

Significance
Conformational dynamics are likely critical to the activity of many multi-step enzymes. However, it is challenging to map the small-amplitude (sub-angstrom) and fast (nanosecond) dynamics that may underlie active site rearrangements. Here, the authors present X-ray diffraction experiments and analyses that allow for the sensitive identification of coupled motions in the enzyme dihydrofolate reductase. These methods resolve rapid structural changes throughout the enzyme that modify the active site and regulate access of water to the substrate, serving as an allosteric switch that organizes the enzyme’s two-step catalytic mechanism. The methods presented here can be used to characterize enzyme dynamics in unprecedented detail.
Keywords: protein dynamics, allostery, enzyme catalysis, X-ray crystallography, DHFR
Abstract
Enzymes catalyze biochemical reactions through precise positioning of substrates, cofactors, and amino acids to modulate the transition-state free energy. However, the role of conformational dynamics remains poorly understood due to poor experimental access. This shortcoming is evident with Escherichia coli dihydrofolate reductase (DHFR), a model system for the role of protein dynamics in catalysis, for which it is unknown how the enzyme regulates the different active site environments required to facilitate proton and hydride transfer. Here, we describe ligand-, temperature-, and electric-field-based perturbations during X-ray diffraction experiments to map the conformational dynamics of the Michaelis complex of DHFR. We resolve coupled global and local motions and find that these motions are engaged by the protonated substrate to promote efficient catalysis. This result suggests a fundamental design principle for multistep enzymes in which pre-existing dynamics enable intermediates to drive rapid electrostatic reorganization to facilitate subsequent chemical steps.
Enzymes selectively enhance the rates of chemical reactions. This catalysis is often explained through precise positioning of substrates and functional groups to stabilize the transition state of a reaction (1, 2). Proteins, however, contain many rotatable bonds with energetic barriers that can be crossed by thermal motion. Therefore, proteins exhibit conformational dynamics best described by an ensemble of structures (3, 4). Since even sub-angstrom changes in important interactions are, in principle, sufficient to impact the energetics of catalytic steps or their allosteric regulation (3, 5–8), conformational changes can be small enough to be overlooked by existing methods, yet key to understanding enzyme function. A central question therefore remains—how do the conformational dynamics of enzymes relate to the chemical reaction coordinate? This question is salient for enzymes with multi-step mechanisms for which the optimal active site arrangement is likely to differ between subsequent steps.
Critical gaps in our understanding of the interplay of conformational dynamics and the chemical steps of enzyme catalysis are evident for even the best-studied enzymes. Dihydrofolate reductase (DHFR) from Escherichia coli (hereafter, ecDHFR) has been studied intensively for decades (9–16). DHFR catalyzes the stereospecific transfer (Fig. 1A) of a hydride ion (H−) from reduced NADPH to dihydrofolate (DHF), yielding NADP+ and tetrahydrofolate (THF), an essential precursor for purine synthesis (10). Kinetic isotope effect measurements support a stepwise catalytic mechanism for ecDHFR in which protonation of DHF at the N5 atom precedes hydride transfer (17) (Fig. 1A). A key active-site loop, the Met20 loop, adopts two different conformations depending on the bound ligands: The closed conformation is associated with the Michaelis complex—the catalytically competent state in which the enzyme is bound to its cofactor and substrate, as shown in Fig. 1B. The occluded conformation is, instead, adopted by-product complexes to promote exchange of the spent NADP+ cofactor (9).
Fig. 1.
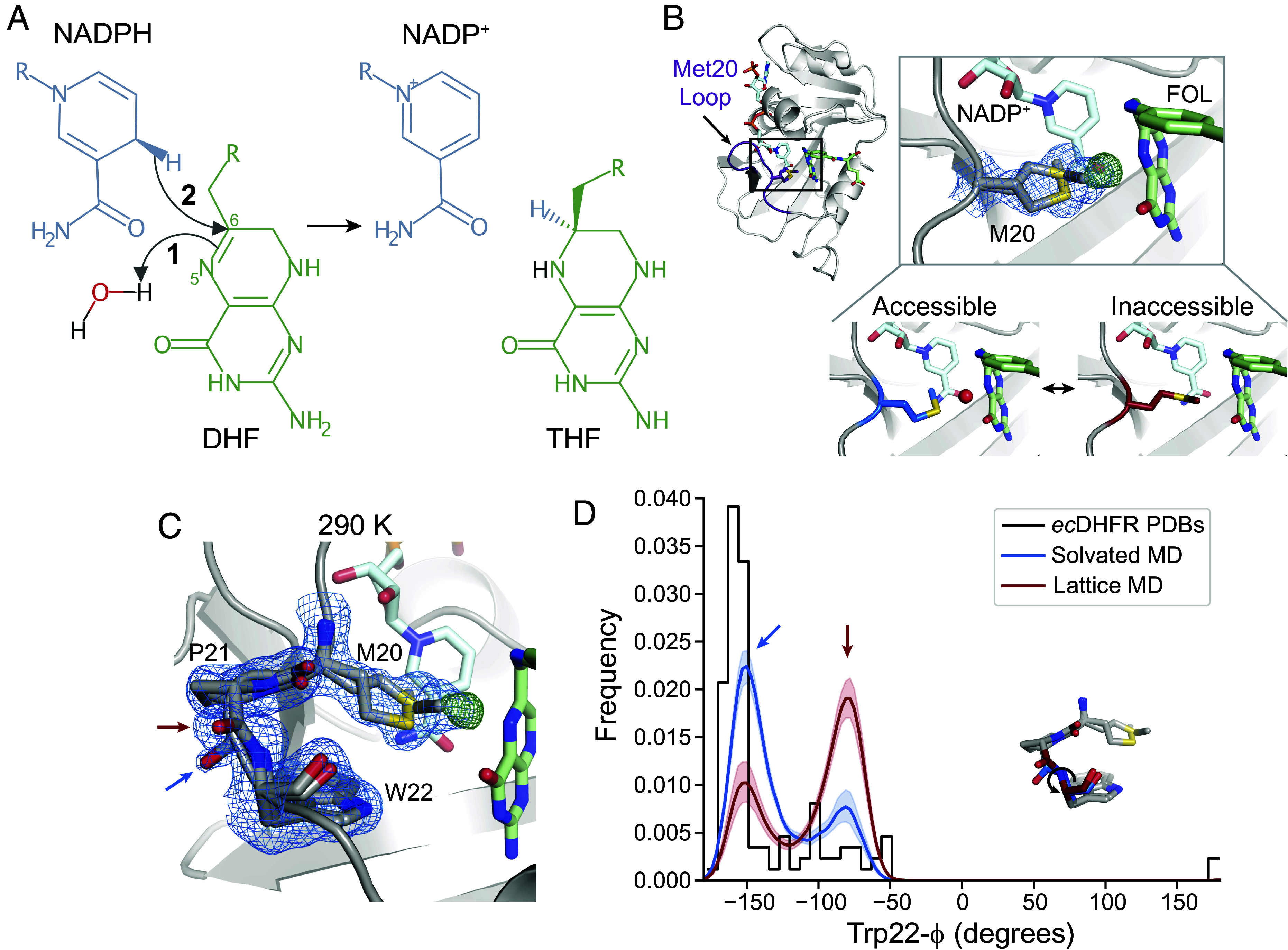
The closed state of the Met20 loop contains two interconverting substates. (A) Schematic of the hydride transfer reaction catalyzed by DHFR. Hydride transfer occurs from NADPH to DHF, yielding NADP+ and THF. The reaction proceeds by a stepwise mechanism: protonation of DHF from water precedes hydride transfer. The N5 nitrogen and C6 carbon of DHF are labeled. (B) and (C) map (blue mesh; 0.7), (green mesh; +4.0), and refined model for a ecDHFR:NADP+:FOL structure at 290 K. (B) The ecDHFR complex adopts the Met20 closed conformation and two rotamer states can be modeled for Met20 (both shown in stick representation), accompanied by unmodeled density. The Bottom panel depicts how this electron density can be interpreted as a superposition of an “accessible” state that allows water into the active site and an “inaccessible” state that occludes water. (C) The region composed of Met20, Pro21, and Trp22 adopts two conformations marked by distinct backbone conformations between Pro21 and Trp22 (blue and red arrows). (D) Kernel density estimates of the Trp22- dihedral from MD simulations in the context of a crystal lattice and a solvated water box, and a histogram of the Trp22- dihedral in deposited structures of ecDHFR. The two states observed in (C) are shown with corresponding blue and red arrows, and the Inset structure indicates the Trp22- dihedral. The and maps shown in (B) and (C) are carved within 1.5 and 3 Å, respectively, of the indicated residues for clarity.
Water is both a dynamic component of the electrostatic environment and a direct participant in the enzymatic mechanism. Although ordered water is not observed in the active site of the Michaelis complex, the rotamer state of Met20 is hypothesized to regulate access of a water molecule to the N5 atom of DHF based on conformational heterogeneity in high-resolution structures (18). Proton transfer directly from the solvent is further supported by molecular dynamics (MD) simulations and neutron diffraction (18–21). Whereas proton transfer requires solvent access to the active site (18, 21), the presence of water near the N5 atom of DHF would destabilize the partial positive charge on the C6 carbon, inhibiting hydride transfer (22). Indeed, transfer of charged groups often depends strongly on electrostatic environment (23). How, then, does the enzyme regulate solvent access and electrostatically retune its active site to promote successive chemical steps with conflicting requirements—protonation which requires solvent access and hydride transfer for which solvent access is inhibitory? Rapid reorganization of active site electrostatics may be a frequent and critical factor in the evolution and rational design of proficient multistep enzymes.
Existing experimental methods have not provided sufficient access to the conformational dynamics of DHFR to answer this question. Here, we apply improved crystallographic methods to resolve conformational changes in ecDHFR. These methods resolve a surprising array of conformational motions—a global hinge motion that constricts the active site cleft and influences the Met20 sidechain, along with local networks of coupled backbone and sidechain motions affecting the active site. Using MD simulations, we find that the protonated intermediate engages these motions by conformational selection to shield the active site from bulk solvent—a rapid rearrangement of the active site that follows substrate protonation to promote hydride transfer. This mechanism explains a “dynamic knockout” mutant of ecDHFR—a mutant for which the effects on hydride transfer rate were proposed to result from altered dynamics alone, and not from a change in ground state structure (24). We also describe how the mechanism appears to have constrained the evolution of the enzyme.
The approach taken here, combining advanced X-ray diffraction experiments with MD simulations identifies global and local conformational dynamics that promote efficient catalysis. We expect that for many natural and designed proteins, this approach will similarly reveal important conformational rearrangements and answer fundamental questions about how these proteins work.
Results
The Closed Met20 Loop Exhibits Distinct Substates.
Structural, kinetic, and computational studies, combined with mutagenesis, have led to a basic understanding of how the active site of ecDHFR supports the chemical steps of catalysis. In this model, the Met20 sidechain regulates solvent access to allow for protonation at the N5 atom of the substrate (Fig. 1B) (17, 18). To begin characterizing the conformational dynamics of the Michaelis complex, we used a widely employed model of the DHFR Michaelis complex with NADP+ and folate (FOL) as cofactor and substrate analogs, respectively, as the true Michaelis complex is not stable for the timescales necessary for crystallization (9). The crystal form we used is also compatible with all steps of the catalytic cycle (9). We first solved a structure of the model Michaelis complex to 1.04 Å at 290 K. Consistent with previous structures (9, 16, 18), the protein adopts the closed Met20 loop conformation, in which FOL and NADP+ are very close (3.2 Å; Fig. 1B). Inspection of the electron density map (blue mesh, ) near the Met20 sidechain shows electron density for two rotamers that differ in their dihedrals and the placement of the terminal methyl group. In addition, there is a large, peak in the difference electron density map between observed data and the refined model (green mesh, ). This peak partially overlaps with one of the Met20 rotamer states (Fig. 1B) and can be identified as the proton-donating water by comparison with a previous X-ray diffraction study (18). Together, these electron density features can be interpreted as a superposition of two Met20 sidechain conformations: an “accessible” Met20 rotamer can let water into the active site and an “inaccessible” rotamer excludes water. This structure supports a solvent-gating role for Met20, and its analysis recapitulates the features observed by Wan et al. (18).
Our data, however, reveal additional conformational heterogeneity in the Met20 loop. The backbone amide between Pro21 and Trp22 adopts two distinct conformations, offset by approximately 90° (arrows, Fig. 1C). These alternate backbone orientations can be thought of as substates of the closed loop conformation and can be classified by the Trp22- dihedral angle with the two states centered at (blue arrow) and (red arrow). Although this heterogeneity has not been previously noted, we find a range of values for Trp22- consistent with these states in published structures of ecDHFR (Fig. 1D). Both backbone configurations are also modeled in structures that used ensemble refinement to represent structural heterogeneity (16).
MD simulations of the crystal lattice and solvated DHFR confirm that the two substates represent dynamic exchange within the closed conformation of the Met20 loop. In these simulations, the substate at is populated approximately twofold more than the other substate in the context of the crystal lattice (Fig. 1D). The relative population of the states is inverted in the solvated simulation, indicating that the crystal lattice biases the thermodynamics between these states by about 0.8 kcal/mol.
A Modified Substrate Analog Resolves the Solvent Gating Mechanism.
The model presented in Fig. 1B suggests that the Met20 sidechain regulates the occupancy of the proton-donating water. To test this hypothesis directly, we sought to bias the rotamer distribution of Met20 with a modified substrate analog, 10-methylfolate (MFOL). This compound has a methyl substituent on the N10 nitrogen (dashed circle in Fig. 2A) that makes close contact with the Met20 sidechain. We determined the structure of the ecDHFR:NADP+:MFOL complex to 1.14 Å (SI Appendix, Table S1). As anticipated, this methyl group alters the Met20 rotamer states (Fig. 2B). This structural change is accompanied by the appearance of an ordered water in the electron density map within 3.6 Å of the N5 nitrogen of MFOL (arrow in Fig. 2B), consistent with the location of the unmodeled difference density in Fig. 1C.
Fig. 2.

Ligand-dependent conformational changes illustrate Met20 solvent gating. (A) Chemical structures of FOL and MFOL. (B) Refined structure and electron density map of the ecDHFR:NADP+:MFOL complex. The 10-methyl group is in close contact with the Met20 sidechain, and a water (red sphere; indicated by an arrow) can be resolved within 3.6 Å of the N5 nitrogen of MFOL. The map is contoured at (blue mesh; carved within 1.5 Å of shown atoms) and (light blue mesh; carved within 1.5 Å of shown water). (C) isomorphous difference map, phased with the MFOL-bound model. The overview shows the difference electron density induced by the 10-methyl substituent (), and the Inset highlights the structural differences observed in the active site (, carved within 3.0 Å of shown atoms). The added methyl group (label a) displaces an ordered water (label b), alters the rotamer distribution of Met20 (label c), rotates the pterin ring (label d), and leads to the introduction of an ordered water near the N5 nitrogen (label e). The 10-methyl substituent is indicated with a dashed circle in each panel.
To identify the structural changes induced by the methyl substituent in more detail, we used the difference map, which can sensitively detect changes in electron density (Fig. 2C). Strong difference density is visible near the added methyl group (Fig. 2C, Inset; labeled ). This 10-methyl group displaces two ordered waters from the FOL-bound structure (labeled ), induces a change in the Met20 rotamer distribution (labeled ), and causes the pterin ring to shift away from the Met20 residue (labeled ). Accompanying these changes, electron density for an ordered water increases near the N5 nitrogen (labeled ). That is, the 10-methyl substituent alters the Met20 rotamer equilibrium, increasing solvent access to the N5 atom of the substrate.
Multi-Temperature Diffraction Resolves a Global Hinge Motion.
The structural changes observed in the MFOL complex validate the solvent-gating role of the Met20 sidechain but were strongly localized near the 10-methyl substituent. Because the FOL-bound structure at 290 K and the MD simulations suggest additional conformational heterogeneity in the active site, we sought to bias the population of states of the enzyme using multi-temperature X-ray diffraction experiments. Because pre-existing equilibria that involve entropic change will be sensitive to temperature, these experiments can uncover correlated motions by observing structural states that change together as a function of temperature.
The earliest diffraction experiments to investigate the dependence of conformational heterogeneity on temperature used atomic displacement parameters as a reporter (25–27). Since those early studies, multi-temperature X-ray crystallography has been applied to probe conformational changes caused by temperature with atomic detail in order to understand the dynamics of enzymes (28–30). However, these experiments often probe a broad range of temperatures—from cryogenic to physiological—which can complicate analysis due to cryocooling artifacts and imperfect isomorphism (16). Here, we collected 23 high-resolution datasets from crystals from 270 to 310 K, in 10 K increments, including multiple datasets at each temperature to assess the uncertainty of any observations (SI Appendix, Tables S2–S6). We also inferred consensus datasets by combining data from the multiple crystals collected at each temperature (Fig. 3A and SI Appendix, Table S7). To identify temperature-dependent structural changes within this physiological range, we adopted an automated refinement strategy yielding consistent models for each dataset. This approach enables detailed biophysical comparison across temperatures.
Fig. 3.

Multi-temperature experiments reveal a global hinge motion and local rearrangements. (A) Schematic of multi-crystal, multi-temperature diffraction experiment. (B and C) The primary structural mode from SVD of the pairwise distances describes 88% of the variance among experimental structures. (B) Plot of the temperature dependence of the first left singular vector. (C) Heatmap of the contribution of each pairwise distance in the first right singular vector. Residues 38 to 88 are indicated with an orange bar and residues 120 to 130 are indicated with a yellow bar. (D) Structure of ecDHFR with arrows to depict displacements greater than 0.1 Å of atoms between 310 and 270 K refined models. The arrows are enlarged 10-fold relative to the corresponding displacements. Residues 38 to 88 are shown in orange, residues 120 to 130 are shown in yellow, and the distance between Asn23- and Pro53- (hinge distance) is shown as a dashed line. (E) Plot of the hinge distance as a function of temperature. Data points are shown for each independent crystal and the mean SE at each temperature. (F) Structure and map for the 290 K consensus structure for Asp127 and Tyr128. The 1.0 isocontour plot of the map in the plane of the backbone carbonyl is shown for the consensus structures at each temperature. (G) Schematic for the single-crystal, multi-temperature diffraction experiment. (H) Overview of the temperature-resolved isomorphous difference map between the 280 and 310 K datasets. (I–K) Insets highlighting regions of the difference map. All maps are carved within 2 Å of the displayed atoms, and arrows highlight the structural changes.
To interpret overall conformational change, we computed the pairwise distances between the atoms in each refined structure for the consensus models at each temperature and used singular value decomposition (SVD) to determine the primary temperature-dependent modes of structural change (see SI Appendix for details). The resulting singular vectors describe the weights of the pairwise distances and temperature dependence for each structural mode. The first singular vector explains 88% of the variance of distances across datasets, and depends monotonically on temperature (Fig. 3B). The corresponding heatmap depicts the weight of each pairwise distance (Fig. 3C) and emphasizes two regions that correspond to residues 38 to 88 (orange bar) and residues 120 to 130 (yellow bar). These regions are colored on the structure of ecDHFR in Fig. 3D: Residues 38 to 88, shown in orange, comprise the adenosine binding subdomain and residues 120 to 130, shown in yellow, span the end of the FG loop.
To visualize the temperature-dependent motion corresponding to the first singular vector, Fig. 3D depicts the displacements in positions between the models refined to the 270 and 310 K datasets. These are rendered as arrows for displacements greater than 0.1 Å and are enlarged 10 relative to the corresponding displacement. The arrows reveal a hinge motion that constricts the active site cleft. One of the strongest features in the pairwise distance heatmap corresponds to the distance between Asn23- and Pro53- (hereafter: hinge distance), which increases with temperature (Fig. 3E). Together, this analysis reveals a dominant, temperature-dependent global hinge motion that constricts the active site cleft by about 0.5 Å Although this is a small-amplitude motion, the largest SE in Fig. 3E is only 0.04 Å among replicate datasets.
In addition to the hinge motion, the region comprising residues 120 to 130 shows significant temperature dependence in Fig. 3C. In this region, Tyr128 adopts two shifted sidechain conformations, marked by distinct states for the amide backbone between Asp127 and Tyr128 (Fig. 3F). Accordingly, the refined electron density maps show a titration of density from one backbone configuration to the other as a function of increasing temperature, reaching equal occupancy at about 290 K (Fig. 3F).
Temperature-Resolved Difference Maps Identify Networks of Correlated Motions.
The analysis of multi-crystal, multi-temperature diffraction experiments above identifies a global hinge-bending motion and shifts in the conformational equilibrium of the loop containing Tyr128. This approach works best to detect such graded shifts of the dominant conformation. Inspired by time-resolved diffraction experiments (31, 32), we sought to improve the detection of excited states by conducting single-crystal perturbation experiments, followed by analysis with isomorphous difference maps. In these experiments, we collected diffraction data at multiple temperatures from the same crystal (Fig. 3G). Difference maps obtained this way showed reproducible and remarkably sensitive results (see SI Appendix, Materials and Methods for details and SI Appendix, Fig. S1, and Tables S8 and S9). We estimate that each dataset had an average diffraction weighted dose of 0.2 MGy, for a cumulative dose of 1.0 MGy per crystal (33, 34). Using repeated experiments with different sequences of temperature changes (SI Appendix, Fig. S1A), we demonstrated that the changes in hinge distance are reversible (SI Appendix, Fig. S1B). We also used interleaved datasets collected at 295 K to assess radiation damage. The relatively flat difference maps between subsequent 295 K datasets suggest that radiation-driven effects were limited (SI Appendix, Fig. S1 C and D). Furthermore, although the multi-temperature, multi-crystal experiments could be used to compute temperature-resolved difference maps, we found the single-crystal experiments yielded more reproducible results (SI Appendix, Fig. S1 E and F).
The temperature-resolved difference maps obtained from single-crystal experiments reveal a range of conformational changes that were not readily detected by the multi-crystal, refinement-based analysis. The to isomorphous difference map is relatively flat in the adenosine binding subdomain, but exhibits regions of paired positive and negative difference density in the loop subdomain (Fig. 3H), which identify networks of temperature-dependent motion propagating through the enzyme, in addition to the large-scale hinge motion.
Three interesting regions of the protein have strong (5) peaks in the to difference map (Fig. 3H). As illustrated in Fig. 3I, the most significant difference map peak () involves the oxidized Cys152 sidechain and the nearby rotamers of Asp116. The paired difference density on the rotamers implies a correlated shift in their occupancy, which can be rationalized based on the corresponding movement of ordered water molecules found between these sidechains. A second network of temperature-dependent changes ( peak) runs through the active site including the Met20 loop (Fig. 3J). Paired difference density on the pterin ring of folate indicates that the ring settles deeper in the binding site with the constriction of the active site cleft. Asp27, which coordinates the pterin ring, shifts accordingly along with an ordered water bridging Asp27 and the Trp22 indole ring. Corresponding motions are observed in the Met20 loop itself, with a small shift in Trp22 and stronger density for the gate-open Met20 rotamer at lower temperature. Finally, the temperature-resolved difference maps show that a second dynamic network reaches into the active site: The region from Phe125 to Tyr128 again shows significant temperature-dependent features in the difference map (; Fig. 3K). The backbone amide between Asp127 and Tyr128 shows strong, paired difference density, consistent with the differences observed during refinement (Fig. 3F). The difference map, however, also allows us to match the backbone carbonyl with the corresponding Tyr128 sidechain conformation based on their shared temperature dependence. Furthermore, strong difference density for Pro126, Phe125, and Tyr100 highlights an extended, contiguous network of temperature-dependent conformational changes that spans about 15 Å to the site of hydride transfer. Previous studies support the significance of these residues in catalysis. Tyr100 plays an important electrostatic role in hydride transfer (35), and the Y100F mutation decreases by 10-fold (17). Similarly, double-mutant studies implicate Phe125 as part of a network of residues coupled to hydride transfer (36, 37).
In summary, single-crystal temperature-resolved diffraction experiments reveal detailed views of three extended networks of correlated motions that propagate throughout the enzyme and involve key active site residues.
Electric-Field-Dependent Constriction of the Active Site Cleft.
Although temperature can effectively bias conformational equilibria to observe correlated changes by X-ray diffraction, it impacts all states that differ entropically, possibly confounding a mechanistic interpretation of observed conformational changes. To further resolve the coupling between observed motions, we used electric-field-stimulated X-ray crystallography (EF-X). In an EF-X experiment, a strong electric field is used to apply force on the charges and local dipoles within a protein crystal to induce motions. These motions can then be observed by X-ray diffraction at room temperature (Fig. 4A). By using X-ray pulses at defined delays after the onset of the electric field, the induced dynamics can be followed with nanosecond resolution. EF-X has been used to study a PDZ domain, and the observed motions were consistent with proposed mechanisms of ligand-induced allostery (38). Here, we used an updated apparatus for EF-X as shown in Fig. 4B and SI Appendix, Fig. S2A (see SI Appendix, Materials and Methods for details). At each orientation of the crystal, we collected three timepoints: an “Off” reference timepoint in the absence of a high-voltage pulse, a 200-ns timepoint during a 3.5-kV pulse, and a 200-ns timepoint during a 3.5-kV pulse. To collect a complete dataset, we then rotated the sample, repeating the timepoints at each angle. This interleaved data collection ensures similar accumulated X-ray exposure for each dataset (Fig. 4C). The data collection statistics are presented in SI Appendix, Table S10.
Fig. 4.
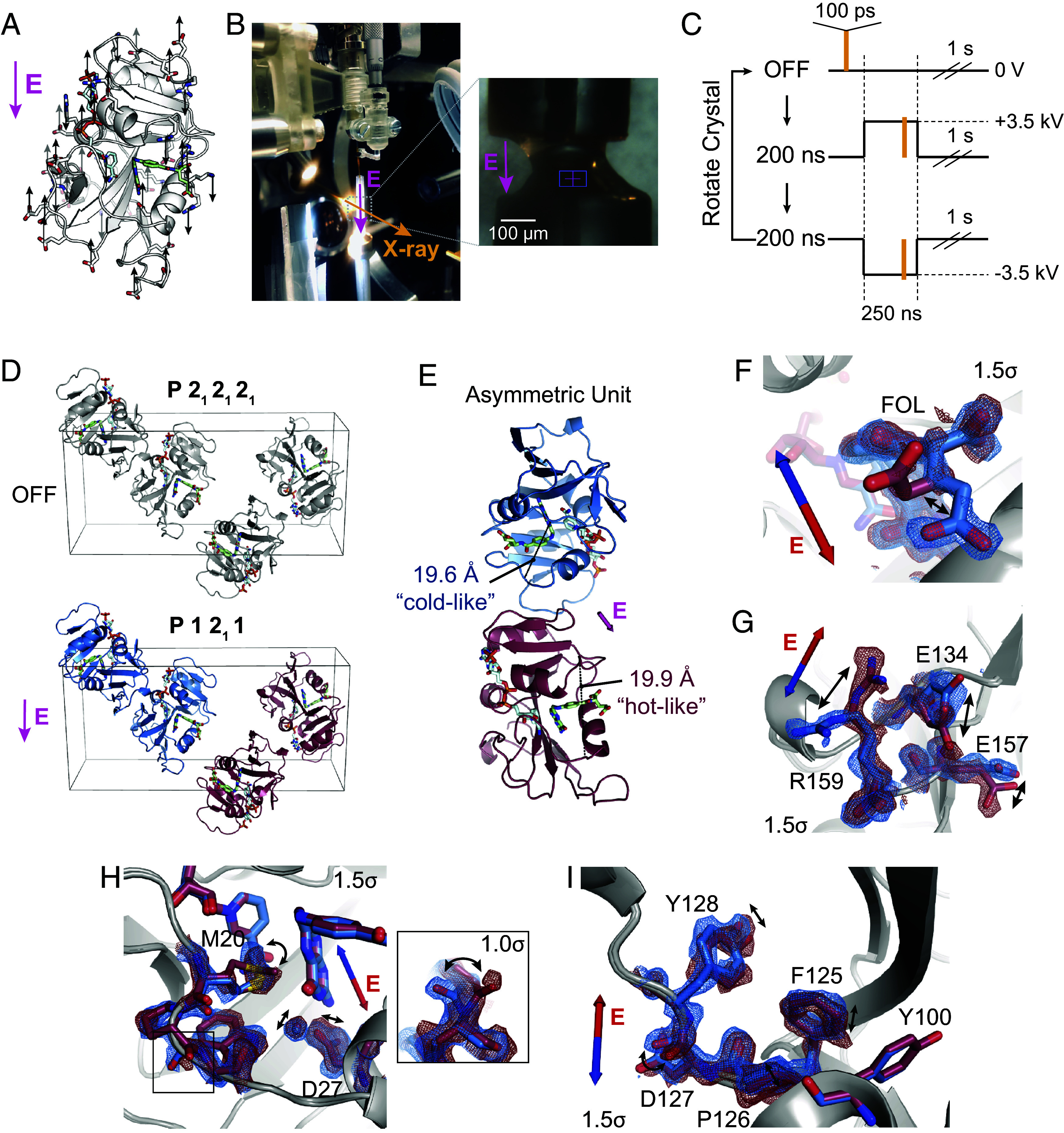
Electric-field-dependent structural changes recapitulate hinge motion and influence on active site residues. (A) Diagram of a possible pattern of force applied by an external electric field (E, in magenta) to ecDHFR based on the distribution of charged residues. (B) Photograph of the experimental apparatus for electric-field-stimulated X-ray crystallography (EF-X) at the BioCARS ID-B beamline; (Inset) zoom-in showing an ecDHFR crystal between two electrodes. (C) Schematic of the data collection strategy, which included three consecutive X-ray pulses at each angle: OFF (no high voltage pulse), 200 ns into a +3.5 kV pulse, and 200 ns into a 3.5 kV pulse. The crystal was rotated after each sequence of three diffraction images in order to collect a complete dataset for each condition. (D) Unit cell of the ecDHFR crystal during the EF-X experiments. During the OFF images, the crystal is in the spacegroup. The applied electric field along the -axis alters the symmetry of the crystal, rendering the crystal in a spacegroup during the high voltage pulses, with two copies in the new asymmetric unit (ASU; copies shown in red and blue). (E) The ASU of the refined excited state model. The two copies in the ASU differ in hinge distance. The different copies of the protein are colored in red and blue as an analogy to the multi-temperature experiment; red represents the expanded active site cleft observed at hotter temperatures, and blue represents the constricted cleft observed at colder temperatures. (F–I) Superposed models and maps from both protein molecules of the excited state ASU highlight electric-field dependent motion of charged groups. Blue and red arrows depict electric field vector for the blue and red models, respectively, and maps are contoured at and carved within 1.5 Å of shown atoms. (F) Carboxylate sidechain of folate and (G) charged sidechains near the C terminus demonstrate electric-field-dependent structural changes consistent with the formal charges of the residues. (H) Active site residues and Pro21 backbone carbonyl (Inset; contoured at ) differ between protein molecules. (I) Conformational changes among residues 125 to 128.
The high-voltage pulse applied in an EF-X experiment is directional. Copies of ecDHFR in the crystal’s unit cell are initially related by the symmetry operations of the P212121 spacegroup. During the pulse, these copies experience the electric field, and therefore patterns of forces, in different orientations (Fig. 4D). In our case, two copies of ecDHFR experience the electric field in nearly the same direction (e.g., both blue copies) while the other two molecules (both red copies) experience the opposite field. The resulting deformations are therefore different for the red and blue copies. Notably, we can use the resulting symmetry breaking to confirm that there is significant signal in the experiment (see SI Appendix for details and SI Appendix, Fig. S2B).
To interpret the structural changes during the high-voltage pulse, we refined models of the induced excited states (see SI Appendix for details and SI Appendix, Table S11). Notably, the copies of the model Michaelis complex seeing the electric field in opposite direction refined to different hinge distances (19.6 Å for the “blue” copy and 19.9 Å for the “red” copy, Fig. 4E and SI Appendix, Fig. S2C). These changes recapitulate the hinge motion observed using multi-temperature diffraction experiments (Fig. 3). Accordingly, we chose the color scheme for the two protein molecules to emphasize the comparison: The constricted copy is colored blue for “cold-like” and the extended copy is colored red for “hot-like”. The resulting electron density maps show clear electric-field dependent effects in which positively charged sidechains, like Arg159, move with the electric field, and negatively charged sidechains, like Glu134, move against the electric field (Fig. 4 F and G), consistent with the expected movement of charge in an applied electric field. We also observe several shifts in the active sites of the two molecules, including motions of Asp27, the ordered water, and the sidechain rotamer of Met20 (Fig. 4H), as well as a flip in the backbone state of Pro21-Trp22 (Fig. 4H, Inset). Because many residues in the Met20 loop lack a formal charge or significant charge dipole, these motions indicate conformational coupling of the Met20 loop with the rest of the enzyme. Furthermore, residues 125 to 128 display induced conformational rearrangements (Fig. 4I), similar to the conformational exchange observed in the multi-temperature experiment. The structural differences in Fig. 4F–I are also supported by composite omit maps, indicating that the results cannot be attributed to model bias (SI Appendix, Fig. S3). Despite the very different perturbations being used, the sets of conformational changes observed in the active site and Tyr128 region for the multi-temperature and electric-field-dependent experiments are consistent in terms of the residues involved and the sign of the influence of the hinge distance. Together, this supports a common mechanism in which the global hinge motion is coupled to local rearrangements throughout the enzyme on the nanosecond timescale.
Allosteric Coupling of Hinge Motion to Active Site Dynamics.
MD simulations provide a means to directly validate the mechanistic model that the hinge motion allosterically regulates the local conformational equilibria in the active site. Specifically, we can bias the hinge distance in simulation using an imposed distance restraint to observe its impact on other observables in the protein. To do so, we applied a distance restraint across the active site cleft with equilibrium values chosen to span the crystallographically observed range (Fig. 5A). We ran 100 independent, 100-ns MD simulations at each hinge distance. These restraints successfully biased the sampled conformations to particular widths of the active site cleft (Fig. 5B). In response, the population of states of the Met20 loop backbone changes monotonically (Fig. 5C, using the Trp22- backbone dihedral as a reporter). Similarly, with increasing hinge distance the Met20 sidechain shifts its rotamer distribution, as reported by a decrease of the population of the dihedral around (Fig. 5D). This change is consistent with the multi-temperature experiment, in which the Met20- of approximately was more populated at lower temperature (shorter hinge distance; Fig. 3J). This is also consistent with the EF-X results, in which the copy with a shorter hinge distance favored the Trp22 backbone and Met20 rotamer states observed in MD (Fig. 4H). These simulation results, therefore, corroborate the crystallographic analysis and confirm that the width of the active site cleft is allosterically coupled to the occupancy of the Met20 loop substates.
Fig. 5.
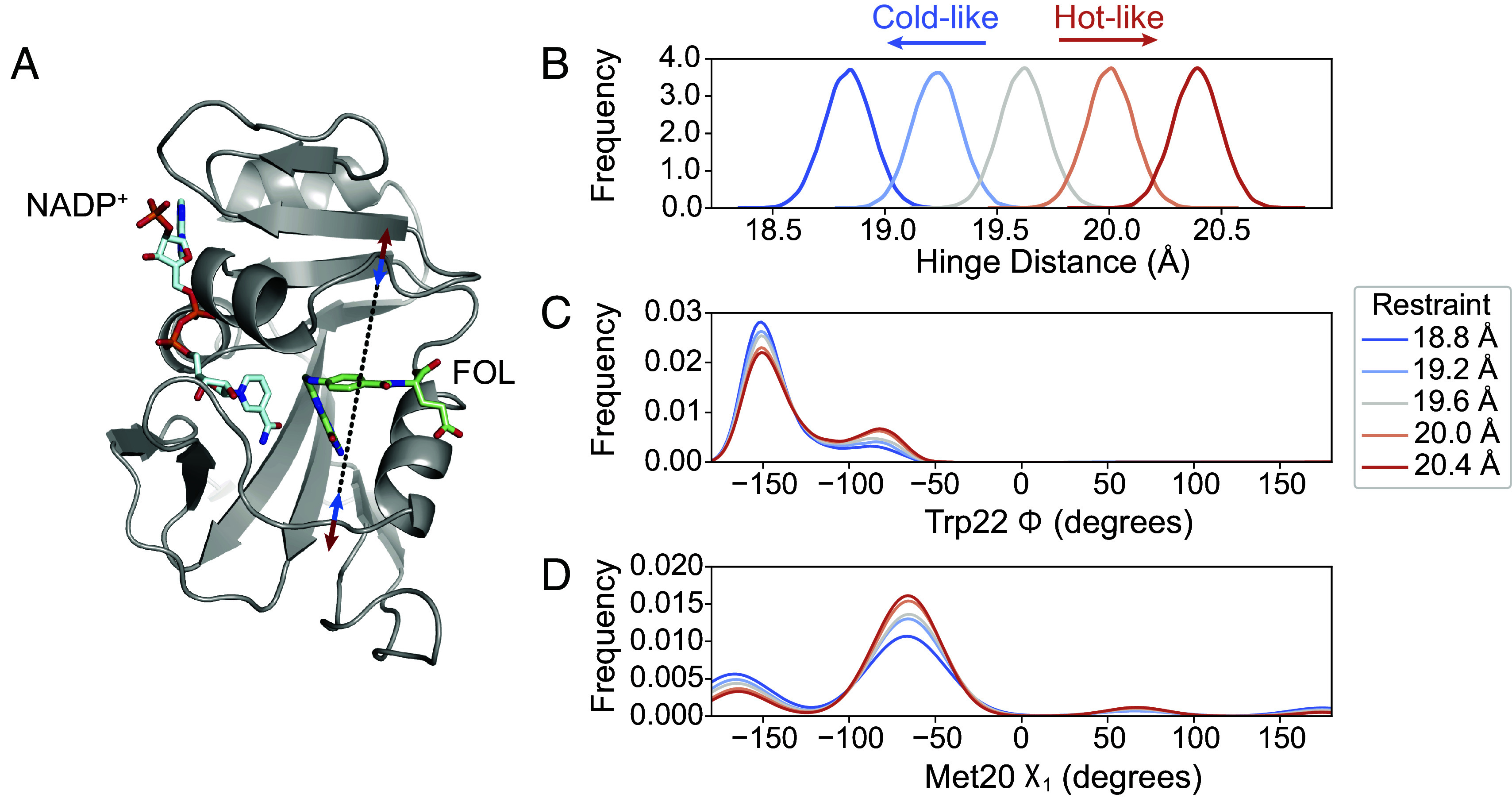
MD simulations validate the influence of hinge motion on the substates of the closed Met20 loop. (A) Simulation model of ecDHFR:NADP+:FOL highlighting the distance restraint applied in MD simulations between Asn23- and Pro53- (black dashed line) to model the effects of constricting (blue arrows, cold-like by analogy to the multi-temperature experiment) or expanding (red arrows, hot-like) the active site cleft. Kernel density estimates of the (B) hinge distance being restrained, (C) the Trp22-, and the (D) Met20- dihedrals. The Trp22- and Met20- dihedrals, which report on the Met20 closed substates, show a monotonic response to the distance restraint. The kernel density estimates were produced from 100 independent simulations of 100-ns duration at each restraint distance.
Substrate Protonation Regulates Active Site Solvent Access.
Do these global and local active site motions impact catalysis? As described, reduction of DHF involves two sequential steps: substrate protonation and hydride transfer—charge-transfer steps that may be strongly coupled to the electrostatics of the active site. To address the effect of protonation on the reactive Michaelis complex (DHFR:NADPH:DHF), we ran MD simulations of the deprotonated and N5-protonated complexes. Statistical distributions of key structural parameters are shown in Fig. 6. Upon protonation, the average hinge distance decreases by approximately 0.5 Å and the Trp22- equilibrium is further shifted towards the state near . This combination of changes recapitulates the allosteric mechanism identified above and indicates that substrate protonation engages this dynamic mode.
Fig. 6.
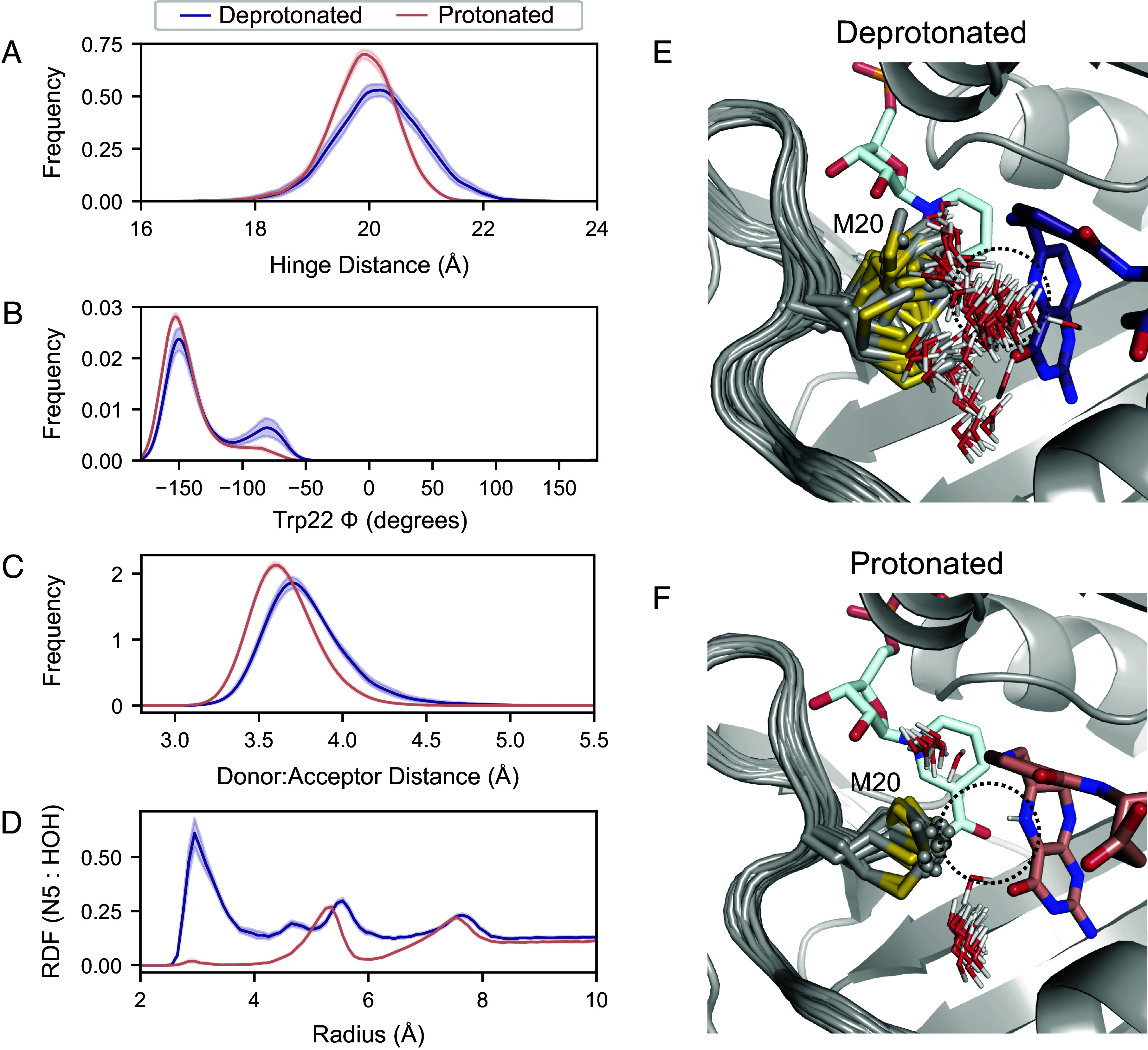
Protonation of the substrate orders the Met20 sidechain in the Michaelis complex. Fifty independent MD simulations of the ecDHFR:NADPH:DHF complex, with and without protonation of the N5 nitrogen, were run for 100 ns each. Kernel density estimates of the (A) hinge distance, (B) Trp22-, (C) donor–acceptor distance for hydride transfer change upon protonation of the substrate. These kernel density estimates were computed for each trajectory independently and the mean and 95% CI is shown for each condition. (D) The density of water around the N5 nitrogen of DHF as a function of distance from the N5 nitrogen RDF mean and 95% CI are shown. The first 50 frames (20 ns) from one trajectory are superimposed for the (E) deprotonated and (F) protonated substrate, depicting the Met20 sidechain and all waters within 4.5 Å of the N5 nitrogen of DHF. Only the initial frame is depicted for DHF and NADPH for visual clarity.
The donor–acceptor distance for hydride transfer also decreases upon protonation (Fig. 6C). This distance is the primary determinant of hydride transfer (39), and the change is consistent with the increase in the partial charge assigned to the C6 of DHF upon protonation. Protonation of the N5 nitrogen also effectively eliminates water from its proximity by ordering the Met20 sidechain. Indeed, the radial distribution function (RDF) of water molecules around the N5 nitrogen indicates very little occupancy of the proton-donating water site after protonation (Fig. 6D), consistent with findings in complementary simulation-based studies (40, 41). To visualize this change in the organization of the active site, we superpose frames from the trajectories. Overlaying 20 ns of one representative trajectory shows heterogeneity in the Met20 rotamer and frequent occupancy of the water site (dashed circle) for the deprotonated substrate (Fig. 6E), whereas the protonated substrate coordinates the Met20 rotamer that occludes the water site (Fig. 6F).
Experimentally, we also observed that the network involving Tyr128, Phe125, and Tyr100 exhibits pronounced temperature dependence (Fig. 3C) and motions extending into the active site (Fig. 3K). This network did not respond to variation in hinge distance in MD simulations (SI Appendix, Fig. S4A) but does respond to substrate protonation in MD (SI Appendix, Fig. S4B). Most likely, then, this network of residues contributes to electrostatic remodeling of the active site in response to protonation independently from the enzyme’s hinge motion.
In summary, before protonation, the active site has a pre-existing equilibrium of states that permits solvent access to the N5 nitrogen. This equilibrium is allosterically coupled to the width of the active site cleft. This dynamic architecture allows the enzyme to quickly reorganize the active site in response to protonation of its substrate. This rearrangement facilitates subsequent hydride transfer by polarizing the C6 carbon, shortening the donor–acceptor distance, and inhibiting the competing deprotonation reaction by excluding bulk solvent, consistent with a proposal by McTigue et al. (22).
Discussion
By a combination of innovative X-ray diffraction methods and analyses, we resolved the correlated motions of an enzyme in atomic detail. Using room-temperature diffraction, we first identified extended conformational heterogeneity in the enzyme’s active site loop (Fig. 1C). We then used a substrate mimetic to demonstrate that the Met20 sidechain directly regulates solvent access to the active site (Fig. 2C). Multi-temperature and EF-X experiments then uncovered a global hinge motion that constricts the enzyme’s active site and local networks of conformational rearrangements throughout the enzyme (Figs. 3 and 4). MD simulations confirmed that the hinge motion has a direct allosteric effect on conformational equilibria within the active site (Fig. 5). This coupling enables the protonated substrate to rapidly select an active site arrangement that favors the subsequent hydride transfer step over deprotonation (Fig. 6). The result is a model of catalysis by ecDHFR in which the product of the first chemical step (a reaction intermediate) drives rapid rearrangements in the active site by conformational selection to favor the second chemical step. That is, the enzyme is wired to undergo conformational change in response to completion of the first chemical step, just like it does after substrate binding, product formation, and product release, a view that naturally extends the notion of a dynamic free energy landscape as the organizing principle of enzyme catalysis (11).
Functional Significance of Solvent Gating in ecDHFR.
Our work validates the proposed solvent-gating role of Met20 and resolves conformational dynamics in ecDHFR that allosterically regulate the organization of the active site in response to substrate protonation. But, how important is proper solvent gating for hydride transfer? An important case study for the role of the Met20 loop in catalysis is the N23PP ecDHFR mutant (and the related N23PP/S148A mutant) that introduces the double proline insertion found in the human enzyme. This mutation decreases the rate of hydride transfer () by approximately 15-fold (Fig. 7A) with little apparent change in the overall structure. Because relaxation–dispersion experiments showed that this variant no longer displays millisecond-dynamics of the Met20 loop, Bhabha et al. concluded that these motions influence the chemical step(s) of catalysis in ecDHFR and classified the mutant as a “dynamic knockout” (24). Adamczyk et al. (42) disputed this conclusion with arguments about the importance of electrostatic preorganization and MD simulations showing no productive relationship between a putative coordinate for millisecond dynamics of the Met20 loop and the energy barrier for hydride transfer. Loveridge et al. (39) found that although the insertion mutant showed a reduced rate of hydride transfer, the corresponding kinetic isotope effect and its temperature dependence were largely unaffected. They interpreted this as evidence that the mutation does not alter direct dynamic contributions to hydride transfer. Based on our work, we believe that the N23PP mutation impedes the solvent-gating activity of ecDHFR: Close inspection of the active site in the published N23PP/S148A ecDHFR structure (24) shows that a water occupies the site typically occluded by Met20 in the wild-type enzyme (Fig. 7B; Protein Databank (PDB: 3QL0). The proline insertion increases the spacing between Met20 and the subsequent -helix by about 0.3 Å (measured from Met20- to Leu28-), such that the methionine sidechain no longer blocks solvent access to the substrate, trapping the protein in an accessible state that is less competent for hydride transfer. In this view, the solvent-gating function of Met20 mechanistically underpins the effect of the N23PP mutation, providing a structural explanation for the prior notion that the insertion disrupts the electrostatic environment of the ecDHFR active site (42).
Fig. 7.

Functional importance and conservation of solvent gating in DHFR. (A) The rate of hydride transfer, , for selected mutants of ecDHFR. (B) The structure of the N23PP/S148A mutant of ecDHFR (PDB: 3QL0) shows well-supported density for an ordered water in the map (blue mesh; 1.5). (C) Structures of human DHFR (PDB: 4M6K and PDB: 2W3M, molecule B) have unmodeled density consistent with partial-occupancy water within 3.5 Å of the N5 nitrogen of FOL and evidence of an alternate rotamer for Phe31 (; green/red mesh; ). A single rotamer is supported for Leu22 in the maps (blue mesh; 1.0) suggesting that Phe31 instead serves as the solvent-gating residue in the human enzyme. Although only molecule B is presented for the 2W3M deposited structure, similar features are observed in both protein molecules of the asymmetric unit.
Consistent with this inference, about six-fold of the catalytic activity of N23PP can be rescued by the point mutation L28F (Fig. 7A), which introduces a larger residue on the adjacent -helix (43) and likely partially restores the capability to shield the substrate from solvent. These results are consistent with a central role for solvent-gating in enhancing the hydride transfer rate of ecDHFR.
Functional Conservation of Dynamic Modes in DHFR.
Evolutionary conservation provides further perspective on the importance of the observed motions as DHFR homologs catalyze the same reaction and face similar challenges. The hinge motion characterized here within the model Michaelis complex resembles the conformational changes observed between substrate and product ternary complexes (44) in terms of its associated changes in pairwise-distance between atoms (Fig. 3C). The latter motion reflects a small (1 Å) hinge motion, and has been described as a subdomain rotation that alters the width of the active site cleft (9). Notably, the human homolog exhibits a substantially larger hinge motion (3 Å) upon product release (44), which was postulated to facilitate cofactor exchange in versions of DHFR with a more rigid Met20 loop (44). Consistently, the occluded state of the Met20 loop, which facilitates cofactor release in ecDHFR, has not been observed in eukaryotic DHFRs (9, 10, 44). These observations suggest that relative motion of the subdomains in the DHFR family readily evolves to maintain a dynamic mode (45) or shear plane (46) that can support multiple aspects of its functional cycle, reminiscent of the observation that a single collective motion observed in the intrinsic dynamics of ubiquitin underlies its different conformations bound to other proteins (47).
Indeed, the Met20 loop of human DHFR does not exhibit the conformational flexibility observed for the E. coli enzyme (9), and the analogous residue to Met20, Leu22, has well-resolved density for a single conformation in models of the human DHFR Michaelis complex (Fig. 7C). Strikingly, however, the differences between the modeled and observed electron density () for two previously deposited structures of human DHFR both show clear evidence of an excited state rotamer of the Phe31 sidechain (Fig. 7C). Accordingly, there is unmodeled positive difference density near the N5 nitrogen of folate, suggesting partial occupancy by a proton-donating water as observed for ecDHFR (Fig. 1B). Together, these observations strongly suggest that in human DHFR Phe31 is functionally analogous to ecDHFR Met20, rather than the structurally homologous Leu22 residue. This functional analogy was first proposed by McTigue et al. (22) and implies that solvent-gating is functionally conserved in the active sites of DHFR enzymes. Considering these structural observations along with partial functional rescue of the N23PP mutation by the L28F mutation in the E. coli enzyme, this suggests a mechanistic basis for the appearance of both mutations at a similar point in evolutionary history (43).
In summary, the work presented here used ligand-, temperature-, and electric-field-dependent X-ray diffraction experiments and MD simulations to resolve a conserved dynamic mode that allosterically influences local conformational equilibria in the active site of ecDHFR. This reveals an enzyme with dynamics primed to respond to the protonation of its substrate, suggesting a general principle for the rational and evolutionary design of enzymes with multi-step catalytic mechanisms: the introduction of specific coupled motions that allow the catalytic intermediate itself to drive active site reorganization. We believe the approach presented here will have broad application. The protein crystals we used are equivalent to those used for decades (e.g., in refs. 9, 15, and 16). However, the advances described here, building on improvements in hardware (38, 48), data collection strategies (7, 49), and analysis methods (50–52), enabled elucidation of the correlated motions of an enzyme in atomic detail. We expect the presented methods and strategy will likewise permit identification of the motions that underlie the function of a wide range of proteins, promoting the development of mechanistic models to explain protein function and its allosteric regulation.
Materials and Methods
The expression, purification, and crystallization of ecDHFR was performed as described previously (49), with the exception of the MFOL complex which required a modified purification scheme to avoid contamination with bound folate. Monochromatic data collection was conducted at the stanford synchrotron radiation lightsource (SSRL) beamline 12-1 at the SLAC National Accelerator Laboratory. All data collection employed a helical acquisition strategy described previously to mitigate radiation damage during room-temperature diffraction experiments (49). The multi-temperature experiments used the 800 Series Cryostream Cooler (Oxford Cryosystems) available at the SSRL 12-1 beamline, which controls the sample temperature to within 0.1 K. Data reduction and scaling was done with DIALS (50, 53), and an automated refinement strategy with phenix.refine (54) was applied for each dataset using a common R-free set to ensure comparable R-factors between datasets. EF-X experiments were conducted at BioCARS (Advanced Photon Source, Argonne National Laboratory) using an experimental apparatus and collection strategy based on previous work (38). All crystallographic analyses and difference electron density maps generated in this work used custom scripts implemented using reciprocalspaceship (51). All MD simulations were run using OpenMM, and a biasing potential was added between Asn23 and Pro53 using a custom distance force to evaluate the allosteric influence of hinge distance. Further details regarding the methods used in this study are available in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Drs. B. Correia, S. Eddy, R. Gaudet, and R. Losick for comments on the manuscript. D.R.H. is supported by the Searle Scholarship Program (SSP-2018-3240), a fellowship from the George W. Merck Fund of the New York Community Trust (338034), and the NIH Director’s New Innovator Award (DP2-GM141000). J.B.G. was supported by the NSF Graduate Research Fellowship under Grant No. DGE1745303. K.M.D. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. M.A.K. is supported by the NSF-Simons Center for Mathematical and Statistical Analysis of Biology at Harvard (award number #1764269) and the Harvard Quantitative Biology Initiative. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (including P41GM103393). In addition, this research used resources of the Advanced Photon Source, a DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of BioCARS was also supported by the National Institute of General Medical Sciences of the NIH under grant number P41 GM118217. The time-resolved set-up at Sector 14 was funded in part through a collaboration with Philip Anfinrud (National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of National Institute of General Medical Sciences or NIH.
Author contributions
J.B.G., K.M.D., and D.R.H. designed research; J.B.G., K.M.D., D.E.B., M.A.K., C.J.S., I.-S.K., R.W.H., S.R., and D.R.H. performed research; J.B.G. contributed new reagents/analytic tools; J.B.G. and D.R.H. analyzed data; and J.B.G. and D.R.H. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All structures determined in this study have been deposited in the Protein Databank with IDs: 8DAI (55), 5SSS (56), 5SST (57), 5SSU (58), 5SSV (59), 5SSW (60), 7FPL (61), 7FPM (62), 7FPN (63), 7FPO (64), 7FPP (65), 7FPQ (66), 7FPR (67), 7FPS (68), 7FPT (69), 7FPU (70), 7FPV (71), 7FPW (72), 7FPX (73), 7FPY (74), 7FPZ (75), 7FQ0 (76), 7FQ1 (77), 7FQ2 (78), 7FQ3 (79), 7FQ4 (80), 7FQ5 (81), 7FQ6 (82), 7FQ7 (83), 7FQ8 (84), 7FQ9 (85), 7FQA (86), 7FQB (87), 7FQC (88), 7FQD (89), 7FQE (90), 7FQF (91), 7FQG (92), 8G4Z (93), and 8G50 (94), as referenced in SI Appendix, Tables 1–11. Python and PyMOL scripts for generating figures, along with (difference) electron density maps are deposited in Zenodo (https://doi.org/10.5281/zenodo.7634123) (95). Crystallographic analyses make use of reciprocalspaceship and rs-booster, which are available from https://rs-station.github.io/ (96, 97). The forcefields, starting models, and scripts for reproducing the molecular dynamics trajectories are included in the Zenodo deposition.
Supporting Information
References
- 1.Lienhard G. E., Enzymatic catalysis and transition-state theory. Science 180, 149–154 (1973). [DOI] [PubMed] [Google Scholar]
- 2.Warshel A., Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 273, 27035–27038 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Henzler-Wildman K., Kern D., Dynamic personalities of proteins. Nature 450, 964–972 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Yabukarski F., et al. , Ensemble-function relationships to dissect mechanisms of enzyme catalysis. Sci. Adv. 8, eabn7738 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karplus M., McCammon J. A., Dynamics of proteins: Elements and function. Annu. Rev. Biochem. 53, 263–300 (1983). [DOI] [PubMed] [Google Scholar]
- 6.Ma B., Kumar S., Tsai C. J., Hu Z., Nussinov R., Transition-state ensemble in enzyme catalysis: Possibility, reality, or necessity? J. Theor. Biol. 203, 383–397 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Fraser J. S., et al. , Hidden alternative structures of proline isomerase essential for catalysis. Nature 462, 669–673 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanoian P., Liu C. T., Hammes-Schiffer S., Benkovic S., Perspectives on electrostatics and conformational motions in enzyme catalysis. Accu. Chem. Res. 48, 482–489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawaya M. R., Kraut J., Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 36, 586–603 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Schnell J. R., Dyson H. J., Wright P. E., Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 33, 119–140 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Boehr D. D., McElheny D., Dyson H. J., Wrightt P. E., The dynamic energy landscape of dihydrofolate reductase catalysis. Science 313, 1638–1642 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Kohen A., Dihydrofolate reductase as a model for studies of enzyme dynamics and catalysis. F1000Research 4, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boehr D. D., McElheny D., Dyson H. J., Wright P. E., Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc. Natl. Acad. Sci. U.S.A. 107, 1373–1378 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oyen D., Fenwick R. B., Stanfield R. L., Dyson H. J., Wright P. E., Cofactor-mediated conformational dynamics promote product release from Escherichia coli dihydrofolate reductase via an allosteric pathway. J. Am. Chem. Soc. 137, 9459–9468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van den Bedem H., Bhabha G., Yang K., Wright P. E., Fraser J. S., Automated identification of functional dynamic contact networks from X-ray crystallography. Nat. Methods 10, 896–902 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keedy D. A., et al. , Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR. Structure 22, 899–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C. T., et al. , Escherichia coli dihydrofolate reductase catalyzed proton and hydride transfers: Temporal order and the roles of Asp27 and Tyr100. Proc. Natl. Acad. Sci. U.S.A. 111, 18231–18236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wan Q., et al. , Toward resolving the catalytic mechanism of dihydrofolate reductase using neutron and ultrahigh-resolution X-ray crystallography. Proc. Natl. Acad. Sci. U.S.A. 111, 18225–18230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shrimpton P., Allemann R. K., Role of water in the catalytic cycle of E. coli dihydrofolate reductase. Prot. Sci. 11, 1442–1451 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rod T. H., Brooks C. L. III, How dihydrofolate reductase facilitates protonation of dihydrofolate. J. Am. Chem. Soc. 125, 8718–8719 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Wan Q., et al. , Capturing the catalytic proton of dihydrofolate reductase: Implications for general acid-base catalysis. ACS Catal. 11, 5873–5884 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McTigue M. A., Davies J. F., Kraut J., Kaufman B. T., Crystal structure of chicken liver dihydrofolate reductase complexed with NADP+ and biopterin. Biochemistry 31, 7264–7273 (1992). [DOI] [PubMed] [Google Scholar]
- 23.Marcus R., Sutin N., Electron transfers in chemistry and biology. Biochim. Biophys. Acta (BBA) - Rev. Bioenerg. 811, 265–322 (1985). [Google Scholar]
- 24.Bhabha G., et al. , A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 332, 234–238 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frauenfelder H., Petsko G. A., Tsernoglou D., Temperature-dependent X-ray diffraction as a probe of protein structural dynamics. Nature 280, 558–563 (1979). [DOI] [PubMed] [Google Scholar]
- 26.Frauenfelder H., et al. , Thermal expansion of a protein. Biochemistry 26, 254–261 (1987). [DOI] [PubMed] [Google Scholar]
- 27.Tilton R. F., Dewan J. C., Petsko G. A., Effects of temperature on protein structure and dynamics: X-ray crystallographic studies of the protein ribonuclease-a at nine different temperatures from 98 to 320 K. Biochemistry 31, 2469–2481 (1992). [DOI] [PubMed] [Google Scholar]
- 28.Keedy D. A., et al. , Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. eLife 4, e07574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keedy D. A., et al. , An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. eLife 7, e36307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebrahim A., et al. , The temperature-dependent conformational ensemble of SARS-CoV-2 main protease (Mpro). IUCrJ 9, 682–694 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Šrajer V., et al. , Photolysis of the carbon monoxide complex of myoglobin: Nanosecond time-resolved crystallography. Science 274, 1726–1729 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Genick U. K., et al. , Structure of a protein photocycle intermediate by millisecond time-resolved crystallography. Science 275, 1471–1475 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Zeldin O. B., Gerstel M., Garman E. F., RADDOSE-3D: Time- and space-resolved modelling of dose in macromolecular crystallography. J. Appl. Crystallogr. 46, 1225–1230 (2013). [Google Scholar]
- 34.Bury C. S., Brooks-Bartlett J. C., Walsh S. P., Garman E. F., Estimate your dose: Raddose-3D. Prot. Sci. 27, 217–228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu C. T., et al. , Probing the electrostatics of active site microenvironments along the catalytic cycle for Escherichia coli dihydrofolate reductase. J. Am. Chem. Soc. 136, 10349–10360 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh P., Sen A., Francis K., Kohen A., Extension and limits of the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. J. Am. Chem. Soc. 136, 2575–2582 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Francis K., Kohen A., Protein motions and the activation of the CH bond catalyzed by dihydrofolate reductase. Curr. Opin. Chem. Biol. 21, 19–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hekstra D. R., et al. , Electric-field-stimulated protein mechanics. Nature 540, 400–405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loveridge E. J., Behiry E. M., Guo J., Allemann R. K., Evidence that a “dynamic knockout’’ in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat. Chem. 4, 292–297 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Mhashal A. R., Vardi-Kilshtain A., Kohen A., Major D. T., The role of the Met20 loop in the hydride transfer in Escherichia coli dihydrofolate reductase. J. Biol. Chem. 292, 14229–14239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mhashal A. R., Pshetitsky Y., Cheatum C. M., Kohen A., Major D. T., Evolutionary effects on bound substrate pKa in dihydrofolate reductase. J. Am. Chem. Soc. 140, 16650–16660 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Adamczyk A. J., Cao J., Kamerlin S. C., Warshel A., Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc. Natl. Acad. Sci. U.S.A. 108, 14115–14120 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C. T., et al. , Functional significance of evolving protein sequence in dihydrofolate reductase from bacteria to humans. Proc. Natl. Acad. Sci. U.S.A. 110, 10159–10164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhabha G., et al. , Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 20, 1243–1249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rouviere E., Ranganathan R., Rivoire O., Emergence of single- versus multi-state allostery. PRX Life 1, 023004 (2023). [Google Scholar]
- 46.Mitchell M. R., Tlusty T., Leibler S., Strain analysis of protein structures and low dimensionality of mechanical allosteric couplings. Proc. Natl. Acad. Sci. U.S.A. 113, E5847–E5855 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lange O. F., et al. , Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320, 1471–1475 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Graber T., et al. , BioCARS: A synchrotron resource for time-resolved X-ray science. J. Synchrot. Rad. 18, 658–670 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greisman J. B., et al. , Native SAD phasing at room temperature. Acta Crystallogr. Sect. D 78, 986–996 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winter G., et al. , DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D 74, 85–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greisman J. B., Dalton K. M., Hekstra D. R., reciprocalspaceship: A Python library for crystallographic data analysis. J. Appl. Crystallogr. 54, 1521–1529 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dalton K. M., Greisman J. B., Hekstra D. R., A unifying Bayesian framework for merging X-ray diffraction data. Nat. Commun. 13, 7764 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beilsten-Edmands J., et al. , Scaling diffraction data in the DIALS software package: Algorithms and new approaches for multi-crystal scaling. Acta Crystallogr. Sect. D 76, 385–399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Afonine P. V., et al. , Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greisman J. B., Brookner D. E., Hekstra D. R., E. coli DHFR complex with NADP+ and 10-methylfolate. Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/8DAI. Deposited 13 June 2022.
- 56.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 270 K (crystal 1). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/5SSS. Deposited 3 July 2022.
- 57.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 270 K (crystal 2). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/5SST. Deposited 3 July 2022.
- 58.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 270 K (crystal 3). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/5SSU. Deposited 3 July 2022.
- 59.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 270 K (crystal 4). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/5SSV. Deposited 3 July 2022.
- 60.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 270 K (multi-crystal). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/5SSW. Deposited 3 July 2022.
- 61.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 280 K (crystal 1). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPL. Deposited 9 September 2022.
- 62.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 280 K (crystal 2). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPM. Deposited 9 September 2022.
- 63.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 280 K (crystal 3). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPN. Deposited 9 September 2022.
- 64.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 280 K (crystal 4). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPO. Deposited 9 September 2022.
- 65.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 280 K (crystal 5). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPP. Deposited 9 September 2022.
- 66.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 280 K (multi-crystal). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPQ. Deposited 9 September 2022.
- 67.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 290 K (crystal 1). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPR. Deposited 10 September 2022.
- 68.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 290 K (crystal 2). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPS. Deposited 10 September 2022.
- 69.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 290 K (crystal 3). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPT. Deposited 10 September 2022.
- 70.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 290 K (crystal 4). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPU. Deposited 10 September 2022.
- 71.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 290 K (crystal 5). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPV. Deposited 10 September 2022.
- 72.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 290 K (multi-crystal). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPW. Deposited 10 September 2022.
- 73.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 300 K (crystal 1). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPX. Deposited 10 September 2022.
- 74.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 300 K (crystal 2). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPY. Deposited 10 September 2022.
- 75.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 300 K (crystal 3). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FPZ. Deposited 10 September 2022.
- 76.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 300 K (crystal 4). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ0. Deposited 10 September 2022.
- 77.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 300 K (crystal 5). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ1. Deposited 10 September 2022.
- 78.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 300 K (multi-crystal). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ2. Deposited 10 September 2022.
- 79.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 310 K (crystal 1). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ3. Deposited 12 September 2022.
- 80.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 310 K (crystal 2). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ4. Deposited 12 September 2022.
- 81.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 310 K (crystal 3). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ5. Deposited 12 September 2022.
- 82.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex at 310 K (multi-crystal). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ6. Deposited 12 September 2022.
- 83.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 1, pass 1, 295 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ7. Deposited 12 September 2022.
- 84.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 1, pass 2, 310 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ8. Deposited 12 September 2022.
- 85.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 1, pass 3, 295 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQ9. Deposited 12 September 2022.
- 86.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 1, pass 4, 280 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQA. Deposited 12 September 2022.
- 87.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 1, pass 5, 295 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQB. Deposited 12 September 2022.
- 88.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 2, pass 1, 295 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQC. Deposited 12 September 2022.
- 89.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 2, pass 2, 280 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQD. Deposited 12 September 2022.
- 90.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 2, pass 3, 295 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQE. Deposited 12 September 2022.
- 91.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 2, pass 4, 310 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQF. Deposited 12 September 2022.
- 92.Greisman J. B., Dalton K. M., Brookner D. E., Hekstra D. R., DHFR:NADP+:FOL complex (crystal 2, pass 5, 295 K). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/7FQG. Deposited 12 September 2022.
- 93.Greisman J. B., Dalton K. M., Brookner D. E., Klureza M. A., Hekstra D.R., E. coli DHFR complex with NADP+ and folate: EF-X off model by Laue diffraction (no electric field). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/8G4Z. Deposited 10 February 2023.
- 94.Greisman J. B., Dalton K. M., Brookner D. E., Klureza M. A., Hekstra D. R., E. coli DHFR complex with NADP+ and folate: EF-X excited state model by Laue diffraction (electric field along b axis; 8-fold extrapolation of structure factor differences). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/8G50. Deposited 10 February 2023.
- 95.Greisman J. B., et al. , Resolving conformational rearrangements in DHFR: Supporting Data (1.0.0). Zenodo. 10.5281/zenodo.7634124. Deposited 26 February 2023. [DOI]
- 96.Greisman J. B., et al. , reciprocalspaceship (v0.10.2). GitHub. https://github.com/rs-station/reciprocalspaceship/tree/v0.10.2. Accessed 11 July 2022.
- 97.Greisman J. B., et al. , rs-booster (v0.1.0). GitHub. https://github.com/rs-station/rs-booster/tree/v0.1.0. Accessed 1 September 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All structures determined in this study have been deposited in the Protein Databank with IDs: 8DAI (55), 5SSS (56), 5SST (57), 5SSU (58), 5SSV (59), 5SSW (60), 7FPL (61), 7FPM (62), 7FPN (63), 7FPO (64), 7FPP (65), 7FPQ (66), 7FPR (67), 7FPS (68), 7FPT (69), 7FPU (70), 7FPV (71), 7FPW (72), 7FPX (73), 7FPY (74), 7FPZ (75), 7FQ0 (76), 7FQ1 (77), 7FQ2 (78), 7FQ3 (79), 7FQ4 (80), 7FQ5 (81), 7FQ6 (82), 7FQ7 (83), 7FQ8 (84), 7FQ9 (85), 7FQA (86), 7FQB (87), 7FQC (88), 7FQD (89), 7FQE (90), 7FQF (91), 7FQG (92), 8G4Z (93), and 8G50 (94), as referenced in SI Appendix, Tables 1–11. Python and PyMOL scripts for generating figures, along with (difference) electron density maps are deposited in Zenodo (https://doi.org/10.5281/zenodo.7634123) (95). Crystallographic analyses make use of reciprocalspaceship and rs-booster, which are available from https://rs-station.github.io/ (96, 97). The forcefields, starting models, and scripts for reproducing the molecular dynamics trajectories are included in the Zenodo deposition.