.; Chatzianastasis M.; Bostrom H.; Vazirgiannis M.. Advancing Antibiotic Resistance Classification with Deep Learning Using Protein Sequence and Structure. bioRxiv; preprint, 2022 10.1101/2022.10.06.511103. [DOI] [Google Scholar]
- Jamasb A. R.; Viñas R.; Ma E. J.; Harris C.; Huang K.; Hall D.; Lió P.; Blundell T. L.. Graphein—A Python Library for Geometric Deep Learning and Network Analysis on Protein Structures and Interaction Networks. bioRxiv; preprint, 2020 10.1101/2020.07.15.204701. [DOI] [Google Scholar]
- Qiu Y.; Wei G.-W. Persistent Spectral Theory-Guided Protein Engineering. Nat. Comput. Sci. 2023, 3, 149–163. 10.1038/s43588-022-00394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirnsberger G.; Pritisanac I.; Oberdorfer G.; Gruber K.. Flattening the Curve—How to Get Better Results with Small Deep-Mutational-Scanning Datasets. bioRxiv; preprint, 2023 10.1101/2023.03.27.534314. [DOI] [PubMed] [Google Scholar]
- Robinson L. C. B.; Atkinson T.; Copoiu L.; Bordes P.; Pierrot T.; Barrett T.. Contrasting Sequence with Structure: Pre-Training Graph Representations with PLMs. bioRxiv; preprint, 2023 10.1101/2023.12.01.569611. [DOI] [Google Scholar]
- Zhong E. D.; Bepler T.; Berger B.; Davis J. H. CryoDRGN: Reconstruction of Heterogeneous Cryo-EM Structures Using Neural Networks. Nat. Methods 2021, 18 (2), 176–185. 10.1038/s41592-020-01049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo-Rocha C. G.; Li A.; D’Amore L.; Hoebenreich S.; Sanchis J.; Lubrano P.; Ferla M. P.; Garcia-Borràs M.; Osuna S.; Reetz M. T.. Pervasive Cooperative Mutational Effects on Multiple Catalytic Enzyme Traits Emerge via Long-Range Conformational Dynamics. Nat. Commun. 2021, 12 ( (1621), ) 10.1038/s41467-021-21833-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adcock S. A.; McCammon J. A. Molecular Dynamics: Survey of Methods for Simulating the Activity of Proteins. Chem. Rev. 2006, 106 (5), 1589–1615. 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Biel J. T.; Fraser J. S. Keep on Moving: Discovering and Perturbing the Conformational Dynamics of Enzymes. Acc. Chem. Res. 2015, 48 (2), 423–430. 10.1021/ar5003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Wang J.; Han W.; Xu D. Neural Relational Inference to Learn Long-Range Allosteric Interactions in Proteins from Molecular Dynamics Simulations. Nat. Commun. 2022, 13 (1), 1661. 10.1038/s41467-022-29331-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbitt G. A.; Rajendran M.; Lynch M. L.; Asare-Bediako R.; Mouli L. T.; Ryan C. J.; Srivastava H.; Phadke K.; Reed M. L.; Moore N.; Ferran M. C.; Fokoue E. P.. ATOMDANCE: Machine Learning Denoising and Resonance Analysis for Functional and Evolutionary Comparisons of Protein Dynamics. bioRxiv; preprint, 2023 10.1101/2023.04.20.537698. [DOI] [Google Scholar]
- Matthews D. M.; Spence M. A.; Mater A. C.; Nichols J.; Pulsford S. B.; Sandhu M.; Kaczmarski J. A. B.; Miton C. M.; Tokuriki N.; Jackson C. J.. Leveraging Ancestral Sequence Reconstruction for Protein Representation Learning. bioRxiv; preprint, 2023 10.1101/2023.12.20.572683. [DOI] [Google Scholar]
- Clements H. D.; Flynn A. R.; Nicholls B. T.; Grosheva D.; Lefave S. J.; Merriman M. T.; Hyster T. K.; Sigman M. S. Using Data Science for Mechanistic Insights and Selectivity Predictions in a Non-Natural Biocatalytic Reaction. J. Am. Chem. Soc. 2023, 145 (32), 17656–17664. 10.1021/jacs.3c03639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaugg J.; Gumulya Y.; Malde A. K.; Bodén M. Learning Epistatic Interactions from Sequence-Activity Data to Predict Enantioselectivity. J. Comput. Aided Mol. Des. 2017, 31 (12), 1085–1096. 10.1007/s10822-017-0090-x. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Zeng T.; Wu R. QM/MM Modeling Aided Enzyme Engineering in Natural Products Biosynthesis. J. Chem. Inf. Model. 2023, 63, 5018 10.1021/acs.jcim.3c00779. [DOI] [PubMed] [Google Scholar]
- Goldman S.; Das R.; Yang K. K.; Coley C. W. Machine Learning Modeling of Family Wide Enzyme-Substrate Specificity Screens. PLOS Comput. Biol. 2022, 18 (2), e1009853 10.1371/journal.pcbi.1009853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Wu J.; Song Y. S.; Mahadevan R.. Enzyme Activity Prediction of Sequence Variants on Novel Substrates Using Improved Substrate Encodings and Convolutional Pooling. In Proceedings of the 16th Machine Learning in Computational Biology meeting; Knowles D. A., Mostafavi S., Lee S.-I., Eds.; Proceedings of Machine Learning Research; PMLR, 2022; Vol. 165, pp 78–87. [PMC free article] [PubMed] [Google Scholar]
- Corso G.; Stärk H.; Jing B.; Barzilay R.; Jaakkola T.. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. arXiv October 4, 2022. http://arxiv.org/abs/2210.01776.
- Qiao Z.; Nie W.; Vahdat A.; Miller T. F. III; Anandkumar A.. State-Specific Protein-Ligand Complex Structure Prediction with a Multi-Scale Deep Generative Model. arXiv April 19, 2023. http://arxiv.org/abs/2209.15171.
- Yang T.; Ye Z.; Lynch M. D. Multiagent” Screening Improves Directed Enzyme Evolution by Identifying Epistatic Mutations. ACS Synth. Biol. 2022, 11 (5), 1971–1983. 10.1021/acssynbio.2c00136. [DOI] [PubMed] [Google Scholar]
- Hie B.; Bryson B. D.; Berger B. Leveraging Uncertainty in Machine Learning Accelerates Biological Discovery and Design. Cell Syst. 2020, 11 (5), 461–477. 10.1016/j.cels.2020.09.007. [DOI] [PubMed] [Google Scholar]
- Greenman K. P.; Amini A. P.; Yang K. K.. Benchmarking Uncertainty Quantification for Protein Engineering. bioRxiv; preprint, 2023 10.1101/2023.04.17.536962. [DOI]
- Qiu Y.; Hu J.; Wei G.-W. Cluster Learning-Assisted Directed Evolution. Nat. Comput. Sci. 2021, 1 (12), 809–818. 10.1038/s43588-021-00168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.; Wei G.-W. CLADE 2.0: Evolution-Driven Cluster Learning-Assisted Directed Evolution. J. Chem. Inf. Model. 2022, 62 (19), 4629–4641. 10.1021/acs.jcim.2c01046. [DOI] [PubMed] [Google Scholar]
- Stanton S.; Maddox W.; Gruver N.; Maffettone P.; Delaney E.; Greenside P.; Wilson A. G.. Accelerating Bayesian Optimization for Biological Sequence Design with Denoising Autoencoders. arXiv July 12, 2022. http://arxiv.org/abs/2203.12742.
- Gruver N.; Stanton S.; Kirichenko P.; Finzi M.; Maffettone P.; Myers V.; Delaney E.; Greenside P.; Wilson A. G.. Effective Surrogate Models for Protein Design with Bayesian Optimization. ICML Workshop on Computational Biology 2021. [Google Scholar]
- Hu R.; Fu L.; Chen Y.; Chen J.; Qiao Y.; Si T. Protein Engineering via Bayesian Optimization-Guided Evolutionary Algorithm and Robotic Experiments. Brief. Bioinform. 2023, 24 (1), bbac570 10.1093/bib/bbac570. [DOI] [PubMed] [Google Scholar]
- Sinai S.; Wang R.; Whatley A.; Slocum S.; Locane E.; Kelsic E. D.. AdaLead: A Simple and Robust Adaptive Greedy Search Algorithm for Sequence Design. ArXiv201002141 Cs Math Q-Bio 2020. [Google Scholar]
- Brookes D.; Park H.; Listgarten J.. Conditioning by Adaptive Sampling for Robust Design. In Proceedings of the 36th International Conference on Machine Learning; Chaudhuri K., Salakhutdinov R., Eds.; Proceedings of Machine Learning Research; PMLR, 2019; Vol. 97, pp 773–782.
- Brookes D. H.; Listgarten J.. Design by Adaptive Sampling. arXiv February 10, 2020. http://arxiv.org/abs/1810.03714.
- Kirjner A.; Yim J.; Samusevich R.; Jaakkola T.; Barzilay R.; Fiete I.. Optimizing Protein Fitness Using Gibbs Sampling with Graph-Based Smoothing. arXiv July 2, 2023. http://arxiv.org/abs/2307.00494.
- Minot M.; Reddy S. T.. Meta Learning Improves Robustness and Performance in Machine Learning-Guided Protein Engineering. bioRxiv; preprint, 2023 10.1101/2023.01.30.526201. [DOI] [Google Scholar]
- Daulton S.; Balandat M.; Bakshy E.. Parallel Bayesian Optimization of Multiple Noisy Objectives with Expected Hypervolume Improvement. arXiv October 26, 2021. http://arxiv.org/abs/2105.08195. [Google Scholar]
- Amin A. N.; Weinstein E. N.; Marks D. S.. Biological Sequence Kernels with Guaranteed Flexibility. arXiv April 6, 2023. http://arxiv.org/abs/2304.03775.
- Gligorijević V.; Berenberg D.; Ra S.; Watkins A.; Kelow S.; Cho K.; Bonneau R.. Function-Guided Protein Design by Deep Manifold Sampling. bioRxiv; preprint, 2021 10.1101/2021.12.22.473759. [DOI] [Google Scholar]
- Aghazadeh A.; Nisonoff H.; Ocal O.; Brookes D. H.; Huang Y.; Koyluoglu O. O.; Listgarten J.; Ramchandran K. Epistatic Net Allows the Sparse Spectral Regularization of Deep Neural Networks for Inferring Fitness Functions. Nat. Commun. 2021, 12 (1), 5225. 10.1038/s41467-021-25371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes D. H.; Aghazadeh A.; Listgarten J. On the Sparsity of Fitness Functions and Implications for Learning. Proc. Natl. Acad. Sci. U. S. A. 2022, 119 (1), e2109649118 10.1073/pnas.2109649118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisonoff H.; Wang Y.; Listgarten J.. Augmenting Neural Networks with Priors on Function Values. arXiv October 14, 2022. http://arxiv.org/abs/2202.04798.
- Ding D.Independent Mutation Effects Enable Design of Combinatorial Protein Binding Mutants. bioRxiv; preprint, 2022 10.1101/2022.10.31.514613. [DOI] [Google Scholar]
- Ding D.; Shaw A.; Sinai S.; Rollins N.; Prywes N.; Savage D. F.; Laub M. T.; Marks D. S.. Protein Design Using Structure-Based Residue Preferences. bioRxiv; preprint, 2022 10.1101/2022.10.31.514613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco S.; Monasson R.; Posani L.. Minimal Epistatic Networks from Integrated Sequence and Mutational Protein Data. bioRxiv; preprint, 2023 10.1101/2023.09.25.559251. [DOI] [Google Scholar]
- Nisonoff H.; Wang Y.; Listgarten J. Coherent Blending of Biophysics-Based Knowledge with Bayesian Neural Networks for Robust Protein Property Prediction. ACS Synth. Biol. 2023, 12, 3242 10.1021/acssynbio.3c00217. [DOI] [PubMed] [Google Scholar]
- Ren Z.; Li J.; Ding F.; Zhou Y.; Ma J.; Peng J.. Proximal Exploration for Model-Guided Protein Sequence Design. In Proceedings of the 39th International Conference on Machine Learning; Chaudhuri K., Jegelka S., Song L., Szepesvari C., Niu G., Sabato S., Eds.; Proceedings of Machine Learning Research; PMLR, 2022; Vol. 162, pp 18520–18536. [Google Scholar]
- Outeiral C.; Deane C. M.. Codon Language Embeddings Provide Strong Signals for Protein Engineering. bioRxiv; preprint, 2022 10.1101/2022.12.15.519894. [DOI] [Google Scholar]
- Minot M.; Reddy S. T. Nucleotide Augmentation for Machine Learning-Guided Protein Engineering. Bioinforma. Adv. 2023, 3 (1), vbac094 10.1093/bioadv/vbac094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang C.; Bates S.; Angelopoulos A. N.; Listgarten J.; Jordan M. I. Conformal Prediction under Feedback Covariate Shift for Biomolecular Design. Proc. Natl. Acad. Sci. U. S. A. 2022, 119 (43), e2204569119 10.1073/pnas.2204569119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang C.; Listgarten J. Is Novelty Predictable?. Cold Spring Harb. Perspect. Biol. 2023, a041469 10.1101/cshperspect.a041469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlberg S. A.; Freschlin C. R.; Heinzelman P.; Romero P. A.. Neural Network Extrapolation to Distant Regions of the Protein Fitness Landscape. bioRxiv; preprint, 2023 10.1101/2023.11.08.566287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. G.; Hu Z.; Salakhutdinov R.; Xing E. P.. Deep Kernel Learning. arXiv November 6, 2015. http://arxiv.org/abs/1511.02222.
- Ober S. W.; Rasmussen C. E.; van der Wilk M.. The Promises and Pitfalls of Deep Kernel Learning. arXiv July 7, 2021. http://arxiv.org/abs/2102.12108.
- Chory E. J.; Gretton D. W.; DeBenedictis E. A.; Esvelt K. M. Enabling High-throughput Biology with Flexible Open-source Automation. Mol. Syst. Biol. 2021, 17 (3), e9942 10.15252/msb.20209942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBenedictis E. A.; Chory E. J.; Gretton D. W.; Wang B.; Golas S.; Esvelt K. M. Systematic Molecular Evolution Enables Robust Biomolecule Discovery. Nat. Methods 2022, 19 (1), 55–64. 10.1038/s41592-021-01348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscher B. A.; Canty R. B.; McDonald M. A.; Greenman K. P.; McGill C. J.; Bilodeau C. L.; Jin W.; Wu H.; Vermeire F. H.; Jin B.; Hart T.; Kulesza T.; Li S.-C.; Jaakkola T. S.; Barzilay R.; Gómez-Bombarelli R.; Green W. H.; Jensen K. F. Autonomous, Multiproperty-Driven Molecular Discovery: From Predictions to Measurements and Back. Science 2023, 382 (6677), eadi1407 10.1126/science.adi1407. [DOI] [PubMed] [Google Scholar]
- Boiko D. A.; MacKnight R.; Gomes G.. Emergent Autonomous Scientific Research Capabilities of Large Language Models. 2023. [DOI] [PMC free article] [PubMed]
- Yu T.; Boob A. G.; Singh N.; Su Y.; Zhao H. In Vitro Continuous Protein Evolution Empowered by Machine Learning and Automation. Cell Syst. 2023, 14 (8), 633–644. 10.1016/j.cels.2023.04.006. [DOI] [PubMed] [Google Scholar]
- Mahjour B.; Hoffstadt J.; Cernak T. Designing Chemical Reaction Arrays Using Phactor and ChatGPT. Org. Process Res. Dev. 2023, 27 (8), 1510–1516. 10.1021/acs.oprd.3c00186. [DOI] [Google Scholar]
- Mahjour B.; Zhang R.; Shen Y.; McGrath A.; Zhao R.; Mohamed O. G.; Lin Y.; Zhang Z.; Douthwaite J. L.; Tripathi A.; Cernak T. Rapid Planning and Analysis of High-Throughput Experiment Arrays for Reaction Discovery. Nat. Commun. 2023, 14 (1), 3924. 10.1038/s41467-023-39531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Ducharme J.; Johnston K. E.; Li F.-Z.; Yue Y.; Arnold F. H. DeCOIL: Optimization of Degenerate Codon Libraries for Machine Learning-Assisted Protein Engineering. ACS Synth. Biol. 2023, 12, 2444 10.1021/acssynbio.3c00301. [DOI] [PubMed] [Google Scholar]
- Wittmann B. J.; Johnston K. E.; Almhjell P. J.; Arnold F. H. evSeq: Cost-Effective Amplicon Sequencing of Every Variant in a Protein Library. ACS Synth. Biol. 2022, 11 (3), 1313–1324. 10.1021/acssynbio.1c00592. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Brookes D. H.; Busia A.; Carneiro A.; Fannjiang C.; Popova G.; Shin D.; Donohue K. C.; Lin L. F.; Miller Z. M.; Williams E. R.; Chang E. F.; Nowakowski T. J.; Listgarten J.; Schaffer D. V.. Optimal Trade-off Control in Machine Learning-Based Library Design, with Application to Adeno-Associated Virus (AAV) for Gene Therapy. Sci. Adv. 2024, 10 (4), , In Press 10.1126/sciadv.adj3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsch D.; Eichenberger M.; Voss M.; Bornscheuer U. T.; Buller R. M. LibGENiE – A Bioinformatic Pipeline for the Design of Information-Enriched Enzyme Libraries. Comput. Struct. Biotechnol. J. 2023, 21, 4488–4496. 10.1016/j.csbj.2023.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Z. A.; Murray R. M.; Thomson M. W.. TRILL: Orchestrating Modular Deep-Learning Workflows for Democratized, Scalable Protein Analysis and Engineering.bioRxiv; preprint, 2023 10.1101/2023.10.24.563881. [DOI] [Google Scholar]