

Abstract
How lipidome changes support CD8+ effector T (Teff) cell differentiation is not well understood. Here we show that, although naive T cells are rich in polyunsaturated phosphoinositides (PIPn with 3–4 double bonds), Teff cells have unique PIPn marked by saturated fatty acyl chains (0–2 double bonds). PIPn are precursors for second messengers. Polyunsaturated phosphatidylinositol bisphosphate (PIP2) exclusively supported signaling immediately upon T cell antigen receptor activation. In late Teff cells, activity of phospholipase C-γ1, the enzyme that cleaves PIP2 into downstream mediators, waned, and saturated PIPn became essential for sustained signaling. Saturated PIP was more rapidly converted to PIP2 with subsequent recruitment of phospholipase C-γ1, and loss of saturated PIPn impaired Teff cell fitness and function, even in cells with abundant polyunsaturated PIPn. Glucose was the substrate for de novo PIPn synthesis, and was rapidly utilized for saturated PIP2 generation. Thus, separate PIPn pools with distinct acyl chain compositions and metabolic dependencies drive important signaling events to initiate and then sustain effector function during CD8+ T cell differentiation.
Metabolic reprogramming during T cell activation is required to support the increased biosynthetic demands of CD8+ Teff cell proliferation, effector function and epigenetic remodeling needed to enforce differentiation1,2. Metabolites also directly modulate the activity of signaling pathways (for example, amino acids regulate mammalian target of rapamycin (mTOR), ATP/AMP regulates AMP-activated protein kinase (AMPK), phosphoenolpyruvate regulates sarco/endoplasmic reticulum Ca2+-ATPase (SERCA))3, tying T cell signaling to metabolite acquisition. While changes in polar metabolites during CD8+ T cell activation have been studied4–7, less is known about lipid metabolism and how polar metabolites impact lipid signaling.
Phosphatidylinositol (PI) is one of eight phospholipids of the phosphoinositide (PIPn) family that differ in their inositol headgroup that can be unphosphorylated (PI), or phosphorylated at one or more specific sites (PI(3)P, PI(4)P, PI(5)P, PI(4,5)P2, PI(3,4)P2, PI(3,5)P2, PI(3,4,5) P3). Mammalian cells have PIPn with a dominant acyl chain composition (38:4), in contrast to most other phospholipids that display a range of chain lengths and saturation8. In 38:4 PIPn, the 38 denotes the numbers of carbons in the acyl chains, and 4 denotes the double bonds, which decrease with increasing saturation. In mammalian cells, 19 kinases and 29 phosphatases dynamically interconvert family members to regulate PIPn signaling9. The introduction of ‘new’ PIPn, which increases the net PIPn pool, occurs through de novo synthesis of unphosphorylated PI. PIP2 and PIP3 represent less than 1% of membrane phospholipids, but are essential for many cellular processes, including cytoskeletal organization, phagocytosis, endocytosis and exocytosis10. T cell antigen receptor (TCR) ligation initiates rapid PIPn interconversion (phosphorylation and dephosphorylation) and salvage, marked by phospholipase C (PLC)-mediated PIP2 hydrolysis and PIP2 phosphorylation to PIP3 by phosphoinositide 3-kinase (PI3K)11. Phospholipase C-γ1 (PLCγ1) is the predominant isoform in T cells and its activity is regulated by the proximal signaling complex that is formed upon TCR ligation10. PIP2 hydrolysis generates the membrane lipid diacylglycerol (DAG) and soluble inositol triphosphate (IP3)12. DAG recruits and activates several proteins, including Ras guanyl nucleotide-releasing proteins, which in turn activate Ras, the kinase Raf and the downstream MEK1/2–ERK1/2 cascade that promotes gene expression important for Teff cell activation and survival13. IP3 binds endomplasmic reticulum (ER) membrane receptors to release stored Ca2+ into the cytosol, initiate store-operated calcium entry, activate downstream target enzymes and transcription factors and regulate remodeling of the filamentous actin (F-actin) cytoskeleton required for T cell migration14,15.
It has been a longstanding question whether discrete PIP2 pools are essential for specific cellular effects16–18. Here we show that Teff cells are equipped with a distinct PIP2 pool with saturated acyl chains (we refer to PIPn with 0–2 double bonds as saturated) that differs from the polyunsaturated PIP2 pool of unactivated T cells (we refer to PIPn with 3–4 double bonds as polyunsaturated). We found that specific T cell programs utilize these different PIPn to drive signaling events. Saturated PIPn mediate Teff cell signaling, while polyunsaturated PIPn exclusively mediate signaling early after T cell activation. Our data also show that glucose is the major substrate of Teff cell de novo PIPn synthesis, implicating PIPn as an important link between extracellular glucose availability, glycolytic metabolism and Teff cell signaling and function.
Results
CD8+ effector T cells synthesize PIPn with saturated acyl chains
We stimulated mouse CD8+ T cells with anti-CD3/CD28 plus interleukin (IL)-2 for 3 d in vitro and compared them to unstimulated CD8+ T cells cultured in IL-7 (Fig. 1a). Using liquid chromatography coupled with triple quadrupole tandem mass spectrometry (LC–QqQ–MS/MS), we detected over 300 lipids from 18 lipid subclasses in stimulated CD8+ T cells (Extended Data Fig. 1a) with a general increase in lipid content (Extended Data Fig. 1b). To potentially identify lipids with important biological functions beyond building biomass, we performed quantile normalization of lipid content before comparing changes in individual lipid species between unstimulated and stimulated CD8+ T cells (Extended Data Fig. 1b). We identified 11 PI species that increased in Teff cells (Fig. 1b) relative to unstimulated T cells—where PI 38:4 was the most abundant (Fig. 1c). PI 38:4 is the predominant PI species (>70%) in most mammalian cells8, and when corrected for cell size changes, it did not increase with stimulation (Fig. 1c). However, in Teff cells, PI species with 0–2 double bonds increased (Fig. 1d), shifting the overall PI composition toward saturated (Fig. 1e). Enhanced saturated PI synthesis in Teff cells increased total PI relative to other lipids (Extended Data Fig. 1c) and increased the PI concentration per cell (Fig. 1f). PIPn signaling is regulated by de novo PI synthesis that introduces new PIPn, and by PIPn phosphorylation or dephosphorylation of the inositol headgroup (Fig. 1g). Although we could not distinguish between PI(4,5)P2, PI(3,4) P2 and PI(3,5)P2, we found that Teff cells had an increase in total PIP2 species with 0–2 double bonds, which shifted the overall PIP2 composition toward saturated (Fig. 1h). Total polyunsaturated PIP2 species (with ≥3 double bonds) did not change between unstimulated T cells and Teff cells (Fig. 1h). Total PI(3,4,5)P3 (PIP3) species with 0–2 double bonds increased in Teff cells and shifted the PIP3 composition toward saturated (Fig. 1i); however, in contrast to PI and PIP2, the total amount of PIP3 decreased in Teff cells, as described for T cells 20 h after activation19.
Fig. 1 |. CD8+ effector T cells synthesize PIPn with saturated acyl chains.
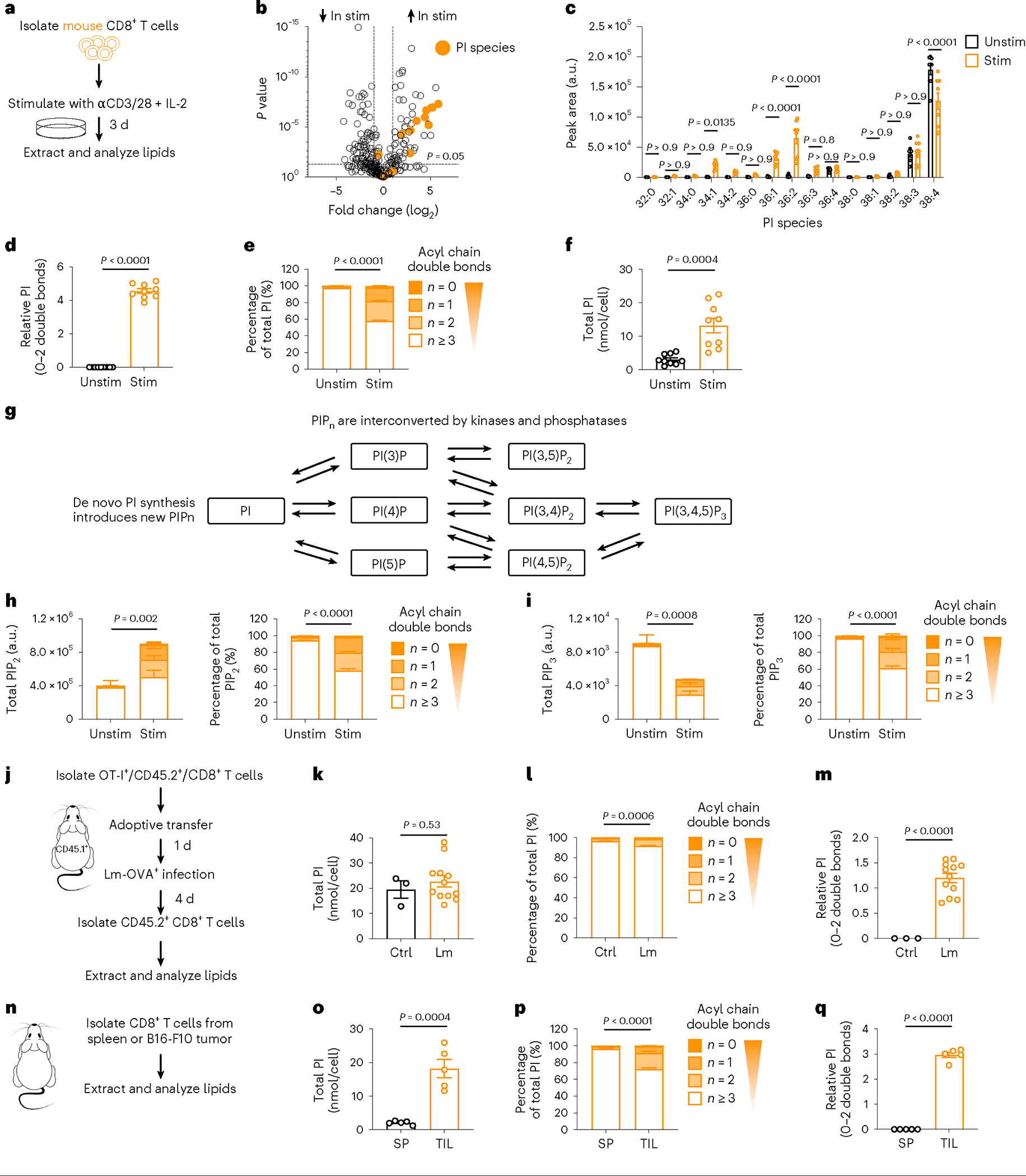
a, Experimental scheme. WT CD8+ T cells were stimulated (Stim) with anti-CD3, anti-CD28 and IL-2, or cultured with IL-7 (Unstim), for three d. b,c, Relative and absolute lipid intensities, respectively. n = 9 biologically independent samples from three independent experiments; ordinary one-way analysis of variance ANOVA (b) or two-way ANOVA (c) corrected for multiple comparisons (Sidak test). d, Relative intensity (log2 fold change) of saturated PI normalized to unstimulated n = 9 biologically independent samples from three independent experiments; one-sample t-test. e, Percentage PI saturation. n = 9 biologically independent samples from three independent experiments; unpaired two-tailed t-test comparing saturated PIs. f, Total PI concentration (nmol/cell). n = 9 biologically independent samples from three independent experiments; unpaired two-tailed t-test. g, PIPn synthesis and interconversion scheme. h, Total (left) and percentage (right) PIP2 saturation. n = 3 biologically independent samples, representative of two independent experiments; unpaired two-tailed t-test comparing the percentage of saturated PIP2; total polyunsaturated PIP2 = not significant. i, Total (left) and percentage (right) PIP3 saturation. n = 3 biologically independent samples, representative of two independent experiments; unpaired two-tailed t-test comparing the total or percentage saturated PIP3; total polyunsaturated PIP3 = P < 0.001. j, Experimental scheme. OT-I CD45.2+ CD8+ T cells (OT-I) were adoptively transferred into CD45.1+ C57BL/6 mice (1 × 106 cells per mouse). One day later mice were infected intravenously (i.v.) with LmOVA. Four d later, WT CD45.2+/CD8+ T cells were isolated from infected (Lm) or uninfected (CTRL) mice. k–m Total PI concentration (nmol/cell), percentage PI saturation and relative intensity (log2 fold change) of saturated PI normalized to CTRL. n = 3 Ctrl and 12 Lm biologically independent samples; unpaired two-tailed t-test: total PI, percentage saturated PI; one-sample t-test: relative saturated PI. n, Experimental scheme. Sex-matched C57BL/6 mice were injected in the right flank with 1 × 106 B16-F10-OVA cells. Tumor growth was measured up to an average of 7 mm in diameter, then CD8+ T cells were isolated from the spleen (SP) or tumor (TIL). o–q, Total PI concentration (nmol/cell), percentage PI saturation and relative intensity (log2 fold change) of saturated PI normalized to SP. n = 5 biologically independent samples; unpaired two-tailed t-test: total PI, percentage saturated PI; one-sample t-test: relative saturated PI. Error bars show the s.e.m. a.u., arbitrary units.
To confirm increased PI saturation in physiologically activated antigen-specific mouse CD8+ T cells, we adoptively transferred OVA-specific (OT-I) CD8+ T cells into recipient mice followed by infection with Listeria monocytogenes expressing OVA (LmOVA, Fig. 1j). CD8+ T cells from LmOVA-infected mice compared to uninfected OT-I mice did not have increased PI per cell (Fig. 1k); however, their overall PI composition shifted toward saturated (Fig. 1l), and the total saturated PI was elevated (Fig. 1m). We made a similar observation when we subcutaneously inoculated mice with B16-F10 melanoma and isolated CD8+ T cells either from the tumor or spleen when the tumors reached 7 mm in diameter (Fig. 1n). Compared to splenic CD8+ T cells, tumor-infiltrating lymphocytes (TILs) had an increased concentration of PI (Fig. 1o) with a shift toward saturated species (Fig. 1p,q). Thus, PIPn species with saturated acyl chains accumulate in mouse CD8+ Teff cells in vitro and in vivo.
We next stimulated human CD8+ T cells with anti-CD3/CD28 and IL-2 for 5 d in vitro and compared them to newly isolated unstimulated CD8+ T cells (Extended Data Fig. 1d). We observed increased saturated PI, and like mouse T cells, PI 38:4 was the predominant PI in human unstimulated T cells and it was not specifically enriched after activation (Extended Data Fig. 1e,f). PI with 0–2 double bonds increased (Extended Data Fig. 1g), shifting the overall PI composition toward saturated (Extended Data Fig. 1h). Total PI increased relative to other lipids (Extended Data Fig. 1i), aligning with an increased PI concentration (Extended Data Fig. 1j). Changes in the amount and degree of PIP2 and PIP3 saturation in human Teff cells also matched the observations in mouse Teff cells (Extended Data Fig. 1k,l). To investigate in vivo, we compared circulating CD8+ T cells from the blood of healthy donors or participants with acute Epstein–Barr virus (EBV) infection (Extended Data Fig. 1m). CD8+ T cells in participants with acute EBV infection extensively proliferated (Extended Data Fig. 1n)20, and their CD8+ T cells had elevated PI species with 0–2 double bonds (Extended Data Fig. 1o) that shifted the PI composition toward saturated (Extended Data Fig. 1p), with a similar PI concentration per cell (Extended Data Fig. 1q). Thus, PIPn species with saturated acyl chains accumulate in in vitro-generated and in vivo-generated mouse and human CD8+ Teff cells and results in saturated PIP2 accumulation.
De novo PIPn synthesis is essential for effector T cell fitness and function
In the final step of de novo PI synthesis, CDP-diacylglycerol-inositol 3-phosphatidyltransferase (CDIPT, Extended Data Fig. 2a)8 transfers free inositol to CDP-diacylglycerol (CDP-DAG). We cultured cells for the last 24 h of activation with vehicle (CTRL) or inostamycin, a CDIPT inhibitor (CDIPTi)21 (Fig. 2a). CDIPTi treatment depleted saturated PI species, but only partially decreased polyunsaturated PI (Fig. 2b). This indicated that saturated PI was mainly synthesized through the de novo pathway, while polyunsaturated PI could also be synthesized through remodeling lysophosphatidylinositol (LPI). Consistently, LPI decreased following CDIPTi treatment (Extended Data Fig. 2a,b). We observed a marked decrease in saturated PIP2, while the polyunsaturated PIP2 pool was unchanged (Fig. 2b). This demonstrated that CDIPT inhibition selectively targeted saturated PIP2 synthesis, and indicated preference for phosphorylation of saturated PI, despite an abundance of polyunsaturated PI (Fig. 2b).
Fig. 2 |. De novo PIPn synthesis is essential for effector T cell fitness and cytotoxic function.
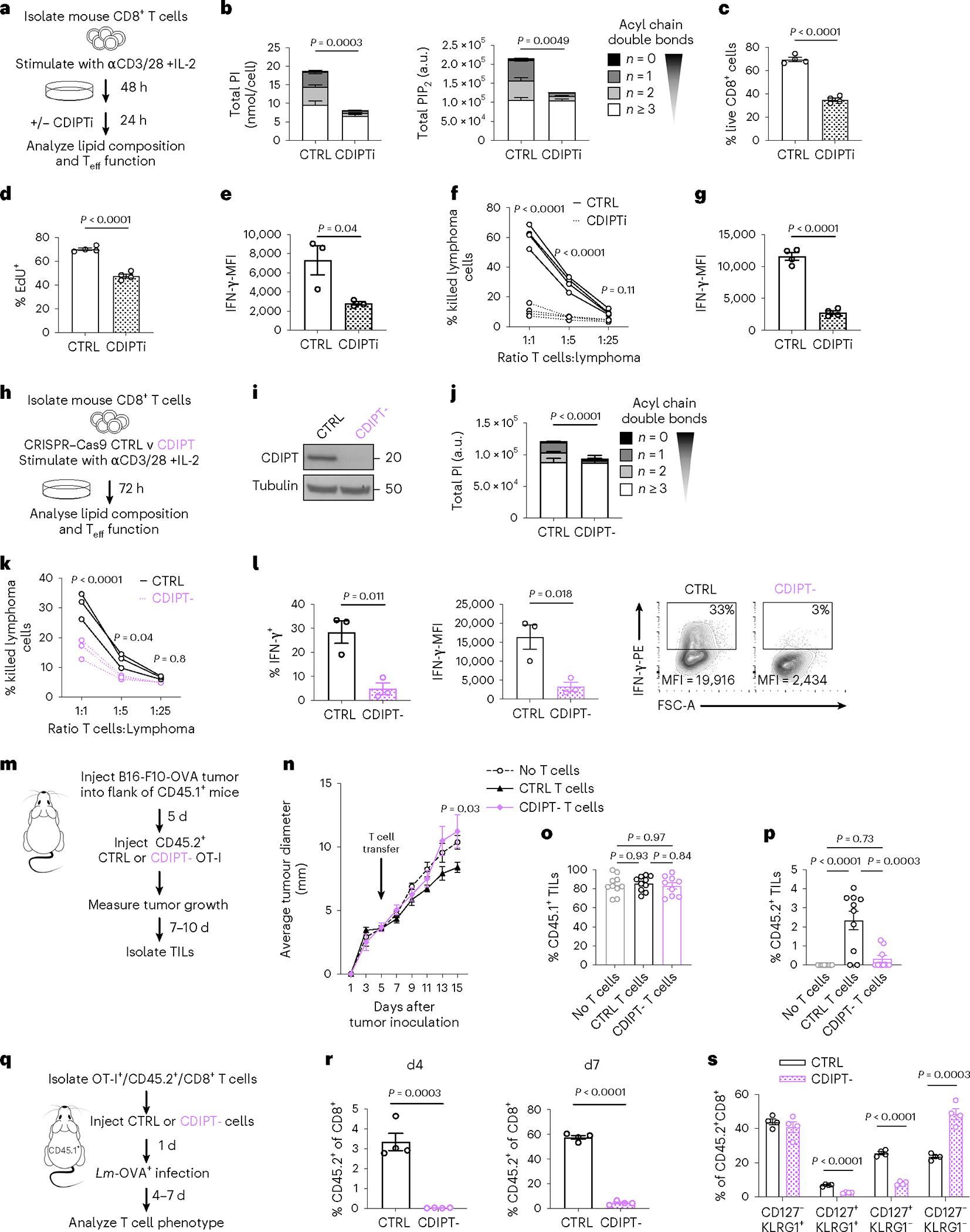
a, Experimental scheme. b, PI (left) or PIP2 (right) acyl chain saturation. n = 4 biologically independent samples, representative of three independent experiments for PI and one experiment for PIP2; unpaired two-tailed t-test comparing saturated PI or PIP2. c, Percentage of live, CD8+ T cells gated on Live/Dead-blue− and CD8-FITC+. d, Incorporation of EdU into newly synthesized DNA. e, Intracellular expression of IFN-γ quantified by flow cytometry. Cells were gated on Live/Dead-IR−, CD8-BV421+. In c–d, n=4 and in e, n = 3 biologically independent samples, representative of three independent experiments; unpaired two-tailed t-test. f,g, OT-I were activated by SIINFEKL peptide with IL-2 for 2 d, treated with CTRL or CDIPTi (plus IL-2), then co-cultured at the indicated ratio with Cell Trace Violet (CTV)-stained EL4-OVA for eight h. f, Percentage killing was determined by CTV+, Live/Dead-IR+ cells. g, Cells co-cultured at a 1:1 ratio in the presence of brefeldin A. IFN-γ expression in Live/Dead-IR− and CD8-APC+ cells. n = 4 biologically independent samples; two-way ANOVA corrected for multiple comparisons (Sidak test) (f) or unpaired two-tailed t-test (g). h, Experimental scheme. i, Protein expression of CDIPT. n = 3 biologically independent samples pooled into a single lane. j, PI saturation. n = 4 biologically independent samples; unpaired two-tailed t-test on saturated PI. k,l, EL4-OVA co-culture as in f and g. n = 3 biologically independent samples; two-way ANOVA corrected for multiple comparisons (Sidak test) (k) or unpaired two-tailed t-test (l). m, Experimental scheme. n, Tumor growth. n = 5 biologically independent samples; two-way ANOVA corrected for multiple comparisons (Dunnett test) comparing no T cell transfer, to transfer of CTRL or CDIPT− T cells at each time point. o,p, Tumors were stained for congenic markers. Cells were gated on LD-Aqua−, CD8-APC-Cy7+ and the percentage of CD45.1-PE-Cy7+ (o) or CD45.2-FITC+ (p) cells is shown. n = 10 represents no T cell transfer and CTRL and n = 9 represents CDIPT− biologically independent samples; one-way ANOVA corrected for multiple comparisons (Tukey test). q, Experimental scheme. r,s, At four and seven d after infection, blood samples from infected mice were analyzed by flow cytometry for the expression of CD8, CD45.1, CD45.2, CD127 and KLRG1. n = 4 biologically independent samples per group; unpaired two-tailed t-test. All error bars show the s.e.m. MFI, mean fluorescence intensity.
CD8+ T cell viability (Fig. 2c and Extended Data Fig. 2c), proliferation (Fig. 2d and Extended Data Fig. 2d) and interferon (IFN)-γ production (Fig. 2e) decreased after CDIPTi treatment. We tested whether restoring PI composition could rescue CDIPT inhibition by treating cells with bovine serum albumin (BSA)-conjugated Soy PI, which comprises only saturated PI species (Extended Data Fig. 2e). Soy PI treatment shifted the intracellular PI pool toward saturated (Extended Data Fig. 2f), and partially restored the reduced IFN-γ expression after CDIPTi treatment (Extended Data Fig. 2g). Substitution with polyunsaturated PI 38:4 shifted the total PI pool toward polyunsaturated (Extended Data Fig. 2h), but it did not restore IFN-γ production upon CDIPT inhibition (Extended Data Fig. 2i). CDIPTi-treated OT-I CD8+ T cells had a reduced ability to kill OVA-expressing EL4 tumor cells and decreased IFN-γ production when co-cultured with tumor cells (Fig. 2f,g). CDIPT inhibition also resulted in lower viability, EdU incorporation and IFN-γ production when treatment was started on day four or six of culture (Extended Data Fig. 2j). Of note, the effects of CDIPTi treatment were reversible as cells recovered viability, proliferation and cytokine production after drug withdrawal (Extended Data Fig. 2k). These data indicate that de novo PI synthesis is required to synthesize saturated PIPn, which are needed for Teff cell fitness and function.
To reinforce our CDIPTi findings, we used CRISPR−Cas9 to delete CDIPT (CDIPT−) in CD8+ T cells before activation (Fig. 2h,i). We observed a specific decrease of saturated PI in CDIPT− cells compared to CTRL (Fig. 2j), which confirmed that CD8+ Teff cells rely on CDIPT-dependent de novo PI synthesis to synthesize saturated PI. Importantly, whether CDIPT was pharmacologically inhibited, or genetically deleted, activated T cells maintained substantial and unperturbed pools of polyunsaturated PI (Fig. 2b,j). CDIPT− cells displayed impaired EL4-OVA cell killing and IFN-γ expression (Fig. 2k,l). To assess in vivo, we injected B16-F10-OVA melanoma cells into the flanks of wild-type (WT) mice, then adoptively transferred CTRL OT-I cells, CDIPT− OT-I cells or no T cells five d after tumor inoculation (Fig. 2m). We observed reduced tumor control in the CDIPT− T cell group relative to the CTRL group, similarly to the no T cell transfer group (Fig. 2n). CDIPT− OT-I cells failed to persist in vivo compared to CTRL OT-I cells, highlighted by decreased percentages of tumor-infiltrating donor-derived CD45.2+ cells (Fig. 2o,p). We further tested CDIPT− T cell fitness by injecting CTRL and CDIPT− OT-I cells in LmOVA-infected mice (Fig. 2q). Expansion of CDIPT− donor T cells was almost completely abolished four and seven d after infection (Fig. 2r). Among the remaining donor T cells, we observed fewer memory precursor effector cells (CD127+ KLRG1−, Fig. 2s)22. Together, these results indicate that saturated PIPn synthesis via CDIPT is required for CD8+ Teff cell fitness and function during cancer and infection.
Saturated PIPn are dispensable for early CD8+ T cell activation
PIPn are critical signaling mediators downstream of the TCR23. Inositol headgroup phosphorylation transduces signals by activating alternate signaling networks, which in turn control cell differentiation24. Given our findings that PIPn acyl chain saturation impacts Teff cell fitness and function, we next asked whether saturated PIPn are important early during T cell activation. The PIP2 pool increased in the first hours after activation (Fig. 3a), but in contrast to Teff cells, this was due to an increase in polyunsaturated PIP2 (Fig. 3b). Over the same time, there was no change in PI abundance or acyl chain saturation, suggesting that immediately after TCR activation PIP2 was generated by phosphorylating polyunsaturated PI present in unactivated T cells (Fig. 3c). We next deleted CDIPT (CDIPT−), and as an additional control we deleted ZAP70 (ZAP70−), a critical tyrosine kinase for T cell activation (Fig. 3d and Extended Data Fig. 3a). CTRL and CDIPT− T cells comprised a similar percentage of cells with an effector phenotype (CD44+CD62L−) 48 h after stimulation, which was higher than unstimulated or ZAP70− cells (Fig. 3e). CTRL and CDIPT− cells also similarly expressed activation markers CD25 (Fig. 3f) and programmed cell death protein-1 (PD-1, Fig. 3g). CTRL and CDIPT− cells comparably utilized extracellular glucose over 48 h (Fig. 3h), indicating that CDIPT− T cells undergo appropriate metabolic remodeling during T cell activation. To determine whether increasing the saturated PI pool impacts early CD8+ T cell activation, we stimulated CD8+ T cells in the presence of Soy PI (Fig. 3i). Soy PI enlarged the intracellular PI pool substantially and shifted the PI composition toward saturated (Fig. 3j). This increased saturated PI content had no effect on the expression of the activation markers CD25 and CD69 48 h after T cell activation (Fig. 3k,l). Soy PI-supplemented T cells comprised similar percentages of CD44+CD62L− cells (Fig. 3m) and had comparable IFN-γ production (Fig. 3n) 48 h after T cell activation. Therefore, while saturated PIPn species are essential for fully differentiated Teff cell fitness and function, hydrolysis of polyunsaturated PIPn abundant in unactivated T cells is sufficient to mediate CD8+ T cell activation early after TCR stimulation.
Fig. 3 |. Saturated PIPn are dispensable for early CD8+ T cell activation.
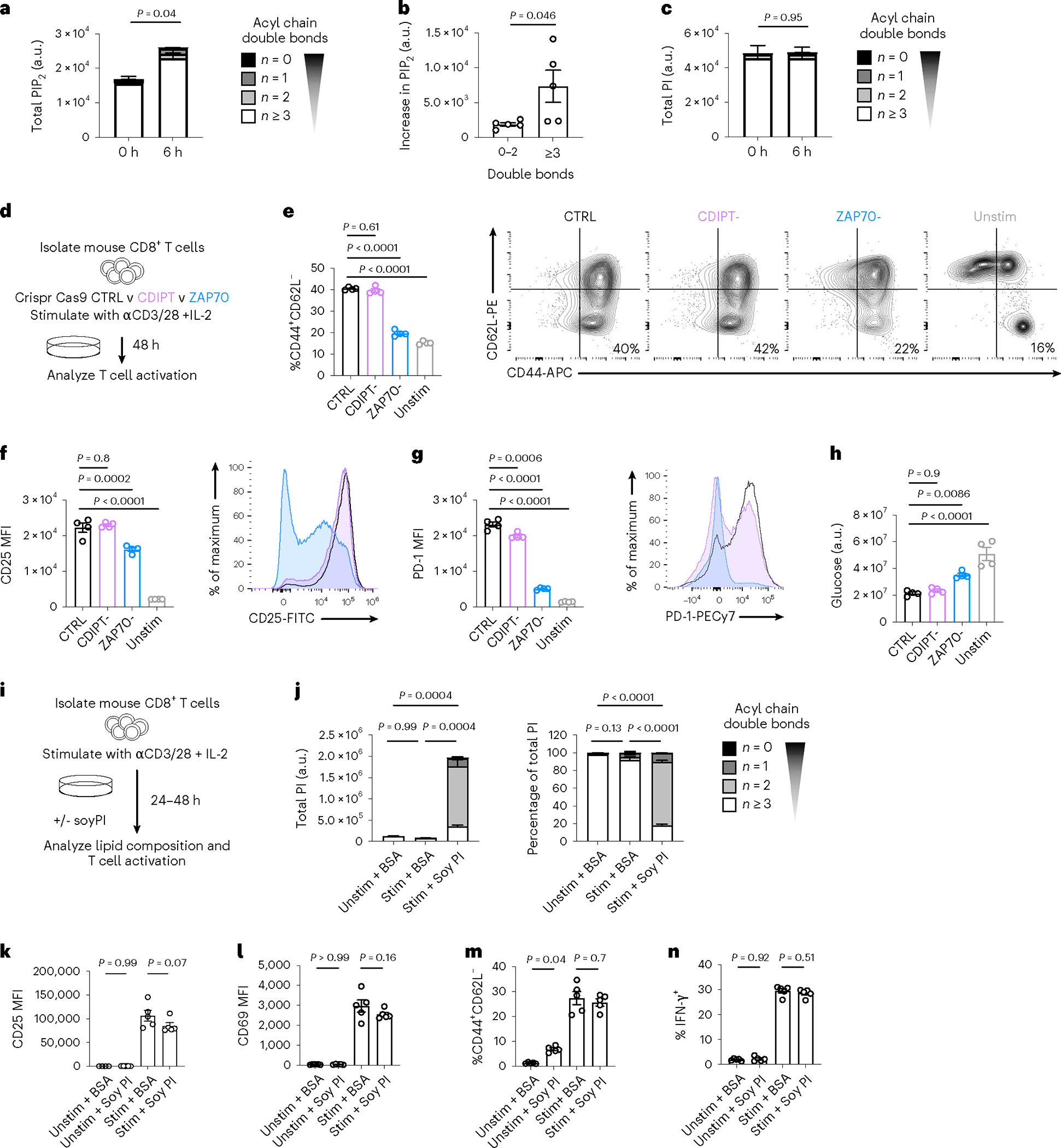
a–c, WT CD8+ T cells were stimulated with anti-CD3, anti-CD28 and IL-2 for 6 h. Intracellular lipids were analyzed by LC–QqQ–MS/MS or liquid chromatography quadrupole time-of-flight mass spectrometry (LC–QTOF–MS/MS). a, Total PIP2 at indicated time points after activation. n = 5 biologically independent samples; unpaired two-tailed t-test comparing total PIP2. b, Increase in PIP2 species with 0–2 versus ≥3 double bonds between zero h and six h after activation. n = 5 biologically independent samples; unpaired two-tailed t-test. c, Total PI at indicated time points after activation. n = 5 biologically independent samples; unpaired two-tailed t-test comparing total PI. d–h, WT CD8+ T cells were isolated from C57BL/6 mice, then CRISPR–Cas9 technology was utilized to delete CDIPT (CDIPT−) compared to control (CTRL) or ZAP70 deletion (ZAP70−). Cells were stimulated with anti-CD3, anti-CD28 and IL-2 for 48 h, or IL-7 for 48 h (unstimulated control) and T cell activation was assessed. e, CD8+ T cells were gated on Live/Dead-aqua−, then CD44+ and CD62L−. f, Expression of CD25 was quantified after gating on Live/Dead-aqua−CD8+. g, Expression of PD-1 was quantified after gating on Live/Dead-aqua− CD8+. h, Polar metabolites were extracted from supernatants and glucose was measured by LC–MS. Depletion of extracellular glucose across conditions is shown, normalized to cell number. In e–h, n = 4 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Dunnett test). i, Experimental scheme. WT CD8+ T cells were stimulated with anti-CD3, anti-CD28 and IL-2 for 24 h (j), or 48 h (k–n) in the presence or absence of Soy PI (100 μM). j, Total (left) and percentage (right) PI saturation. n = 3 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Tukey test) comparing saturated PI. k,l, Expression of CD25 (k) and CD69 (l) in Live/Dead-IR−, CD8-Brilliant Violet 421+ cells. n = 4 unstimulated and n = 5 stimulated biologically independent samples, representative of three independent experiments; one-way ANOVA corrected for multiple comparisons (Sidak test). m, CD8+ T cells were gated on Live/Dead-aqua−, then CD44+ and CD62L−. n, Intracellular expression of IFN-γ was quantified by flow cytometry. Cells were gated on Live/Dead-IR−, CD8-Brilliant Violet 421+. In m and n, n = 5 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Sidak test). Error bars show the s.e.m.
PIPn acyl chain saturation potentiates late effector T cell signaling
While polyunsaturated PIP2 exclusively supported early activation (Fig. 3a,b), saturated PIP2 was essential for maintaining late Teff cell function (Fig. 2c–g). Polyunsaturated PIP2 remained abundant during CDIPT inhibition or deletion, but it was not sufficient to maintain Teff cell function, highlighting the essential role of saturated PIPn in Teff cells. PIP2 at the plasma membrane is continuously hydrolyzed by PLCγ1 to maintain sufficient levels of the second messengers DAG and IP3 important for Teff cell function25 (Fig. 4a). To study whether depletion of the saturated PIP2 pool would abrogate downstream signaling in Teff cells, we analyzed PLCγ1-mediated signaling pathway activity upon CDIPT inhibition. Phosphorylated PLCγ1 expression or activity did not change in Teff cells or in Jurkat cells (Fig. 4b and Extended Data Fig. 4a). In contrast, total DAG decreased (Fig. 4c). Downstream of DAG, p-Raf, p-MEK1/2 and p-ERK1/2 expression levels were decreased in CDIPTi-treated cells compared to CTRL cells (Fig. 4d,e). We were not able to measure IP3 by LC–MS, but assessed downstream markers of IP3 signaling. While intracellular basal Ca2+ levels were maintained, expression of F-actin was reduced in CDIPTi compared to CTRL cells (Fig. 4f,g). No difference in AKT phosphorylation suggests that PIP3 signaling was unaffected by CDIPTi (Fig. 4h). These results support that saturated PIPn is necessary to maintain PLCγ1-mediated signaling in Teff cells.
Fig. 4 |. PIP2 saturation potentiates effector T cell signaling.
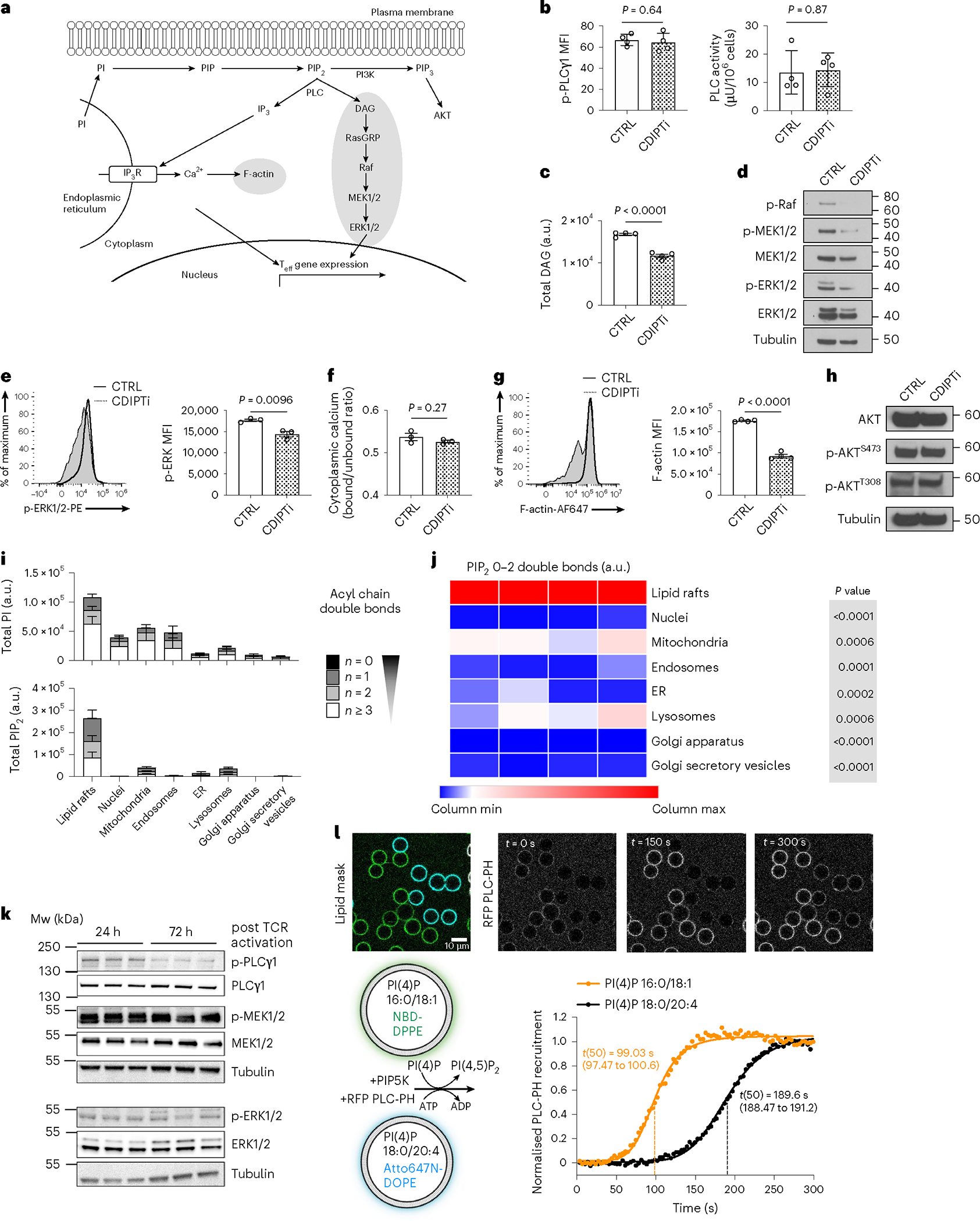
a, Schematic of PIP2 signaling in Teff cells. b–g, WT CD8+ T cells were stimulated with anti-CD3, anti-CD28 and IL-2 for two d, treated with CTRL or CDIPTi (plus IL-2) for one d and then readouts of downstream signaling were measured. b, Expression of phosphorylated PLCγ1 (left) and activity of PLC enzyme (right). n = 4 biologically independent samples from two independent experiments; unpaired two-tailed t-test. c, Total DAG. n = 4 biologically independent samples representative of three independent experiments; unpaired two-tailed t-test. d, Protein expression of the Raf–MEK1/2–ERK1/2 signaling pathway. e, Relative intensity of p-ERK1/2-PE. n = 3 biologically independent samples; unpaired two-tailed t-test. f, Basal cytoplasmic calcium was calculated using the indo-AM dye bound/unbound ratio. n = 3 biologically independent samples; unpaired two-tailed t-test. g, Relative intensity of Phalloidin-AF647. n = 4 biologically independent samples; unpaired two-tailed t-test. h, Protein expression of the PI3K signaling pathway. i,j, WT CD8+ T cells were stimulated with anti-CD3, anti-CD28 and IL-2 for two d, then a further two d in IL-2. Organelle compartments were enriched and PIPn were extracted and analyzed by LC–QqQ–MS/MS or LC–QTOF–MS/MS. i, PI (upper) or PIP2 (lower) saturation across organelles. n = 4 biologically independent samples. j, Heat map of the relative amount of saturated PIP2 across organelles. n = 4 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Dunnett test) comparing all groups with the lipid rafts. k, WT CD8+ T cells were stimulated with anti-CD3, anti-CD28 and IL-2 for 24 h or 72 h (the final 24 h of which were only with IL-2). Expression of total and phosphorylated PLCγ1, MEK1/2 and ERK1/2 with tubulin as the loading control. n = 4 biological replicates pooled into one lane. l, Lower left, experimental schematic. Membrane-coated beads were generated by mixing liposomes (harboring 16:0–18:1 PI(4)P or 18:0–20:4 PI(4) together with 0.1% Atto647N-DOPE or NBD-DPPE) and silica beads. Purified PLC-PH fused to RFP was added, followed by PIP5K1C immediately before acquisition. Upper microscopy images. Lower right, recruitment of PLC measured by the normalized MFIs of the RFP signal to either of the membranecoated bead populations, segmented using the NBD or Atto647N signal. n = 40–50 individual beads. Error bars show the s.e.m.
We questioned how saturated PIPn specifically maintains Teff cell signaling, and speculated that distinct PIPn pools might exist in different cellular compartments. We enriched distinct compartments from Teff cells, and measured their lipid content. While PI localized to numerous organelles, PIP2 was strongly enriched in lipid rafts (Fig. 4i,j). Of note, there was no compartment-specific difference in PIP2 acyl chain saturation (Fig. 4i and Extended Data Fig. 4b), suggesting that preferential cellular localization was not an important driver of Teff cell dependency on saturated PIPn. To investigate how changes in PIPn compare to changes in structural lipids in rafts, we measured the abundance of PI, phosphatidylserine (PS), sphingomyelin (SM) and gangliosides in Teff cells and unstimulated CD8+ T cells. We found that PI species and the ganglioside GM1 (but not GM2 or GM3) strongly increased, whereas PS species varied in expression and SM species decreased (Extended Data Fig. 4c). This suggested that increased PIP2 in lipid rafts was not simply due to a general increase in all lipid components in rafts.
Lipid rafts are central to TCR signaling26. We questioned whether saturated PIPn depletion affected raft formation. We observed decreased staining with cholera toxin subunit B, which binds to GM1 in lipid rafts27, following CDIPTi in both Teff cells and Jurkat cells (Extended Data Fig. 4d,e). Next, we used the fluorescent dye di-4-ANEPPDHQ28 to examine membrane lipid order in Teff cells treated with CDIPTi. CDIPT inhibition resulted in a higher emission at 630 nm, which correlated with a decreased fraction of cells with high lipid order and an increased fraction with low lipid order in both Teff cells and Jurkat cells (Extended Data Fig. 4f,g). The generalized polarization (GP) value—a normalized intensity ratio of the two spectral emissions commonly used to quantify membrane lipid order—decreased in Teff cells treated with CDIPTi (Extended Data Fig. 4h). We questioned whether changes in lipid raft formation and membrane lipid order were an immediate effect of saturated PIP2 depletion or a secondary effect resulting from reduced PIP2 signaling. At lower CDIPTi concentrations, cholera toxin B binding was less affected than MEK phosphorylation and IFN-γ production (Extended Data Fig. 4i). These data suggest that de novo synthesis of saturated PIP2 is primarily required to maintain downstream PIP2 signaling and thus CD8+ Teff cell function. Over longer times, synthesis of other lipid species and membrane lipid order are affected by CDIPTi as a consequence of diminished Teff cell signaling.
PLCγ1 is activated by phosphorylation that occurs rapidly upon TCR ligation. We hypothesized that initial TCR stimulation would strongly activate PLCγ1, but that its activity would wane over time and in late Teff cells, as signals from TCR stimulation dissipate, similar to what has been shown for other signaling pathways downstream of the TCR19. Supporting this idea, we observed that PLCγ1 phosphorylation decreased 72 h after activation, compared to 24 h after activation (Fig. 4k). In contrast, p-MEK1/2 and p-ERK1/2 expression was maintained (Fig. 4k), indicating that although PLCγ1 was less activated in late Teff cells, downstream signaling was sustained. We hypothesized that this could be due to saturated PIPn being a superior enzyme substrate compared to their polyunsaturated counterparts. We studied this in an in vitro phosphoinositide conversion assay, using supported lipid bilayers and purified components as previously described29. Briefly, membrane-coated beads comprising either PI(4)P 16:0/18:1 or PI(4)P 18:0/20:4 were distinctly labeled with NBD-DPPE or Atto647N-DOPE, respectively, and mixed. The PLC family recognizes its substrate, PI(4,5) P2, through a conserved pleckstrin homology (PH) domain, which was fluorescently labeled and added to the membrane-coated beads. The main source of cellular PI(4,5)P2 results from phosphorylation of PI(4)P into PI(4,5)P2 by phosphatidylinositol 4-phosphate 5-kinases (PIP5K). To reconstitute this metabolic transition, recombinant PIP5K was added to the system at catalytic amounts, resulting in conversion of PI(4)P to PI(4,5)P2 and subsequent recruitment of PLC-PH to the beads. PI conversion and subsequent PLC-PH recruitment to beads coated with bilayers containing PI(4)P 16:0/18:1 occurred almost twice as fast as to beads coated with PI(4)P 18:0/20:4 (Fig. 4l), indicating that PIP5K-mediated phosphorylation and/or PLC recruitment occurred more efficiently with saturated PIPn compared to polyunsaturated PIPn (Fig. 4l). The data from this assay show that saturated PIPn are either superior enzyme substrates for PIP5K, or PLCγ1 recruitment to them is faster, as compared to polyunsaturated PIPn. This ‘preferential usage’ of saturated PIPn suggests why their presence is needed to maintain T cell signaling and function in late Teff cells with waning PLCγ1 phosphorylation.
Effector T cell PIPn synthesis is dependent on glycolytic metabolism
To understand which metabolic events drive synthesis of saturated PIPn in Teff cells, we measured PI every 24 h from 0–72 h after stimulation. Concentration of polyunsaturated PI species (predominantly PI 38:4) increased from 0 h to 48 h; however, after 48 h, those with 0–2 double bonds increased and shifted the PI composition toward saturated (Fig. 5a). De novo PI synthesis requires three substrates—two fatty acyl chains, a glycerol phosphate backbone and an inositol head group (Fig. 5b)—that may be directly acquired from the extracellular milieu or synthesized de novo from glucose. Following de novo synthesis, PI may be further remodeled, which involves cleavage of a saturated acyl chain to form LPI, then addition of a polyunsaturated fatty acid, which most commonly is arachidonic acid (Fig. 5b). We first assessed if differential enzyme expression in the de novo and remodeling pathways led to saturated PI accumulation. CDIPT levels did not change during the 72-h activation course (Fig. 5c), and two important enzymes in the remodeling pathway, lysocardiolipin acyltransferase (LYCAT, encoded by Lclat1)30,31 and lysophosphatidylinositol acyltransferase (LPIAT, encoded by Mboat7)32 showed elevated transcription after activation, suggesting that increased saturated PI was not due to a deficiency in the remodeling pathway machinery (Extended Data Fig. 5a). We also queried whether saturated PI accumulated due to a deficiency in linoleic acid (LA), an essential substrate for arachidonic acid synthesis and therefore polyunsaturated PI; however, LA treatment 48–72 h after stimulation did not reduce the proportion of PI with 0–2 double bonds (Fig. 5d). Stearoyl-CoA desaturase (SCD) synthesizes monounsaturated fatty acids (with one double bond). SCD inhibition decreased the proportion of saturated PI, which was rescued by oleic acid (OA; 18:1) addition (Fig. 5d). SCD inhibition also impaired T cell proliferation and cytokine production (Extended Data Fig. 5b). These findings indicate that saturated PI synthesis occurs between 48 and 72 h after T cell activation, depends on the de novo synthesis of fatty acids with one double bond, and is not caused by a deficiency in PI remodeling pathway enzymes or substrate.
Fig. 5 |. Effector T cell PIPn synthesis depends on glycolytic metabolism.
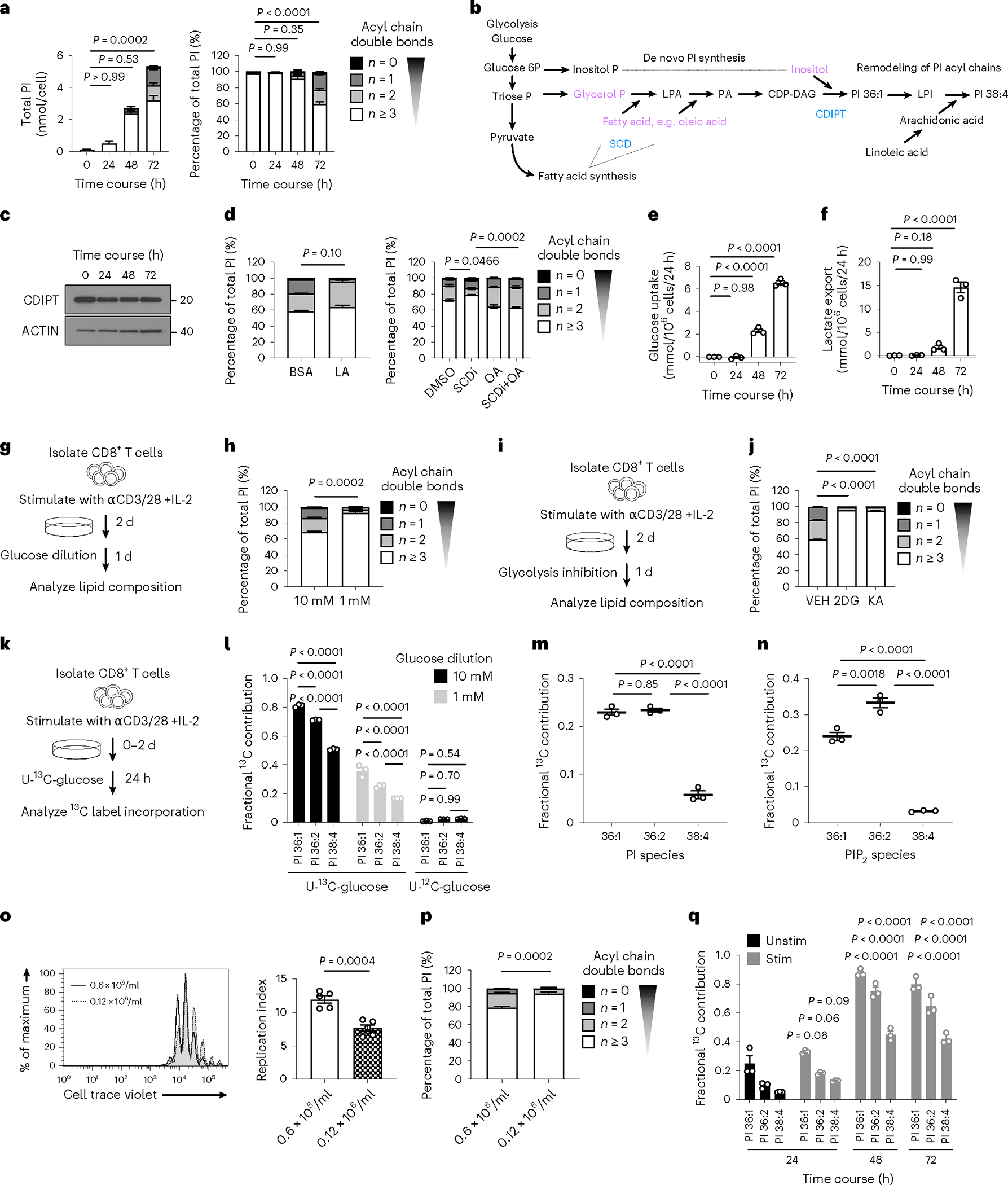
a, PI saturation (left) and percentage PI saturation (right) during WT CD8+ T cell stimulation. n = 3 biologically independent samples representative of 3 independent experiments; one-way ANOVA corrected for multiple comparisons (Dunnett test) comparing saturated PI with the 0 h time point. b, Schematic of de novo PI synthesis. c, Protein expression of CDIPT, as in a. n = 3 biologically independent samples pooled into one sample. d, WT CD8+ T cells were stimulated for 2 d, then for a further 24 h with BSA or BSA-conjugated LA (50 μM; left), or A939572 (SCDi; 100 nM), BSA-conjugated OA (100 μM), or a combination of both (right; plus IL-2), and PI saturation is shown. n = 4 or n = 3 biologically independent samples, respectively; unpaired two-tailed t-test or one-way ANOVA corrected for multiple comparisons (Tukey test) respectively on saturated PI. e,f, Glucose uptake and lactate export respectively, as in a. n = 3 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Dunnett test) comparing each time point with the 0-h time point. g, Experimental schematic. h, Total PI saturation. n = 3 biologically independent samples; unpaired two-tailed t-test on saturated PI. i, Experimental schematic. j, Total PI saturation. n = 3 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Dunnett test) comparing all groups with the vehicle control. k, Experimental schematic. l, Fractional 13C incorporation into major PI species. n = 3 biologically independent samples; two-way ANOVA corrected for multiple comparisons (Sidak test). m,n, As in k with 6-h labeling. Fractional 13C incorporation into major PI (m) or PIP2 (n) species from U-13C-glucose normalized to unlabeled glucose. n = 3 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Tukey test). o,p, WT CD8+ T cells were stained with CTV, activated as in a, and plated at 0.6 × 106 cells/ml or 0.12 × 106 cells/ml. o, Left, CTV dilution after 72 h of activation. Right, replication index. n = 5 biologically independent samples; unpaired two-tailed t-test. p, Total PI saturation. n = 5 biologically independent samples; unpaired two-tailed t-test comparing saturated PI. q, Fractional contribution of 13C-glucose-derived carbon to PI major species as in a, with the addition of 100% U-13C-glucose 24 h before lipid analysis at each time point. n = 3 biologically independent samples; two-way ANOVA corrected for multiple comparisons (Dunnett test) comparing respective PIs within each group with unstimulated cells. Error bars show the s.e.m.
Glucose is an important substrate for de novo fatty acid synthesis. Saturated PI species appeared late during CD8+ T cell activation (Fig. 5a), a time that correlated with increased glucose uptake (Fig. 5e) and glycolytic metabolism, as determined by elevated lactate export (Fig. 5f). To test whether extracellular glucose was critical for de novo synthesis of saturated PI, we cultured mouse CD8+ T cells in 10 mM or 1 mM glucose during the final 24 h of activation (Fig. 5g). PI composition shifted toward polyunsaturated in low glucose (Fig. 5h), indicating that glucose availability is important for de novo PI synthesis. With the same experimental setup (Fig. 5i), we inhibited hexokinase with 2-deoxy-glucose (2-DG) or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) with koningic acid (KA), and observed a similar shift in PI toward polyunsaturated (Fig. 5j). Interestingly, GAPDH inhibition would still allow inositol and glycerol phosphate synthesis from glucose, but not de novo fatty acid synthesis, highlighting the importance of de novo fatty acid synthesis in saturated PI generation.
To show that glucose is a substrate for PI synthesis, we cultured cells in 10 mM or 1 mM uniformly labeled 13C (U-13C)-glucose for the last 24 h of culture (Fig. 5k). Using this long 24-h labeling time, we could determine the maximum fractional contribution of glucose carbon to PI: ~80% for PI 36:1 and ~70% for PI 36:2, which was reduced in 1 mM U-13C-glucose, and was <1% in unlabeled glucose (Fig. 5l). U-13C-glucose also contributed carbons to PI 38:4 (Fig. 5l), indicating that Teff cells also actively maintain PI acyl chain remodeling (Fig. 5b,h). To compare the dynamics of PI and PIP2 synthesis, we traced U-13C-glucose into Teff cells for 6 h (Fig. 5m,n). 13C incorporation into PI 36:1 and PI 36:2 was ~5 times higher than into PI 38:4 (Fig. 5m). 13C incorporation into PIP2 36:1 and PIP2 36:2 was ~8 times and ~11 times higher than into PIP2 38:4, respectively (Fig. 5n). These data confirm that in Teff cells, de novo saturated PI and PIP2 synthesis occurs more rapidly than polyunsaturated PI and PIP2. Increased PIPn synthesis may be critical for maintaining downstream Teff cell signaling during Teff cell expansion, when high PIPn amounts are required to support intensive proliferation. To test whether rapid proliferation is directly linked to a saturated PI pool, we activated CD8+ T cells for 72 h at two different cell concentrations: 0.6 × 106 cells/ml, which allowed for sufficient cell–cell contact to support robust Teff cell expansion, and at a fivefold lower concentration, where cell contact was inhibited (Fig. 5o). This approach allowed us to limit proliferation without potentially toxic pharmacological intervention that may also rewire cellular metabolism. Reduced proliferation aligned with a substantially decreased percentage of saturated PI (Fig. 5p), suggesting that rapid Teff cell proliferation drives the de novo saturated PIPn synthesis.
We next tested when glucose contributes to de novo PI synthesis. We measured incorporation of U-13C-glucose into PI species every 24 h after CD8+ T cell stimulation. During the first 24 h, we found no difference in the fractional contribution of U-13C-glucose to PI production between unstimulated and stimulated CD8+ T cells (Fig. 5q). However, at 48 and 72 h after activation, U-13C-glucose became a major substrate for PI synthesis and contributed to ~80% of PI 36:1 (Fig. 5q), confirming that de novo PI synthesis occurs late during T cell activation, after Teff cells have engaged glycolytic metabolism (Fig. 5e,f). These data also indicate that the turnover of PI species occurs at least every 24 h during the second and third day after CD8+ T cell activation. Teff cells are dependent on glycolytic metabolism to support their proliferation and function, but they reduce glycolytic rates during IL-15-driven differentiation into memory T (TM) cells33. We compared the PI 36:2 and PIP2 pools of Teff cells and TM cells differentiated in vitro and found that TM cells decreased saturated PI and PIP2 and had a similar PIPn composition to unstimulated T cells (Extended Data Fig. 5c,d). Upon stimulation, TM cells engage glycolytic metabolism and effector function more rapidly than naive T cells33. Following stimulation, TM cells rapidly increased PI 36:2 (Extended Data Fig. 5c). Furthermore, TM incorporated U-13C-glucose into PI within the first 24 h of secondary stimulation with anti-CD3/CD28 plus IL-2 (Extended Data Fig. 5e). Collectively, these data show that de novo saturated PI synthesis depends on extracellular glucose and enhanced glycolytic metabolism, linking nutrient availability to Teff cell signaling. Notably, glucose carbon incorporation into saturated PI and PIP2 occurs faster than into polyunsaturated counterparts, indicating that reaction speed might be critical to maintain the PIPn pool and support signaling in rapidly expanding Teff cells.
Immunotherapy-boosted tumor-infiltrating lymphocytes synthesize saturated PI
Expression of the inhibitory receptors PD-1 and cytotoxic T lymphocyte-associated protein 4 (CTLA4) decreases CD8+ T cell glucose uptake and glycolysis34. We previously demonstrated that PD-1 blockade reverses glucose restriction in CD8+ TILs, which restores IFN-γ expression and antitumor function35. Here, we show that glucose is an essential substrate for de novo saturated PIPn synthesis (Fig. 5g–q). We hypothesized that TILs with restored glycolytic metabolism and boosted function after checkpoint inhibitor therapy would synthesize saturated PI. We compared TILs from mice bearing B16-F10-OVA melanoma tumors treated with three rounds of isotype controls (IgG1 + IgG2) or combined checkpoint inhibitor therapy anti-PD-1 plus anti-CTLA4 (αPD-1 + αCTLA4; Fig. 6a). αPD-1 + αCTLA4-treated mice had smaller tumors compared to IgG1 + IgG2-treated mice (Fig. 6b), and their CD8+ TILs expressed more IFN-γ and granzyme B (Extended Data Fig. 6a–c). CD8+ TILs isolated from αPD-1 + αCTLA4-treated mice 12 d after tumor injection had increased saturated PI composition compared to IgG1 + IgG2-treated mice (Fig. 6c). The total amount of PI was equal between groups (Extended Data Fig. 6d). Next, we questioned whether tumors with different sensitivity to checkpoint blockade have specific changes in the PI saturation of their TILs. We treated mice bearing B16-F10-OVA melanoma tumors with three rounds of anti-PD-1 antibody alone and found that tumor size was not reduced (Extended Data Fig. 6e) and the TILs did not show a shift in PI saturation (Extended Data Fig. 6f), indicating that only CD8+ TILs with improved antitumor function following checkpoint inhibitor therapy are marked by increased saturated PI.
Fig. 6 |. Immunotherapy-boosted tumor-infiltrating lymphocytes synthesize saturated PI.
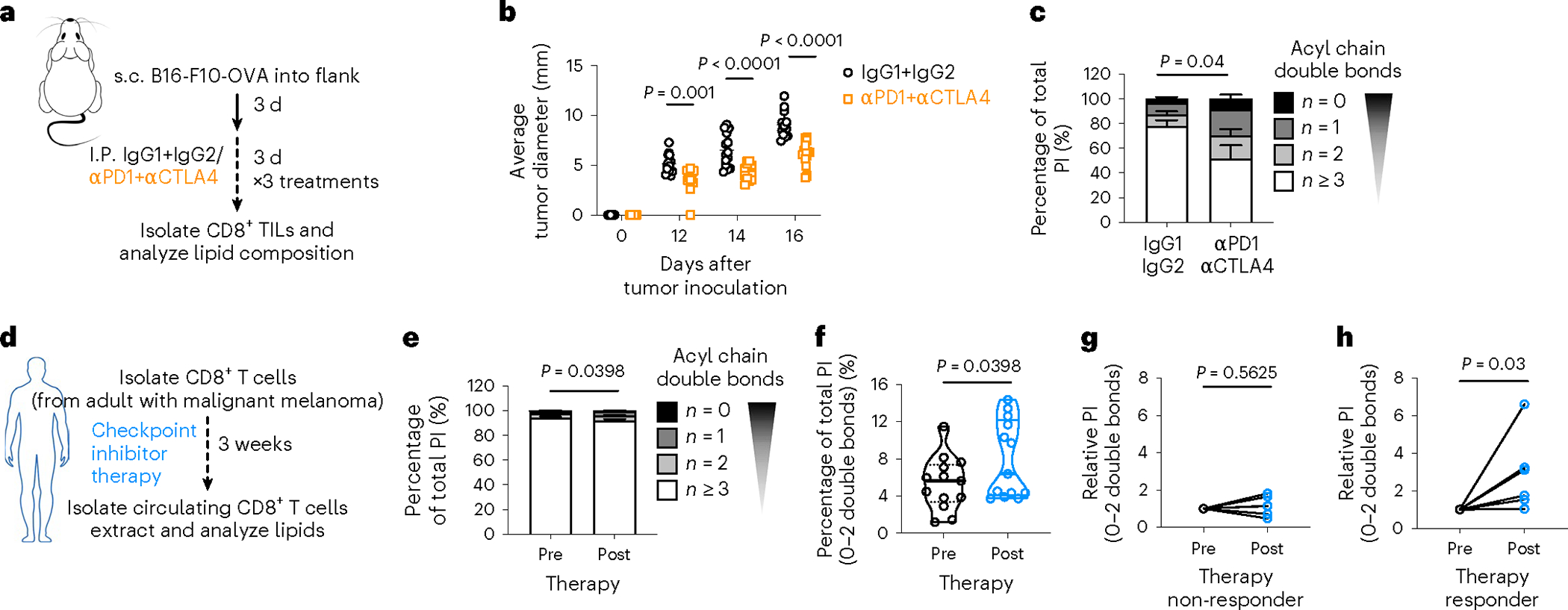
a–c, Sex-matched C57BL/6 mice were injected in the right flank with 1 × 106 B16-F10-OVA cells. After three d, mice were given intraperitoneal injections of anti-PD-1 and anti-CTLA4 (aPD1 + aCTLA4) or matching isotype controls (IgG1 + IgG2) every three d for three rounds in total. Tumor growth was measured up to day 16 after tumor injection and CD8+ T cells were isolated on day 12 after tumor injection to measure T cell function and lipid composition. b, Average tumor diameter at indicated time points. n = 14 biologically independent animals; two-way ANOVA corrected for multiple comparisons (Sidak test) comparing the two groups at each time point across the dataset. c, Lipids were analyzed from TILs on day 12 after tumor injection. Saturation of acyl chains is expressed as a percentage of total PI. Statistical analysis was carried out on the summed percentage of saturated PI on n = 7 IgG1 + IgG2 and n = 8 aPD1 + aCTLA4 biologically independent samples using an unpaired two-tailed t-test. d–h, CD8+ T cells were isolated from the blood of adult participants with histologically confirmed stage III or IV malignant melanoma (n = 13) on the day of immune checkpoint inhibitor treatment initiation (before therapy, “pre”) and then when the participant presented for the second administration of treatment (after therapy, “post”). Long-term disease response (non-responder versus responder) to checkpoint inhibitor treatment was assessed (median follow-up time of 318 d). Lipids were extracted and analyzed by LC–QqQ–MS/MS. e,f, Lipids were analyzed before and after therapy. Saturation of acyl chains is expressed as a percentage of total PI. Statistical analysis was carried out on the summed percentage of saturated PI using a two-tailed Wilcoxon matched-pairs signed-rank test (n = 13). g,h, Lipids were analyzed and grouped by response to therapy. Relative intensity of saturated PI species normalized to pre-therapy level is shown. Statistical analysis was carried out using a two-tailed Wilcoxon matched-pairs signed-rank test (n = 6 responders and n = 6 non-responders). Error bars show the s.e.m. s.c., subcutaneous.
Immune checkpoint inhibitors are a frontline treatment for patients with advanced melanoma36. We questioned whether an increase in saturated PI could be detected in peripheral blood CD8+ T cells of participants undergoing immunotherapy. We isolated circulating CD8+ T cells from adults with stage III or stage IV melanoma before and after the first round of checkpoint inhibitor therapy (Fig. 6d). We measured CD8+ T cell lipids and observed a shift in PI composition toward saturation after therapy (Fig. 6e,f). We then analyzed the clinical and radiological assessments of disease courses with a median follow-up time of 318 d (range 184–484 d). Per the radiologist’s assessment, we divided participants into non-responder (stable or progressive disease) and responder (partial or complete remission) groups. In the non-responder group, we observed no relative increase in saturated PI species (Fig. 6g), while in the responder group we observed a statistically significant increase (Fig. 6h). Therefore, early changes in PI composition may correlate with successful long-term response to checkpoint inhibitor therapy in patients with advanced melanoma.
Discussion
We analyzed the lipidome and found that PI with a distinct acyl chain composition marked by a reduced number of double bonds accumulated in Teff cells. We discovered that two separate PIPn pools—one saturated with 0–2 double bonds, and one polyunsaturated with ≥3 double bonds—drive important signaling events at specific stages of Teff cell differentiation. Sustained Teff cell signaling is driven by saturated PIPn, while polyunsaturated PIPn are exclusively utilized early during T cell activation. Saturated PIPn synthesis only occurred late during Teff cell differentiation, which might be necessary to sustain DAG production and downstream signaling as TCR-driven PLCγ1 activation waned. Teff cell PIPn signaling was maintained (1) by the rapid and continuous de novo saturated PI synthesis from glucose, facilitated by Teff cell glycolytic metabolism; and (2) by the preference of PIP5K and/or PLC for more saturated PIPn, or their association with more saturated PIPn, even in the presence of abundant polyunsaturated PIPn. The dependence on extracellular glucose for PIPn synthesis directly ties the nutrient environment with Teff cell signaling. Finally, T cells with improved antitumor function following checkpoint inhibitor therapy synthesized more saturated PIPn in mouse and human melanoma.
Rapid interconversion and salvage of polyunsaturated PIPn following TCR ligation and co-stimulation is critical for initiating signaling and T cell activation25,37. However, how PIPn signaling is maintained during differentiation into Teff cells with increased biosynthetic demands, along with the diminution of TCR signaling38,39 is unknown. Mammalian PIPn is unique among other lipids in its characteristic fatty acid profile enriched in PIPn 38:4, with stearic acid (18:0) at the sn-1 position and arachidonic acid (20:4) at the sn-2 position8. We found that in contrast to the first hours after activation, when polyunsaturated PIP2 is prevalent, CD8+ Teff cells augmented saturated PIPn synthesis 24–48 h after stimulation, which increased the total PI and PIP2 pool, while the polyunsaturated PIP2 pool remained constant. Increased PIPn saturation has been observed in transformed cell lines40,41, which may indicate that saturated PIPn synthesis drives PIPn signaling in cell types with increased biosynthetic demands.
Teff cell function is dependent on metabolic reprogramming involving increased glucose metabolism42,43. Teff cell glycolytic metabolism was previously linked to downstream PIP2 signaling, as increased phosphoenolpyruvate synthesis inhibits SERCA-dependent calcium reuptake, thus maintaining increased cytoplasmic calcium and NFAT1 signaling44. Here we report that glycolytic metabolism directly regulates PIP2 signaling, as glucose is the predominant substrate for de novo saturated PIPn synthesis, meaning that PIP2 signaling is dependent on glucose availability and enhanced Teff cell glycolytic metabolism. We measured the de novo synthesis of PI with different acyl chain compositions using U-13C-glucose and found that saturated PI synthesis occurs more rapidly, in keeping with recent findings45, as does saturated PI phosphorylation to PIP2 by PIP5K. Notably, PIP5K exhibits acyl chain preference. The Michaelis constant, which denotes the substrate concentration at which an enzyme reaches half its maximum rate, is lower for PIP 36:1 compared to PIP 38:446, supporting our findings that Teff cells synthesize saturated PIP2 more rapidly than polyunsaturated PIP2. We measured the conversion of PI(4)P 16:0/18:1 versus 18:0/20:4 to PI(4,5)P2, and concomitant recruitment of PLC and demonstrated that this process occurred almost twice as fast for PI(4)P 16:0/18:1 as for PI(4)P 38:4. Thus, increased de novo synthesis of saturated PIPn from glucose directly links PIPn signaling to Teff cell glycolytic metabolism, and is critical to maintain a higher rate of PIPn turnover in Teff cells.
CDIPT exchanges inositol for CMP on CDP-DAG in the final step of de novo PI synthesis47. CDIPT is not known to have acyl chain selectivity48, suggesting that the regulation of PIPn acyl chain composition occurs during de novo fatty acid synthesis. Supplementing Teff cells with LA did not increase polyunsaturated PI synthesis, indicating that saturated PI synthesis does not occur due to a lack of substrate for arachidonic acid synthesis. Furthermore, our data suggest that expression of enzymes in the PI remodeling pathway are intact30–32. CDIPT inhibition or deletion specifically inhibited de novo saturated PI and PIP2 synthesis. PLCγ1 expression and activity were maintained after CDIPT inhibition, but downstream of PIP2 there was decreased DAG, p-Raf, p-MEK1/2 and p-ERK1/2. Interestingly, while the polyunsaturated PI pool was maintained during CDIPT inhibition, likely due to remodeling of LPI to PI 38:4, polyunsaturated PI was not phosphorylated to PIP2, and the total PIP2 content was halved. Thus, de novo PI synthesis in Teff cells predominantly supports the saturated pool, while the polyunsaturated pool is maintained even when CDIPT is inhibited or deleted.
We also found that PIP2 exhibited a strong preference for lipid rafts, potentially because in this compartment phospholipids with more saturated acyl chains are able to pack closer with the saturated acyl chains of sphingolipids49, and hence can be efficiently hydrolyzed by PLCγ1. PLCγ1 depletes PIP2 and produces DAG more rapidly in lipid domains with high order versus low order50, and PLCγ1 activation is regulated by lipid rafts51. CDIPT inhibition reduced lipid order and lipid raft formation in Teff cells. A decrease in lipid raft formation might be caused directly by disturbed cytoskeleton remodeling in the absence of PIP2. In fact, PIP5Kβ, one of the main PIP2-generating enzymes, regulates actin reorganization required for lipid raft recruitment at the immunological synapse of CD4+ T cells52. Notably, PLCγ1 phosphorylation was lower in Teff cells 72 h after activation compared to 24 h, and we believe that the efficient synthesis and hydrolysis of saturated PIP2 is essential to sustain signaling in late Teff cells. The functional consequence of a diminished saturated PIP2 pool in Teff cells was reduced viability, proliferation and effector function. Therefore, glycolysis-driven de novo saturated PIPn synthesis is central to Teff cell function.
CD8+ Teff cells can kill malignant cells; however, tumor-imposed metabolic restrictions that affect Teff cell glycolytic metabolism can hamper CD8+ T cell antitumor function and permit tumor progression35,44. We found that saturated PI synthesis was increased in CD8+ TILs, and CDIPT was required for CD8+ TIL antitumor fitness and function. A common feature of TILs is increased inhibitory checkpoint receptor (for example, PD-1 and CTLA4) expression that impairs TCR signaling and glycolytic metabolism53. Checkpoint inhibitor therapy has shown promise in many advanced cancers36,54. We observed increased PI saturation after immunotherapy in both mouse TILs and circulating CD8+ T cells isolated from participants with melanoma who responded to checkpoint inhibitor treatment, indicating that saturated PI may be a characteristic of functional Teff cells in a clinical setting.
In summary, diverse pools of PIPn with differing acyl chain saturation are critical regulators of distinct CD8+ T cell differentiation programs. Polyunsaturated PIP2 (≥3 double bonds) sufficiently supports T cell signaling early after TCR stimulation when cells have limited proliferation rates and recent TCR activation triggers abundant PLCγ1 phosphorylation. As CD8+ T cells commit to their full Teff cell function marked by intense proliferation and an increased demand for second messengers in the light of waning PLCγ1 activity, rapid synthesis and conversion of saturated PIPn from glucose becomes essential for sustained Teff cell signaling. As such, inhibition of saturated PIPn synthesis impairs Teff cell fitness and function in infection and cancer (Fig. 7). Thus, fatty acid chain saturation in PIPn is a new mechanism that governs T cell signaling at different stages of CD8+ T cell differentiation. Given the central role of Teff cells in antitumor immunity and in autoimmune diseases, our findings may provide an avenue for modulating PIPn saturation as a means to balance Teff cell activity.
Fig. 7 |. PIP2 saturation defines early activation versus late effector T cell signaling.
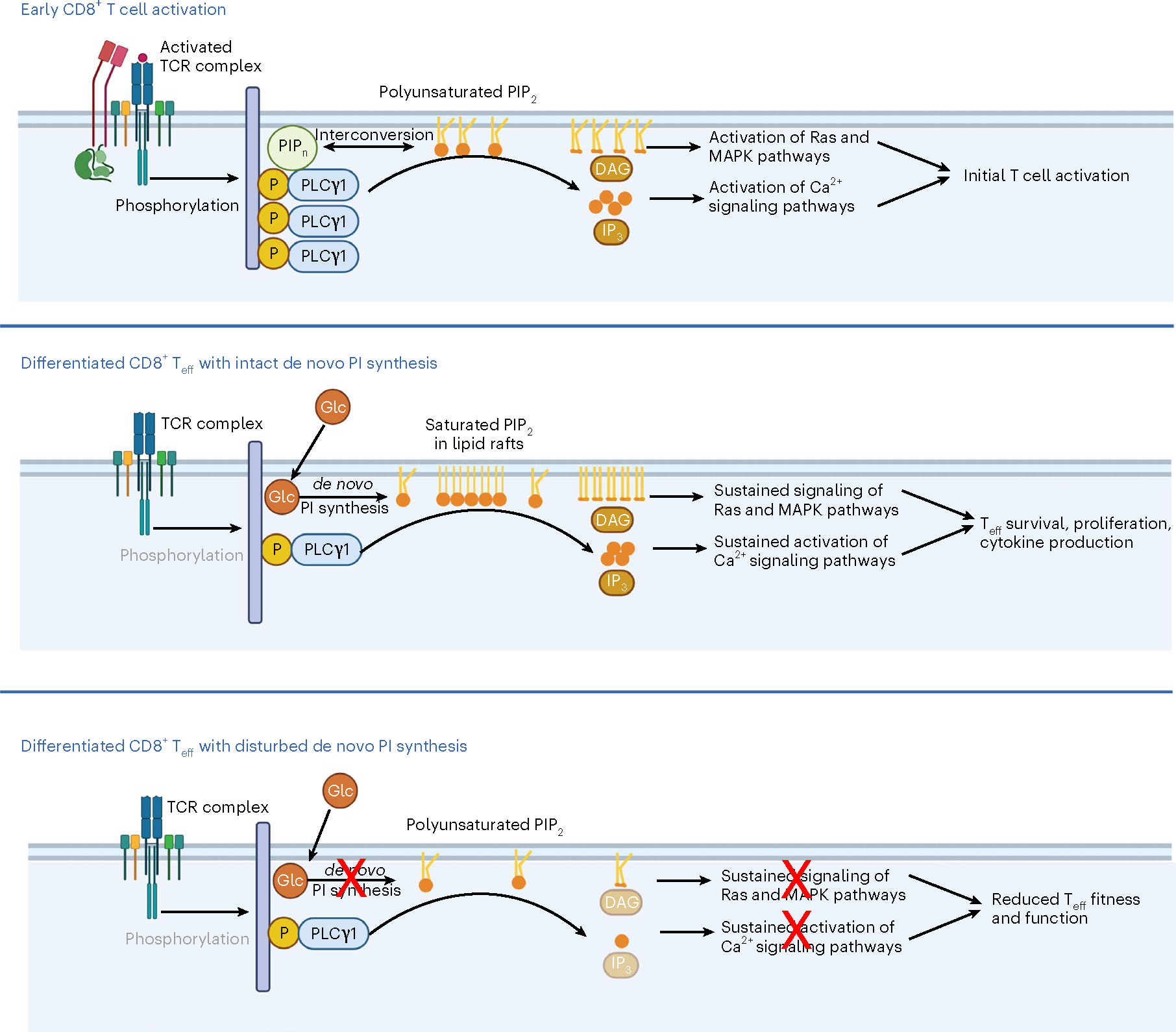
Early CD8+ T cell activation is marked by a robust phosphorylation of PLCγ1, which cleaves polyunsaturated PIP2 to generate the second messengers DAG and IP3. In fully differentiated CD8+ Teff cells, PLCγ1 phosphorylation decreases as TCR signals dissipate; and a lipid raft-accumulated saturated PIP2 pool, synthesized de novo from glucose, becomes essential for sustained signaling and Teff cell survival, proliferation and cytokine production. When de novo PI synthesis is inhibited, the saturated PIP2 pool is specifically depleted, and this results in disturbed downstream signaling and reduced Teff cell fitness and function. Glc, glucose; MAPK, mitogen-activated protein kinase; Teff, CD8+ Teff cell. Created with BioRender.com.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41590-023-01419-y.
Methods
Mice
C57BL/6J (RRID: IMSR_JAX:000664), major histocompatibility complex (MHC) class I-restricted OVA-specific TCR OT-I transgenic mice (RRID: IMSR_JAX:003831) and CD45.1+ C57BL/6J (B6.SJL-Ptprca Pepcb/BoyJ; Jax, 002014) mouse strains were purchased from The Jackson Laboratory. Male and female mice (8–12 weeks old, age-matched and sex-matched between experimental conditions) were used in all experiments. All mice were maintained in the animal facilities at the Max Planck Institute for Immunobiology and Epigenetics or at the Johns Hopkins University under specific-pathogen-free conditions and following institutional animal use and care guidelines. Mice were exposed to a 14-h/10-h light/dark cycle and fed ad libitum (Ssniff, V1185–300 or Envigo 2018SX) with acidified water (pH 2.5–3.3). The room temperature and humidity were maintained and monitored. The animal protocols were approved by the Federal Ministry for Nature, Environment and Consumer Protection of the state of Baden-Württemberg, Germany and by the Johns Hopkins University Animal Care and Use (G-16/129, G-17/71 and MO19M71, MO22M15).
Primary T cell cultures
CD8+ T cells were isolated from the spleens and lymph nodes of 8- to 12-week-old age-matched and sex-matched C57BL/6 mice using the EasySep CD8+ T cell isolation kit (Stem Cell Technologies, 19753) according to the manufacturer’s instructions. CD8+ T cells were activated using plate-bound anti-CD3 (5 μg ml−1; InVivoMab anti-mouse CD3, BioXCell, BE0002) and soluble anti-CD28 (0.5 μg ml−1; InVivoMab anti-mouse CD28, BioXcell, BE0015) in 1640 media (Invitrogen) supplemented with 10% fetal calf serum (Gibco), 4 mM l-glutamine, 100 U ml−1 penicillin–streptomycin, 100 U ml−1 IL-2 (Peprotech), 55 μM beta-mercaptoethanol in a humidified incubator at 37 °C, atmospheric oxygen supplemented with 5% CO2. Cells were stimulated for 48 h or 72 h as indicated in the figure, and then further expanded in IL-2 only. For the generation of TM cells, IL-2 was replaced by 100 U ml−1 IL-15 starting from 72 h after activation for three d. Unstimulated cells were maintained as indicated in 100 U ml−1 IL-7 (Peprotech). For OT-I cultures, single-cell suspensions of splenocytes were stimulated for 48 h in IL-2-containing media as above in the presence of SIINFEKL peptide. At 48 h after activation, media was replaced with expansion media containing IL-2 only.
Cell lines
The mouse E.G7 lymphoblast cell line expressing OVA (EL4-OVA) was purchased from the American Type Culture Collection (ATCC no. CRL-2113; RRID: CVCL_3505). The mouse melanoma cell line B16-F10-OVA was a kind gift from D. Zehn (parental cell line B16-F10 from ATCC, CRL-6475). The human T lymphocyte cell Jurkat (ATCC, TIB-152) was a kind gift from J. Powell. Further authentication of the cell lines was not performed. Cells were maintained in 1640 medium (EL4-OVA and Jurkat) or DMEM (B16-F10-OVA) supplemented with 10% fetal calf serum, 4 mM l-glutamine, 100 U ml−1 penicillin–streptomycin and 55 μM beta-mercaptoethanol at 37 °C in a humidified incubator containing atmospheric oxygen supplemented with 5% CO2. For EL4-OVA co-cultures, sub-confluent EL4-OVA cells were pre-stained with Cell-Trace Violet Cell Proliferation Kit (Invitrogen) and plated in fresh media at 50,000 cells per well in 100 μl.
L. monocytogenes infection studies
In total, 1 × 104 OT1+ CD45.2+ CD8+ cells per mouse from donor splenocytes were transferred i.v. into congenic (CD45.1+) age-matched recipient mice. After one d, mice were injected i.v. with a sublethal dose of 1 × 106 colony-forming units (CFUs) of recombinant L. monocytogenes expressing OVA deleted for actA (LmOVA ΔactA). On day four after infection, CD45.2+ CD8+ T cells were negatively isolated from recipient splenocytes using a BioLegend antibody cocktail comprising: CD11b (101204), CD11c (117304), CD19 (115504), CD45R (B220; 103204), CD49b (DX5) (108904), anti-MHC Class II (I-A/I-E) (107604), Ter-119 (116204), TCRγ/δ (118103), CD4 (100404) and CD45.1 (110704), all at a 600× dilution. Uninfected OT1+ CD45.2+ CD8+ cells from donor splenocytes were used as a control. Lipids were extracted and analyzed by LC–QqQ–MS/MS. Flow cytometry analysis of the expansion of donor T cells was performed on day four and day seven after infection in peripheral blood.
Mouse melanoma model
Mice were shaved and 1 × 106 B16-F10-OVA cells in 100 μl were injected into the right flanks. For immunotherapy experiments, mice were randomized into two groups. On days three, six and nine after tumor inoculation, mice received intraperitoneal injections of either a combination of 0.2 mg per mouse of anti-CTLA-4 antibody (BioXCell, BE0032) and 0.2 mg per mouse of anti-PD-1 antibody (BioXCell, BE0146) or a combination of the respective IgG control antibodies (BioXCell, BE0091 and BE0089). The injection volume was 100 μl. Tumor sizes were measured with calipers every other day. The maximal allowed tumor size was 20 mm in diameter and animals were humanely euthanized if this size was reached. Mouse body condition was monitored over the whole experimental period. Mice were euthanized if the tumor diameter reached 20 mm or if there was tumor ulceration or bleeding. At the indicated time point, mice were humanely euthanized and the tumors were excised. Tumors were digested by shaking at 37 °C with 1 mg ml−1 collagenase IA (Sigma, C9891) and 50 μg ml−1 DNase I (Roche, 10104159001) for one h. Digested tumors were passed through a 70-μm filter then the lymphocyte fraction was obtained using a Percoll gradient. CD8+ T cells were isolated by positive selection using CD8 TIL MicroBeads (Miltenyi Biotec, 130–116-478) according to the manufacturer’s instructions.
For adoptive T cell transfer, 1 × 106 B16-F10-OVA cells in 100 μl were injected into the right flanks of mice. Tumor sizes were measured with calipers every other day. On day five after tumor inoculation, mice received an i.v. injection of 5 × 106 CTRL or CDIPT− OT-I T cells that had been activated for three d in vitro with the SIINFEKL peptide in the presence of IL-2. Tumor growth was measured until day 15. For analysis of TIL composition, the mice were euthanized when tumors reached 10 mm in diameter, the tumors were excised and digested as described above and TILs were analyzed by flow cytometry.
For lipidome analysis of TILs, mice were injected with 1 × 106 B16-F10-OVA cells in 100 μl into the right flanks. Tumors were measured until they reached seven mm in diameter, and were then excised and digested as described above. CD8+ T cells were isolated by positive selection using CD8 TIL MicroBeads according to the manufacturer’s instructions. CD8+ T cells enriched from the spleens of the same mice were used as a control.
Human T cell analysis
Buffy coats for in vitro CD8+ T cell experiments were kindly provided by the Institute for Transfusion Medicine and Gene Therapy, Medical Center – University of Freiburg (donor consent, anonymized), and processed as described below.
Human melanoma study
All procedures involving human participants were approved by the Ethics Committee of the Medical Center – University of Freiburg (protocol no. 310/18) and were conducted according to the Declaration of Helsinki. The study was registered at the German Clinical Trial Register (DRKS00023625). Adults with histologically confirmed malignant melanoma stage III or IV (male and female, n = 13, median age 55 years) were recruited with a standardized procedure based on assessment of inclusion and exclusion criteria. Inclusion criteria were a histologically confirmed malignant melanoma, no previous treatment with an immune checkpoint inhibitor and age >18 years. Participants with a previously diagnosed immunodeficiency syndrome were excluded. Study participants gave their written informed consent before study enrollment. Collection of biological material was performed at the Department of Dermatology, Medical Center – University of Freiburg. No financial compensation was provided to participants. Peripheral blood mononuclear cells (PBMCs) were collected at two time points. The first time point was on the day of immune checkpoint inhibitor treatment initiation. The second time point was when the participant presented for the second administration of treatment. All samples were de-identified and analyzed in a pseudonymized manner. Disease response to checkpoint inhibitor treatment was assessed on a regular basis by 18F positron emission tomography combined with computer tomography, computer tomography alone and/or magnetic resonance imaging. Responders were defined as participants who reached a partial or complete remission according to the radiologist’s assessment. Partial remission was defined as a decrease in size and tumor metabolism of the majority of lesions. Complete remission was defined as the absence of malignant lesions. One participant was not eligible for response assessment. The median follow-up time was 318 d (range 184–484 d). Information on demographic characteristics and disease course was provided by the medical records at the Medical Center – University of Freiburg (Supplementary Table 1). Blood was collected in EDTA-coated tubes and stored at 4 °C before processing. Processing was performed within three h of blood collection. Blood was diluted at a 1:1 ratio with PBS containing 2% FCS and 1 mM EDTA. PBMCs were isolated using a density gradient method. Lymphoprep solution (Stem Cell) was pipetted to the bottom of SepMate-50 tubes (Stem Cell), and diluted blood carefully layered on top. After 10 min of centrifugation at 1,200g, the upper phase containing mononuclear cells was transferred to a new tube. PBMCs were washed twice with PBS with 2% FCS and 1 mM EDTA. Isolation of CD8+ T cells was performed using the EasySep Human CD8+ T Cell Isolation Kit (Stem Cell Technologies, 17953) according to the manufacturer’s instructions.
Human Epstein–Barr virus study
Participants (male and female, n = 3, age unknown) with acute EBV infection were recruited based on clinical symptoms and positivity for EBV-VCA-IgM. All participants gave informed consent. The study was conducted according to the Declaration of Helsinki and approved by Ethics Committees of the University of Freiburg (protocol no. 282/11). No financial compensation was provided to participants. Circulating CD8+ T cells were enriched and processed as described above.
CRISPR knockout studies
Primary mouse CD8+ T cells were electroporated with gRNA-Cas9 ribonucleoproteins (RNPs) using the Amaxa 4D-Nucleofector system (Lonza). RNPs were prepared by complexing 180 pmol of gRNAs (IDT) with 60 pmol of Cas9 nuclease (IDT). Electroporation was performed in P4 Primary Cell solution using the program DS137. The nucleotide sequences for gRNA are listed in Supplementary Table 2.
Western blot analysis
For western blot analysis, cells were washed with ice-cold PBS and lysed in lysis buffer (Cell Signaling Technology) supplemented with 1 mM phenylmethyl sulfonyl fluoride (PMSF). Samples were frozen and thawed three times, followed by centrifugation at 20,000g for 10 min at 4 °C. Cleared protein lysate was denatured with LDS loading buffer for 10 min at 70 °C and loaded on precast 4–12% Bis–Tris protein gels (Life Technologies). Proteins were transferred onto nitrocellulose membranes using the iBLOT 2 system (Life Technologies) according to the manufacturer’s protocols. Membranes were blocked with 5% wt/vol milk or BSA and 0.1% Tween-20 in TBS and incubated with the appropriate antibodies in 5% wt/vol BSA in TBS with 0.1% Tween-20 at 4 °C overnight. The antibodies were from Cell Signaling and used at a dilution of 1:1,000 unless otherwise stated: PLCγ1 (2822), Phospho-PLCγ1 (Tyr783) (2821), Phospho-c-Raf (Ser338) (Clone 56A6, 9427), Phospho-MEK1/2 (Ser217/221) (Clone 41G9, 9154), MEK1/2 (4694), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Clone D13.14.4E, 4370), p44/42 MAPK (Erk1/2) (Clone L34F12, 4696), α-tubulin (2125S; 1:5,000 dilution), β-actin (Clone 13E5, 4970S), ZAP70 (Clone 99F2, 2705 S), Phospho-Akt (Thr308; clone D25E6, 13038S), Phospho-Akt (Ser743) (clone D9E, 4060S), Akt (4685S) and PIS1 (Santa Cruz, sc-514255). All primary antibody incubations were followed by incubation with secondary horseradish peroxidase-conjugated antibody (goat anti-rabbit IgG, 31460 or goat anti-mouse IgG, 31430, Pierce/Thermo Scientific) in 5% milk or BSA and 0.1% Tween-20 in TBS (1:5,000–1:20,000 dilution) and visualized on radiosensitive film (Amersham) or a ChemiDoc imaging system (Bio-Rad) using chemiluminescent substrate (SuperSignal West Pico or Femto, Pierce).
Flow cytometry
Extracellular staining was performed in 2% FBS/PBS for 30 min on ice, and dead cells were excluded with the LIVE/DEAD Fixable Dead Cell Stain Kit (Thermo Fisher Scientific). For intracellular cytokine staining, cells were reactivated with PMA (50 ng ml−1; Sigma) and ionomycin (500 ng ml−1; Sigma) in the presence of brefeldin A (0.1%; BioLegend) for five h before fixation using Cytofix Cytoperm (BD Biosciences). For the co-culture experiments, brefeldin A was added directly into the co-culture (without extra restimulation) five h before staining. For intracellular p-ERK1/2, p-PLCγ1 and phalloidin (Thermo Fisher Scientific, A22287) staining, cells were fixed using Cytofix Cytoperm (BD Biosciences) before incubation with the antibody for 30 min. For intracellular p-MEK1 staining, cells were fixed with the Phosphoflow Fix Buffer I (BD Biosciences), followed by permeabilization with Phosphoflow Perm/Wash Buffer I (BD Biosciences). The following fluorochrome-conjugated monoclonal antibodies were used (dilution 1:200 unless otherwise specified): For surface staining of mouse cells: anti-CD8α (clone 53–6.7, BioLegend, 100706, 100712, 100713, 100714, 100738), anti-CD45.1 (clone A20, eBioscience, 25–0453-82), anti-CD45.2 (clone 104, BD, 553772), anti-CD127 (clone A7R34, BioLegend, 135020), anti-KLRG1 (clone MAFA, BioLegend, 138418), anti-CD44 (clone IM7, BioLegend, 103027, 103056), anti-CD62L (clone MEL-14, BioLegend, 104407, 104438), anti-CD25 (clone 3C7, BioLegend, 101907 and clone PC61, BioLegend, 102008), anti-PD-1 (clone RMP1–30, BioLegend, 109110 and clone 29F.1A12, BioLegend, 109110) and anti-CD69 (clone H1.2F3, BioLegend, 104512); for surface staining of human cells: anti-CD4-APC-Cy7 (clone RPA-T4, BioLegend, 300518), anti-CD8-KrO (clone B9.11, Beckman Coulter, B00067), anti-HLA-DR (clone G46–6, eBioscience, 556643); for intracellular staining of mouse cells: anti-IFN-γ (clone XMG1.2, BioLegend, 505808, 505810 and 505806), anti-Granzyme B (clone GB11, BioLegend, 515406 and clone NZGB, eBioscience, 12–8898-82), anti-phospho-PLCγ1 (clone A17025A, BioLegend, 612403, dilution 1:25), anti-phospho-ERK1/2 (clone 6B8B69, BioLegend, 369506), anti-phospho-MEK1 (clone A16117B, BioLegend, 610608, dilution 1:25); for intracellular staining of human cells: anti-Ki67 (clone MIB-1, Agilent Dako, F726801–8).
EdU incorporation was measured using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay kit according to the provided instructions (Thermo Fisher Scientific, C10424). Cell proliferation was quantified using the CellTrace Violet Cell Proliferation kit for flow cytometry (Thermo Fisher Scientific, 34557). Flow cytometry analyses were performed on an LSRFortessa flow cytometer, FACSymphony and FACSCelesta flow cytometer (BD Biosciences) or CytoFLEX platform (Beckmann Coulter) and data were analyzed with FlowJo 10 software (BD Biosciences).
Analysis of membrane lipid order and lipid rafts
Cultured Teff cells or Jurkat cells were first stained with the LIVE/DEAD Fixable Dead Cell Stain Kit (Thermo Fisher Scientific), followed by incubation with 5 μM di-4-ANEPPDHQ (Thermo Fisher Scientific, D36802) for 30 min in the dark at 37 °C, 5% CO2. Flow cytometry analysis was performed on a FACSymphony flow cytometer (BD Biosciences) immediately without washing or fixing the cells. High-order cells emit at ~570 nm and low-order cells emit at ~630 nm, allowing the gating of two separate populations. The GP value was calculated as a quantitative measure for lipid order as previously described28:
Lipid rafts were quantified by measuring fluorescently labeled cholera toxin B subunit (CTB), which binds to glycosphingolipid GM1, an abundant component of lipid rafts. Cultured Teff cells or Jurkat cells were first stained with the LIVE/DEAD Fixable Dead Cell Stain Kit (Thermo Fisher Scientific), followed by incubation with 25 μg ml−1 Alexa Fluor 488-conjugated recombinant Cholera Toxin Subunit B (Thermo Fisher Scientific, C34775) for 30 min in the dark at 4 °C. Cells were washed twice with staining buffer before flow cytometry acquisition.
Cytolysis assay
OT-I splenocytes were stimulated with plate-bound anti-CD3 (5 μg ml−1) (InVivoMab anti-mouse CD3, BioXCell, BE0002), soluble anti-CD28 (0.5 μg ml−1; InVivoMab anti-mouse CD28, BioXcell, BE0015) and 100 U ml−1 IL-2 (Peprotech) for 48 h, then cultured for a further 24 h in IL-2 plus vehicle or CDIPTi. OT-I cells were then washed and plated with CellTrace Violet pre-stained EL4-OVA cells over a dilution series. The percentage of dead EL4-OVA cells was determined after 8 h by flow cytometry. For assessment of cytokine production by OT-I T cells, Brefeldin A was added for the final 5 h of co-culture.
Pharmacological inhibitors
Inostamycin (CDIPTi) was a kind gift of M. Imoto and E. Tashiro21. Inostamycin was dissolved at a concentration of 1 mg ml−1 in methanol and used in cell culture at the indicated concentrations. Next, 2-DG (Sigma, L07338.14) was used in cell culture at 5 mM. KA (Cayman Chemical, 14079) was used in cell culture at 5 μM, and A939572 (SCDi) was used in cell culture at 100 nM (Sigma, SML2356).
Phospholipase C activity assay
PLC activity was measured using a colorimetric assay kit (Abcam, ab273343) according to the manufacturer’s instructions. In total, 2 × 106 primary Teff cells or Jurkat cells were used per sample. Data were collected on a SpectraMax M3 instrument with the SoftaMax Pro 7.1.2 software.
Calcium measurement
Basal calcium was measured using the Indo-1 AM dye according to the manufacturer’s instructions (Thermo, I1223). In brief, cells were pelleted and resuspended in RPMI 1640 with 0.25 mM Indo-1 AM for 30 min at 37 °C. Cells were then washed and acquired by flow cytometry using the following parameters: unbound = 530/30, bound = 405/20. The bound/unbound ratio was calculated as a measure of basal calcium.
Organelle enrichment for lipidomic analysis
CD8+ T cells were isolated and activated as described above for 96 h. Samples of 20 × 106 cells per organelle compartment were harvested. Organelle enrichment was performed using the following kits from Invent Biotechnologies according to the manufacturer’s manuals: Minute Total Lipid Raft Isolation Kit for Mammalian Cells and Tissues (LR-039), Minute Mitochondria Isolation Kit for Mammalian Cells and Tissues (MP-007), Minute Lysosome Isolation Kit for Mammalian Cells/Tissues (LY-034), Minute Endosome Isolation and Cell Fractionation Kit (ED-028), Minute Golgi Apparatus Enrichment kit (GO-037), Minute ER Enrichment kit (ER-036) and Minute Cytoplasmic & Nuclear Extraction Kit for Cells (SC-003). Lipid extraction and PIPn measurement were performed as described below.
Stable isotope labeling studies
To measure the contribution of U-13C-glucose-derived carbon into PI synthesis, RPMI 1640 media (supplemented as above) was changed for RPMI 1640 no glucose media (supplemented as above) containing the indicated concentration of U-13C-glucose or unlabeled glucose for 6 h or 24 h as indicated.
Preparation of lipids for cell culture
Soy-derived PI (Avanti, 840044P), 18:0–20:4 PI (Avanti, 850144) and LA (Sigma, L1376) were dissolved at a final concentration of 10 mM in 20:9:1 (vol/vol/vol) chloroform:methanol:water. One hundred μl of dissolved lipid was dried per 2-ml glass vial using a Genevac EZ2 speed vac and stored at −20 °C until the day of use. Fatty-acid-free BSA (Roche, 3117057001) was dissolved in RPMI 1640 at 10 mg ml−1 (10 min, 37 °C), and then conjugated to PI or LA (1 h, 37 °C). BSA-conjugated PI was used in cell culture at a final concentration of 100 μM (1 mg ml−1 BSA). OA was purchased pre-conjugated to BSA and used at a final concentration of 100 μM (Sigma, 03008).
Lipid extraction
In general, lipids were extracted using a biphasic Methyl tert-butyl ether (MTBE) extraction protocol (adapted from Matyash et al.55). In brief, cells were resuspended in 100 μl cold PBS in 8-ml glass vials. Cold methanol (750 μl), MTBE (2 ml) and water (625 μl) were added sequentially with vortexing. Samples were centrifuged to separate phases, and the upper organic phase was taken into a 2-ml glass vial using a glass stripette. Samples were dried using a Genevac EZ2 speed vac. Dried samples were stored at 4 °C, then resuspended in 50 μl 2:1:1 (vol/vol) 2-propanol:acetonitrile:water and transferred to a sample vial with conical glass insert before analysis.
For measurements of glycosphingolipids, lipid extraction was performed with a Folch extraction protocol adapted from ref. 56. Briefly, cells were resuspended in 150 μl water, followed by addition of 400 μl methanol and 200 μl chloroform. Cells were then sonicated and after a centrifugation step, the clear supernatant was further processed. Phase separation was performed by addition of 100 μl 0.1 M KCl and the water/methanol-rich upper layer containing glycosphingolipids was collected. Solid phase extraction was performed using Bond Elut C8 cartridges (Agilent). Samples were dried using a Genevac EZ2 speed vac. Dried samples were stored at 4 °C, then resuspended in 50 μL 2:1:1 (vol/vol) 2-propanol:acetonitrile:water and transferred to a sample vial with conical glass insert before analysis.
Phosphatidylinositol phosphate extraction
PI phosphates were extracted and derivatized as described by Clark et al.57. In brief, cells were resuspended in 150 μl cold PBS in 2-ml tubes and quenched with 750 μl quench mixture (484 ml methanol, 242 ml CHCl3 and 23.55 ml 1 M HCl). Phases were separated by adding 725 μl CHCl3 and 170 μl 2 M HCl, and centrifuging at 12,500g for two min. The lower phase was collected and washed with 700 μl pre-derivatization wash (240 ml CHCl3, 120 ml methanol and 90 ml 0.01 M HCl). Samples were centrifuged at 12,500g for two min and the lower phase was collected. Extracts were derivatized by adding 50 μl 2 M TMS-diazomethane in hexane for 10 min at room temperature, before quenching with 6 μl glacial acetic acid. Samples were washed twice with 700 μl post-derivatization wash (240 ml CHCl3, 120 ml methanol and 90 ml H2O), and the lower phase was collected in a 2-ml glass vial. Then, 100 μl 9:1 (vol/vol) methanol:water was added to each sample before drying under a stream of nitrogen gas. Just before the sample was completely dry, extracts were resuspended in 50 μl 9:1 (vol/vol) methanol:water ready for acquisition by LC–QqQ–MS/MS.
Lipid measurement by LC–MS/MS
Chromatographic separation was performed on an Agilent 1290 infinity II UHPLC system using an Agilent Zorbax Eclipse Plus C18 column (100 × 2 mm, 1.8-μm particles). Buffer A was 10 mM ammonium formate in 60:40 acetonitrile:water. Buffer B was 10 mM ammonium formate in 90:10 2-propanol:acetonitrile. The gradient profile was: 0 min, 30% B, 0.2 ml min−1; 0.5 min, 30% B, 0.25 ml min−1; 0.6 min, 30% B, 0.4 ml min−1; 1.2 min, 30% B, 0.4 ml min−1; 5.2 min, 68% B, 0.4 ml min−1; 21.2 min, 75% B, 0.4 ml min−1; 21.7 min, 97% B, 0.4 ml min−1; 26.0 min, 97% B, 0.4 ml min−1; 26.5 min, 30% B, 0.4 ml min−1; stop time, 28 min. Column temperature was 35 °C and autosamper temperature was 5 °C.
For LC–QqQ–MS/MS analysis, the LC system was coupled to an Agilent 6495 Triple Quad QQQ-MS. Data were acquired with the Mass-Hunter LC/MS Data Acquisition Software (Agilent, version B.08.02). Lipids were identified by fragmentation and retention time (as predetermined by standards for each subclass), and peak area was determined using Agilent Mass Hunter software. To determine the concentration of PI, peak areas were normalized to the peak area of PI 8:0/8:0 (Avanti, 850181 P) that was added to samples before extraction.
For LC–QTOF–MS analysis, the LC system was coupled to a Bruker impact II QTOF MS or a Bruker timsTOF MS equipped with an ion-Booster ESI source. The mass spectrometer was operated in negative mode without Auto MS/MS. The mass axis was calibrated at the beginning of every sample run. Data were acquired with the Compass Hystar software (Bruker, version 5.1.8.2). Lipids were identified by accurate mass, isotope distribution and by matching retention time to LC–QqQ–MS/MS data. Lipid peak areas were determined using the R package AssayR58 or the software Metaboscape and TASQ (Bruker).
Phosphatidylinositol phosphate measurement by LC–MS
Targeted quantification of derivatized PIP2 and PIP3 species was carried out using an Agilent 1290 Infinity II UHPLC in line with an Agilent 6495 QQQ-MS operated in MRM mode. MRM settings were optimized for a pure standard of PIP2 16:10/16:0 and propagated to other PIP2 and PIP3 species in accordance with the transitions described by Clark et al.57. LC separation was on a Waters BSH C4 column (100 × 2 mm, 1.7-μm particles) using a solvent gradient of 40% buffer A (0.1% formic acid in milliQ-H2O) to 100% buffer B (0.1% formic acid in acetonitrile). The flow rate was 400 μl min−1, autosampler temperature was 5 °C and injection volume was 10 μl. Data processing was performed using Agilent MassHunter software.
Phosphoinositide conversion assay
For the PI conversion assay, 16:0–18:1 PI(4)P and 18:0–20:4 PI(4) were purchased from Avanti and liposomes were produced by mixing 85 mol % 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 10 mol % phosphatidylserine (POPS) with 5 mol % of the respective PI together with 0.1% Atto647N-DOPE or NBD-DPPE. The mixtures were evaporated under nitrogen flow and dried overnight in a vacuum. Dried lipids were resuspended in buffer containing 20 mM HEPES, 150 mM NaCl and 0.5 mM TCEP at 37 °C and subjected to six cycles of freeze thawing in liquid nitrogen. Liposomes were extruded to 100 nm and aliquoted, frozen in liquid nitrogen and stored at −20 °C. Membrane-coated beads were generated by mixing liposomes and 10-μm silica beads (Whitehouse Scientific) in 200 mM NaCl. Beads were washed twice with 20 mM HEPES and resuspended in buffer containing 20 mM HEPES, 150 mM NaCl and 0.5 mM TCEP.
Phosphoinositide conversion assay was carried out as previously described29. Membrane-coated beads were washed into 100 μl kinase buffer (20 mM HEPES (pH 7.0), 150 mM NaCl, 5 mM MgCl2, 0.5 mM EGTA, 200 μg ml−1 β-casein, 20 mM BME and 20 mM glucose) in the presence of 1 mM ATP. Samples were applied to an imaging chamber and purified PLCδ-PH fused to RFP was added to a final concentration of 150 nM. Immediately before acquisition, reconstituted PIP5K1C was added to the beads at a final concentration of 50 nM. The reaction was monitored using a Leica SP8 Confocal Microscope with a Leica HC PL APO CS2 ×63/1.40 oil-immersion objective at 0.75 base zoom with 1,024 × 1,024-pixel scan at 3.4 s per frame. PLCδ-PH fused to RFP was purified from BL21 cells (CMC0014, Scientific Laboratory Supplies-reagents) and PIP5K1C was purified from Sf9 insect cells (B82501, Thermo Fisher) as described in detail elsewhere29. Recruitment of PLCδ was determined by measuring the MFIs on the beads by segmentation of either the NBD or the Atto647N signal as mask using ImageJ.
Data collection
Data collection and analysis were not performed blind to the conditions of the experiments. For in vitro experiments, T cells were equally distributed into multi-well plates and the treatment condition was randomly applied. For in vivo animal experiments, no conscious biases were used to assign experimental groups. Mice were randomly assigned between groups and analyzed along age-matched and sex-matched controls. For human participants, the allocation to a certain group was based on the clinical condition that was used as an experimental model (for example, healthy donor versus participant with an acute EBV infection). The study of humans was observational and not interventional, so that no randomization to different treatment groups was possible/necessary. No data were excluded.
Statistics and normalization
No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications35,44,59. Statistical analysis was performed using Prism 9 software (GraphPad) unless otherwise indicated. Data distribution was assumed to be normal but this was not formally tested. Results are represented as the mean ± s.e.m. Comparisons of two groups were calculated using an unpaired two-tailed Student’s t-test; when one group was used for normalization and thus all values in this group were equal (for example, 0 or 1), a one-sample t-test was applied. Comparisons of more than two groups were calculated using ordinary one-way ANOVA or two-way ANOVA with the indicated appropriate test for multiple comparisons. Comparisons of matched participant samples were calculated using a two-tailed Wilcoxon matched-pairs signed-rank test. The selection of sample size was based on extensive experience with metabolic and in vivo tumor immunology assays. Quantile normalization of lipidomic data was carried out in R using the R package ‘preprocessCore’ (ref. 60). Statistical analysis of whole lipidomic datasets was carried out in R using a one-way ANOVA.
Extended Data
Extended Data Fig. 1 |. Human CD8+ TE accumulate PIPn with saturated acyl chains.
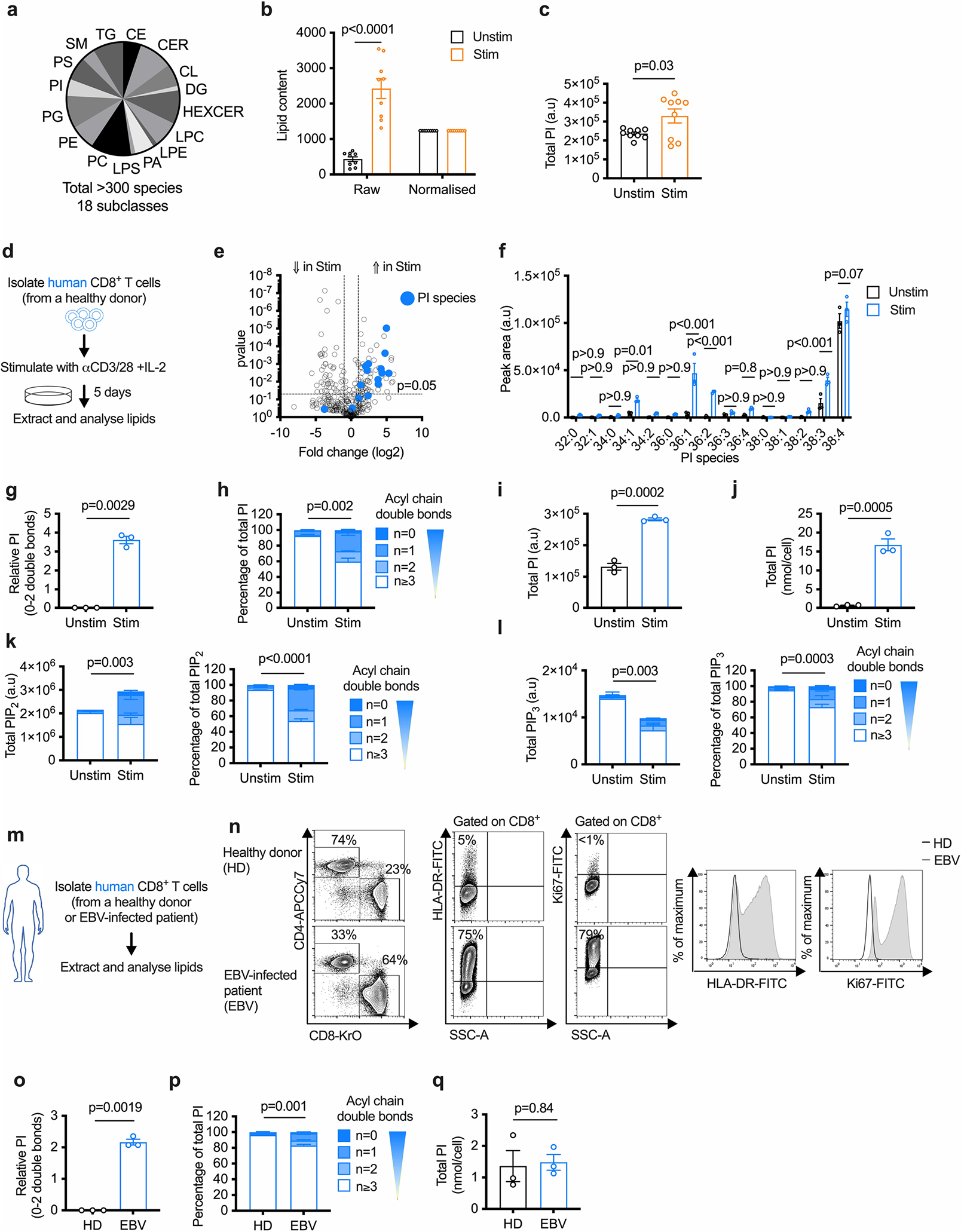
a, Pie chart showing the number of lipid species measured by LC-QqQ-MS/MS in each of the 18 lipid subclasses. b-c, WT CD8+ T cells were stimulated (Stim) with anti-CD3, anti-CD28 and IL-2 or cultured with IL-7 (unstim) for 3 days. b, Median lipid content per million cells before (raw) and after (normalized) quantile normalization is shown. n = 9 biologically independent samples across 3 independent experiments; unpaired two-tailed t-test. c, Relative total PI level calculated from quantile normalized lipid peak areas. n = 9 biologically independent samples across 3 independent experiments; unpaired two-tailed t-test. d, Experimental schematic. e-f, Volcano plot shows the log2 fold change (e) or total PI (f) between Unstim and Stim conditions using quantile normalized lipid peak areas. n = 3 biologically independent samples; one-way ANOVA or two-way ANOVA corrected for multiple comparisons (Sidak test). g, Relative intensity of saturated PI normalized to Unstim (log2 fold change). n = 3 biologically independent samples; one sample t-test. h, PI saturation percentage. n = 3 biologically independent samples; unpaired two-tailed t-test comparing saturated PI. I, Total PI. n = 3 biologically independent samples; unpaired two-tailed t-test. j, Total PI concentration (nmol/cell). n = 3 biologically independent samples; unpaired two-tailed t-test. k, Saturation of acyl chains in human TE is expressed as a relative proportion of the total PIP2 (left panel) and as a percentage of total PIP2 (right panel). n = 5 biologically independent samples; unpaired two-tailed t-test comparing the total or percentage saturated PIP2. Total polyunsaturated PIP2 was not significantly different. l, Saturation of acyl chains in human TE is expressed as a relative proportion of the total PIP3 (left panel) and as a percentage of PIP3 (right panel). n = 5 biologically independent samples; unpaired two-tailed t-test comparing the total or percentage saturated PIP3. Total polyunsaturated PIP3 was also significantly different (p < 0.001). m, Experimental schematic. n, Percentage CD8+ cells, activation marker HLA-DR+ cells, and proliferation marker Ki67+ cells are shown. MFI of HLA-DR and Ki67 are also shown. o, Relative intensity of saturated PI normalized to HD (log2 fold change). n = 3 HD and n = 3 EBV biologically independent samples; one sample t-test. p, PI saturation. n = 3 HD and n = 3 EBV biologically independent samples; unpaired two-tailed t-test comparing the summed percentage of saturated PI. q, Total PI concentration (nmol/cell). n = 3 HD and n = 3 EBV biologically independent samples; unpaired two-tailed t-test. Error bars show the standard error of the mean. a.u., arbitrary units.
Extended Data Fig. 2 |. CDIPT inhibition or deletion impairs TE fitness and function.
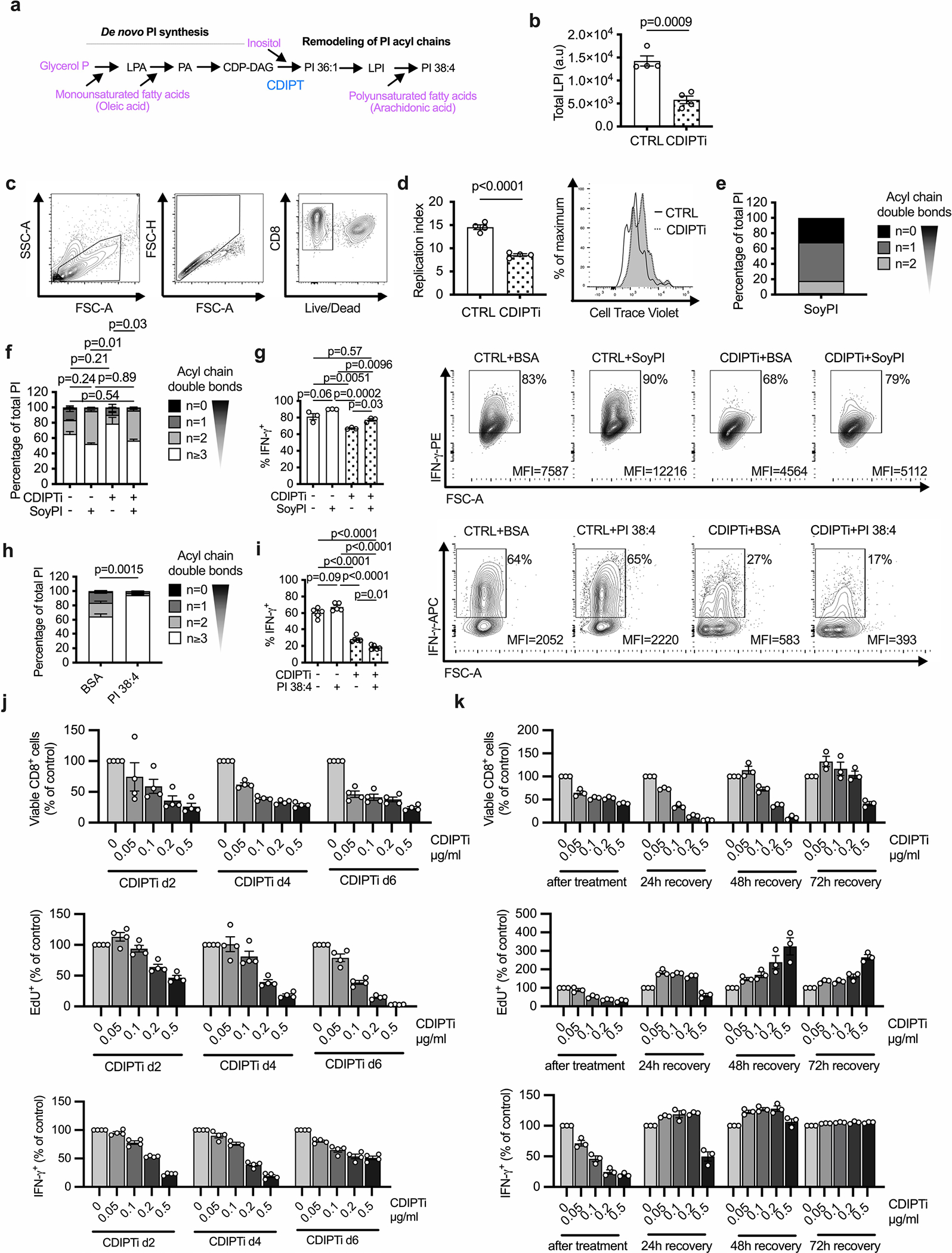
a, Simplified overview of PI de novo synthesis and remodeling. b, WT CD8+ T cells were activated and treated as in Fig. 2a. Total lysophosphatidlyinositol (LPI) content is shown. n = 4 biologically independent samples; unpaired two-tailed t-test. c, Gating strategy for flow cytometry analysis. d, Replication Index based on cell trace violet dilution was calculated using FlowJo software. n = 4 biologically independent samples; unpaired two-tailed t-test. e, PI species present in PI derived from Soy expressed by degree of acyl chain saturation. f-g, Cells were prepared as in (b), with the addition of BSA (10 mg/ml) control or BSA conjugated to Soy-derived PI (SoyPI, 100 μM) for the final day. f, Saturation of acyl chains expressed as a percentage of total PI. n = 3 biologically independent samples, one-way ANOVA corrected for multiple comparisons (Tukey test) comparing saturated PI. g, Percentage IFN-γ+ cells are shown, gated on Live/Dead-aqua− and CD8-Brilliant Violet 421+. n = 3 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Tukey test). h-I, Cells were prepared as in (b), with the addition of BSA (10 mg/ml) control or BSA conjugated to PI 38:4 (100 μM) for the final day. h, Saturation of acyl chains expressed as a percentage of total PI. n = 3 biologically independent samples; unpaired two-tailed t-test comparing saturated PI. I, Percentage IFN-γ+ cells are shown, gated on Live/Dead-near-IR− and CD8-Brilliant Violet 421+. n = 5 biologically independent samples representative of three experiments; one-way ANOVA corrected for multiple comparisons (Tukey test). j, WT CD8+ T cells were activated as in Fig. 2a and the CDIPT inhibitor inostamycin was added at the indicated concentrations for 24 h starting either after either two, four or six days of activation. Upper panel: percentage of viable CD8+ cells. Cells were gated on FSC and SSC and single cells. Middle panel: percentage of EdU+ cells. Cells were gated on Live/Dead-aqua− CD8-APC-Cy7+. Lower panel: percentage of IFN-γ+ cells. Cells were gated on Live/Dead-aqua− CD8-APC-Cy7+. For each time point, the values were normalized to the values of the control samples (0 μg/ml CDIPTi). n = 4 biologically independent samples representative of two independent experiments. k, WT CD8+ T cells were activated and treated as in Fig. 2a. After 24 h of CDIPTi treatment, the cells were cultured in media without CDIPTi for the indicated time intervals. Upper panel: percentage of viable CD8+ cells. Cells were gated on FSC and SSC and single cells. Middle panel: percentage of EdU+ cells. Cells were gated on Live/Dead-aqua− CD8-APC-Cy7+. Lower panel: percentage of IFN-γ+ cells. Cells were gated on Live/Dead-aqua− CD8-APC-Cy7+. For each time point, the values were normalized to the values of the control samples at the respective time point (0 μg/ml CDIPTi). n = 3 biologically independent samples representative of two independent experiments. Error bars show s.e.m.
Extended Data Fig. 3 |. CRISPR-Cas9 deletion efficiency for CDIPT and ZAP70.
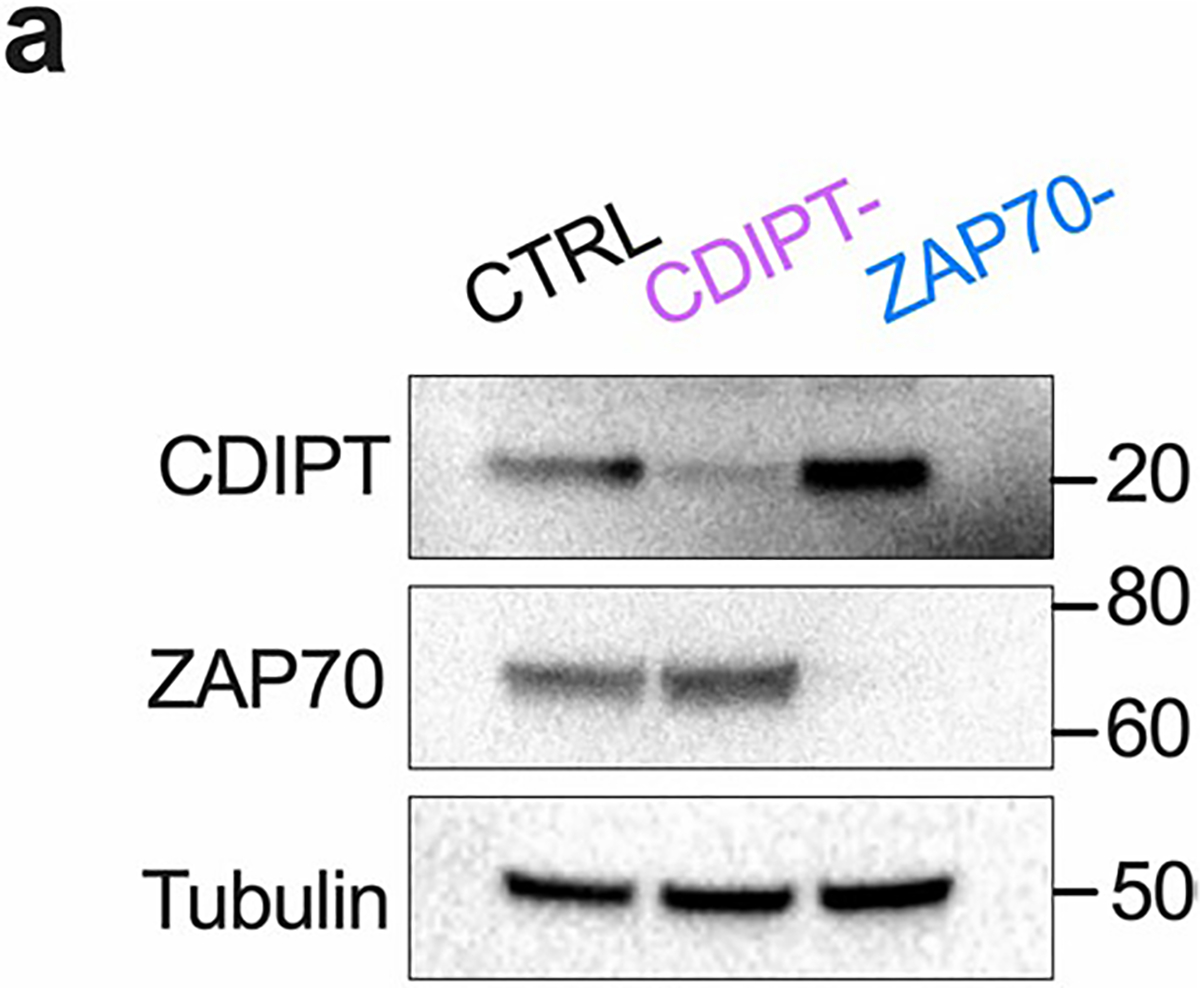
a, The efficiency of CRISPR-Cas9 for CDIPT and ZAP70 was verified by measuring protein expression.
Extended Data Fig. 4 |. Inhibition of CDIPT changes membrane lipid order and impairs lipid raft formation.
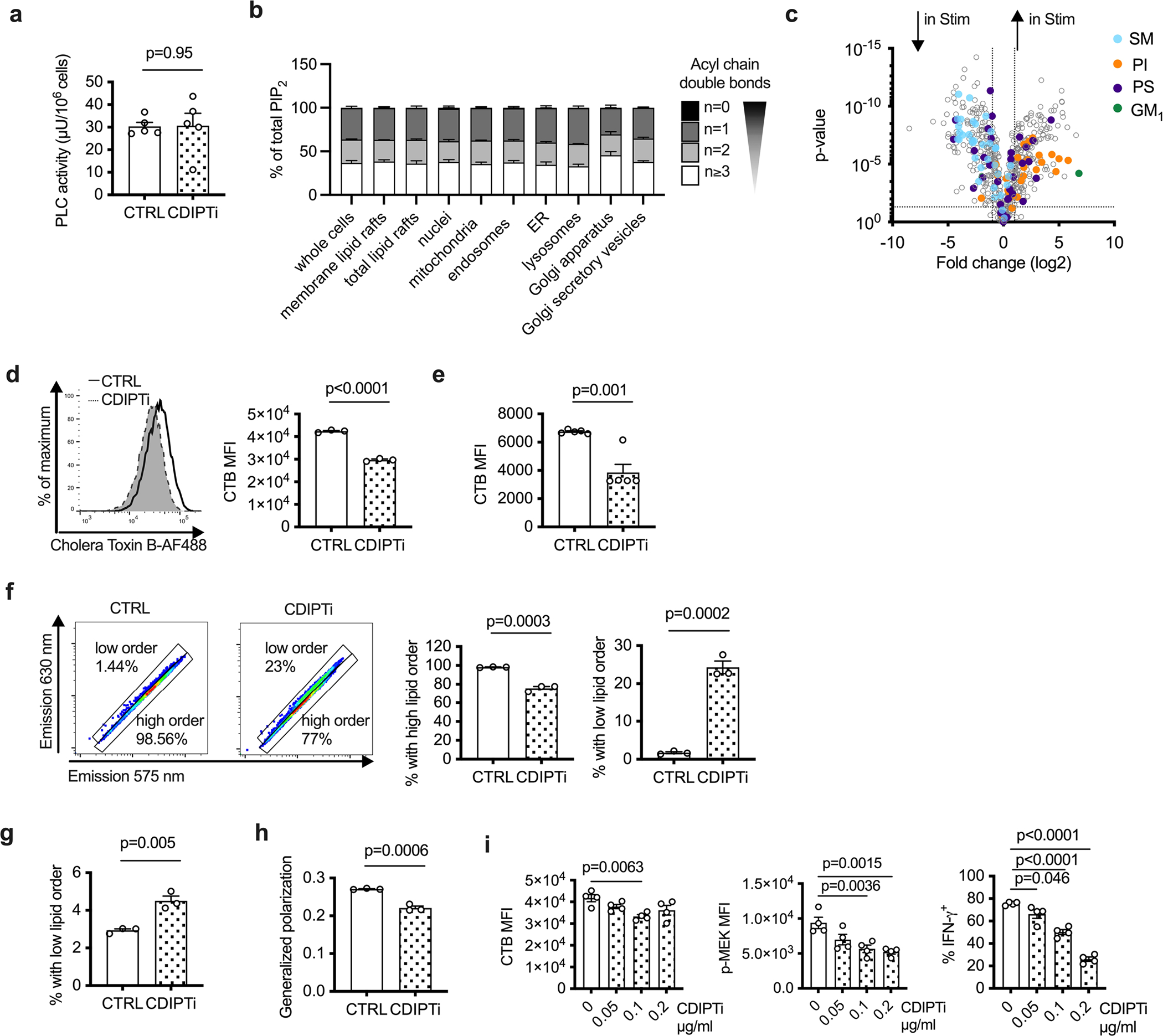
a, Phospholipase C activity in Jurkat T cells cultured in the presence or absence of 0.5 μg/ml CDIPT inhibitor and activated for 30 min with 50 ng/ml PMA and 500 ng/ml ionomycin. n = 5 biologically independent samples representative of 2 independent experiments; unpaired two-tailed t-test. b, WT CD8+ T cells were activated and cellular compartments were enriched as in Fig. 4i. Saturation of acyl chains expressed as a percentage of total PIP2 across organelles. n = 4 biologically independent samples. c, WT CD8+ T cells were isolated and activated or cultured in IL-7 as in Fig. 1a. Volcano plot shows the log2 fold change between Unstim and Stim using quantile normalized lipid peak areas. n = 5 biologically independent samples across 2 independent experiments; one-way ANOVA. Lipids from the following subclasses are highlighted: sphingomyelin (SM) in blue, phosphatidylinositol (PI) in orange, phosphatidylserine (PS) in purple and ganglioside GM1 in green. d, Cholera toxin subunit B measurement on primary CD8+ TE activated and treated as in Fig. 2a. n = 3 biologically independent samples representative of 3 independent experiments; unpaired two-tailed t-test. e, Cholera toxin subunit b measurement on Jurkat T cells treated as in (a). n = 5 biologically independent samples representative of 2 independent experiments; unpaired two-tailed t-test. f, WT CD8+ T cells were activated and treated as in Fig. 2a. Membrane lipid order was assessed by a di-4-ANEPPDHQ staining. Percentage of cells with high lipid order (di-4-ANEPPDHQ emission at 630 nm) and low lipid order (di-4-ANEPPDHQ emission at 570 nm). n = 3 biologically independent samples representative of 3 independent experiments; unpaired two-tailed t-test. g, Percentage of Jurkat T cells with low lipid order as determined by di-4-ANEPPDHQ flow cytometry staining after treatment as in (a). n = 3 biologically independent samples representative of two independent experiments; unpaired two-tailed t-test. (h) Generalized polarization (GP) index as a normalized intensity ratio of the two spectral emissions for CD8+ TE analyzed as in (e). n = 3 biologically independent samples representative of 3 independent experiments; unpaired two-tailed t-test. (I) WT CD8+ T cells were activated and treated as in Fig. 2a except that the CDIPT inhibitor was used at the indicated concentrations. Cells were gated on Live/Dead-aqua− CD8-APC-Cy7+. Left panel: Cholera toxin subunit B binding. Middle panel: mean fluorescence intensity of phosphorylated MEK1. Right panel: Percentage of IFN-γ+ cells. n = 4 biologically independent samples representative of two independent experiments; one-way ANOVA with Dunnett’s multiple comparisons test comparing all groups to ‘0 μg/ml’. Error bars show the standard error of the mean.
Extended Data Fig. 5 |. TE PIPn synthesis depends on glycolytic metabolism.
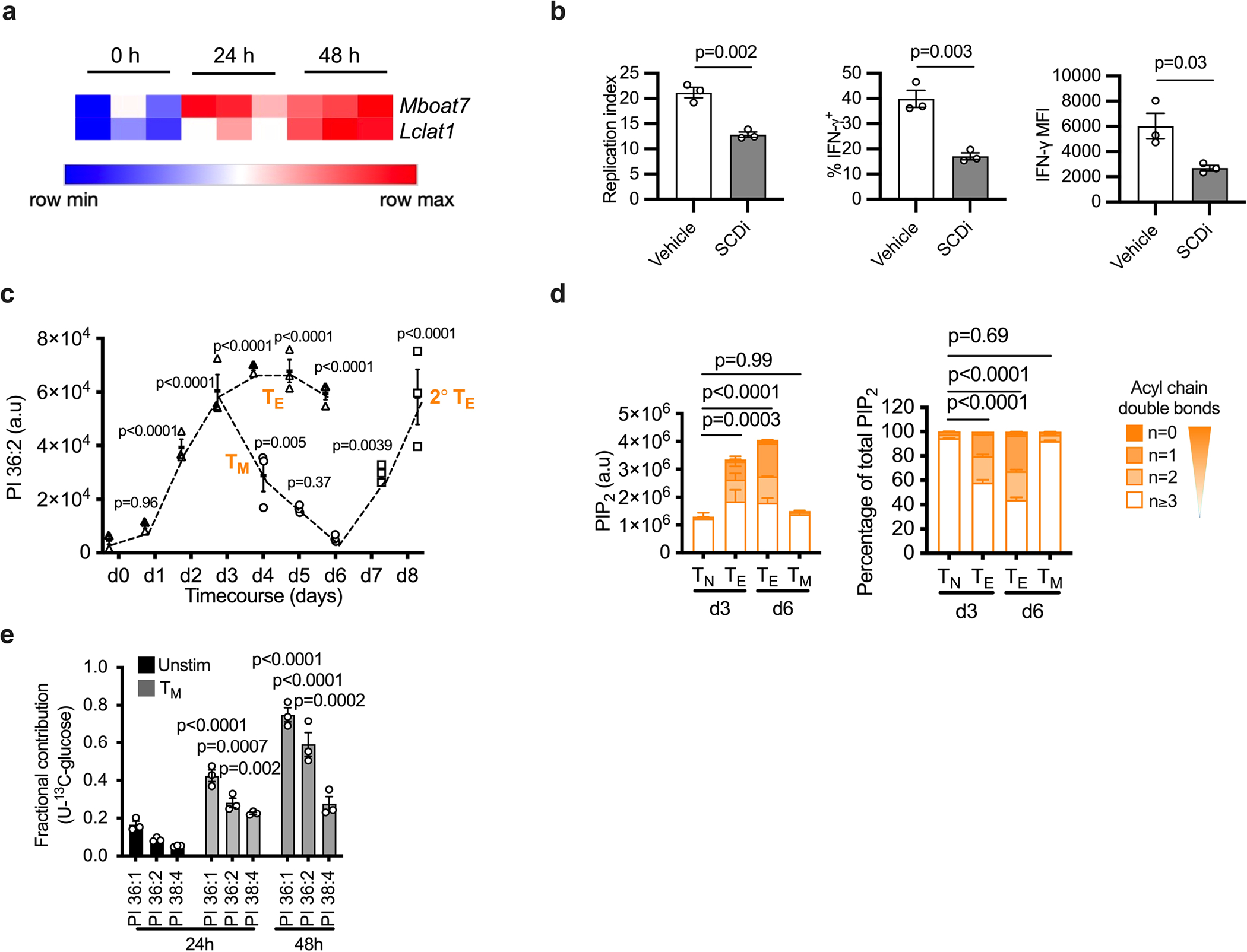
a, WT CD8+ T cells were isolated from spleens and lymph nodes of C57BL/6 mice then stimulated with plate-bound anti-CD3 (5 μg/ml), soluble anti-CD28 (0.5 μg/ml) and IL-2 (100 U/ml) for 2 days. Samples for RNA sequencing were harvested every 24 h. Heatmap depicting the gene expression levels of Lclat1 and Mboat7 in n = 3 biologically independent samples. b, WT CD8+ T cells were isolated and activated as in (a), followed by a treatment for 24 h with the Stearoyl-CoA-desaturase inhibitor A939572 (SCDi, 100 nM) in the presence of IL-2. Left panel: Replication Index based on Cell Trace Violet dilution calculated using FlowJo software. Middle and right panel: intracellular expression of IFN-γ. Cells were gated on Live/Dead-IR−, CD8-APC+. n = 3 biologically independent samples; unpaired two-tailed t-test. c, d WT CD8+ T cells were isolated and activated as in (a), followed by culture in IL-2 for 24 h. On d3, cells were either switched to IL-15 (100 U/ml, TM) or maintained in IL-2 until d6. TM were re-activated on d6 by stimulation with plate-bound anti-CD3 (5 μg/ml), soluble anti-CD28 (0.5 μg/ml) and IL-2 (100 U/ml) for 2 days to generate secondary TE. c, Relative amount of PI 36:2 measured every 24 h. n = 3 biologically independent samples; one-way ANOVA corrected for multiple comparisons (Dunnett test) compared all groups to 0 h timepoint. d, Saturation of acyl chains expressed as a proportion of total PIP2 (left panel) and as a percentage of PIP2 (right panel). Statistical analysis of the saturated PI on n = 5 independent biological samples; one-way ANOVA with Dunnett’s multiple comparisons’s test compared all groups to ‘Tn d3’. e, WT CD8+ T cells were differentiated into TM as in (c), with the addition of 100% U-13C-glucose 24 h before lipid analysis at each timepoint. Unstimulated cells were cultured in IL-7 with 100% U-13C-glucose. Fractional contribution of 13C-glucose-derived carbon to PI species was calculated. n = 3 biologically independent samples, two-way ANOVA with correction for multiple comparisons (Sidak’s test) comparing PI species in each group with the relevant 24 h unstimulated PI. Error bars show the standard error of the mean.
Extended Data Fig. 6 |. Elevated numbers and cytokine production by CD8+ T cells after immunotherapy.
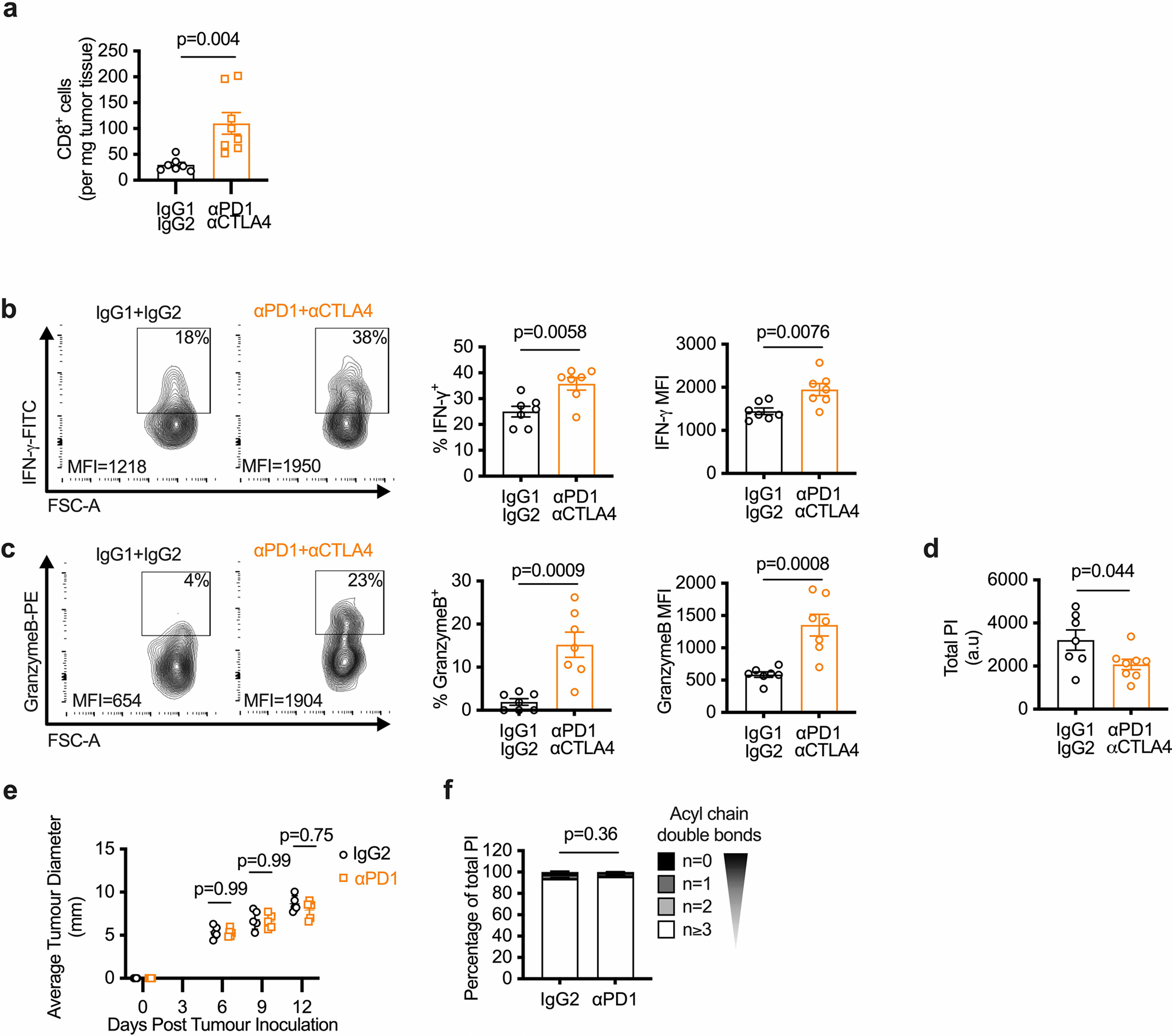
a–d, Sex-matched C57BL/6 mice were injected in the right flank with 1 × 106 B16-F10-OVA cells. After 3 days, mice were given i.p. injections of anti-PD1 and anti-CTLA4 (aPD1 + aCTLA4) or matching isotype controls (IgG1 + IgG2) every 3 days for 3 rounds in total. Tumor growth was measured up to day 16 post-tumor injection and CD8+ T cells were isolated on day 12 post-tumor injection to measure T cell function and lipid composition. a, CD8+ tumor infiltrating lymphocytes (TILs) on day 12 post-tumor injection normalized to tumor weight. n = 7 IgG1 + IgG2 and n = 8 aPD1 + aCTLA4 biologically independent samples; unpaired two-tailed t-test. b–c, TILs were isolated day 12 post-tumor injection and restimulated with PMA/ionomycin plus brefeldin A for 5 h in vitro. n = 7 biologically independent samples per group, unpaired two-tailed t-test. b, Representative contour plots of IFN-γ expression (left panel), percentage IFN-γ+ (middle panel) and IFN-γ expression (right panel) are shown. c, Representative contour plots of Granzyme B expression (left panel), percentage Granzyme B+ (middle panel) and Granzyme B expression (right panel) are shown. d, Relative total PI calculated from quantile normalized lipid peak areas. n = 7 IgG1 + IgG2 and n = 8 aPD1 + aCTLA4 biologically independent samples; unpaired two-tailed t-test. e-f, Sex-matched C57BL/6 mice were injected in the right flank with 1×106 B16-F10-OVA cells. After 3 days, mice were given i.p. injections of anti-PD1 antibody or matching isotype control (IgG2) every 3 days for 3 rounds in total. Tumor growth was measured up to day 12 post-tumor injection and CD8+ T cells were isolated on day 12 post-tumor injection to measure T cell function and lipid composition. e, Average tumor diameter measured at the indicated time points. n = 5 biologically independent samples per group; two-way ANOVA corrected for multiple comparisons (Sidak test) comparing the two groups at each time point across the dataset. f, Lipids were analyzed from TILs on day 12 post-tumor injection. Saturation of acyl chains is expressed as a percentage of total PI. n = 5 biologically independent samples per group; unpaired two-tailed t-test comparing saturated PI. Error bars show the standard error of the mean.
Supplementary Material
Acknowledgements
The authors thank A. Shaw and Z. Katz for insightful discussion. This work was supported by the Max Planck Society, the Leibniz Prize, and the National Institutes of Health R01AI156274 (to E.L.P.), two Bloomberg Distinguished Professorships (to E.L.P. and E.J.P.), a research grant from Bristol Myers Squibb, a Marie-Sklodowska-Curie actions Individual Fellowship (to F.B.), a Sir Henry Wellcome Fellowship (to D.J.P.), two Alexander von Humboldt Fellowships (to M.V. and M.C.) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—project no 492259164 (to P.A.) and SFB-1479 project no. 441891347 (to R.Z.).
Footnotes
Competing interests
E.L.P. is an SAB member of ImmunoMet Therapeutics and E.L.P. and E.J.P are Founders and Scientific Advisors to Rheos Medicines. The other authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41590-023-01419-y.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41590-023-01419-y.
Peer review information Nature Immunology thanks Michael Dustin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: N. Bernard, in collaboration with the Nature Immunology team.
Reprints and permissions information is available at www.nature.com/reprints.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA sequencing data were accessed in the Gene Expression Omnibus under accession number GSE17124559. Source data are provided with this paper.
References
- 1.Waickman AT & Powell JD mTOR, metabolism, and the regulation of T cell differentiation and function. Immunol. Rev. 249, 43–58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacIver NJ, Michalek RD & Rathmell JC Metabolic regulation of T lymphocytes. Annu Rev. Immunol. 31, 259–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shyer JA, Flavell RA & Bailis W Metabolic signaling in T cells. Cell Res 30, 649–659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carr EL et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 185, 1037–1044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang R et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maciver NJ et al. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 84, 949–957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cham CM & Gajewski TF Glucose availability regulates IFN-γ production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol. 174, 4670–4677 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Barneda D, Cosulich S, Stephens L & Hawkins P How is the acyl chain composition of phosphoinositides created and does it matter? Biochem. Soc. Trans. 47, 1291–1305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sasaki T et al. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 48, 307–343 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Czech MP PIP2 and PIP3: complex roles at the cell surface. Cell 100, 603–606 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Huang YH & Sauer K Lipid signaling in T cell development and function. Cold Spring Harb. Perspect. Biol. 2, a002428 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berridge MJ Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 1793, 933–940 (2009). [DOI] [PubMed] [Google Scholar]
- 13.D’Souza WN, Chang CF, Fischer AM, Li M & Hedrick SM The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol. 181, 7617–7629 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joseph N, Reicher B & Barda-Saad M The calcium feedback loop and T cell activation: how cytoskeleton networks control intracellular calcium flux. Biochim. Biophys. Acta 1838, 557–568 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Dandekar RD, Mao YS, Yin HL & Wulfing C Phosphatidylinositol (4,5) bisphosphate controls T cell activation by regulating T cell rigidity and organization. PLoS One 6, e27227 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vickers JD & Mustard JF The phosphoinositides exist in multiple metabolic pools in rabbit platelets. Biochem. J. 238, 411–417 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakrabarti P et al. A dPIP5K dependent pool of phosphatidylinositol 4,5 bisphosphate (PIP2) is required for G-protein-coupled signal transduction in Drosophila photoreceptors. PLoS Genet. 11, e1004948 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita A, Cheng J, Tauchi-Sato K, Takenawa T & Fujimoto T A distinct pool of phosphatidylinositol 4,5-bisphosphate in caveolae revealed by a nanoscale labeling technique. Proc. Natl Acad. Sci. USA 106, 9256–9261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costello PS, Gallagher M & Cantrell DA Sustained and dynamic inositol lipid metabolism inside and outside the immunological synapse. Nat. Immunol. 3, 1082–1089 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Latour S & Fischer A Signaling pathways involved in the T cell-mediated immunity against Epstein–Barr virus: lessons from genetic diseases. Immunol. Rev. 291, 174–189 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Imoto M, Taniguchi Y & Umezawa K Inhibition of CDP-DG: inositol transferase by inostamycin. J. Biochem. 112, 299–302 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Bengsch B et al. Analysis of CD127 and KLRG1 expression on hepatitis C virus-specific CD8+ T cells reveals the existence of different memory T cell subsets in the peripheral blood and liver. J. Virol. 81, 945–953 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward SG & Cantrell DA Phosphoinositide 3-kinases in T lymphocyte activation. Curr. Opin. Immunol. 13, 332–338 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Hawse WF & Cattley RT T cells transduce T cell receptor signal strength by generating different phosphatidylinositols. J. Biol. Chem. 294, 4793–4805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang JR, Byeon Y, Kim D & Park SG Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 52, 750–761 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janes PW, Ley SC, Magee AI & Kabouridis PS The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 12, 23–34 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Holmgren J, Lonnroth I & Svennerholm L Tissue receptor for cholera exotoxin: postulated structure from studies with GM1 ganglioside and related glycolipids. Infect. Immun. 8, 208–214 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waddington KE, Pineda-Torra I & Jury EC Analyzing T cell plasma membrane lipids by flow cytometry. Methods Mol. Biol. 1951, 209–216 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Maib H & Murray DH A mechanism for exocyst-mediated tethering via Arf6 and PIP5K1C-driven phosphoinositide conversion. Curr. Biol. 32, 2821–2833 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Chen YQ, Li S, Konrad RJ & Cao G The microsomal cardiolipin remodeling enzyme acyl-CoA lysocardiolipin acyltransferase is an acyltransferase of multiple anionic lysophospholipids. J. Lipid Res. 50, 945–956 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bone LN et al. The acyltransferase LYCAT controls specific phosphoinositides and related membrane traffic. Mol. Biol. Cell 28, 161–172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HC et al. Caenorhabditis elegans mboa-7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mol. Biol. Cell 19, 1174–1184 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Windt GJ et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl Acad. Sci. USA 110, 14336–14341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bengsch B et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 45, 358–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang CH et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolchok JD et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 369, 122–133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hawse WF, Boggess WC & Morel PA TCR signal strength regulates Akt substrate specificity to induce alternate murine Th and T regulatory cell differentiation programs. J. Immunol. 199, 589–597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee KH et al. T cell receptor signaling precedes immunological synapse formation. Science 295, 1539–1542 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Lee KH et al. The immunological synapse balances T cell receptor signaling and degradation. Science 302, 1218–1222 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Traynor-Kaplan A et al. Fatty-acyl chain profiles of cellular phosphoinositides. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 513–522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naguib A et al. p53 mutations change phosphatidylinositol acyl chain composition. Cell Rep. 10, 8–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Windt GJ & Pearce EL Metabolic switching and fuel choice during T cell differentiation and memory development. Immunol. Rev. 249, 27–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frauwirth KA et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Ho PC et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barneda D et al. Acyl chain selection couples the consumption and synthesis of phosphoinositides. EMBO J. 41, e110038 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shulga YV, Anderson RA, Topham MK & Epand RM Phosphatidylinositol-4-phosphate 5-kinase isoforms exhibit acyl chain selectivity for both substrate and lipid activator. J. Biol. Chem. 287, 35953–35963 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blunsom NJ & Cockcroft S Phosphatidylinositol synthesis at the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cel. Biol. Lipids 1865, 158471 (2020). [DOI] [PubMed] [Google Scholar]
- 48.D’Souza K & Epand RM The phosphatidylinositol synthase-catalyzed formation of phosphatidylinositol does not exhibit acyl chain specificity. Biochemistry 54, 1151–1153 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Pike LJ Lipid rafts: bringing order to chaos. J. Lipid Res. 44, 655–667 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Myeong J, Park CG, Suh BC & Hille B Compartmentalization of phosphatidylinositol 4,5-bisphosphate metabolism into plasma membrane liquid-ordered/raft domains. Proc. Natl Acad. Sci. USA 118, e2025343118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veri MC et al. Membrane raft-dependent regulation of phospholipase Cγ−1 activation in T lymphocytes. Mol. Cell. Biol. 21, 6939–6950 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kallikourdis M et al. Phosphatidylinositol 4-phosphate 5-kinase beta controls recruitment of lipid rafts into the immunological synapse. J. Immunol. 196, 1955–1963 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Parry RV et al. CTLA-4 and PD-1 receptors inhibit T cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Callahan MK, Postow MA & Wolchok JD Targeting T cell co-receptors for cancer therapy. Immunity 44, 1069–1078 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A & Schwudke D Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 49, 1137–1146 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong M, Xu G, Park D, Barboza M & Lebrilla CB Intact glycosphingolipidomic analysis of the cell membrane during differentiation yields extensive glycan and lipid changes. Sci. Rep. 8, 10993 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark J et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat. Methods 8, 267–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wills J, Edwards-Hicks J & Finch AJ AssayR: a simple mass spectrometry software tool for targeted metabolic and stable isotope tracer analyses. Anal. Chem. 89, 9616–9619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Sullivan D et al. Fever supports CD8+ effector T cell responses by promoting mitochondrial translation. Proc. Natl Acad. Sci. USA 118, e2023752118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bolstad B preprocessCore: a collection of pre-processing functions. R package version 1.42.0 https://github.com/bmbolstad/preprocessCore [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data were accessed in the Gene Expression Omnibus under accession number GSE17124559. Source data are provided with this paper.