

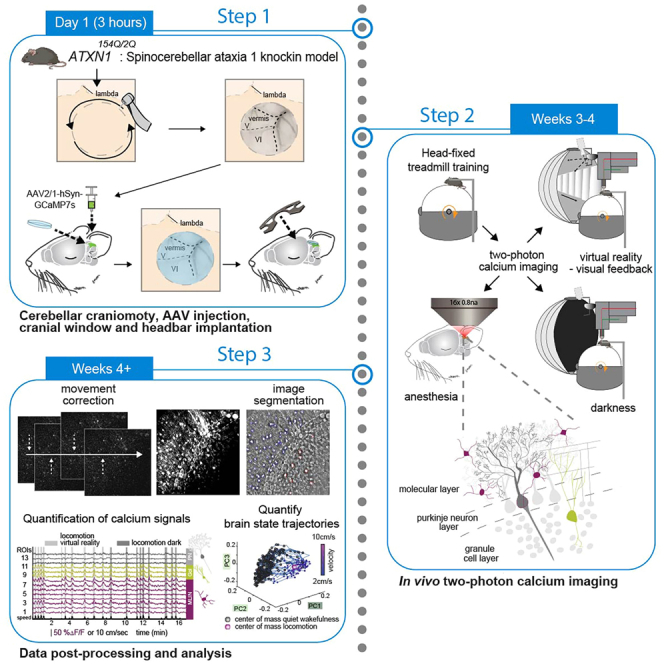
Summary
Purkinje neuron degeneration characterizes spinocerebellar ataxia type 1, yet the comprehension of the impact on the broader cerebellar circuit remains incomplete. We here detail simultaneous in vivo two-photon calcium imaging of diverse neuronal populations in the cerebellar cortex of Sca1 mice while they are navigating a virtual environment. We outline surgical procedures and protocols to chronically record from identical neurons, and we detail data post-processing and analysis to delineate disease-related alterations in neuronal activity and sensorimotor-driven response properties.
For complete details on the use and execution of this protocol, please refer to Pilotto et al.1
Subject areas: Health Sciences, Neuroscience, Cognitive Neuroscience, Behavior
Graphical abstract
Highlights
-
•
Simultaneous 2P calcium imaging of diverse neuronal populations in cerebellar cortex
-
•
Detailed description of surgery, in vivo imaging in behaving mice, and image processing
-
•
Characterization of single cell and network alterations in Sca1 tg mice
-
•
Impact of chemogenetic MLIN inhibition on neuronal activity in cerebellar cortex
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Purkinje neuron degeneration characterizes spinocerebellar ataxia type 1, yet the comprehension of the impact on the broader cerebellar circuit remains incomplete. We here detail simultaneous in vivo two-photon calcium imaging of diverse neuronal populations in the cerebellar cortex of Sca1 mice while they are navigating a virtual environment. We outline surgical procedures and protocols to chronically record from identical neurons, and we detail data post-processing and analysis to delineate disease-related alterations in neuronal activity and sensorimotor-driven response properties.
Before you begin
The degeneration of Purkinje neurons (PNs) in the cerebellar cortex is a hallmark feature of spinocerebellar ataxia type 1 (Sca1). However, there is mounting evidence that neuronal and network dysfunction precede overt degeneration in many neurodegenerative diseases (NDs), such as Huntington’s disease,2 Alzheimer’s disease,3,4,5 Amyotrophic lateral sclerosis,6,7 Parkinson’s disease8 and Spinal muscular atrophy.9 Across these NDs it remains incompletely understood, how different circuit elements are functionally altered in a disease-stage specific manner, and how such functional changes would affect information processing in neural networks, or even contribute to the degenerative process per se. In Sca1, earlier research in mouse models of the disease has shown presymptomatic disruption of PN action potential firing rates, in combination with altered excitatory and inhibitory inputs to PN.10 Unravelling the development of such dysfunction across the cerebellar cortex circuit, including the elements that are seemingly unaffected by the degenerative process, could be critical in identifying novel therapeutics targets in Sca1. Here we outline a protocol for in vivo two-photon calcium imaging in the cerebellar cortex of Sca1 mice. We demonstrate the simultaneous imaging of distinct neuronal populations across all layers of the cerebellar cortex in behaving mice, allowing a comprehensive insight into their functional alterations in the Sca1 model. The protocol describes step by step the surgical procedure, in vivo two-photon imaging data acquisition, and subsequent examples of imaging data post-processing and quantification. Using this pipeline, we recently identified hyperactive molecular layer interneurons (MLIN) as a critical driver of PN dysfunction and degeneration in Sca1.1 Using chemogenetics to reduce MLIN activity, we demonstrate that MLIN inhibition can mitigate PN dysfunction, slow down PN degeneration and motor symptom deficits and thus serve as a potential therapeutic strategy. Furthermore, this method can be applied to investigate basic cerebellar function or other cerebellar disorders.
For complete details on the use and execution of this protocol, please refer to Pilotto et al., 2023.1
Institutional permissions
Animal care, housing, ethical usage, and procedures were approved by the government of upper Bavaria in accordance with the required guidelines.
Experimenters following this protocol must gain permission from and abide by their local authorities.
Preparation of materials for cerebellar craniotomy, AAV injection, and cranial window implantation
Timing: 1 h
Completing these steps will allow the experimenter to have all the equipment prepared for surgical procedures, along with the virus loaded in the pipette for cerebellar cortex injections.
-
1.Gather supplies for surgical procedures (Figure 1).
-
a.Gather the following items for surgery: stereotaxic frame and micromanipulator, heating pad, dental drill, hypodermic needles (30G 12 mm & 25G 25 mm), spring scissors (straight 2 mm), iris scissors fine pointed, Dumont forceps (#5 & #3), circular borosilicate glass coverslips 3 mm in diameter, custom designed open headbars (see Figures 1B and 1C) glass micropipette, butterfly wings connection 3-way stopcock, 15 mL syringe, Sugi Sponge Points Saugtupfer, Gelfoam, Venus Diamond Flow UV curable glue, Histoacryl, sterile saline solution, eye cream, Paladur powder and solution for dental cement, with a suitable mixing dish (e.g., a 48-well plate).
-
b.Cut Gelfoam into small pieces (e.g., < 0.5 cm2) for wiping away blood, soak in saline.
-
c.Load larger syringe (e.g., 1–15 mL) with saline.
-
d.Prepare anesthesia and analgesics so that they can be administered in legally approved doses.
-
e.Ensure necessary tools, the coverslip, headbar, micromanipulator, stereotaxic frame and surgery area is sterilized in order to achieve aseptic surgical environment (e.g., 70% ethanol or autoclaving the appropriate tools).
-
a.
CRITICAL: Ensuring surgical environment is aseptic, will reduce risk of infection or strong immune response under the cranial window, which is important for both post-surgery recovery and window quality.
-
2.Prepare AAV2/1-hSyn-GCaMP7s-WPRE (viral stock titer: 2.2 × 10ˆ13 GC/mL, Addgene, #104487).
-
a.Pull micropipettes (10 μL pipettes, Blaubrand), to produce tip with diameter of approximately 1 μm, cut tip (approximately 5 mm from end) with iris scissors.
-
b.Thaw stock AAV aliquot on ice, dilute in saline or Milli-Q water with a ratio of 1:10 for GCaMP7s (final titer: 2 × 10ˆ12 GC/mL).
-
c.In preparation for stereotaxic injections into cerebellar cortex (lobule V & VI), load diluted virus into pulled glass pipette.
-
i.The pipette can be connected to a syringe with winged infusion set tubing and stop-cock (Figure 1A), thus allowing the experimenter to manually control pressure in order to draw the desired volume into the pipette.
-
ii.We recommend loading the pipette with sufficient volume of virus to complete all injections for the current mouse.
-
iii.Alternatively a microinjector can also be used to regulate the air pressure.
-
i.
-
a.
Figure 1.

Supplies for craniotomy, cortical injection, cranial window and headbar implantation
(A) General surgical equipment.
(B) 3 mm circular coverslip and custom designed headbar.
(C) Custom designed open headbar. All measurements in mm, ⌀ indicates diameter, R indicates the radius of curvature.
Preparation of experimental mice
Anesthetize the mouse in preparation for surgical procedures.
-
3.
Select animals 3–4 weeks younger than the desired age for imaging experiments, allowing appropriate time for recovery, viral expression and habituation. In order to perform imaging experiments around postnatal day (P) 60, we performed surgery at ∼ P40 in the Sca1 model, Atxn1154Q/2Q knock-in mice (B6.129S-Atxn1tm1Hzo/J), or age-matched wild type littermates for control experiments.
-
4.Administer Meloxicam (10 mg/kg) and Metamizol (200 mg/kg) orally for pre-emptive analgesia 30 min prior to start of surgery, followed by intraperitoneal injection of Fentanyl (0.05 mg/kg), Midazolam (5 mg/kg), and Metedomidin (0.5 mg/kg) for anesthesia.
-
a.Check sufficient anesthesia depth by pinching the tail or paw. Procedures were approved by the local authorities for sufficient induction of analgesia and anesthesia.
-
a.
Note: Age of mice during window implantation is ideally not younger than 4 weeks old (∼P30), to allow for full development of the skull.
-
5.
Shave, clean, and apply local anesthetic to surgical region.
-
6.
Once the mouse is sufficiently anesthetized, apply eye ointment cream (Bepanthen) to the eyes to prevent drying over the course of the procedure.
-
7.
Shave posterior area of head with a razor or hair removal cream.
-
8.
Using a cotton swap or Sugi, wipe away excess loose hair and clean the area to reduce risk of infection (sterile saline solution, small amounts of Braunol or 70% ethanol can also be used).
-
9.
Apply local anesthetic (Xylocaine Gel 2%, Aspen) to region, wait 3–5 min before cutting the fur.
-
10.
The mouse can now be moved to the aseptic surgical environment to proceed with the following steps.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| AAV2/1-hSyn-GCaMP7S-WPRE | Dana et al., 201911 | Addgene: 104487-AAV2/1 |
| AAV2/8-hSyn-DIO-hM4D(Gi)-mCherry | Krashes et al., 201112 | Addgene: 44362-AAV2/8 |
| Experimental models: Organisms/strains | ||
| Atxn154Q/2Q (B6.129S-Atxn1tm1Hzo/J) Figures 6A–6C include data from 6 Sca1 mice (5 female, 1 male) and 8 wild-type (WT) littermates (6 female, 2 male), at ∼ P60 (see Before you begin, step 3). Figure 6E includes data from 5 Sca1 (4 female, 1 male) mice and 8 WT (6 female, 2 male) (see Pilotto et al., 2023).1 |
Jackson Laboratory | RRID: IMSR_JAX:005601 |
| PV-IRES-cre (B6;129P2-Pvalbtm1(cre)Arbr/J) | Jackson Laboratory | RRID: IMSR_JAX:008069 |
|
Sca1::PV and wild-types littermates (WT::PV) were obtained through crossing male Atxn1154Q/2Q with PV-IRES-cre homozygous females. Figure 7 includes data from 5 Sca1::PV (3 female, 2 male) and 5 WT::PV (2 female, 3 male), at ∼ P110 (see Pilotto et al., 2023).1 |
||
| Software and algorithms | ||
| MATLAB R2018a version | MathWorks, Inc. https://de.mathworks.com/ |
RRID:SCR_001622 |
| Custom-built software on LabVIEW 2018 SP1, 2020 SP1, National Instruments | Keller et al.13 | https://www.ni.com/de-de/shop/software/products/labview.html |
| SciScan 1.4 - Imaging acquisition software | Scientifica UK | N/A |
| Other | ||
| IR camera | Imaging source | DMK 22BUC03 |
| Ti:Sapphire laser with a DeepSee pre-chirp unit | Spectra-Physics Mai Tai HP | N/A |
| Two-photon microscope, equipped with an 8 kHz resonant scanner | HyperScope, Scientifica | N/A |
| ×16 water-immersion objective (0.8 NA) | Nikon | N/A |
| Filter (dichroic beamsplitter) | Olympus | U- MF2 |
| Emission filter | G 525/50nm | |
| Optical computer mouse | Logitech | MX-518 |
| Sugi Sponge Points Saugtupfer | Kettenbach-medical | N/A |
| Histoacryl (Braun) | B. Braun | N/A |
| Dental drill | NSK Presto II | N/A |
| Dental drill tips | Meisinger, HM 1 Round RA | N/A |
| Stereotaxic frame and micromanipulator | RWD Life Science | Model: 68513 |
| Thermometer | RWD Life Science | Model: 69002 |
| 48 well plate | Sigma-Aldrich | N/A |
| Vertical micropipette puller | Sutter Instrument Co. | Model: P-30 |
| Headbar | e.g. https://laserjob.de/ | Custom design, see Figure 1C. |
| 10 μL glass pipettes | Blaubrand | N/A |
| D263 Coverslip/coverglass | Warner Instruments | CS-3R |
| Forceps Dumont (#3/#5) | Dumont SA | N/A |
| Hypodermic needle (25G 25 mm) | B. Braun | N/A |
| Hypodermic needle (30G 12 mm) | B. Braun | N/A |
| NaCl 0.9% (saline) | B. Braun | N/A |
| Ringer’s solution | B. Braun | N/A |
| GELFOAM | Pfizer | N/A |
| Venus Diamond Flow (UV curable glue) | Kulzer | N/A |
| UV light (BA Optima 10) | BA International | B2-BASES50 |
| Paladur liquid & powder | Kulzer | N/A |
| Eye ointment | Bepanthen | N/A |
| Braunol | B. Braun | N/A |
| Xylocaine gel 2% | Aspen | N/A |
| SONOSID ultraschall-gel | Asid Bonz | N/A |
| 3-way stopcock | B. Braun | N/A |
| 15 mL syringe | B. Braun | N/A |
| Winged infusion sets | B. Braun | N/A |
| Isoflurane Baxter vet 1000 mg/g | Baxter | HDG9623V |
| Meloxicam (1.5 mg/mL, 5 mg/mL) | Ceva Meloxidyl | N/A |
| Medetomidine (1 mg/mL) | Alfavet Dormilan | N/A |
| Fentanyl (Fentadon) | Dechra Pharmaceuticals | N/A |
| Midazolam (500 mg/mL) | Roche Dormicum | N/A |
| Naloxone (0.4 mg/mL) | Ratiopharm | N/A |
| Atipamezol (5 mg/mL) | Prodivet pharmaceuticals | N/A |
| Flumazenil Kabi (0.5 mg/ 5 mL, 5 × 5 mL) | Fresenius Kabi | N/A |
| Physiological monitoring system for mouse (10 g–100 g) with 8” tablet | Harvard Apparatus | 75-1500 |
| Isoflurane vaporizer | Harvard Apparatus | N/A |
Step-by-step method details
Cerebellar craniotomy
In this step a circular part of the skull overlaying the cerebellum is removed to expose the desired imaging region, thus allowing the stereotaxic injection of AAVs into the target area. The removed part of the skull matches the shape and size of the experimenter’s cranial window, to allow the implantation of a glass cover slip to provide chronic optical access.
-
1.Cut the fur along the midline above the skull and gently remove the periosteum to expose the skull over the cerebrum and cerebellum.
-
a.Fix the mouse’s head securely in the stereotaxic frame (Figure 2A).
-
b.Perform a final reflex check (e.g., pinch of the toe or tail) to ensure mouse is appropriately anesthetized.
-
c.Cut the fur overlaying the skull using sterilized scissors.
-
d.Pull skin apart to fully expose the skull (Figure 2B).
-
e.Gently remove the periosteum using sterile sugis (Sugi Sponge Points).
-
f.Use fine spring scissors and forceps to gently loosen the occipital muscles to expose the interparietal and rostral portion of the occipital bone.
-
i.Take care when removing the rostral parts of the muscles, overlaying the interparietal bone, tissue further caudal, overlaying the occipital bone contains larger blood vessels and severing these can result in excess bleeding.
-
ii.Removing the rostral muscle tissue exposes more bone, which provides a larger surface area for the dental cement to bind to, when sealing the window and positioning the headbar in place.
-
i.
-
a.
-
2.Mark the location of a 3 mm diameter circular craniotomy via gentle scratching of the skull (Figure 2C).
-
a.Coordinates from bregma, centered at −6.5 mm antero-posterior (AP), 0.5 mm medio-lateral (ML).
-
b.Place a 3mm coverslip (Warner Instruments, thickness 0.15 ± 0.02 mm) centered on the marked position. Gently scratch the skull using the sharp edge of a hypodermic needle (25G 25 mm, Braun), to mark the outline of the cranial window as a reference for drilling.
-
a.
Note: based on preference one could scratch a circle directly around the coverslip or a square as shown in Figure 2C).
-
3.Using a dental drill (drill: NSK Presto II, tips: Meisinger, HM 1 Round RA), gently drill a 3 mm diameter circle into the skull, guided by the scratch marks as a reference and remove the bone flap to complete the craniotomy. Check with a sample 3 mm diameter coverslip as you drill to ensure the shape of your circle is precise. The craniotomy should allow a tight-fitting area for the window.
-
a.Move the drill evenly around the edge of the circle to gradually thin the skull in this shape. Apply saline solution to the thinned skull as you move deeper, the underlying cerebellar vasculature should become visible through the drilled skull, following saline application.Note: When drilling along the outer perimeter of the coverslip the resulting cranial window will be slightly bigger than 3 mm due to the size of the groove formed by the drill head, which will allow for a complete insertion of the coverslip.
-
b.Use a hypodermic needle (25G) to carefully ease the center circle of the skull upwards away from the cortex, pressing the tip of the needle lightly into the side of the exposed central bone flap, without damaging or removing the dura.
-
i.Flush with saline to aid removal, the bone flap should separate from the surrounding skull. After the separation, flush again with saline to drain any blood (Figure 2D).Note: The size of the craniotomy should be as tight-fitting as possible to prevent unnecessary exposure of the brain surface and allow for sufficient window stability.
-
i.
-
c.Gently place a small Gelfoam piece that has been soaked in sterile saline onto the exposed cerebellar cortex to absorb blood and prevent the meninges and brain tissue from drying out prior to AAV injection.CRITICAL: While drilling do not apply pressure towards the cerebellar cortex. This is vital to avoid damage to the superficial layers of the cerebellar cortex. Run the drill head horizontally to carefully thin the skull in a circle.CRITICAL: Avoid drilling directly over the transverse sinus and take extra care when drilling this area (towards lambda). Rupturing these sinuses will lead to severe bleeding.
-
a.
Figure 2.

Steps for craniotomy, stereotaxic injection, window and headbar implantation
(A) Following anesthesia, the mouse head is shaved and placed in a stereotaxic frame.
(B) The fur is cut along the midline and the periosteum gently removed, exposing both bregma, lambda, the interparietal (IP) and occipital bones (OB). Note rostral portion of muscle lying on the occipital bone towards where it meets the interparietal has been removed, exposing a larger area of the skull.
(C) The area for the craniotomy can then be marked out on the skull, through scratching with a hypodermic needle.
(D) Circular craniotomy exposes the cerebellar vermis and hemisphere.
(E) Stereotaxic injection of AAV2/1-hSyn-GCaMP7s into the cerebellar cortex of lobules V and VI.
(F) A glass coverslip placed over the craniotomy and sealed flush to the surrounding skull using UV curable glue.
(G) Prior to headbar implantation, the skull should be scratched to facilitate a strong bond between the headbar and the skull when sealing together. The image shows a diagonal cross section scratched across the skull.
(H) The headbar is initially attached using Histoacryl glue, ensuring that when the headbar is horizontal, the head of the mouse is resting in a natural position, and that the edges of the headbar are away from the window to provide optical access.
(I) Once the headbar is positioned correctly, it can be sealed in place by application of dental cement across the skull.
(J) Side view of completed headbar implantation.
(K) Front view of completed headbar implantation.
(L) Mouse recovered in cage following surgery. Scale bars in (C), (D) and (F) 1 mm.
Stereotaxic injection of Adeno-associated viruses
Inject the AAV2/1-hSyn-GCaMP7s-WPRE to allow for expression of the genetically encoded calcium indicator GCaMP7s (or any other version) for functional imaging of distinct neuronal populations in the cerebellar cortex.
-
4.Inject ∼300 nL of pre-diluted AAV2/1-hSyn-GCaMP7s-WPRE into the cerebellar cortex (Figure 2E).
-
a.Position the pipette tip above the injection site using the AP/ML coordinates for the desired region (for expression in areas of lobule V & VI we performed 2 injections, one at −6.8 mm AP, 0.5 mm ML, and the other at –6.5 mm AP, 1 mm ML).
-
b.Gently position the pipette tip onto the cortical surface and set the depth to ‘0’ on the micromanipulator.
-
c.Lower the pipette tip into cerebellar cortex, inject 150 nL at a depth of 250 μm at a rate of 50 nL/min, wait 5 min and draw pipette tip up to 150 μm, inject a further 150 nL again at 50 nL/min. Wait 5–10 min and then slowly draw up the pipette tip to remove from the cortex.
-
d.Repeat for following injection sites.
-
a.
Note: Keep brain surface hydrated with sterile saline solution. Place small pieces of saline soaked Gelfoam on the edge of the craniotomy. If there is any bleeding, use the Gelfoam to cover the craniotomy until the bleeding stops, then gently flush or wipe away any excess blood again using sterile saline or sterile saline soaked Gelfoam.
Note: Waiting before withdrawing the pipette tip following injection prevents backflow and minimizes spread of the virus away from desired imaging area.
Cerebellar window and headbar implantation
In this step the craniotomy is sealed to the surrounding skull tissue, using a glass coverslip to create a cranial window, to allow optical access to the cerebellar cortex. A metal headbar allows for head-fixation underneath the objective and thus stable positioning during two-photon imaging in behaving head-fixed mice.
-
5.Fit a borosilicate glass coverslip (Warner Instruments) into the craniotomy to cover the exposed cerebellar cortex and seal it, using UV-curable glue to attach it to the surrounding skull (Figure 2F).
-
a.Place the coverslip gently on the cerebellar cortex; apply light pressure so that the window is level with the skull.
-
b.To fix the coverslip in place, apply UV-curable glue (Venus Diamond Flow, Heraeus Kulzer GmbH) initially on 2–4 points around the window (use tip of a hypodermic needle to apply small controlled volumes) and apply UV light (BA Optima 10, 420–480 nm, 1200 mw/cm2, for 4–6 s) to cure the glue, ensuring the window is secured level with skull and in contact with the cortex beneath.
-
c.After the window is secured, carefully move around the coverslip perimeter with UV-curable glue to ensure the window is sealed flush to the surrounding skull; apply UV light once entire perimeter is covered to solidify the seal.
-
d.Apply small amounts of Histoacryl (1–2 μL at a time) to the outer edge of the UV- curable filling connected to the skull, to provide more stable sealing between the curable glue and the skull. Do not apply Histoacryl directly onto the window. Otherwise gently wipe or scratch it off using Sugis or a thin hypodermic needle.
-
a.
-
6.Secure a headbar to the skull.
-
a.Slightly scratch the surface of the entire skull using a hypodermic needle (25G), so that grid-like marks are clearly visible. This increases surface area for the subsequently applied glue to allow for a more stable headbar implantation (Figure 2G).
-
b.Place the headbar on the skull around bregma, roughly away from the cerebellar window to ensure that there is space for an objective to be lowered above the cranial window within working distance appropriate to the imaging setup. We thus use a custom-designed headbar (Figures 1B and 1C) to facilitate such placement without potentially impeding objective movement, but care must be taken to avoid limitations to optical access.
-
c.Fix the headbar in place, initially using a small volume of Histoacryl (Braun) (roughly 10–30 μL should be sufficient) (Figure 2H).
-
a.
Pause point: Check the headbar is appropriately positioned for natural head positioning and maximal comfort of the mouse during head-fixation. If the headbar position is off, head-fixed imaging in awake mice is strongly hampered as the mouse will be uncomfortable and movements on the ball will be strongly affected. To check the position, remove the mouse from the stereotaxic frame and verify that the headbar is positioned horizontally (parallel to the bench surface, Figure 2J) without larger tilts or any rotation . If initially uncertain about the correct implantation angle, it is recommended to mount the anesthetized mouse briefly underneath the objective and assess head and body position upon head fixation and correct if necessary. This procedure should take 3–5 min and mice remain anesthetized during that time.
-
7.Seal headbar and all exposed skull with dental cement (Figures 2H–2K).
-
a.Prepare dental acrylic (Paladur, Heraeus Kulzer GmbH), using e.g., a 48-well plate with a volume of 0.5 mL per well. Fill half a well with the Paladur powder and cover with the Paladur liquid, stir in and check viscosity as you stir to a suitable viscosity, where one can apply it in a controlled manner to the skull and spread to desired areas, without it running onto skin or the window.
-
b.Apply to the skull, sealing the area surrounding the window, sealing the skin to the skull.
-
c.Wait until cement has dried to a level where the headbar cannot be moved, which usually takes around 10–20 min, then the mouse can be awoken from anesthesia.
-
a.
Post-operative care
The following steps outline methods to ensure the mouse recovers as comfortably as possible following the surgical procedure.
-
8.Antagonism of anesthetics and immediate care.
-
a.At the end of the surgery, administer Ringer’s solution (∼25 mL/kg, however experimenter should check if this is in line with local regulations) subcutaneously (s.c.) to aid recovery.
-
b.At the end of the procedure the anesthesia is antagonized through the i.p. application of Atipamezol (2.5 mg/kg), Flumazenil (0.5 mg/kg), and Naloxone (1.2 mg/kg) used in line with local regulations.
-
c.Monitor the mouse in a warmed recovery chamber, separate to the usual home cage. Ensure that the mouse is awake and responsive before returning to the home cage. Implanted mice can be housed in groups; however, they must be checked regularly in line with local project licensing.
-
a.
-
9.Post-operative care over following weeks.
-
a.Administer the analgesic Meloxicam (10 mg/kg) orally every 12 h over the first 72 h following surgery in line with local regulations.
-
b.The mouse should be behaving and responding as usual within 24–72 h following surgery. Assess appearance, motor skills, and general behavior in line with local regulations.
-
c.After 10–14 days of continued assessment, if the mouse is healthy, training on air-supported treadmill can begin. For head fixation during the initial training, a light very brief anesthesia with isoflurane can reduce stress. Place the mouse in an isoflurane chamber (2.5% isoflurane, flow rate 0.5 L/min) and wait until the animal stops moving (around 10–15 s). Immediately remove the mouse and secure head-fixed on the treadmill, the mouse should wake up after 30–60 s.
-
d.Check on the quality of the window, vasculature should be clearly visible, without patches of dried blood. If the window appears “cloudy” this is likely a sign of dural thickening, infection or bone regrowth, which will compromise the image quality and resolution.
-
e.Three - four weeks following the AAV injection, GCaMP7s expression should be sufficiently large and the initial inflammatory response should have ceased14 to enable imaging.
-
a.
Two-photon calcium imaging in awake and anesthetized mice
The following steps allow a comprehensive characterization of neuronal activity levels and response properties, through recording calcium signals from various neuronal populations in the cerebellar cortex in behaving animals. The awake recording sessions include distinct phases in either darkness, or with visual feedback provided through a virtual reality, in order to probe, amongst other features, sensorimotor integration.
Note: Alterations to neuronal activity in a disease model that occur in one condition may not persist in others as they may hinge on a particular signal integration or the lack thereof in disease cases.15 For instance, it was shown that the same motor action can drive differential neuronal responses depending on whether the animal is in a light or dark environment.16 In addition, given that light volatile anesthesia more vigorously diminishes neuronal inhibition compared to excitation,17 monitoring the same set of neurons under light anesthesia can potentially ‘unmask’ alterations in intrinsic excitability or neuronal excitation, which are otherwise covered during wakefulness.
Note: Hardware and software preparation of the two-photon scanning microscope, air supported treadmill, virtual reality environment and pupil tracking follows a previous description by Leinweber et al., 2014.18 Design of the virtual reality apparatus and air supported treadmill was adapted from Dombeck and colleagues.19,20
-
10.Description of two-photon microscope and air supported treadmill setup (for more detailed description, see Leinweber et al., 201418).
-
a.Two-photon microscope (HyperScope, Scientifica), equipped with an 8 kHz resonant scanner, yielding frame rates of 30 Hz at 512 × 512 pixel resolution.
-
b.Light source was a Ti:Sapphire laser with a DeepSee pre-chirp unit (Spectra Physics Mai Tai eHP).
-
c.A custom designed 3D printed ball holder is connected to an air supply tube (approximately 10 mm diameter).
-
i.The polystyrene ball (20 cm diameter) can be placed in the ball holder and air flow should be set to a suitable rate allowing smooth rotation of the ball.
-
ii.Use a pin or hypodermic needle (25G) inserted from either side into the ball, through a metal plate attached to the ball holder to restrain rotation to forward and backward motion.Note: A freely rotating ball can also be used to allow movement in x and y.
-
i.
-
d.Movement of the ball can be recorded using an optical computer mouse to track forward or backward (and lateral) motion.
-
a.
-
11.Virtual reality environment setup, as described previously.18,20,21
-
a.Virtual environments can be generated using the open-source software Wings3D.
-
b.LED projector with integrated gating circuit for fast flickering throughout imaging sessions.
-
c.Wings3D supports nonlinear transformation of the environment, allowing the projection onto the toroidal screen through standard reflective and angular amplification mirrors (Figures 3A–3D).
-
a.
-
12.Imaging awake mice, navigating a virtual reality environment (Figures 3E–3H).
-
a.When mice are suitably habituated to the treadmill and running comfortably when head restrained, they can be placed on the ball, and gently positioned in the head restraints without anesthesia.CRITICAL: Comfort of the mouse is absolutely critical here, if the mouse expresses signs of stress or discomfort the experiment must be ceased.
-
b.Once head-restrained, clean the window by application of Milli-Q water and absorb the solution clear with Sugi Saugtupfer.
-
c.Apply Ultrasound-Gel (Sonosid ultraschall-gel, Asid Bonz) diluted 1:1 with Milli-Q water as an immersion medium onto the cranial window. Note: spin down the gel before usage to remove air bubbles and carefully check upon application on to the window for air bubbles.
-
d.Lower the 16× water-immersion objective (Nikon) into the immersion solution over the window. Our two-photon microscope also allows fluorescence imaging through the same objective with a LED light source and camera, allowing the visualization of green wavelength band (505–545 nm) fluorescence signal and vasculature across the window.Note: The experimenter can adjust the position of the objective to the desired imaging location with emitted fluorescence signal from the GCaMP. We use prominent blood vessels as fiduciary landmarks for alignment in chronic imaging experiments. Place black tape around the objective, surrounding the window to reduce light leak.
-
e.Position the virtual reality screen as near as possible to the mouse, so that the toroidal screen is almost surrounding the mouse’s view.
-
f.Briefly, check all signals recorded in the software and ensure the virtual reality is properly running.
-
i.The eye camera should be positioned lateral to the mouse to focus on the pupil, whilst covering parts of the snout area.
-
ii.Check the optical mouse sensor is active (sampling rate is 1000 Hz) and feeding back the rotation of the treadmill to the virtual reality.
-
i.
-
g.Switch to two-photon imaging setup. The LED light from the projector is synchronized to the resonant scanner and will only switch on in the “turnaround periods” of the scan to further minimize light leak during the recording (for detailed description see18).
-
h.GCaMP7s can be excited at 910 nm, emitted photons are detected by a GaAsP PMT with a bandpass filter in front (525/50 nm). Gradually move up the laser power (10–30 mW should be sufficient to excite neurons in the cerebellar cortex expressing GCaMP7s with a high-quality window) until neurons can be visualized.
-
i.Select appropriate field of view dimensions to image your desired cell types. We found 453 × 453 μm (at 512 × 512 pixels resolution) appropriate to simultaneously image molecular layer interneurons, Purkinje neurons, and putative Golgi cells.Note: Typically this was at a cortical depth of 150 μm–250 μm. However, imaging depth varies due to the curvature of the cerebellar cortex and position in the lobule. Record a z-stack to gain an overview of the area.
-
j.View the z-stack and identify the area that yields you a large number of cells with high quality resolution.
-
k.Record a time-stack. We here recorded 15,000 frames corresponding to approximately 8 min.
-
l.Ensure an example mean projection of each field of view (FOV) is saved in the software (SciScan) for re-alignment of the same neurons over time in case of chronic imaging experiments.Note: SciScan software has a built-in “Position Save Module”, which allows multiple positions to be stored with their x,y,z position coordinates relative to a landmark considered the origin. These saved coordinates and mean projection images facilitate straightforward re-alignment of the current live FOV to previous ones through overlaying the images. ImageJ can also be a useful tool to overlay and align FOVs.
-
a.
-
13.Imaging the same neurons in darkness.
-
a.Switch virtual reality off, allow at least 10–15 min for the mouse to adjust to being in complete darkness. Make sure all light sources are eliminated, e.g., tape LEDs of instruments etc.
-
b.During this time, check alignment of cells, ensure the FOV position is identical to previous recordings.
-
c.Repeat imaging sequence with same length as with visual feedback provided by the virtual reality, ensuring that the cells are aligned with previous recording.
-
d.Repeat for further spots, typically 2–3 field of views (FOV) per mouse.
-
a.
Note: Switching off the virtual reality environment often causes increased and prolonged locomotion in the mice. The pause allows animals to adjust to the change in the environment. Observation of the mouse is critical during this time. Should a mouse not adapt to the change in light condition and continue to run relentlessly, consider to extent the waiting period, make running on the ball more strenuous by reducing the air flow or consider repeating the experiment after a resting period of a couple of hours.
Figure 3.

Two-photon imaging set-up for imaging of head restrained, behaving mice
(A) Metal frame to support toroidal screen.
(B) Toroidal screen with air supported spherical styrofoam treadmill and two-photon microscope fitted with 16× objective (0.8 NA, Nikon).
(C) A projector positioned above the screen projects the virtual reality display onto the initial mirror, which reflects display onto the angular amplification mirror, which in turn reflects the light across the toroidal screen. Arrows indicate light path.
(D) View from above showing light path from projector to reflecting mirror to angular amplification mirror toroidal screen.
(E) Infrared camera (IR) is used to record behaviors, such as whisking pupil size, and position. The movement of the spherical treadmill is recorded through an optical mouse sensor on the front. Metal plates and screws from each side allow head fixation of the mouse on the treadmill.
(F) Mouse head-fixed on the treadmill. Blue tube to the right side of the mouse delivers an air puff.
(G) Mouse engaging in virtual reality linear track.
(H) Pupil diameter tracking through infra-red (IR) camera (DMK 22BUC03, ImagingSource) (left image: view from IR camera with delineated areas to measure alterations to pupil diameter and facial movement associated with whisking, right: Pupil tracking, recording of diameter depicted with inner dark blue circle of dots, through a custom-written algorithm in LabVIEW (National Instruments) as described previously (Liebscher et al., 2016).
(I) Mouse anesthetized on a physiological monitoring system for anesthetized two-photon imaging session.
(J) Volatile anesthetics (e.g., isoflurane) are delivered through a tube positioned at the snout.
On the following day, or after an appropriate rest period according to local regulations, the same neurons can be imaged again under anesthesia.
-
14.Imaging the same neurons under anesthesia (Figures 3J and 3K).
-
a.Replace the polystyrene ball with a stable stage, carrying the physiological monitoring system apparatus to allow for maintenance of the body temperature and continuous measurement of the respiratory rate. The latter of which also allows to track the anesthesia depth. Consistent anesthesia depth is crucial to maintaining stable recordings and thus identifying alterations in calcium signals in the Sca1 model in comparison to the wild type littermates.
-
b.Ensure the oxygen tube is connected to the isoflurane vaporizer, and that isoflurane can be delivered through the tubing at a given flow rate and percentage.
-
c.Lightly anesthetize the mouse in an isoflurane chamber (2.5 vol % isoflurane, flow rate 0.5 L/min) and wait until the animal stops moving (around 10–15 s). Immediately remove the mouse and secure head-fixed on the physiological monitoring system.
-
d.Quickly position the nozzle of the tube in front of the mouse’s snout while head-fixed. Deliver isoflurane at 2 vol % in pure oxygen, with a flow rate of 0.5 L/min.
-
e.Wait 60 s to ensure the mouse remains motionless. Then gradually reduce anesthesia to 1.5 vol % and keep the flow rate constant.
-
f.Check body temperature, heart rate and breathing rate are being recorded. Reduce isoflurane concentration so that the breathing rate is between 110–150 breaths per minute. Keep stable for a minimum of 30 min before beginning recording. For most mice we apply a final isoflurane concentration of 1.25 vol %.
-
g.During this stabilization period, apply the diluted ultrasound gel to the window, ensuring that the window is clean. The objective can then be lowered and the imaging region relocated and realigned to the previous imaging spot used in the awake session using epifluorescence illumination to visualize the cerebellar cortex vasculature.
-
h.When the vasculature is aligned to the previous FOV, switch to two-photon excitation. Ensure that the cells in the current FOV can also be overlaid and aligned with the awake two-photon recording FOVs.
-
i.Once the current FOV is thoroughly aligned (see "Note” from step 12l) to the FOV of the awake recordings, record a time-series of identical length (here 15,000 frames) in the anesthetized mouse.
-
a.
Note: It is critical to achieve comparable anesthesia depth across individual mice and over time. To this end, we not only monitor breathing and heart rate, but also record pupil position and width as well as whisking. Experiments with indications of mice waking up are excluded from analysis.
Note: Because the FOV may appear different under anesthesia (altered fluorescence intensity due to changes in calcium levels aka neuronal activity of individual neurons), overlay epifluorescence images of the vasculature on ImageJ to make sure the imaged region is aligned precisely and use two-photon z-stacks to also compare orientation.
Processing and analysis of calcium signals from neuronal populations of the cerebellar cortex in behaving and anesthetized mice
The following steps outline methods for processing and analyzing the recorded calcium signals acquired through the previous two-photon imaging steps.
Note: Simultaneous patch-clamp recordings and calcium imaging of PNs revealed that distinct calcium transients emerge due to pauses in the simple spike train.22 Ramirez and Stell showed that although the pacemaker firing of PNs is too fast for genetically encoded calcium indicators to resolve individual action potentials, longer trains of action potentials do indeed increase the amplitude of the recorded calcium signal.22 To account for the limitations in temporal resolution inherent to the use of genetically encoded calcium indicators but acknowledging that the fluorescent signal scales with the level of neuronal activity, we computed the area under the curve (AUC, that is the sum of the 0-centered and thresholded calcium trace) of these calcium transients to approximate neuronal activity. As both MLINs and Golgi cells also display fast spiking properties, we thus opt to utilize the AUC to quantify the activity of all neuronal cell types.
-
15.Calcium imaging data preprocessing.
-
a.Full frame images are corrected for potential x and y brain displacement, using custom written MATLAB scripts, as is all following data processing and analysis.
-
b.Imaging data segmentation is done in a semi-automated fashion using both the mean and maximum projection of all frames.
-
i.Regions of interest (ROIs), such as cell bodies or dendrites of distinct neuronal populations, are semi-automatically selected based on a chosen threshold for fluorescence intensity and adjusted manually based on visual assessment of morphological features (Figure 4).
-
ii.Relying on both the mean and the maximum intensity projection enables us to not only select highly active ROIs but also neurons with little to no detectable activity.
-
iii.Alternatively, or in combination to this approach, we also use routines based on constrained non-negative matrix factorization to segment imaging data.23Note: Be careful when segmenting the somata of Purkinje neurons to not include the perisomatic synapses (appearing as puncta or ring-like structure around the Purkinje neurons soma) and the pinceaux (depending on orientation of optical plane appearing as triangular structure next to the Purkinje neuron soma) provided by molecular layer interneurons.
-
i.
-
c.From each ROI, the fluorescence intensity of all pixels within the area is averaged per frame, resulting in time series (calcium trace), which can subsequently be processed further, e.g., we perform a low pass filtering at 10 Hz and smooth over 3 frames.
-
d.To address potential neuropil signal contamination, for instance by neighboring PNs dendrites, we also sample the fluorescent intensity in the immediate surrounding of the selected ROI (for details see(3)). The following equation is employed:(FROI_comp, neuropil-compensated fluorescence of the ROI; FROI, initial fluorescence signal of the ROI; Fneuropil, signal from the neuropil) (modified after Chen et al., 201324; Liebscher et al., 20163 and Scekic-Zahirovic et al., 20217).For ROIs with Fneuropil > FROI a neuropil compensation factor of 0.7 is used instead of 0.8.
-
e.Next to estimate the baseline (F0) and the noise of the calcium traces, we subtract the 8th percentile of all values in a given very short sliding window (e.g., of 1 s) to obtain a trace (noise band), in which transients are largely eliminated.
-
f.The standard deviation of this trace divided by its median and after subtracting 1, yields the noise of each trace. F0 corresponds to the median of all values lower than the 70% percentile of the noise band.
-
g.Analyses can then be performed on (FROI_comp/F0)-1 traces, reflecting the ΔF/F.
-
h.The area under the curve is the sum of the ΔF/F, which is thresholded at 2× the standard deviation of the noise band, thus minimizing the contributions of noise fluctuations.
-
a.
-
16.Quantification of spontaneous activity, running responses, and air puff responses.
-
a.Quiet wakefulness is defined as the behavioral state when the mouse is stationary on the ball. We defined spontaneous activity in these periods as the AUC/min of quiet wakefulness. Only experiments (recordings) with a heuristically set total of at least 4,000 frames (approximately 2.7 min), spent in quiet wakefulness are used for the analysis. All other experiments are not included.
-
b.To assess the neuronal responses to locomotion, we compare the fluorescence intensity of all neurons in a given FOV as a function of running speed. To this end, we downsample the data by averaging both the ΔF/F trace and the running speed over 5 frames (equaling ∼170 ms) and compute a population response curve.
-
c.We, furthermore, quantify the proportion of running responsive neurons per FOV.
-
i.Running responsive neurons are characterized by a significantly higher neuronal activity level (that is the median ΔF/F) during locomotion compared to quiet wakefulness.
-
ii.We thus derive the ratio of the median ΔF/F during locomotion and during quiet wakefulness. This ratio must be larger than the 95 percentile of a shuffled control data set.
-
iii.We here randomly circularly shift the ΔF/F trace with respect to the running vector and calculate the corresponding ratio of average neuronal activity of the circularly shifted trace during locomotion and quiet wakefulness.
-
iv.We repeat this procedure 1000 times and define the 95th percentile of the resulting ratios as a cut-off for running responsive neurons.
-
i.
-
d.Puffs of pressurized air are applied to the trunk of the mouse during experiments, to examine the responses of calcium signals to the air puff stimulation. To avoid signal contamination whilst quantifying air puff responses, we only include air puff events that are not followed by locomotion within a window of 3 s.
-
a.
-
17.Population activity dimensionality reduction and manifold analysis.
- a.
-
b.We perform a multivariate linear regression on the first three principal components and the various recorded behaviors (for example: locomotion, pupil width, whisking, and air puff and all measured behavior combined), to determine the correlation of the population response properties and the given behavior.
-
c.A PCA-based manifold can then be derived, by projecting the high dimensional population response pattern trajectories into 3-dimensional PC space with its associated behavioral states.
-
d.This allows the recognition of a manifold subspace, associating certain behavioral states with population response patterns.
-
e.Once the manifold subspace for a given behavioral state has been established, the center of mass for the respective subspace (here locomotion vs. quiet wakefulness) is calculated as the mean of the coordinates within each subspace.
-
f.The Euclidean distance within a given subspace corresponds to the average length of each data point to the center of mass.
-
g.The Euclidean distance between two subspaces reflects the distance between the center of mass of each subspace.
-
h.At this point, a discrimination between wild type and Sca1 mice can be made for different behavioral states.
Figure 4.

Representative in vivo two-photon images and image segmentation
(A) Individual frames (30 Hz frame rate) of two-photon raw data displaying fluorescence emitted by the genetically encoded calcium indicator GCaMP7s expressed across distinct neuronal populations of the cerebellar cortex. Raw frame images are corrected for potential x and y brain displacement in order to account for small changes in the x and y planes across the frames in the time-series.
(B) Maximum intensity projection of a 15.000 frames time-series.
(C) Mean fluorescence intensity projection of same time series shown in (B). Regions of interest (ROIs) are selected and assigned to distinct cellular populations. Molecular layer interneuron cell body (blue), Purkinje neuron somata or dendrites (white), putative Golgi cell somata (red), ROI present in former time points but absent and thus not included for analysis in current FOV recording (black). Scale bar in (A) and (B) 100 μm, scale is consistent in (B) and (C). Dashed lines in (A) and (B) delineate cerebellar cortex layers, ML, Molecular layer; PNL, Purkinje neuron layer; GCL, Granular cell layer.
Functional imaging to probe impact of chemogenetic inhibition of MLINs in Sca1 tg mice
These steps outline how simple adaptations to the above sections can allow the assessment of the impact of chemogenetic modulation of MLINs on their activity levels in awake and anesthetized mice.
-
18.
To selectively target MLIN, cross a PV-cre mouse line (B6;129P2-Pvalbtm1(cre)Arbr/J, expressing the Cre-recombinase in parvalbumin positive neurons) with the Sca1 line. For example, we crossed Atxn1154Q/2Q males with PV-cre homozygous females (PV::Sca1). This produced both Sca1 offspring and wild type littermates, but all expressing the Cre-recombinase in parvalbumin-positive neurons.
-
19.
Prepare as described above in the “Preparation of materials for cerebellar craniotomy, AAV injection and cranial window implantation” section, with the addition of preparation the virus AAV2/8-hSyn-DIO-hDM4Gi-mCherry-WPRE alongside the GCaMP. This virus allows for Cre-recombinase-dependent expression of the receptor hM4D(Gi), with AAV serotype 2/8 to supporting the expression in MLINs.1 A member of the “Designer Receptors Exclusively Activated by Designer Drugs” (DREADDs) family, hM4D(Gi) is coupled to a Gαi/o G-protein, thereby inhibiting neuronal excitability, when activated.
-
20.
Complete steps as described for “Preparation of Experimental Animal” using the PV::Sca1 mice. Perform the “cerebellar craniotomy" section as described, however center the window along the midline (−6.5 AP, 0.0 ML).
-
21.
Shifting the window centrally will support bilateral injections of 300 nL of AAV2/8-hSyn-DIO-hDM4Gi-mCherry-WPRE (stock titer: 4.4 × 10ˆ12 vg/mL, dilution to 1:6 in NaCl/Milli-Q), in addition to the functional calcium indicator injected at a singular imaging site. This can be completed in accordance with the steps outlined in the section “stereotaxic injection of Adeno-associated viruses”.
-
22.
An identical set of neurons in each mouse can be imaged across two sessions in the conditions outlined in the “two-photon calcium imaging in awake and anesthetized mice” sections of the protocol. The first session provides a baseline recording, the second session can then be conducted following an intraperitoneal injection of clozapine-N-oxide (CNO, 3 mg/kg), administered 45 min prior to imaging. Calcium signals can then be recorded across the distinct neuronal populations during MLIN inhibition.
-
23.
Processing and analysis of these calcium signals can be completed as described in the remaining sections above, one can then compare the signals of the same distinct cell types with and without CNO mediated inhibition of MLINs.
Expected outcomes
This protocol is used to record calcium signals to approximate activity from distinct neuron types across the cerebellar cortex circuit in unison, in a mouse model of spinocerebellar ataxia type 1. Calcium traces are calculated for each individual region of interest (Figure 4), representing neuronal somata or dendrites based on the protocol. The area under the curve (AUC) is computed as a measure of neuronal activity. By monitoring calcium signals of the same neurons as a readout for neuronal activity under distinct conditions and behaviors (Figure 5), one can gain novel insight into the pathophysiology of Sca1. The protocol provides detailed methods to allow researchers to implant a chronic cerebellar cranial window in order to study neurons of the cerebellar cortex in unison, in distinct conditions and over time. In combination with the calcium imaging “tools” we outline, the protocol can be used to establish in which cell types alterations to neuronal function emerge in disease models, across a variety of behaviors and environments, and how alterations progress over the disease course. Unravelling neuronal dysfunction is providing much needed novel insight into a variety of diseases,2,3,6,8,9 this protocol can therefore provide a valuable tool for understanding SCA1 and other cerebellar disorders .
Figure 5.

Example calcium traces from awake and anesthetized recordings
(A) Average projection of an episode of quiet wakefulness (QW, left) and during locomotion (loco, right) for the same field of view (FOV) for a healthy wild type mouse (WT). Images reproduced from Pilotto et al., 2023.
(B) Exemplary calcium traces of selected molecular layer interneurons (MLIN, magenta), Golgi cells (Gol, yellow) and Purkinje neurons (PN, gray) in a feedback session (fb) and in darkness in a WT mouse. Shaded gray areas indicate periods of locomotion.
(C) Same as in (A) for a Sca1 mouse.
(D) Same as in (B) for a Sca1 mouse.
(E) Representative anesthetized example traces of all 3 cell types in a WT mouse.
(F) Same as in (E) for a Sca1 mouse. Scale bar in (A) 100 μm, scale is consistent across images. Dashed lines in (A) and (B) delineate cerebellar cortex layers, ML, Molecular layer; PNL, Purkinje neuron layer; GCL, Granular cell layer.
Here we show examples of these findings, displaying elevated spontaneous neuronal activity (AUC/min) in MLINs of Sca1 mice, during anesthetized recordings (Figure 6A). In awake mice, MLINs displayed increased activity during quiet wakefulness (Figure 6B) and elevated population responses to locomotion (Figure 6C). In addition, to tackle one of the caveats of population studies, given large number of cells including different types captured simultaneously in the network, we applied PCA-based dimension reduction to separate typical brain activity patterns in order to assess the coding accuracy of different behavioral states. Manifolds formed by the subspace separation as a function of locomotion speed were found to be more pronounced in WT mice in comparison to Sca1 (Figures 6D and 6E). Utilizing this protocol we have identified that molecular layer interneurons turn hyperactive, causing alterations in their response properties, as well as a perturbed coding accuracy of behavioral state of the network, already in early symptomatic Sca1 mice.1 Thanks to recent advances in the development of genetically encoded indicators and sensors, the protocol can readily be used to also assess other aspects of circuit function, such as the release of other neurotransmitters or neuromodulators and the quantification intracellular signaling molecules. We also applied the procedures to probe the efficacy of a cell type – specific chemogenetic intervention.1 We here show an example of such modification, where we selectively target MLIN, a parvalbumin (PV) positive population with chemogenetics. This was achieved by crossing a PV-cre mouse line with the Sca1 line, in combination with injecting a floxed AAV serotype 2/8 to express the hM4D(Gi) alongside the genetically encoded calcium indicator. A member of the “Designer Receptors Exclusively Activated by Designer Drugs” (DREADDs) family, hM4D(Gi) is coupled to a Gαi/o G-protein, thereby inhibiting inhibit neuronal signaling when activated, thus reducing MLIN activity in this context (Figure 7). We probe the resulting impact on the activity levels of MLIN using the above described two-photon imaging approach. Histological post-mortem analysis showed our protocol resulted in approximately ∼70% of MLINs within the injection site being transduced by the AAV2/8 as identified by the expression of mCherry.1 Interestingly, we found that only approximately 47% of MLINs displayed reduced in activity (Figure 7B). The discrepancy in transduction vs. activity reduction is likely due to the diverse interactions of these interneurons in vivo. MLINs provide reciprocal inhibition to other MLINs, therefore chemogenetically reducing activity in some, likely increases activity in other MLIN. Manipulating the activity of MLINs allowed us to establish the functional relevance of our initially descriptive findings, revealing that hyperactive MLINs directly contribute to the reduced network coding accuracy of behavioral state.1
Figure 6.

Two-photon imaging reveals hyperexcitability of molecular layer interneurons and perturbed encoding of behavioral state in Sca1 mice
(A) Average neuronal activity of MLIN under anesthesia (AUC across the entire recording of ∼8 min) (p < 10−7, KS test, WT 1022, Sca1 702 MLIN).
(B) Quantification of spontaneous neuronal activity of MLIN (AUC/min) during quiet wakefulness (p = 0.0005, KS test, WT 1386 MLIN, Sca1 714 MLIN).
(C) Average population activity of MLIN as a function of instantaneous running velocity (group effect: F(1,2092) = 71.89, p < 0.001, 2-way repeated measures ANOVA, superimposed by sigmoidal fit).
(D) Example of an input matrix consisting of 0-centered and z-scored ΔF/F traces used for dimensionality reduction aligned with behavioral parameters (lower traces) derived from all cells in one FOV of a feedback session of a WT mouse. Cell types are indicated by colored boxes on the left side (magenta – MLIN, molecular layer interneurons; gray – P, Purkinje neurons; yellow – G, Golgi cells).
(E) Representative manifold upon dimensionality reduction using principal component analysis of quiet wakefulness and active states (as a function of locomotion speed) for WT and Sca1 mice. In WT, PC space emerges as distinct structures dependent on behavioral state (top), whereas the two subspaces are less segregated in Sca1 (bottom). All images reproduced from Pilotto et al., 2023. Data in (A) and (B) presented as box plots displaying the median and interquartile range. Data in (C) is mean ± SEM.
Figure 7.

Impact of chemogenetic inhibition on MLIN activity levels
(A) Example maximum intensity projection of an awake recording in a Sca1::PV mouse before (left) and after CNO application (intraperitoneal injection 3 mg/kg, right).
(B) Proportional change in neuronal activity in Sca1:: PV MLIN upon CNO i.p. application. scale bar in (A) 50 μm. All images reproduced from Pilotto et al., 2023.
Limitations
Kinetics of genetically encoded calcium indicators
Purkinje neurons, Golgi cells, and molecular layer interneurons all display particularly fast firing properties.27,28 PNs in particular produce action potential trains of 20–70 Hz.27 Considering the kinetics of GCaMP7s (time to rise for a single AP 50% ΔF/F is ∼70 ms, half decay time ∼1,690 ms11) and, to the best of our knowledge, the lack of suitable deconvolution algorithms to infer underlying action potential firing rates, it is therefore not possible to resolve individual action potentials in PNs with this protocol. Despite this limitation, ΔF/F does scale with neuronal AP discharge (∼320% ΔF/F at 10 Hz to 620% ΔF/F at 160 Hz11). Therefore, such cerebellar calcium imaging will detect strong changes in firing frequency, as has been shown previously with simple spike pauses,22 but more subtle alterations may be undetectable.
Movement artifacts
If the window and headbar is not completely secure, locomotion of the animal will cause shifts in the field of view during imaging. The registration process can account for smaller changes in x and y planes, however, if changes are larger or the changes in position occur in z this cannot be corrected and will induce artificial alterations to the recorded calcium signal in response to locomotion. In this case, locomotion response signals from this animal should not be included in the analysis.
Atxn1154/2Q model
There are a number of mouse models depicting different features of the disease Sca1. Each experimenter should assess what element of the disorder they want to investigate, and select their model based on their specific research questions and hypothesis. The protocol outlined in this paper can be applied to a wide range of models, replicating different symptoms of the spinocerebellar ataxias, and other disorders of the cerebellum.
Troubleshooting
Problem 1
-
•
Rupturing the transverse sinus while drilling the anterior edge of the craniotomy ("cerebellar craniotomy" step 3).
Potential solution
-
•
Drill slightly more posterior at first for this edge of the circle, the craniotomy can be enlarged by gradually shaving away more of the skull to fit the window in once the flap has been removed and the experimenter is confident they will not damage the transverse sinus.
-
•
The position of the craniotomy can also be moved posterior. However, we found, as there is a sharp ventral curve of the cerebellum, the window cannot be in contact with the entire exposed cortex when the entire window is centered too far posterior. If the craniotomy is performed and there are parts of the cortex that cannot be covered by the window, we recommend using extra inert UV curable dental glue to fill the gaps to connect the window to the skull.
Problem 2
-
•
Mouse not running on treadmill or prolonged locomotion (the latter may occur when changing from virtual reality feedback to darkness ("two-photon calcium imaging in awake and anesthetized mice" steps 12 & 13).
Potential solution
-
•
If a mouse is not running, it requires more training sessions on the treadmill before imaging can take place. Using the virtual reality feedback when training can stimulate the mice and encourage locomotion. The experimenter can also gently move the ball one turn to help the mouse see the environment will move as it move forward. Check the position of the headbar and of the treadmill to ensure the mouse is in a comfortable position.
-
•
If the mouse is running constantly, you should not record until it has resumed to normal intermittent levels of locomotion. This can commonly occur after switching off the virtual reality for the darkness sessions, allowing 10–15 min before recording in the new condition should suffice.
Problem 3
-
•
Varying depth of anesthesia during recording results in artificial alterations to the calcium signals that will mask any differences between wild type mice and the disease model ("two-photon calcium imaging in awake and anesthetized mice" step 14).
Potential solution
-
•
Once the breathing rate has reached the desired level, leave for 30 min to ensure this is stable. As well as tracking heart rate and breathing rate, record pupil position and width as well as whisking. Alterations in these measurements can give indications of mice waking up, which can then lead to the experiment being excluded from the analysis.
Problem 4
-
•
Expression of GCaMP7 in various neuron types in the cerebellar cortex results in high background signal, particularly in the molecular layer due to expression in Purkinje neuron dendrites ("processing and analysis of calcium signals from neuronal populations of the cerebellar cortex in behaving and anesthetized mice" step 15).
Potential solution
-
•
The increased bleed through of signal from PN dendrites and parallel fibers within the molecular layer lead to a high background signal that could result the surrounding neuropil signal to be brighter than the selected cell body within a ROI. To adjust our analysis for this we increased the neuropil subtraction to 0.8 from the typical established factor of 0.7.24 Unless the median intensity of the neuropil was larger than the median value of the selected ROI. In that case a factor of 0.7 was used.
Problem 5
-
•
Too few PN express GCaMP7s ("processing and analysis of calcium signals from neuronal populations of the cerebellar cortex in behaving and anesthetized mice" step 15).
Potential solution
-
•
Position the tip of injection needle right within the Purkinje neuron layer (Typically ∼150 μm from surface in lobules V & VI, the experimenter should check exact PN layer depth for their desired location site) to increase local AAV concentration at this point.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sabine Liebscher (Sabine.Liebscher@med.uni-muenchen.de, sabine.liebscher@uk-koeln.de).
Technical contact
Sabine Liebscher (Sabine.Liebscher@med.uni-muenchen.de, sabine.liebscher@uk-koeln.de).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology - EXC 2145 SyNergy - ID 390857198 (S.L.), the Emmy Noether Programme (S.L.), and the Graduate School for Systemic Neurosciences GSN-LMU (C.D.).
Author contributions
C.D. and S.L. wrote the manuscript. C.D. and S.L. devised the surgical and imaging protocols. C.D. and C.T. performed the experiments. C.D., X.Y., and S.L. performed data processing and analysis.
Declaration of interests
The authors declare no competing interests.
References
- 1.Pilotto F., Douthwaite C., Diab R., Ye X., Al Qassab Z., Tietje C., et al. Early molecular layer interneuron hyperactivity triggers Purkinje neuron degeneration in SCA1. Neuron. 2023;111:2523–2543. doi: 10.1016/j.neuron.2023.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braz B.Y., Wennagel D., Ratié L., de Souza D.A.R., Deloulme J.C., Barbier E.L., Buisson A., Lanté F., Humbert S. Treating early postnatal circuit defect delays Huntington's disease onset and pathology in mice. Science. 2022;377 doi: 10.1126/science.abq5011. [DOI] [PubMed] [Google Scholar]
- 3.Liebscher S., Keller G.B., Goltstein P.M., Bonhoeffer T., Hübener M. Selective Persistence of Sensorimotor Mismatch Signals in Visual Cortex of Behaving Alzheimer's Disease Mice. Curr. Biol. 2016;26:956–964. doi: 10.1016/j.cub.2016.01.070. [DOI] [PubMed] [Google Scholar]
- 4.Busche M.A., Eichhoff G., Adelsberger H., Abramowski D., Wiederhold K.H., Haass C., Staufenbiel M., Konnerth A., Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 5.Palop J.J., Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016;17:777–792. doi: 10.1038/nrn.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunes Z.I., Kan V.W.Y., Ye X., Liebscher S. Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology. Front. Neurosci. 2020;14:573. doi: 10.3389/fnins.2020.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scekic-Zahirovic J., Sanjuan-Ruiz I., Kan V., Megat S., De Rossi P., Dieterlé S., Cassel R., Jamet M., Kessler P., Wiesner D., et al. Cytoplasmic FUS triggers early behavioral alterations linked to cortical neuronal hyperactivity and inhibitory synaptic defects. Nat. Commun. 2021;12:3028. doi: 10.1038/s41467-021-23187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGregor M.M., Nelson A.B. Circuit Mechanisms of Parkinson's Disease. Neuron. 2019;101:1042–1056. doi: 10.1016/j.neuron.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Imlach W.L., Beck E.S., Choi B.J., Lotti F., Pellizzoni L., McCabe B.D. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151:427–439. doi: 10.1016/j.cell.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edamakanti C.R., Do J., Didonna A., Martina M., Opal P. Mutant ataxin1 disrupts cerebellar development in spinocerebellar ataxia type 1. J. Clin. Invest. 2018;128:2252–2265. doi: 10.1172/JCI96765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dana H., Sun Y., Mohar B., Hulse B.K., Kerlin A.M., Hasseman J.P., Tsegaye G., Tsang A., Wong A., Patel R., et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods. 2019;16:649–657. doi: 10.1038/s41592-019-0435-6. [DOI] [PubMed] [Google Scholar]
- 12.Krashes M.J., Koda S., Ye C., Rogan S.C., Adams A.C., Cusher D.S., Maratos-Flier E., Roth B.L., Lowell B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller G.B., Bonhoeffer T., Hübener M. Sensorimotor mismatch signals in primary visual cortex of the behaving mouse. Neuron. 2012;74:809–815. doi: 10.1016/j.neuron.2012.03.040. [DOI] [PubMed] [Google Scholar]
- 14.Holtmaat A., Bonhoeffer T., Chow D.K., Chuckowree J., De Paola V., Hofer S.B., Hübener M., Keck T., Knott G., Lee W.C.A., et al. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg D.S., Houweling A.R., Kerr J.N.D. Population imaging of ongoing neuronal activity in the visual cortex of awake rats. Nat. Neurosci. 2008;11:749–751. doi: 10.1038/nn.2140. [DOI] [PubMed] [Google Scholar]
- 16.Helmchen C., Büttner U. Saccade-related Purkinje cell activity in the oculomotor vermis during spontaneous eye movements in light and darkness. Exp. Brain Res. 1995;103:198–208. doi: 10.1007/BF00231706. [DOI] [PubMed] [Google Scholar]
- 17.Haider B., Häusser M., Carandini M. Inhibition dominates sensory responses in the awake cortex. Nature. 2013;493:97–100. doi: 10.1038/nature11665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leinweber M., Zmarz P., Buchmann P., Argast P., Hübener M., Bonhoeffer T., Keller G.B. Two-photon calcium imaging in mice navigating a virtual reality environment. J. Vis. Exp. 2014;84 doi: 10.3791/50885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dombeck D.A., Khabbaz A.N., Collman F., Adelman T.L., Tank D.W. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 2007;56:43–57. doi: 10.1016/j.neuron.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dombeck D.A., Harvey C.D., Tian L., Looger L.L., Tank D.W. Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat. Neurosci. 2010;13:1433–1440. doi: 10.1038/nn.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harvey C.D., Collman F., Dombeck D.A., Tank D.W. Intracellular dynamics of hippocampal place cells during virtual navigation. Nature. 2009;461:941–946. doi: 10.1038/nature08499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramirez J.E., Stell B.M. Calcium Imaging Reveals Coordinated Simple Spike Pauses in Populations of Cerebellar Purkinje Cells. Cell Rep. 2016;17:3125–3132. doi: 10.1016/j.celrep.2016.11.075. [DOI] [PubMed] [Google Scholar]
- 23.Pnevmatikakis E.A., Soudry D., Gao Y., Machado T.A., Merel J., Pfau D., Reardon T., Mu Y., Lacefield C., Yang W., et al. Simultaneous Denoising, Deconvolution, and Demixing of Calcium Imaging Data. Neuron. 2016;89:285–299. doi: 10.1016/j.neuron.2015.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen T.W., Wardill T.J., Sun Y., Pulver S.R., Renninger S.L., Baohan A., Schreiter E.R., Kerr R.A., Orger M.B., Jayaraman V., et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato S., Kaplan H.S., Schrödel T., Skora S., Lindsay T.H., Yemini E., Lockery S., Zimmer M. Global brain dynamics embed the motor command sequence of Caenorhabditis elegans. Cell. 2015;163:656–669. doi: 10.1016/j.cell.2015.09.034. [DOI] [PubMed] [Google Scholar]
- 26.Lanore F., Cayco-Gajic N.A., Gurnani H., Coyle D., Silver R.A. Cerebellar granule cell axons support high-dimensional representations. Nat. Neurosci. 2021;24:1142–1150. doi: 10.1038/s41593-021-00873-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arancillo M., White J.J., Lin T., Stay T.L., Sillitoe R.V. In vivo analysis of Purkinje cell firing properties during postnatal mouse development. J. Neurophysiol. 2015;113:578–591. doi: 10.1152/jn.00586.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prestori F., Mapelli L., D'Angelo E. Diverse Neuron Properties and Complex Network Dynamics in the Cerebellar Cortical Inhibitory Circuit. Front. Mol. Neurosci. 2019;12:267. doi: 10.3389/fnmol.2019.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.