

Abstract
Metastatic BRAFV600E colorectal cancer (CRC) carries an extremely poor prognosis and is in urgent need of effective new treatments. While the BRAFV600E inhibitor encorafenib in combination with the EGFR inhibitor cetuximab (Enc+Cet) was recently approved for this indication, overall survival is only increased by 3.6 months and objective responses are observed in only 20% of patients. We have found that a limitation of Enc+Cet treatment is the failure to efficiently induce apoptosis in BRAFV600E CRCs, despite inducing expression of the pro-apoptotic protein BIM and repressing expression of the pro-survival protein MCL-1. Here, we show that BRAFV600E CRCs express high basal levels of the pro-survival proteins MCL-1 and BCL-XL, and that combining encorafenib with a BCL-XL inhibitor significantly enhances apoptosis in BRAFV600E CRC cell lines. This effect was partially dependent on the induction of BIM, as BIM deletion markedly attenuated BRAF plus BCL-XL inhibitor-induced apoptosis. As thrombocytopenia is an established on-target toxicity of BCL-XL inhibition, we also examined the effect of combining encorafenib with the BCL-XL -targeting PROTAC DT2216, and the novel BCL-2/BCL-XL inhibitor dendrimer conjugate AZD0466. Combining encorafenib with DT2216 significantly increased apoptosis induction in vitro, while combining encorafenib with AZD0466 was well tolerated in mice and further reduced growth of BRAFV600E CRC xenografts compared to either agent alone. Collectively, these findings demonstrate that combined BRAF and BCL-XL inhibition significantly enhances apoptosis in pre-clinical models of BRAFV600E CRC and is a combination regimen worthy of clinical investigation to improve outcomes for these patients.
Subject terms: Colon cancer, Targeted therapies
Introduction
Mutations in the BRAF oncogene (predominantly BRAFV600E) occur in approximately 10% of metastatic CRCs and drive tumorigenesis by constitutive activation of the MAPK/ERK signalling pathway [1–4]. In metastatic CRC, BRAFV600E mutations are associated with a particularly poor prognosis [5–7].
Despite inducing objective responses in ~50% of melanoma patients [8], monotherapy using BRAF inhibitors such as vemurafenib, dabrafenib and encorafenib only induce objective responses in ~5% of BRAFV600E metastatic CRCs [9]. This was attributed to relief of a negative feedback loop between ERK and the EGFR, leading to re-activation of MAPK/ERK signalling [10, 11]. Based on this observation, combination treatment with encorafenib (Enc) and the EGFR inhibitor cetuximab (Cet) were tested and found to significantly improve overall survival in BRAFV600E metastatic CRC patients [12, 13], and is the current standard of care for these patients. While the Enc+Cet regimen represents a major advance in the treatment of BRAFV600E CRC, the improvement in overall survival (OS) is <4 months and objective response rates remain below 20% [13], warranting the need for further refinement of this treatment.
One limitation of BRAF and MAPK/ERK pathway inhibitors in general is their failure to induce substantial levels of apoptosis in CRC and other tumour cell types [11, 14]. Notably, this is despite these agents inducing the expression of the pro-apoptotic proteins BIM [15–17], BMF [17] and PUMA [15, 17], and repressing expression of the pro-survival protein MCL-1 [16, 18].
Notably, CRC cells have been reported to express high basal levels of pro-survival proteins, particularly BCL-XL [19]. Herein, we considered the possibility that the high basal level of expression of pro-survival proteins may establish a high apoptotic threshold in CRC cells that cannot be overcome by BRAF inhibitors alone. To overcome this, we investigated the combinatorial use of BRAF inhibitors with BH3 mimetics, a class of anti-cancer compounds that mimic the function of BH3-only proteins to block the activity of pro-survival proteins [20]. We found that both MCL-1 and BCL-XL are highly expressed in BRAFV600E CRC cell lines and that combining BRAF inhibitors with BCL-XL inhibitors significantly enhances apoptosis in these cell lines. We also demonstrate that this effect is at least partially dependent on the induction of BIM. Importantly, we further confirm this finding using two emerging strategies of BCL-XL inhibition, first using the BCL-XL targeting PROTAC DT2216 and second using the novel BCL-2/BCL-XL inhibitor dendrimer conjugate AZD0466 [21]. Our pre-clinical findings demonstrate that the addition of BCL-XL inhibitors to BRAF inhibitor treatment is worthy of clinical investigation as a means of improving outcomes for BRAFV600E CRC patients.
Methods
Cell culture
The BRAFV600E colorectal cancer cell lines were sourced as follows; COLO 201, COLO 205, HT29, SW1417 and RKO (American Type Culture Collection, Manassas, VA, USA), LIM2551 and LIM2405 cells (Ludwig Institute for Cancer Research), CO115 [22], VACO432 [23], LS411 [24] and VACO5 [25]. All cell lines were maintained in Dulbecco’s Minimal Essential Media/F12 (DMEM/F12, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 5% FBS (v/v)(Moregate, Queensland, Australia) at 37 °C with 5% CO2. Cell line authentication was performed by short tandem repeat (STR) profiling using the GenePrint 10 system (Promega, Madison, WI, USA), and all lines were found to be exact matches to published profiles. Mycoplasma testing was performed every 3-6 months as part of routine monitoring in our laboratory.
Chemicals
Encorafenib, vemurafenib, A-1331852, S63845, ABT-199 (all from Assay Matrix, Melbourne, Australia), DT2216 (Axon Medchem, Netherlands) and AZD4320 (AstraZeneca, Cambridge, UK) were dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO, USA). Cetuximab (Erbitux, Lilly, Indianapolis, IN, USA) was obtained from the Austin Health Pharmacy. AZD0466 was obtained from AstraZeneca.
Western blot
Whole cell lysates were prepared using NP-40 lysis buffer (50 mM Tris-HCL pH 7.5, 150 mM NaCl, 1% NP-40 (v/v), 1 mM EDTA pH 8) supplemented with cOmplete protease inhibitor and PhosSTOP phosphatase inhibitor cocktails (Roche, Basel, Switzerland). A total of 30 μg of protein per sample was resolved on NuPAGE 4-12% Bis-Tris pre-cast polyacrylamide gels (Novex, Thermo Fisher Scientific), transferred onto iBLOT2 Polyvinylidene Difluoride (PVDF) membranes (Invitrogen, Thermo Fisher Scientific) and blocked using Odyssey blocking buffer (LiCor, Lincoln, NE, USA). Primary antibodies used in western blot analysis were BIMS/L/EL (ALX-804-527, Enzo Life Sciences, New York, NY, USA), MCL-1 (5453S, Cell Signalling Technologies, Danvers, MA, USA), BCL-XL (2764, Cell Signalling Technologies), Cleaved Caspase 3 (9661, Cell Signalling Technologies), and β-tubulin (ab6046, Abcam, Cambridge, UK). Secondary antibodies used were IRDye®680RD Goat anti-rat IgG (H + L)(926-68076, LiCor) and IRDye®800CW Goat anti-rabbit IgG (H + L)(926-32211, LiCor). Full western blot images can be found in the Supplementary File.
Flow cytometry
Cells were seeded in 24 well plates and treated with drugs for 24 or 72 h. Both adherent and non-adherent cells were collected at completion of drug treatment and incubated overnight at 4 °C in 50 μg/mL propidium iodide diluted in sodium citrate buffer (0.1% sodium citrate (w/v) and 0.1% Triton X-100 (v/v)) (all from Sigma-Aldrich). Samples were then analysed on a BD FACSymphony A3 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA), by analysis of 10,000 events. Apoptotic cells were defined as having a sub-diploid DNA content and quantified using FlowJo V8.0 (FlowJo LLC, Ashland, OR, USA). Cell cycle analysis was performed using ModFit LTTM version 2.0 (Verity Software House, Topsham, ME, USA).
Clonogenic survival assays
Cells were seeded at 500 cells per well in 12-well plates and treated the following day with encorafenib (1 nM or 100 nM) alone and in combination with either A-1331852 (10 nM), S63845 (1 μM) or ABT-199 (1 μM). Colonies were then allowed to form over 10 days, at this point cells were fixed with 10% formalin for 5 min and stained with 0.1% crystal violet solution (Sigma-Aldrich) for 15 min at room temperature, washed with PBS and allowed to air dry. Analysis of clonogenic survival was performed using ImageJ (Java).
Generation of BIM knockout COLO 201 and LIM2551 cells
The COLO 201 and LIM2551 BIM knockout cell line was generated using the previously described doxycycline-inducible CRISPR-Cas9 lentiviral system [26]. Briefly, cells were stably transduced with lentivirus expressing Cas9-mCherry and FACS sorted for mCherry positivity. Cells were subsequently transduced with lentivirus expressing GFP and the doxycycline-inducible sgRNA sequence targeting exon 3 of the human BIM gene (5′-GCCCAAGAGTTGCGGCGTAT-3′). Cells were double sorted for mCherry and GFP and subsequently maintained in vehicle or doxycycline (1 μg/ml).
Xenograft study
Six-week-old female NOD Scid gamma (NSG) mice were obtained from a colony maintained at the Austin Health BioResource Facility (Melbourne, Australia) and housed in specific pathogen free (SPF) microisolators. Mice were subcutaneously injected with 2 × 106 COLO 201 cells into the left and right flanks in a 1:1 mixture of matrigel matrix (75 μL)(Corning): DMEM/F12 (75 μL). Once tumours became palpable (~100 mm3), mice were randomised to receive either vehicle control (n = 8 mice, 16 tumours), encorafenib (20 mg/kg, bi-daily, oral gavage) (n = 8 mice, 16 tumours), AZD0466 (103 mg/kg, once weekly, tail vein injection) (n = 8 mice, 16 tumours) or encorafenib plus AZD0466 (n = 8 mice, 14 tumours) for a total of 3 weeks. Tumours from one mouse were excluded from the combination arm due to an adverse event unrelated to treatment. Allocation of mice into groups was not blinded by the investigator. Sample sizes of the cohorts were based on previous experience, however no formal power analyses were performed prior to experimentation. Calliper measurements blinded to treatment group were performed a minimum of three times per week to measure tumour size. Investigators were not blinded at endpoint for tumour collection. Encorafenib was prepared in 50% PhosalPG (v/v), 27.5% PEG400 (v/v), 10% Ethanol (v/v) and 2.5% DMSO (v/v). AZD0466 was prepared in citrate/phosphate buffer. The study was approved by the Austin Health Animal Ethics Committee (A2018_05584).
Microarray analysis
Gene expression profiling was performed on 11 BRAF-mutant colorectal cancer cell lines treated with vemurafenib (5 μM) or DMSO control for 6 h, using the Affymetrix HG-U-133-Plus-2-Human Genome Array platform at the Peter MacCallum Cancer Centre genomics core facility (Melbourne, Australia). Affymetrix microarray. CEL files were imported into the Partek Genomics Suite version 6.6 (Partek, Chesterfield, MO, USA) and normalised using the Robust Multiarray Averaging (RMA) method. Normalised data was analysed using the Limma package. Affymetrix probes were determined to be differentially expressed between DMSO control and vemurafenib treatments if the adjusted p-value was <0.05.
Immunohistochemistry
Formalin-fixed paraffin-embedded sections (4 μm) were de-paraffinized and rehydrated through serial washes in xylene and ethanol. Sections were rinsed in H2O and quenched in 3% H2O2 (Chemsupply) for 10 min. Antigen retrieval was performed by heating in Citrate buffer (pH 6.0) for 2 min at 1.0 power and 8 min at 0.2 power in the microwave. Slides were probed and incubated with anti-BIM (1:100, Cell Signalling Technology, 2933S) primary antibody at 4 °C overnight. Slides were then washed and incubated with Labeled polymer HRP-anti Rabbit (K4003, Dako) for 1-h at room temperature. Slides were washed and chromagen was developed using the DAB (3, 3-diaminobenzide) reagent (Dako). Sections were counter-stained using pre-filtered Mayer’s hematoxylin (Amber Scientific, Australia) then dehydrated through serial ethanol and xylene washes prior to mounting using DPX mounting solution (Sigma-Aldrich). BIM expression was analysed using HALO software (Indica Labs, NM, USA).
Statistics
Statistical analyses were performed using GraphPad Prism v8.0 software (GraphPad Software, La Jolla, CA, USA). Groups were compared using either Student’s t test with Welch’s correction or One-way ANOVA with Tukey’s multiple comparison, unless stated otherwise. In all cases: ns (not significant), *(p ≤ 0.05), **(p ≤ 0.01), ***(p ≤ 0.001) and ****(p ≤ 0.0001).
Results
BRAF inhibitors inhibit proliferation but have minimal effects on apoptosis in BRAFV600E CRC cell lines
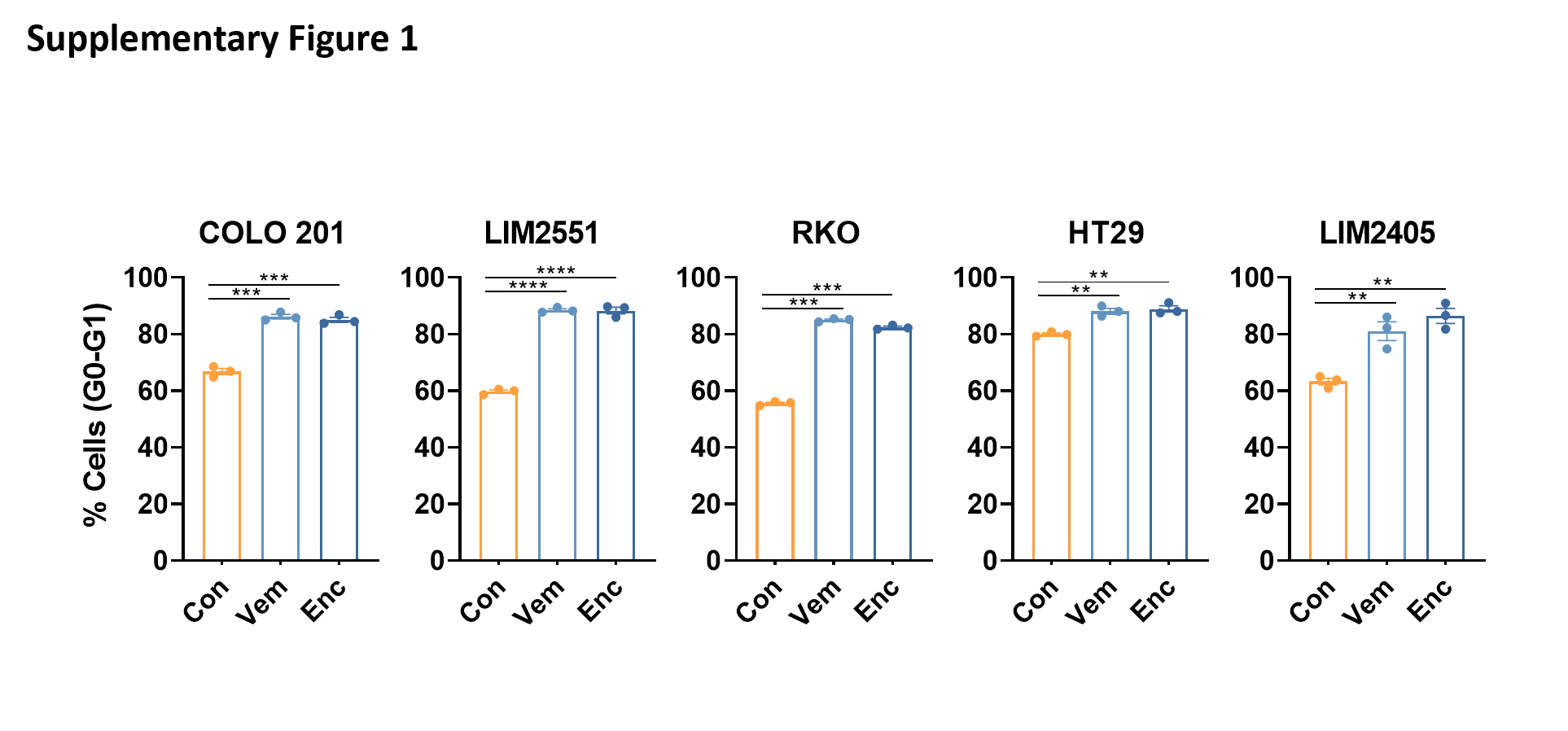
BRAF inhibitor monotherapy induces objective responses in only ~5% of metastatic BRAFV600E CRC patients [9]. While combination BRAF plus EGFR inhibitor treatment improves this to ~20%, overall survival is increased by only 4 months. One limitation of MAPK pathway inhibitors in general is that they predominantly induce cytostatic effects, and often fail to induce extensive levels of apoptosis [11, 14]. To investigate if similar effects occur upon BRAF inhibitor treatment of BRAFV600E CRC cells, we treated five BRAFV600E CRC cell lines with the BRAF inhibitors vemurafenib or encorafenib. Both inhibitors induced minimal to modest (<25%) apoptosis across all cell lines (Fig. 1A). Comparatively, both agents inhibited cell cycle progression in all five BRAFV600E CRC lines tested, illustrated by the significant reduction in the percentage of cells in S-phase and increase in the percentage of cells in the G0-G1 phase (Fig. 1B, Supplementary Fig. 1).
Fig. 1. BRAF inhibitors induce predominantly cytostatic effects in BRAFV600E CRC cell lines.

A, B BRAFV600E CRC cell lines were treated with vemurafenib (Vem, 5 μM) or encorafenib (Enc, 100 nM) for 72 h and stained with propidium iodide (PI) and analysed by FACS to determine (A) apoptosis induction (percentage of cells with sub-diploid DNA content), and (B) percentage of cells in S-phase. Values shown are mean ± SEM from a representative experiment performed in technical triplicate. Similar results were obtained in two additional independent experiments. Groups were compared using an unpaired Student’s t-test using Welch’s correction; *(p ≤ 0.05), **(p ≤ 0.01) and ***(p ≤ 0.001). C BRAFV600E CRC cell lines were treated with encorafenib (100 nM) plus cetuximab (10 μg/mL) (Enc+Cet) or irinotecan (10 μM)(Irino) for 72 h and apoptosis levels determined by PI staining as above. Values shown are mean ± SEM from a representative experiment performed in technical triplicate. Similar results were obtained in 2 additional independent experiments. Differences were compared using one-way ANOVA with Tukey’s multiple comparison testing; ns (not significant), *(p ≤ 0.05), **(p ≤ 0.01) and ****(p ≤ 0.0001). D, E Western blot analysis for BIMS/L/EL, MCL-1 and BCL-XL in BRAFV600E CRC cell lines treated with (D) encorafenib (100 nM) or vemurafenib (5 μM) or (E) encorafenib (100 nM) plus cetuximab (10 μg/mL) for 6 h. β-tubulin was used as a loading control.
To determine if the limited effect of BRAF inhibitors on apoptosis extended to the clinically relevant encorafenib+cetuximab (Enc+Cet) combination, we treated the five BRAFV600E CRC cell lines with Enc+Cet. As observed for single agent BRAF inhibitor treatment, the combination regimen also induced minimal to modest (<25%) apoptosis across all cell lines (Fig. 1C), contrasting with the effect of the chemotherapeutic agent, irinotecan, which induced >50% apoptosis in two of five lines and >25% apoptosis in the remaining cell lines (Fig. 1C).
BRAF inhibitors prime BRAFV600E CRC cells for apoptosis
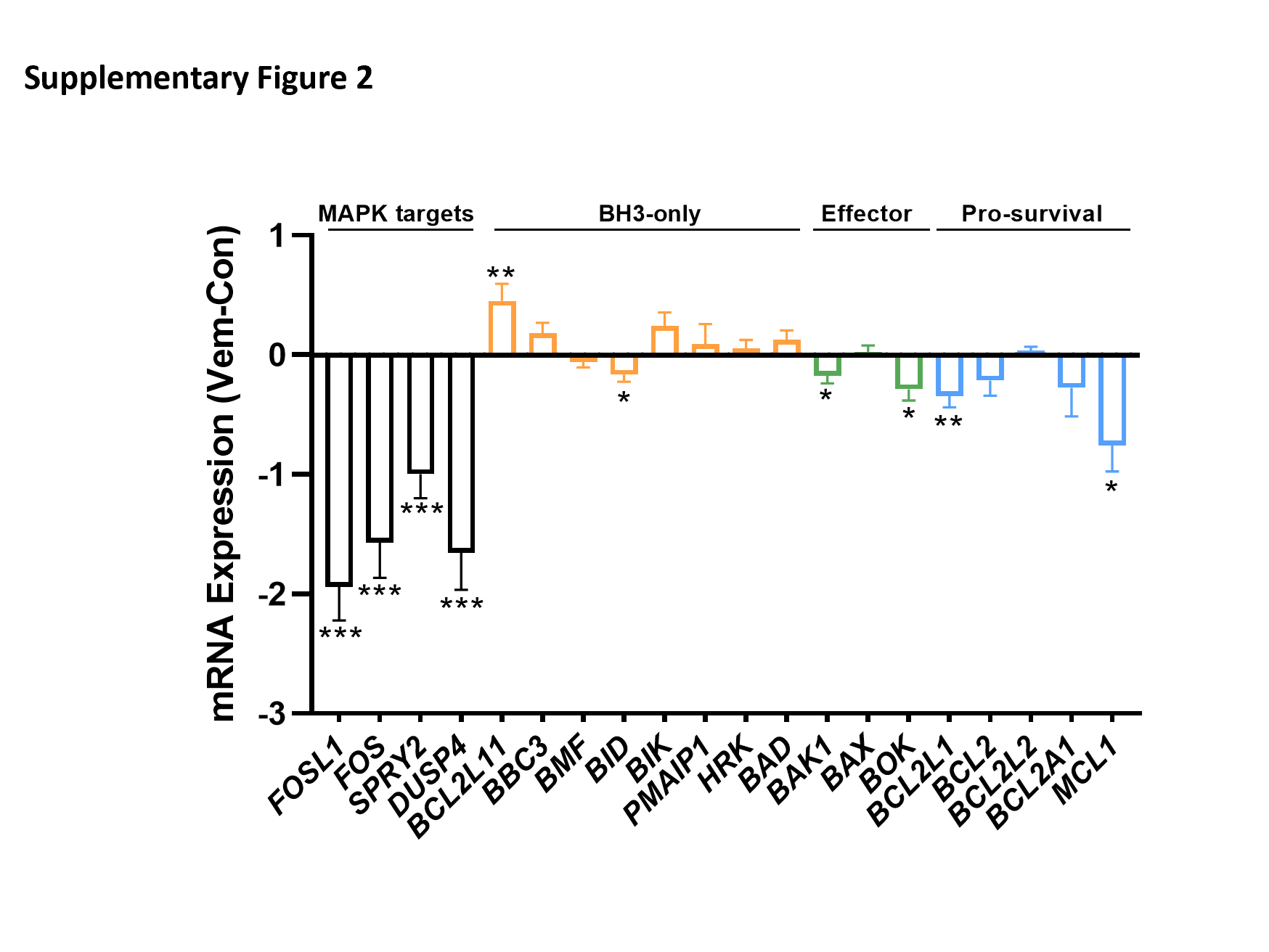
Inhibition of MAPK/ERK signalling has been shown to alter the apoptotic rheostat in tumour cells by inducing expression of several pro-apoptotic proteins and suppressing expression of several pro-survival proteins of the intrinsic apoptotic pathway [15, 27]. To determine if BRAF inhibitors induce similar effects in BRAFV600E CRC cells, we profiled gene expression changes in 12 BRAFV600E CRC cell lines treated with vemurafenib. As expected, vemurafenib treatment significantly reduced expression of the MAPK target genes FOSL1, FOS, SPRY2 and DUSP4 (Supplementary Fig. 2). Vemurafenib also significantly induced expression of the pro-apoptotic gene BCL2L11 (BIM) and repressed expression of the pro-survival gene MCL1, as well as a more modest suppression of BCL2L1 (BCL-XL) (Supplementary Fig. 2). Expression of the pro-apoptotic gene BID and the apoptosis effector genes BAK1 and BOK were also downregulated by vemurafenib treatment, although the magnitude of repression was relatively modest. The induction of BIM and repression of MCL-1 were further confirmed at the protein level in the majority of the BRAFV600E CRC cell lines tested (Fig. 1D, Supplementary Fig. 2). The induction of BIM and suppression of MCL-1 protein was also observed following treatment with Enc+Cet (Fig. 1E). Collectively, these findings suggest that despite their relatively modest effects on apoptosis, BRAF inhibitors may prime BRAFV600E CRC cell lines to undergo apoptosis by altering the apoptotic rheostat.
BRAFV600E CRCs express high levels of MCL-1 and BCL-XL
As apoptosis is regulated by the balance of expression between pro-apoptotic and pro-survival proteins, we postulated that the magnitude of BIM induction and MCL-1 repression induced by BRAF inhibition may be insufficient to trigger apoptosis (BAX/BAK activation), if CRC cells express high levels of one or more pro-survival proteins. To investigate this, we interrogated the TCGA COAD RNA-seq dataset [28], as well as RNA-seq data from a large panel of CRC cell lines [29] to examine the basal expression levels of the major pro-survival proteins in CRCs. These analyses revealed higher expression of MCL1 and BCL2L1 compared to BCL2, BCL2L2 and BCL2A1 in both primary CRCs (Fig. 2A) and CRC cell lines (Fig. 2B). Analysis of the BRAFV600E subset of primary CRCs and CRC cell lines within these datasets also revealed the same overall trend of high basal mRNA expression of MCL1 and BCL2L1 compared to other pro-survival proteins (Fig. 2C, D, blue). Furthermore, comparison of BRAFV600E versus BRAFWT CRCs in these datasets revealed that MCL1 mRNA expression was also significantly higher in BRAFV600E tumours (Figs. 2C and 2D, blue). This finding suggests that MCL-1 is further upregulated by mutant BRAF signalling, which is also consistent with the repression of MCL-1 following BRAF inhibitor treatment (Fig. 1D, S2). BCL2 and BCL2A1 levels were also higher in BRAFV600E compared to BRAFWT tumours (Fig. 2C, D, blue), although the overall level of expression was considerably lower than that of MCL1 and BCL2L1. Collectively these findings reveal that BCL-XL and MCL-1 are the most highly expressed pro-survival factors expressed in BRAF-mutant CRCs, and that MCL-1 is further upregulated in BRAFV600E tumours.
Fig. 2. CRCs express high basal expression of BCL-XL and MCL-1.

A Basal mRNA expression levels of pro-survival genes in human CRCs. Data obtained from the COAD cohort profiled by the cancer genome atlas (TCGA)(n = 266). B Basal mRNA expression of pro-survival genes in CRC cell lines. mRNA expression was extracted from n = 58 CRC cell lines profiled by RNA-seq analysis as previously reported [29]. C Relative mRNA expression of pro-survival genes in primary BRAFWT versus BRAFV600E CRCs. Data obtained from the TCGA COAD cohort (WT, n = 236, BRAF-mutant, n = 30). D Relative mRNA expression of pro-survival genes in BRAFWT versus BRAFV600E CRC cell lines. mRNA expression was extracted from n = 47 BRAFWT and n = 11 BRAFV600E CRC cell lines profiled by RNA-seq analysis by [29].
Combining BRAF inhibitors with a BCL-XL inhibitor induces extensive apoptosis in BRAFV600E CRC cell lines
To determine if the high basal levels of MCL-1 and BCL-XL in CRC cells may restrict their sensitivity to BRAF inhibitor (encorafenib) induced apoptosis, we tested the effect of combining encorafenib with BH3 mimetics targeting either MCL-1 (S63845), BCL-XL (A-1331852), or BCL-2 (ABT-199) on apoptosis in BRAFV600E CRC cell lines. Combining encorafenib with an MCL-1 inhibitor induced only a modest increase in apoptosis, surpassing 20% in only one (LIM2551) of the five cell lines tested (Fig. 3A). Comparatively, combination treatment of encorafenib with a BCL-XL inhibitor enhanced apoptosis to >50% in 3/5 BRAFV600E cell lines (Fig. 3B). Finally, consistent with the low levels of BCL-2 expression in BRAFV600E CRC cells and thus their likely independence on BCL-2 for survival, combination treatment with encorafenib and a BCL-2 inhibitor failed to enhance apoptosis to >25% in any of the cell lines (Fig. 3C).
Fig. 3. Effect of combining BRAF inhibitors with BH3 mimetics on apoptosis in BRAFV600E CRC cell lines.

BRAFV600E CRC cell lines were treated with encorafenib (100 nM) alone and in combination with either (A) the MCL-1 inhibitor S63845 (1 μM), (B) the BCL-XL inhibitor A-1331852 (10 nM) or (C) the BCL-2 inhibitor ABT-199 (1 μM) for 72 h and apoptosis induction determined by PI staining and FACS analysis. Values shown are mean ± SEM from a representative experiment performed in technical triplicates. Differences between groups were compared using one-way ANOVA with Tukey’s multiple comparison testing; ns (not significant), *(p ≤ 0.05), ***(p ≤ 0.001) and ****(p ≤ 0.0001).
Notably, HT29 and RKO cells were refractory to all BH3 mimetic + encorafenib combinations. While this may be due to the relatively low level of encorafenib-induced expression of pro-apoptotic proteins (e.g BIM) in these lines (Fig. 1D), examination of basal mRNA expression of pro-survival factors revealed that RKO cells also expressed higher levels of BCL2 and BCL2A1 relative to the other cell lines, which may also contribute to its relative drug resistance (Supplementary Fig. 3).
Finally, to confirm these findings in longer term assays, we assessed the efficacy of these drug combinations in clonogenic survival assays in LIM2551 cells over 2 weeks. Consistent with the findings of the apoptosis assays, combining low-dose encorafenib with the BCL-XL inhibitor A-1331852 further reduced colon forming capacity in LIM2551 cells, whereas combining encorafenib with S63845 or ABT-199 had minimal additional effect (Supplementary Fig. S4A, B).
These findings reveal that combining a BRAF inhibitor with a BCL-XL inhibitor is a highly effective means of inducing apoptosis in BRAFV600E CRC cells.
BIM is required for BRAF + BCL-XL inhibitor-induced apoptosis
As BRAF inhibitor treatment also significantly enhanced BIM expression in BRAFV600E CRC cells (Fig. 1, Supplementary Fig. 2), we next assessed whether BIM was required for BRAF + BCL-XL inhibitor-induced apoptosis. To address this, we engineered a doxycycline-inducible CRISPR/Cas9 cell line to delete BIM in BRAFV600E COLO 201 and LIM2551 cells, and confirmed effective deletion of BIM following doxycycline treatment (Fig. 4A/Supplementary Fig. 5A). Deletion of BIM significantly attenuated BRAF + BCL-XL inhibitor-induced apoptosis in COLO201 cells, as assessed by propidium iodide staining (Fig. 4B) and cleaved caspase 3 levels (Fig. 4C). A rescue of the apoptotic effect was also observed in LIM2551 cells albeit to a lesser extent (Supplementary Fig. 5B). While this finding demonstrates a direct role for BIM induction in BRAF + BCL-XL inhibitor-induced apoptosis, it also suggests that other factors are likely to be involved.
Fig. 4. BRAF + BCL-XL inhibitor induced apoptosis requires BIM induction.

A Validation of CRISPR-mediated deletion of BIM in COLO 201 cells. Cells were maintained in 1 μg/mL of doxycycline (+DOX) to induce BIM deletion and subsequently treated with encorafenib (100 nM) for 6 h to induce BIM expression. BIM expression [extra-long (BIMEL), Long (BIML) and short (BIMS) forms] was assessed by western blot with β-tubulin used as a loading control. B Control and BIM-deleted COLO 201 cells were treated with encorafenib (100 nM) and A-1331852 (10 nM) alone and in combination for 72 h and apoptosis determined by PI staining and FACS analysis. Values shown are mean ± SEM from a representative experiment performed in technical triplicate. Similar results were obtained in a second independent experiment. One-Way ANOVA, with Tukey’s multiple comparison testing; ****(p≤0.0001). C Western blot analysis of cleaved caspase 3 (Cl. Caspase 3) induction following treatment of control and BIM-deleted COLO 201 cells with encorafenib (100 nM) plus A-1331852 (10 nM) for 12 h. β-tubulin was used as a loading control.
Combining encorafenib with next generation BCL-XL inhibitors additively enhances apoptosis in BRAFV600E CRC cells in vitro and suppresses tumour growth in vivo
While our findings demonstrate that combining BCL-XL inhibitors with BRAF inhibitors robustly induces apoptosis in BRAFV600E CRC cells in vitro, the clinical use of BCX-XL inhibitors is currently limited by their induction of thrombocytopenia [30], an on-target toxicity driven by the high dependency of platelets on BCL-XL for their survival [31]. Therefore, we sought to determine the efficacy of combining BRAF inhibitors with two recently emergent strategies for BCL-XL inhibition, designed to circumvent this limitation. First, we utilized DT2216, a proteolysis-targeting chimera (PROTAC) to target BCL-XL for degradation. DT2216 is a conjugate of the BH3 mimetic ABT263 and a ligand for the Von Hippel-Lindau (VHL) E3 ubiquitin ligase, which has minimal expression in platelets [32]. The capacity of DT2216 to target BCL-XL for degradation in a dose-dependent manner was validated in the panel of five BRAFV600E cell lines (Fig. 5A). Consistent with previous results, combinatorial treatment of DT2216 with encorafenib significantly enhanced apoptosis in a dose-dependent manner compared to either agent alone in COLO201, LIM2551 and LIM2405 cells whilst RKO and HT29 remained refractory to treatment (Fig. 5B).
Fig. 5. Effect of combining the BCL-XL -targeting PROTAC, DT2216, with encorafenib in BRAFV600E CRC cells.

A Western blot analysis of BCL-XL expression following treatment of BRAFV600E CRC cells with DT2216 (10, 100 and 1000 nM) for 24 h. β-tubulin was used as a loading control. B BRAFV600E CRC cells were treated with encorafenib (100 nM) and DT2216 (10, 100 or 1000 nM) alone and in combination for 72 h and apoptosis determined by PI staining and FACS analysis. Values shown are mean ± SEM from 3 independent experiments, except for LIM2405 (values shown are mean ± SEM from 2 independent experiments). Differences were compared using one-way ANOVA with Tukey’s multiple comparison testing; ns (not significant), *(p ≤ 0.05), **(p ≤ 0.01), ***(p ≤ 0.001), and ****(p ≤ 0.0001).
Second, we determined the apoptotic effect of combining encorafenib with the recently developed BCL2/BCLXL inhibitor AZD4320 [33], and its drug dendrimer conjugate AZD0466, designed to minimise toxicity by gradually releasing AZD4320 by hydrolysis thus resulting in lower peak plasma levels, as well as through increased dendrimer retention within the tumour [21]. Combination treatment of COLO 201 cells with encorafenib and AZD4320 in vitro, significantly enhanced apoptosis compared to either agent alone (Fig. 6A). To determine the efficacy of this combination in vivo, COLO 201 cells were grown as xenografts and mice treated with encorafenib (20 mg/kg, bid, oral gavage) and AZD0466 (103 mg/kg, once weekly, tail vein injection) alone or in combination for 22 days. The combination significantly suppressed tumour growth compared to vehicle control or either agent alone and also resulted in tumour regression from treatment commencement (Fig. 6B–D). No significant change in body weight was observed with combination treatment over the duration of the experiment (Fig. 6E), collectively demonstrating that combining a BRAF inhibitor with the BCL-XL inhibitor AZD0466 may be an effective and tolerable treatment for BRAFV600E CRC. Consistent with our in vitro findings, immunohistochemical staining of resected tumours revealed an increase in BIM expression in mice treated with Encorafenib alone or in combination with AZD0466 (Fig. 6F, G). Somewhat surprisingly, analysis for apoptosis using TUNEL staining failed to reveal a significant difference between treatment groups, which may be due to tumour resection occurring 72-h post the final AZD0466 treatment, a period in which apoptotic cells may have been cleared from the tumour (data not shown). Collectively these findings demonstrate that combining a BRAF inhibitor with the BCL-XL inhibitor AZD0466 may be an effective and tolerable treatment for BRAFV600E CRC.
Fig. 6. Anti-tumour effects of encorafenib plus AZD4320/AZD0466 on BRAFV600E CRC cells in vitro and in vivo.

A COLO 201 cells were treated with encorafenib (100 nM) and AZD4320 (500 nM) alone and in combination for 72 h in vitro and apoptosis determined by PI staining and FACS analysis. Values shown are mean ± SEM from 3 independent experiments. Differences were compared using one-way ANOVA with Tukey’s multiple comparison testing; ****(p ≤ 0.0001). B Effect of encorafenib and AZD0466 on the growth of COLO 201 xenografts. NOD scid gamma mice (n = 8 per group) were subcutaneously injected with 2 × 106 COLO 201 cells into the left and right flanks (n = 16 tumours per group except the combination treatment group where n = 14). Mice were then randomised to receive either vehicle control, encorafenib (20 mg/kg, b.i.d, og), AZD0466 (103 mg/kg, once weekly, I.V) or the combination, for 22 days. Tumour volume was determined by caliper measurements every second day and normalised to the volume at day 1 of treatment. Values shown are mean ± SEM. (C) Weight (g) and (D) representative images of excised tumours at experimental endpoint. Hash (#) depicts one less mouse (two less tumours) for the combination treatment group. E Relative change in mouse body weight from treatment commencement. Values shown are mean ± SEM. F Representative images of immunohistochemical staining of BIM in resected tumours and corresponding annotation performed using HALO software depicting areas of low (yellow), moderate (orange) and strong (red) BIM staining. G Quantification of BIM staining (percentage of positively stained tumour cells) in resected tumours. Values shown are mean ± SEM. Differences were compared using one-way ANOVA with Tukey’s multiple comparison testing; *(p≤0.05), **(p≤0.01), ***(p≤0.001) and ****(p≤0.0001).
Discussion
Metastatic BRAFV600E CRC carries an extremely poor prognosis and is in urgent need of better treatments. In this regard, a recent breakthrough was the approval of encorafenib plus cetuximab for treatment of chemorefractory BRAFV600E metastatic CRC patients [13]. However, objective responses only occur in ~20% of patients and overall survival is only increased by ~4 months, warranting the search for strategies to enhance the efficacy of this treatment regime. Herein, we demonstrate that a limitation of BRAF inhibitors when used either alone or in combination with EGFR inhibitors, is that they fail to effectively induce apoptosis in BRAFV600E CRC cell lines. Notably, despite their failure to induce apoptosis, we found that BRAF inhibitors induced expression of the pro-apoptotic protein BIM and repressed expression of the pro-survival protein MCL-1, suggesting these agents may prime BRAFV600E cells to undergo apoptosis.
An important element of this study was the investigation of the mechanism underpinning apoptotic resistance of BRAFV600E CRC cells to BRAF inhibitors, which was investigated by transcriptomic profiling of basal expression levels of pro-survival genes. This analysis revealed high basal expression of both MCL-1 and BCL-XL, and lower expression of BCL-2, BCL-w and BFL1 in all CRCs including the BRAFV600E subset. Notably, despite the high basal expression of both BCL-XL and MCL-1, a greater enhancement of apoptosis was observed when encorafenib was combined with a BCL-XL inhibitor compared to an MCL-1 inhibitor. While this finding may reflect differences in target inhibition between current BCL-XL and MCL-1 inhibitors, a further explanation may be the capacity of the encorafenib+BCL-XL combination to inhibit both BCL-XL as well as MCL-1, the latter through encorafenib-mediated transcriptional and/or post-translational effects (Fig. 1). Comparatively, when encorafenib is combined with an MCL-1 inhibitor, the apoptotic threshold may remain elevated due to the sustained presence of high BCL-XL. In addition, we also demonstrate a key role for BIM induction in encorafenib+BCL-XL inhibitor-induced apoptosis. The encorafenib-induced transcriptional and/or post-translational stabilization of BIM [34–37], and suppression of MCL-1 expression, combined with a BCL-XL inhibitor, may result in sufficient levels of BAX and BAK being released from pro-survival proteins allowing BIM to activate BAX and BAK, and initiate apoptosis [38].
While these findings reveal that combining BRAF and BCL-XL inhibitors represents a promising therapeutic approach for BRAFV600E CRC, the clinical use of BCL-XL inhibitors is currently limited by their on-target toxicity of thrombocytopenia [30, 31]. We found that combining encorafenib with the BCL-XL degrading PROTAC DT2216 and AZD0466, a novel BCL-2/BCL-XL inhibitor-dendrimer conjugate [21, 39], significantly enhanced apoptosis. Furthermore, combination of encorafenib with AZD0466 significantly reduced tumour growth in vivo and was generally well tolerated in mice. AZD0466 is currently being tested in phase I/II clinical trials for the treatment of haematological malignancies (ClinicalTrials.gov identifier: NCT05205161 and NCT04865419), and our findings suggest that combining encorafenib with AZD0466 may also be a promising approach for treating BRAFV600E tumours.
The concept of combining MAPK pathway inhibitors with BH3 mimetics has been explored in colorectal cancer and other tumour types. Specifically, a pre-clinical study in BRAFV600E melanoma demonstrated synergistic induction of apoptosis when BRAF or MEK inhibitors were combined with a MCL-1 inhibitor, which aligned with MCL-1 being the predominant pro-survival protein expressed in melanoma cells [17]. A further study in KRAS-mutant NSCLC and other KRAS-mutant solid tumour cell lines also reported synergistic cell killing when MAPK pathway inhibitors were combined with BH3 mimetics [40]. Excitingly, these studies have prompted the initiation of a clinical trial of the MEK inhibitor trametinib in combination with the BCL-2/BCL-XL inhibitor navitoclax in patients with advanced or metastatic solid tumours harbouring KRAS or NRAS mutations (Clinicaltrials.gov identifier: NCT02079740).
The high expression of multiple pro-survival proteins in CRC cells suggests alternative strategies for inducing apoptosis could be through the combined inhibition of BCL-XL and MCL-1. However, pre-clinical studies of these combinations have demonstrated acute liver toxicity, which is consistent with the cooperative role of BCL-XL with MCL-1 in hepatocyte survival [41, 42]. The current approach of using a targeted therapy (BRAF inhibitor) in combination with a BCL-XL inhibitor therefore has the potential to minimize these toxicities, while providing the added benefit of inducing pro-apoptotic proteins such as BIM.
In summary, our findings demonstrate that BRAF inhibitors alone and in combination with the EGFR inhibitor cetuximab fail to induce extensive levels of apoptosis in BRAFV600E CRC cells. Importantly, we reveal that this can be overcome by combining a BRAF inhibitor with a BCL-XL inhibitor, in both in vitro and in vivo models of BRAFV600E CRC, suggesting this combination regimen is worthy of clinical validation.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This project was supported by a Tour de Cure Vic Discovery research grant and the Operational Infrastructure Support Program, Victorian Government, Australia. LJ Jenkins was supported by La Trobe University Australian Postgraduate Awards. JM Mariadason (1046092) and OM Sieber (1136119) were supported by National Health and Medical Research Council (NHMRC) Senior Research Fellowships. EFL and FC were supported by Victorian Cancer Agency Mid and Early Career Fellowships, respectively (MCR19045, ECRF21035).
Author contributions
LJ Jenkins: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing-original draft, writing-reviewing and editing. IY Luk: Data curation and validation. F Chionh: Data curation. T Tan: Data Curation and editing. K Needham: Data curation. J Ayton: Data Curation and analysis. CM Reehorst: Formal data analysis. N Vukelic: Data curation and editing. OM Sieber: Resources and editing. D Mouradov: Data analysis. P Gibbs: Resources and editing. DS Williams: Formal data analysis and resources. NC Tebbutt: Conceptualization and supervision. J Desai: Conceptualization and editing. F Hollande: Resources and editing. AS Dhillon: Conceptualization, supervision and editing. EF Lee: Resources, writing-review and editing. D Merino: Resources, editing. WD Fairlie: Resources, writing-review and editing. JM Mariadason: Conceptualization, supervision, data curation, formal analysis, investigation, visualization, methodology, writing-original draft, writing-reviewing and editing.
Data availability
Raw data for this study were generated at the Olivia Newton-John Cancer Research Institute, Melbourne, Australia. The data/datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
AZD0466 and AZD4320 were provided by AstraZeneca.
Ethics approval
The mouse experiment performed was approved by the Austin Health Animal Ethics Committee (A2018_05584). Human ethics is not applicable to this study.
Footnotes
Edited by Professor Massimiliano Agostini
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-024-06478-z.
References
- 1.Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50:307–12. doi: 10.1002/gcc.20854. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 3.Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B. BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol Cancer. 2006;5:2. doi: 10.1186/1476-4598-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seligmann JF, Fisher D, Smith CG, Richman SD, Elliott F, Brown S, et al. Investigating the poor outcomes of BRAF-mutant advanced colorectal cancer: analysis from 2530 patients in randomised clinical trials. Ann Oncol. 2017;28:562–8. doi: 10.1093/annonc/mdw645. [DOI] [PubMed] [Google Scholar]
- 5.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–9. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 6.Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14:749–59. doi: 10.1016/S1470-2045(13)70163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price TJ, Hardingham JE, Lee CK, Weickhardt A, Townsend AR, Wrin JW, et al. Impact of KRAS and BRAF gene mutation status on outcomes from the phase III AGITG MAX trial of capecitabine alone or in combination with bevacizumab and mitomycin in advanced colorectal cancer. J Clin Oncol. 2011;29:2675–82. doi: 10.1200/JCO.2010.34.5520. [DOI] [PubMed] [Google Scholar]
- 8.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33:4032–8. doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 11.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–35. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381:1632–43. doi: 10.1056/NEJMoa1908075. [DOI] [PubMed] [Google Scholar]
- 13.Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON sStudy. J Clin Oncol. 2021;39:273–84. doi: 10.1200/JCO.20.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenkins LJ, Luk IY, Fairlie WD, Lee EF, Palmieri M, Schoffer KL, et al. Genotype-tailored ERK/MAPK pathway and HDAC inhibition rewires the apoptotic rheostat to trigger colorectal cancer cell death. Mol Cancer Ther. 2023;22:52–62. doi: 10.1158/1535-7163.MCT-22-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohrbeck L, Gong JN, Lee EF, Kueh AJ, Behren A, Tai L, et al. Hepatocyte growth factor renders BRAF mutant human melanoma cell lines resistant to PLX4032 by downregulating the pro-apoptotic BH3-only proteins PUMA and BIM. Cell Death Differ. 2016;23:2054–62. doi: 10.1038/cdd.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao Y, Aplin AE. BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ. 2012;19:2029–39. doi: 10.1038/cdd.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sale MJ, Minihane E, Monks NR, Gilley R, Richards FM, Schifferli KP, et al. Targeting melanoma’s MCL1 bias unleashes the apoptotic potential of BRAF and ERK1/2 pathway inhibitors. Nat Commun. 2019;10:5167. doi: 10.1038/s41467-019-12409-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunda V, Sarosiek KA, Brauner E, Kim YS, Amin S, Zhou Z, et al. Inhibition of MAPKinase pathway sensitizes thyroid cancer cells to ABT-737 induced apoptosis. Cancer Lett. 2017;395:1–10. doi: 10.1016/j.canlet.2017.02.028. [DOI] [PubMed] [Google Scholar]
- 19.Luo MJ, Palmieri M, Riffkin CD, Sakthianandeswaren A, Djajawi TM, Hirokawa Y, et al. Defining the susceptibility of colorectal cancers to BH3-mimetic compounds. Cell Death Dis. 2020;11:735. doi: 10.1038/s41419-020-02815-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, Kelly GL. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. 2022;22:45–64. doi: 10.1038/s41568-021-00407-4. [DOI] [PubMed] [Google Scholar]
- 21.Patterson CM, Balachander SB, Grant I, Pop-Damkov P, Kelly B, McCoull W, et al. Design and optimisation of dendrimer-conjugated Bcl-2/xL inhibitor, AZD0466, with improved therapeutic index for cancer therapy. Commun Biol. 2021;4:112. doi: 10.1038/s42003-020-01631-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrel S, Sordat B, Merenda C. Establishment of a cell line (Co-115) from a human colon carcinoma transplanted into nude mice. Cancer Res. 1976;36:3978–84. [PubMed] [Google Scholar]
- 23.Willson JK, Bittner GN, Oberley TD, Meisner LF, Weese JL. Cell culture of human colon adenomas and carcinomas. Cancer Res. 1987;47:2704–13. [PubMed] [Google Scholar]
- 24.Suardet L, Gaide AC, Calmes JM, Sordat B, Givel JC, Eliason JF, et al. Responsiveness of three newly established human colorectal cancer cell lines to transforming growth factors beta 1 and beta 2. Cancer Res. 1992;52:3705–12. [PubMed] [Google Scholar]
- 25.McBain JA, Weese JL, Meisner LF, Wolberg WH, Willson JK. Establishment and characterization of human colorectal cancer cell lines. Cancer Res. 1984;44:5813–21. [PubMed] [Google Scholar]
- 26.Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10:1422–32. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Kawakami H, Huang S, Pal K, Dutta SK, Mukhopadhyay D, Sinicrope FA. Mutant BRAF upregulates MCL-1 to confer apoptosis resistance that is reversed by MCL-1 antagonism and cobimetinib in colorectal cancer. Mol Cancer Ther. 2016;15:3015–27. doi: 10.1158/1535-7163.MCT-16-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014;74:3238–47. doi: 10.1158/0008-5472.CAN-14-0013. [DOI] [PubMed] [Google Scholar]
- 30.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–59. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–86. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 32.Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25:1938–47. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balachander SB, Criscione SW, Byth KF, Cidado J, Adam A, Lewis P, et al. AZD4320, a dual inhibitor of Bcl-2 and Bcl-xL, induces tumor regression in hematologic cancer models without dose-limiting thrombocytopenia. Clin Cancer Res. 2020;26:6535–49. doi: 10.1158/1078-0432.CCR-20-0863. [DOI] [PubMed] [Google Scholar]
- 34.Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–48. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–25. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–6. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 37.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–93. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 38.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 39.Arulananda S, O’Brien M, Evangelista M, Jenkins LJ, Poh AR, Walkiewicz M, et al. A novel BH3-mimetic, AZD0466, targeting BCL-XL and BCL-2 is effective in pre-clinical models of malignant pleural mesothelioma. Cell Death Discov. 2021;7:122. doi: 10.1038/s41420-021-00505-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology. 2009;50:1217–26. doi: 10.1002/hep.23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weeden CE, Ah-Cann C, Holik AZ, Pasquet J, Garnier JM, Merino D, et al. Dual inhibition of BCL-XL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene. 2018;37:4475–88. doi: 10.1038/s41388-018-0268-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data for this study were generated at the Olivia Newton-John Cancer Research Institute, Melbourne, Australia. The data/datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.