

Abstract
Background
Chronic kidney disease (CKD) patients exhibit a heightened cardiovascular (CV) risk which may be partially explained by increased medial vascular calcification. Although gut‐derived uremic toxin trimethylamine N‐oxide (TMAO) is associated with calcium‐phosphate deposition, studies investigating phenylacetylglutamine's (PAG) pro‐calcifying potential are missing.
Methods
The effect of TMAO and PAG in vascular calcification was investigated using 120 kidney failure patients undergoing living‐donor kidney transplantation (LD‐KTx), in an observational, cross‐sectional manner. Uremic toxin concentrations were related to coronary artery calcification (CAC) score, epigastric artery calcification score, and markers of established non‐traditional risk factors that constitute to the ‘perfect storm’ that drives early vascular aging in this patient population. Vascular smooth muscle cells were incubated with TMAO or PAG to determine their calcifying effects in vitro and analyse associated pathways by which these toxins may promote vascular calcification.
Results
TMAO, but not PAG, was independently associated with CAC score after adjustment for CKD‐related risk factors in kidney failure patients. Neither toxin was associated with epigastric artery calcification score; however, PAG was independently, positively associated with 8‐hydroxydeoxyguanosine. Similarly, TMAO, but not PAG, promoted calcium‐phosphate deposition in vitro, while both uremic solutes induced oxidative stress.
Conclusions
In conclusion, our translational data confirm TMAO's pro‐calcifying effects, but both toxins induced free radical production detrimental to vascular maintenance. Our findings suggest these gut‐derived uremic toxins have different actions on the vessel wall and therapeutically targeting TMAO may help reduce CV‐related mortality in CKD.
Keywords: kidney failure, oxidative stress, phenylacetylglutamine, TMAO, vascular calcification
1. INTRODUCTION
Chronic kidney disease (CKD) is a progressive, debilitating disease, affecting up to 13% of the world's population, 1 and is predicted to become the fifth highest cause of years of life lost globally by 2040. 2 Currently, 7.6% of cardiovascular disease (CVD)‐related deaths are associated with presence of CKD, 3 while 40%–50% of deaths in advanced CKD patients are a result of CVD. As traditional risk factors are not sufficient in explaining this elevated risk, non‐traditional risk factors have been attributed 4 —these include inflammation, oxidative stress, 5 retention of uremic toxins 6 , and vascular calcification. 7 Medial calcification is the dominant form of calcification in CKD patients; a sequelae associated with poor clinical outcomes such as left ventricular hypertrophy and heart failure. 8 This is best exemplified by coronary artery calcification, a hallmark of early vascular aging (EVA) in CKD, which improved CV risk prediction in CKD. 9
Once thought to be passive process, ectopic calcium‐phosphate deposition in the medial layer is now known to be a highly active and complex process that differs from intimal calcification. 10 Osteo/chondrogenic differentiation of vascular smooth muscle cells (VSMCs) is primarily driven by hyperphosphatemia. 8 Uremic toxins may also promote calcium‐phosphate deposition; protein‐bound solutes indoxyl sulphate (IS) and p‐cresyl sulphate (pCS) have both been studied extensively and have been shown to drive calcification in CKD rats, 11 while clinical studies suggest both toxins are independently associated with mortality in CKD populations. 12 , 13 Gut‐derived toxins trimethylamine N‐oxide (TMAO) and phenylacetylglutamine (PAG), however, are less well studied.
Trimethylamine N‐oxide (TMAO) is formed from its precursor, trimethylamine, generated from choline, phosphatidylcholine (lecithin), or L‐carnitine by gut microbial metabolism. The plasma level of TMAO is determined by multiple factors, such as diet and gut microbial flora. 14 In CKD patients, TMAO is independently associated with all‐cause mortality and calcification, 15 , 16 while TMAO promotes calcium‐phosphate deposition in vitro. 16 , 17
Phenylacetylglutamine (PAG) is also generated in the liver via phenylacetic acid metabolism, derived from phenylalanine. 18 Similar to IS and pCS, PAG relies on tubular secretion to be excreted from the circulation, which becomes compromised as renal function declines. As a result, pre‐dialysis serum levels of PAG can be more than 100 times higher than in healthy subjects. 19 While PAG is independently associated with CVD in non‐CKD cohorts, 20 , 21 evidence of a link between PAG and all‐cause mortality is less convincing. 22 , 23 Findings from the HEMO study found no association between uremic solutes (which included IS, pCS and PAG) and CV outcomes (sudden cardiac death, cardiac death, first CV event) in prevalent haemodialysis patients. 22 The inconclusiveness indicates that further research is needed to better understand the pathological effects of uremic solutes. To the best of our knowledge, a study investigating the relationship between PAG and calcification, clinically or in vitro, has not yet been performed. Furthermore, the relationship between TMAO and CAC score, or epigastric calcification, has not been fully elucidated in kidney failure patients.
Thus, in the present study, we hypothesised that TMAO and PAG are promoters of vascular calcification in kidney failure patients. We studied the relationship between toxins and measures of vascular calcification (CAC score and epigastric artery calcification score) and investigated links to known drivers of vascular calcification in CKD, such as oxidative stress and advanced glycation end products (AGEs). Lastly, to implement a translational approach, we used in vitro assays to incubate VSMCs with TMAO and PAG to investigate their potential pro‐calcifying effects and analyse pathways by which these toxins may induce vascular calcification.
2. METHODS
2.1. Patient cohort
This cohort is based on an ongoing study, with all adult patients undergoing living‐donor kidney transplantation (LD‐KTx) at the Department of Transplantation Surgery, Karolinska University Hospital between 2009 and 2022. Exclusion criteria: <18 years of age, acute kidney injury, signs of overt infection, and unwillingness to participate. Informed consent was obtained from each patient and the study protocols were approved by the Swedish Ethical Review Authority in Stockholm; all subsequent experiments were performed in accordance with the Declaration of Helsinki. Data were collected immediately before LD‐KTx, thus the present work is an observational, cross‐sectional study.
2.2. CAC score and epigastric artery calcification score assessment
All kidney failure patients underwent a non‐contrast multi‐detector cardiac CT scan (LightSpeed VCT or Revolution CT; GE Healthcare, Milwaukee, WI, USA) to determine their coronary artery calcium score. CAC‐scoring details were previously described. 24 CAC score was assessed as a lesion with an area >1 mm2 and peak intensity >130 Hounsfield units (HU) based on the Agatston method and expressed in Agatston units. 25 For statistical analysis, strata were formed based on presence of CAC versus no CAC, that is, CAC >0 and CAC = 0. In addition, epigastric arteries and blood were obtained at LD‐KTx and stored for future analysis. In short, epigastric arteries were scored for calcification by a trained pathologist on tissue sections 1–2 μm thick. Vessels were stained with von Kossa (silver nitrate plus nuclear fast red) before the extent of medial calcification was graded from 0 (no calcification) to 3 (severely calcified). The final study size was based on the number of patients in the Kärl‐tx cohort that had both CAC and epigastric artery calcification scores available, as summarised in Figure 1.
FIGURE 1.
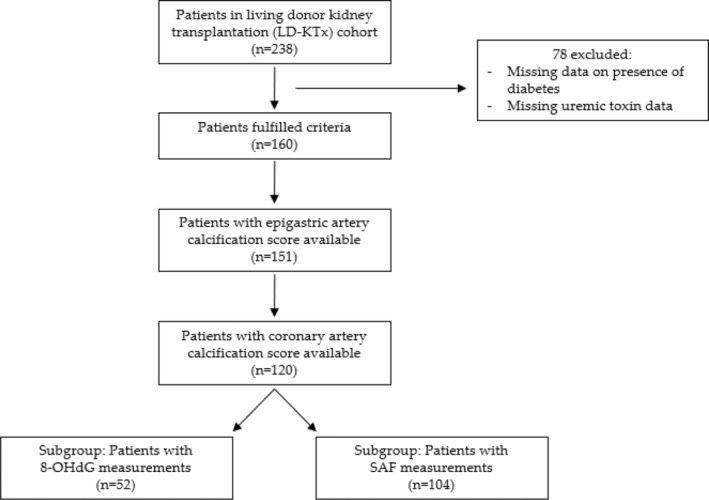
Flowchart depicting study design. 8‐OHdG, 8‐hydroxydeoxyguanosine; SAF, skin autofluorescence.
2.3. Uremic toxin quantification
Serum concentrations of TMAO and PAG were centrally quantified by a previously described method. 26 In brief, serum samples were deproteinised with acetonitrile after addition of an internal standard (stable isotope labelled analogues) and then filtered over a 96‐well Ostro plate (Waters, Zellik, Belgium). After drying with nitrogen and redissolving in MilliQ water, samples were analysed using ultra‐performance liquid chromatography–tandem mass spectrometry with alternating positive and negative electrospray ionisation, for TMAO and PAG, respectively.
2.4. Biochemical analysis/clinical parameters
Blood specimens were routinely drawn at LD‐KTx. Subjects were fasted before blood collection. Routine clinical parameters quantified include measurements of high sensitivity C‐reactive protein (hsCRP), albumin, total cholesterol, high‐density lipoprotein (HDL) cholesterol, and triglycerides. Skin autofluorescence (SAF), a validated marker of AGE accumulation in the skin, was also obtained using the AGE reader device (Diagnoptics, Groningen, Netherlands), as per the manufacturer's instructions. Serum 8‐hydroxydeoxyguanosine (8‐OHdG) was determined by competitive enzyme‐linked immunosorbent assay (ELISA) using the High Sensitivity 8‐OHdG ELISA Assay Kit (JaICA, Shizuoka, Japan). Relevant demographics, comorbidities, and routine biochemistry data were extracted from medical electronic records.
2.5. Cell culture
Human aortic VSMCs (ATCC) from a healthy male donor were incubated between passages 4–7 with one of the following conditions: control (DMEM), high phosphate (2.5 mmol/L sodium phosphate), TMAO (2.5 mmol/L sodium phosphate + 100/300 μM TMAO), or PAG (2.5 mmol/L sodium phosphate + 100/300 μM PAG). Cells were incubated for 7 days (37°C, 5% CO2). On Day 7, cells were harvested for subsequent analysis: calcium content assay (IRDye® 800CW BoneTag™, Odyssey® CLx Infrared Imaging System, LI‐COR Biosciences, NE, USA), qPCR analysis, or Alizarin Red S staining (Sigma‐Aldrich, St Louis, USA).
2.6. Calcification assay
Twenty‐four hours before cells were harvested (Day 6), cells were incubated with IRDye® 800 CW BoneTag™ for 24 h in a Blackwell 96‐well plate. On Day 7, cells were washed three times and fluorescent signals were detected using the Odyssey CLx Infrared imaging system (LI‐COR Biosciences, NE, USA). Readouts were normalised for protein content using BCA protein assay (Abcam, Amsterdam, Netherlands).
2.7. Gene expression analysis
Cultured cells were incubated in 6‐well plates to ensure sufficient RNA was extracted. Total RNA extraction was performed using RNeasy kit (Qiagen, Hilden, Germany) and analysed using NP80 Nanophotometer (Implen, Munich, Germany). Maxima First Strand cDNA Synthesis Kit (ThermoFisher Scientific, Walham, MA, USA) was used before qPCR was performed using PerfeCTa SYBR® Green SuperMix Low ROX (Quantabio, Beverly, MA, USA). Data were analysed using QuantStudio 7 Flex Real‐Time PCR System (Applied Biosystems, Waltham, MA, USA) and relative expression was calculated according to the 2ΔΔCt method using GAPDH as a reference gene. Gene expression for osteogenic marker Runt‐related transcription factor 2 (Runx2) was analysed.
2.8. Alizarin red
Cultured cells, incubated in 12‐well plates, were fixed with 4% formaldehyde in phosphate‐buffered saline (PBS) for 45 min at 4°C. Samples were then washed in distilled water and exposed to Alizarin Red S (2% aqueous, Sigma‐Aldrich, St Louis, USA) for 5 min. Finally, the cells were washed with distilled water and observed using Axio‐observer Z1 microscope (Zeiss, Jena, Germany).
2.9. Reactive oxygen species generation assay
Reactive oxygen species (ROS) production was determined using H2DCFDA assay (Abcam, Amsterdam, Netherlands) as per the manufacturer's instructions. In short, VSMCs were seeded in quadruplets in a Blackwell 96‐well plate at a density of 30,000 cells/well. After 24 h, media was removed and 100 μL/well of diluted DCFDA Solution was added for 45 mins in the dark. Compounds of interest, TMAO or PAG, were then added for 4 h before intracellular ROS was quantified by measuring the fluorescence intensity using a microplate reader (Infinite F200 PRO, Tecan Group Ltd., Männedorf, Switzerland).
2.10. Statistical analysis
Data are expressed as median (IQR) or N [percentage], as appropriate. Differences between two groups were assessed by non‐parametric Mann–Whitney U test. Comparisons between three groups were assessed with one‐way ANOVA followed by group‐wise comparisons using Tukey's post hoc test. Chi‐squared test was used for categorical variables. Spearman rank correlation analysis was used to determine correlations between TMAO, PAG, 8‐OHdG, and SAF. To identify independent associations for significant correlations, multivariate linear or logistic regression analyses were performed. Covariates included in the multivariate models were age at study entry, sex, and presence of diabetes mellitus (DM). In all multivariate models, non‐normally distributed variables were log10 transformed and missing values were handled using a pairwise exclusion process. A p‐value <.05 was deemed statistically significant. Statistical analyses were performed using Stata 17 (Stata Corp, College Station, TX, USA), SAS 9.4 M7 (SAS Institute, Cary, NC, USA), and SPSS (v.27.0, IBM, USA); data visualisation was performed using GraphPad Prism 8 (Dotmatics, Boston, MA, USA).
3. RESULTS
3.1. Baseline characteristics
Baseline patient characteristics at before LD‐KTx are presented in Table 1, stratified by presence of CAC score. Age, presence of DM, weight, BMI, hsCRP, and SAF, that is, presence of AGEs, were all significantly increased in patients with CAC. The study design is depicted in Figure 1.
TABLE 1.
Baseline characteristics of the study cohort, stratified by presence of CAC.
| Total | CAC = 0 | CAC >0 | p‐value | |
|---|---|---|---|---|
| N = 120 | N = 55 | N = 65 | ||
| Age (years) | 47 (32–58) | 32 (24–45) | 54 (48–61) | <.001 |
| Sex (% males) | 78 [65%] | 34 [62%] | 44 [68%] | .501 |
| Presence of DM | 11 [9%] | 1 [2%] | 10 [15%] | .010 |
| Dialysis | 79 [66%] | 35 [64%] | 44 [68%] | .641 |
| Weight (kg) | 74 (63–83) | 71 (61–81) | 77 (68–85) | .010 |
| BMI (kg/m2) | 24.2 (22.3–26.4) | 23.1 (21.0–25.1) | 25.2 (23.5–27.8) | <.001 |
| hsCRP (mg/L) | 1.0 (0.5–2.2) | 0.6 (0.3–1.3) | 1.2 (0.6–3.1) | <.001 |
| Albumin (g/L) | 35 (33–38) | 35 (33–39) | 35 (32–38) | .608 |
| Triglycerides (mmol/L) | 1.3 (1.0–1.8) | 1.2 (1.0–1.8) | 1.4 (0.9–1.8) | .752 |
| Total cholesterol (mmol/L) | 4.3 (3.7–5.0) | 4.3 (3.8–4.9) | 4.3 (3.6–5.1) | .660 |
| HDL‐cholesterol (mmol/L) | 1.4 (1.1–1.6) | 1.4 (1.1–1.6) | 1.4 (1.0–1.6) | .763 |
| SAF (AU) | 3.1 (2.6–3.5) | 2.9 (2.4–3.1) | 4.3 (2.9–3.8) | <.001 |
| 8‐OHdG (mg/mL) | 0.17 (0.12–0.27) | 0.16 (0.12–0.24) | 0.18 (0.12–0.31) | .435 |
| CAC score (HU) | 3 (0–184) | – | 152 (14–970) | – |
Note: Data are presented as median (interquartile range) for continuous measures, N [percentage] for categorical measures. Differences between strata were assessed using non‐parametric Mann–Whitney U test for continuous parameters or chi‐squared test for categorical variables. Significant p‐values (p < .05) are depicted in bold. Missing values: Weight = 2; BMI = 2; hsCRP = 2; albumin = 2; SAF = 16; 8‐OHdG = 68.
Abbreviations: 8‐OHdG, 8‐hydroxyguanosine; BMI, body mass index; CAC, coronary artery calcium; HDL, high‐density lipoprotein; hsCRP, high‐sensitivity C‐reactive protein; PAG, phenylacetylglutamine; SAF, skin autofluorescence; TMAO, trimethylamine N‐oxide.
3.2. TMAO/PAG versus calcification
Firstly, TMAO and PAG were investigated in relation to CAC score in the LD‐KTx patient cohort (n = 120) after stratifying patients into two groups based on CAC score: no CAC (n = 55) versus presence of CAC (n = 65), that is, CAC = 0 versus CAC >0. TMAO was significantly higher in patients with CAC (p < .001; Figure 2A). No significant difference was observed for PAG between patients with and without CAC (p > .05; Figure 2B), although there was a small tendency for increased PAG concentrations in the presence of CAC. Uremic toxin concentrations were then studied in relation to epigastric artery calcification score to assess whether trends obtained with CAC score were comparable. Representative images of epigastric artery calcification scores are shown in Figure 3. Contrary to CAC score, neither TMAO or PAG concentrations significantly differed between patients with non‐calcified/mildly calcified vessels (n = 78) versus patients with moderately/severely calcified vessels (n = 42) (Figure 2C,D; both p > .05). However, a higher epigastric artery calcification score was related to increased CAC score (p < .001; Figure 4), suggesting that patients with calcification in one region are prone to calcification in another, i.e., a systemic sequelae.
FIGURE 2.
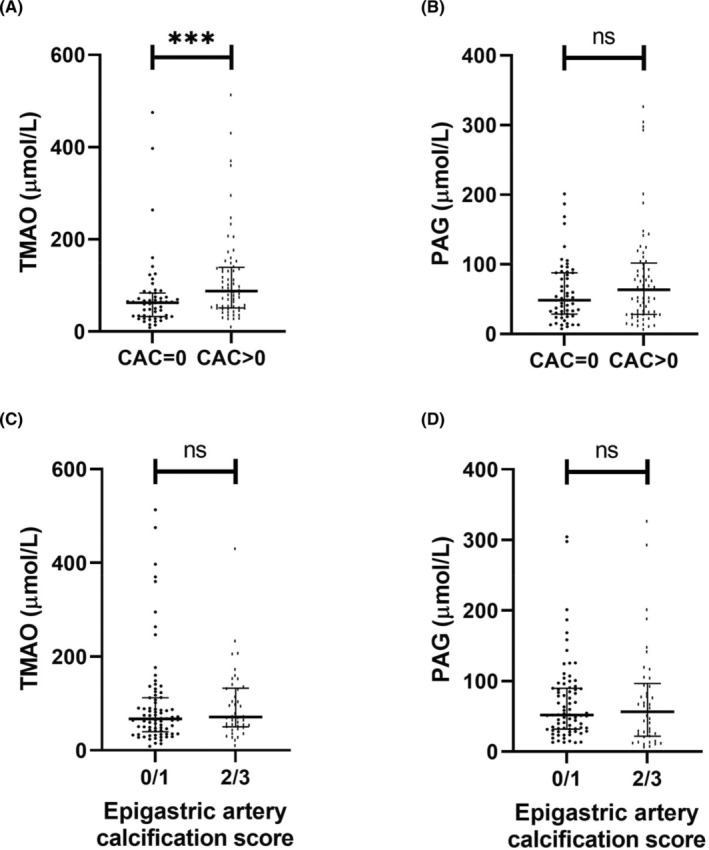
Comparisons between TMAO and PAG versus coronary artery calcification or epigastric artery calcification in a living‐donor kidney transplantation patient cohort (n = 120). (A) Serum TMAO concentration in study population stratified by presence of CAC. (B) Serum PAG concentration in study population stratified by presence of CAC. For (A and B), CAC score was quantified in Agatston units using non‐contrast multi‐detector cardiac CT scan. n = 55 and n = 65 for CAC = 0 and CAC >0 groups, respectively. (C) Serum TMAO concentration in relation with epigastric artery calcification score. (D) Serum PAG concentration in relation with epigastric artery calcification score. For (C and D), epigastric arteries were obtained at living‐donor kidney transplantation and scored for calcification by a trained pathologist on tissue sections 1–2 μm thick. Vessels were stained with von Kossa (silver nitrate plus nuclear fast red) and the extent of medial calcification was graded from 0 (no calcification) to 3 (severely calcified). Groups were formed based on the degree of calcification, that is, 0/1 corresponds to non‐calcified and mildly calcified vessels (n = 78), and 2/3 corresponds to moderately and severely calcified vessels (n = 42). Data are presented as mean ± standard error of mean. Differences between groups were assessed by non‐parametric Mann–Whitney U test. ***p < .001. CAC, coronary artery calcium; PAG, phenylacetylglutamine; TMAO, trimethylamine N‐oxide.
FIGURE 3.
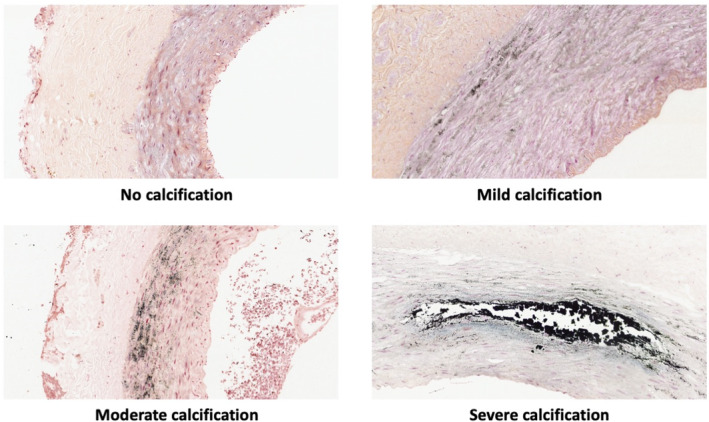
Representative epigastric artery calcification score images. Arteries were isolated at living‐donor kidney transplantation from kidney failure patients and scored by a trained pathologist. Photos taken by Magnus Söderberg.
FIGURE 4.
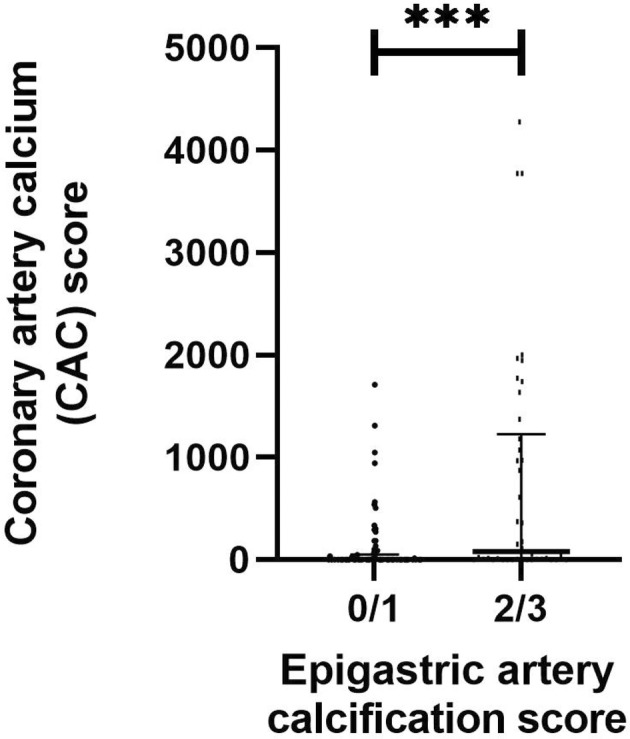
Comparisons between CAC score and epigastric artery calcification in the living‐donor kidney transplantation patient cohort (n = 120). CAC score was quantified in Agatston units using non‐contrast multi‐detector cardiac CT scan. Epigastric arteries were obtained at living‐donor kidney transplantation and scored for calcification by a trained pathologist on tissue sections 1–2 μm thick. Vessels were stained with von Kossa (silver nitrate plus nuclear fast red) before the extent of medial calcification was graded from 0 (no calcification) to 3 (severely calcified). Groups were formed based on degree of calcification, that is, 0/1 (n = 78) corresponds to non‐calcified and mildly calcified vessels, and 2/3 (n = 42) corresponds to moderately and severely calcified vessels. Data are presented as mean ± standard error of mean. Differences between groups were assessed by non‐parametric Mann–Whitney U test. ***p < .001.
To identify independent associations between TMAO and CAC score, multivariate logistic regression analysis was performed with adjustment for age, sex, and presence of DM. This revealed that TMAO was independently associated with CAC score (odds ratio: 3.18; p < .05; Table 2).
TABLE 2.
Multivariate logistic regression analysis between TMAO groups (low + middle vs. high) and coronary artery calcification score in the living‐donor kidney transplantation cohort (n = 120) adjusted for age, sex, and presence of DM.
| Odds ratio | p‐value | [95% confidence interval] | ||
|---|---|---|---|---|
| Age | 17.37 | <.001 | 6.35 | 47.49 |
| Sex | 2.69 | .070 | 0.92 | 7.85 |
| Presence of DM | 5.26 | .242 | 0.33 | 84.92 |
| TMAO | 3.18 | .030 | 1.12 | 9.05 |
Note: Significant associations (p < .05) are depicted in bold. Reference groups: Age – ref <47 years of age versus >47 years of age; sex – ref females versus males; presence of DM – ref no versus yes; TMAO – low/middle tertiles versus high tertile.
Abbreviations: DM, diabetes mellitus; TMAO, trimethylamine N‐oxide.
3.3. TMAO/PAG versus oxidative stress
Calcification in CKD patients has previously been associated with non‐traditional risk factors such as increased oxidative stress and accumulation of AGEs. Thus, it was hypothesised that these mechanisms may promote TMAO‐induced calcification in our cohort. TMAO was not correlated with 8‐OHdG (rho: 0.071; p > .05; Figure 5A), a marker of oxidative stress (n = 52), or AGE accumulation (rho: 0.150; p > .05; Figure 5B), measured through SAF (n = 104). On the contrary, PAG positively correlated with both 8‐OHdG (rho: 0.377; p < .01; Figure 5C) and SAF (rho: 0.329; p < .01; Figure 5D). After adjustment for age, sex, and presence of DM, PAG was independently, positively associated with 8‐OHdG (standardised beta: 0.294; p < .05; Table 3), but not independently associated with SAF (standardised beta: 0.165; p > .05; Table 3).
FIGURE 5.
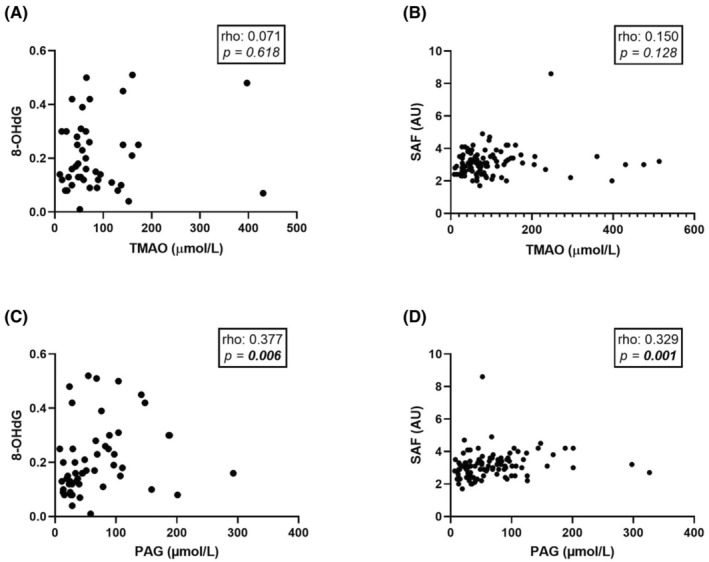
Correlations between TMAO and PAG versus 8‐OHdG and SAF in the living‐donor kidney transplantation cohort (n = 120). (A) TMAO correlated with 8‐OHdG. (B) TMAO correlated with SAF. (C) PAG correlated with 8‐OHdG. (D) PAG correlated with SAF. Univariate correlations were assessed using Spearman's rank correlation. Significant associations (p < .05) are depicted in bold. 8‐OHdG, 8‐hydroxydeoxyguanosine; PAG, phenylacetylglutamine; SAF, skin autofluorescence; TMAO, trimethylamine N‐oxide.
TABLE 3.
Multivariate linear regression analyses between PAG and 8‐OHdG, and skin autofluorescence (SAF), adjusted for age, sex and presence of DM.
| 8‐OHdG | SAF | |||
|---|---|---|---|---|
| Standardised beta | p‐value | Standardised beta | p‐value | |
| Age | 0.385 | .005 | 0.403 | <.001 |
| Sex | 0.009 | .947 | −0.132 | .117 |
| Presence of DM | −0.125 | .398 | 0.161 | .071 |
| PAG | 0.294 | .047 | 0.165 | .062 |
Note: A multivariate model was calculated only for correlations for which a significant univariate correlation was found (Figure 5). Non‐normally distributed variables were log10 transformed prior to analysis. Standardised β coefficients and p‐values are given for each model. Significant associations (p < .05) are depicted in bold.
Abbreviations: 8‐OHdG, 8‐hydroxydeoxyguanosine; DM, diabetes mellitus; PAG, phenylacetylglutamine; SAF, skin autofluorescence.
3.4. TMAO, but not PAG, promotes vascular smooth muscle cell calcification in vitro
Next, to implement a translational study design, healthy VSMCs were incubated with TMAO or PAG for 7 days, in the background of high phosphate media. TMAO promoted calcium‐phosphate deposition at 100 and 300 μM (Figure 6A, p < .01 for TMAO 100/300 μM vs. osteogenic control). This finding was further supported by significantly increased Runx2 gene expression (Figure 6C, p < .05 vs. osteogenic control) and elevated alizarin red staining (Figure 5D), both at 300 μM.
FIGURE 6.
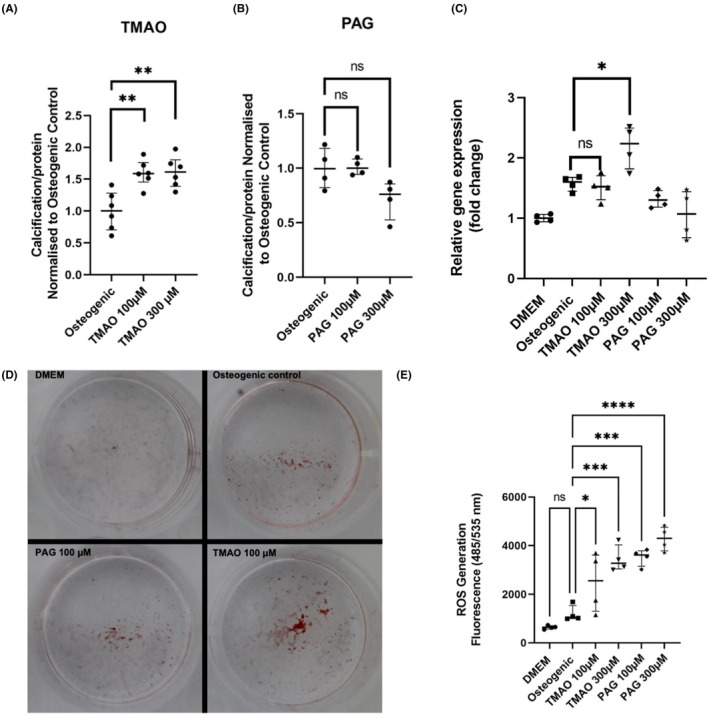
In vitro experiments investigating the effect of TMAO and PAG in VSMC calcification and oxidative stress. (A) TMAO‐induced calcification assay using BoneTag Optical Dye (n = 6 per group). (B) PAG‐induced calcification assay using BoneTag Optical Dye. Aortic VSMCs were incubated with uremic toxins for 7 days in the background of high phosphate (2.5 mM phosphate). For both experiments, readouts were normalised for protein content using BCA assay (n = 4 per group). (C) mRNA expression analysis of osteogenic marker Runx2. Relative gene expression was normalised to DMEM control group (n = 4 per group). (D) Representative images of alizarin red staining for specified conditions. (E) Reactive oxygen species assay for specified conditions using DCFDA/H2DCFDA assay (n = 4 per group). Readout taken 4 h after addition of toxins. Statistics: data presented as mean ± SEM. Groups were compared using one‐way ANOVA followed by group‐wise comparisons using Tukey's post hoc test. p‐values <.05 were deemed statistically significant. *p < .05, **p < .01, ***p < .001, ****p < .0001. PAG, phenylacetylglutamine; ROS, reactive oxygen species; TMAO, trimethylamine N‐oxide.
Similar to in vivo findings, PAG failed to promote calcification in VSMCs in the background of high phosphate, assessed with the calcification assay (Figure 6B, p > .05 for PAG 100/300 μM vs. osteogenic control), Runx2 gene expression (Figure 6C, p > .05 vs. osteogenic control) and alizarin red staining (Figure 6D).
3.5. TMAO and PAG induce oxidative stress in vitro
Finally, since PAG was independently associated with oxidative stress in vivo, TMAO/PAG's pro‐oxidative potential was studied in vitro. Using the H2DCFDA assay, both TMAO and PAG, at 100 and 300 μM, induced oxidative stress in VSMCs after 4 h incubation (Figure 5E, p < .05 for TMAO 100 μM vs. osteogenic control; p < .001 for TMAO 100 μM/PAG 100 μM/PAG 300 μM vs. osteogenic control).
4. DISCUSSION
In the present study, we found distinct effects of two uremic toxins, TMAO and PAG, on vascular dysfunction, both of which accumulate with reduced eGFR and are independent risk factors for mortality and CVD. 23 , 27 While TMAO was directly linked to the development of calcification (a confirmatory finding) and generation of reactive oxygen species in vitro, PAG had detrimental effects on vascular maintenance through free radical production leading to increased presentation of oxidative stress. We are the first to report PAG‐induced oxidative stress, both in vivo and in vitro. Our findings illustrate the complexity of triggering factors related to uremia‐induced EVA.
Altered biomolecules from uremic toxin‐induced carbamylation, oxidative stress, or covalent adduct formation are linked to CVD and mortality in CKD. 28 , 29 Clinical studies have reported a link between TMAO and mortality, 15 , 29 and CVD. 30 Zhang et al. found that TMAO promotes vascular calcification by activation of the NLRP3 inflammasome and NF‐κB signalling pathway, 16 while higher TMAO is an independent risk factor for abdominal aortic calcification in haemodialysis patients. 31 In addition to validating previously reported findings, our study sheds additional light on mechanisms involved, and suggests that calcification is mediated through free radical production. Our in vitro findings are supportive of TMAO‐induced ROS driving an osteochondrogenic phenotype switch of VSMCs and subsequent calcification. 32 Five other uremic toxins—tumour necrosis factor alpha, interleukin (IL)‐1 beta, IL‐8, 100 calcium‐binding protein A12 and salusin‐beta—have been implicated as promoters of oxidative stress‐induced vascular calcification. 33 Uremic toxin‐induced oxidative stress has been well documented in multiple cell types especially in relation to IS and pCS. Hippuric acid, indole‐3‐acetic acid, and phenyl acetic acid promote free radical production in endothelial cells. 34 , 35 , 36 Indeed, excessive ROS generation contributes to the activation of certain osteochondrogenic signal transduction pathways, thereby accelerating osteochondrogenic trans‐differentiation of VSMCs. 37 , 38
Contrary to our in vitro data, however, we did not observe a correlation between TMAO and 8‐OHdG, a surrogate marker of DNA oxidation. 39 This suggests that TMAO may act locally on VSMCs, which cannot be captured with systemic oxidative stress readouts. Our data also indicates that TMAO has differential pro‐calcifying effects on muscular arteries in different regions. As the in vivo study design is largely observational and our experimental work only uses aortic VSMCs, it is difficult to ascertain specific reasons for the observed discrepancy. A logical explanation is that TMAO promotes atherosclerotic calcification, that is, intimal vascular calcification, as CAC score is a compound of atherosclerotic and uremic calcification. 40 Additionally, as calcification is a highly complex and multifactorial process, 7 we cannot rule out the possibility that some vessels are more tolerant to TMAO toxicity than others.
Phenylacetylglutamine is independently associated with CVD in CKD stages 1–5 24 and different non‐CKD populations, 20 , 21 , 41 while also associated with increased arterial stiffness in middle‐aged women. 42 Poesen et al. found that PAG is associated with CVD and mortality in CKD stages 1–5. 23 On the contrary, we did not find an association between PAG and CAC score or epigastric artery calcification score. Uremia, age, and sex differences may account for these contrasting data, while arterial stiffness is influenced by a variety of factors in addition to medial vascular calcification. 43 A meta‐analysis found that 16 toxins have calcification‐inducing effects, 13 have no effect, and 3 solutes have an inhibitory effect in vitro 33 —PAG has not been previously investigated. Although Nemet et al. elegantly demonstrated that PAG may promote CVD through adrenergic receptors and increased thrombosis, 44 experimental data concerning the link between PAG and CVD is scarce.
While we found that PAG was independently, positively associated with 8‐OHdG in vivo, and promoted reactive oxygen species production in vitro, neither were linked with vascular calcification. Our findings present a novel pathogenetic mechanism by which PAG links the prooxidative environment with CV dysfunction. Numerous studies have shown elevated oxidative stress leads to CV complications in kidney failure. 45 AGEs, the result of irreversible glycation modification of amino acids or proteins by reducing sugars, are elevated in the uremic milieu and often exacerbated in the presence of DM. 46 In fact, some AGEs, such as pentosidine, glyoxal and glucosepane, constitute as protein‐bound uremic toxins. 28 Indeed, pentosidine is associated with extensive coronary artery calcification in HD patients. 46 Despite losing statistical significance after adjustment, the positive correlation observed between PAG and AGE accumulation is not surprising considering the bidirectional relationship between AGEs and oxidative stress—AGEs interact with the receptor for advanced glycation end products (RAGE) to activate oxidative stress, while reactive oxygen species (ROS) also promote AGE formation. 47
Our findings suggest that modulation of pathways related to free radical production may ameliorate medial calcification. Interventional therapies have yet to be developed; however, the fact that free radical generation persists after generation of both toxins is of clinical importance. Using a novel animal model of 5/6 nephrectomised rats, we reported dysregulated NRF2 is linked to ectopic vascular calcification. 48 Hence therapeutically targeting the NRF2 pathway using NRF2 agonists may have positive clinical implications. However, further experimental studies are needed to determine the optimal window of intervention. 49 Furthermore, dietary intervention could lead to reduced levels of TMAO in CKD, as recently comprehensively reviewed by Zixin et al. 50
Strengths of the current study include a reasonably large kidney failure cohort that was carefully phenotyped. We also implemented a translational approach to test our hypothesis in a clinical cohort and an in vitro calcification model, as recapitulating uremic components in vitro has been difficult. 48 In addition, we assessed the magnitude of calcification in two separate locations of the vascular tree—coronary and epigastric arteries. While the Agatston score is a standardised computed tomography calcium scoring method in clinical practice for quantification of coronary calcium, it is an indirect measurement of calcium deposition, does not reflect a pathophysiologic variable and cannot differentiate between intima and media calcification. 51 , 52 , 53 On the other hand, in epigastric artery biopsies, media and intima calcification are separated. 53 We also acknowledge limitations in our study, such as the cross‐sectional study design and the absence of intervention to ameliorate TMAO‐associated vascular calcification. Our findings may also only be relevant to our patient cohort—a selected group of kidney failure patients that are younger, healthier, and with lower prevalence of DM than the typical kidney failure cohort.
In summary, we show that both TMAO and PAG may affect CVD complications related to calcification and increased oxidative stress in uremic patients. Their actions differ, since TMAO is more prone to affect calcification and free radical production, while PAG is solely associated with oxidative stress. Further studies are warranted to assess the potential additive effects of these two toxins currently emerging as leading retention solutes related to the EVA phenotype in uremic patients.
AUTHOR CONTRIBUTIONS
Conceptualisation: Karolina Kublickiene and Peter Stenvinkel; methodology: Sam Hobson, Jetty de Loor, Jonaz Ripswedan and Lars Wennberg; validation: Jetty de Loor, Sam Hobson, Jonaz Ripswedan and Lars Wennberg; resources: Karolina Kublickiene, Peter Stenvinkel, Jonaz Ripswedan and Lars Wennberg; data curation: Sam Hobson, Jonaz Ripswedan and Lars Wennberg; writing—original draft preparation: Sam Hobson; writing—review and editing: Sam Hobson, Abdul Rashid Qureshi, Jonaz Ripswedan, Lars Wennberg, Henriette de Loor, Thomas Ebert, Magnus Söderberg, Pieter Evenepoel, Peter Stenvinkel and Karolina Kublickiene; supervision: Karolina Kublickiene, Peter Stenvinkel, Thomas Ebert and Pieter Evenepoel; project administration: Karolina Kublickiene, Peter Stenvinkel and Sam Hobson; funding acquisition: Karolina Kublickiene, Peter Stenvinkel and Pieter Evenepoel. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
This research was funded by EU RESEARCH FRAMEWORK PROGRAMME: Innovative Training Network ‘CaReSyAn’ (H2020‐MSCA‐ITN‐764474), Heart and Lung Foundation (20160384), Njurfonden, Swedish Medical Research Council (Vetenskapsrådet 2018‐00932 GOING‐FWD; 2021‐01102) and CIMED.
CONFLICT OF INTEREST STATEMENT
P.S. reports serving in advisory or leadership roles for Astellas, AstraZeneca, Baxter, Fresenius Medical Care (FMC), GlaxoSmithKline, Reata and Vifor; receiving honoraria from Astellas, AstraZeneca, Baxter, FMC, Novo Nordisk, Boerhinger Ingelheim and Pfizer/Bristol Square Myers (BSM); receiving research funding from AstraZeneca and Bayer; serving on advisory boards for AstraZeneca, Baxter, FMC, GlaxoSmithKline, Reata and Vifor. All other authors of this manuscript have nothing to declare.
INSTITUTIONAL REVIEW BOARD STATEMENT
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Swedish Ethical Review Authority in Stockholm, Sweden.
INFORMED CONSENT STATEMENT
Informed consent was obtained from all subjects involved in the study.
Hobson S, Qureshi AR, Ripswedan J, et al. Phenylacetylglutamine and trimethylamine N‐oxide: Two uremic players, different actions. Eur J Clin Invest. 2023;53:e14074. doi: 10.1111/eci.14074
REFERENCES
- 1. Hill NR, Fatoba ST, Oke JL, et al. Global prevalence of chronic kidney disease – a systematic review and meta‐analysis. PLoS One. 2016;11:e0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kovesdy CP. Epidemiology of chronic kidney disease: an update 2022. Kidney Int Suppl (2011). 2022;12:7‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bikbov B, Purcell CA, Levey AS, et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2020;395:709‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation. 2021;143:1157‐1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kooman JP, Kotanko P, Schols AMWJ, Shiels PG, Stenvinkel P. Chronic kidney disease and premature ageing. Nat Rev Nephrol. 2014;10:732‐742. [DOI] [PubMed] [Google Scholar]
- 6. Liabeuf S, Drüeke TB, Massy ZA. Protein‐bound uremic toxins: new insight from clinical studies. Toxins (Basel). 2011;3:911‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Demer LL, Tintut Y. Vascular calcification. Circulation. 2008;117:2938‐2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nelson AJ, Raggi P, Wolf M, Gold AM, Chertow GM, Roe MT. Targeting vascular calcification in chronic kidney disease. JACC Basic Transl Sci. 2020;5:398‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matsushita K, Sang Y, Ballew SH, et al. Subclinical atherosclerosis measures for cardiovascular prediction in CKD. J Am Soc Nephrol. 2015;26:439‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lanzer P, Hannan FM, Lanzer JD, et al. Medial arterial calcification. J Am Coll Cardiol. 2021;78:1145‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Opdebeeck B, Maudsley S, Azmi A, et al. Indoxyl sulfate and p‐Cresyl sulfate promote vascular calcification and associate with glucose intolerance. J Am Soc Nephrol. 2019;30:751‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barreto FC, Barreto DV, Liabeuf S, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4:1551‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liabeuf S, Barreto DV, Barreto FC, et al. Free p‐cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol Dial Transplant. 2010;25:1183‐1191. [DOI] [PubMed] [Google Scholar]
- 14. Velasquez M, Ramezani A, Manal A, Raj D. Trimethylamine N‐Oxide: the good, the bad and the unknown. Toxins (Basel). 2016;8:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang WHW, Wang Z, Kennedy DJ, et al. Gut microbiota‐dependent trimethylamine N‐oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116:448‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang X, Li Y, Yang P, et al. Trimethylamine‐N‐oxide promotes vascular calcification through activation of NLRP3 (nucleotide‐binding domain, leucine‐rich‐containing family, pyrin domain‐Containing‐3) Inflammasome and NF‐κB (nuclear factor κB) signals. Arterioscler Thromb Vasc Biol. 2020;40:751‐765. [DOI] [PubMed] [Google Scholar]
- 17. Li J, Zeng Q, Xiong Z, et al. Trimethylamine N‐oxide induces osteogenic responses in human aortic valve interstitial cells in vitro and aggravates aortic valve lesions in mice. Cardiovasc Res. 2022;118:2018‐2030. [DOI] [PubMed] [Google Scholar]
- 18. Rysz J, Franczyk B, Ławiński J, Olszewski R, Ciałkowska‐Rysz A, Gluba‐Brzózka A. The impact of CKD on uremic toxins and gut microbiota. Toxins (Basel). 2021;13:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. London GM, Safar ME, Pannier B. Aortic aging in ESRD: structural, hemodynamic, and mortality implications. J Am Soc Nephrol. 2016;27:1837‐1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ottosson F, Brunkwall L, Smith E, et al. The gut microbiota‐related metabolite phenylacetylglutamine associates with increased risk of incident coronary artery disease. J Hypertens. 2020;38:2427‐2434. [DOI] [PubMed] [Google Scholar]
- 21. Romano KA, Nemet I, Prasad Saha P, et al. Gut microbiota‐generated phenylacetylglutamine and heart failure. Circ Heart Fail. 2023;16:e009972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shafi T, Sirich TL, Meyer TW, et al. Results of the HEMO study suggest that p‐cresol sulfate and indoxyl sulfate are not associated with cardiovascular outcomes. Kidney Int. 2017;92:1484‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poesen R, Claes K, Evenepoel P, et al. Microbiota‐derived Phenylacetylglutamine associates with overall mortality and cardiovascular disease in patients with CKD. J Am Soc Nephrol. 2016;27:3479‐3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mukai H, Dai L, Chen Z, et al. Inverse J‐shaped relation between coronary arterial calcium density and mortality in advanced chronic kidney disease. Nephrol Dial Transplant. 2020;35:1202‐1211. [DOI] [PubMed] [Google Scholar]
- 25. Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M Jr, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. 1990;15:827‐832. [DOI] [PubMed] [Google Scholar]
- 26. de Loor H, Poesen R, de Leger W, et al. A liquid chromatography – tandem mass spectrometry method to measure a selected panel of uremic retention solutes derived from endogenous and colonic microbial metabolism. Anal Chim Acta. 2016;936:149‐156. [DOI] [PubMed] [Google Scholar]
- 27. Zhou Z, Jin H, Ju H, Sun M, Chen H, Li L. Circulating trimethylamine‐N‐oxide and risk of all‐cause and cardiovascular mortality in patients with chronic kidney disease: a systematic review and meta‐analysis. Front Med (Lausanne). 2022;9:828343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pieniazek A, Bernasinska‐Slomczewska J, Gwozdzinski L. Uremic toxins and their relation with oxidative stress induced in patients with CKD. Int J Mol Sci. 2021;22:6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ebert T, Tran N, Schurgers L, Stenvinkel P, Shiels PG. Ageing – oxidative stress, PTMs and disease. Mol Aspects Med. 2022;86:101099. [DOI] [PubMed] [Google Scholar]
- 30. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He L, Yang W, Yang P, Zhang X, Zhang A. Higher serum trimethylamine‐N‐oxide levels are associated with increased abdominal aortic calcification in hemodialysis patients. Ren Fail. 2022;44:2029‐2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao M‐M, Xu MJ, Cai Y, et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high‐phosphate‐induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79:1071‐1079. [DOI] [PubMed] [Google Scholar]
- 33. Holmar J, de la Puente‐Secades S, Floege J, Noels H, Jankowski J, Orth‐Alampour S. Uremic toxins affecting cardiovascular calcification: a systematic review. Cell. 2020;9:2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morita M, Yano S, Yamaguchi T, Yamauchi M, Sugimoto T. Phenylacetic acid stimulates reactive oxygen species generation and tumor necrosis factor‐α secretion in vascular endothelial cells. Ther Apher Dial. 2011;15:147‐150. [DOI] [PubMed] [Google Scholar]
- 35. Dou L, Sallée M, Cerini C, et al. The cardiovascular effect of the uremic solute Indole‐3 acetic acid. J Am Soc Nephrol. 2015;26:876‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Itoh Y, Ezawa A, Kikuchi K, Tsuruta Y, Niwa T. Protein‐bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal Bioanal Chem. 2012;403:1841‐1850. [DOI] [PubMed] [Google Scholar]
- 37. Tóth A, Balogh E, Jeney V. Regulation of vascular calcification by reactive oxygen species. Antioxidants. 2020;9:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang M, Zheng L, Xu H, et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J Mol Cell Cardiol. 2020;138:256‐268. [DOI] [PubMed] [Google Scholar]
- 39. Valavanidis A, Vlachogianni T, Fiotakis C. 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG): a critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C. 2009;27:120‐139. [DOI] [PubMed] [Google Scholar]
- 40. Shreya D, Zamora DI, Patel GS, et al. Coronary artery calcium score – a reliable indicator of coronary artery disease? Cureus. 2021;13:e20149. doi: 10.7759/cureus.20149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Y, Liu S, Zhao Z, Song X, Qu H, Liu H. Phenylacetylglutamine is associated with the degree of coronary atherosclerotic severity assessed by coronary computed tomographic angiography in patients with suspected coronary artery disease. Atherosclerosis. 2021;333:75‐82. [DOI] [PubMed] [Google Scholar]
- 42. Menni C, Mangino M, Cecelja M, et al. Metabolomic study of carotid–femoral pulse‐wave velocity in women. J Hypertens. 2015;33:791‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cecelja M, Chowienczyk P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc Dis. 2012;1:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nemet I, Saha PP, Gupta N, et al. A cardiovascular disease‐linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180:862‐877.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Daenen K, Andries A, Mekahli D, van Schepdael A, Jouret F, Bammens B. Oxidative stress in chronic kidney disease. Pediatr Nephrol. 2019;34:975‐991. [DOI] [PubMed] [Google Scholar]
- 46. Taki K, Takayama F, Tsuruta Y, Niwa T. Oxidative stress, advanced glycation end product, and coronary artery calcification in hemodialysis patients. Kidney Int. 2006;70:218‐224. [DOI] [PubMed] [Google Scholar]
- 47. Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5:194‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laget J, Hobson S, Muyor K, et al. Implications of senescent cell burden and NRF2 pathway in uremic calcification: a translational study. Cell. 2023;12:643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD. Modulating NRF2 in disease: timing is everything. Annu Rev Pharmacol Toxicol. 2019;59:555‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zixin Y, Lulu C, Xiangchang Z, et al. TMAO as a potential biomarker and therapeutic target for chronic kidney disease: a review. Front Pharmacol. 2022;13:929262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sandfort V, Bluemke DA. CT calcium scoring. History, current status and outlook. Diagn Interv Imaging. 2017;98:3‐10. [DOI] [PubMed] [Google Scholar]
- 52. Berman DS, Arnson Y, Rozanski A. Coronary artery calcium scanning. JACC Cardiovasc Imaging. 2016;9:1417‐1419. [DOI] [PubMed] [Google Scholar]
- 53. Erlandsson H, Qureshi AR, Ripsweden J, et al. Scoring of medial arterial calcification predicts cardiovascular events and mortality after kidney transplantation. J Intern Med. 2022;291:813‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]