

Abstract
Pancreatic ductal adenocarcinoma (PDAC) has extremely poor prognosis, with a 5-year survival rate of approximately 11%. Yes-associated protein (YAP) is a major downstream effector of the Hippo-YAP pathway and plays a pivotal role in regulation of cell proliferation and organ regeneration and tumorigenesis. Activation of YAP signaling has been associated with PDAC progression and drug resistance. Verteporfin (VP) is a photosensitizer used for photodynamic therapy and previous work showed that it can function as a YAP inhibitor. The efficacy of VP on human cancer are being tested in several trials. In this study, we examined the effect of VP on reactive oxygen species (ROS) and lipid peroxidation in pancreatic cancer cells, by using fluorescent molecular probes and by measuring the levels of malondialdehyde, a metabolic byproduct and marker of lipid peroxidation. We found that VP causes rapid increase of both overall ROS and lipid peroxide levels, independent of light activation. These effects were not dependent on YAP, as knockdown of YAP did not cause ROS or lipid peroxidation or enhance VP-induced ROS production. Temoporfin, another photodynamic drug, did not show similar activities. In addition, VP treatment led to loss of cell membrane integrity and reduction of viability. Notably, the activity of VP to induce lipid peroxidation was neutralized by ferroptosis inhibitors ferrostatin-1 or liproxstatin-1. VP treatment also reduced the levels of glutathione peroxidase 4 (GPX4), an enzyme that protects against lipid peroxidation. These results indicate that VP can induce lipid peroxidation and ferroptosis in the absence of light activation. Our findings reveal a novel mechanism by which VP inhibits tumor growth and provide insights into development of new therapeutic strategies for the treatment of pancreatic cancer.
Keywords: Verteporfin, reactive oxygen species, lipid peroxidation, ferroptosis, pancreatic cancer
Graphical Abstract

Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the US, with an overall five-year survival rate of 11% [1]. The vast majority of PDAC cases exhibit resistance to the currently available chemotherapeutic regimens, such as FOLFIRINOX and gemcitabine with nab-paclitaxel [2, 3]. It is a major challenge to develop more effective methods for the treatment of PDAC. Yes-associated protein (YAP) is the core component of the Hippo-YAP signaling pathway and plays an important role in pancreatic tumorigenesis [4–12]. Activation of YAP signaling has also been associated with other forms of human malignancy [13–20]. YAP acts as a transcriptional co-activator by association with TEAD and other DNA binding transcription factors and mediates expression of genes involved in aspects of cell proliferation and tumorigenesis [7–12]. YAP can be negatively regulated by a series of phosphorylation events [21–23], which leads to either protein degradation [24–26] or cytoplasmic retention [27, 28]. Verteporfin (VP) has been used in the clinics as a photosensitizer for photodynamic therapy with activation of red laser light [29]. VP was identified as an inhibitor of YAP in a screen of small molecule library [30]. Although the structural basis of its activity is not clear, VP was shown to disrupt the YAP-TEAD interaction and inhibit YAP-mediated transcriptional events without light activation [30]. VP treatment can also suppress YAP-mediated liver overgrowth in vivo [30]. In addition, VP was shown to block YAP function by causing its retention to the cytoplasm [31]. Moreover, VP has been reported to downregulate the expression levels of YAP in a variety of cell lines, by unknown mechanisms [31–34].
Ferroptosis is a recently described form of programmed cell death that results from iron-dependent lipid peroxidation [35]. Lipid peroxidation occurs as a chain reaction of free radicals with unsaturated fatty acids in cell membrane and accumulation of lipid peroxidation can lead to loss of membrane integrity [36]. Ferroptosis is mediated by metabolic pathways that impact on the balance of intracellular reactive oxygen species (ROS) and antioxidants. For example, the extracellular source of cysteine is required for the synthesis of the antioxidant peptide glutathione (GSH), and cysteine deprivation or inhibition of the cystine-glutamate transporter (also known as the system Xc−) by the small molecule erastin can cause iron-dependent, oxidative death [35, 37]. The glutathione-peroxidase 4 (GPX4) catalyzes the conversion of phospholipid hydroperoxides to lipid alcohol and can protect the cells from lipid oxidative damage [38, 39]. Inhibition of GPX4 by the small molecule inhibitor RSL3 can lead to increase of lipid peroxidation and ferroptosis that can be prevented by iron chelation [35, 40, 41]. The small molecules ferrostatin-1 and liproxstatin-1 can specifically inhibit ferroptosis [35].
In this study, we examined the effect of VP on oxidative stress and tested the hypothesis that VP may cause lipid peroxidation and ferroptosis in pancreatic cancer cells.
Results
Verteporfin induces ROS production and lipid peroxidation in pancreatic cancer cells
We examined the effect of VP (Fig. 1A) on pancreatic cancer cell proliferation and viability. VP showed dose-dependent inhibition of viability of both PANC-1 and BxPC3 cells (Fig. 1B). VP treatment induced moderate delay in the G1 phase of the cell cycle (Supplemental Fig. S1). In addition, we observed morphological changes in pancreatic cancer cells upon VP treatment, with a significant reduction in cell volume and increase in cell membrane density (Fig. 1C), which is consistent with the morphological features of ferroptosis[35]. VP also reduced the migratory capacities of the pancreatic cancer cells (Fig. 1D).
Figure 1.

Dose-dependent inhibition of pancreatic cancer cells by verteporfin.
(A) CCK8 cell viability assay of PANC-1 and BxPC3 cells treated with VP at various concentration for 24 hours; (B) Morphology of PANC-1 and BxPC3 cells (20X magnification); Wound-healing assay of PANC-1 (C) or BxPC3 (D) cell migration following treatment with VP for 24 hours. (* p<0.01; *** p<0.001).
We used DCFH-DA and C11-BODIPY as the fluorescent probes to detect intracellular ROS and lipid peroxidation, respectively, by flow cytometry. Our results showed an increase in both ROS and lipid peroxidation levels following VP treatment (Fig. 2A and 2B).
Figure 2.

Verteporfin induces rapid increase of ROS and lipid peroxidation in pancreatic cancer cells.
(A) PANC-1 or BxPC3 cells were treated with VP at the indicated concentration for 24 hours, followed by flow cytometry analysis of DCF and C11-BODIPY fluorescence, for ROS and lipid peroxidation, respectively; (B) Quantification of flow cytometry analysis shown in (A);
(C) MDA adduct ELISA of PANC1 or BxPC3 cells following treatment with VP at the indicated concentration for 24 hours;
(D) PANC1 or BxPC3 cells were treated with VP at the indicated concentration for 2 hours, followed by flow cytometry analysis of C11-BODIPY fluorescence. Quantification of flow cytometry analysis is shown;
(E) PANC1 or BxPC3 cells were transfected with control (siNC) or siRNAs targeting YAP (siYAP1 and siYAP2), followed by flow cytometry analysis of C11-BODIPY fluorescence. Quantification of flow cytometry analysis is shown;
(F) MDA adduct ELISA of PANC-1 or BxPC3 cells following transfection with control (siNC) or siRNAs targeting YAP (siYAP1 and siYAP2);
(G) Temoporfin does not affect lipid peroxidation levels. PANC-1 or BxPC3 cells were treated with temoporfin at the indicated concentration for 24 hours, followed by flow cytometry analysis of C11-BODIPY fluorescence. Quantification of flow cytometry analysis is shown;
(H) MDA levels in PANC1 or BxPC3 cells following treatment with temoporfin at the indicated concentration for 24 hours. (*** p<0.001; ns, not significant).
To confirm the effect of VP on lipid peroxidation with an independent method, we used the thiobarbituric acid reactive substances (TBARS) assay to measure the level of malondialdehyde (MDA), a byproduct of the chain reaction that generates lipid peroxide being widely used as a marker for lipid peroxidation [42, 43]. Because the TBARS assay has limited specificity for MDA [44], we also employed an ELISA method to detect the levels of MDA-protein adduct, which has also been used to as a surrogate for MDA levels in ferroptotic cells [45–47]. Our results of both the TBARS (Supplemental Fig. S4) and MDA adduct-ELISA assays (Fig. 2C) support the notion that VP increased MDA levels in the cells, which is consistent with our findings using the fluorescent probes.
The induction of ROS and lipid peroxidation by VP is rapid, as it can be readily detected upon 2 hours of treatment (Fig. 2D). Furthermore, knockdown of YAP does not alter VP-induced ROS or lipid peroxidation (Fig. 2E). Interestingly, temoporfin, another porphyrin-derived molecule that is also used as a photosensitizer in PDT, does not cause any lipid peroxidation under similar experimental conditions (Fig.2H).
Verteporfin treatment causes ferroptosis
Next, we investigated whether VP induced oxidative stress can lead to ferroptosis. We employed two ferroptosis inhibitors, namely ferrostatin-1 (Fer-1) and liporstatin-1 (Lip-1), which have been identified as antioxidant molecules that can prevent ferroptosis [35, 48]. We found that both Fer-1 and Lip1 can reduce VP-induced lipid peroxidation, as shown by the C11-BODIPY staining (Fig. 3A) and measurement of MDA protein adduct (Fig. 3C) and TBARS levels (Supplemental Fig. S4). Importantly, VP-induced lipid ROS can be blocked by deferoxamine (DFO) (Fig 3A and 3C), an iron chelator that has been shown to prevent ferroptosis by deprivation of iron supply [35, 49]. Moreover, we found that VP-induced lipid peroxide levels were also decreased in the presence of the antioxidant N-acetyl-l-cysteine (NAC) (Fig. 3B and 3D). Furthermore, our study showed that VP-induced cytotoxicity can also be reduced by Fer-1, Lip-1, DFO, or NAC (Fig. 3E), but not by the inhibitors of necrosis or apoptosis (Fig. 3F). These findings support the hypothesis that VP can cause ferroptosis in the pancreatic cancer cells.
Figure 3.

Verteporfin-induced lipid peroxidation is reduced by ferroptosis inhibitors and iron chelator.
(A) PANC-1 or BxPC3 cells were treated with VP, either alone or in combination with ferroptosis inhibitors ferrostatin- (Fer-1), liproxstatin-1 (Lip-1), or iron chelator desferoxamine (DFO) at the indicated concentration for 24 hours, followed by flow cytometry analysis of C11-BODIPY fluorescence;
(B) PANC1 or BxPC3 cells were treated with VP and/or N-acetyl-l-cysteine (NAC) at the indicated concentration for 24 hours, followed by flow cytometry analysis of C11-BODIPY fluorescence;
(C) MDA adduct ELISA of PANC1 or BxPC3 cells following treatment with VP in combination with Fer-1, Lip-1 or DFO for 24 hours;
(D) MDA adduct ELISA of PANC-1 or BxPC3 cells following treatment with VP and/or NAC, for 24 hours.
(E) CCK8 cell viability assay of PANC-1 or BxPC3 cells treated with VP in combination with NAC, Fer-1, Lip-1 or DFO for 24 hours;
(F) CCK8 cell viability assay of PANC-1 or BxPC3 cells treated with VP in combination with necrosis (Necro) or apoptosis inhibitor (z-VAD) for 24 hours. (**p<0.01; *** p<0.001; ns, not significant).
Verteporfin disrupts mitochondrial functions
To study the effect of VP on mitochondria, we used the fluorescent probe mitoSOX Red, which specifically detects mitochondrial ROS [50, 51]. VP treatment increase mitochondrial ROS in both PANC-1 and BxPC3 cells (Fig. 4A). Notably, mito-Tempo, a mitochondria-targeted antioxidant [52], decreased VP-induced mitochondrial ROS (Fig.4B). However, mito-Tempo showed negligible effect on VP-induced lipid peroxidation (Fig.4C). We also performed JC-1 staining to evaluate the effect of VP on mitochondria membrane potential. We found that VP decreased mitochondrial membrane potential, to an extent comparable to that caused by the mitochondria uncoupling agent carbonyl cyanide 3-chlorophenyl hydrazone (CCCP) (Fig.4D). Finally, by the Seahorse mitochondrial stress test, we found that VP treatment led to drastic loss of mitochondrial functions within 2 hours (Fig. 4E and Supplemental Fig. S2).
Figure 4.

Effect of VP treatment on mitochondrial function.
(A) Flow cytometry analysis of mitochondrial ROS (mito ROS) by MitoSox Red staining in PANC-1 or BxPC3 cells treated with VP at the indicated concentration for 24 hours;
(B) MitoSox Red staining of PANC-1 or BxPC3 cells treated with VP and/or mitochondrial antioxidant mitoTempo at the indicated concentration for 24 hours;
(C) Flow cytometry analysis of lipid ROS by C11-BODIPY staining in PANC-1 or BxPC3 cells treated with VP and/or Mito Tempo at the indicated concentration for 24 hours;
(D) Flow cytometry analysis of PANC1 or BxPC3 cells using the mitochondrial membrane potential probe JC-1, following treatment with mitochondrial uncoupling agent carbonyl cyanide 3-chlorophenyl hydrazone (CCCP) or VP;
(E) JC-1 staining of PANC-1 or BxPC3 cells following treatment with VP in combination with NAC, mitoTempo, Fer-1, Lip-1 or DFO;
(F) Seahorse mitochondrial stress test of PANC-1 and BXPC-3 cells treated with VP (1μg/ml and 2.5μg/ml) for 2 or 24 hours. (*p<0.05; *** p<0.001; ns, not significant).
Ferroptosis contributes to VP-induced cytotoxicity
We performed propidium iodide (PI) exclusion assay to assess the effect of VP on cell membrane integrity. By flow cytometry, we found that VP treatment for 24 hours led to an increased percentage of cells that lost cell membrane integrity, which can be partially prevented by the presence of Fer-1, Lip-1 or DFO (Fig. 5A). Similarly, prolonged treatment with VP reduced pancreatic cancer cell proliferation, as measured by colony formation assay, which can also be partly reverted by co-treatment with the ferroptosis inhibitors or the iron chelator (Fig. 5B).
Figure 5.

The cytotoxicity of verteporfin can be reduced by ferroptosis inhibitors and iron chelator.
(A) Flow cytometry analysis of cell membrane integrity by PI staining in PANC-1 or BxPC3 cells following treatment with VP in combination with Fer-1, Lip-1 or DFO for 24 hours;
(B) Colony formation assay of PANC1 or BxPC3 cells following treatment with VP in combination with Fer-1, Lip-1 or DFO.
VP treatment down regulates GPX4, a mediator of lipid peroxidation and ferroptosis
We explored the mechanism involved in VP-induced ROS and ferroptosis. Our western blot studies show that VP treatment caused rapid decrease in GPX4 levels in a dose dependent manner (Fig. 6A). The reduction of GPX4 was only partially reversed by inhibitor of ferroptosis, such as Fer-1, Lip-1 or DFO (Fig.6B); by the antioxidant NAC (Fig.6C); or by mitoTEMPO (Fig.6D). However, the treatment with these inhibitors and antioxidants showed little effects when VP was used at a higher concentration, under the conditions that we tested (Fig. 6B, 6C and 6D). This is consistent with the finding that these molecules can only partially protect the cells from VP-induced cytotoxicity.
Figure 6.
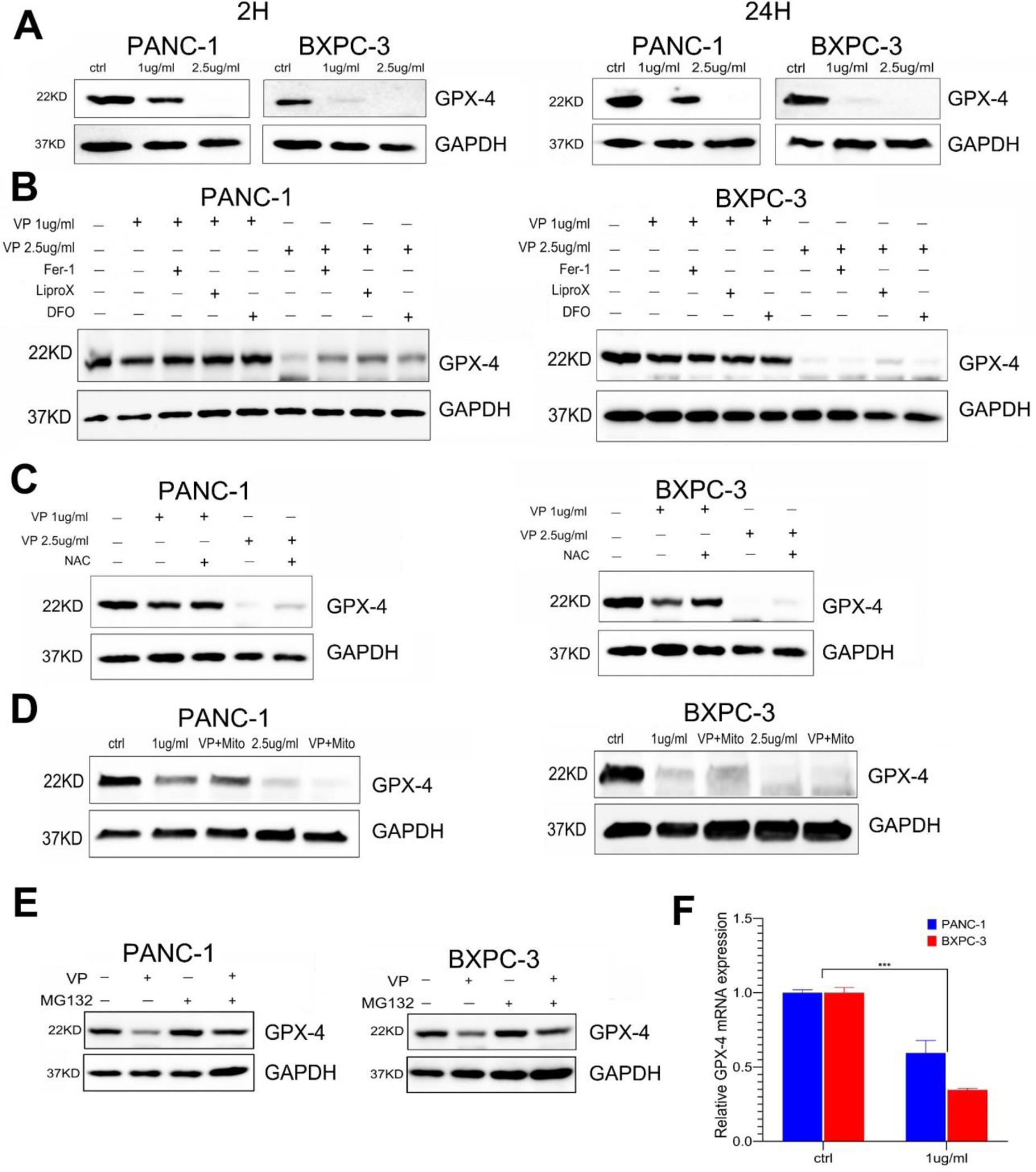
VP treatment causes rapid down regulation of mediator of ferroptosis GPX4
(A) Western blot analysis of GPX4 and GAPDH in PANC-1 or BxPC3 following treatment with VP for 2 hours or 24 hours;
(B) Western blot analysis of GPX4 following treatment with VP in combination with Fer-1, Lip-1 or DFO, for 24 hours;
(C) Western blot analysis of GPX4 following treatment with VP and/or NAC for 24 hours;
(D) Western blot analysis of GPX4 following treatment with VP and/or mitoTempo for 24 hours;
(E) Western blot analysis of GPX4 or GAPDH in PANC-1 or BxPC3 following treatment with VP and/or the proteosome inhibitor MG132.
(F) qPCR analysis of GPPX4 mRNA levels in PANC1 or BxPC3 cells following treatment with VP. The relative expression levels are calculated using GAPDH levels as normalization control. (*** p<0.001).
We found that VP-induced decrease of GPX4 protein levels can be blocked in the presence of the proteosome inhibitor MG132 (Fig. 6E). This result supports the idea that VP can down regulate GPX4 by affecting its protein stability. In addition, GPX4 mRNA levels were moderately reduced by VP treatment (Fig. 6F).
We performed RNA-seq analysis using pancreatic cancer cell lines treated with VP and identified ATF-3 as a gene that is significantly up regulated by VP treatment in pancreatic cancer cell lines (Fig. 7A and Supplemental Fig. S3). In the work presented here, we focus on ATF-3 because it is implicated in playing a role in cellular adaptive response and tumorigenesis, including in pancreatic cancer [53–55]. Our western blot analysis showed that the ATF-3 protein levels were indeed increased by VP, but knockdown of ATF3 has little effect on GPX4 levels (Fig 7B). Moreover, knockdown of ATF-3 led to reduction of VP-induced lipid ROS (Fig 7C). Interestingly, knockdown of ATF-3 decreased overall viability of pancreatic cancer cells (Fig 7D). These observations support the notion that ATF-3 has a protective role in pancreatic cancer cells and negatively regulates ferroptosis (Fig 7E).
Figure 7.

VP induces ATF3 expression levels.
(A) qPCR analysis of ATF-3 mRNA levels in PANC1 or BxPC3 cells following treatment with VP. The relative expression levels are calculated using GAPDH levels as normalization control;
(B) Western blot analysis of ATF-3, GPX4, or GAPDH in PANC-1 or BxPC3 following transfection with control (siNC) or siRNA targeting ATF-3 (siATF-3);
(C) PANC1 or BxPC3 cells were transfected with control (siNC) or siRNA targeting ATF-3 (siATF-3), followed by flow cytometry analysis of lipid peroxidation levels (C11-BODIPY staining);
(D) CCK8 analysis of PANC1 or BxPC3 cell viability following transfection with control (siNC) or siRNA targeting ATF-3 (siATF-3);
(E) Schematic representation showing the effect of VP on ROS, lipid peroxidation, ferroptosis and mitochondria functions. (*** p<0.001).
Discussion
In this study, we have shown that VP can cause rapid and robust production of ROS and lipid peroxidation. This is accompanied with loss of cell membrane integrity, reduced proliferation, and cell death that is morphologically distinct from apoptosis and unaffected by inhibitors of apoptosis or necrosis. Moreover, the VP induced effects on lipid peroxidation and cell viability can be reversed, at least partly under the experimental conditions we employed, by the antioxidant NAC, the ferroptosis inhibitors Fer-1 and Lip-1, or the iron chelator DFO. These effects are comparable to those caused by RSL-3, an established inducer of ferroptosis [41]. Furthermore, we also observed downregulation of several genes that are known for their role in ferroptosis, such GPX4 and SLC7A11, following treatment with VP. Collectively, these findings support the notion that VP causes ferroptosis in the pancreatic cancer cells.
VP is a benzoporphyrin derivative that has been used clinically as a photosensitizer of PDT, which involves laser light activation at the wavelength of around 690 nm [29, 56, 57]. Our results show that VP can induce ROS and lipid ROS without additional light activation. Indeed, we have minimized the exposure to light in our experimental procedures. Of note, temoporfin, a structurally related porphyrin molecule used also as a photosensitizer in PDT, does not exhibit the ability to generate ROS or lipid peroxidation under similar conditions. The molecular basis for VP to induce ROS without light activation is not clear and merits further investigation.
Since its identification as a YAP inhibitor, VP has been widely used in the study of YAP signaling and its impact on cancer cell phenotypic changes have been primarily attributed to its effects on YAP function as a transcriptional cofactor, such as disrupting the YAP-TEAD interaction or causing YAP retention in the cytoplasm[30, 31]. Our findings that VP can induce rapid increase of ROS and lipid peroxidation provide new insights into the mechanism by which VP inhibit cancer cell proliferation.
YAP has been found to modulate cancer cell sensitivity to ferroptosis by regulating the expression of several genes involved in ROS and ferroptotic pathways [58–62]. However, our results show that YAP is not required for VP-induced lipid peroxidation. Nor did knockout of YAP alter the levels of ROS or lipid peroxidation in the pancreatic cancer cell lines we evaluated. These findings indicate that VP can inflict cytotoxicity by causing excessive ROS that can lead to ferroptosis, in a manner that is independent of its previously reported role as a YAP inhibitor.
Our data show that VP treatment caused a rapid decrease of GPX4, which is known to play a critical role in protecting cells from excessive lipid peroxidation and ferroptosis [41]. We found that VP-induced reduction of GPX4 protein levels can be partially reversed by treatment with proteosome inhibitors, which suggest that VP affects the stability of GPX4. It would be of interest to investigate further the mechanism by which VP reduces GPX4.
We found that VP treatment increases mitochondrial ROS and rapid loss of mitochondrial potential and respiration function. However, the presence of the mitochondria-directed antioxidant Mito-tempo can reduce mitochondrial ROS levels but effect minor changes in lipid peroxidation levels. These findings are consistent with the notion that VP induced lipid peroxidation is not dependent on mitochondria generated ROS. Conversely, the impact of VP on mitochondria was not reduced by treatment with NAC, iron chelator or the ferroptosis inhibitors, (and thus independent of lipid peroxidation). Further investigation is needed to clarify the role mitochondria in VP-induced ferroptosis.
The ferroptotic pathway has emerged as a potential target for the development of new cancer drugs [63]. To date, VP has primarily been used as a photosensitizer for PDT for the treatment of choroidal neovascularization [56, 57]. More recently, VP-based PDT has been used for the treatment of localized pancreatic cancer [64]. Our findings that VP can induce ROS and ferroptosis independent of laser light activation provide additional rationale for its use as a systemic chemotherapy drug. It should be noted that our current work is limited to in vitro assays and additional studies using animal models are needed to verify the effects of VP treatment of ROS and lipid peroxidation in vivo. Nevertheless, it is conceivable that the therapeutic capacity of VP can be maximized when combined with additional drugs. In this regard, several studies indicate that VP could increase sensitivity to targeted or chemotherapy drugs [32, 65–68]. Understanding the molecular basis for light independent VP induction of ROS will provide the opportunity to optimize the efficacy and bioavailability of the drug.
In summary, we have demonstrated that VP can induced rapid lipid peroxidation and ferroptosis in pancreatic cancer cells without laser light activation. These findings provide insights into development of new therapeutic strategies that target ferroptosis.
Material and Methods
Cell lines
The human pancreatic cancer cell lines PANC1 and BxPC3 were obtained from ATCC (Manassas, VA). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) or DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS).
Antibodies and Chemicals
The antibodies used in this study include Anti-YAP (13584-1-AP), anti-GAPDH (60004-1-Ig), anti-GPX4 (14432-1-AP), anti-SLC7A11 (26864-1-AP) and anti-ACSL-4 (22401-1-AP) primary antibodies from Proteintech (Rosement, IL); Anti-ATF-3 (E9J4N), HRP-conjugated secondary antibodies against mouse (7076) or rabbit (7074) from Cell Signaling Technology (Danvers, MA). The following chemicals were used Verteporfin (SML0534), N-Acetyl-L-cysteine (NAC, A9165) from Sigma, (129497-78-5); ferrostatin-1 (A4371) from APExBIO; liproxstatin-1 (s7699) from Selleck Chemicals (Houston, TX), Temoporfin (17333), Deferoxamine (14595), and TBARS (TCA Method detection of MDA) Assay Kit (700870) from Cayman Chemical; Carbonyl cyanide 3-chlorophenylhydrazone (CCCP, HY-100941), Z-VAD (HY-16658) and Mito-TEMPO (HY-112879) from MedChemExpress (Monmouth Junction, NJ); JC-1 (70011) from Biotium (Fremont, CA); C11 BODIPY (D3861), MitoSOX Red (M36008) from ThermoFisher.
Cell proliferation and viability assay
CCK-8 assay (APExBIO, Houston, Texas) was performed according to manufacturer’s instruction. Briefly, cells were seeded in 96-well plates (100μl/well) for 24 hours, followed by treatment with drugs as indicated in each experiment. CCK-8 solution (10μl/well) was added and incubated at 37°C for 2 hours. Absorbance at 450 nm was measured with SpectraMax M3 microplate reader (Molecular Devices, San Jose, CA).
Flow cytometry analysis
The polyunsaturated butadienyl fluorescent probe C11-BODIPY 581/591 was used for detection of lipid peroxidation[69]. C11-BODIPY was added to the cell culture at a final concentration of 5μM and incubated for 2 hours at 37°C. 2′,7′-Dichlorofluorescein diacetate (DCFH-DA, Sigma, 35845) was used to detect overall intracellular ROS levels. DCFH-DA was added to the cells at a final concentration of 10 μM and incubated for 2 hours. MitoSOX Red (ThermoFisher; M36008) was used at a final concentration of 5μM for detection of mitochondrial superoxide levels. JC-1 (ThermoFisher; T3168) was used at a final concentration of 2μM to probe mitochondrial membrane potential; Cells treated with CCCP were used as negative control. Propidium Iodide (PI, Sigma, P4170) staining was performed to evaluate membrane integrity and viability of unfixed cells. For cell cycle analysis, the cells were fixed in methanol and stained with PI in the presence of DNAse-free RNAse (ThermoFisher), as described previously[70]. The stained cells were analyzed using BD FACSymphony™ A5 SE Cell Analyzer (BD Biosciences, San Jose, CA).
MDA measurements - TBARS Assay and MDA ELISA
TBARS (Thiobarbituric Acid Reactive Substances) Assay Kit (Cayman Chemical, 10009055) according to manufacturer’s instruction. Briefly, cell lysates were prepared by sonication on ice. MDA (Cayman Chemical, 10009055) was used to generate standard curve. The colorimetric reaction product was measured with SpectraMax M3 microplate reader (Molecular Devices; absorbance at 530 nm). The MDA Assay Kit (competitive ELISA; Abcam, Cambridge, UK; ab238537) was used to measure MDA adduct levels by following manufacturer’s instruction. The cell lysates were added in an MDA conjugate coated 96-well plate. MDA-BSA was used to generate standard curve. Following incubation with anti-MDA adduct and HRP-conjugate secondary antibodies and substrate reaction, the absorbance at 450 nm was measured using SpectraMax M3 microplate reader.
Seahorse mitochondria stress test
The Mitochondria stress test was performed using Agilent Seahorse Xfe96 and XF Cell Mito Stress Test Kit (Agilent Technologies, Santa Clara, CA) at the Cedars-Sinai Proteomics & Metabolomics Core.
Colony formation assay
Cells were plated at a density of 500 cells per well in a six-well plate and incubate for 14 days, changing the culture medium every 5 days. The cells were fixed in formalin for 30 minutes, followed by crystal violet staining. The clone formation rate was calculated. Clone formation rate = (number of clones/number of inoculated cells) × 100%.
Cell migration assay
Wound-healing assay was used to evaluate cell migration. Silicone Insert (ibidi, Fitchburg, WI) were used to generate cell-free gaps in confluent cell culture. After removing the inserts, the cells were treated with drugs as indicated in each experiment. The images were captured over time using the Nikon Eclipse Ti Microscope (Nikon Instruments, Melville, NY).
Western blot
Cell lysates were prepared using RIPA buffer supplemented with protease inhibitors and phosphatase inhibitors (Thermo Fisher Scientific, #78440). The samples were resolved by 4–20% Tris-HCl SDS-PAGE. The nitrocellulose membrane (Bio-Rad) was used for transfer and blocked in 2% nonfat milk in Tris-buffered saline with 0.05% Tween-20.
RNA isolation and analysis
RNA samples were prepared using the Qiagen RNeasy mini kit (QIAGEN, Venlo, Netherlands) or the TRIzol agent (ThermoFisher Scientific). RNA was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative real-time PCR (qPCR) was performed using SYBR Green and iCycler (Bio-Rad). The qPCR primers are shown in Table 1.
Table 1.
Primers used for qPCR
| GPX4 | Forward:GAGGCAAGACCGAAGTAAACTAC Reverse:CCGAACTGGTTACACGGGAA |
| ACSL4 | Forward: CATCCCTGGAGCAGATACTCT Reverse: TCACTTAGGATTTCCCTGGTCC |
| ATF3 | Forward: TGCTCAGAGAAGTCGGAA Reverse:TGGCACAAAGTTCATAGGGCA |
| SLC7A11 | Forward: TCTCCAAAGGAGGTTACCTGC Reverse: AGACTCCCCTCAGTAAAGTGAC |
| GAPDH | Forward: 5’-CTGGGCTACACTGAGCACC-3’ Reverse:5’-AAGTGGTCGTTGAGGGCAATG-3’ |
RNA sequencing (RNA-seq) was conducted at the Cedars-Sinai Applied Genomics Core. The details of RNA quality control, the procedures to prepare and sequence the library, and sequence analysis were described previously[70]. The expression counts for each gene (i.e., transcripts per million) were normalized based on the sequencing depth.
RNA interference
The siRNAs specific for YAP1 (D-012200-01-0005; D-012200-02-0005), ATF-3 (J-008663-05-0002) or control siRNA (D-001210-01-05) were purchased from Horizon Discovery (Lafayette, CO). siRNA was transfected using Lipofectamine 3000 (Invitrogen). The effects of knockdown on gene expression levels were confirmed by qPCR and western blot.
Statistical analysis
Prism 10 (GraphPad Software, San Diego, CA) was used for Student’s t-test analysis (unpaired, two-sided). p values of less than 0.05 were defined as statistically significant.
Supplementary Material
Highlights.
Verteporfin induces lipid peroxidation and ferroptosis-like cell death independent of light activation
Verteporfin disrupts mitochondrial functions
Verteporfin-induced ROS and mitochondrial changes are rapid and independent of YAP
Verteporfin treatment decreases GPX4 and increase ATF3 expression levels
Funding
National Institutes of Health grant 5P01CA233452 (SJP);
National Center for Advancing Translational Sciences UCLA CTSI Grant UL1TR001881 (QW); Wu Jieping Medical Foundation Grant for Clinical Research 320.6750.2023-19-14 (WZ).
Abbreviations:
- PDAC
pancreatic ductal adenocarcinoma
- VP
verteporfin
- ROS
reactive oxygen species
- GPX4
glutathione peroxidase 4
Footnotes
Declaration of competing interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, et al. , Cancer statistics, 2022. CA Cancer J Clin, 2022. 72(1): p. 7–33. [DOI] [PubMed] [Google Scholar]
- 2.Yachida S and Iacobuzio-Donahue CA, The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med, 2009. 133(3): p. 413–22. [DOI] [PubMed] [Google Scholar]
- 3.Neoptolemos JP, et al. , Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol, 2018. 15(6): p. 333–348. [DOI] [PubMed] [Google Scholar]
- 4.Kapoor A, et al. , Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell, 2014. 158(1): p. 185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shao DD, et al. , KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell, 2014. 158(1): p. 171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morvaridi S, et al. , Role of YAP and TAZ in pancreatic ductal adenocarcinoma and in stellate cells associated with cancer and chronic pancreatitis. Sci Rep, 2015. 5: p. 16759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao B, et al. , Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev, 2011. 25(1): p. 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang JM, et al. , YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene, 2013. 32(17): p. 2220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai J, et al. , The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev, 2010. 24(21): p. 2383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lian I, et al. , The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev, 2010. 24(11): p. 1106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cordenonsi M, et al. , The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell, 2011. 147(4): p. 759–72. [DOI] [PubMed] [Google Scholar]
- 12.Zhao B, et al. , The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev, 2010. 24(9): p. 862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zender L, et al. , Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell, 2006. 125(7): p. 1253–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Overholtzer M, et al. , Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A, 2006. 103(33): p. 12405–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinhardt AA, et al. , Expression of Yes-associated protein in common solid tumors. Hum Pathol, 2008. 39(11): p. 1582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KP, et al. , The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A, 2010. 107(18): p. 8248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Pasolli HA, and Fuchs E, Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A, 2011. 108(6): p. 2270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall CA, et al. , Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res, 2010. 70(21): p. 8517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, et al. , The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene, 2011. 30(25): p. 2810–22. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, et al. , Overexpression of yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci, 2010. 101(5): p. 1279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, et al. , The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell, 2005. 122(3): p. 421–34. [DOI] [PubMed] [Google Scholar]
- 22.Udan RS, et al. , Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol, 2003. 5(10): p. 914–20. [DOI] [PubMed] [Google Scholar]
- 23.Chan EH, et al. , The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene, 2005. 24(12): p. 2076–86. [DOI] [PubMed] [Google Scholar]
- 24.Zhao B, et al. , A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev, 2010. 24(1): p. 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu CY, et al. , The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFbeta-TrCP E3 ligase. J Biol Chem, 2010. 285(48): p. 37159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, et al. , TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem, 2009. 284(20): p. 13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanai F, et al. , TAZ: a novel transcriptional co-activator regulated by interactions with 14–3-3 and PDZ domain proteins. Embo j, 2000. 19(24): p. 6778–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vassilev A, et al. , TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev, 2001. 15(10): p. 1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michels S and Schmidt-Erfurth U, Photodynamic therapy with verteporfin: a new treatment in ophthalmology. Semin Ophthalmol, 2001. 16(4): p. 201–6. [DOI] [PubMed] [Google Scholar]
- 30.Liu-Chittenden Y, et al. , Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev, 2012. 26(12): p. 1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, et al. , Verteporfin inhibits YAP function through up-regulating 14–3-3sigma sequestering YAP in the cytoplasm. Am J Cancer Res, 2016. 6(1): p. 27–37. [PMC free article] [PubMed] [Google Scholar]
- 32.Song S, et al. , The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res, 2015. 21(11): p. 2580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ciamporcero E, et al. , YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene, 2016. 35(12): p. 1541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei L, et al. , Verteporfin reverses progestin resistance through YAP/TAZ-PI3K-Akt pathway in endometrial carcinoma. Cell Death Discov, 2023. 9(1): p. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon SJ, et al. , Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell, 2012. 149(5): p. 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ayala A, Munoz MF, and Arguelles S, Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev, 2014. 2014: p. 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dolma S, et al. , Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell, 2003. 3(3): p. 285–96. [DOI] [PubMed] [Google Scholar]
- 38.Schuckelt R, et al. , Phospholipid hydroperoxide glutathione peroxidase is a selenoenzyme distinct from the classical glutathione peroxidase as evident from cDNA and amino acid sequencing. Free Radic Res Commun, 1991. 14(5–6): p. 343–61. [DOI] [PubMed] [Google Scholar]
- 39.Ursini F, Maiorino M, and Gregolin C, The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta, 1985. 839(1): p. 62–70. [DOI] [PubMed] [Google Scholar]
- 40.Yang WS and Stockwell BR, Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol, 2008. 15(3): p. 234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang WS, et al. , Regulation of ferroptotic cancer cell death by GPX4. Cell, 2014. 156(1–2): p. 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Armstrong D and Browne R, The analysis of free radicals, lipid peroxides, antioxidant enzymes and compounds related to oxidative stress as applied to the clinical chemistry laboratory. Adv Exp Med Biol, 1994. 366: p. 43–58. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, et al. , CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature, 2019. 569(7755): p. 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin H and Porter NA, Specificity of the ferrous oxidation of xylenol orange assay: analysis of autoxidation products of cholesteryl arachidonate. Anal Biochem, 2003. 313(2): p. 319–26. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, et al. , Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol, 2019. 26(5): p. 623–633 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lung CC, et al. , Immunochemical properties of malondialdehyde-protein adducts. J Immunol Methods, 1990. 128(1): p. 127–32. [DOI] [PubMed] [Google Scholar]
- 47.Yamada S, et al. , Immunochemical detection of a lipofuscin-like fluorophore derived from malondialdehyde and lysine. J Lipid Res, 2001. 42(8): p. 1187–96. [PubMed] [Google Scholar]
- 48.Friedmann Angeli JP, et al. , Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol, 2014. 16(12): p. 1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan HF, et al. , Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther, 2021. 6(1): p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kauffman ME, et al. , MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React Oxyg Species (Apex), 2016. 2(5): p. 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson KM, et al. , Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A, 2006. 103(41): p. 15038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trnka J, et al. , A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic Biol Med, 2008. 44(7): p. 1406–19. [DOI] [PubMed] [Google Scholar]
- 53.Azizi N, et al. , Loss of activating transcription factor 3 prevents KRAS-mediated pancreatic cancer. Oncogene, 2021. 40(17): p. 3118–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ku HC and Cheng CF, Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front Endocrinol (Lausanne), 2020. 11: p. 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolford CC, et al. , Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J Clin Invest, 2013. 123(7): p. 2893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller JW, et al. , Photodynamic therapy with verteporfin for choroidal neovascularization caused by age-related macular degeneration: results of a single treatment in a phase 1 and 2 study. Arch Ophthalmol, 1999. 117(9): p. 1161–73. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt-Erfurth U, et al. , Photodynamic therapy with verteporfin for choroidal neovascularization caused by age-related macular degeneration: results of retreatments in a phase 1 and 2 study. Arch Ophthalmol, 1999. 117(9): p. 1177–87. [DOI] [PubMed] [Google Scholar]
- 58.Zhu G, et al. , O-GlcNAcylation enhances sensitivity to RSL3-induced ferroptosis via the YAP/TFRC pathway in liver cancer. Cell Death Discov, 2021. 7(1): p. 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin Y, et al. , Oncogenic Activation of YAP Signaling Sensitizes Ferroptosis of Hepatocellular Carcinoma via ALOXE3-Mediated Lipid Peroxidation Accumulation. Front Cell Dev Biol, 2021. 9: p. 751593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu J, et al. , Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature, 2019. 572(7769): p. 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao R, et al. , YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med, 2021. 13(12): p. e14351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, et al. , MITD1 Deficiency Suppresses Clear Cell Renal Cell Carcinoma Growth and Migration by Inducing Ferroptosis through the TAZ/SLC7A11 Pathway. Oxid Med Cell Longev, 2022. 2022: p. 7560569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stockwell BR, Jiang X, and Gu W, Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol, 2020. 30(6): p. 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huggett MT, et al. , Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer, 2014. 110(7): p. 1698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao X, et al. , A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett, 2017. 402: p. 61–70. [DOI] [PubMed] [Google Scholar]
- 66.Guo L, et al. , Knockdown of TAZ modifies triple-negative breast cancer cell sensitivity to EGFR inhibitors by regulating YAP expression. Oncol Rep, 2016. 36(2): p. 729–36. [DOI] [PubMed] [Google Scholar]
- 67.Ma K, et al. , Nuclear accumulation of Yes-Associated Protein (YAP) maintains the survival of doxorubicin-induced senescent cells by promoting survivin expression. Cancer Lett, 2016. 375(1): p. 84–91. [DOI] [PubMed] [Google Scholar]
- 68.Song J, et al. , Role of YAP in lung cancer resistance to cisplatin. Oncol Lett, 2018. 16(3): p. 3949–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Drummen GP, et al. , C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic Biol Med, 2002. 33(4): p. 473–90. [DOI] [PubMed] [Google Scholar]
- 70.Li C, et al. , Epithelial cell transforming 2 is regulated by Yes-associated protein 1 and mediates pancreatic cancer progression and metastasis. Am J Physiol Gastrointest Liver Physiol, 2021. 320(3): p. G380–G395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.