

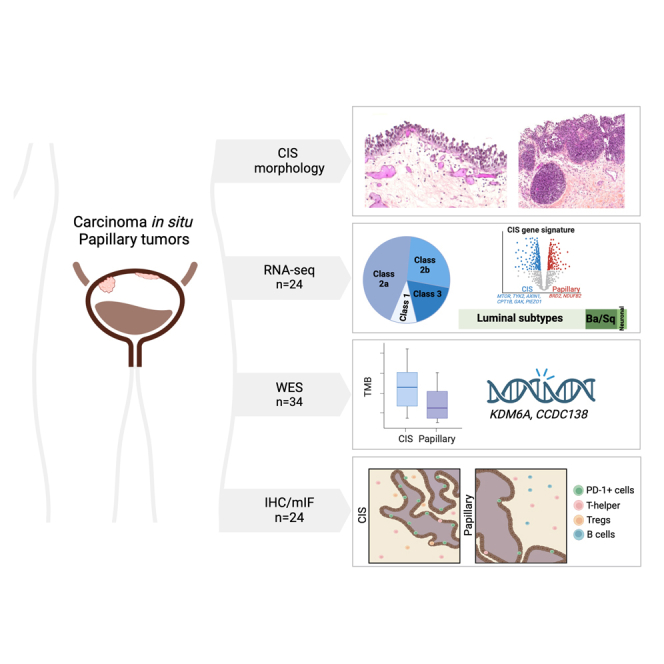
Summary
Urothelial carcinoma in situ (CIS) is an aggressive phenotype of non-muscle-invasive bladder cancer. Molecular features unique to CIS compared to high-grade papillary tumors are underexplored. RNA sequencing of CIS, papillary tumors, and normal urothelium showed lower immune marker expression in CIS compared to papillary tumors. We identified a 46-gene expression signature in CIS samples including selectively upregulated known druggable targets MTOR, TYK2, AXIN1, CPT1B, GAK, and PIEZO1 and selectively downregulated BRD2 and NDUFB2. High expression of selected genes was significantly associated with CIS in an independent dataset. Mutation analysis of matched CIS and papillary tumors revealed shared mutations between samples across time points and mutational heterogeneity. CCDC138 was the most frequently mutated gene in CIS. The immunological landscape showed higher levels of PD-1-positive cells in CIS lesions compared to papillary tumors. We identified CIS lesions to have distinct characteristics compared to papillary tumors potentially contributing to the aggressive phenotype.
Subject areas: Immunology, Cancer, Omics
Graphical abstract
Highlights
-
•
CIS of the bladder is most frequently luminal and UROMOL Class 2a and 2b
-
•
FOXA1 was over-expressed in CIS while basal and immune markers were lower
-
•
A 46-gene signature segregated CIS from papillary and normal bladder tissue
-
•
Immune cell infiltration is high in CIS and papillary stroma with high PD-1 in CIS
Immunology; Cancer; Omics
Introduction
Urothelial carcinoma in situ (CIS) is a flat malignant lesion comprising high-grade malignant urothelial cells confined to the urothelium of the bladder, urethra, or upper urinary tract. On cystoscopic evaluation, CIS may be confused with cystitis or other inflammatory conditions of the bladder making the diagnosis challenging. CIS is not visible with white light cystoscopy in up to 50% of lesions; however, more CIS lesions are detectable with enhanced cystoscopy technologies such as hexaminolevulinate and narrow-band imaging.1 CIS is grouped with the non-muscle-invasive bladder cancers but has the potential to progress to muscle-invasive disease2 and also shares molecular features with subsets of muscle-invasive bladder cancers (MIBCs).3 The pathologic diagnosis of CIS is challenging because of its wide histologic spectrum and potential benign mimics (Figure 1).4,5 It may occur as an isolated pathological finding or may be associated with non-invasive papillary and/or invasive urothelial carcinomas.6 Moreover, the clinical behavior of CIS is highly variable with differences attributed to focality, association with concomitant or prior papillary tumors, and treatment resistance.
Figure 1.

Morphologic spectrum and subtype heterogeneity of urothelial carcinoma in situ (CIS)
(A) Full thickness, partially detached CIS with prominent nuclear pleomorphism and hyperchromasia, architectural disorder, and lack of polarity (10x); (B) prominent denudation and discohesion of CIS with a single layer of tumor attached to the basement membrane. Note the prominent vasculature in the lamina propria (block arrows); (C) High-grade UC involving von Brunn’s nests, which may mimic invasion (4x). The surface tumor is both papillary (shown) and flat (not shown); (D) variable tree showing site, gender, and subtypes for 19 high-quality samples.
Intravesical immunotherapy with Bacillus Calmette-Guérin (BCG) is the primary treatment for CIS following resection of all visible disease in the absence of muscle-invasive cancer. BCG induction and maintenance therapy reduces the risk of recurrence and progression of CIS with initial complete response rates ranging between 70% and 84%; however, later disease recurrences/progression are common.7,8,9 There is a need to understand the biological mechanisms associated with response or failure to BCG treatment.10 At the molecular level, mutations in TP53 and the TERT promoter region are common genetic events in CIS,10,11 but comprehensive genomic studies addressing the molecular biology of CIS are largely lacking. The Cancer Genome Atlas (TCGA) offers the most comprehensive molecular analysis of MIBC,3 and studying the mutational landscape and expression profiles of CIS will help establish how MIBC may evolve from its most common precursor, CIS. A significant barrier to genomic and transcriptomic profiling of CIS is the challenge of getting nucleic acids of sufficient quantity and quality from small formalin-fixed paraffin-embedded (FFPE) specimens.
In this project, we tested the hypothesis that comprehensive analysis of the molecular biology of CIS and comparison to papillary tumors and tumors that progressed to MIBC will help define progression pathways and BCG treatment response/resistance mechanisms in bladder cancer. We describe methodology for extracting mRNA and DNA from small FFPE CIS samples and RNA- and whole-exome sequencing (WES) for expression profiling, pathway analysis, mutation characterization, and association with outcomes. Furthermore, we performed immunohistochemical (IHC) and multiplex immunofluorescence (mIF) analysis to delineate the spatial organization of immune cells in CIS lesions and matched papillary tumors. Finally, we performed exploratory analyses of CIS and its micro-environment and specific signatures of CIS.
Results
Histology and complexities of profiling CIS
As expected, we observed diverse histology in our CIS samples. Figure 1 shows hematoxylin and eosin-stained CIS tissues reflecting the morphological and pathological heterogeneity among CIS samples. Intraepithelial urothelial carcinoma cells with large hyperchromatic nuclei, prominent nuclear pleomorphism, and high nucleus/cytoplasm ratio with partial detachment of mucosal basement membrane are also shown (Figures 1A and 1B). A CIS lesion with partial discohesion of lesional cells and dilated vasculature below the basement membrane in the superficial lamina propria is noted in Figure 1B, and von Brunn’s nests with CIS may be confused with lamina propria invasion (Figure 1C).
Cohort A
The bulk RNA sequencing cohort (cohort A) consisted of a total of 32 samples, comprising 15 CIS, 9 adjacent papillary tumors, and 8 normal samples. The overall aligned read distribution of CIS was lower for CIS samples as compared to papillary tumor or normal tissue samples (Figure S1A). While there was no statistically significant difference in DV200 (measure of RNA stability) across CIS, papillary tumors, and normal samples (average score 33.5, 39.7, and 35.4, respectively), we identified five CIS samples with a low number of aligned reads (Figure S1B). These samples, four of which were from male and one from female subjects, were excluded from further analysis. Hence, the total analytical cohort consisted of 10 CIS, 9 papillary tumor (P), and 8 normal tissue (N) samples. We performed subtyping of tumor samples using multiple methods as shown in Figure S1C including non-muscle invasive bladder cancer (NMIBC) UROMOL,12 TCGA,13 and consensus14 classifiers to CIS and papillary tumor (P) (Figure 1D). The majority of the samples were characterized as the high-risk classes 2a and 2b (six and four out of fifteen, respectively), indicating that CIS lesions share characteristics with high-risk NMIBC.
Ten CIS tumors were predicted to be of Class 2 and three as Class 3 (Figure 1D). These results are consistent with an earlier report showing that CIS tumors were more frequently overlapped with Class 2 tumors.15
Urothelial cancer marker and differential pathway analysis
We used normalized mRNA expression data to investigate published molecular gene signatures13 categorized into luminal, extracellular matrix (ECM) and smooth muscle, EMT and claudin, basal, squamous, immune, neuronal differentiation, CIS surrounding, and Sonic hedgehog markers (Figure 2A). FOXA1 was the only luminal marker that was significantly over-expressed (p = 0.02) in CIS tumors as compared to papillary tumors. On the contrary, basal markers including CD44 and KRT14 were significantly downregulated in CIS tumors (p = 0.001, 0.01 respectively). The immune marker CXCL11 and neuronal differentiation markers APLP1 and MSI1 were also downregulated in CIS tumors. Investigating gene-expression-based immune cell scores, we found Th2 subset of CD4+ T cells and myeloid dendritic cells to be significantly downregulated in CIS tumors (Figure S2A). CIS tumors showed significantly low expression of the “CIS surrounding” markers16 CTSE and MSN. Interestingly, cumulative scores for “CIS surrounding up” genes were significantly lower in CIS tumors compared to papillary tumors (Figure 2B). To further explore the systematic characterization and biological pathways associated with CIS lesions in contrast to papillary tumors, we performed gene set enrichment analysis (GSEA) using differentially expressed genes as input for the KEGG pathway analysis. The Notch signaling, Fanconi anemia, and lysine degradation pathways were significantly enriched in CIS lesions as compared to papillary tumors (Figure 2C). Individual enrichment plots for Notch signaling and lysine degradation pathways are shown in Figures S2B and S2C, respectively.
Figure 2.

CIS gene signature and pathway analysis
(A) Gene signature categorizing samples into luminal, ECM and smooth muscle, EMT and claudin, basal, squamous, Immune, neuronal differentiation, CIS surrounding, and Sonic hedgehog markers.
(B) Composite signature scores shown as boxplots for CIS and tumor samples (Wilcoxon rank-sum test derived p-value shown) (C) Differentially expressed hallmark pathways specific to CIS tumors. Upregulated and downregulated pathways are shown in red and blue, respectively.
CIS-specific gene signature
Identification of differentially regulated pathways prompted us to apply an additional filter to differentially expressed genes that were over- or under-expressed when compared not just to papillary tumors but also to normal samples. CIS-specific gene signature was identified by employing a combination of two statistical filtering criteria including Wilcoxon rank-sum test (p < 0.05) comparing CIS and papillary tumors, followed by an ANOVA test comparing CIS, papillary Ta/T1, and normal samples (p < 0.05). This resulted in a set of 46 differentially expressed up- and downregulated genes. Unsupervised hierarchical clustering showed clear segregation of CIS samples from other samples (Figure 3A). Interestingly, the upregulated gene set included six druggable targets consisting of MTOR, TYK2, AXIN1, CPT1B, GAK, and PIEZO1. Differential levels of druggable kinases MTOR, GYK1, and TYK2 across sample types are shown (Figure S3A). To assess the robustness of these markers in an independent dataset from GSE3167 (9) consisting of 5 CIS and 55 other tumor samples, we found expression information for 36 out of 46 genes. Principal-component analysis (PCA) plot shows samples colored by their type based on these genes (Figure S3B). Average expression of these 36 genes was used as input for area under curve (AUC) calculation. Five CIS samples were treated as cases and the 55 other samples as control. The 36-gene signature was able to accurately predict the CIS samples by receiver operating characteristic (ROC) analysis (AUC = 0.90; Figure 3B). At individual gene levels, we validated selective overexpression of MTOR, GUSBP11, KMT2D, and URB1 and under-expression of HMGB1, RPS7, NDUFB4, BRD2, HNRNPK, CANX, and HSP90B1 in CIS samples (Figure 3C).
Figure 3.

CIS gene signature in CIS and papillary tumors with validation at individual gene level
(A) CIS-specific gene signature composed of 46 genes (up- and downregulated) were identified. Unsupervised clustering shows clear segregation of CIS samples from other samples.
(B) AUC curve showing performance of CIS-specific gene signature average score in predicting CIS samples from an independent cohort (GSE3167).
(C) volcano plot showing fold change and p value for individual gene validation from the signature in GSE3167 dataset. Genes passing both Log2 fold change cutoff of 0.5 and Wilcoxon rank-sum test p value cutoff of 0.05 are shown as red dots.
Cohort B
Mutational analysis of CIS lesions and intrapatient similarity
In total, DNA from 34 CIS lesions and 33 tumor samples was analyzed using WES. CIS lesions were sequenced to a mean coverage of 142X. Mutations in CIS lesions were observed at significantly lower-variant allele frequencies (VAFs) compared to mutations observed in other tumor samples (p < 0.0001; mean = 0.08 vs. 0.2; Figure S4A). The majority of single-nucleotide variants observed in the samples were C>T alterations (Figure S4B) with increasing relative contribution with increasing VAFs (Figure S4C). In papillary tumors, mutation VAFs were found to vary between alteration types, being highest for C>G and C>T mutations. In CIS samples, VAFs were lower and more uniform, potentially due to low carcinoma cell content (Figure S4D). The read orientation phred-scaled quality (ROQ) score describes the possibility of read orientation artifacts. Artifacts related to FFPE sampling processes will likely have a strand or read orientation bias toward one of the directions, whereas true mutations should have an equal distribution of forward and reverse strands.17 We filtered out mutations with ROQ scores below or equal to 30 to remove potential FFPE artifacts. Of the remaining mutations, C>T alterations had the lowest ROQ scores in CIS samples, compatible with the fact that this mutation type is often introduced during formalin fixation of the tissue (Figure S4E). In papillary tumors, C>A mutations had the lowest ROQ scores. C>A mutations have been associated with the COSMIC single-base signature (SBS) 4, which is associated with tobacco smoking. Double-base signature (DBS) 2 has been shown to be associated with SBS4 and showed transcriptional strand bias, potentially explaining the low ROQ scores for these mutations.18 The number of mutations in the samples varied but was generally high (Figure S4F) with median TMBs for CIS and papillary of 44 mutations/Mb and nine mutations/Mb, respectively.
Because of the relatively small cohort size, we investigated mutations in genes frequently mutated in high-risk NMIBC patients11 (Figure 4). Generally, when analyzing mutations within patients, we observed that the same alteration types were present in the same genes across multiple tumor samples from different time points and tumor types, indicating clonality. Additionally, many alterations were detected both before and after BCG treatment. KDM6A was the most frequently mutated gene across CIS and papillary tumor samples. When analyzing CIS samples alone, CCDC138 was the most frequently mutated gene (Figure S5). We did not observe any significant differences when analyzing the mutational landscape of CIS and papillary tumors based on BCG outcome, progression status, or sampling time in relation to BCG treatment.
Figure 4.

Mutational landscape of CIS and papillary tumor samples (moderate and high impact SNVs and indels)
Columns correspond to unique samples. Groups of columns correspond to all analyzed samples from a single patient. Top: the number of the four mutation types in every sample. Middle: oncoplot showing the four mutation types in selected genes, sorted by mutation frequency across patients. Frequencies of patients with mutations in the given gene are indicated to the left. Bars to the right indicate the numbers of the different mutation types within each gene across the samples in the cohort. Bottom: clinical and sample information. BCG outcome: high-grade disease within two years after BCG treatment or progression to muscle-invasive bladder cancer (MIBC) progression to MIBC. BCG Timing: sample timing in relation to BCG treatment.
Immune contexture of CIS and papillary tumors
Using mIF and IHC, we investigated the immunological landscape of 24 CIS and papillary tumors from 15 patients estimating the levels of cytotoxic T cells (CTLs), T helper cells, Tregs, B cells, M1 and M2 macrophages, and cells expressing PD-1 and PD-L1 from cohort B. Tissue biopsies were separated into epithelial (normal urothelium, CIS, and papillary tumor) and stromal (normal stroma, CIS stroma, papillary tumor stroma, and deep stroma) regions of interest (ROIs) using digital pathology, and the immune cells within each were counted (Figure 5A). A detailed overview of sample types and ROIs within each is provided in Figure S6A. We did not observe any significant differences when analyzing the immune landscape of normal tissue, CIS, and papillary tumors, and more immune cells were observed in stromal regions compared to epithelial areas (Figure 5B) and significantly higher fractions of T helper cells, regulatory T cells (p = 0.0063), and B cells (p = 0.0039) in all stromal areas combined (Figure 5C). When analyzing all stromal regions separately, we observed that T helper cell and B cell fractions were high in all stromal regions, although not significant in most cases for B cells (Figure S6B). There were no significant differences between immune cell infiltration in CIS stroma and papillary stroma (Figure S6B). Additionally, there was a significantly larger fraction of PD-1-expressing cells in CIS regions and in stroma compared to papillary regions (p = 0.031 and p = 0.014; Figure 5C).
Figure 5.

Immunofluorescence and immunohistochemistry workflow and immune cell characterization
(A) Workflow of the mIF and IHC methods. Multiple visits from the same patients are marked with different letters, and numbers following letters mark samples collected from the same clinical visit. Sections stained with different mIF panels or antibodies are shown in the order it was applied. Examples of analyzed regions of interest (ROIs) are shown.
(B) From top bar to bottom bar: percentage of immune cells in ROIs. Immune cell proportions in regions where all stainings were available. Heatmap indicating the levels of immune cells and markers in different ROIs/patients. Bottom annotation includes sample information with patient of origin, sample types, and ROIs as well as clinical information with prior BCG treatment, post-BCG outcome (CR: complete response at 6 months. Failure: any persistent high-grade cancer), and progression status (T-stage progression).
(C) Comparisons of the levels of immune cell types and markers in different ROIs. All stromal areas have been combined to one ROI. Adjusted p values are indicated when below-significance level of 0.05 (Wilcoxon rank-sum test).
Discussion
Whole transcriptomic profiling of bladder cancer with RNA sequencing is widely available, and protocols are well established for doing this with larger FFPE tumors. CIS is a flat, intraepithelial high-grade cancer with histologic features similar to invasive cancer and is often a precursor to invasive bladder cancer. It has both a variable morphology and treated natural history, may be focal or diffuse, and may occur as isolated CIS or associated with papillary non-invasive cancer or muscle-invasive cancer. To understand the variable biology and behavior of CIS, molecular subtyping has been performed using targeted gene panels, and two broad subtypes, luminal and basal, have been identified.19 A transition from luminal to basal subtype may occur in the context of progression to MIBC;15 however, subtype calls may also be determined by the overall tissue contexture associated with CIS and MIBC. CIS presents a significant challenge for standard whole-transcriptome RNA sequencing due to very small tumor sizes, an inability to reliably detect it with white light cystoscopy, and often low RNA quality from FFPE-derived tissues. While enhanced cystoscopy with hexaminolevulonic acid and narrow-band imaging have improved our ability to detect CIS, we are still limited by sampling challenges to obtain fresh tissue to optimize RNA sequencing. In the present study, we determined that established dual nucleic acid extraction protocols for FFPE samples were not feasible due to small biopsy sample sizes and that protocols should be optimized for either RNA or DNA. We therefore curated a convenience cohort of patients with previously untreated FFPE CIS samples with a subset with matched papillary (Ta or T1) and normal tissues and tested the hypothesis that we could obtain RNA samples with adequate quality for whole transcriptome profiling to demonstrate proof of concept of feasibility, determine subtypes, and identify potential pathways associated with risk of progression. Furthermore, WES was performed in separate cohort of CIS samples from BCG-treated patients (BCG-naive and post-BCG CIS lesions).
We found that most CIS samples were luminal (TCGA) or Class 2a/2b (UROMOL) with increased expression of the luminal (FOXA1) marker and low expression of basal markers (CD44, KRT14), immune (CXCL11), and neuronal (APLP1, MS1) markers. Most high-grade papillary tumors were also Class 2a or 2b (85%), which is associated with the least favorable prognosis due to late cell-cycle activation with TP53-, ERBB2-, and APOBEC-related pathways.12 UROMOL Class 2B has immune-infiltrative features that indicate a potential poor response to BCG. Applying the TCGA classifier, 7/9 tumors of Class 2A tumors were of luminal subtypes, supporting previous findings of Class 2A being primarily luminal.12 A minority of CIS and papillary tumors were Basal or Class 3 which has a dormant luminal MIBC type with higher long noncoding RNA (lncRNA) associated with MIBC progression.12
We identified the Notch, Fanconi anemia, and lysine degradation signaling pathways to be significantly associated with CIS compared to adjacent papillary tumors. Notch activation has been described previously and affects cell-cycle regulation and stemness.20 The Fanconi anemia pathway is characterized by a defect in DNA cross-link repair, and mutations in FANC are associated with response to cisplatin-based chemotherapy.21 Lysine can be modified by acetylation, methylation, and ubiquination and can lead to aberrant dysregulated histone acetylation.22 At the individual gene level, we identified a 46-gene expression signature that differentiates CIS samples from papillary tumors and normal urothelium samples and validated most expression changes in an independent patient cohort. Because of small sample size and lack of uniform treatment of the cases, we were unable to observe any association with BCG response.
However, this signature includes increased expression of Mammalian target of rapamycin (MTOR), TYK2, and GAK kinases which may be potential actionable targets for CIS treatment. Although the work has no immediate implications for therapy, it could inform the design of tests to track treatment response for targeted therapy including clinical trials focused on Rapamycin (MTOR inhibitor) including but not limited to NCT04375813.
In a separate cohort of synchronous and metachronous matched CIS and papillary tumors, we focused on DNA-based genomic profiling and described the mutation and immune profiles of CIS and paired papillary tumors. TMB was higher in CIS compared to papillary tumors supporting the preference for immunotherapy with BCG as the primary treatment for CIS. KDM6A was the most frequently mutated gene across CIS and papillary tumors, and CCDC138 was the most frequently mutated gene across CIS alone. KDM6A, a lysine-specific demethylase, is a tumor suppressor that alters chromosomal stability, decreases cell growth,3 and influences natural killer cell signaling and cisplatin sensitivity.23 Previous studies identified KDM6A mutations as a prognostic factor for immune tumor escape in bladder cancer24 and found more KDM6A mutations in females with non-invasive cancer.25 The role of the CCDC138 gene product is not yet fully understood, but it is known to interact with ubiquitin C, a molecule involved in ubiquitination and degradation of proteins.26 High TMB has been associated with response to both chemotherapy and immunotherapy in patients with muscle-invasive or metastatic disease.
Our analysis of paired CIS and papillary tumors using mIF and immunohistochemistry showed a higher immune cell density—including T helper cells, T reg, and B cell fractions—in stromal areas compared to CIS and papillary tumors. Moreover, when CIS regions were compared with papillary tumor, T cells were the only type of immune cells detected in significant numbers. We also identified a larger fraction of PD-1-expressing cells in CIS and surrounding stroma compared to papillary tumors providing a biologic explanation for the utility of checkpoint inhibition targeting CIS as shown in early studies of BCG-treated patients.27 This may represent a high level of PD-1-positive T cells in and around CIS lesions, likely due to the high-grade features of CIS lesions and genomic alterations, potentially leading to immune cell exhaustion. However, this needs to be analyzed in larger studies.
Conclusion
This study met its primary objective of proof of concept for whole transcriptomic profiling of CIS and genomic profiling of matched CIS and papillary tumors and determination of spatial orientation of immune cells associated with these cancers. There are several potential limitations including curated and retrospective cohorts, modest sample sizes, and FFPE specimens failing RNA quality control. This research confirms that CIS is largely luminal and of the UROMOL class 2a/2b and suggests that CIS and papillary tumors may share a similar evolutionary pathway with common genomic alterations and actionable targets. The large fraction of PD-1-expressing cells in CIS and in stroma supports the rationale for use of drugs targeting checkpoint inhibition for treatment of CIS. Validation in larger cohorts and ongoing clinical trials will further our understanding of the biology of CIS toward the goal of identifying predictive and prognostic biomarkers.
Limitations of the study
Overall, though we have validated CIS-specific 46-gene signature findings in an independent patient cohort, small sample size in such an analysis is an inherent issue. Hence, limitations of the study include small cohort sizes and lack of overlay between genomics and transcriptomic analyses. The findings from the study warrant further validation in larger cohorts. Due to small cohort sizes, it was not possible to correlate results to sex, gender, and age.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Cohort A | This paper | See STAR methods: Cohort description and analysis overview |
| Cohort B | This paper and Strandgaard et al.11 | See STAR methods: Cohort description and analysis overview |
| Chemicals, peptides, and recombinant proteins | ||
| PD-L1 monoclonal antibody (Rabbit) | Ventana | Clone: Sp263 |
| PD-1 monoclonal antibody (Mouse) | Ventana | Clone: NAT105 |
| Pan-cytokeratin monoclonal antibody (Mouse) | Dako | Clone: AE1/AE3 |
| CD3 monoclonal antibody (Rabbit) | Ventana | Clone: 2GV6 |
| CD8 monoclonal antibody (Mouse) | Dako | Clone: C8/144B |
| FOXP3 monoclonal antibody (Mouse) | Thermo Fisher | Clone: SP97 |
| CD68 monoclonal antibody (Mouse) | Dako | Clone: PG-M1 |
| CD163 monoclonal antibody (Mouse) | Ventana | Clone: MRQ-26 |
| CD20 monoclonal antibody (Mouse) | Ventana | Clone: L26 |
| Critical commercial assays | ||
| GeneRead FFPE Kit | Qiagen | Cat# 180134 |
| Twist Library Preparation | Twist Bioscience | Cat# 101058 |
| Twist Human Core Exome capture kit | Twist Bioscience | Cat# 100795 |
| Twist Human RefSeq Panel | Twist Bioscience | Cat# 101023 |
| Gentra Puregene Tissue Kit (Proteinase K, RNase A Solution, Cell Lysis Solution, Protein Precipitation Solution, ATE buffer) | Qiagen | Cat# 19131, 158922, 158906, 1045701, 1067929, respectively |
| Qiasymphony DSP DNA midi kit | Qiagen | Cat# 937255 |
| Deposited data | ||
| RNAseq data | This paper | Table S1 |
| Whole Exome Sequencing Data | This paper and a subset from Strandgaard et al.11 | Processed and non-sensitive data provided upon request |
| Immune contexture | This paper | Processed and non-sensitive data provided upon request |
| Software and algorithms | ||
| STAR (2.7.1a) | Dobin et al.28 | https://github.com/alexdobin/STAR |
| RSEM (v 1.3.1) | Li and Dewey29 | https://github.com/deweylab/RSEM |
| WebGestalt | Liao et al.30 | http://www.webgestalt.org |
| MSigDB | Subramanian, Tamayo et al.31 | https://www.gsea-msigdb.org/gsea/msigdb |
| R v4.1.2 | R Core Team 2020 | https://www.r-project.org |
| Visiopharm (Version 2018.9.5.59.52) | Visiopharm A/S, Hørsolm, Denmark | |
| PurBayes | Plummer et al.32 | https://cran.r-project.org/web/packages/PurBayes/index.html |
| bwa mem v.0.7.17. | Li33 (https://arxiv.org/abs/1303.3997) | https://github.com/lh3/bwa |
| MuTect2 | Benjamin et al.34 (doi: https://doi.org/10.1101/861054) | https://github.com/broadinstitute/gatk |
| SnpEffv4.3i | Cingolani et al.35 | http://pcingola.github.io/SnpEff/ |
Resource availability
Materials are not available. No unique reagents were generated for this project.
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Seth P. Lerner, MD (slerner@bcm.edu).
Data and code availability
-
•Data:
-
○Cohort A (Bulk RNA-seq cohort): The raw sequencing data is not available as majority of the patients were consented at Paris. Processed gene expression matrix and data that support the findings of this study are available from the corresponding authors upon reasonable request.
-
○Cohort B (WES and mIF/IHC cohort): The raw sequencing data are not publicly available as this compromise patient consent and ethics regulations in Denmark. Processed non-sensitive data are available upon reasonable request from the corresponding authors.
-
○
-
•Code:
-
○No custom code other than standard coding was used.
-
○
Study participant details
Ethics approval and consent to participate
The study was approved by the Institutional Review Board of the Baylor College of Medicine (H-14435, H-42629) and The Danish National Committees on Health Research Ethics (#1708266), and all study protocols were performed in accordance with the approved guidelines and regulations. Relevant informed consent was obtained from all subjects.
Cohort description and analysis overview
Cohort A (bulk RNA-seq cohort)
For the study of mRNA, 24 patients with newly diagnosed NMIBC were enrolled. Their tissues were obtained from either transurethral resection or cystoscopic biopsies of bladder tumor under general anesthesia. Three matched samples of bladder including normal mucosa, papillary tumor, and CIS lesions were obtained in each patient. Relevant histologic sections were re-reviewed by a genitourinary pathologist for histologic confirmation and identification of the area(s) of interest. A total of 32 samples from 24 patients were selected to enter the analysis. All were adults and 22 were male; 6 were female; sex was not recorded in 4.
Cohort B (WES and mIF/IHC cohort)
For WES, a separate cohort of 19 patients (13 males and six females) with matched CIS and papillary tumor samples was analyzed (nCIS = 34 and npapillary = 33). Using mIF/IHC analyses, 24 samples from 15 patients (8 of these overlapping with WES-analyzed patients) were analyzed for the presence of cytotoxic T-cells (CTLs), T-helper cells, regulatory T-cells (Tregs), B-cells, M1 and M2 macrophages and PD-1 and PD-L1-expressing cells. All were adults and ten were male and five were female. Detailed long-term follow-up was available for cohort B.
Method details
RNA - Sequencing
Transcriptome data was generated for 32 formalin fixed paraffin embedded (FFPE) samples in this study. For this, strand-specific, poly-A+ RNA-seq libraries for sequencing on the Illumina platform similar to prior work described by Peters et al.36 Briefly, poly-A+ mRNA was extracted from 1 μg total RNA, followed by fragmentation and first strand cDNA synthesis. The resultant cDNA was end-repaired, A-tailed and ligated with Illumina Dual barcode adapters. Libraries were sequenced on NovaSeq 6000 instruments using the S4 reagent kit (300 cycles) to generate 2 × 150 bp paired-end reads. Between 59.96 and 112.62M total reads were generated for these 32 samples. The average strand-specificity and rRNA rate was 97.04% and 1.79% respectively.
mRNA expression and subtyping
The paired-end reads were mapped to the human genome version GRCh38.d1.vd1 (from GDC) using STAR-2.7.1a. The gene expression estimation was performed using RSEM-1.3.129 and the RSEM value was normalized using the gene-centering/upper quantile method. TCGA subtyping was performed using the R script provided by Dr Jaegil Kim upon request. The details of the methods have been earlier published.37 UROMOL12 and Consensus14 classification was also applied.
DNA extraction, WES, and data processing
CIS samples were obtained from FFPE specimens and DNA was extracted from 20 serial sections of 10 μm using GeneRead FFPE Kit (Qiagen) with an extra deparaffinization solution (320 μL). DNA from tumor was extracted from 20 to 25 serial cryosections of 20 μm using Gentra Puregene Tissue Kit. Leukocyte DNA from peripheral blood (for germline reference) was extracted using Qiasymphony DSP DNA midi kit (Qiagen).11 Carcinoma cell percentage was estimated for CIS and papillary tumors using PurBayes 17. CIS samples had a mean carcinoma cell percentage of 17% (range: 7%–45%; missing info for one sample). Papillary tumor samples had a mean carcinoma cell percentage of 72% (range: 8%–100%).
Libraries for WES and subsequent capture were prepared using Twist Enzymatic Fragmentation Library prep and Human Core Exome capture kit (Twist Bioscience). To lower error rates, UMI adapters were applied. CIS samples were sequenced to a median coverage of 138 X (Range: 19X-359X) and papillary tumor samples to a median of 153X (range: 72X-196X). Reads were mapped against the hg38 genome using bwa mem v.0.7.17.33 After mapping, consensus reads were generated from UMIs with at least three identical UMIs supporting each consensus mutation call. Mutations were called using MuTect2.34 All somatic alterations were annotated using SnpEffv4.3i35 and hg38 build. Samples were sequenced using Illumina platforms.
To avoid potential erroneous calls, only mutations with a frequency above 0.02, ROQ (read orientation phred-scaled quality) scores above 30 and mutations where the frequency of the alternate allele was more than four times the frequency of the reference allele were kept for further analysis.
Multiplex immunofluorescence and immunohistochemistry
CIS samples were analyzed for the presence and spatial distribution of immune cells and immune evasion markers similar to prior work from our lab in Taber et al.38,39 In short, two sections of 3 μm were stained from each tissue block with two different panels of mIF antibodies using a tyramide signal amplification strategy. The first antibody panel targeted T-cells (CD3, CD8 and FOXP3) and the second targeted B-cells and macrophages (CD20, CD68 and CD163). Following the mIF staining, the sections were stained with a chromogenic antibody targeting pan-cytokeratin to separate stromal areas from epithelial areas. Two additional sections were stained against PD-1 and PD-L1 using standard bright field immunohistochemistry and aligned to a final section stained against pan-cytokeratin.
Stained sections were scanned and images processed using the Visiopharm software version 2018.9.5.59.52 (Visiopharm A/S, Hørsolm, Denmark). Guided by the pan-cytokeratin staining, the tissues were separated into epithelial and stromal areas by the software. Hereafter, a manual subdivision of the areas into epithelial and stromal regions of interest (ROIs) was performed. ROIs annotated in the tissues included normal urothelium, CIS and papillary tumor as epithelial ROIs and stromal areas adjacent to the epithelial areas, normal stroma, CIS stroma and papillary stroma as well as deep stroma. Expert pathologists made histological evaluations of the sections before analysis and annotated the cell regions in the tissue. Due to technical issues with sample processing and staining, it was not possible to determine the level of all immune cells and evasion markers in all samples, and not all cell types were assessed in all samples.
Quantification and statistical analysis
Statistical analysis
Gene set enrichment analysis (GSEA) was performed using signed -log10 p value of differential expression of genes in high quality CIS samples as compared against the rest of the tumor samples using WebGestalt.40,41 For GSEA, we utilized Hallmark and Reactome geneset within the Molecular Signatures Database (MSigDB) to compare sample groups. A false discovery rate (FDR) of 0.05 was utilized to define the statistical significance of GSEA in this study.
For identification of a CIS specific gene signature, a combination of two statistical filtering criteria were applied. These included Wilcoxon rank-sum test (p < 0.05) comparing CIS and papillary tumors, followed by an ANOVA test comparing CIS, papillary Ta/T1 and normal samples (p < 0.05). This resulted in a set of 46 genes that were observed to be differentially expressed in CIS (including up and downregulated genes) compared to remaining tumor samples. The R was used for all the analyses.
Receiver operating characteristic (ROC) curve analysis was applied to the RNA sequencing data of tumor samples from the GSE3167 cohort.16 R package pROC was used to draw the ROC curves and calculate the area under curve (AUC). Only 36 out of 46 genes were found in the microarray expression set for this cohort and average expression of these 36 genes was used as input for AUC calculation. A higher AUC value means the gene can better distinguish between CIS samples (case) as compared to other bladder samples (control).
Acknowledgments
This study is supported by the Leo & Anne Albert Institute for Bladder Cancer Care and Research (“Molecular Characterization of Urothelial Carcinoma in situ [USCIS]”) grant # 53549-I.
The graphical abstract was created using BioRender.com.
Author contributions
S.P.L. and L.D. designed the study. M.A., T.S., and A.T. conducted the analysis. S.P.L., E.C., J.B.J., and L.D. were involved in sample acquisition. M.A.,T.S., Y.D., A.T., and I.N. were involved in sample handling and data generation and processing. M.A., S.P.L., T.S., A.T., L.D., H.A.-A., and B.A.I. were involved in the interpretation of the results. M.A., T.S., A.T., S.H.K., S.P.L., and L.D. drafted the manuscript. All authors reviewed and approved the final version of the manuscript.
Declaration of interests
L.D. has sponsored research agreements with C2i Genomics, Natera, AstraZeneca, Photocure, and Ferring and has an advisory/consulting role at Ferring, MSD, and UroGen. L.D. has received speaker honoraria from AstraZeneca, Pfizer, and Roche and received travel support from MSD. L.D. is a board member at BioXpedia.
S.P.L. has funding for clinical trials from Aura Bioscience, FKD, JBL (SWOG), Genentech (SWOG), Merck (Alliance), QED Therapeutics, Surge Therapeutics, Vaxiion, and Viventia; is a consultant/Advisory Board member for Aura Bioscience, BMS, Pfizer/EMD Serono, Protara, Surge Therapeutics, Vaxiion, and Verity; has a patent for the TCGA classifier; and received honoraria from Grand Rounds Urology and UroToday.
J.B.J. is a member of Advisory Boards at Ferring, Roche, Cepheid, Urotech, Olympus, AMBU, Janssen, and Cystotech; is a speaker at medac, Olympus, Intuitive Surgery, and Photocure ASA; and has research collaborations with Medac, Photocure ASA, Roche, Ferring, Olympus, Intuitive Surgery, Astellas, Cepheid, Nucleix, Urotech, Pfizer, AstraZenica, MeqNordic, Laborie, VingMed, AMBU, and Cystotech.
H.A.-A. provides consultation to AstaZeneca and Paige.AI.
B.I. runs clinical trials or scientific studies in collaboration with FKD Therapies, Taris Biomedical, CG Oncology, Genentech Inc, Janssen, Medtronic, and Profound Medical and has consultant or advisory roles at Seattle Genetics, Combat Medical, and Johnson & Johnson.
Published: February 9, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109179.
Contributor Information
Lars Dyrskjøt, Email: lars@clin.au.dk.
Seth P. Lerner, Email: slerner@bcm.edu.
Supplemental information
This table contains upper quartile normalized RSEM values median centered by gene.
References
- 1.von Rundstedt F.C., Lerner S.P. New imaging techniques for nonmuscle invasive bladder cancer. Curr. Opin. Urol. 2014;24:532–539. doi: 10.1097/MOU.0000000000000093. [DOI] [PubMed] [Google Scholar]
- 2.Lamm D.L., van der Meijden P.M., Morales A., Brosman S.A., Catalona W.J., Herr H.W., Soloway M.S., Steg A., Debruyne F.M. Incidence and treatment of complications of bacillus Calmette-Guerin intravesical therapy in superficial bladder cancer. J. Urol. 1992;147:596–600. doi: 10.1016/s0022-5347(17)37316-0. [DOI] [PubMed] [Google Scholar]
- 3.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2018;174:1033. doi: 10.1016/j.cell.2018.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenney J.K., Gomez J.A., Desai S., Lee M.W., Amin M.B. Morphologic expressions of urothelial carcinoma in situ: a detailed evaluation of its histologic patterns with emphasis on carcinoma in situ with microinvasion. Am. J. Surg. Pathol. 2001;25:356–362. doi: 10.1097/00000478-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Epstein J.I., Amin M.B., Reuter V.R., Mostofi F.K. The World Health Organization/International Society of Urological Pathology consensus classification of urothelial (transitional cell) neoplasms of the urinary bladder. Bladder Consensus Conference Committee. Am. J. Surg. Pathol. 1998;22:1435–1448. doi: 10.1097/00000478-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Netto G.J., Amin M.B., Berney D.M., Compérat E.M., Gill A.J., Hartmann A., Menon S., Raspollini M.R., Rubin M.A., Srigley J.R., et al. The 2022 World Health Organization Classification of Tumors of the Urinary System and Male Genital Organs-Part B: Prostate and Urinary Tract Tumors. Eur. Urol. 2022;82:469–482. doi: 10.1016/j.eururo.2022.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Sylvester R.J., van der Meijden A., Witjes J.A., Jakse G., Nonomura N., Cheng C., Torres A., Watson R., Kurth K.H. High-grade Ta urothelial carcinoma and carcinoma in situ of the bladder. Urology. 2005;66:90–107. doi: 10.1016/j.urology.2005.06.135. [DOI] [PubMed] [Google Scholar]
- 8.Lamm D.L., Blumenstein B.A., Crissman J.D., Montie J.E., Gottesman J.E., Lowe B.A., Sarosdy M.F., Bohl R.D., Grossman H.B., Beck T.M., et al. Maintenance bacillus Calmette-Guerin immunotherapy for recurrent TA, T1 and carcinoma in situ transitional cell carcinoma of the bladder: a randomized Southwest Oncology Group Study. J. Urol. 2000;163:1124–1129. [PubMed] [Google Scholar]
- 9.Oddens J., Brausi M., Sylvester R., Bono A., van de Beek C., van Andel G., Gontero P., Hoeltl W., Turkeri L., Marreaud S., et al. Final results of an EORTC-GU cancers group randomized study of maintenance bacillus Calmette-Guerin in intermediate- and high-risk Ta, T1 papillary carcinoma of the urinary bladder: one-third dose versus full dose and 1 year versus 3 years of maintenance. Eur. Urol. 2013;63:462–472. doi: 10.1016/j.eururo.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 10.Chade D.C., Shariat S.F., Godoy G., Savage C.J., Cronin A.M., Bochner B.H., Donat S.M., Herr H.W., Dalbagni G. Clinical outcomes of primary bladder carcinoma in situ in a contemporary series. J. Urol. 2010;184:74–80. doi: 10.1016/j.juro.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strandgaard T., Lindskrog S.V., Nordentoft I., Christensen E., Birkenkamp-Demtröder K., Andreasen T.G., Lamy P., Kjær A., Ranti D., Wang Y.A., et al. Elevated T-cell Exhaustion and Urinary Tumor DNA Levels Are Associated with Bacillus Calmette-Guerin Failure in Patients with Non-muscle-invasive Bladder Cancer. Eur. Urol. 2022;82:646–656. doi: 10.1016/j.eururo.2022.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Lindskrog S.V., Prip F., Lamy P., Taber A., Groeneveld C.S., Birkenkamp-Demtröder K., Jensen J.B., Strandgaard T., Nordentoft I., Christensen E., et al. An integrated multi-omics analysis identifies prognostic molecular subtypes of non-muscle-invasive bladder cancer. Nat. Commun. 2021;12:2301. doi: 10.1038/s41467-021-22465-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171:540–556.e25. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamoun A., de Reyniès A., Allory Y., Sjödahl G., Robertson A.G., Seiler R., Hoadley K.A., Groeneveld C.S., Al-Ahmadie H., Choi W., et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020;77:420–433. doi: 10.1016/j.eururo.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hedegaard J., Lamy P., Nordentoft I., Algaba F., Høyer S., Ulhøi B.P., Vang S., Reinert T., Hermann G.G., Mogensen K., et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell. 2016;30:27–42. doi: 10.1016/j.ccell.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Dyrskjøt L., Kruhøffer M., Thykjaer T., Marcussen N., Jensen J.L., Møller K., Ørntoft T.F. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–4048. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- 17.Diossy M., Sztupinszki Z., Krzystanek M., Borcsok J., Eklund A.C., Csabai I., Pedersen A.G., Szallasi Z. Strand Orientation Bias Detector to determine the probability of FFPE sequencing artifacts. Brief. Bioinform. 2021;22 doi: 10.1093/bib/bbab186. [DOI] [PubMed] [Google Scholar]
- 18.Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akhtar M., Al-Bozom I.A., Ben Gashir M., Taha N.M., Rashid S., Al-Nabet A.D.M.H. Urothelial Carcinoma In Situ (CIS): New Insights. Adv. Anat. Pathol. 2019;26:313–319. doi: 10.1097/PAP.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C., Berndt-Paetz M., Neuhaus J. A Comprehensive Bioinformatics Analysis of Notch Pathways in Bladder Cancer. Cancers. 2021;13 doi: 10.3390/cancers13123089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plimack E.R., Dunbrack R.L., Brennan T.A., Andrake M.D., Zhou Y., Serebriiskii I.G., Slifker M., Alpaugh K., Dulaimi E., Palma N., et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer. Eur. Urol. 2015;68:959–967. doi: 10.1016/j.eururo.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L., Miao Y., Liu M., Zeng Y., Gao Z., Peng D., Hu B., Li X., Zheng Y., Xue Y., et al. Pan-Cancer Analysis Reveals the Functional Importance of Protein Lysine Modification in Cancer Development. Front. Genet. 2018;9:254. doi: 10.3389/fgene.2018.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramakrishnan S., Granger V., Rak M., Hu Q., Attwood K., Aquila L., Krishnan N., Osiecki R., Azabdaftari G., Guru K., et al. Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death Differ. 2019;26:2100–2114. doi: 10.1038/s41418-019-0278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X., Lin X., Pang G., Deng J., Xie Q., Zhang Z. Significance of KDM6A mutation in bladder cancer immune escape. BMC Cancer. 2021;21:635. doi: 10.1186/s12885-021-08372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurst C.D., Alder O., Platt F.M., Droop A., Stead L.F., Burns J.E., Burghel G.J., Jain S., Klimczak L.J., Lindsay H., et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell. 2017;32:701–715.e7. doi: 10.1016/j.ccell.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drew K., Lee C., Huizar R.L., Tu F., Borgeson B., McWhite C.D., Ma Y., Wallingford J.B., Marcotte E.M. Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 2017;13:932. doi: 10.15252/msb.20167490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inman B.A., Sebo T.J., Frigola X., Dong H., Bergstralh E.J., Frank I., Fradet Y., Lacombe L., Kwon E.D. PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer. 2007;109:1499–1505. doi: 10.1002/cncr.22588. [DOI] [PubMed] [Google Scholar]
- 28.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plummer M. Penalized loss functions for Bayesian model comparison. Biostatistics. 2008;9:523–539. doi: 10.1093/biostatistics/kxm049. [DOI] [PubMed] [Google Scholar]
- 33.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013;4 doi: 10.48550/arXiv.1303.3997. Preprint at. [DOI] [Google Scholar]
- 34.Benjamin D., Sato T., Cibulskis K., Getz G., Stewart C., Lichtenstein L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv. 2019;4 doi: 10.1101/861054. Preprint at. [DOI] [Google Scholar]
- 35.Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters T.L., Kumar V., Polikepahad S., Lin F.Y., Sarabia S.F., Liang Y., Wang W.L., Lazar A.J., Doddapaneni H., Chao H., et al. BCOR-CCNB3 fusions are frequent in undifferentiated sarcomas of male children. Mod. Pathol. 2015;28:575–586. doi: 10.1038/modpathol.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J., Kwiatkowski D., McConkey D.J., Meeks J.J., Freeman S.S., Bellmunt J., Getz G., Lerner S.P. The Cancer Genome Atlas Expression Subtypes Stratify Response to Checkpoint Inhibition in Advanced Urothelial Cancer and Identify a Subset of Patients with High Survival Probability. Eur. Urol. 2019;75:961–964. doi: 10.1016/j.eururo.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 38.Taber A., Prip F., Lamy P., Agerbæk M., Jensen J.B., Steiniche T., Dyrskjøt L. Immune Contexture and Differentiation Features Predict Outcome in Bladder Cancer. Eur. Urol. Oncol. 2022;5:203–213. doi: 10.1016/j.euo.2022.01.008. [DOI] [PubMed] [Google Scholar]
- 39.Taber A., Christensen E., Lamy P., Nordentoft I., Prip F., Lindskrog S.V., Birkenkamp-Demtröder K., Okholm T.L.H., Knudsen M., Pedersen J.S., et al. Molecular correlates of cisplatin-based chemotherapy response in muscle invasive bladder cancer by integrated multi-omics analysis. Nat. Commun. 2020;11:4858. doi: 10.1038/s41467-020-18640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J., Duncan D., Shi Z., Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table contains upper quartile normalized RSEM values median centered by gene.
Data Availability Statement
-
•Data:
-
○Cohort A (Bulk RNA-seq cohort): The raw sequencing data is not available as majority of the patients were consented at Paris. Processed gene expression matrix and data that support the findings of this study are available from the corresponding authors upon reasonable request.
-
○Cohort B (WES and mIF/IHC cohort): The raw sequencing data are not publicly available as this compromise patient consent and ethics regulations in Denmark. Processed non-sensitive data are available upon reasonable request from the corresponding authors.
-
○
-
•Code:
-
○No custom code other than standard coding was used.
-
○