

Abstract
Vertebrate poly(A) polymerase (PAP) contains a catalytic domain and a C-terminal Ser-Thr-rich regulatory region. Consensus and nonconsensus cyclin-dependent kinase (cdk) sites are conserved in the Ser-Thr-rich region in vertebrate PAPs. PAP is phosphorylated by cdc2-cyclin B on these sites in vitro and in vivo and is inactivated by hyperphosphorylation in M-phase cells, when cdc2-cyclin B is active. In the experiments described here, we undertook a genetic approach in chicken DT40 cells to study the function of PAP phosphorylation. We found that PAP is highly conserved in chicken and is essential in DT40 cells. While cells could tolerate reduced levels of PAP, even modest overexpression of either wild-type PAP or a mutant PAP with two consensus cdk sites mutated (cdk− PAP) was highly deleterious and at a minimum resulted in reduced growth rates. Importantly, cells that expressed cdk− PAP had a significantly lower growth rate than did cells that expressed similar levels of wild-type PAP, which was reflected in increased accumulation of cells in the G0-G1 phase of the cell cycle. We propose that the lower growth rate is due to the failure of hyperphosphorylation and thus M-phase inactivation of cdk− PAP.
Since the discovery of poly(A) polymerase (PAP) almost 40 years ago (9), much progress has been made toward the understanding of the function of poly(A) tails, as well as the machinery that carries out polyadenylation (for reviews, see references 6 and 37). The polyadenylation machinery is composed of multiple factors. There are two coupled reactions in polyadenylation, the endonucleolytic cleavage of the pre-mRNA and the synthesis of the poly(A) tail onto the cleaved mRNA. Cleavage and polyadenylation specificity factor is required for both the cleavage and the poly(A) synthesis phases of the reaction. It is composed of four subunits of 160, 100, 73, and 30 kDa (e.g., references 2 and 22). CPSF-160 recognizes the polyadenylation signal AAUAAA (23). Cleavage stimulation factor (CstF) is required for efficient cleavage. It is composed of three subunits of 77, 64, and 50 kDa (31). CstF-64 binds the GU-rich region found just downstream of the cleavage site in many pre-mRNAs (20, 32, 34). Cleavage factors I and/or II (CF I and CF II [30]) are likely directly involved in cleavage of the pre-mRNA, which occurs about 10 to 30 nucleotides downstream of AAUAAA. CF I appears to consist of three subunits with molecular masses of 68, 59, and 25 kDa (26, 27). PAP synthesizes the poly(A) tail onto precleaved mRNA and is targeted to the mRNA by interaction with CPSF (23, 24, 36).
The cloning of components of the polyadenylation machinery has facilitated an understanding of cellular regulation of pre-mRNA processing. For example, CPSF was detected associated with transcription factor TFIID and in the RNA polymerase II (Pol II) holoenzyme, which revealed a link between transcription initiation and elongation by Pol II and processing of the 3′ end of the mRNA (8, 21). The level of CstF-64 was shown to increase during B-cell maturation, causing a switch from expression of membrane-bound to secreted-form immunoglobulin M (IgM). This reflects an increase in intact CstF, which results in enhanced usage of a weak upstream polyadenylation signal, which in turn enhances synthesis of the secreted-form IgM mRNA (33).
PAP itself can be a target of regulation. Multiple forms of PAP mRNA exist in vivo in vertebrates. The “full-length” PAPs (PAP I, II, and IV) arise by alternative splicing of 3′ exons. They all contain a functional catalytic region and a C-terminal serine-threonine-rich (S/T-rich) region (19, 24). The “short-form” mRNAs (PAP III, V, and VI) are produced by competition between polyadenylation and splicing, and the proteins they would produce (which have to date not been detected) would be truncated in the middle of the catalytic region (40). The function of the short forms are unknown, as are the functional differences, if any, between the full-length PAPs. The full-length PAPs contain consensus and nonconsensus cyclin-dependent kinase (cdk) sites in the S/T-rich region (24), which are phosphorylated by cdc2-cyclin B in vitro and in vivo (5, 7). PAP is hyperphosphorylated in Xenopus M-phase oocytes and in mitotic HeLa cells when cdc2-cyclin B is active. PAP preparations from either mitotic HeLa cells or Sf9 insect cells coinfected with PAP, p34cdc2, and cyclin B baculoviruses showed significant reductions in activity, and this repression could be reversed by treatment with phosphatase (5). This indicates that PAP is inactivated by hyperphosphorylation, likely by cdc2-cyclin B, in the M phase of mitosis and meiosis. Complete phosphorylation of both the consensus and nonconsensus sites, which are conserved throughout metazoans, is required for PAP inactivation (7). In Xenopus oocytes, PAP consensus sites are phosphorylated prior to the nonconsensus sites during maturation (7). This and other results indicate that differential phosphorylation of consensus and nonconsensus sites could result in a temporal control of hyperphosphorylation, and thus PAP activity, during cell cycle progression.
In this study, we undertook a genetic analysis of PAP in chicken DT40 cells (38) in an effort to understand the functional significance of PAP regulation. We first attempted to disrupt the two endogenous PAP alleles in the presence of a conditional PAP allele in chicken DT40 cells. However, this was not possible, indicating that PAP both is essential and must be very tightly controlled in DT40 cells. We also developed conditions that allowed establishment of cell lines modestly overexpressing wild type (wt) or cdk− PAP II. The phenotypes of these cells with respect to cell growth and cell cycle progression were studied, and both types were found to display defects relative to wt cells, with the cdk− PAP-expressing cells being the most severely affected. These results together indicate that PAP levels must be highly regulated during the cell cycle and support the hypothesis that down-regulation of PAP by cdc2-cyclin B phosphorylation is important for normal cell growth.
MATERIALS AND METHODS
Library screening.
A chicken fibroblast cDNA library (Stratagene) was screened with bovine PAP II cDNA (24) as a probe. Ten positive clones were isolated from 106 plaques. In vivo excision of the pBluescript phagemid from the uni-ZAP vector was carried out following the manufacturer’s instructions, and cDNAs from 10 clones were sequenced. All clones contained a poly(A) tail and a stop codon but lacked a start codon. Nine cDNAs encode the chicken homolog of PAP II, the longest starting from nucleotide (nt) 380 (compared with bovine PAP II) in the open reading frame. The other cDNA represented PAP III, starting from nt 360 in the open reading frame.
Chicken PAP III cDNA was used as a probe to screen a chicken genomic library (Stratagene). Six clones were isolated and analyzed by Southern blot assays. A 9-kb fragment that covers exons 2 to 5 was excised and subcloned into pBluescript vector (pGenePAP). Part of pGenePAP was sequenced, and exon 2 (encoding amino acids 4 to 60) of the chicken PAP gene was identified.
In vitro mutagenesis.
Two rounds of PCR with four primers were carried out to mutate the two consensus cdk sites in chicken PAP II cDNA. Primer se175 corresponds to nt 491 through 514 in chicken PAP II cDNA. Primer cdk2 is complementary to nt 811 through 842, with the three codons that would encode serine (SSPHK and SPKK) mutated to codons that would encode glycine. Primer cdk3 corresponds to nt 821 through 850, also with the three codons that would encode serine mutated to codons that would encode glycine. Primer chUTR is complementary to nt 1104 through nt 1126 in the 3′ UTR region of chicken PAP II. Two sets of PCRs were carried out separately, one with se175 and cdk2 as the primers and the other with cdk3 and ch3UTR as the primers. The chicken PAP II cDNA was used as a template for both reactions. The two PCRs give rise to two products, a 352-bp product from the first reaction (Fgcdk2) and a 295-bp product from the second reaction (Fgcdk3). The two PCR products overlap by 22 nt. Fgcdk2 and Fgcdk3 were purified from agarose gel, and 20 ng of each was used in the second round of PCR, with se175 and ch3UTR as the primers. A 635-bp fragment was amplified and digested with XbaI and BglII, yielding a fragment of 303 bp that contains the two mutated cdk sites. The 303-bp fragment was swapped into the XbaI and BglII sites in pTFPAP, yielding pTFPAPcdk−. Successful mutagenesis was verified by sequencing.
Plasmid constructs.
The bacterial neomycin- and hygromycin-resistant genes under control of the chicken β-actin promoter (38) were inserted into the PstI site in exon 2 of the chicken PAP gene by blunt-end ligation, yielding the constructs neo-PAP and hygro-PAP. The fusion PAP II cDNA (bcPAP II) was constructed by swapping the S/T-rich region of bovine PAP II (24) with that of chicken PAP II. The conserved SpeI sites present in the S/T-rich region of both PAPs were used for swapping. A three-fragment ligation of the Tetr-flu element, in which the flu epitope is downstream of the tetracycline-resistant (Tetr) element (XbaI and blunt [16a]), bcPAP II (blunt and BamHI), and pBluescript (SpeI and BamHI) was carried out. The junction region between the flu epitope and bcPAP II was sequenced to confirm the open reading frame (pTFPAP). The histidinol (hisD)-resistant gene under the control of the chicken β-actin promoter was ligated into pTFPAP (XhoI and BamHI sites) to make pTFPAP-hisD. Flu-tagged wt or cdk− bcPAP II was excised from pTFPAP or pTFPAPcdk− with SacII and BamHI and then ligated into the polylinker site in the PA vector (36), yielding (pFPAP-puro and pFPAPcdk−puro).
Cell cultures and transfections.
DT40 cells were maintained in RPMI 1640 media with 10% fetal bovine serum (Hyclone) and 1% chicken serum (Sigma) at 37°C and 5% CO2. Transfections were carried out as described previously (38). The plasmids neo-PAP, hygro-PAP, tTA, pFPAP-puro, and pFPAPcdk−puro were linearized with NotI. The plasmid pTFPAP was linearized with BamHI.
Southern blot analyses and RNase protection assays.
Genomic DNA was isolated as described, and random priming was used to label the probes (29). In the Southern blot shown in Fig. 2, a DNA fragment of 320 bp prepared from mouse PAP VI cDNA (40) and containing exons 8 and 9 was used as a probe. Isolation of total RNA, preparation of labeled RNA probes, and RNase protection assays were performed as described previously (40). Probe 1 in the RNase protection assays was prepared from chicken PAP III DNA: a fragment of 268 nt that contains 191 nt from exon 12 and 77 nt from intron 12 (40) was amplified by PCR and subcloned into a pBluescript vector; the vector was linearized by BssHII and in vitro transcribed with T7 RNA polymerase. Probe 2 was prepared from chicken PAP II cDNA: the last 400 bp in the open reading frame of chicken PAP II cDNA was amplified by PCR and subcloned into a pBluescript vector; the plasmid was linearized with XbaI and in vitro transcribed with T7 RNA polymerase (Promega).
FIG. 2.
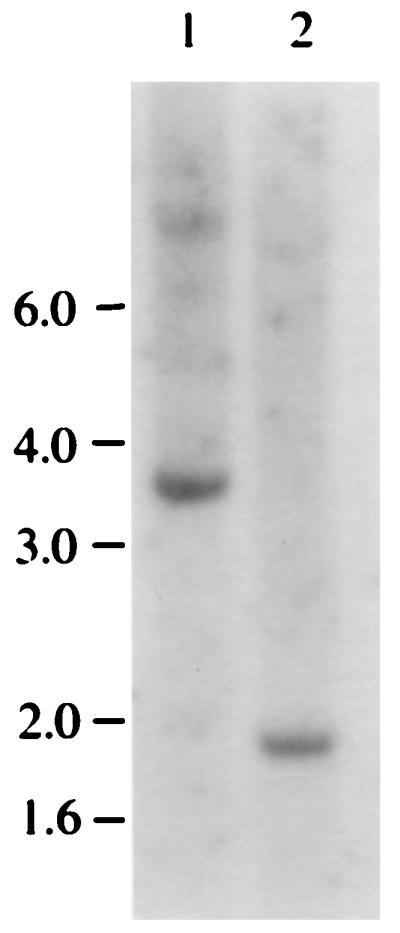
PAP is a single-copy gene. A Southern blot analysis of genomic DNA isolated from DT40 cells. DNA (30 μg) was digested with EcoRI (lane 1) or HindIII (lane 2) and separated on a 7% agarose gel. The positions of the DNA size markers are indicated in kilobases on the left.
Western blot analyses.
DT40 whole-cell lysates were prepared as described previously (29). Bromophenol blue was added to the lysates after the protein concentration determination, which was done by the Bradford method (Bio-Rad). The lysates were separated in a sodium dodecyl sulfate (SDS)-polyacrylamide gel and blotted onto nitrocellulose. The filter was probed with a 1:20 dilution of anti-flu antibody (11) or affinity-purified polyclonal anti-PAP antibody (40), followed by a 1:2,000 dilution of secondary antibody (horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibody [Cappel]). The signal was detected with the ECL kit from Amersham.
FACS assays.
Cells in log phase (2 × 105 to 4 × 105/ml) were centrifuged, resuspended in 300 μl of ice-cold phosphate-buffered saline (PBS), and kept on ice for at least 10 min. The resuspended cells were vortexed at low speed while 5 ml of −20°C methanol was added dropwise and then were kept at −20°C for >40 min. The fixed cells were then centrifuged, resuspended in 2 ml of ice-cold PBS, and kept at 4°C for >1 h. Before they were applied to the fluorescence-activated cell sorter (FACS) (FACSCalibur; Becton Dickson), the cells were centrifuged, resuspended in 1 ml of staining solution containing 50 μg of RNase A and 0.5 μg of propidium iodide per ml in PBS, and kept at room temperature for >10 min. The FACSCalibur program was used to sort and count cells, and the Modfit program was used to calculate the percentage of cells in the G0/G1, S, and G2/M phases.
RESULTS
A number of questions regarding PAP regulation are unsolved. For example, what are the functions of the multiple PAP isoforms? Is a single form of full-length PAP sufficient for cell viability? Are the short forms functional? Is hyperphosphorylation of PAP in M phase essential for cell cycle progression? To address these issues, we wished to undertake a genetic approach utilizing chicken DT40 cells (38). DT40 is a chicken B-cell line that has a short generation time and very high frequency of homologous recombination. If PAP is an essential gene in chicken, as it is in yeast (18), then we would be unable to obtain a homozygous PAP knockout cell line (PAP−/−). However, if we introduced an exogenous source of PAP, it would then in principle be feasible to disrupt the two endogenous PAP alleles. This approach has been used successfully for the splicing factor ASF/SF2 (38) and the polyadenylation factor CstF-64 (29b). The strategy combines two techniques: Tet-repressible expression of an exogenous gene (13) and high-frequency homologous recombination (4). Briefly, we wished to introduce a cDNA encoding a functional PAP that is under control of the Tet-repressible (Tetr) element into DT40 cells and then to inactivate the two alleles of PAP by homologous recombination.
Characterization and expression of the PAP gene in chicken DT40 cells.
To begin our genetic analysis of PAP, we first screened a chicken spinal fibroblast cDNA library with bovine PAP II cDNA (24) as a probe. Ten positive clones were obtained from the screening and were sequenced. Nine cDNAs encode chicken PAP II, and one represents chicken PAP III (see Materials and Methods). However, none of the clones was complete, as all lacked the start codon. Figure 1 shows the alignment of full-length sequences of human, bovine, mouse, and frog PAP II and the partial sequence of chicken PAP II. Residues 3 to 60 of chicken PAP II were deduced from the sequence of exon 2 of the chicken PAP gene (see below). The sequences of residues 1 to 3 and 61 to 119 in chicken PAP II are not known and are indicated by dots. Consistent with previous observations (1, 19), the homology between chicken, frog, and mammalian PAPs is high in the catalytic region, 97% identical from amino acids (aa) 120 to 540, and 86% identical (95% similar) from aa 4 to 60 between chicken and cow. The homology is lower in the S/T-rich region, especially the region corresponding to sequences encoded by exons 20 and 21 in the mouse, which are involved in alternative splicing (40). It is noteworthy, however, that sequences corresponding to exon 22, which is also subject to alternative splicing, are very conserved (31 of 32 residues are identical between chicken and cow sequences). PAP II has been reported to interact with the U1A protein, which results in inhibition of polyadenylation of U1A’s own pre-mRNA (3, 14). The interaction region in PAP II was mapped to the carboxy-terminal 20 residues (15), which are encoded by exon 22. The high homology of this region among human, bovine, mouse, chicken, and frog cells indicates that the potential for an interaction between PAP II and U1A is conserved throughout vertebrates. In the S/T-rich region, clustered consensus (S/TPXK/R) and nonconsensus (S/TPXX) sites are conserved (indicated in boldface in Fig. 1). There are eight nonconsensus and two consensus cdk sites in the S/T-rich region of chicken PAP II. Among the eight nonconsensus sites, two of them are adjacent (TPSPVT). The two consensus sites are conserved in human, bovine, mouse, and chicken PAPs.
FIG. 1.


High evolutionary conservation of PAP. Full-length aa sequence alignment of human, bovine, mouse, and frog PAP II and the partial sequence of chicken PAP II. Asterisks indicate residue identity in all five sequences; colons (:) indicate residue similarity in the five sequences. The potential cdk sites are in boldface. The unknown sequences in chicken PAP II (from residues 1 to 3 and 62 to 119) are indicated by dots. Deletion of residues in homologous sequences are indicated by line breaks.
To determine whether PAP is a single-copy gene in chicken cells, we performed a Southern blot analysis (Fig. 2). The genomic DNA isolated from DT40 cells was digested with EcoRI (lane 1) or HindIII (lane 2), separated on an agarose gel, and probed with a cDNA fragment prepared from mouse PAP VI cDNA (see Materials and Methods). As shown in Fig. 2, hybridization gave a single band of 3.7 kb in lane 1 and a single band of 1.9 kb in lane 2, indicating that PAP is a single-copy gene in DT40 cells.
Previous studies have shown that multiple forms of PAP mRNA, produced by alternative splicing, exist in human cells and mouse tissues (see reference 40 for details). To determine if this is the case in DT40 cells, we performed RNase protection assays. Total RNA was isolated from DT40 cells. An antisense probe (probe 1) was made from chicken PAP III cDNA (see Materials and Methods), which will protect a fragment of 268 nt for PAP III mRNA, a fragment of 191 nt for the full-length PAP mRNA(s), and a fragment of 106 nt for PAP V mRNA (Fig. 3A). As shown in Fig. 3B, both the full-length and the PAP III mRNAs were detected, but PAP V mRNA was not. The probe used in this experiment would not detect PAP VI. To determine whether multiple forms of full-length PAP mRNA are expressed in DT40 cells, another antisense probe (probe 2) was made from chicken PAP II cDNA (see Materials and Methods). Probe 2 will protect a fragment of 400 nt for PAP II, fragments of 153 and 72 nt for PAP I, and fragments of 153 and 183 nt for PAP IV (Fig. 3C). Another alternatively spliced form of full-length PAP was isolated from human cells (35a), and probe 2 will protect fragments of 153 and 111 nt from it (diagram not shown). As shown in Fig. 3D, PAP II mRNA, but not the other full-length forms, was detected. However, we cannot exclude the possibility that these other forms are expressed at low levels that are beyond the sensitivity of our experiments.
FIG. 3.

PAP II and PAP III mRNAs are expressed in chicken DT40 cells. (A) Diagram of RNase protection assays with probe 1. The open boxes indicate sequences encoded by PAP exons. The dotted box indicates sequences encoded by intron 12, which are unique to PAP III (40). The solid lines indicate probe 1 or the protected fragments. (C) Diagram of RNase protection assays with probe 2. The solid, horizontal-striped, dotted, and slant-striped boxes indicate sequences encoded by exons 19, 20, 21, and 22, respectively (40). In both panels A and C, the expected sizes of the probes and protected fragments are indicated on the right. (B) Total RNA isolated from DT40 cells was analyzed with probe 1. (D) An RNase protection assay was performed with probe 2. In both panels B and D, the RNase protection products (lanes 2) were separated on a 5% polyacrylamide-urea gel. Labeled MspI-digested pBR322 DNA was used as a DNA size marker (nt, lane 1). The positions of probes and different forms of PAP are indicated by arrows.
Targeted disruption of one PAP allele.
From the above-described studies, we conclude that PAP is a single-copy gene in DT40 cells and that PAP II is the major and perhaps the only functional form expressed. If PAP is an essential gene in DT40 cells, it might be possible to introduce an exogenous PAP II under control of the Tetr element and then disrupt both alleles of PAP. In this case, we could create a cell line that contains a single, conditional PAP allele.
To begin this approach, we first attempted to establish a heterozygous cell line (PAP+/−). To this end, a chicken genomic library was screened, and a 9-kb clone that hybridized to a chicken PAP III cDNA probe was isolated (see Materials and Methods). Southern blot analyses indicated that this genome fragment covers approximately exon 2 to exon 5, and partial sequencing confirmed the identity of exon 2. Figure 4A shows a diagram of a portion of the chicken PAP locus. Bacterial hygromycin- and neomycin-resistant genes were inserted into exon 2, yielding the knockout constructs hygro-PAP and neo-PAP (see Materials and Methods). DT40 cells were first transfected with linearized hygro-PAP and grown in medium containing hygromycin. Genomic DNAs isolated from the resulting drug-resistant clones were digested with EcoRI, and Southern blot analysis was performed with a probe that flanks the knockout constructs 5′ (see Fig. 4A). This will give a DNA fragment of 12 kb for the wt locus and a fragment of 4.5 kb for the hygro-PAP locus. DNAs from 14 hygromycin-resistant clones were tested by Southern blot analysis, and four were shown to have undergone homologous recombination. The Southern blot analysis of DNAs isolated from wt cells and from a heterozygous cell line, kh12, is shown in Fig. 4B.
FIG. 4.

Construction of heterozygous (PAP+/−) DT40 cell lines. (A) A restriction map of the chicken PAP locus and the knockout constructs neo-PAP and hygro-PAP. The bacterial neomycin- or hygromycin-resistant genes were inserted into exon 2 (indicated by a solid box) of the PAP gene. The restriction sites are indicated by arrowheads and a single capitalized letter: E (EcoRI), K (KpnI), and X (XbaI). (B and C) Southern blot analyses of genomic DNA isolated from wt, kh12 (hygro-PAP targeted), and kn1 (neo-PAP targeted) cells. DNA (30 μg) was digested with EcoRI (B) or EcoRI and XbaI (C) and then separated on a 7% agarose gel. The positions of the DNA size markers are indicated in kilobases on the left. (D) Western blot analyses of whole-cell extracts isolated from wt, kh12, and kn1 cells. Affinity-purified PAP polyclonal antibodies were used (upper panel). The same filter was probed with mAb104 monoclonal antibodies (recognizing splicing factor SRp75 and other proteins) to confirm equal loading of extracts in each lane. The positions of PAP and SRp75 are indicated by arrows. The positions of protein size markers are indicated in kilodaltons on the left.
The homologous recombination efficiency of construct neo-PAP was also analyzed. Genomic DNAs isolated from G418-resistant clones were digested with EcoRI and XbaI. Hybridization with the probe indicated in Fig. 4A will give a signal of 6 kb for the wt locus and a signal of 9 kb for the neo-PAP locus. Nineteen clones were analyzed and five displaying homologous recombination were identified. Southern blot analysis of one heterozygote, kn1, is shown in Fig. 4C. The frequencies of the homologous recombinations obtained with the hygro-PAP and neo-PAP constructs were 4 of 14 and 5 of 19, respectively. The PAP level in heterozygous cells was studied by Western blot analysis and compared with that in the wt cells. Affinity-purified anti-cow PAP polyclonal antibodies (40) were used as primary antibodies. As shown in the upper panel of Fig. 4D, the PAP levels in kh12 or kn1 cells was decreased to about half of that in the wt cells, indicating that disruption of one PAP allele led to reduced expression of the endogenous PAP. A monoclonal antibody recognizing SR protein splicing factors (mAb 104 [25]) was used as a control to confirm equal loadings of proteins in the lanes, and the blotting of SRp75 is shown in the lower panel of Fig. 4D. The growth properties of kh12 and kn1 cells appear normal; these results thus indicate that DT40 cells can tolerate reduced levels of PAP.
Expression of Tet-repressible PAP II cDNA.
Next we introduced an epitope-tagged PAP II cDNA under control of the Tetr element into a heterozygous PAP+/− cell line. As mentioned above, we have been unable to obtain a full-length chicken PAP II cDNA. However, since the homology between the chicken and bovine PAPs is extremely high throughout the entire catalytic region (∼97% identity, 99% similarity; see above), we constructed a fusion protein, bcPAP II, in which the N-terminal S/T-rich region (aa 1 to 540) is from bovine cells and in which the C-terminal part (aa 540 to 740) is from chicken cells. The fusion protein was flu epitope tagged at its N terminus, and the cDNA was placed under control of the Tetr element (pTFPAP; see Materials and Methods). To test if the fusion protein was functional, bcPAP II was produced by in vitro translation in a rabbit reticulocyte lysate, tested by in vitro nonspecific poly(A) synthesis assays, and compared to in vitro-translated bovine PAP II. The specific activity of bcPAP II was equivalent to bovine PAP II (data not shown), indicating that the fusion protein is properly folded and functional. The two PAPs also accumulated identically in transiently transfected HeLa cells (not shown). A hisD-resistant gene was then inserted into pTFPAP to make the pTFPAP-hisD construct (see Materials and Methods).
To establish a heterozygous cell line that expresses PAP II in a Tet-repressible manner, we cotransfected one of the heterozygous cell lines (kh12) with tTA (which bears DNA encoding the tet-VP16 chimeric activator and puromycin resistance [38]) and pTFPAP-hisD. Six clones that grew in the presence of puromycin and hisD were tested by Western blot analysis. After growth in the presence or absence of Tet, whole-cell lysates were prepared, and Western blot analysis was carried out with a monoclonal antibody against the flu epitope (see Materials and Methods). Three clones, A2, B4, and B7, expressed a protein of about 110 kDa in a Tet-repressible manner (Fig. 5A). The 110-kDa species is likely one or more phosphorylated form of PAP II (5).
FIG. 5.

Western blot analyses of endogenous and exogenous PAP II in kn12 cells. Drug-resistant clones were grown in the presence (+) or absence (−) of Tet. Whole-cell lysates (100 μg) were separated on an SDS-polyacrylamide gel, and Western blot analyses were performed. (A) Anti-flu epitope antibodies were used as the primary antibody. (B) Affinity-purified antibodies raised against bovine PAP I were used as the primary antibody. In both panels A and B, the cell lines are indicated on the top of the panels. The positions of protein size markers are indicated in kilodaltons on the left. The positions of PAP II are indicated by arrows on the right.
To compare the expression level of exogenous PAP II with that of the endogenous enzyme, we performed Western blot analysis with affinity-purified antibodies raised against bovine PAP I (40). As shown in Fig. 5B, after withdrawal of Tet from the medium, the expression level of PAP was about twice that in cells grown in the presence of Tet, indicating that the expression level of exogenous PAP II was similar to that of the endogenous PAP. The total accumulation of PAP was thus comparable to that found in the parental DT40 cells.
The second PAP allele cannot be disrupted.
We next tried to inactivate the other PAP allele in A2, B4, and B7 cells. Cells were transfected with the neo-PAP construct and grown in medium with G418 and hygromycin. Here, we used a two-step screening. The G418 and hygromycin-resistant clones were divided into medium with Tet and medium without Tet. If PAP is an essential gene in DT40 cells, clones with the second PAP allele inactivated by homologous recombination will die in the presence of Tet, since the only source of PAP (the PAP II cDNA) is repressed. Table 1 presents a summary of the second allele knockout attempts. A total of 34, 40, and 108 double-resistant clones were obtained from cotransfection in A2, B4, and B7 cells, respectively. All the drug-resistant clones from B4 cells grew both with and without Tet. Only two clones that grew in the absence of Tet but stopped growing in the presence of Tet were obtained, one from A2 cells and one from B7 cells. DNAs from the two clones were subject to Southern blot analysis. However, neither one contained a disruption of the second PAP allele (results not shown). To exclude the possibility that it may take a long time to diminish the exogenous PAP (cells were grown in medium with Tet for 2 to 6 days) and that some clones that grew in the presence of Tet may therefore be homozygotes, we performed Southern blot analysis on additional clones. As summarized in Table 1, a total of 34, 40, and 34 clones from A2, B4, and B7 cells, respectively, were tested by Southern blot analysis, and no homozygote was obtained. As summarized above, the homologous recombination efficiency of both hygro-PAP and neo-PAP was about 26%. In an effort to knock out the second PAP allele in heterozygous cells that express exogenous PAP II, we screened 182 drug-resistant clones by testing whether they fail to grow in the presence of Tet; we also analyzed 108 clones (of the 182 clones) by Southern blotting, from which no homozygote was obtained. These results indicate that PAP is an essential gene in DT40 cells. We do not know why the PAP II-encoding cDNA did not allow disruption of the second allele, but possible reasons for this are discussed below.
TABLE 1.
Frequency of homologous recombination in A2, B4, and B7 cells
| Characteristic | Recombination frequency for:
|
||
|---|---|---|---|
| A2 cells | B4 cells | B7 cells | |
| Clones failed to grow in medium with Tet | 1/30 | 0/40 | 1/108 |
| No. with second allele disrupted | 0/30 | 0/40 | 0/34 |
Construction and characterization of heterozygous cells lines expressing exogenous wt and a cdk− PAP.
Previous studies have shown that PAP is inactivated by hyperphosphorylation in the M phase of mitosis or meiosis (5, 7). We wanted to study the physiological function of this event, e.g., whether it is essential for normal cell growth or division. Since we were unable to obtain a cell line with a single conditional PAP allele, it was impossible to determine whether a cell line that expresses only cdk− PAP, which is resistant to hyperphosphorylation, would be viable. We therefore chose to take another approach: to express cdk− PAP in DT40 cells and to ask whether cdk− PAP has any dominant effect. Previous studies have shown that complete phosphorylation of all consensus and nonconsensus sites is required for PAP inactivation (7), so we chose to mutate the two consensus cdk sites in pTFPAP, with Ser changed to Gly (pTFPAPcdk−; see Materials and Methods). These mutations will prevent hyperphosphorylation, and hence the inhibition of PAP activity should not occur (5, 7). Flu-tagged PAP II and cdk− PAP II were subcloned from pTFPAP or pTFPAPcdk− into the polylinker site of the PA vector (38; see also Materials and Methods), which is downstream of the constitutive chicken β-actin promoter (pFPAP and pFPAPcdk−).
We first set out to obtain cell lines overexpressing wt and cdk− PAP. Previous experiments employing mammalian cell lines (24a, 29a) failed to obtain PAP-overexpressing cell lines. We attempted to introduce an exogenous, Tet-repressible PAP II allele into wt DT40 cells and obtained seven drug-resistant clones. Four of them expressed a protein of about 40 kDa in a Tet-repressible manner, likely representing the products of degraded PAP II, and the other three failed to express exogenous PAP (results not shown). These results are consistent with the idea that PAP levels must be tightly controlled and that overexpression can be toxic. Since the PAP level is reduced in heterozygous cells and since we had obtained heterozygous cell lines expressing Tet-repressible wt PAP II (see above), we decided to use such cells in these experiments with constitutively expressed wt and cdk− PAP. To this end, 107 kn1 cells were transfected with 25 μg of pFPAP or pFPAPcdk−. Colonies became visible 6 days after plating, and those that appeared within 20 days were counted (colonies that appeared 20 days after plating were not counted because they were no longer viable after being transferred to new media with selecting drugs). A total of 30 colonies were obtained from cells transfected with pFPAP, but only 6 colonies were obtained from cells transfected with pFPAPcdk−. These results suggest that expression of cdk− PAP may be deleterious to cell growth. To verify this, the experiment was repeated with another sample of cells. This time, a total of 87 clones were detected in the wt PAP II transfection, but only 23 clones were detected in the cdk− PAP II transfection. These results indicate that, under identical conditions, about fourfold more colonies arose from cells transfected with the plasmid expressing wt PAP II than from those expressing cdk− PAP II. To exclude the possibility that this phenomenon was specific to kn1 cells, we repeated the experiments with another heterozygous cell line, kh12. As shown in Table 2, a total of 104 colonies were detected in the wt PAP II transfection, while only 25 colonies were obtained from the parallel cdk− PAP II transfection. In total, the number of clones expressing cdk− PAP was 24% of the number of clones expressing wt PAP. Together, these results suggest that heterozygous DT40 cells are less able to tolerate overexpression of cdk− PAP II than is wt PAP II.
TABLE 2.
Number of clones from parallel transfections with plasmids expressing wt or cdk− PAP II
| Expt | No. of clones expressing:
|
|
|---|---|---|
| wt PAP II | cdk− PAP II | |
| 1 | 30 | 6 |
| 2 | 87 | 23 |
| 3 | 104 | 25 |
To extend these results, we examined expression of PAP in cell lines transformed with wt or cdk− PAP II expression vectors. Six puromycin-resistant clones from the pFPAPcdk− transfection and six from the pFPAP transfection in kn1 cells were analyzed by Western blot analysis. Whole-cell lysates were prepared from the 12 clones, and anti-flu antibodies were used for detection (results not shown). From the 12 clones tested, 2 of 6 expressed detectable levels of wt PAP II (wt1 and wt6) and 3 of 6 expressed cdk− PAP II (cdk3, cdk5, and cdk6). We do not know why only five clones expressed detectable PAP, but we suspect that expression was silenced because PAP overexpression is toxic. When similar experiments were performed with vectors expressing the SR protein splicing factor ASF/SF2, 80 to 90% of the drug-resistant clones expressed the exogenous protein (38; unpublished data).
The PAP-expressing clones were reanalyzed and compared to untransfected cells (Fig. 6A). In all the cell lines that expressed exogenous PAP, the anti-flu antibodies detected a major species of ∼110 kDa (indicated by arrow 1 in Fig. 6A). In wt1 (lane 1) and wt6 (lane 2), a species with greater mobility (ca. 100 kDa), likely representing a hypophosphorylated form of PAP, was also detected (Fig. 6A, arrow 2). This species was not detected in the early experiments with pTFPAP, probably because it is expressed at much lower levels than the 110-kDa species. More PAP species were detected in cells that express cdk− PAP II. Two species of about 105 and 100 kDa (indicated by arrow 2 in Fig. 6A) and one species of about 90 kDa (indicated by arrow 3) were detected in cdk3 (lane 3), cdk5 (lane 4), and cdk6 (lane 5). A species of about 80 kDa (indicated by arrow 4) was detected in cdk3 cells only. These lower-molecular-weight forms likely represent hypo- or unphosphorylated isoforms, or possibly breakdown products, but it is notable that they were detected only in cdk− PAP. To provide evidence that cdk− PAP is indeed underphosphorylated, an SDS–5% polyacrylamide gel was used to separate wt (from wt1) and cdk− (from cdk5) PAP II. The Western blot of this gel (Fig. 6B) reveals that the 110-kDa form of cdk− PAP II (lane 2) displayed slightly greater mobility than did that of wt PAP II (lane 1). The other species were visible after longer exposure, and the 100-kDa species from wt PAP II and the 105-kDa species from cdk− PAP II actually resolved into two species (data not shown). It is intriguing that the cdk− PAP was resolved into more isoforms than wt PAP (Fig. 6A). Possible explanations for this are discussed below.
FIG. 6.

Western blot analysis of wt or cdk− PAP II under the control of the chicken β-actin promoter in kn1 cells. (A) Whole-cell lysates from wt1 (lane 1), wt6 (lane 2), cdk3 (lane 3), cdk5 (lane 4), cdk6 (lane 5), and kn1 (lane 6) cells were separated on an SDS–5% polyacrylamide gel. The positions of protein size markers are indicated in kilodaltons on the left. The arrows on the right indicate the different species of PAP detected by the flu antibody. (B) Whole-cell lysate from wt1 (lane 1) and cdk5 (lane 2) were separated on an SDS–5% polyacrylamide gel, and the 56-kDa protein size marker was run off the gel. The positions of protein size markers are indicated in kilodaltons on the left.
Overexpression of exogenous PAP results in a reduced growth rate.
To investigate whether overexpression of PAP affects cell growth, we compared growth curves of kn1, wt1, wt6, cdk3, and cdk5 cells. Cell samples from each line were each passaged into fresh medium at a density of 105/ml, and the cells were counted every 12 h. Although all of the cells began at the same density, it was obvious that the concentration of kn1 cells exceeded those of cells expressing exogenous PAP after only 24 h (Fig. 7A). At 72 h, kn1 cells had reached confluence (3.4 × 106/ml), but the other cells were at much lower densities, with wt1 cells being at the highest density (1.4 × 106/ml). Transfection of the PA vector alone did not have any effect on growth (38).
FIG. 7.

Growth curves of kn1 cells and kn1 cells expressing wt or cdk− PAP II. (A) Growth curves of kn1, wt1, wt6, cdk3, and cdk5 cells. Cells (105) were passaged into new medium, and the numbers of cells were counted every 12 h, until the cells reached confluence. (B) Growth curves of B7 cells. (C) Growth curves of TFcdk6 cells. In both panels B and C, cells were maintained in medium containing Tet. Cells (105) were passaged into new medium with Tet (Tet+) or without Tet (Tet−), and cells were counted every 12 or 24 h. The growth curves were repeated three times, and similar results were obtained.
The lower growth rate observed in cell lines that express wt or cdk− PAP II seems likely due to the overexpression of exogenous PAP, but it could also have been due to the insertion of transfected DNA into the chicken genome or some other secondary effect. To exclude this second possibility, we next compared the growth curves of cells expressing wt PAP II in a Tet-repressible manner (A2, B4, and B7 cells; see above) in the presence or absence of Tet. Tet itself does not affect the growth rate of DT40 cells (29b, 38). Cells previously grown in the presence of Tet were split into fresh medium at a density of 105/ml with or without Tet. The growth curves of B7 cells are shown in Fig. 7B. After 72 h, the density of cells grown in the presence of Tet significantly exceeded that of cells grown in the absence of Tet. Cells grown in the absence of Tet reached confluence 12 h later than those grown with Tet. The growth curves of A2 and B4 cells in the presence or absence of Tet were also compared, and similar results were obtained (data not shown).
To extend this analysis to cdk− PAP, a conditional allele of cdk− PAP was constructed and introduced into kh12 cells by cotransfection of tTA and pTFPAPcdk− hisD (see Materials and Methods). Three cell lines that express cdk− PAP II in a Tet-repressible manner were obtained (these cell lines expressed similar levels of repressible PAP as A2, B4, and B7 cells [data not shown]). The growth curves of these three cell lines in the presence or absence of Tet were compared. Similar to the results with B7 cells, the three cell lines grew slower in medium with Tet. The growth curve of one cell line, TFcdk6, is shown in Fig. 7C. After 60 h, cells that were grown in the presence of Tet showed a greater number of cells than those grown in the absence of Tet. Those grown in the presence of Tet reached confluence (2.7 × 106/ml) by 84 h, while those grown in the absence of Tet reached a density of only 1.8 × 106/ml, and at 96 h, their density actually dropped to 1.2 × 106/ml. These results indicate that overexpression of cdk− PAP II results in a reduced growth rate.
The cdk− PAP II overexpressing cells had a more pronounced growth defect than did wt PAP II overexpressing cells (Fig. 7A). As shown in Fig. 6A, wt1 expressed levels of exogenous PAP similar to those of cdk3, but wt1 grew significantly faster than cdk3 (expression levels were carefully quantitated in a shorter exposure of the Western blot). Cell line wt6 expressed a higher level of exogenous PAP than did cdk5, but its growth curve was also similar to that of cdk5. Cell line cdk6 expressed a level of exogenous PAP similar to that of cdk5, and its growth curve was similar to that of wt6 (data not shown). These results indicate that at the same protein levels, cdk− PAP II has a more significant effect on growth rate than does wt PAP II and provides an explanation for why we obtained fewer colonies in the cdk− PAP II transfections than in the wt PAP II transfections.
The low growth rate of cells expressing exogenous PAP could be due to cell death or to a slowed-down cell cycle. To study the percentage of cells in each phase of the cell cycle, we performed FACS analysis with kn1, wt1, and cdk3 cells. The profiles are shown in Fig. 8A. No increase of wt1 or cdk3 cells in sub-G1/G0 phase was observed, suggesting that the slower growth was not due to cell death. The percentage of cells in the G0/G1, S, and G2/M phases was calculated (see Materials and Methods) and compared in kn1, wt1, and cdk3 cells, and the results are shown in Fig. 8B. The percentage of cells in the G0/G1 phase was highest in cdk3 cells (42%), followed by wt1 cells (34%) and kn1 cells (28%). The increased number of cells in G0/G1 phase was accompanied by a corresponding decrease of cells in the S and G2/M phases, indicating that the low growth rate observed in cells overexpressing exogenous PAP was due to a delay or arrest of cells in the G0/G1 phase. Possible explanations for this finding are discussed below.
FIG. 8.

FACS analysis of kn1, wt1, and cdk5 cells. (A) Profiles of the numbers of cells in each phase of the cell cycle. The two arrowheads indicate the DNA content of G0/G1 cells (left) and of G2/M cells (right). The vertical axis indicates the number of cells. (B) Percentage of cells in G0/G1, S, and G2 phases. The FACS analysis was repeated two times with essentially identical results.
DISCUSSION
The initial aim of the experiments described here was to establish a chicken DT40 cell line in which the only source of PAP was from an exogenous conditional PAP allele. While we succeeded in disrupting one PAP allele and in introducing a conditional allele, we were unable to disrupt the second allele. The fact that we could not disrupt both alleles of PAP strongly suggests that PAP is an essential gene in DT40 cells. This is not surprising, both because PAP has been shown to be essential in budding yeast (18) and because polyadenylation has been suggested to influence virtually all aspects of mRNA metabolism, including mRNA stability, translational efficiency, and transport of processed mRNA from the nucleus to the cytoplasm (for recent reviews, see references 17, 28, and 39).
There are several possible explanations as to why we were unable to disrupt the second PAP allele in heterozygous cells expressing exogenous PAP II. Although we did not detect full-length forms of PAP other than PAP II, it is possible that one or more of these isoforms are expressed at very low levels that are beyond our detection abilities yet are essential for viability of DT40 cells. One of the short forms of PAP mRNA (1, 12, 40), PAP III, is expressed at the mRNA level in DT40 cells. Although the short PAPs, when produced as recombinant proteins, are not active in polyadenylation (40) and are not detected at the protein level in HeLa cells (35, 40) or frog oocytes (1, 12), the short PAPs or their mRNAs might have some function that is essential for viability. For example, we suggested previously that the short forms are the products of autoregulation (40), and perhaps this regulation is essential. PAP II cDNA cannot give rise to any of these alternatively spliced forms and if one or more performs an essential function, then cells expressing only PAP II would not be viable. An alternative possibility reflects the fact that the exogenous PAP II introduced into DT40 cells is a fusion protein of bovine and chicken PAP II. Although the fusion protein is as active as bovine PAP II in vitro and the substituted sequences are nearly identical, we cannot rigorously exclude the possibility that the fusion protein cannot substitute completely for chicken PAP II in vivo. Finally, it is also possible that the expression level of PAP II must be transcriptionally regulated throughout the cell cycle and that the Tetr element cannot properly control expression.
Our experiments indicate that DT40 cells can tolerate moderate reduction of PAP levels. Heterozygous cells express about half the PAP level of wt cells yet have a normal growth rate. The level of another polyadenylation factor, CstF-64, can be reduced by ∼10-fold with no significant effect on cell growth (29b). In contrast, the SR protein splicing factor ASF/SF2 seems to up-regulate its mRNA and protein level, which is reflected by similar ASF/SF2 protein levels in wt and heterozygous cells (37a). As discussed by Takagaki et al. (33), perhaps polyadenylation is not a rate-limiting step in gene expression and thus cells can tolerate reduced levels of PAP and other polyadenylation factors.
In contrast, our results indicate that the upper-level limit of PAP must be very tightly controlled within cells, which was likely reflected in the difficulties encountered in the overexpression experiments. As noted above, we were unable to obtain wt DT40 cells overexpressing intact PAP, and similar results have been obtained with mammalian cell lines. This likely indicates that even modest overexpression of PAP can be toxic. Even in the heterozygous lines, expression of exogenous PAP was always equivalent to or, at most, slightly elevated relative to the levels of endogenous PAP. Similar experiments with the SR protein splicing factor ASF/SF2 (38) or with CstF-64 (33) allowed expression of either protein at higher levels than the corresponding endogenous protein. Indeed, levels of CstF-64 can be increased to as much as 50 times the endogenous levels without detectably affecting cell growth (33). This indicates that, in contrast to PAP, CstF-64 overexpression is not toxic to the cell, which is consistent with the fact that CstF-64 levels are regulated during B-cell differentiation and can modulate poly(A) site choice in IgM heavy-chain pre-mRNA (33). It is intriguing that the levels of one polyadenylation factor (CstF-64) can fluctuate widely, while those of another (PAP) must be very tightly controlled.
More PAP isoforms were detected with cdk− than with wt PAP II-expressing cells. One possibility is that the presence of the consensus sites in some way affects the phosphorylation status of the nonconsensus sites. This contrasts with observations from previous experiments, in which bovine wt or cdk− PAP mRNAs were injected into the Xenopus oocyte or in which insect cells were infected with recombinant baculoviruses encoding wt or cdk− PAP II (5, 7). In these experiments, equivalent numbers of wt and cdk− PAP II species were detected, the only apparent difference being that each species in cdk− cells had a greater mobility than the corresponding ones in wt PAP II. One possibility is that this reflects the differences in the S/T-rich region between bovine and chicken PAP II. However, another and perhaps more likely possibility is that our experiments involved expression in stably transformed growing cells. It may be that under these conditions the cell adapts to the presence of the toxic cdk− PAP by differentially modifying it.
In M phase of the cell cycle, there is a general repression of RNA and protein synthesis (10, 16), and inhibition of PAP activity likely contributes to this (5). Expression of exogenous, unregulatable wt PAP II may result in an excess of PAP activity in M phase. Expression of cdk− PAP II, which cannot be inactivated by hyperphosphorylation, will contribute even more PAP activity in M phase. This is probably why the expression of cdk− PAP II reduced cell growth more dramatically than did comparable levels of wt PAP II. In M phase, the disassembly of the nuclear envelope causes relocation of PAP to the cytoplasm. In cells with excess PAP activity, this may result in the inappropriate stabilization of some mRNAs, which in turn in G1 phase could cause a delay in S-phase onset or even bring about an exit from the cell cycle into G0 phase. This offers an explanation for why cells overexpressing wt or cdk− PAP II showed an increased cell number in G0/G1 phase relative to wt cells. It is also possible that phosphorylation of the consensus cdk sites is important for other aspects of regulation besides hyperphosphorylation-mediated inactivation of PAP. In any event, the failure to phosphorylate the consensus cdk sites in cdk− PAP results in a reduced growth rate, confirming that this phosphorylation plays a physiologically important role.
ACKNOWLEDGMENTS
We thank Tsuyoshi Kashima for providing the Tetr flu construct; Lin Ge for help in sequencing; and Diana F. Colgan, Gareth Bond, Jin Wang, Zheng Chen, Moonkyoung Um, and other members of the Manley lab for helpful discussions.
This work was supported by NIH grant GM 28983 to J.L.M.
REFERENCES
- 1.Ballantyne S, Bilger A, Astrom J, Virtanen A, Wickens M. Poly(A) polymerases in the nucleus and cytoplasm of frog oocytes: dynamic changes during oocyte maturation and early development. RNA. 1995;1:64–78. [PMC free article] [PubMed] [Google Scholar]
- 2.Bienroth S, Wahle E, Satler-Crazzolara C, Keller W. Purification of the cleavage and polyadenylation factor involved in 3′ processing of mRNA precursors. J Biol Chem. 1991;266:19768–19776. [PubMed] [Google Scholar]
- 3.Boelens W C, Jansen E, Venrooij W, Stripecke R, Mattaj I, Gunderson S I. The human U1 snRNP-specific U1A protein inhibits polyadenylation of its own pre-mRNA. Cell. 1993;72:881–892. doi: 10.1016/0092-8674(93)90577-d. [DOI] [PubMed] [Google Scholar]
- 4.Buerstedde J M, Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell. 1991;67:179–188. doi: 10.1016/0092-8674(91)90581-i. [DOI] [PubMed] [Google Scholar]
- 5.Colgan D F, Murthy K, Prives C, Manley J L. Cell-cycle related regulation of poly(A) polymerase by phosphorylation. Nature. 1996;384:282–285. doi: 10.1038/384282a0. [DOI] [PubMed] [Google Scholar]
- 6.Colgan D F, Manley J L. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 1997;11:2755–2766. doi: 10.1101/gad.11.21.2755. [DOI] [PubMed] [Google Scholar]
- 7.Colgan D F, Murthy K G K, Zhao W, Prives C, Manley J L. Inhibition of poly(A) polymerase requires p34cdc2/cyclin B phosphorylation of multiple consensus and nonconsensus sites. EMBO J. 1998;17:1053–1062. doi: 10.1093/emboj/17.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dantonel J, Murthy K G K, Manley J L, Tora L. Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature. 1997;389:399–402. doi: 10.1038/38763. [DOI] [PubMed] [Google Scholar]
- 9.Edmonds M, Abrams R J. Polynucleotide biosynthesis: formation of a sequence of adenylate units from adenosine triphosphate by an enzyme from thymus nuclei. J Biol Chem. 1960;235:1142–1149. [PubMed] [Google Scholar]
- 10.Fan H, Penman S. Regulation of protein synthesis in mammalian cells. II. Inhibition of protein synthesis at the level of initiation during mitosis. J Mol Biol. 1970;50:655–670. doi: 10.1016/0022-2836(70)90091-4. [DOI] [PubMed] [Google Scholar]
- 11.Field J, Nikawa J I, Broek D, MacDonald B, Rodgers L, Wilson I A, Lerner R, Wigler M. Purification of a RAS-responsive adenyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gebauer F, Richter J D. Cloning and characterization of a Xenopus poly(A) polymerase. Mol Cell Biol. 1995;15:1422–1430. doi: 10.1128/mcb.15.3.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:3547–3551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunderson S I, Beyerm K, Martin G, Keller W, Boelens W, Mattaj I W. The human U1A snRNP protein regulates polyadenylation via a direct interaction with poly(A) polymerase. Cell. 1994;76:531–541. doi: 10.1016/0092-8674(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 15.Gunderson S I, Vagner S, Polycarpou-Schwarz M, Mattaj I W. Involvement of the carboxy terminus if vertebrate poly(A) polymerase in U1A autoregulation and in coupling of splicing and polyadenylation. Genes Dev. 1997;11:761–773. doi: 10.1101/gad.11.6.761. [DOI] [PubMed] [Google Scholar]
- 16.Kanki J P, Newport J W. The cell cycle dependence of protein synthesis during Xenopus laevis development. Dev Biol. 1991;146:198–213. doi: 10.1016/0012-1606(91)90460-k. [DOI] [PubMed] [Google Scholar]
- 16a.Kashima, T., and J. L. Manley. Unpublished results.
- 17.Lewis, J., S. Gunderson, and I. M. Mattaj. 1995. The influence of 5′ and 3′ end structures on pre-mRNA metabolism. J. Cell. Sci. 19(Suppl.):13–19. [DOI] [PubMed]
- 18.Linger J, Kellermann J, Keller W. Cloning and expression of the essential gene for poly(A) polymerase from S. cerevisiae. Nature. 1991;354:496–498. doi: 10.1038/354496a0. [DOI] [PubMed] [Google Scholar]
- 19.Martin G, Keller W. Mutational analysis of mammalian poly(A) polymerase identifies a region for primer binding and a catalytic domain, homologues to the family X polymerases, and to other nucleotidyltransferases. EMBO J. 1996;15:2593–2603. [PMC free article] [PubMed] [Google Scholar]
- 20.MacDonald C C, Wilusz J, Shenk T. The 64-kilodalton subunit of the CstF polyadenylation factor binds to pre-mRNA downstream of the cleavage site and influences cleavage site location. Mol Cell Biol. 1994;14:6647–6654. doi: 10.1128/mcb.14.10.6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson S D, Wickens M, Bentley D L. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- 22.Murthy K G K, Manley J L. Characterization of the multisubunit cleavage polyadenylation specificity factor from calf thymus. J Biol Chem. 1992;267:14804–14811. [PubMed] [Google Scholar]
- 23.Murthy K G K, Manley J L. The 160kD subunit of human cleavage-polyadenylation specificity factor coordinates pre-mRNA 3′ end formation. Genes Dev. 1995;9:2672–2683. doi: 10.1101/gad.9.21.2672. [DOI] [PubMed] [Google Scholar]
- 24.Raabe T, Bollum F J, Manley J L. Primary structure and expression of bovine poly(A) polymerase. Nature. 1991;353:229–234. doi: 10.1038/353229a0. [DOI] [PubMed] [Google Scholar]
- 24a.Raabe, T., and J. L. Manley. Unpublished data.
- 25.Roth M B, Zahler A M, Stolk J A. A conserved family of nuclear phosphoproteins localized to the sites of polymerase II transcription. J Cell Biol. 1991;115:587–596. doi: 10.1083/jcb.115.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruegsegger U, Beyer K, Keller W. Purification and characterization of human cleavage factor I involved in the 3′ end processing of messenger RNA precursors. J Biol Chem. 1996;271:6107–6113. doi: 10.1074/jbc.271.11.6107. [DOI] [PubMed] [Google Scholar]
- 27.Ruegsegger U, Blank D, Keller W. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol Cell. 1998;1:243–254. doi: 10.1016/s1097-2765(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 28.Sachs A B, Sarnow P, Hentze M W. Starting at the beginning, middle and end: translation initiation in eukaryotes. Cell. 1997;89:831–838. doi: 10.1016/s0092-8674(00)80268-8. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 29a.Sonnenberg, N., and J. L. Manley. Unpublished data.
- 29b.Takagaki, Y., and J. L. Manley. Unpublished data.
- 30.Takagaki Y, Ryner L C, Manley J L. Four factors are required for 3′-end cleavage of pre-mRNA. Genes Dev. 1989;3:1711–1724. doi: 10.1101/gad.3.11.1711. [DOI] [PubMed] [Google Scholar]
- 31.Takagaki Y, Manley J L, MacDonald C C, Wilusz J, Shenk T. A multisubunit factor, CstF, is required for polyadenylation of mammalian pre-mRNAs. Genes Dev. 1990;4:2112–2120. doi: 10.1101/gad.4.12a.2112. [DOI] [PubMed] [Google Scholar]
- 32.Takagaki Y, MacDonald C C, Shenk T, Manley J L. The human 64-kD polyadenylation factor contains a ribonucleoprotein-type RNA binding domain and unusual auxiliary motifs. Proc Natl Acad Sci USA. 1992;89:1403–1407. doi: 10.1073/pnas.89.4.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takagaki Y, Seipelt R L, Petrson M L, Manley J L. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell. 1996;87:941–952. doi: 10.1016/s0092-8674(00)82000-0. [DOI] [PubMed] [Google Scholar]
- 34.Takagaki Y, Manley J L. RNA recognition by the human polyadenylation factor CstF. Mol Cell Biol. 1997;17:3907–3914. doi: 10.1128/mcb.17.7.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thuresson A, Astrom J, Astrom A, Gronvik K, Virtanen A. Multiple forms of poly(A) polymerases in human cells. Proc Natl Acad Sci USA. 1994;91:979–983. doi: 10.1073/pnas.91.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35a.Thuresson, A., and A. Virtanen. Personal communication.
- 36.Wahle E, Martin G, Schilts E, Keller W. Isolation and expression of cDNA clones encoding mammalian poly(A) polymerase. EMBO J. 1991;10:421–425. doi: 10.1002/j.1460-2075.1991.tb05003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wahle E, Keller W. The biochemistry of polyadenylation. Trends Biochem Sci. 1996;21:247–250. [PubMed] [Google Scholar]
- 37a.Wang, J., and J. L. Manley. Unpublished data.
- 38.Wang J, Takagaki Y, Manley J L. Targeted disruption of an essential vertebrate gene: ASF/SF2 is required for cell viability. Genes Dev. 1996;10:2588–2599. doi: 10.1101/gad.10.20.2588. [DOI] [PubMed] [Google Scholar]
- 39.Wickens M P, Anderson P, Jackson R J. Life and death in the cytoplasm: messages from the 3′ end. Curr Opin Genet Dev. 1997;7:220–232. doi: 10.1016/s0959-437x(97)80132-3. [DOI] [PubMed] [Google Scholar]
- 40.Zhao W, Manley J L. Complex alternative RNA processing generates an unexpected diversity of poly(A) polymerase isoforms. Mol Cell Biol. 1996;16:2378–2386. doi: 10.1128/mcb.16.5.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]