

SUMMARY
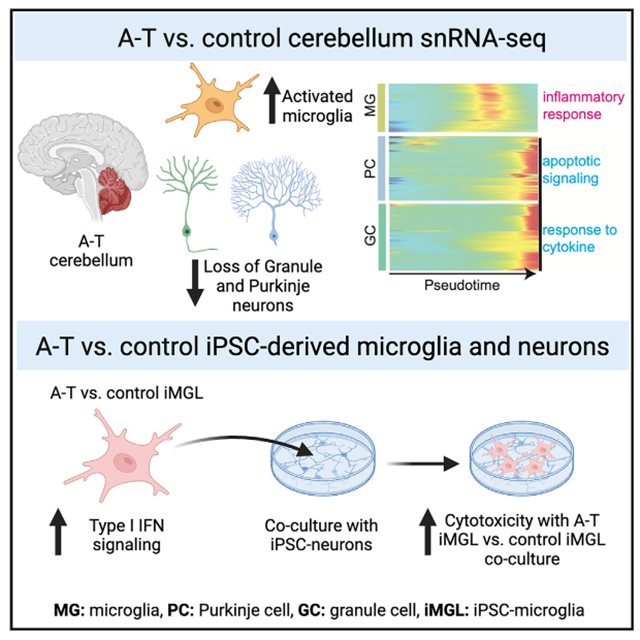
While ATM loss of function has long been identified as the genetic cause of ataxia-telangiectasia (A-T), how it leads to selective and progressive degeneration of cerebellar Purkinje and granule neurons remains unclear. ATM expression is enriched in microglia throughout cerebellar development and adulthood. Here, we find evidence of microglial inflammation in the cerebellum of patients with A-T using single-nucleus RNA sequencing. Pseudotime analysis revealed that activation of A-T microglia preceded upregulation of apoptosis-related genes in granule and Purkinje neurons and that microglia exhibited increased neurotoxic cytokine signaling to granule and Purkinje neurons in A-T. To confirm these findings experimentally, we performed transcriptomic profiling of A-T induced pluripotent stem cell (iPSC)-derived microglia, which revealed cell-intrinsic microglial activation of cytokine production and innate immune response pathways compared to controls. Furthermore, A-T microglia co-culture with either control or A-T iPSC-derived neurons was sufficient to induce cytotoxicity. Taken together, these studies reveal that cell-intrinsic microglial activation may promote neurodegeneration in A-T.
Graphical abstract
In brief
Lai et al. characterize cell-type-specific transcriptional changes in ataxia-telangiectasia (A-T) using single-nucleus RNA sequencing of patient brain tissue and find that cell-intrinsic activation of microglia in A-T promotes cytotoxicity. These findings provide mechanistic insight into the role of microglia on cerebellar degeneration in A-T.
INTRODUCTION
Selective degeneration of the cerebellum occurs in genetic ataxias such as ataxia-telangiectasia (A-T). However, the mechanisms underlying cerebellar degeneration remain poorly understood. A-T is an autosomal recessive multi-system disorder caused by mutations in the gene ATM.1 It affects as many as 1 in 40,000 live births and typically presents in early childhood with a median age of diagnosis of 6 years.2 Clinical features of A-T include progressive impairment of gait and coordination due to cerebellar neurodegeneration, immunodeficiency, and increased predisposition for cancers. There are currently no therapies to slow neurodegeneration in A-T.2
ATM is a multi-functional kinase known for its role in the DNA damage response to double-strand breaks (DSBs).1,3 DNA damage leads to ATM activation, which phosphorylates downstream regulators of cell-cycle arrest, DNA repair, and apoptosis.4 Additionally, ATM plays a role in cellular metabolism. ATM can form a dimer that mediates mitochondrial redox sensing and regulates antioxidant capacity.5 Moreover, ATM deficiency leads to upregulation of autophagy and perinuclear accumulation of lysosomes, implicating ATM in lysosomal trafficking.6
While ATM has roles in diverse cellular functions, it remains unclear how ATM loss of function leads to selective and progressive degeneration of Purkinje and granule neurons in the cerebellum. ATM-null mice do not recapitulate the cerebellar degeneration found in human patients with A-T,7 but bulk transcriptomic analyses of human brain tissue and neuronal models have begun to offer some insights into dysregulated pathways in A-T. Expression of ITPR1, a calcium channel that is highly expressed in Purkinje cells and is associated with spinocerebellar ataxias, is significantly altered in A-T cerebellum.8 Transcriptome analyses of induced pluripotent stem cell-derived cerebellar-like neurons from individuals with A-T and controls have also revealed alterations in pathways related to synaptic vesicle cycling, oxidative stress, and insulin secretion.9 However, it remains unresolved whether bulk transcriptomic changes reflect loss of certain cell types in A-T or perturbation of cellular functions in specific cell types.
Emerging evidence implicates dysfunctional microglia in the pathogenesis of A-T. Human cellular models of microglia with ATM deficiency reveal dysfunctional phagocytosis of neuronal processes.10 However, whether microglia activation is present in A-T patient brains, whether the microglia are reactive to cell-intrinsic or -extrinsic signals, and their role in neurodegeneration remain unknown. Characterization of microglia and their transcriptional signatures in human A-T brain may provide further insight into mechanisms that underlie cerebellar degeneration in A-T.
Here, we present a single-nucleus transcriptomic atlas of the adult human cerebellar vermis in A-T. We profiled 126,356 nuclei from the postmortem human cerebellar vermis of six individuals with genetically confirmed A-T and seven matched control individuals. In addition, we profiled 86,354 nuclei from postmortem human prefrontal cortex (PFC) from two A-T and two matched control individuals. We annotated major cell types in the human cerebellum and PFC and identified cell-type proportion changes between A-T and controls. We also demonstrated cell-type-specific expression of cerebellar-ataxia-associated disease genes, some of which were significantly dysregulated in A-T Purkinje neurons, supporting a convergence in disease pathophysiology underlying several hereditary ataxias. We analyzed the cell-type-specific molecular pathways perturbed in A-T across these brain regions, revealing prominent and widespread activation of pro-inflammatory pathways in A-T microglia. Pseudotime analysis revealed that activation of A-T microglia preceded upregulation of apoptosis-related genes in granule and Purkinje neurons, while ligand-receptor analysis suggested that microglia have increased neurotoxic cytokine signaling to granule and Purkinje neurons in A-T. We experimentally interrogated the role of microglia in neurodegeneration using A-T patient and control induced pluripotent stem cell (iPSC)-derived microglia (iMGLs) and neurons (iNs). Transcriptomic profiling of A-T patient iMGLs revealed cell-intrinsic microglial activation of pro-inflammatory pathways, and A-T patient iMGLs were sufficient to induce elevated cytotoxicity of co-cultures with control and A-T iNs. Overall, our data suggest that activated microglia are central to A-T pathophysiology and cerebellar degeneration. Our data are publicly available for exploration at the Broad Single Cell Portal (https://singlecell.broadinstitute.org; Study IDs: SCP1300 and SCP1302).
RESULTS
Single-nucleus RNA-seq profiling of A-T human cerebellum and PFC
A-T presents with prominent loss of motor coordination associated with early selective atrophy of the cerebellum. To investigate cell-type- and region-specific perturbations in A-T, we performed single-nucleus RNA sequencing (snRNA-seq) of human postmortem tissues from the cerebellar vermis (the region with the most prominent atrophy in A-T11) and the PFC (Figure 1A). We profiled six cerebella from neuropathologically and genetically confirmed A-T cases and seven control cerebella, as well as matched A-T and control PFC from two cases each (Table S1). The ages of the A-T cases ranged from 19 to 50 years old (mean age: 28 years). Biallelic ATM variants (with evidence of pathogenicity in ClinVar12,13,14) were confirmed in all six A-T cases by whole-genome sequencing (WGS) (Table S1). All cases were balanced for age, sex, RNA integrity number (RIN), and postmortem interval (PMI) (Table S1). After quality control and doublet filtering,15–17 there were 126,356 cerebellar nuclei (51,297 A-T; 75,059 control) and 86,354 PFC nuclei (16,979 A-T; 69,375 control) for downstream analysis (Figures S1A–S1D).
Figure 1. Dissecting cell-type-specific contributions to neurodegeneration in ataxia-telangiectasia using single-nucleus RNA sequencing of postmortem brain and patient iPSC-derived cultures.
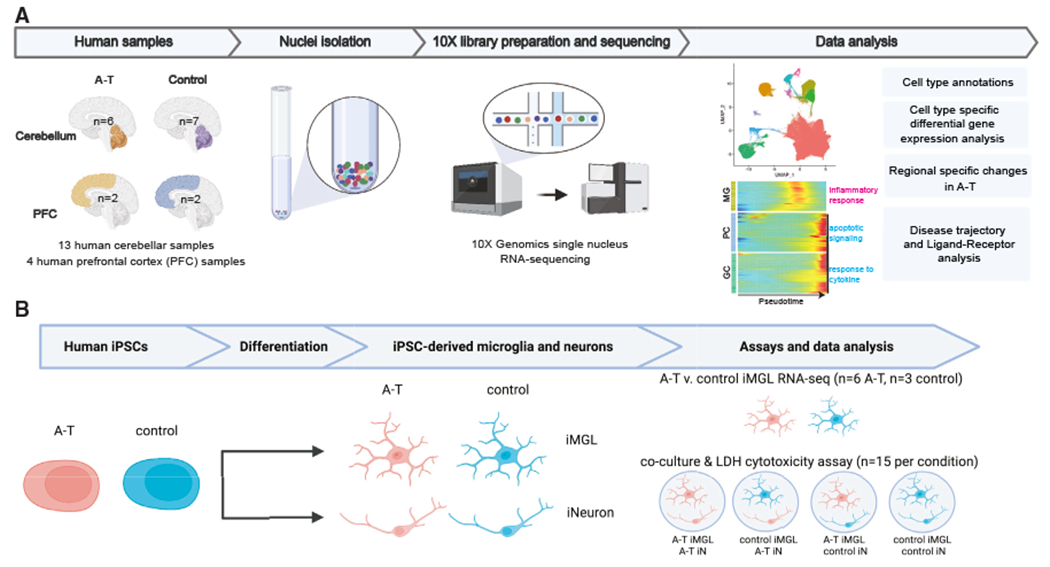
(A) Schematic overview of single-nucleus RNA sequencing (snRNA-seq) experimental design and computational analyses.
(B) Schematic overview of ataxia-telangiectasia (A-T) patient and control human iPSC-derived experimental design. n represents the number of independent culture wells. MG, microglia; PC, Purkinje cells; GC, granule cells; iMGL, iPSC-derived microglia; iN, iPSC-derived neurons.
snRNA-seq recapitulates neuropathological hallmarks and reveals increased glial populations in A-T cerebellum
To distinguish cell types in the cerebellum, we used Seurat15 to perform graph-based clustering with the Leiden algorithm and annotated clusters based on the expression of canonical marker genes and existing single-cell atlases of the cerebellum.13,14 Ten major cell types were identified, including cerebellum-specific granule cells (RIMS1, GRM4), Purkinje cells (ITPR1, CALB1), and Bergmann glia (TUBB2B, AQP4), as well as interneurons (GAD1, PVALB), astrocytes (TTN, AQP1), oligodendrocytes (PLP1 ,MBP), oligodendrocyte precursor cells (OPCs) (PDGFRA, OLIG1), microglia (CD74, CSF1R), endothelial cells (CLDN5, VWF), and fibroblasts (DCN, APOD) (Figures 2A–2C). We annotated PFC cell types by reference-based mapping with SingleR using an existing human PFC snRNA-seq dataset as the reference.18,19 There were thirteen cell types identified, including astrocytes, microglia, endothelial cells, oligodendrocytes, OPCs, and multiple subtypes of excitatory and inhibitory neurons (Figure S2A).
Figure 2. Cell types resolved in A-T and control human cerebellum by snRNA-seq.
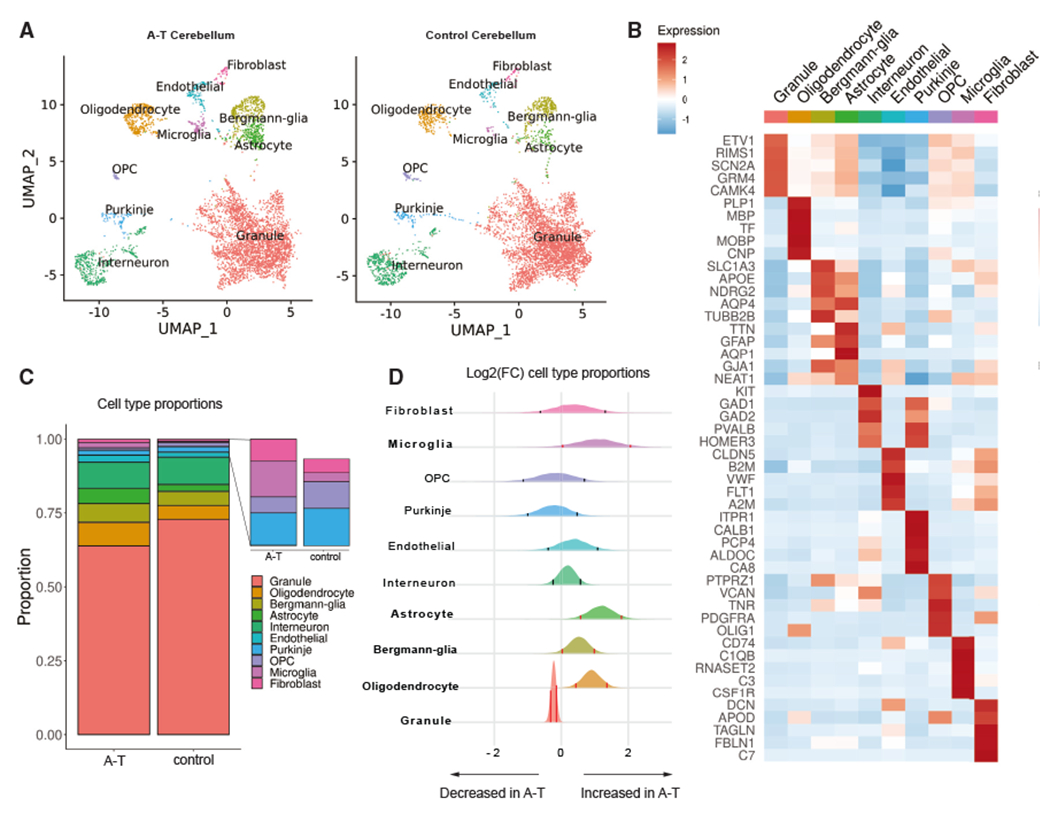
(A) Uniform manifold approximation and projection (UMAP) dimensionality reduction plot of major cell types identified in A-T and control human cerebellum, downsampled to 10,000 cells for each condition.
(B) Top five marker genes for each major cell type in control cerebellum. Heatmap depicts centered and scaled log-normalized expression values.
(C) Cell-type proportions in A-T and control cerebellum.
(D) Relative abundance of cell types in A-T versus control cerebellum, shown as the posterior distribution of log2(proportion in A-T/proportion in control) with 89% credible interval. Red bars highlight credible intervals that do not overlap 0. Bolded cell-type labels indicate a significant difference in relative abundance.
Next, to understand the patterns of cerebellar cell loss in A-T, we examined the cell-type proportions in A-T compared to controls. Due to the negative covariance structure of compositional data–as one feature increases, the other features must decrease–we implemented Dirichlet multi-nomial modeling using Hamiltonian Monte Carlo (DMM-HMC) to estimate the cell-type proportions in A-T and control.20 We generated multi-nomial distributions of cell-type proportions for each sample based on the nuclei counts (Table S1) and used these to construct a Dirichlet distribution of cell-type proportions in A-T and controls. To determine the relative shift in abundance of each cell type in A-T versus control, we subtracted the control posterior probability distribution of proportions from the A-T distribution (Figure 2D). Granule cells were significantly decreased in A-T (Figure 2D; 89% credibility interval). Purkinje cells were reduced in abundance, although not significantly, possibly due to individual sample variability and decreased power for detecting changes in rare cell types. In addition, astrocytes, Bergmann glia, microglia, and oligodendrocytes showed significant increases in A-T compared to control (Figure 2D; 89% credibility interval). In contrast, permutation testing of disease labels (A-T versus control) showed no significant differences for any cell type, as expected (permutation n = 500; Figure S1E). Unlike A-T cerebellum, there was no evidence of neuronal loss in A-T PFC (Figure S2B). However, oligodendrocytes were significantly decreased in A-T PFC compared to control (Figure S2B), consistent with reports of cortical white matter degeneration in A-T.21,22 Taken together, these results are consistent with the neuropathology autopsy reports from individuals with A-T, which describe granule and Purkinje cell atrophy and gliosis in the cerebellum, while the PFC is grossly unremarkable (Table S1).
Monogenic cerebellar disease genes are expressed in specific cell types
Next, we characterized the expression of ATM across cell types in the cerebellum and PFC. ATM had the highest expression in microglia in adult control human cerebellum and PFC (Figure S3A). To investigate human developmental expression patterns, we profiled the expression of ATM in an snRNA-seq dataset of human control fetal cerebellum,23 which also revealed the highest expression of ATM in microglia (Figure S3B). Taken together, these data suggest an important role for ATM in microglia during normal cerebellar development.
Given that ATM is enriched in microglia, we asked whether additional hereditary ataxia genes are enriched in specific cerebellar cell types. We curated a list of genes implicated in human hereditary ataxias using the Online Mendelian Inheritance in Man (OMIM) database (Table S2) and profiled their expression in control cells. We found that like ATM, most hereditary ataxia disease genes demonstrate cell-type-specific patterns of expression (Figure S3C; Bonferroni-adjusted p < 0.05, t test). Cerebellar disease gene expression was most commonly enriched in Purkinje cells, including the potassium- and calcium-ion-related genes KCND3, PRKCG, CACNA1G, ITPR1, TRPC3, and KCNC3. Microglia also showed enriched expression of several ataxia disease genes (ATM, TPP1, SETX, and VPS13D) (Figure S3C). These gene expression patterns implicate Purkinje cells and microglia in the pathogenesis of cerebellar ataxias.
Transcriptional dysregulation across cell types in A-T
To investigate the cell types and molecular pathways that are most perturbed in A-T cerebellum, we performed differential gene expression analysis between A-T and control for each cell type (Table S2). Astrocytes, microglia, and oligodendrocytes had remarkably high numbers of differentially expressed genes (DEGs; n = 1,631, 1,577, and 1,563, respectively) while granule cells had the least (n = 265). These data suggest that while granule cells degenerate in A-T, their gene expression program is less dysregulated than other cerebellar cell types such as glia.
Next, we performed Gene Ontology (GO) analysis on the DEGs found in each cerebellar cell type to gain insight into the biological processes dysregulated in A-T. Consistent with the loss of Purkinje and granule cells in A-T, both cell types had significant upregulation of genes involved in apoptotic signaling (Figures 3A and 3B; Table S2). A-T Purkinje and granule cells also upregulated genes involved in ribosome assembly and biogenesis, which has been observed in several studies of aging brains.24,25 A-T microglia specifically demonstrated upregulation of immune-response-related genes involved in microglial cell activation, phagocytosis, and cytokine production (Figure 3C).
Figure 3. Gene Ontology (GO) analysis of differentially expressed genes in A-T cerebellum.
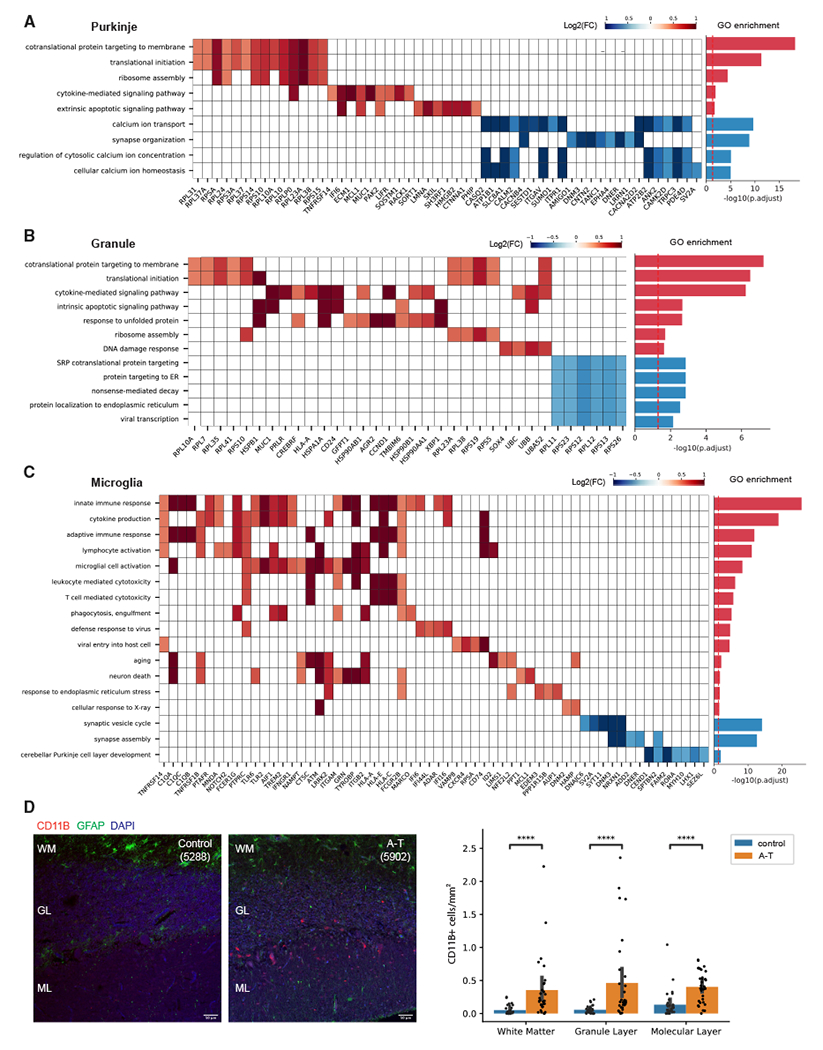
(A–C) Select enriched GO biological processes among DEGs in (A) A-T Purkinje neurons, (B) granule neurons, and (C) microglia. Select genes in each pathway and their log2 fold changes (A-T/control) shown in the heatmap. Bar plot shows significance of pathway enrichment among upregulated genes (red) and downregulated genes (blue).
(D) CD11B immunostaining of postmortem cerebellar cortex from A-T and control. WM, white matter; GL, granule layer; ML, molecular layer. Quantification of CD11B+ cells across cerebellar cortex layers in A-T and control. Error bar represents 95% confidence interval. ****Bonferroni-adjusted p < 1e—04, Mann-Whitney U test. Scale bar, 50 μm.
Activated microglia associate with sites of injury and pathological changes across various neurodegenerative diseases26 and can induce local neurodegeneration by phagocytosis of synapses and secretion of neurotoxic cytokines.27 We thus sought to characterize the spatial distribution and number of microglia in A-T and control cerebellum by immunostaining for CD11B, a microglia marker that is upregulated in activated microglia,28 in four A-T cerebellum and three matched control cerebellum samples (Table S1). In A-T cerebellum, there were increased CD11B+ cells throughout the white matter, the granule layer, and the molecular layer compared to control cerebellum (Figure 3D). Across all layers, the number of CD11B+ microglia per area was significantly higher in A-T compared to control (Bonferroni-adjusted p < 1e–04, Mann-Whitney U test) (Figure 3D). These data confirm that A-T cerebellum has a widespread increased presence of activated microglia.
Dysregulated calcium signaling in Purkinje neurons in A-T
GO analysis of genes downregulated in A-T revealed significant enrichment of calcium-related pathways (calcium ion transport, regulation of cytosolic calcium ion concentration, cellular calcium ion homeostasis) in A-T Purkinje cells (Figure 3A; Table S2). Among these genes was ITPR1, an endoplasmic reticulum (ER) calcium channel that causes spinocerebellar ataxias in an autosomal-dominant manner29,30 (Figure 3A). We asked whether additional cerebellar disease genes are differentially expressed in A-T. Hereditary ataxia genes with enriched expression in Purkinje cells (i.e., KCNC3, PRKCG, ITPR1, KCND3, CACNA1G) were significantly downregulated in A-T Purkinje cells (Figure S4A). These disease genes were also enriched for calcium processes (calcium ion transport and regulation of cytosolic calcium ion concentration) (Figure S4B). These results suggest that ATM may be an upstream activator of calcium signaling and point to converging underlying mechanisms of a subset of hereditary ataxias in Purkinje cells. HDAC4 is a known negative regulator of calcium ion genes (ITPR1, DAB1),31 and ATM is an inhibitor of HDAC4. Loss of ATM leads to nuclear accumulation of HDAC4, which could lead to dysregulated expression of HDAC4 target genes.32 In support of this, there was significant overlap between HDAC4 targets and A-T downregulated genes (p = 6.08e–22, Fisher’s exact test; Figure S4C). Taken together, these findings support that abnormal calcium ion homeostasis may be a consequence of ATM loss of function in Purkinje neurons, which may lead to aberrant firing patterns and ataxic phenotypes.33,34
Activated microglia in A-T cerebellum share transcriptomic signatures with aging and neurodegeneration-associated microglia
Microglia in A-T cerebellum express activation markers, including strong upregulation of the complement genes C1QA, C1QB, C1QC, and C3 (Figure 4A). Increased expression of complement is associated with microglial activation in neurodegenerative diseases such as Alzheimer’s disease (AD),35 where they have been shown to mediate synaptic loss and dysfunction.36,37 To assess whether A-T cerebellar microglia share additional features associated with microglia in aging and other neurodegenerative disorders, we compared the upregulated genes in A-T cerebellar microglia with three gene sets: markers of aged human microglia, AD-associated microglia markers from a human snRNA-seq study of AD, and markers of disease-associated microglia (DAMs) from a murine model of AD.38,39,40 We found significant overlaps between A-T cerebellar microglia upregulated genes and these three gene sets, with the highest overlap being with aging microglia (Fisher’s exact test p < 1e–13; Figures 4B and 4C). TREM2, TSPO, MS4A6A, complement components, and major histocompatibility complex (MHC) class II genes were among the overlapping genes, all of which have been associated with neuroinflammation and microglia activation41 (Figure 4C). GO enrichment analysis of the overlapping genes and A-T microglia-specific upregulated genes revealed common enrichment in immune-response-and RNA-processing-related pathways and A-T-specific enrichment of cytoskeletal organization, ER stress, and regulation of neuron projection development (Figure 4D). Overall, these data show that A-T cerebellar microglia share transcriptomic signatures found in aging and other neurodegenerative diseases.
Figure 4. A-T cerebellar microglia share transcriptomic signatures with aging and neurodegenerative microglia.
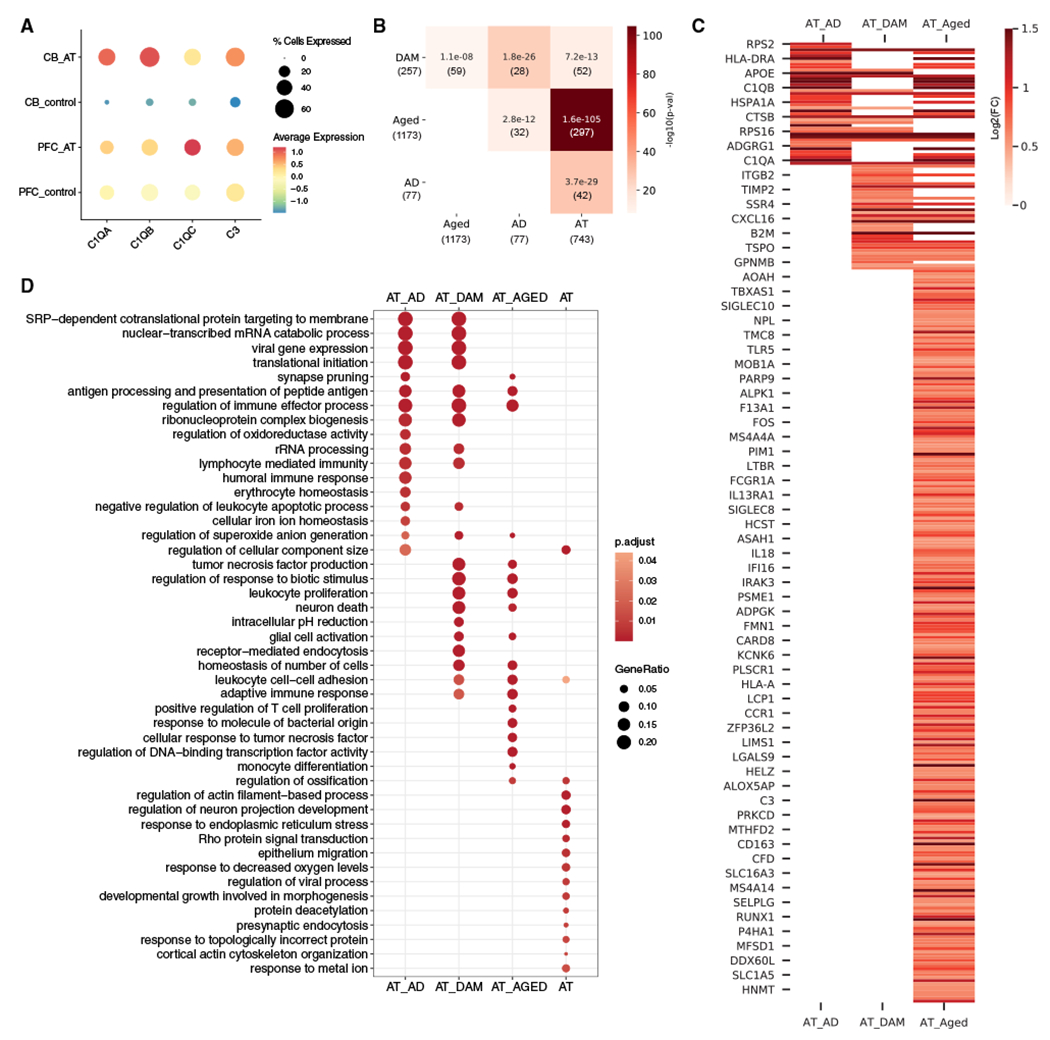
(A) Average scaled expression of complement components C1QA, C1QB, C1QC, and C3 in microglia from A-T and control cerebellum (CB) and prefrontal cortex (PFC).
(B) Overlap between A-T microglia upregulated genes and human Alzheimer’s disease (AD) microglia markers,38 disease-associated microglia (DAM) markers,40 and human aging microglia markers (Aged).39 Overlap p values from Fisher’s exact test shown in each cell. Color represents —log10(overlap p value). Number of genes in each set and intersection are shown in parentheses.
(C) Heatmap of A-T microglia log2 fold changes (false discovery rate [FDR] < 0.05) for overlapping microglia markers. AT_AD, A-T and Alzheimer’s disease microglia overlapping genes; AT_DAM, A-T and DAM overlapping genes; AT_AGED, A-T and human aging microglia overlapping genes.
(D) GO biological process enrichment of overlapping microglia markers. AT, A-T microglia-only upregulated genes. Pathways with FDR <0.05 shown.
Differential activation of microglia in A-T cerebellum versus PFC
To systematically compare gene expression changes in A-T cerebellum with A-T PFC, we identified genes and pathways that were more significantly changed in A-T cerebellum than in A-T PFC in cell types common to the two brain regions (microglia, OPCs, oligodendrocytes, astrocytes, endothelial cells) (Figure S5; Tables S3 and S4). While both cerebellar and PFC microglia showed upregulation of genes involved in immune response in A-T, microglia were more strongly activated in the cerebellum compared to the PFC. Cerebellar A-T microglia had significantly higher upregulation of genes related to inflammatory processes (adaptive immune response, leukocyte proliferation, glial cell activation, tumor necrosis factor superfamily cytokine production) and neuronal death than A-T PFC microglia (Figure 5A).
Figure 5. Stronger activation of microglia in A-T CB compared to PFC.
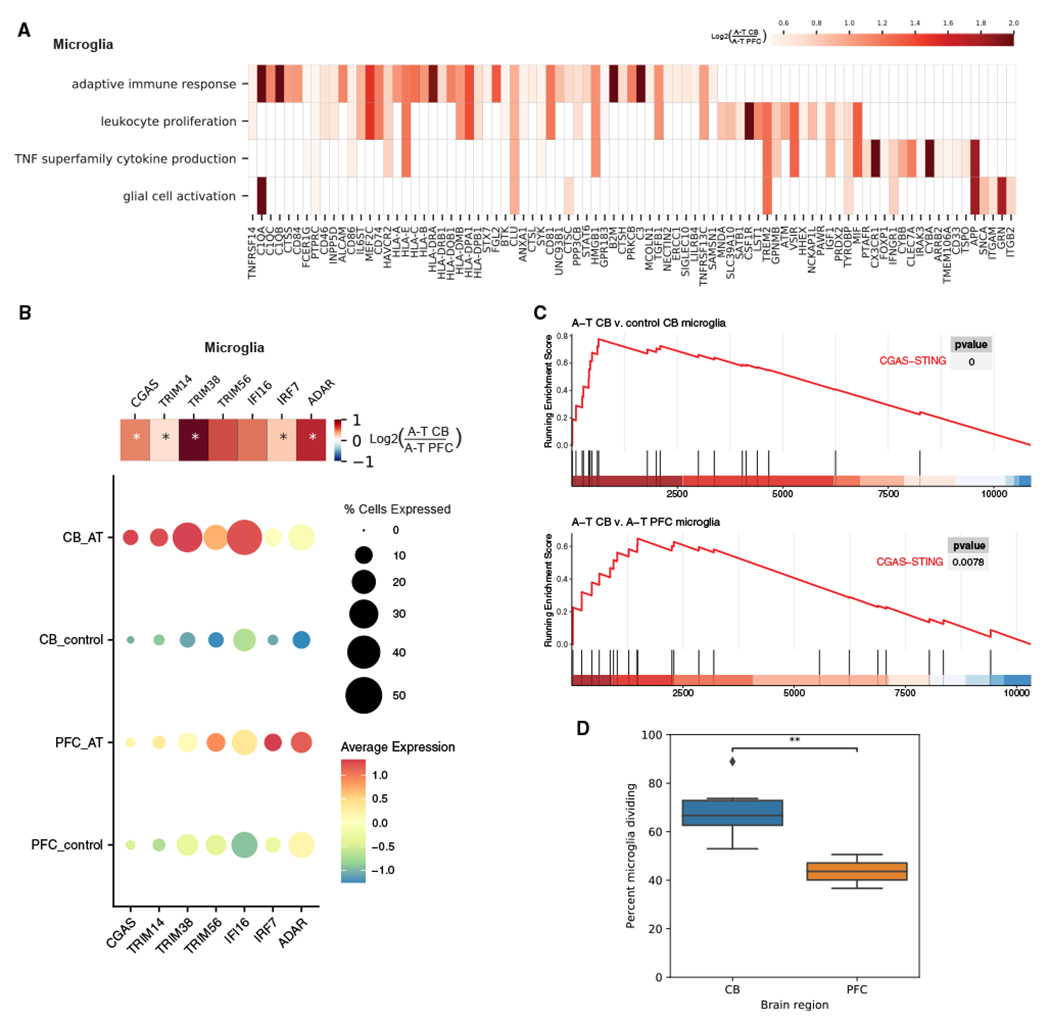
(A) Heatmap showing enriched GO biological processes and log2 fold changes of significant DEGs in each pathway with greater upregulation in A-T CB microglia than A-T PFC microglia.
(B) Dot plot of average scaled expression of CGAS-STING pathway genes in microglia of the CB and PFC in A-T and control. Heatmap shows log2 fold change of CGAS-STING pathway genes in A-T cerebellar microglia versus A-T PFC microglia. *FDR < 0.05.
(C) Gene set enrichment analysis (GSEA) plot for the CGAS-STING pathway in A-T CB microglia versus control microglia (top) and A-T CB versus A-T PFC microglia (bottom).
(D) Percentage of microglia putatively in replicating phase in control CB and PFC. **p < 0.01, t test.
Microglia can be activated by neuronal death, a mechanism common to multiple neurodegenerative conditions.42 However, since PFC neurons are generally spared in A-T, we hypothesized that A-T microglia in the PFC and cerebellum are cell-intrinsically activated. One potential mechanism of intrinsic activation is the accumulation of DNA damage due to ATM deficiency. Deficits in DNA repair caused by ATM deficiency are associated with increased cytosolic DNA.43 Cytosolic DNA is recognized by the CGAS-STING pathway, which triggers an innate immune response by inducing type I interferons and inflammatory cytokines.41 We found that the CGAS-STING pathway, including CGAS and activators of CGAS signaling – TRIM14, TRIM38, and TRIM5644–46–were upregulated specifically in A-T microglia from the cerebellum and PFC, suggesting the presence of cytosolic DNA (Figures 5B and 5C). Moreover, the CGAS-STING pathway showed significant enrichment among genes upregulated in A-T cerebellum versus A-T PFC, suggesting that CGAS-STING activation was more prominent in cerebellar microglia (Figure 5C). CGAS-STING pathway genes (TRIM14, TRIM38, IFI16, IRF7) were also upregulated in aged human microglia but not AD or DAM gene sets, implicating DNA damage and cytoplasmic DNA as common underlying causes of microglial activation in A-T and aging microglia, while AD microglia may be activated by another mechanism, such as extrinsic stimuli (Table S5). Taken together, activation of the CGAS-STING pathway suggests that cytosolic DNA contributes to microglial activation that is more pronounced in A-T cerebellum than PFC.
Self-DNA enters the cytosol in a mitosis-dependent manner through the formation and rupture of DNA-damage-induced micronuclei.47,48 Thus, we hypothesized that underlying differences in proliferation may explain the differential activation of CGAS-STING in cerebellar microglia compared to PFC microglia. We tested whether microglia in the cerebellum and PFC differ in proliferation rate by inferring the cell-cycle status of each cell by the expression of canonical markers of G1, S, and G2 phases. We found that cerebellar microglia have an increased percentage of cells in replicating phases compared to cortical microglia (p < 0.01, t test) (Figure 5D). This is consistent with previous reports that cerebellar microglia have a higher turnover rate than cortical microglia, which may make them more vulnerable to accumulation of DNA damage due to ATM deficiency.49,50 The differences in proliferation may underlie our observation that cerebellar microglia have a stronger CGAS-STING-mediated immune response in A-T than cortical microglia.
A-T patient iPSC-derived microglia demonstrate cell-intrinsic activation of NF-κB and type I interferon signaling
To understand whether microglia might contribute to neurodegeneration in A-T, we sought to determine when cerebellar microglia become activated in disease progression relative to Purkinje and granule cell death. We employed pseudotime analysis to understand the cell-type-specific cascades of molecular events across disease progression in A-T.51 For each cell type, cells were ordered along a continuous trajectory that progressed from the control to the diseased state (Figure S5A). We performed differential analysis to identify genes that change as a function of pseudotime and clustered these genes by their pseudotime expression patterns. We then applied GO analysis to determine the enriched biological functions of each gene cluster. For microglia, the beginning of pseudotime (healthy state) had high expression of genes involved in cellular respiration, oxidative phosphorylation, and synaptic vesicle exocytosis, consistent with a high metabolism gene expression profile characteristic of cerebellar microglia52 (Figure S5B). Over disease progression, upregulation of CGAS-STING pathway genes (i.e., CGAS, TRIM14, TRIM38, TRIM56) was followed by leukocyte activation, cytokine production, and innate immune response (Figures S5B and S5C). In Purkinje neurons, disease progression was characterized first by a decline in genes related to calcium ion homeostasis (i.e., ITPR1), followed by a cluster of genes related to immune response and response to cytokines (i.e., HSPA1A, B2M, ISG15) and the apoptotic signaling pathway (i.e., XBP1, BCL2, CASP2) (Figure S5D). Granule neurons first demonstrated a decrease in chromatin organization (i.e., HDAC4, HDAC2, CHD5) and glutamatergic synaptic signaling genes (i.e., NRXN1, RELN, SYT1), followed by upregulation genes related to oxidative phosphorylation (i.e., PINK1, NDUFS5, UQCRH), and then upregulation of genes related to response to cytokine (i.e., SP100, STAT6, ISG15), response to unfolded protein (i.e., HSPA1A, HSPE1, DNAJB1), and programmed cell death (i.e., IFI6, BCL2, XBP1) at the end of pseudotime, suggesting that synaptic and metabolic dysfunction precedes granule neuron death in A-T (Figure S5E).
We then compared molecular events between microglia, Purkinje cells, and granule cells along disease progression by aligning their pseudotime trajectories using dynamic time warping.53 This revealed that the upregulation of inflammatory response and cytokine production genes in microglia (i.e., C1QA, IL18, IFI16, CGAS, LTBR, TNFSF10) occurs earlier in disease progression pseudotime than the onset of response to cytokine and apoptosis genes (XBP1, MUC1, TFAP2A) in Purkinje and granule cells (Figures 6A and 6B). These findings suggest that microglial activation and inflammation precede Purkinje and granule cell death in A-T and support a model of cell-intrinsic microglial activation in A-T.
Figure 6. A-T patient iPSC-derived microglia reveal cell-intrinsic activation and increased cytotoxicity in neuronal co-cultures.
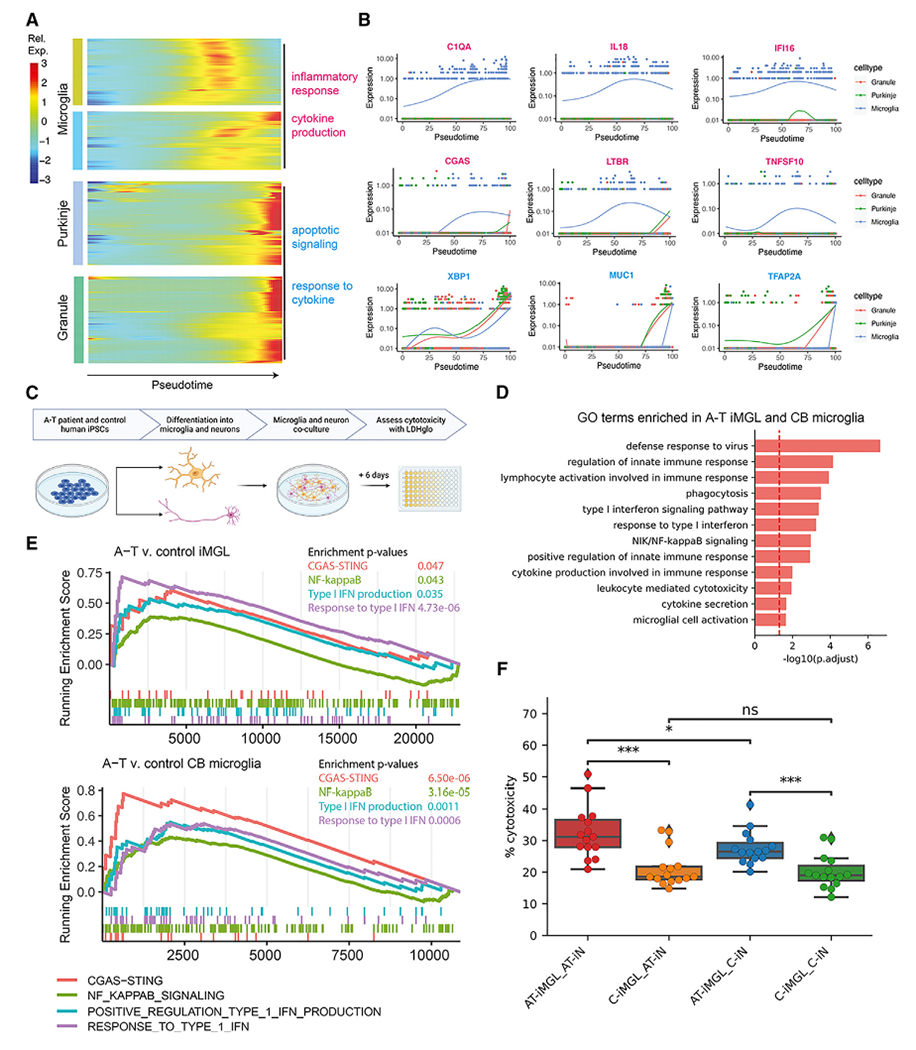
(A) Heatmap of genes that significantly change over aligned pseudotime in microglia, Purkinje neurons, and granule neurons clustered by pseudotemporal expression patterns. Each cluster is annotated with enriched GO terms (FDR < 0.05).
(B) Expression of select inflammatory genes (magenta) and apoptotic signaling and response to cytokine genes (cyan) over aligned pseudotime in microglia, Purkinje neurons, and granule neurons.
(C) Schematic of A-T and control iPSC-microglia (iMGLs) and iPSC-neuron (iNs) co-culture experiment.
(D) GO terms enriched among genes upregulated in A-T iMGL and A-T cerebellar microglia versus control.
(E) GSEA plots for CGAS-STING, NF-κB, type I interferon (IFN) production, and response to type I IFN pathways in A-T versus control iMGLs (top) and A-T versus control CB microglia (bottom).
(F) Percentage of cytotoxicity of iMGL and iN co-cultures based on LDH-based cytotoxicity assay (bottom). C, PGP1 control. *p < 0.05 and ***p < 0.001, ns, not significant, Mann-Whitney U test.
To experimentally characterize microglial activation in A-T, we developed iPSC-based experimental models using patients with A-T and controls (Figure 1B and S6A). We generated human iPSCs from fibroblasts of an individual with A-T with compound heterozygous splice-altering variants in ATM (hg38 chr11:g.108332838C>T; chr11:g.108347266_108353792del27) and used the well-characterized PGP1 iPSC line as a control. We then differentiated microglia-like cells (iMGLs) from the A-T patient and control iPSCs through a hematopoietic progenitor intermediate54 (Figure S6A). The resulting iMGLs were a highly homogeneous population, with >85% cells expressing microglia markers CD45 and CD11B as assessed by flow cytometry (Figure S6B). Profiling of the iMGLs by RNA-seq showed that they expressed canonical markers of microglia (AIF1, CX3CR1, CSF1R, SPI1, TREM2) but did not express myeloid lineage (MPO, KLF2), neuronal (MAP2, SOX2), or pluripotency (POU5F1) marker genes Figure S6C). Principal-component analysis with transcriptomic profiles from A-T and control iMGLs along with our snRNA-seq data from human adult cerebellum and published snRNA-seq data from human fetal cerebellum23 confirmed that iMGLs cluster more closely with microglia from adult and fetal human cerebellum than with other brain cell types (Figure S6D).
We performed differential gene expression analysis of A-T versus control iMGLs, revealing upregulation of genes involved in cytokine secretion, phagocytosis, nuclear factor κB (NF-κB) signaling, and type I interferon signaling (Figure S6E; Table S5). To compare A-T iMGLs and A-T cerebellar microglia, we obtained genes upregulated in both and identified enrichment of immune-related GO terms, including microglial cell activation, cytokine secretion, NF-κB, and type I interferon signaling pathways (Figure 6D; Table S5). These results confirmed that A-T microglia grown in isolation take on an activated phenotype, demonstrating that this is a cell-intrinsic property of microglia in A-T.
We hypothesized that cell-intrinsic activation of NF-κB and type I interferon signaling in A-T microglia could be due to cytoplasmic DNA triggering CGAS-STING since a major consequence of CGAS-STING activation is initiation of NF-κB signaling and production of type I interferon-related cytokines.55,56 In support of cytoplasmic DNA as a potential trigger for microglia activation in A-T, A-T iMGLs also showed positive enrichment of a CGAS-STING gene set (Figure 6E; Table S5). The similar enrichment of CGAS-STING, NF-κB, and type I interferon pathways among genes upregulated in A-T iMGLs and A-T postmortem microglia supports that A-T microglia are activated by a cell-intrinsic mechanism that may involve cytoplasmic DNA and CGAS-STING (Figure 6E; Table S5).
In silico and in vitro models of microglia-neuron interactions reveal that A-T microglia increase cytotoxicity
A-T Purkinje and granule neurons showed enrichment of response to cytokine genes late in disease progression, raising the possibility that cytokines produced by microglia induce molecular changes in Purkinje and granule neurons in A-T (Figure 6A). Consistently, ligand-receptor analysis revealed increased cytokine signaling (IL1B, IL7, IL4, IL15), type I interferon signaling (IFNA2), CD40 signaling (CD40LG), and complement signaling (C3, C4) from microglia to granule and Purkinje neurons in A-T versus control (Figure S6F). Notably, several of these pro-inflammatory cytokines, including IL1B and IFNA2, have been shown to induce neurotoxicity.57–59 This suggests that activated microglia in A-T have increased secretion of pro-inflammatory cytokines that may have neurotoxic consequences.
To test this hypothesis experimentally, we differentiated A-T patient and control iPSCs into neurons using overexpression of Ngn260 and co-cultured them with A-T patient and control iMGLs (Figure 6C). iNs transcriptionally correlated with human midgestational cerebellum, making them a relevant model for studying microglia-neuron interactions in A-T (Figure S6G). We co-cultured iMGLs and iNs for 6 days and assessed cytotoxicity by lactate dehydrogenase (LDH) release. We found that co-culturing control iNs with A-T iMGLs was sufficient for increased cytotoxicity compared to co-culturing with control iMGLs (Figure 6C; p < 0.001, Mann-Whitney U test). Moreover, A-T iMGLs co-cultured with A-T iNs demonstrated increased cytotoxicity compared to control iMGLs co-cultured with A-T iNs (Figure 6F; p < 0.001, Mann-Whitney U test). A-T iMGLs co-cultured with A-T iNs had the highest levels of cytotoxicity, suggesting that A-T iNs are more vulnerable to A-T iMGLs. Taken together, these results indicate that A-T microglia have increased pro-inflammatory interactions with neurons and lead to increased cytotoxicity.
DISCUSSION
In this study, we present an atlas of human cerebellar transcriptomes in health and degeneration at single-cell resolution. We identified major neuronal and glial cell types in the cerebellum and transcriptomic signatures in A-T. We resolved cell-type-specific transcriptional perturbations in A-T, including activation of pro-inflammatory pathways in microglia that was more pronounced in the cerebellum versus the PFC. The activation of microglia in A-T PFC despite a lack of cortical atrophy or neuronal loss suggests that A-T microglia are activated by a cell-intrinsic mechanism rather than in response to neurodegeneration, with brain-region-specific factors leading to stronger activation in the cerebellum. We found that genes in the CGAS-STING pathway, including CGAS itself, were significantly upregulated specifically in A-T microglia, with preferential activation in the cerebellum. Moreover, A-T patient iPSC-derived microglia cultured in the absence of neurons also demonstrated upregulation of genes involved in CGAS-STING, NF-κB, and type I interferon signaling, further suggesting a cell-intrinsic mechanism of activation. The CGAS-STING pathway is activated in response to cytosolic DNA, suggesting the presence of cytosolic DNA in A-T microglia. These results are consistent with recent reports of cytosolic DNA and CGAS-STING activation in microglia from rodent models of A-T.61,62 Potential sources of cytosolic DNA include release of nuclear self-DNA,43,41 mitochondrial DNA released by defective mitochondria,63–65 reverse-transcribed transposable elements as observed in aging-related disorders,66 and phagocytosis of nucleic acids from cellular debris. It will be of great interest for future work to identify the source of cytosolic DNA in A-T microglia as a potential therapeutic avenue for modulating inflammation in A-T. Reducing inflammation in rodent models of A-T with ibuprofen led to decreased microglial activation and neuronal apoptosis, further highlighting microglia as a therapeutic target in A-T.67
Activated microglia have been intensely studied in the context of neurodegenerative disorders including AD, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS).68 Our study highlights the involvement of microglia in human cerebellar degeneration as well. Through pseudotime analyses, we found that the rise in pro-inflammatory genes in A-T microglia precedes the onset of cytokine response and apoptosis-related genes in A-T Purkinje and granule neurons, and ligand-receptor analysis indicated increased pro-inflammatory signaling from microglia to neurons in A-T. These findings support a model of pathogenesis whereby cell-intrinsically activated microglia release neurotoxic cytokines and factors that trigger Purkinje and granule cell death. Recent work in human microglial cell lines demonstrated that persistent DNA damage due to loss of ATM leads to microglial activation and excessive phagocytosis of neurites, providing further validation for a cell-intrinsic mechanism of microglia activation that contributes to neuronal damage.10 Rodent models of A-T also show evidence of neuroinflammation and microglial activation that lead to secretion of neurotoxic cytokines,61,62,69 but why these models do not fully recapitulate cerebellar degeneration remains unknown. Potential reasons may be that the rodent cerebellum has an increased numerical density (number/mm3) of Purkinje cells compared to human cerebellum70 or that Purkinje cell transcriptomic differences between rodent and human may underlie the different phenotypes.71,72 Here, our data from human patients with A-T and patient-derived iPSC models provides critical support that microglial activation, previously demonstrated in vitro and in rodent models, is also found in human A-T patient brains and may contribute to A-T disease mechanisms. The pathological nature of pro-inflammatory signaling from activated microglia is also supported by clinical observations that treatment with glucocorticoids reduced ataxia symptoms in A-T.73–76 The presence of activated microglia in A-T PFC without cortical neuron loss suggests that cerebellar Purkinje and granule neurons are more vulnerable to activated microglia. Moreover, the increased cytotoxicity of A-T iNs cocultured with A-T iMGLs suggests that A-T iNs have increased vulnerability to microglia. It is possible that cell-autonomous and non-cell-autonomous effects of ATM loss in cerebellar neurons and microglia interact to cause A-T pathology.
This study identifies cell-type-specific changes in A-T across multiple brain regions and serves as a resource as an adult human cerebellum vermis dataset in health and degeneration. While granule and Purkinje neurons have been the most studied cell types in A-T due to their selective degeneration, this study highlights the central role of microglia in A-T cerebellar degenerative pathogenesis.
Limitations of the study
Limitations of this study include that the iPSC lines used were derived from a single patient with A-T and an unrelated control. The iPSC-derived findings presented here represent a preliminary study on the interaction between microglia and neurons in A-T disease pathogenesis. It will be important for future experiments to expand this work to include isogenic control lines or at least three independent A-T patient iPSC lines and control iPSC lines. Another limitation is that the iPSC-derived neuronal model employed in this study does not develop the same morphological complexity characteristic of Purkinje neurons. An exciting area of future work includes co-culture of iPSC-derived microglia with iPSC-derived cerebellar organoids or Purkinje and granule neurons as the most relevant models for A-T. Given that microglia and other brain-resident macrophages share several transcriptomic markers, another limitation is that the microglia cluster may include a small percentage of other brain-resident macrophages. As markers of subtypes of brain-resident macrophages become elucidated, future studies will be able to better distinguish these cell types in the brain. Moreover, it is possible that some of the transcriptional alterations identified in certain cell types reflect processes other than degeneration, such as compensatory changes in surviving cells. It will be important for future work to further dissect the developmental onset, contribution of cell-type interactions, and cellular responses to cerebellar degeneration in A-T. Taken together, our findings suggest that cell-intrinsic microglial activation may play a critical role in A-T, setting the stage for future experiments that will further illuminate the precise mechanisms by which microglia-neuron interactions may contribute to selective cerebellar degeneration in A-T.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Eunjung Alice Lee (ealee@childrens.harvard.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
Single-nucleus RNA-sequencing and whole genome sequencing data from postmortem individuals and RNA-sequencing data from human iPSC lines generated in this study have been deposited at dbGaP: phs003005.v2.p1 and are publicly available as of date of publication. Single-nucleus RNA-seq data have also been deposited at the Broad Single Cell Portal (https://singlecell.broadinstitute.org; Study ID SCP1300 and SCP1302). Accession numbers for all deposited data are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD11B/Integrin Alpha M | Proteintech | Cat# 66519-1-Ig; RRID:AB_2881882 |
| Anti-GFAP | Millipore Sigma | Cat# HPA056030 |
| Anti-CD32 | STEMCELL Technologies | Cat# 60012; RRID:AB_2925215 |
| Anti-CD45-APC | STEMCELL Technologies | Cat# 60018AZ |
| Anti-CD34-PE | STEMCELL Technologies | Cat# 60119PE |
| Anti-CD11B-FITC | STEMCELL Technologies | Cat# 60040FI |
| Biological samples | ||
| Healthy adult BA9 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5288 |
| Healthy adult BA46 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4638 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB6052 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5988 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4643 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5887 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5350 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5288 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5559 |
| Ataxia-Telangiectasia adult BA9 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4874 |
| Ataxia-Telangiectasia adult BA9 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB1459 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB6018 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5902 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4663 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB1459 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4874 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB1485 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human BDNF | PeproTech | Cat# 450-02 |
| Recombinant Human NT-3 | PeproTech | Cat# 450-03 |
| Laminin | Thermo Fisher Scientific | Cat# 23017-015 |
| Doxycycline | Clontech | Cat# 631311 |
| 2AraC | Sigma-Aldrich | Cat# C1768 |
| Y-27632 | Cayman | Cat# 10005583 |
| Corning Matrigel hESC-Qualified Matrix, LDEV-Free | Corning | Cat# 354277 |
| Gentle Cell Dissociation Reagent | STEMCELL Technologies | Cat# 100-0485 |
| mTeSR-plus | STEMCELL Technologies | Cat# 100-1130 |
| OneComp eBeads | Themro Scientific | Cat# 01111142 |
| Accutase | STEMCELL Technologies | Cat# 07920 |
| Critical commercial assays | ||
| Chromium Single Cell 3′ v3 kit | 10X Genomics | Cat# PN-1000268 |
| STEMdiff Microglia Differentiation Kit | STEMCELL Technologies | Cat# 100-0019 |
| STEMdiff Hematopoietic Kit | STEMCELL Technologies | Cat# 05310 |
| STEMdiff Microglia Maturation Kit | STEMCELL Technologies | Cat# 100-0020 |
| PureLink RNA Mini Kit | Life Technologies | Cat# 12183018A |
| DNA Mini Kit | Qiagen | Cat# 51304 |
| LDH-Glo Cytotoxicity Assay | Promega | Cat# J2380 |
| Deposited data | ||
| snRNA-seq data | This study | dbGaP: phs003005.v2.p1 |
| WGS data | This study | dbGaP: phs003005.v2.p1 |
| RNA-seq data | This study | dbGaP: phs003005.v2.p1 |
| Experimental models: Cell lines | ||
| PGP1-SV1 iPSC line | Synthego | PGP1 |
| A-T patient iPSC line | This paper | This paper |
| Software and algorithms | ||
| CellRanger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| CellBender | Fleming et al., 202217 | https://github.com/broadinstitute/CellBender |
| DoubletFinder | McGinnis et al., 201916 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Seurat | Hao et al., 202115 | https://github.com/satijalab/seurat/ |
| RStan | Stan Development Team | http://mc-stan.org/. |
| EdgeR | Robinson et al., 201077 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| DESeq2 | Love et al., 201478 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| CellChat | Jin et al., 202179 | https://github.com/sqjin/CellChat |
| Monocle2 | Qiu et al., 201751 | https://cole-trapnell-lab.github.io/monocle-release/ |
| GATK4 | Poplin et al., 201880 | https://github.com/broadinstitute/gatk |
| ANNOVAR | Wang et al., 201081 | https://github.com/WGLab/doc-ANNOVAR |
| clusterProfiler | Yu et al., 201282 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| ZEN Black Software (2012) | Zeiss | https://www.micro-shop.zeiss.com/en/us/softwarefinder/software-categories/zen-black/zen-black-system/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human tissue
All human postmortem brain tissue samples were obtained from the NIH NeuroBioBank. Samples were matched as closely as possible for age, sex, RIN, and PMI. All available neuropathology reports and medical records for each sample were obtained. Detailed sample information is in Table S1. All cerebellar vermis samples are from the same anatomical section (section 1) and include the entire vermis, and all cortex samples are from BA9/BA46, which are adjacent Brodmann areas. Sectioning was performed as described by the University of Maryland Brain and Tissue Bank Sectioning Protocol: “LEFT HEMISPHERE: CEREBRUM A cut is made just posterior to the cerebral peduncle and the midbrain/pons/cerebellum are removed as a unit from the left hemisphere (Cut #3). The remaining cerebrum is sectioned coronally, at approximate 1 cm intervals beginning from the frontal pole apex and proceeding caudally. As each section is isolated, it is gently rinsed with water, blotted dry, assigned a sequential numeric identifier (odd numbers only!), and placed in the freezing bath. The handling of sections is best aided by the use of a plastic spatula. Each frozen section is placed into individual plastic bags appropriately labeled and sealed. All bags are then stored in a −80°C freezer prior to shipping. Frozen sections of the cerebrum are identified as.
LEFT HEMISPHERE: CEREBELLUM The remaining cerebellum is placed in a vertical plane (its normal anatomic position) and sectioned at 0.5 to 0.6 cm intervals beginning from the medial surface (vermis) and moving laterally. Each resulting section is assigned a sequential identifier (odd numbers only!). Frozen sections of the cerebellum are identified as,9” (https://www.medschool.umaryland.edu/btbankold/Brain-Protocol-Methods/Brain-Sectioning–Protocol-Method-2/).
Human cell lines
Fibroblasts from an individual with A-T (2 years old, female) were reprogrammed into iPSCs using non-integrative Sendai virus. Control iPSCs from Personal Genome Project participant 1 (PGP1, 53 years old, male) were used as the control line (Synthego).
METHOD DETAILS
Isolation of single nuclei from fresh frozen tissue samples
The nuclei isolation protocol was adapted from two previous publications.83,84 Specifically, the protocol was performed as follows: all procedures were performed on ice or at 4°C. Approximately 100 mg of fresh frozen samples were processed using a Dounce homogenizer in 5 mL of tissue lysis buffer (0.32M sucrose, 5mM CaCl2, 3mM MgAc2, 0.1mM EDTA, 10mM Tris-HCl (pH 8), 0.1% Triton X-100, 1mM DTT). The homogenized solution was loaded on top of a sucrose cushion (1.8M sucrose, 3mM MgAc2, 10mM Tris-HCl (pH 8), 1mM DTT) and spun in an ultracentrifuge in an SW28 rotor (13,300 RPM, 2hrs, 4°C) to separate nuclei. After spinning, supernatant was removed and nuclei were resuspended (1% BSA in PBS plus 25 μL 40U/μL RNAse inhibitor) then filtered through a 40 μm cell strainer. After filtration, nuclei were counted and diluted to a concentration of 1000 cells/μL.
Droplet-based snRNA-seq
Droplet-based libraries were generated using the Chromium Single Cell 3’ v3 reagent kits (10x Genomics Cat# PN-1000268) according to manufacturer’s instructions. Resulting libraries were sequenced on an Illumina Novaseq at 150 paired-end reads with depth 20,000 reads/nucleus.
snRNA-seq analysis
snRNA-seq data for each individual was preprocessed and aligned to the GRCh38–3.0.0 reference genome using CellRanger count. After alignment, ambient/background RNA was removed from the count matrix using CellBender remove-background (v0.2.0).17 The resulting count matrices were used to create Seurat objects per individual using Seurat v4.15 Further quality control removed cells with less than 200 genes per cell, greater than 5000 genes per cell, and greater than 30% mitochondrial content. DoubletFinder16 was used to detect and remove doublets with an expected multiplet rate of 7.6% for ~16,000 cells loaded per sample based on the 10x user guide.
SCTransform was used to perform normalization of expression values per sample.85 After normalization, samples were integrated to align common clusters across individual datasets using Seurat’s integration method.15 Dimensionality reduction was performed using RunPCA and RunUMAP. A shared nearest neighbor graph was constructed using FindNeighbors with dims 1:30, k.param 100, and cosine metric. Clusters were then identified using the Leiden algorithm at a resolution of 1.5.
Marker genes for each cluster were obtained using FindAllMarkers with a minimum log2 fold-change of 0.25. Cluster markers were used to assign cerebellar cell type identities based on known literature cell type markers. For annotating clusters in the prefrontal cortex, SingleR was used for reference-based annotation since reference datasets from the human prefrontal cortex are available.19 Velmeshev et al., 2019 was used as the prefrontal cortex ref.18.
To identify changes in cell type proportions in AT compared to controls, we used a Bayesian statistics approach to compositional data analysis previously described in the context of microbial ecology.20 Compositional data has a negative covariance structure that is accounted for by the multinomial and Dirichlet probability distributions. We implemented Dirichlet multinomial modeling (DMM) using Hamiltonian Monte Carlo (HMC) using the R packages rstan and bayestestR (Stan Development Team (2020). “RStan: the R interface to Stan.” R package version 2.21.2, http://mc-stan.org/.).86 The input to the model was a matrix of counts where the rows correspond to replicates (individual samples) and the columns correspond to cell types. We then modeled the replicates using the multinomial distribution for the probability of each cell type per individual. The Dirichlet distribution was used to model the multinomial parameters. The prior on the Dirichlet parameters was another Dirichlet distribution with a fixed parameter alpha 10−7, which gives uniform cell type proportions in expectation. The resulting posterior distributions were Dirichlet distributions of the cell type proportions in AT and control. We then subtracted the posterior probability distribution (89% credible interval) of control from AT to see whether there are significant differences in relative cell type composition. A cell type shift in log2 abundance ratio (AT/control) was considered significant if the 89% credible did not include 0.
Differential gene expression and Gene Ontology analysis
Differential gene expression analysis was performed on each cell type using edgeR.77 The integrated Seurat object was subset to obtain Seurat objects for each cell type. The read counts were modeled and normalized using a negative binomial distribution with the trimmed mean of M-values (TMM) normalization method. To ensure robust signals, only genes that were expressed in at least 5% of one cell type were included in the analysis. The design matrix formula was ~ disease.status + sex + age + cellular detection rate. Differentially expressed genes between AT and control were identified using the quasi-likelihood approach (glmQLF). Genes with FDR <0.05 and a log2 fold-change greater than 0.50 were used for downstream analysis.
Pseudobulk differential gene expression analysis was performed using DESeq2.78 To generate pseudobulk data, we took the sum of raw counts for each gene over all cells in each sample, resulting in a gene by sample counts matrix. The design matrix was specified as ~ sex + age + disease.status. Differentially expressed genes between AT and controls were identified after running lfcShrink. Genes with FDR <0.05 were considered statistically significant.
Gene Ontology (GO) enrichment analysis was performed on the upregulated and downregulated differentially expressed genes in each cell type using clusterProfiler.82 GO biological processes with FDR <0.05 were considered significant.
Gene Set Enrichment Analysis was performed on differentially expressed genes ordered by log2 fold-change (descending order) with the function GSEA() with default settings from clusterProfiler. Human gene sets of interest were obtained from the Molecular Signatures Database (MSigDB)87,88 and are listed in Supplementary Table 21.
CellChat79 was used to perform ligand-receptor analysis in order to identify differential cell-cell communication in AT compared to control. Differentially active signaling pathways with FDR<0.05 were considered significant.
Cell cycle scores
AddModuleScore calculates the average expression of a gene set in each cell, subtracted by the average expression of a control gene set, as previously described.89 A positive score means the module of genes is expressed more highly in a particular cell than expected by chance. Cell cycle phase scores were calculated using this method based on canonical cell cycle markers using the Seurat function CellCycleScoring, which then outputs the predicted classification (G1, S, or G2M phase) for each cell.
Disease gene analysis
Known monogenic cerebellar disease genes were obtained from the Online Mendelian Inheritance in Man (OMIM) database. The mouse cerebellum data was obtained from Saunders et al., 2018.90 To determine the correlation of cerebellar disease gene expression between humans and mice, cosine similarity was calculated between the human and mouse expression vectors of the genes for each cell type.
Pseudotime analysis
Pseudotime trajectory analysis was performed using Monocle2.51 First, Seurat objects for each cell type (Microglia, Purkinje, and granule cells) were converted into the Monocle2 compatible cell dataset form using as.CellDataSet. Cells were clustered in an unsupervised manner using clusterCells. The top 1000 differentially expressed genes between AT and control in each cell type were used as the ordering genes to order cells along the pseudotime trajectory. The root state of the trajectory was defined as the cluster with the highest proportion of control cells. Genes that change as a function of pseudotime were identified using differentialGeneTest with the model formula specified as ~ sm.ns(Pseudotime), which fits a natural spline to model gene expression as a smooth nonlinear function of pseudotime. Genes that change over pseudotime with an FDR<0.05 were considered significant. These genes were then clustered according to their pseudotime expression profiles. GO enrichment analysis was performed on each cluster using cluster-Profiler to identify the biological processes that co-vary across pseudotime.
To align the pseudotime trajectories between cell types, the method dynamic time warping (DTW) was applied as previously described.53 DTW aligns similar temporal processes that differ in length by calculating the optimal matching between points in each sequence. For each cell type, ordered cells in pseudotime were binned into 100 “pseudocells” and the average expression of each gene was calculated within each pseudocell. The resulting gene x pseudocell matrices were the input for the dtw package in R. The Purkinje cell matrix was set as the reference and the other cell type was the query. The cost matrix was calculated as 1-corr(M1, M2), where corr(M1, M2) is the cosine similarity matrix between cell type 1 matrix (M1, reference) and cell type 2 matrix (M2, query). The query cell type pseudotime values were then warped into the reference to obtain aligned pseudotimes.
Whole genome sequencing and variant Calling
DNA was isolated using the Qiagen DNA Mini kit (Qiagen Cat# 51304) according to the protocol for tissues. Approximately 25mg of fresh frozen tissue was minced into small pieces. Tissue was transferred to a 1.5mL microcentrifuge tube and 180μL of Buffer ATL as added. 20ul of proteinase K was added prior to 4 h of agitation at 56°C on a thermomixer (1400 RPM). DNA isolation proceeded as written in the protocol with the inclusion of the optional RNase A step. Libraries were prepared with the TruSeq DNA PCR-free library kit (Illumina). Libraries were sequenced at 30X coverage with Illumina NovaSeq 6000 S4, 2 x 150 bp. WGS data for each individual was processed using GATK4.80 Specifically, we used the DRAGEN-GATK whole genome germline pipeline for variant discovery with the WGS_Maximum_Quality mode on Terra (terra.bio). Reads were aligned using hg38 as the reference genome. Resulting VCF files were annotated using ANNOVAR.81 Sample information and variants are listed in Table S1.
Immunostaining and Imaging
Postmortem human formalin fixed vermis samples from four subjects diagnosed with Ataxia-Telangiectasia and three age matched controls were obtained from the University of Maryland Brain and Tissue Bank (Table S1). The samples were embedded in paraffin by the UMCCTS Biospecimen, Tissue and Tumor Bank, and sectioned on a LEICA RM2125 microtome. Sections were cut at 20 μm thickness, placed on uncharged untreated Fisherfinest premium microscope slides (Cat# 12-544-7), and dried overnight at 37°C. Eight sections (four control and four with AT pathology) were selected at random, deparaffinized with xylene and rehydrated in a graded ethanol wash series. This was followed by antigen retrieval, done by heating citrate buffer (pH 6.2) to 90°C and leaving the slides to soak for 45 min, then allowing the solution and slides to cool to room temperature. The samples were then subjected to brief PBS washes before application of a primary antibody solution consisting of blocking solution, 10% Triton in 10x PBS for permeabilization, and primary antibody. Primary antibodies used included Anti-CD11B/Integrin Alpha M Monoclonal Antibody (Proteintech, Cat# 66519-1-Ig, concentration 1:100), and Anti-GFAP antibody produced in rabbit (Millipore Sigma Cat# HPA056030, concentration 1:250). The samples were incubated at 4°C overnight. The following day the samples were washed for an hour and a half in cold 1x PBS before application of secondary antibody solution consisting of blocking solution, 10% Triton, and Donkey anti-Mouse Cross-Adsorbed Secondary Antibody, Alexa Fluor plus 555 (ThermoFisher Scientific, Invitrogen, Cat# A32773, concentration 1:125) and Donkey anti-Rabbit Cross-Adsorbed Secondary Antibody Alexa Fluor plus 488 (ThermoFisher Scientific, Invitrogen, Cat# A32790, concentration 1:125). After incubating overnight in the dark at 4°C the samples were washed in 1x PBS for an hour and a half in the dark. The tissue was stained for DAPI (ThermoFisher Scientific, Cat# 62248, concentration 1:10,000 in 1x PBS), washed and then treated with TrueBlack lipofuscin autofluorescence quencher (Biotium, Cat# 23007) according to the manufacturer’s protocol. The samples were subjected to 30 min of washes in Barnstead water before being allowed to air dry. The tissue was mounted in Everbrite (Biotium, Cat# 23003) with Platinum Line cover glass (FisherScientific, Cat# 15-183-89) and left to cure overnight. Imaging was performed on a Zeiss LSM700 inverted microscope using ZEN Black 2012 Software.
Maintenance and culture of iPSCs
Fibroblasts from an individual with A-T (2 years old, female) were reprogrammed into iPSCs using non-integrative Sendai virus. The patient had the following compound-heterozygous variants in ATM: hg38 chr11:g.108332838C>T; chr11:g.108347266_108353792del27. Control iPSCs from Personal Genome Project participant 1 (PGP1) were used as the control line. Both iPSC lines were mycoplasma negative, karyotypically normal, and expressed pluripotency markers OCT4, SOX2, NANOG, SSEA4, TRA-1-60. iPSCs were maintained in 6-well or 10 cm plates (Corning) coated with LDEV-free hESC-qualified Matrigel (Corning Cat# 354277) in feeder-free conditions with complete mTeSR-plus medium (STEMCELL Technologies Cat# 100–1130) in a humidified incubator (5% CO2, 37°C). iPSCs were fed fresh media daily and passaged every 3–4 days.
Differentiation of iPSCs to hematopoietic progenitor Cells
Human iPSC-derived hematopoietic progenitors (HPC) were generated using the STEMdiff Hematopoietic Kit (STEMCELL Technologies Cat# 05310) according to the manufacturer instructions based on a protocol by Abud et al., 2017.54 Briefly, iPSCs were dissociated into aggregates using Gentle Cell Dissociation Reagent (STEMCELL Technologies Cat# 100–0485) and 40–80 aggregates were plated per well on Matrigel coated 12-well plates. Cells were maintained in Medium A to induce a mesoderm-like state until day 3. From day 3 to day 12, cells were maintained in Medium B with half-media changes every 2 days to promote further differentiation into hematopoietic progenitor cells. To confirm the differentiation from iPSC to HPC, we performed flow cytometry on day 12 at the end of HPC differentiation. Cells were harvested and incubated with CD45-APC (1 μmL per 50,000 cells in 100 μL, STEMCELL Technologies Cat# 60018AZ) and CD34-PE (1 μL per 50,000 cells in 100 μL, STEMCELL Technologies Cat# 60119PE) antibodies for 15 min in the dark at 4°C. In addition to double-stained samples, we included unstained samples, Fluorescence Minus One (FMO) controls, and compensation single-stain controls using OneComp eBeads (Thermo Scientific Cat# 01111142). Samples were washed twice with FACS buffer (Phosphate Buffered Saline (PBS) with 2% FBS) and transferred to 5 mL Round Bottom Polystyrene FACS tubes. Flow cytometry was performed at the HSCI-BCH Flow Cytometry Research Facility using the BD FACSAria II. Analysis was performed using FlowJo software.
Differentiation of hematopoietic progenitor cells to Microglia
Human iPSC-derived microglia (iMGL) were generated from human iPSC-derived hematopoietic progenitors using the STEMdiff Microglia Differentiation Kit (STEMCELL Technologies Cat# 100-0019) and STEMdiff Microglia Maturation Kit (STEMCELL Technologies Cat# 100-0020) according to manufacturer instructions. Briefly, hematopoietic progenitor cells from day 12 of the STEMdiff Hematopoietic Kit protocol were collected and transferred to a Matrigel coated 6-well plate (200,000 cells per well) with STEMdiff Microglia Differentiation Medium. From day 0–24, 1 mL Microglia Differentiation Medium was added to each well every other day. After day 24, cells were maintained in STEMdiff Microglia Maturation Medium with half-medium volume added every other day. To confirm the differentiation from HPC to iMGL, we performed flow cytometry on day 24 at the end of microglia differentiation. Cells were harvested and blocked with CD32 antibody (2 μL per 50,000 cells in 100 μL, STEMCELL Technologies Cat# 60012) for 15 min at 4°C. Samples were then incubated with CD45-APC (1 μL per 50,000 cells in 100 μL, STEMCELL Technologies Cat# 60018AZ) and CD11B-FITC (1 μL per 50,000 cells in 100 μL, STEMCELL Technologies Cat# 60040FI) antibodies for 15 min in the dark at 4°C. In addition to double-stained samples, we included unstained samples, FMO controls, and compensation single-stain controls using OneComp eBeads (Thermo Scientific Cat# 01111142). Samples were washed twice with FACS buffer (Phosphate Buffered Saline (PBS) with 2% FBS) and transferred to 5 mL Round Bottom Polystyrene FACS tubes. Flow cytometry was performed at the HSCI-BCH Flow Cytometry Research Facility using the BD FACSAria II. Analysis was performed using FlowJo software.
RNA-sequencing of A-T and control iMGL
RNA was harvested from day 29 iMGL samples using the PureLink RNA Mini kit (Life Technologies Cat# 12183018A). Libraries were prepared with the Illumina Stranded mRNA prep. Sequencing was performed on Illumina HiSeq, 2x150bp configuration at approximately 20X depth per sample. Reads were aligned to hg38 using STAR v2.7.9a followed by processing with featureCounts to obtain a gene-level counts matrix. Differential gene expression analysis was performed using DESeq2 with control set as the reference condition and results coefficient set as ‘condition_AT_vs_control’.
Differentiation of iPSCs to neurons
Human iPSC-derived neurons (iNs) were generated from iPSCs transduced with the lentiviruses Tet-O-Ngn2-Puro and FUW-M2rtTA based on a previously published protocol.60 On day −1 of differentiation, iPSC colonies were dissociated into single-cells using Accutase (STEMCELL Technologies Cat# 07920) and 4–8 million cells were plated on Matrigel coated 10 cm dishes with complete mTeSR plus medium supplemented with Y-27632 (10 μM). On day 0, cells were fed N2 media (DMEM/F-12 media, 1X N2, 1X Nonessential Amino Acids) supplemented with doxycycline (2 μg/mL), BDNF (10 ng/mL), NT3 (10 ng/mL), and laminin (0.2 μg/mL). On day 1, media was replaced with N2 media with puromycin (1 μg/mL) in addition to the supplements listed above. On day 2, cells were fed B27 media (Neurobasal media, 1X B27, 1X Glutamax) supplemented with puromycin (1 μg/mL), doxycycline (2 μg/mL), Ara-C (2 μM), BDNF (10 ng/mL), NT3 (10 ng/mL), and laminin (0.2 μg/mL). On day 3, cells were dissociated into single cells using Accutase and replated onto polyethylenimine/laminin coated 96-well plates (10,000 cells/well) with B27 media supplemented with Y-27632 (10 μM), doxycycline (2 μg/mL), Ara-C (2 μM), BDNF (10 ng/mL), NT3 (10 ng/mL), and laminin (0.2 μg/mL). On day 5, cells were fed with Conditioned Sudhof Neuronal Growth Medium (1:1 ratio of Astrocyte Conditioned Media and Neurobasal Media, 1X B27, Glutamax, NaHCO3, and transferrin) supplemented with BDNF (10 ng/mL), NT3 (10 ng/mL), and laminin (0.2 μg/mL).
Co-culture of iMGL and iNs
Day 29 iMGL were added to Day 6 iNs plated in a 96-well plate at a 1:5 ratio (2,000 iMGL:10,000 neurons per well) with 15 independent wells per co-culture condition (A-T iMGL/A-T iN; A-T iMGL/control iN; control iMGL/A-T iN; control iMGL/control iN). The co-cultures were maintained for 6 days.
Cytotoxicity assay
Cytotoxicity of co-cultures were assessed with the LDH-Glo Cytotoxicity Assay (Promega Cat# J2380) according to the manufacturer instructions. 5 μL of medium from each co-culture well was diluted in 95 μL LDH Storage Buffer (200 mM Tris-HCl pH 7.3, 10% Glycerol, 1% BSA). LDH activity was measured by combining 10 μL LDH Detection Reagent with 10 μL diluted sample in a 384-well white opaque-walled assay plate (Greiner Bio-One Catalog # 784080) after incubating for 45 min at room temperature. LDH standards were prepared according to manufacturer instructions. Fresh culture medium was used as a no-cell control. To generate a maximum LDH release control, 2 μL of 10% Triton X-100 was added per 100 μL to 96-wells containing iNs for 10 min followed by sample collection in LDH storage buffer as described above. Plate was read using the Spectramax iD5 plate reader with the luminescence read mode. Percent cytotoxicity was calculated according to the kit’s protocol as % Cytotoxicity = 100 × (Experimental LDH Release – Medium Background)/(Maximum LDH Release Control – Medium Background).
Quantification and statistical analysis
No statistical methods were used to predetermine sample sizes. All statistical tests were performed in R (v4.0.1) or Python (v3.8) with multiple hypothesis correction using the Benjamini-Hochberg procedure (FDR<0.05) unless otherwise specified. Statistical tests and sample sizes are noted in figure legends, and include Fisher’s exact test, Student’s t-test and Mann-Whitney U-test.
Supplementary Material
Highlights.
A-T microglia show signatures of aging and disease-associated microglia
A-T cerebellum has greater microglia activation compared to cortex
A-T patient iPSC-derived microglia (iMGLs) exhibit cell-intrinsic activation
A-T iMGLs are sufficient to increase cytotoxicity of neuronal co-cultures
ACKNOWLEDGMENTS
We thank the donors, patients, and NIH NeuroBioBank for providing human tissues used in this study. We would like to thank Brad Margus from the A-T Children’s Project for helpful scientific discussion. We also acknowledge Allison R. McLean and Jeffery R. Gulcher from Genuity Sciences for their assistance with snRNA-seq data coordination, Ronald Mathieu from the HSCI-BCH Flow Cytometry Research Facility for his assistance with flow cytometry, and the HSCI iPS Core Facility for their assistance with iPSC derivation. Illustrations were created with BioRender.com. This work was supported by National Institutes of Health grants K01 AG051791 (E.A.L.), DP2 AG072437 (E.A.L.), and R00 AG054748 (M.A.L.); the A-T Children’s Project (E.A.L., M.A.L.); the Suh Kyungbae Foundation (E.A.L.); the Paul G. Allen Family Foundation (E.A.L.); the American Federation for Aging Research (M.A.L. and A.M.J.); and the Charles H. Hood Child Health Foundation (E.A.L., M.A.L.). J.L. was supported by award T32GM007753 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113622.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- 2.Petley E, Yule A, Alexander S, Ojha S, and Whitehouse WP (2022). The natural history of ataxia-telangiectasia (A-T): A systematic review. PLoS One 17, e0264177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavin MF, and Shiloh Y (1997). The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol 15, 177–202. [DOI] [PubMed] [Google Scholar]
- 4.Watters D, Khanna KK, Beamish H, Birrell G, Spring K, Kedar P, Gatei M, Stenzel D, Hobson K, Kozlov S, et al. (1997). Cellular localisation of the ataxia-telangiectasia (ATM) gene product and discrimination between mutated and normal forms. Oncogene 14, 1911–1921. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Lee JH, Paull TT, Gehrke S, D’Alessandro A, Dou Q, Gladyshev VN, Schroeder EA, Steyl SK, Christian BE, and Shadel GS (2018). Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci. Signal 11, eaaq0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng A, Tse KH, Chow HM, Gan Y, Song X, Ma F, Qian YXY, She W, and Herrup K (2021). ATM loss disrupts the autophagy-lysosomal pathway. Autophagy 17, 1998–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavin MF (2013). The appropriateness of the mouse model for ataxia-telangiectasia: neurological defects but no neurodegeneration. DNA Repair 12, 612–619. [DOI] [PubMed] [Google Scholar]
- 8.Jiang D, Zhang Y, Hart RP, Chen J, Herrup K, and Li J (2015). Alteration in 5-hydroxymethylcytosine-mediated epigenetic regulation leads to Purkinje cell vulnerability in ATM deficiency. Brain 138, 3520–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nayler SP, Powell JE, Vanichkina DP, Korn O, Wells CA, Kanjhan R, Sun J, Taft RJ, Lavin MF, and Wolvetang EJ (2017). Human iPSC-Derived Cerebellar Neurons from a Patient with Ataxia-Telangiectasia Reveal Disrupted Gene Regulatory Networks. Front. Cell. Neurosci 11, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourseguin J, Cheng W, Talbot E, Hardy L, Lai J, Jeffries AM, Lodato MA, Lee EA, and Khoronenkova SV (2022). Persistent DNA damage associated with ATM kinase deficiency promotes microglial dysfunction. Nucleic Acids Res. 50, 2700–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tavani F, Zimmerman RA, Berry GT, Sullivan K, Gatti R, and Bingham P (2003). Ataxia-telangiectasia: the pattern of cerebellar atrophy on MRI. Neuroradiology 45, 315–319. [DOI] [PubMed] [Google Scholar]
- 12.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, and Zhang K (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozareva V, Martin C, Osorno T, Rudolph S, Guo C, Vanderburg C, Nadaf N, Regev A, Regehr WG, and Macosko E (2021). A transcriptomic atlas of mouse cerebellar cortex comprehensively defines cell types. Nature 598, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGinnis CS, Murrow LM, and Gartner ZJ (2019). Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleming SJ, Chaffin MD, Arduini A, Akkad A-D, Banks E, Marioni JC, Philippakis AA, Ellinor PT, and Babadi M (2022). Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Preprint at bioRxiv, 10.1101/791699. [DOI] [PubMed] [Google Scholar]
- 18.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison JG, Calder WJ, Shastry V, and Buerkle CA (2020). Dirichlet-multinomial modelling outperforms alternatives for analysis of microbiome and other ecological count data. Mol. Ecol. Resour 20, 481–497. [DOI] [PubMed] [Google Scholar]
- 21.Sahama I, Sinclair K, Fiori S, Pannek K, Lavin M, and Rose S (2014). Altered corticomotor-cerebellar integrity in young ataxia telangiectasia patients. Mov. Disord 29, 1289–1298. [DOI] [PubMed] [Google Scholar]
- 22.Tse K-H, and Herrup K (2017). DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech. Ageing Dev 161, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aldinger KA, Thomson Z, Phelps IG, Haldipur P, Deng M, Timms AE, Hirano M, Santpere G, Roco C, Rosenberg AB, et al. (2021). Spatial and cell type transcriptional landscape of human cerebellar development. Nat. Neurosci 24, 1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, et al. (2007). AGEMAP: a gene expression database for aging in mice. PLoS Genet. 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, Mayweather BA, Nguyen L, Niziolek Z, Ozek C, et al. (2019). Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci 22, 1696–1708. [DOI] [PubMed] [Google Scholar]
- 26.Song WM, and Colonna M (2018). The identity and function of microglia in neurodegeneration. Nat. Immunol 19, 1048–1058. [DOI] [PubMed] [Google Scholar]
- 27.Nakayama H, Abe M, Morimoto C, Iida T, Okabe S, Sakimura K, and Hashimoto K (2018). Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nat. Commun 9, 2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy A, Fung YK, Liu X, and Pahan K (2006). Up-regulation of Microglial CD11b Expression by Nitric Oxide. J. Biol. Chem 281, 14971–14980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn-Hardy K, Fung HC, Lin X, et al. (2007). Deletion at ITPR1 Underlies Ataxia in Mice and Spinocerebellar Ataxia 15 in Humans. PLoS Genet. 3, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang L, Chardon JW, Carter MT, Friend KL, Dudding TE, Schwartzentruber J, Zou R, Schofield PW, Douglas S, Bulman DE, and Boycott KM (2012). Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis 7, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sando R, Gounko N, Pieraut S, Liao L, Yates J 3rd, and Maximov A (2012). HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151, 821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, and Herrup K (2012). Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat. Med 18, 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasan G, and Sharma A (2020). Regulation of neuronal physiology by Ca2+ release through the IP3R. Current Opinion in Physiology 17, 1–8. [Google Scholar]
- 34.Hisatsune C, Miyamoto H, Hirono M, Yamaguchi N, Sugawara T, Ogawa N, Ebisui E, Ohshima T, Yamada M, Hensch TK, et al. (2013). IP3R1 deficiency in the cerebellum/brainstem causes basal ganglia-independent dystonia by triggering tonic Purkinje cell firings in mice. Front. Neural Circuits 7, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang TM, et al. (2019). Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 28, 2111–2123.e6. [DOI] [PubMed] [Google Scholar]
- 36.Jafari M, Schumacher AM, Snaidero N, Ullrich Gavilanes EM, Neziraj T, Kocsis-Jutka V, Engels D, Jürgens T, Wagner I, Weidinger JDF, et al. (2021). Phagocyte-mediated synapse removal in cortical neuroinflammation is promoted by local calcium accumulation. Nat. Neurosci 24, 355–367. [DOI] [PubMed] [Google Scholar]
- 37.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, Piehowski P, Kapasi A, Nejad P, Cimpean M, et al. (2018). A transcriptomic atlas of aged human microglia. Nat. Commun 9, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 41.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marín-Teva JL, Cuadros MA, Martín-Oliva D, and Navascués J (2011). Microglia and neuronal cell death. Neuron Glia Biol. 7, 25–40. [DOI] [PubMed] [Google Scholar]
- 43.Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, et al. (2015). DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 42, 332–343. [DOI] [PubMed] [Google Scholar]
- 44.Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, Zhou Y, Chen Y, Huang J, Wang RF, and Cui J (2016). TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 64, 105–119. [DOI] [PubMed] [Google Scholar]
- 45.Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, and Jung JU (2018). TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun 9, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu M-M, and Shu H-B (2017). Multifaceted roles of TRIM38 in innate immune and inflammatory responses. Cell. Mol. Immunol 14, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stowell RD, Wong EL, Batchelor HN, Mendes MS, Lamantia CE, Whitelaw BS, and Majewska AK (2018). Cerebellar microglia are dynamically unique and survey Purkinje neurons in vivo. Dev. Neurobiol 78, 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tay TL, Sagar Dautzenberg J, Grüm D, Grüm D, and Prinz M (2018). Unique microglia recovery population revealed by single-cell RNAseq following neurodegeneration. Acta Neuropathol. Commun 6, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, and McColl BW (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci 19, 504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cacchiarelli D, Qiu X, Srivatsan S, Manfredi A, Ziller M, Overbey E, Grimaldi A, Grimsby J, Pokharel P, Livak KJ, et al. (2018). Aligning Single-Cell Developmental and Reprogramming Trajectories Identifies Molecular Determinants of Myogenic Reprogramming Outcome. Cell Syst. 7, 258–268.e3. [DOI] [PubMed] [Google Scholar]
- 54.Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. (2017). iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 94, 278–293.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong C, Tijhuis AE, and Foijer F (2019). The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells 8, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Z, and Hornung V (2022). cGAS-STING signaling. Curr. Biol 32, R730–R734. [DOI] [PubMed] [Google Scholar]
- 57.Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, Qian G, and Zheng JC (2013). IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J. Neurochem 125, 897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mantovani A, Dinarello CA, Molgora M, and Garlanda C (2019). Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 50, 778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kessing CF, and Tyor WR (2015). Interferon-α Induces Neurotoxicity Through Activation of the Type I Receptor and the GluN2A Subunit of the NMDA Receptor. J. Interferon Cytokine Res 35, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quek H, Luff J, Cheung K, Kozlov S, Gatei M, Lee CS, Bellingham MC, Noakes PG, Lim YC, Barnett NL, et al. (2017). A rat model of ataxia-telangiectasia: evidence for a neurodegenerative phenotype. Hum. Mol. Genet 26, 109–123. [DOI] [PubMed] [Google Scholar]
- 62.Song X, Ma F, and Herrup K (2019). Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J. Neurosci 39, 6378–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, Garcia FJ, Therrien M, Novis HS, Gao F, et al. (2020). Cell Type-Specific Transcriptomics Reveals that Mutant Huntingtin Leads to Mitochondrial RNA Release and Neuronal Innate Immune Activation. Neuron 107, 891–908.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI, Duffy D, Tamby C, et al. (2018). Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sprenger H-G, MacVicar T, Bahat A, Fiedler KU, Hermans S, Ehrentraut D, Ried K, Milenkovic D, Bonekamp N, Larsson NG, et al. (2021). Cellular pyrimidine imbalance triggers mitochondrial DNA–dependent innate immunity. Nat. Metab 3, 636–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. (2019). L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hui CW, Song X, Ma F, Shen X, and Herrup K (2018). Ibuprofen prevents progression of ataxia telangiectasia symptoms in ATM-deficient mice. J. Neuroinflammation 15, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hickman S, Izzy S, Sen P, Morsett L, and El Khoury J (2018). J. Microglia in neurodegeneration. Nat. Neurosci 21, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Levi H, Bar E, Cohen-Adiv S, Sweitat S, Kanner S, Galron R, Mitiagin Y, and Barzilai A (2022). Dysfunction of cerebellar microglia in Ataxia-telangiectasia. Glia 70, 536–557. [DOI] [PubMed] [Google Scholar]
- 70.Korbo L, and Andersen BB (1995). The distributions of Purkinje cell perikaryon and nuclear volume in human and rat cerebellum with the nucleator method. Neuroscience 69, 151–158. [DOI] [PubMed] [Google Scholar]
- 71.Buchholz DE, Carroll TS, Kocabas A, Zhu X, Behesti H, Faust PL, Stalbow L, Fang Y, and Hatten ME (2020). Novel genetic features of human and mouse Purkinje cell differentiation defined by comparative transcriptomics. Proc. Natl. Acad. Sci. USA 117, 15085–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu X, Stoyanova EI, Lemiesz AE, Xing J, Mash DC, and Heintz N (2018). Species and cell-type properties of classically defined human and rodent neurons and glia. Elife 7, e37551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zannolli R, Buoni S, Betti G, Salvucci S, Plebani A, Soresina A, Pietrogrande MC, Martino S, Leuzzi V, Finocchi A, et al. (2012). A randomized trial of oral betamethasone to reduce ataxia symptoms in ataxia telangiectasia. Mov. Disord 27, 1312–1316. [DOI] [PubMed] [Google Scholar]
- 74.Menotta M, Biagiotti S, Bianchi M, Chessa L, and Magnani M (2012). Dexamethasone Partially Rescues Ataxia Telangiectasia-mutated (ATM) Deficiency in Ataxia Telangiectasia by Promoting a Shortened Protein Variant Retaining Kinase Activity. J. Biol. Chem 287, 41352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chessa L, Leuzzi V, Plebani A, Soresina A, Micheli R, D’Agnano D, Venturi T, Molinaro A, Fazzi E, Marini M, et al. (2014). Intra-Erythrocyte Infusion of Dexamethasone Reduces Neurological Symptoms in Ataxia Teleangiectasia Patients: Results of a Phase 2 Trial. Orphanet J. Rare Dis 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saberi-Karimian M, Beyraghi-Tousi M, Jamialahmadi T, and Sahebkar A (2022). The positive short-term effect of dexamethasone on ataxia symptoms in a patient with ataxia-telangiectasia: A case report. Clin. Case Rep 10, e05895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun 12, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Auwera G.A.V. der, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, et al. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at bioRxiv, 10.1101/201178. [DOI] [Google Scholar]
- 81.Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matevossian A, and Akbarian S (2008). Neuronal nuclei isolation from human postmortem brain tissue. J. Vis. Exp 914, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, and Frisén J (2005). Retrospective birth dating of cells in humans. Cell 122, 133–143. [DOI] [PubMed] [Google Scholar]
- 85.Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Makowski D, Ben-Shachar M, and Lüdecke D (2019). Describing Effects and their Uncertainty, Existence and Significance within the Bayesian Framework. J. Open Source Softw 4, 1541. [Google Scholar]
- 87.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdöttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-nucleus RNA-sequencing and whole genome sequencing data from postmortem individuals and RNA-sequencing data from human iPSC lines generated in this study have been deposited at dbGaP: phs003005.v2.p1 and are publicly available as of date of publication. Single-nucleus RNA-seq data have also been deposited at the Broad Single Cell Portal (https://singlecell.broadinstitute.org; Study ID SCP1300 and SCP1302). Accession numbers for all deposited data are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD11B/Integrin Alpha M | Proteintech | Cat# 66519-1-Ig; RRID:AB_2881882 |
| Anti-GFAP | Millipore Sigma | Cat# HPA056030 |
| Anti-CD32 | STEMCELL Technologies | Cat# 60012; RRID:AB_2925215 |
| Anti-CD45-APC | STEMCELL Technologies | Cat# 60018AZ |
| Anti-CD34-PE | STEMCELL Technologies | Cat# 60119PE |
| Anti-CD11B-FITC | STEMCELL Technologies | Cat# 60040FI |
| Biological samples | ||
| Healthy adult BA9 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5288 |
| Healthy adult BA46 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4638 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB6052 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5988 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4643 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5887 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5350 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5288 |
| Healthy adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5559 |
| Ataxia-Telangiectasia adult BA9 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4874 |
| Ataxia-Telangiectasia adult BA9 brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB1459 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB6018 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB5902 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4663 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB1459 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB4874 |
| Ataxia-Telangiectasia adult cerebellar vermis brain tissue | NIH NeuroBioBank; https://neurobiobank.nih.gov/ | Cat# UMB1485 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human BDNF | PeproTech | Cat# 450-02 |
| Recombinant Human NT-3 | PeproTech | Cat# 450-03 |
| Laminin | Thermo Fisher Scientific | Cat# 23017-015 |
| Doxycycline | Clontech | Cat# 631311 |
| 2AraC | Sigma-Aldrich | Cat# C1768 |
| Y-27632 | Cayman | Cat# 10005583 |
| Corning Matrigel hESC-Qualified Matrix, LDEV-Free | Corning | Cat# 354277 |
| Gentle Cell Dissociation Reagent | STEMCELL Technologies | Cat# 100-0485 |
| mTeSR-plus | STEMCELL Technologies | Cat# 100-1130 |
| OneComp eBeads | Themro Scientific | Cat# 01111142 |
| Accutase | STEMCELL Technologies | Cat# 07920 |
| Critical commercial assays | ||
| Chromium Single Cell 3′ v3 kit | 10X Genomics | Cat# PN-1000268 |
| STEMdiff Microglia Differentiation Kit | STEMCELL Technologies | Cat# 100-0019 |
| STEMdiff Hematopoietic Kit | STEMCELL Technologies | Cat# 05310 |
| STEMdiff Microglia Maturation Kit | STEMCELL Technologies | Cat# 100-0020 |
| PureLink RNA Mini Kit | Life Technologies | Cat# 12183018A |
| DNA Mini Kit | Qiagen | Cat# 51304 |
| LDH-Glo Cytotoxicity Assay | Promega | Cat# J2380 |
| Deposited data | ||
| snRNA-seq data | This study | dbGaP: phs003005.v2.p1 |
| WGS data | This study | dbGaP: phs003005.v2.p1 |
| RNA-seq data | This study | dbGaP: phs003005.v2.p1 |
| Experimental models: Cell lines | ||
| PGP1-SV1 iPSC line | Synthego | PGP1 |
| A-T patient iPSC line | This paper | This paper |
| Software and algorithms | ||
| CellRanger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| CellBender | Fleming et al., 202217 | https://github.com/broadinstitute/CellBender |
| DoubletFinder | McGinnis et al., 201916 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Seurat | Hao et al., 202115 | https://github.com/satijalab/seurat/ |
| RStan | Stan Development Team | http://mc-stan.org/. |
| EdgeR | Robinson et al., 201077 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| DESeq2 | Love et al., 201478 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| CellChat | Jin et al., 202179 | https://github.com/sqjin/CellChat |
| Monocle2 | Qiu et al., 201751 | https://cole-trapnell-lab.github.io/monocle-release/ |
| GATK4 | Poplin et al., 201880 | https://github.com/broadinstitute/gatk |
| ANNOVAR | Wang et al., 201081 | https://github.com/WGLab/doc-ANNOVAR |
| clusterProfiler | Yu et al., 201282 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| ZEN Black Software (2012) | Zeiss | https://www.micro-shop.zeiss.com/en/us/softwarefinder/software-categories/zen-black/zen-black-system/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |