

Abstract
Cannabis use disorder (CUD) remains a significant public health issue globally, affecting up to one in five adults who use cannabis. Despite extensive research into the molecular underpinnings of the condition, there are no effective pharmacological treatment options available. Therefore, we sought to further explore genetic analyses to prioritise opportunities to repurpose existing drugs for CUD. Specifically, we aimed to identify druggable genes associated with the disorder, integrate transcriptomic/proteomic data and estimate genetic relationships with clinically actionable biochemical traits. Aggregating variants to genes based on genomic position, prioritised the phosphodiesterase gene PDE4B as an interesting target for drug repurposing in CUD. Credible causal PDE4B variants revealed by probabilistic finemapping in and around this locus demonstrated an association with inflammatory and other substance use phenotypes. Gene and protein expression data integrated with the GWAS data revealed a novel CUD associated gene, NPTX1, in whole blood and supported a role for hyaluronidase, a key enzyme in the extracellular matrix in the brain and other tissues. Finally, genetic correlation with biochemical traits revealed a genetic overlap between CUD and immune‐related markers such as lymphocyte count, as well as serum triglycerides.
Keywords: Cannabis use disorder, Genetics, Pharmacotherapy
The prevalence of cannabis use disorder is increasing; however, there remain no approved medications for the treatment of this condition. We leveraged data from the largest genetic study of cannabis use disorder to date to prioritise treatment targets for the disorder.
1. INTRODUCTION
Cannabis is currently one of the most widely used psychoactive substances worldwide, with an estimated 200 million users in 2018 (UNODC World Drug Report 2020) 1 . However, one in five users meet diagnostic criteria for CUD, 2 which is characterised by increased compulsivity, physical dependence and difficulty achieving abstinence. 3 Despite these issues, the validity of cannabis dependence and withdrawal was previously not well recognised. This can be seen through the Global Burden of Disease (GBD) excluding cannabis dependence in 1990 4 and cannabis withdrawal not being included in the DSM until the Fifth Edition in 2013 and International Classification of Diseases until the Tenth Revision (ICD‐10) in 2015. 5
The largest published CUD genome‐wide association study (GWAS) to date was performed by Johnson et al in 2020. 6 This study identified two genome‐wide significant loci on chromosome 7 (FOXP2) and chromosome 8 (near CHRNA2 and EPHX2), with an estimated liability scale SNP‐heritability (h2 SNP) for CUD, depending on the estimated population prevalence, as 6.7%–12.1%. 6 Twin studies have provided insight into the total heritability (h2) of cannabis use and dependence, with the h2 for lifetime cannabis use (ever vs. never) shown to be around 45% and cannabis dependence as high as 78%. 7 , 8 The high heritability of CUD suggests a strong genetic component, which could be leveraged to inform new treatment options. Drug repurposing, where existing compounds are used in a new indication, is an attractive option to expediate changes in CUD clinical practice, as investment and success in de novo drug development for psychiatry remains comparatively limited.
Despite the potential of genetics, the tremendous heterogeneity among individuals with CUD makes current drug development challenging. To date, no pharmacotherapy has been clearly effective, and there are no approved medications for CUD treatment. 5 , 9 Various pharmacological approaches have been tested to assist people with CUD in reducing their cannabis use by addressing withdrawal symptoms, craving and other cognitive factors. However, most of these interventions have not progressed beyond small pilot trials. 10 , 11
Brezing and Levin's report highlights the need to consider individual patient characteristics for the treatment of CUD. Complex, polygenic disorders such as CUD do not have a one‐size‐fits‐all approach, 5 and research over the past 20 years has shown that not everyone who uses cannabis is affected adversely in the same way. Emerging vulnerability factors, including certain genes and personality characteristics, are being identified, although the mechanisms underlying the negative effects of cannabis use are not fully understood. 12 Given the heritability of CUD, genetics may provide a means to identify and prioritise novel treatment opportunities with greater specificity. Gene‐based enrichment approaches, functional genomics and genetic correlation and causality analyses could be used to identify and refine opportunities for pharmacological interventions. Furthermore, genetic analyses of therapeutically actionable traits such as serum blood biomarkers could also inform drug repurposing opportunities. 13 In the present study, we explore these approaches to inform drug repurposing opportunities, expand on current literature and identify new therapeutic targets for those with CUD.
2. MATERIALS AND METHODS
2.1. GWAS
GWAS of cannabis use disorder (CUD) summary statistics on unrelated genotyped individuals of European ancestry (N cases = 14 080, N controls = 343 726) were obtained from the Psychiatric Genomics Consortium (PGC; https://pgc.unc.edu). These summary statistics are the combination of samples from the PCG Substance Use Disorders working group, iPSYCH and deCODE. The iPSYCH cohort consists of individuals born in Denmark between 1981 and 2005. CUD cases were defined using ICD10 codes (F12.1‐12.2). 14 Controls were individuals who did not have ICD10 codes related to CUD. DeCODE cases were drawn from the largest addiction treatment centre in Iceland, the SAA‐National Centre of Addiction Medicine. CUD diagnoses in this treatment cohort were made by clinicians using the Diagnostic and Statistical Manual of Mental Disorders (DSM) system (DSM‐IIIR, DSM‐IV and DSM‐5 criteria). 15 The cases in the PGC Substance Use Disorders working groups all met DSM‐IV diagnostic criteria.
2.2. Gene‐based analyses to identify druggable targets associated with CUD
Gene‐based association analysis for CUD GWAS summary statistics 6 was performed using MAGMA version 1.09 (https://ctg.cncr.nl/software/magma). MAGMA maps SNPs to genes by aggregating common variants (MAF > 0.01) at the gene level, which increases discovery power by using the linear combination of SNP‐wise P‐values as test statistic, reducing the burden of multiple testing correction seen in univariate GWAS. We used the 1000 genomes phase 3 European reference panel population for LD estimation and mapped variants to the NCBI hg19 genome assembly, which contained 18 297 autosomal protein‐coding genes with SNPs mapped within the defined coordinates of 5 kb upstream and 1.5 kb downstream. Any genes from the major histocompatibility complex (MHC, chr6:28477797–33448354) on chromosome 6 were removed. Multiple testing correction was done using the Bonferroni method, where we divided the alpha level by the number of comparisons and set P < 2.7 × 10−6 as the P‐value required for significance.
2.3. Probabilistic finemapping
Given the association signal may be therapeutically actionable and to further support the MAGMA findings that PDE4B as the most significant gene in this locus, we subjected the PDE4B region to further analysis to evaluate whether it is a causal association. Firstly, we leveraged probabilistic finemapping to prioritise putatively causal genetic variation in this region. Specifically, we used a conventional Bayesian approach that was applied to all variants in the CUD GWAS within 3 MB of the defined PDE4B genic boundaries. Asymptotic Bayes' factors (ABF) for each SNP were approximated using Wakefield's method, assuming a prior variance of 0.2, 2 as outlined extensively elsewhere. 16 ABF were summed to define credible sets given that Bayes' factors are proportional to the posterior probability (PP) for causality for each variant. In other words, to define a 95% credible set of variants in this region, which contains a causal variant with 95% probability, variant‐wise PP were summed in descending order until 0.95 is exceeded. This method assumes a single causal variant so that we did not have to account for LD between variants, which has been demonstrated to be susceptible to false positives in finemapping studies with a prior of multiple causal variants that use references external to the GWAS or not otherwise very well matched at a population level. 17
2.4. PDE4B phenome‐wide association study
We then wished to further investigate the phenotypic relevance for the finemapped CUD association signal in PDE4B. For the variant with the highest PP, we performed a phenome‐wide association study (pheWAS) using IEU open GWAS project database (https://gwas.mrcieu.ac.uk/phewas/) and the FinnGen release 7 resource (https://r7.finngen.fi/about). The concept underlying this is to assess other traits to which this variant is linked.
2.5. Colocalisation of the PDE4B association signals with expression quantitative trait loci
We then attempted to infer using expression quantitative trait loci (eQTL) data whether upregulation or down‐regulation of PDE4B was associated with liability to CUD and whether CUD and PDE4B expression displayed statistical colocalisation. PDE4B eQTLs were sourced from the multi‐tissue eQTL catalogue resource (https://fivex.sph.umich.edu/). 18 We used cortical eQTL data assembled by the MetaBrain consortium (N = 2970) for colocalisation analyses via the coloc method. 19 The coloc approach infers the PP of five competing hypotheses (H) for a given region: H 0 = the region is associated with neither trait, H 1 = the region is associated with trait one, H 2 = the region is associated with trait two, H 3 = the region is associated with both traits but with a different underlying causal variant, and H 4 = both traits share a causal variant. This was implemented using default priors via version 4 of the coloc R package. We also performed a sensitivity analysis where we varied the SNP priors between (1 × 10−8 and 1 × 10−4), as implemented by the sensitivity function in the coloc package.
2.6. Functional genomics analyses of CUD using transcriptome‐ and proteome‐wide association studies
To further refine our understanding of CUD associated genes that may present opportunities for repurposing, we performed a transcriptome‐wide association study (TWAS) and a proteome‐wide association study (PWAS) via the FUSION framework. 20 We achieved this through leveraging genetically imputed models of mRNA and protein expression. For TWAS, we used tissue samples of whole blood (GTEx v7) and post‐mortem brain (GTEx v7, PsychENCODE), 20 , 21 whereas protein expression weights of post‐mortem brain and whole blood were yielded from Religious Orders Study and Memory and Aging Project (ROS/MAP) 22 and NHLBI's Atherosclerosis Risk in Communities (ARIC), 23 respectively. As this method integrates SNP effects from the model of genetically predicted expression with the effects of the same SNPs on CUD, TWAS/PWAS Z‐scores, after accounting for linkage disequilibrium (LD), can be a conceptualised measure of genetic covariance between mRNA or protein expression of the gene and the GWAS trait of interest. As a result, the sign of the TWAS/PWAS Z score is informative as to which direction of genetically predicted mRNA or protein expression is associated with increased odds of CUD. To ensure we captured only the most confidently associated genes that could constitute drug repurposing candidates, we utilised a conservative method for multiple‐testing correction whereby the Bonferroni methodology was implemented to divide the alpha level (0.05) by the total number of significantly cis‐heritable models of genetically regulated expression (GReX) tested from any brain tissue considered or whole blood. Finally, we implemented probabilistic finemapping of TWAS Z scores using FOCUS v0.6.10 to refine potential causal genes for which predicted expression is associated with CUD, as outlined extensively elsewhere. 24 , 25 Using the default prior and prior variance, we estimated marginal posterior inclusion probabilities (PIP) for membership of 90% credible set for each gene in the region in and around the implicated hyaluronidase gene cluster given its potential therapeutic relevance.
2.7. Genetical correlation between CUD and biochemical traits
LD‐score regression analysis (LDSR; v1.0.1) (https://github.com/bulik/ldsc) 26 was used to estimate genetic correlation between CUD and GWAS on 50 biochemical traits from the UK Biobank (UKBB) as outlined previously by our group for other psychiatric disorders. 13 As the mode of action of many existing drugs involves modulating biochemical traits, for example, lipids and blood glucose, shared biology that may be indexed by genetic correlation could be informative as drug repurposing opportunities. In LDSR, the genetic covariance is estimated by regressing SNP‐wise χ2, the product of the marginal SNP effects from both traits, on its LD score. The SNP heritability estimates for both traits are used to normalise the genetic covariance to obtain genetic correlation (r g), which can be accurately estimated in the presence of any sample overlap only affects the LDSR intercept and not the slope. Bonferroni multiple testing correction was used for the 50 biochemical traits tested to define a significant rg. The CUD and biochemical trait GWAS summary statistics were cleaned to ensure they contain HapMap3 SNPs outside the MHC with minor allele frequency >0.05 for consistency. To evaluate evidence of a causal relationship, we used the latent causal variable (LCV) on all genetically correlated CUD and biochemical trait pairs as demonstrated elsewhere and in our previous work. 13 , 27 To estimate partial genetic causality, the LCV framework leverages the bivariate genome‐wide distribution of marginal SNP effects on both CUD and each of the biochemical traits and outputs the posterior mean genetic causality proportion metric (GCP), with GCP > 0 implying partial genetic causality of trait one on trait two, and vice versa. To calculate the GCP metric, the LCV model utilises the genome‐wide SNP–trait association Z scores for two traits and the mixed fourth moments (cokurtosis) of the respective distributions to assess whether there is evidence for a causal effect of one trait on the other. To guard against false positives, partial genetic causality was defined using the recommended threshold of a significantly non‐zero|GCP| > 0.6. 27
3. RESULTS
3.1. Exploring drug interactions of CUD‐associated genes
Using aggregated gene‐level association (MAGMA), we observed two genes that were associated with CUD—PDE4B (P = 2.09 × 10−6) and FOXP2 (P = 9.30 × 10−7) after Bonferroni correction (P < 2.7 × 10−6; Table S1). These genes have been previously reported in Johnson et al. 6 Further analysis using the Drug Gene Interaction Database (DGIdb) on the MAGMA significant genes for CUD revealed the highest interaction score for PDE4B was with dyphylline, a vasodilator and bronchodilator agent. Dyphylline is a phosphodiesterase 4 inhibitor clinically used for the prevention of bronchial asthma or other respiratory diseases. 28 It has similar pharmacological actions and safety profiles as other xanthine derivatives, such as caffeine and theobromine. 29 According to DrugBank, the mechanism of action of dyphylline in humans is a cAMP‐specific 3′,5′‐cyclic phosphodiesterase 4A, 4B, 4C and 4D inhibitor, and an adenosine receptor A1 and A2a antagonist. 30 , 31 , 32 , 33 No drug interactions for FOXP2 were observed in DrugBank or in DGIdb. Interestingly, the PDE3 inhibitor, dipyridamole, also targets PDE4B and has been investigated therapeutically for bipolar disorders. 34
Using probabilistic finemapping, we prioritised one variant in the 95% credible set with a posterior probability greater than 40% (rs1392816, PP = 0.534), out of a total of 34 variants within the credible set. This variant was an intronic variant in the PDE4B gene but was not associated with PDE4B mRNA expression in any of the eQTL catalogue studies or in the MetaBrain cortical eQTL dataset. A pheWAS in the IEU GWAS database of this variant revealed that it was associated with mostly inflammatory [e.g. leukocyte counts, C‐reactive protein (CRP)] and anthropometric (e.g. fat mass, waist‐to‐hip ratio) measures using a conventional phenome‐wide significance threshold (P < 1 × 10−5) (Tables S2 and S3), However, it also demonstrated an association with smoking behaviours. In the FinnGen GWAS database, which only considers binary phenotypes collected from linked hospital inpatient records, it was associated with alcohol use disorder and other related substance abuse phenotypes, supporting its phenotypic relevance for CUD. Statistical colocalisation between PDE4B expression in the cortex and CUD supported the third colocalisation hypothesis that this region is associated with both PDE4B expression and CUD, but both traits are influenced by a different causal variant (PP H3 = 0.93). This conclusion remained consistent with different priors. Indeed, there was no strong evidence to support the CUD‐associated variants mapped to PDE4B as acting as eQTLs, suggesting that the effect of genetic variation in this region is mediated through another modality or it influences expression in a tissue or cell type not considered.
3.2. Dysregulated hyaluronidase enzyme expression associated with liability to CUD through integration of transcriptomic and proteomic data
We utilised TWAS and PWAS methods to integrate genomic information into functionally relevant units that map to genes or proteins and their expression. Next, we assessed the statistical associations between CUD and predicted gene expression. Although TWAS/PWAS associations do not necessarily imply a causal relationship, this approach can be implemented to identify candidate genes located at loci with an inferred mechanistic underpinning through expression. 35 In multi‐brain region and blood TWAS of CUD, significantly down‐regulated genetically proxied expression of the hyaluronidase gene HYAL3 was associated with the disorder in the whole blood, as well as several brain regions including the amygdala (Z = −4.294, P = 1.75 × 10−5), anterior cingulate cortex (Z = −5.103, P = 3.34 × 10−7), hippocampus (Z = −5.103, P = 3.34 × 10−7), hypothalamus (Z = −5.185, P = 1.05 × 10−7), nucleus accumbens (Z = −5.552, P = 2.82 × 10−8) and putamen (Z = −5.433, P = 5.51 × 10−8). The TWAS also revealed significantly up‐regulated expression of NAT6, also known as HYAL1, in the cortex (Figure 1A).
FIGURE 1.
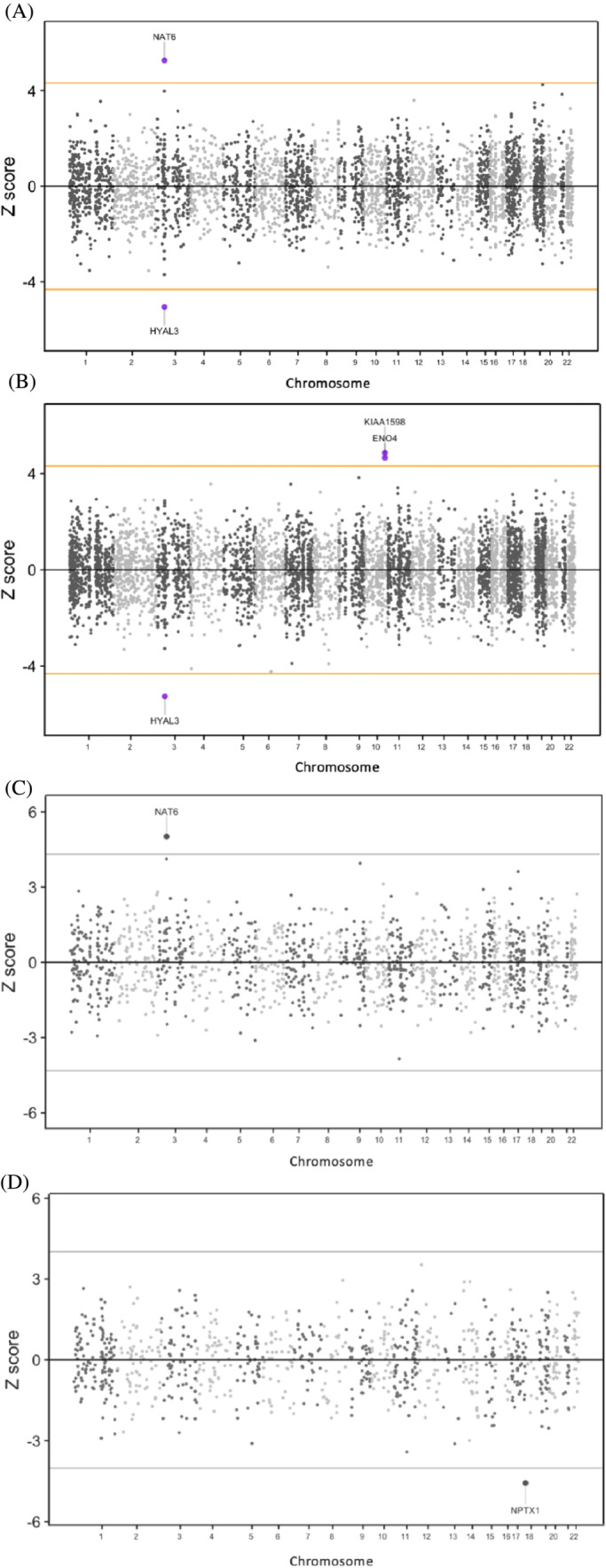
Miami plot of cannabis use disorder (CUD) transcriptome‐wide association (TWA) analysis of brain cerebral cortex (A) and whole blood (B) and proteome‐wide association (PWA) analysis of brain (C) and whole blood (D). The orange horizontal lines are the significance threshold after Bonferroni correction for the total number of imputated models of expression (alpha/n).
NAT6 is a hyaluronidase that is proximally located on chromosome 3, 36 similar to HYAL3. However, HYAL1 is considered to be a more canonically active hyaluronidase compared to HYAL3. 37 , 38 These two transcripts were previously reported in the Johnson et al CUD GWAS using a different statistical method for TWAS (S‐PrediXcan). Additionally, a third gene TTC3 was identified using expression weights from the 3p21.3 putamen region (Z = 4.31, P = 1.63 × 10−5). Overexpression of TTC3 has been associated with negative effects on cognitive function and neuronal health. 39 , 40 As a result, this 3p21.3 region contains several plausible CUD risk genes, and future in silico and experimental follow‐up is warranted to disentangle true effects. In whole blood, ENO4 (Z = 4.8618, P = 3.26 × 10−6) and KIAA1598 (Z = 4.8618, P = 1.16 × 10−7), genes previously reported from European gene‐wise association analysis, were shown to be up‐regulated (Figure 1B). KIAA1598 is also known as SHTN1, with Shootin‐1 protein isoform switch from long isoform (Shtn1L) to short isoform (Shtn1S) known to play an important role in axonogenesis 41 , 42 (Table S4).
We expanded our analysis beyond mRNA expression by conducting a PWAS. In the brain, we again found that NAT6/HYAL1 was significantly up‐regulated in terms of protein abundance (Z = 5.019, P = 5.19 × 10−7) (Figure 1C). In the whole blood, we observed a novel significant association between predicted protein expression of NPTX1 and CUD (Z = −4.57, P = 4.88 × 10−6), which is critical for early human development 43 (Figure 1D). Although NPTX1 down‐regulation was also detected in the TWAS, it did not reach statistical significance after correction for mRNA expression (Table S5).
3.3. Exploring the therapeutic potential of hyaluronidase‐1 enzyme inhibition
The TWAS/PWAS analyses revealed HYAL1 as the most promising target for drug repurposing, as it can be modified by some existing nutraceuticals and investigational compounds (Table 1). However, the other TWAS/PWAS associations did not have current approved therapies that target them, although PDE4B (from the MAGMA analyses) is an existing drug target. If hyaluronidase dysregulation is indeed associated with CUD, as suggested by the TWAS/PWAS, it may represent a promising opportunity for therapeutic investigation. Hyaluronic acid, also known as hyaluronan, is a crucial component of the extracellular matrix and is known to play a role in the regulation of inflammatory processes and embryonic development. 44 In the central nervous system (CNS), hyaluronic acid is a major component of perineuronal nets. These are mesh‐like structures that form around specific neuronal cell bodies and proximal dendrites and have a crucial role in synaptic stabilisation and plasticity. 45 An increase in the expression of NAT6/HYAL1, which is associated with CUD, suggests that there may be abnormal breakdown of hyaluronic acid in individuals with this disorder, as hyaluronidases catalyse the degradation of hyaluronic acid. This abnormal breakdown could potentially increase the risk of developing CUD if the NAT6/HYAL1 gene has a direct causal effect. However, this interpretation is conflicted by the association we found between down‐regulation of another proximal hyaluronidase, HYAL3, and CUD. This requires further investigation, although there is evidence HYAL3 does not directly contribute to the metabolism of hyaluronic acid, 37 but rather it is believed to do so by augmenting the activity of HYAL1. 38 If hyaluronic acid catabolism were dysregulated in CUD, this finding may suggest the potential use of oral hyaluronic acid supplementation, which has been previously trialled for conditions like osteoarthritis. Additionally, the use of ascorbyl palmitate (L‐ascorbic acid 6‐hexadecanoate), an antioxidant that also inhibits hyaluronidase activity, may have therapeutic benefits as a neuroprotective agent. However, it is important to note that ascorbyl palmitate is not currently registered with the Food and Drug Administration (FDA) for therapeutic use (Table 1). 46 , 47 , 48
TABLE 1.
Brief overview of compounds under investigation for inhibition of hyaluronidase‐1.
| Name | IC50 | |
|---|---|---|
| Ascorbyl palmitate | 4.2 ± 0.13 (SagHL) 46 | Potent hyaluronidase inhibitor in vitro. Esterified form of vitamin C. 46 Globally approved antioxidant food additive (E304) |
| Chebulanin | 132 μM (EcH1) 49 | Anti‐inflammatory and anti‐arthritic agent that inhibits NF‐κB and MAPK signalling pathway activation 50 , 51 |
| Chicoric acid | 171 μM (EcH1) 52 | Dicaffeoyl ester with properties that include anti‐inflammatory and anti‐aging properties and glucose and lipid metabolism regulation 53 |
| Glycyrrhizic acid | 177 μM (EcH1) 54 | Known anti‐allergic, anti‐viral and anti‐inflammatory, anti‐lipidaemic and anti‐hyperglycaemic properties. 55 FDA‐approved food additive |
| Testosterone propionate | 124 ± 1.1 μM (EcH1) 52 | Slow‐release anabolic steroid. Shown to be neuroprotective in animal models of Parkinson's disease 56 |
Abbreviations: EcH1, Escherichia coli F470 cells expressing Hyal1; SagHL, Streptococcus agalactiae hyaluronate lyase.
To attempt to resolve the role of hyaluronic acid catabolism in CUD via effects at this locus, we applied probabilistic finemapping of TWAS test statistics on the region located at 3p21.3 that harbours both HYAL1 and HYAL3. Using a combined database of SNP weights from all GTEx tissues, as well some brain and blood datasets, HYAL1 was prioritised as the most likely causal gene for CUD, with a PIP of being in the 90% credible set of 89.3%, compared to 1.51% for HYAL3. However, probabilistic finemapping applied to the PsychENCODE SNP weight set, wherein HYAL1 did not have a model of genetically predicted expression available, prioritised HYAL3 instead (PIP = 78.3%). Given this disparity, further work is still required to reconcile how hyaluronidase activity may be involved in the aetiology of CUD.
3.4. CUD genetically correlated with clinically significant metabolites and immune markers
We tested the genetic correlation between CUD and a panel of blood‐based biomarkers from the UKBB to gain further insight into drug repurposing opportunities by exploring the interplay between circulating biochemical factors and the pathophysiology of CUD. After Bonferroni correction, we found that CUD was genetically correlated with 12 of the 50 blood‐based biomarkers we tested. This included alanine aminotransferase (r g = 0.185, SE = 0.034, P = 7.25 × 10−7), CRP (r g = 0.206, SE = 0.55, P = 2.0 × 10−4), eosinophil count (r g = 0.122, SE = 0.032, P = 9.75 × 10−5), gamma glutamyltransferase (r g = 0.193, SE = 0.043, P = 7.94 × 10−6), lymphocyte count (r g = 0.178, SE = 0.033, P = 6.04 × 10−8), triglycerides (r g = 0.146, SE = 0.039, P = 2.0 × 10−4) and white blood cell (WBC) count (r g = 0.155, SE = 0.032, P = 1.89 × 10−6). It is worth nothing that inflammation of the CNS has long been linked to psychiatric disorders, including schizophrenia. 57 Although we did not find direct evidence of a causal effect, the shared biology between CUD and the biomarkers we tested using LDSC is still informative for future treatment opportunities (Tables S6 and S7).
4. DISCUSSION
CUD has become the primary use disorder for which individuals seek treatment for globally, surpassing all other substances (UNODC, 2017). In this study, we used genetic approaches to explore drug repurposing opportunities for CUD, which currently has no approved treatments. Using gene‐level association, we observed that SNPs localising at PDE4B had the greatest association with CUD. The causal variant in PDE4B, which was found using probabilistic finemapping and is associated with inflammation and substance use in a phenome‐wide analysis, is also believed to have a role in modulating the immune response of monocytes and neutrophils, given that PDE4B is the predominant isoform of the PDE4 protein family. 58 , 59 A role for inflammation in CUD is further supported by its positive genetic correlation with inflammatory markers like CPR and leukocyte count, although a causal relationship could not be confirmed explicitly in LCV models. This suggests that a more complex relationship may exist, perhaps through specific cytokine repertoires that were not able to be considered in this study. While these genetic correlations with CUD are not necessarily causal, they provide insight into potential shared biological mechanisms between biochemical traits that are targeted by drugs that warrant further investigation.
It is also plausible that PDE4B influences CUD more specifically in the brain given its high expression in the tissue. Inhibitors of type 4 phosphodiesterase (PDE) are known to have anti‐inflammatory effects in various cells, including glia, by increasing cAMP and reducing inflammatory signalling 60 . One such PDE inhibitor, ibudilast, can cross the blood–brain barrier and suppresses TNF‐alpha production and astrocyte and microglial activation, making it a potential treatment option for CUD. 61 , 62 , 63
We also conducted PWAS analyses that suggested that increasing genetically predicted neuronal pentraxin 1 (NPTX1) protein expression was protective for CUD. As a member of the pentraxin superfamily, NPTX1 shares structural homology with CRP. 64 NPTX1 is predominantly expressed in the brain and plays an important role in the regulation of synaptic strength and plasticity, as well as neurodegeneration. 65 , 66 Abnormal expression of neuronal pentraxins has been linked to various psychiatric disorders, including schizophrenia and bipolar disorder. 67 , 68 Interestingly, NPTX1 is a negative regulator of excitatory synapses, and its knock‐down has been shown to increase the number of excitatory synapses, while elevated levels of NPTX1 in the plasma are linked to mild cognitive impairment. 69 , 70 If increased NPTX1 expression is protective for CUD, this could reflect a compensatory mechanism in response to the increased number of excitatory synapses in the brain, as previous research has shown that administration of addictive drugs enhances excitatory synaptic strength. 64 Further studies are needed to determine the causal relationship between NPTX1 expression and CUD, as well as to investigate the potential therapeutic implications of targeting NPTX1 in CUD treatment.
In this study, we extended the previously implicated role of HYAL1 and HYAL3 in CUD 6 by demonstrating that an increased genetically proxied protein expression of HYAL1 plausibly increases the risk for CUD beyond a mere association with mRNA expression. Our finemapping analyses provided some support that HYAL1, one of the two proximally located hyaluronidase genes, is more likely a candidate causal gene, although this was not definitive. The role of HYAL1 is particularly interesting because it down‐regulates the expression of hyaluronic acid. A lack of hyaluronic acid in the brain has been shown to cause a reduction in extracellular space (ECS) volume and induce an epileptic phenotype in mice. 71 , 72 This effect could be because HYAL1 digests high molecular weight hyaluronic acid into low molecular weight fragments intracellularly, and these small fragments are important for activating pathways involved in endothelial cell proliferation, adhesion and migration. 73
In addition to the repurposing opportunities provided by oral hyaluronic acid itself or the hyaluronidase inhibitor ascorbyl palmitate, inhibition of hyaluronic acid synthesis using the FDA‐approved prescription drug 4‐methylumbelliferone (4‐MU) or genetic deletion of hyaluronan synthase genes has been shown to improve glucose homoeostasis and is already being explored for the treatment of obesity and diabetes. 74 This is particularly salient given the mounting evidence that metabolic dysfunction is an important component of substance use disorders. 75 However, additional research is needed to clarify the role of hyaluronic acid biology in CUD and to evaluate the potential of pharmacological agents for repurposing as CUD treatments. This could include in vivo investigation of these compounds in suitable animal models of addiction‐associated behaviours, as well as characterising additional evidence of dysregulated hyaluronic acid biology in CUD using resources such as post‐mortem brain. Moreover, follow‐up studies should also be conducted to validate the potential risk genes prioritised in this study including functional investigations.
This study has several limitations. Firstly, we used European‐only summary statistics, and research has shown that predominantly European‐ancestry studies do not translate well to other ancestries and may give an incomplete picture of the genetic underpinnings of CUD. Second, the GWAS was performed using multiple cohorts from the PGC substance use disorders working group, as well as the iPSYCH and deCODE cohorts. Each cohort used different diagnostic criteria, with iPSYCH CUD cases defined using ICD10 codes (F12.1‐12.2) and deCODE cases used DSM‐IIIR, DSM‐IV and DSM‐V criteria for case phenotyping. The cohorts from the PGC substance use disorders working group also had limited homogeneity, with controls for some studies defined as people who simply did not meet criteria for cannabis abuse or dependence, and co‐morbid diagnoses retained in case samples. Therefore, it is crucial that future GWAS of CUD focus predominantly on homogeneity in sample populations.
Finally, while common variant associations with CUD only explain a small amount of phenotypic variance, the effect of risk alleles on molecular traits, like gene expression, are arguably large enough to warrant therapeutic intervention, particularly as these genetics informed targets tend to have a high level of statistical confidence. 76 , 77 Larger sample size panels of expression studies to estimate QTLs and GReX, as well as more diverse neurological tissues and cell‐type data, will further increase discovery power in these approaches in future studies.
AUTHOR CONTRIBUTIONS
Laura A. Greco, William R. Reay and Murray J. Cairns designed the study. Laura A. Greco performed the primary analyses. William R. Reay performed the finemapping and pheWAS. Christopher V. Dayas and Murray J. Cairns supervised the project. Laura A. Greco drafted the manuscript with input from William R. Reay. All authors contributed to the interpretation of the results and the final manuscript.
CONFLICT OF INTEREST STATEMENT
M.J.C. and W.R.R. are directors at PolygenRx Pty Ltd. The remaining authors declare no competing financial interests.
Supporting information
Table S1: Gene based multivariate association individual cannabis use disorder GWAS.
Table S2: Phenome‐wide association study (PheWAS) of the PDE4B variant with the highest posterior proability in the 95% credible set – IEUGWASdb.
Table S3: Phenome‐wide association study (PheWAS) of the PDE4B variant with the highest posterior proability in the 95% credible set ‐ FinnGen release 7.
Table S4: TWAS results for cannabis use disorder.
Table S5: PWAS results for cannabis use disorder.
Table S6: LD‐Score Regression of CUD and 50 biochemical traits.
Table S7: Latent Causal Variable (LCV) modelling of CUD and LDSR significant traits.
ACKNOWLEDGMENTS
Laura A. Greco is supported by a University of Newcastle Research Scholarship (UNRS). William R. Reay is supported by an NHMRC Ideas Grant (1188493). Murray J. Cairns was supported by an NHMRC Senior Research Fellowship (1121474). Open access publishing facilitated by The University of Newcastle, as part of the Wiley ‐ The University of Newcastle agreement via the Council of Australian University Librarians.
Greco LA, Reay WR, Dayas CV, Cairns MJ. Exploring opportunities for drug repurposing and precision medicine in cannabis use disorder using genetics. Addiction Biology. 2023;28 (8): e13313. doi: 10.1111/adb.13313
Contributor Information
Laura A. Greco, Email: laura.greco@newcastle.edu.au.
William R. Reay, Email: william.reay@newcastle.edu.au.
Murray J. Cairns, Email: murray.cairns@newcastle.edu.au.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are publicly available and accessible online and can be found within the article and its supplementary material. The CUD summary statistics can be found here: 10.6084/m9.figshare.14842692.
REFERENCES
- 1. Connor JP, Stjepanovic D, Le Foll B, Hoch E, Budney AJ, Hall WD. Cannabis use and cannabis use disorder. Nat Rev Dis Primers. 2021;7(1):16. doi: 10.1038/s41572-021-00247-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leung J, Chan GCK, Hides L, Hall WD. What is the prevalence and risk of cannabis use disorders among people who use cannabis? A systematic review and meta‐analysis. Addict Behav. 2020;109:106479. doi: 10.1016/j.addbeh.2020.106479 [DOI] [PubMed] [Google Scholar]
- 3. Wiskerke J, Pattij T, Schoffelmeer AN, De Vries TJ. The role of CB1 receptors in psychostimulant addiction. Addict Biol. 2008;13(2):225‐238. doi: 10.1111/j.1369-1600.2008.00109.x [DOI] [PubMed] [Google Scholar]
- 4. Degenhardt L, Ferrari AJ, Calabria B, et al. The global epidemiology and contribution of cannabis use and dependence to the global burden of disease: results from the GBD 2010 study. PLoS ONE. 2013;8(10):e76635. doi: 10.1371/journal.pone.0076635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brezing CA, Levin FR. The current state of pharmacological treatments for cannabis use disorder and withdrawal. Neuropsychopharmacology. 2018;43(1):173‐194. doi: 10.1038/npp.2017.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnson EC, Demontis D, Thorgeirsson TE, et al. A large‐scale genome‐wide association study meta‐analysis of cannabis use disorder. Lancet Psychiatry. 2020;7(12):1032‐1045. doi: 10.1016/S2215-0366(20)30339-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verweij KJ, Zietsch BP, Lynskey MT, et al. Genetic and environmental influences on cannabis use initiation and problematic use: a meta‐analysis of twin studies. Addiction. 2010;105(3):417‐430. doi: 10.1111/j.1360-0443.2009.02831.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Polderman TJ, Benyamin B, de Leeuw CA, et al. Meta‐analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47(7):702‐709. doi: 10.1038/ng.3285 [DOI] [PubMed] [Google Scholar]
- 9. Mariani JJ, Pavlicova M, Jean Choi C, et al. Quetiapine treatment for cannabis use disorder. Drug Alcohol Depend. 2021;218:108366. doi: 10.1016/j.drugalcdep.2020.108366 [DOI] [PubMed] [Google Scholar]
- 10. Nielsen S, Sabioni P, Gowing L, Le Foll B. Pharmacotherapies for cannabis use disorders: clinical challenges and promising therapeutic agents. Handb Exp Pharmacol. 2020;258:355‐372. doi: 10.1007/164_2019_258 [DOI] [PubMed] [Google Scholar]
- 11. Nielsen S, Gowing L, Sabioni P, Le Foll B. Pharmacotherapies for cannabis dependence. Cochrane Database Syst Rev. 2019;1(3):CD008940. doi: 10.1002/14651858.CD008940.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Atakan Z. Cannabis, a complex plant: different compounds and different effects on individuals. Ther Adv Psychopharmacol. 2012;2(6):241‐254. doi: 10.1177/2045125312457586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reay WR, Kiltschewskij DJ, Geaghan MP, et al. Genetic estimates of correlation and causality between blood‐based biomarkers and psychiatric disorders. Sci Adv. 2022;8(14):eabj8969. doi: 10.1126/sciadv.abj8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pedersen CB, Bybjerg‐Grauholm J, Pedersen MG, et al. The iPSYCH2012 case‐cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry. 2018;23(1):6‐14. doi: 10.1038/mp.2017.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tyrfingsson T, Thorgeirsson TE, Geller F, et al. Addictions and their familiality in Iceland. Ann N Y Acad Sci. 2010;1187(1):208‐217. doi: 10.1111/j.1749-6632.2009.05151.x [DOI] [PubMed] [Google Scholar]
- 16. Wellcome Trust Case Control Consortium , Maller JB, McVean G, et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet. 2012;44(12):1294‐1301. doi: 10.1038/ng.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benner C, Havulinna AS, Jarvelin MR, Salomaa V, Ripatti S, Pirinen M. Prospects of fine‐mapping trait‐associated genomic regions by using summary statistics from genome‐wide association studies. Am J Hum Genet. 2017;101(4):539‐551. doi: 10.1016/j.ajhg.2017.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kerimov N, Hayhurst JD, Peikova K, et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat Genet. 2021;53(9):1290‐1299. doi: 10.1038/s41588-021-00924-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi: 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gusev A, Ko A, Shi H, et al. Integrative approaches for large‐scale transcriptome‐wide association studies. Nat Genet. 2016;48(3):245‐252. doi: 10.1038/ng.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Consortium GT . The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45(6):580‐585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimers Dis. 2018;64(s1):S161‐S189. doi: 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. The atherosclerosis risk in communities (ARIC) study: design and objectives. The ARIC investigators. Am J Epidemiol. 1989;129(4):687‐702. [PubMed] [Google Scholar]
- 24. Mancuso N, Freund MK, Johnson R, et al. Probabilistic fine‐mapping of transcriptome‐wide association studies. Nat Genet. 2019;51(4):675‐682. doi: 10.1038/s41588-019-0367-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reay WR, Geaghan MP, Atkins JR, Carr VJ, Green MJ, Cairns MJ. Genetics‐informed precision treatment formulation in schizophrenia and bipolar disorder. Am J Hum Genet. 2022;109(9):1620‐1637. doi: 10.1016/j.ajhg.2022.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bulik‐Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236‐1241. doi: 10.1038/ng.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Connor LJ, Price AL. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet. 2018;50(12):1728‐1734. doi: 10.1038/s41588-018-0255-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morais JA, Neves MC, Silva JR, Avila R. Absorption of intra‐bronchial diprophylline‐methodology and preliminary results. Eur J Drug Metab Pharmacokinet. 1992;17(3):187‐193. doi: 10.1007/BF03190144 [DOI] [PubMed] [Google Scholar]
- 29. Basnet RM, Zizioli D, Guarienti M, Finazzi D, Memo M. Methylxanthines induce structural and functional alterations of the cardiac system in zebrafish embryos. BMC. Pharmacol Toxicol. 2017;18(1):72. doi: 10.1186/s40360-017-0179-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Overington JP, Al‐Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993‐996. doi: 10.1038/nrd2199 [DOI] [PubMed] [Google Scholar]
- 31. Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5(10):821‐834. doi: 10.1038/nrd2132 [DOI] [PubMed] [Google Scholar]
- 32. Iancu L, Shneur A, Cohen H. Trials with xanthine derivatives in systemic treatment of psoriasis. Dermatologica. 1979;159(1):55‐61. doi: 10.1159/000250562 [DOI] [PubMed] [Google Scholar]
- 33. Hariton C. Ocular hypotension induced by topical dopaminergic drugs and phosphodiesterase inhibitors. Eur J Pharmacol. 1994;258(1–2):85‐94. doi: 10.1016/0014-2999(94)90060-4 [DOI] [PubMed] [Google Scholar]
- 34. Machado‐Vieira R, Soares JC, Lara DR, et al. A double‐blind, randomized, placebo‐controlled 4‐week study on the efficacy and safety of the purinergic agents allopurinol and dipyridamole adjunctive to lithium in acute bipolar mania. J Clin Psychiatry. 2008;69(8):1237‐1245. doi: 10.4088/jcp.v69n0806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li B, Ritchie MD. From GWAS to gene: transcriptome‐wide association studies and other methods to functionally understand GWAS discoveries. Front Genet. 2021;12:713230. doi: 10.3389/fgene.2021.713230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ji L, Minna JD, Roth JA. 3p21.3 tumor suppressor cluster: prospects for translational applications. Future Oncol. 2005;1(1):79‐92. doi: 10.1517/14796694.1.1.79 [DOI] [PubMed] [Google Scholar]
- 37. Atmuri V, Martin DC, Hemming R, et al. Hyaluronidase 3 (HYAL3) knockout mice do not display evidence of hyaluronan accumulation. Matrix Biol. 2008;27(8):653‐660. doi: 10.1016/j.matbio.2008.07.006 [DOI] [PubMed] [Google Scholar]
- 38. Hemming R, Martin DC, Slominski E, et al. Mouse Hyal3 encodes a 45‐ to 56‐kDa glycoprotein whose overexpression increases hyaluronidase 1 activity in cultured cells. Glycobiology. 2008;18(4):280‐289. doi: 10.1093/glycob/cwn006 [DOI] [PubMed] [Google Scholar]
- 39. Zhou X, Chen X, Hong T, Zhang M, Cai Y, Cui L. TTC3‐mediated protein quality control, a potential mechanism for cognitive impairment. Cell Mol Neurobiol. 2022;42(6):1659‐1669. doi: 10.1007/s10571-021-01060-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berto G, Camera P, Fusco C, et al. The Down syndrome critical region protein TTC3 inhibits neuronal differentiation via RhoA and citron kinase. J Cell Sci. 2007;120(Pt 11):1859‐1867. doi: 10.1242/jcs.000703 [DOI] [PubMed] [Google Scholar]
- 41. Baba K, Yoshida W, Toriyama M, et al. Gradient‐reading and mechano‐effector machinery for netrin‐1‐induced axon guidance. Elife. 2018;7:e34593. doi: 10.7554/eLife.34593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang M, Ergin V, Lin L, Stork C, Chen L, Zheng S. Axonogenesis is coordinated by neuron‐specific alternative splicing programming and splicing regulator PTBP2. Neuron. 2019;101(4):690‐706.e10. doi: 10.1016/j.neuron.2019.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boles NC, Hirsch SE, Le S, et al. NPTX1 regulates neural lineage specification from human pluripotent stem cells. Cell Rep. 2014;6(4):724‐736. doi: 10.1016/j.celrep.2014.01.026 [DOI] [PubMed] [Google Scholar]
- 44. Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4(7):528‐539. doi: 10.1038/nrc1391 [DOI] [PubMed] [Google Scholar]
- 45. Deepa SS, Carulli D, Galtrey C, et al. Composition of perineuronal net extracellular matrix in rat brain: a different disaccharide composition for the net‐associated proteoglycans. J Biol Chem. 2006;281(26):17789‐17800. doi: 10.1074/jbc.M600544200 [DOI] [PubMed] [Google Scholar]
- 46. Botzki A, Rigden DJ, Braun S, et al. L‐Ascorbic acid 6‐hexadecanoate, a potent hyaluronidase inhibitor. X‐ray structure and molecular modeling of enzyme‐inhibitor complexes. J Biol Chem. 2004;279(44):45990‐45997. doi: 10.1074/jbc.M406146200 [DOI] [PubMed] [Google Scholar]
- 47. Katarzyna Greda A, Nowicka D. Hyaluronidase inhibition accelerates functional recovery from stroke in the mouse brain. J Neurochem. 2021;157(3):781‐801. doi: 10.1111/jnc.15279 [DOI] [PubMed] [Google Scholar]
- 48. Administration USFUFaD . Dietary supplements. 2005. http://www.fda.gov/food/dietarysupplements/default.htm
- 49. Addotey JN, Lengers I, Jose J, Hensel A. Hyal‐1 inhibitors from the leaves of Phyllanthus muellerianus (Kuntze) Excell. J Ethnopharmacol. 2019;236:326‐335. doi: 10.1016/j.jep.2019.03.022 [DOI] [PubMed] [Google Scholar]
- 50. Liu F, Liu Y, Zhan S, et al. Chebulanin exerts its anti‐inflammatory and anti‐arthritic effects via inhibiting NF‐kappaB and MAPK activation in collagen‐induced arthritis mice. Int Immunopharmacol. 2020;88:106823. doi: 10.1016/j.intimp.2020.106823 [DOI] [PubMed] [Google Scholar]
- 51. Lee WJ, Moon JS, Kim SI, et al. Inhibition of the calcineurin pathway by two tannins, chebulagic acid and chebulanin, isolated from Harrisonia abyssinica Oliv. J Microbiol Biotechnol. 2014;24(10):1377‐1381. doi: 10.4014/jmb.1405.05030 [DOI] [PubMed] [Google Scholar]
- 52. Lengers I, Herrmann F, Le Borgne M, Jose J. Improved surface display of human Hyal1 and identification of testosterone propionate and chicoric acid as new inhibitors. Pharmaceuticals (Basel). 2020;13(4). doi: 10.3390/ph13040054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peng Y, Sun Q, Gao R, Park Y. AAK‐2 and SKN‐1 are involved in chicoric‐acid‐induced lifespan extension in Caenorhabditis elegans . J Agric Food Chem. 2019;67(33):9178‐9186. doi: 10.1021/acs.jafc.9b00705 [DOI] [PubMed] [Google Scholar]
- 54. Orlando Z, Lengers I, Melzig MF, Buschauer A, Hensel A, Jose J. Autodisplay of human hyaluronidase Hyal‐1 on Escherichia coli and identification of plant‐derived enzyme inhibitors. Molecules. 2015;20(9):15449‐15468. doi: 10.3390/molecules200915449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li JY, Cao HY, Liu P, Cheng GH, Sun MY. Glycyrrhizic acid in the treatment of liver diseases: literature review. Biomed Res Int. 2014;2014:872139. doi: 10.1155/2014/872139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bispo JMM, Melo JEC, Gois AM, et al. Testosterone propionate improves motor alterations and dopaminergic damage in the reserpine‐induced progressive model of Parkinson's disease. Brain Res Bull. 2022;187:162‐168. doi: 10.1016/j.brainresbull.2022.06.018 [DOI] [PubMed] [Google Scholar]
- 57. Na KS, Jung HY, Kim YK. The role of pro‐inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2014;48:277‐286. doi: 10.1016/j.pnpbp.2012.10.022 [DOI] [PubMed] [Google Scholar]
- 58. Wang ZZ, Zhang Y, Zhang HT, Li YF. Phosphodiesterase: an interface connecting cognitive deficits to neuropsychiatric and neurodegenerative diseases. Curr Pharm des. 2015;21(3):303‐316. doi: 10.2174/1381612820666140826115559 [DOI] [PubMed] [Google Scholar]
- 59. Wang P, Wu P, Ohleth KM, Egan RW, Billah MM. Phosphodiesterase 4B2 is the predominant phosphodiesterase species and undergoes differential regulation of gene expression in human monocytes and neutrophils. Mol Pharmacol. 1999;56(1):170‐174. doi: 10.1124/mol.56.1.170 [DOI] [PubMed] [Google Scholar]
- 60. Blednov YA, Benavidez JM, Black M, Harris RA. Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Front Neurosci. 2014;8:129. doi: 10.3389/fnins.2014.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Snider SE, Vunck SA, van den Oord EJ, Adkins DE, McClay JL, Beardsley PM. The glial cell modulators, ibudilast and its amino analog, AV1013, attenuate methamphetamine locomotor activity and its sensitization in mice. Eur J Pharmacol. 2012;679(1–3):75‐80. doi: 10.1016/j.ejphar.2012.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kolahdouzan M, Futhey NC, Kieran NW, Healy LM. Novel molecular leads for the prevention of damage and the promotion of repair in neuroimmunological disease. Front Immunol. 2019;10:1657. doi: 10.3389/fimmu.2019.01657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999;837(1–2):203‐212. doi: 10.1016/s0006-8993(99)01666-2 [DOI] [PubMed] [Google Scholar]
- 64. Luscher C, Malenka RC. Drug‐evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69(4):650‐663. doi: 10.1016/j.neuron.2011.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Z, Wang X, Zou H, et al. The basic characteristics of the pentraxin family and their functions in tumor progression. Front Immunol. 2020;11:1757. doi: 10.3389/fimmu.2020.01757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kovacs RA, Vadaszi H, Bulyaki E, et al. Identification of neuronal pentraxins as synaptic binding partners of C1q and the involvement of NP1 in synaptic pruning in adult mice. Front Immunol. 2020;11:599771. doi: 10.3389/fimmu.2020.599771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Manchia M, Piras IS, Huentelman MJ, et al. Pattern of gene expression in different stages of schizophrenia: down‐regulation of NPTX2 gene revealed by a meta‐analysis of microarray datasets. Eur Neuropsychopharmacol. 2017;27(10):1054‐1063. doi: 10.1016/j.euroneuro.2017.07.002 [DOI] [PubMed] [Google Scholar]
- 68. Rajkumar AP, Christensen JH, Mattheisen M, et al. Analysis of t(9;17)(q33.2;q25.3) chromosomal breakpoint regions and genetic association reveals novel candidate genes for bipolar disorder. Bipolar Disord. 2015;17(2):205‐211. doi: 10.1111/bdi.12239 [DOI] [PubMed] [Google Scholar]
- 69. Ma QL, Teng E, Zuo X, et al. Neuronal pentraxin 1: a synaptic‐derived plasma biomarker in Alzheimer's disease. Neurobiol Dis. 2018;114:120‐128. doi: 10.1016/j.nbd.2018.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Figueiro‐Silva J, Gruart A, Clayton KB, et al. Neuronal pentraxin 1 negatively regulates excitatory synapse density and synaptic plasticity. J Neurosci. 2015;35(14):5504‐5521. doi: 10.1523/JNEUROSCI.2548-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tan JX, Wang XY, Li HY, et al. HYAL1 overexpression is correlated with the malignant behavior of human breast cancer. Int J Cancer. 2011;128(6):1303‐1315. doi: 10.1002/ijc.25460 [DOI] [PubMed] [Google Scholar]
- 72. Perkins KL, Arranz AM, Yamaguchi Y, Hrabetova S. Brain extracellular space, hyaluronan, and the prevention of epileptic seizures. Rev Neurosci. 2017;28(8):869‐892. doi: 10.1515/revneuro-2017-0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Colombaro V, Jadot I, Decleves AE, et al. Hyaluronidase 1 and hyaluronidase 2 are required for renal hyaluronan turnover. Acta Histochem. 2015;117(1):83‐91. doi: 10.1016/j.acthis.2014.11.007 [DOI] [PubMed] [Google Scholar]
- 74. Grandoch M, Flogel U, Virtue S, et al. 4‐Methylumbelliferone improves the thermogenic capacity of brown adipose tissue. Nat Metab. 2019;1(5):546‐559. doi: 10.1038/s42255-019-0055-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cervantes M, Lewis RG, Della‐Fazia MA, Borrelli E, Sassone‐Corsi P. Dopamine D2 receptor signaling in the brain modulates circadian liver metabolomic profiles. Proc Natl Acad Sci U S A. 2022;119(11):e2117113119. doi: 10.1073/pnas.2117113119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Reay WR, Cairns MJ. Advancing the use of genome‐wide association studies for drug repurposing. Nat Rev Genet. 2021;22(10):658‐671. doi: 10.1038/s41576-021-00387-z [DOI] [PubMed] [Google Scholar]
- 77. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101(1):5‐22. doi: 10.1016/j.ajhg.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Gene based multivariate association individual cannabis use disorder GWAS.
Table S2: Phenome‐wide association study (PheWAS) of the PDE4B variant with the highest posterior proability in the 95% credible set – IEUGWASdb.
Table S3: Phenome‐wide association study (PheWAS) of the PDE4B variant with the highest posterior proability in the 95% credible set ‐ FinnGen release 7.
Table S4: TWAS results for cannabis use disorder.
Table S5: PWAS results for cannabis use disorder.
Table S6: LD‐Score Regression of CUD and 50 biochemical traits.
Table S7: Latent Causal Variable (LCV) modelling of CUD and LDSR significant traits.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are publicly available and accessible online and can be found within the article and its supplementary material. The CUD summary statistics can be found here: 10.6084/m9.figshare.14842692.