

Summary
In normal intestines, a fetal/regenerative/revival cell state can be induced upon inflammation. This plasticity in cell fate is also one of the current topics in human colorectal cancer (CRC). To dissect the underlying mechanisms, we generated human CRC organoids with naturally selected genetic mutation profiles and exposed them to two different conditions by modulating the extracellular matrix (ECM). Among tested mutation profiles, a fetal/regenerative/revival state was induced following YAP activation via a collagen type I-enriched microenvironment. Mechanistically, YAP transcription was promoted by activating AP-1 and TEAD-dependent transcription and suppressing intestinal lineage-determining transcription via mechanotransduction. The phenotypic conversion was also involved in chemoresistance, which could be potentially resolved by targeting the underlying YAP regulatory elements, a potential target of CRC treatment.
Subject areas: Classification Description, Cancer, Stem cell plasticity, Transcriptomics
Graphical abstract
Highlights
-
•
CRC shows a fetal/regenerative/revival state following YAP activation
-
•
YAP transcription is promoted by activation of AP-1 and TEAD via mechanotransduction
-
•
CRC cells in a fetal/regenerative/revival state shows chemoresistance
-
•
Chemoresistance can be resolved by targeting key modules such as TEAD and TNF in CRC
Classification Description: Cancer; Stem cell plasticity; Transcriptomics
Introduction
Cell plasticity can be displayed through a variety of processes such as epithelial-mesenchymal transition (EMT), dedifferentiation, and transdifferentiation.1,2 In colorectal cancer (CRC) biology, cell plasticity has been widely recognized to be involved in the generation of distant metastasis and acquisition of chemoresistance, which leads to a poor prognosis.2 For instance, in vivo settings, CRC cells in the invasive front undergo EMT.3,4 More recently, cell fate changes toward a fetal/regenerative/revival state are particularly focused on acquiring better understanding of the cell plasticity of CRC cells. These include a cell fate conversion between LGR5 negative and positive CRC cells5 or a presence of CRC cells in a fetal progenitor-like state.6 Importantly, CRC cells in a regenerative state have also been reported to show chemoresistance.7 However, the underlying mechanisms of cell fate conversion toward a fetal/regenerative/revival state in human CRC have not been fully elucidated yet. It has been reported that cell fate change is inhibited in cancer cells that harbor specific mutations,8 raising questions about the relationship between cancer mutation profiles and cell plasticity.
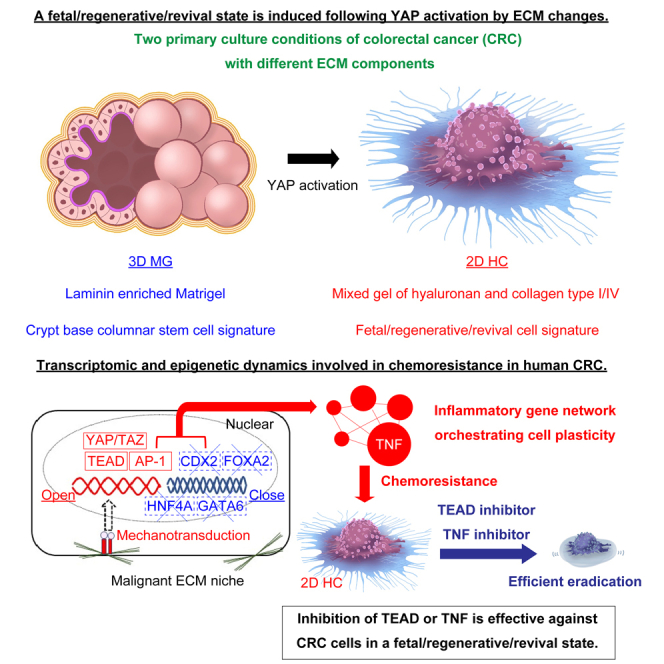
We and others have previously reported that cell fate conversion toward a fetal/regenerative/revival state is induced in the normal intestinal epithelium 9,10,11 upon inflammation and tissue regeneration. The process is orchestrated, at least partially, by collagen type I deposition in the mesenchyme following YAP/TAZ activation during tissue inflammation.9,12 Accordingly, the organoid culture system of normal colonic epithelium in purified collagen type I13 faithfully recapitulates the process.9,12 This enables the establishment of cystic organoids in a fetal/regenerative/revival state driven by the cooperative activation of several transcriptional factors such as YAP/TAZ-TEAD, AP-1, and RUNX2.12
In the current study, we hypothesize that the ECM niche plays a crucial role in understanding cell plasticity in human CRC. To test this hypothesis, we exposed human CRC organoids with naturally selected genetic mutation profiles to two conditions with different ECM components. Primary cancer cells cultured on a gel predominantly composed of collagen type I showed a transcriptomic signature closer to a fetal/regenerative/revival state with activated YAP transcription, at least within tested genetic mutation constellations. Mechanistically, YAP transcription was enhanced through the activation of AP-1 and TEAD-dependent transcription, coupled with the suppression of intestinal lineage-determining transcription factors such as CDX2, HNF4A, GATA6, and FOXA2.14 Interestingly, specific genomic mutations, such as RAS mutations, seemed to be associated with the enhancement of the YAP activation.
These results suggest that CRC cells with genetic mutations maintain their epithelial pattern in response to the surrounding ECM niche. This, in turn, forms the basis for a cancer-specific reaction with a loss of colonic epithelial determinants and gain of a fetal/regenerative/revival signature. The present study also confirmed that an induced fetal/regenerative/revival state is correlated with chemoresistance to typical cytotoxic anticancer agents in CRC cells. However, CRC cells in an induced fetal/regenerative/revival state showed a clear sensitivity to the inhibition of TNF, which was identified as the largest hub gene in the gene network of these cells. Effective suppression of these cells was also achieved through the inhibition of crucial modules in YAP transcription by using a TEAD inhibitor. By using two in vitro models with different ECM components, it would be possible to set up a novel platform to formulate a unique approach to CRC research.
Results
Identifying conditions that induce a fetal/regenerative/revival state in established colorectal cancer cells from primary human tumors
Invasion of CRC is known to be characterized by a transient loss of differentiation.3,15,16 A histopathological analysis of CRC surgical specimens validated the heterogeneous distribution of tumor cells in terms of their degree of differentiation (Figure 1A). Differentiated and polarized tumor glands were observed in the tumoral center. In contrast, poorly differentiated tumor cells were observed occasionally in the invasive front (Figure 1B). Subsequently, we analyzed the distribution of major ECM components in human CRC tissues to identify conditions that induce a fetal/regenerative/revival state in human CRC. Among various types of collagen, which is one of the most prevalent ECM scaffolding proteins in the mesenchyme,17 collagen type I is known to be especially expressed in the mesenchyme with an increase in the invasive front of CRC, where poorly differentiated cancer cells are present.3 IHC analysis of CRC surgical specimens confirmed that collagen type I expression was higher in the invasive front than in the central differentiated area, particularly with an abundant fibril formation in the mesenchyme (Figure 1B). It was also revealed that hyaluronan was expressed with a marked increase in the mesenchymal compartment of the invasive front compared to the tumoral center (Figure 1B). As for a component of basement membranes, laminin surrounded differentiated cancer cells in the tumoral center. Notably, its expression was lost in the invasive front (Figure 1B), as previously reported.3 The distribution of collagen type IV was slightly upregulated in the mesenchymal compartment of the invasive front compared to the tumoral center (Figure 1B).
Figure 1.

Identifying conditions that induce a fetal/regenerative/revival state in established colorectal cancer cells from primary human tumors
(A) A tiled image of a colorectal cancer surgical specimen is shown. Scale bar, 500 μm. Regions indicated with squares are enlarged in (B) for both the tumoral center and invasive front.
(B) Differentiated and polarized tumor glands are observed in the tumoral center (top), whereas de-differentiated tumor cells are observed in the invasive front (bottom) after H.E. staining. Immunohistochemical images of collagen type I, hyaluronan, laminin, and collagen type IV (all green) are shown in the tumoral center (top) and invasive front (bottom). Images are counterstained with E-cadherin (red). For collagen type I and type IV, magnified images of the regions indicated with squares are shown in adjacent panels. Scale bars, 100 μm. Col type I; collagen type I, Col type IV; collagen type IV, E-cad; E-cadherin.
(C) A diagram of different culture methods is presented. Matrigel cancer organoid (3D MG) is cultured in laminin-enriched Matrigel under L-WRN conditioned medium (upper panel). 2D HC is cultured on the mixed gel composed of collagen type I, collagen type IV, and hyaluronan under a serum-free ENRW medium (lower panel).
(D) Both 3D MG (upper panel) and 2D HC (lower panel) show a propagation over time. Scale bars, 200 μm (lower panel) and 100 μm (lower panel).
(E) Phalloidin staining (gray) reveals that the apical F-actin belt is well formed in 3D MG (upper panel), whereas a massive formation of actin stress fiber is observed in 2D HC (lower panel). Images are counterstained with DAPI (blue). Scale bars, 50 μm.
(F) The mutational fingerprint of the nine CRC surgical specimens used for primary culture for the most common alteration, as determined by next-generation sequencing assay employing the Illumina AmpliSeq Cancer HotSpot Panel v2, is displayed. The color code indicates the type of genetic alteration, distinguishing between missense mutation, nonsense mutation, frameshift mutation, and splice mutation. See also Figure S1, and Videos S1, S2, S3, S4, and S5.
We then explored whether combining collagen type I, hyaluronan, and collagen type IV allows CRC cells to expand in vitro on the synthetic gel (HC gel). Primary cancer cells showed a certain propagation as monolayer sheets, referred to as 2D HC, while standard budding CRC organoids were formed in laminin-enriched Matrigel (3D Matrigel; 3D MG) (Figures 1C and 1D). The composition ratio of collagen type I, hyaluronan, and collagen type IV was 70%v/v, 15%v/v, and 15%v/v respectively. Notably, the HC gel did not allow for the 3D expansion of CRC cells (Figure S1A).
Ki-67 positive CRC cells were found in 2D HC, indicating that the proliferation of CRC cells was rigorously maintained in vitro (Figure S1B). Time-lapse imaging showed active mitotic cell divisions in the central part of 2D HC (Video S1 and S2). Upon detailed inspection, multipolar cell division reflecting chromosomal instability18 (Video S3) or a lamellipodia-like structure19 was well observed in 2D HC (Video S4). In addition, the single-cell amoeboid-like cell displaying a rounded morphology20 was actively moving on the gel (Video S5). Considering that these motilities are accompanied by the reorganization of the actin cytoskeleton,21 we investigated the intracellular actin distribution. In 3D MG, the apical F-actin belt was well formed, indicating that cellular polarity was maintained (Figure 1E). In contrast, 2D HC showed a massive formation of actin stress fibers (Figure 1E).
Intriguingly, 2D HC converted efficiently into 3D MG with budding formation when transferred to Matrigel. Conversely, 3D MG could be passaged as 2D HC when seeded on the HC gel (Figure S1C). The morphological reversibility observed between 3D MG and 2D HC suggests that cancer cell phenotype could flexibly shift in an environment-dependent manner, rather than different culture conditions leading to the selection of a specific subset of CRC cells.
In the current study, the organoids were established from nine cases of left-sided differentiated CRC, including eight cases of rectal CRC (sample from #1–#8) and one case of descending CRC (sample #9) (Table 1). The mutational fingerprint of these CRC surgical specimens used for primary culture was evaluated using a next-generation sequencing assay, employing the Illumina AmpliSeq Cancer HotSpot Panel v2. Regarding major significantly mutated genes in non-hypermutated CRC,22 the positivity for APC, TP53, KRAS, PIK3CA, and FBXW7 mutation was observed in 77.8% (7/9 cases), 88.9% (8/9 cases), 33.3% (3/9 cases), 11.1% (1/9 cases), and 22.2% (2/9 cases), respectively. These percentages suggest that the case selection process did not introduce any obvious bias (Figure 1G).
Table 1.
Clinical information of colorectal cancer (CRC) surgical specimens for the establishment of 3D MG and 2D HC
| Cases | Gender | Location | Pathological Diagnosis | TNM classification | Pathological stage |
|---|---|---|---|---|---|
| #1 | Male | Rectum | tub1>tub2>por | pT3N0M0 | ⅡA |
| #2 | Male | Rectum | tub1>tub2>por | pT2N0M0 | Ⅰ |
| #3 | Male | Rectum | tub2>tub1>por | pT2N0M0 | Ⅰ |
| #4 | Male | Rectum | tub1>tub2 | pT3N0M0 | ⅡA |
| #5 | Male | Rectum | tub1>tub2 | pT2N0M0 | Ⅰ |
| #6 | Male | Rectum | tub2>tub1 | pT3N0M0 | ⅡA |
| #7 | Female | Rectum | tub1>tub2>por | pT2N0M0 | ⅡA |
| #8 | Female | Rectum | tub1>tub2 | pT4aN2a.m.1a | IVA (Liver) |
| #9 | Male | Descending | tub2>por>tub1 | pT3N0M0 | ⅡA |
CRC staging in the current study is performed using the TNM classification system based on guidelines of the 8th edition of the Union for International Cancer Control (UICC).
tub1, well differentiated tubular adenocarcinoma; tub2, moderately differentiated tubular adenocarcinoma; por, poorly differentiated adenocarcinoma.
RNA sequencing revealed the acquisition of fetal/regenerative/revival signature in 2D HC
To compare the transcriptional profile of 2D HC and 3D MG established from nine CRC surgical specimens mentioned above, the original 3D MG lines and their derived 2D HCs were subjected to bulk RNA sequencing analysis. Differentially expressed genes (DEG) (log2FC ≥ 1, p value < 0.05) were selected following DESeq2 analysis (Table S1). A total of 865 genes were identified as upregulated in 2D HC (2D HC gene set) and 501 genes were upregulated in 3D MG (3D MG gene set). In the GO TERM (molecular function) analysis, signaling receptor activator activity, cytokine activity, extracellular matrix (ECM) structural constituent, and heparin binding were related to the 2D HC gene set (Figure 2A). In the KEGG enrichment analysis, mechanotransduction-related pathways,23 such as ECM-receptor interaction, focal adhesion, and PI3K-Akt signaling pathway, and inflammatory-related pathways, such as cytokine-cytokine receptor interaction and TNF signaling pathway, were related to the 2D HC gene set (Figure 2B). The gene set enrichment analysis (GSEA) of the 2D HC gene set revealed significant enrichment in hallmarks such as “TNFA-SIGNALING-VIA-NFKB” (normalized enrichment score (NES) = 2.98, the false discovery rate (FDR) < 0.01), “EPITHELIAL-MESENCHYMAL-TRANSITION” (NES = 2.83, FDR<0.01), “INTERFERON-GAMMA-RESPONSE” (NSE = 2.61, FDR<0.01), “INTERFERON-ALPHA-RESPONSE” (NES = 2.48, FDR<0.01), “INFLAMMATORY-RESPONSE” (NES = 2.40, FDR<0.01) (Figure 2C). Moreover, GSEA with a reported fetal signature24 showed a highly significant enrichment in 2D HC (NES = 2.09, FDR<0.01) (Figure 2C). Furthermore, GSEA with a reported tumor budding signature, which consists of upregulated genes in tumor budding cells compared to those of the tumoral center16 showed a highly significant enrichment in 2D HC (NES = 1.89, FDR<0.01) (Figure 2C). In short, 2D HC demonstrated significant enrichment in inflammation-related genes, epithelial mesenchymal transition, and fetal signature. These phenotypes, indicative of cellular plasticity, suggest that the ECM-mediated mechanotransduction could be one of the drivers of cell fate conversion in human CRC.
Figure 2.

RNA sequencing analysis revealed the acquisition of a fetal/regenerative/revival signature in 2D HC
(A) The top 10 GO-TERM (molecular function) of the upregulated gene set in 2D HC compared to 3D MG (2D HC gene set) is shown.
(B) The top 20 KEGG pathways enriched in the 2D HC gene set are shown.
(C) The GSEA of hallmark gene sets, fetal gene signature, and tumor budding signature enriched in 2D HC gene set are shown. NES; normalized enrichment score, FDR; the false discovery rate.
(D) The heatmap displays Z score-transformed relative expression levels of genes for crypt base columnar (CBC) signature (LGR5, SMOC2, OLFM4, LRIG1 and EPHB3), regenerative stem cell (RSC) signature (TNF, TNFAPI3, CXCL2, ITGA2, RHOD, ICAM1, PLAU and PLAUR), and representative upregulated genes in normal COL sphere (TACSTD2, ANXA1, LY6D and CYR61) in nine cultures of 3D MG and 2D HC. COL; collagen.
(E) A volcano plot of differentially expressed genes included in 3D MG and 2D HC gene sets is indicated. Representative genes are indicated in a plot. See also Table S1.
According to a previous report,7 the heterogeneous cell population in CRC tissues was broadly classified into the Lgr5 positive crypt base columnar stem cell signature (CBC signature)25 and the Lgr5 negative regenerative stem cell signature (RSC signature).9,26 Although there were some variations in each sample, the expression of CBC signature genes such as LGR5, SMOC2, OLFM4, LRIG1, EPHB3, and IRS1 were generally upregulated in 3D MG. In contrast, the expression of RSC signature genes such as TNF, TNFAIP3, CXCL2, ITGA2, RHOD, ICAM1, PLAU, and PLAUR were generally upregulated in 2D HC (Figure 2D). Representative genes enriched in the collagen sphere of normal intestines, such as TACSTD2, ANXA1, LY6D, and CYR61,9 were also generally upregulated in 2D HC (Figure 2D). The results suggest that the phenotypic differences between 3D MG and 2D HC reflect the two significant characteristics of CRC tissues, and such phenotypical shifts are induced in the ECM niche-dependent manner. Considering that both SMOC2 and OLFM4 expression decreases in CRC cells at the invasive front,27,28 the differences in transcriptional profiles between 3D MG and 2D HC may reflect the heterogeneity caused by the spatial distribution of ECM components.
The 2D HC gene set includes ECM-related genes such as COL1A1, COL4A1, COL4A2, COL6A1, and FN1; collagen receptor genes such as ITGA2; mechanosensor-relative genes such as CD31; fetal marker genes such as TACSTD2, ANXA1, LY6D, and CYR61, which are enriched in the collagen sphere9; cytoskeleton-related genes such as KRT80, AHNAK2, and AKAP12; ECM degradation-related genes such as PLAU, PLAUR, SERPINE1, and TIMP2; mucin secretion-related genes such as MUC2, MUC16, and TFF2; TNF signaling pathway-related genes such as TNF, CCL2, CXCL2, TNFAIP3, ICAM1, IL1B, CSF2, and MMP14; and members of the AP-1 transcriptional factor family such as BATF, ATF3, and MAFK (Figure 2E).
The expression of some 2D HC markers in the invasive front of CRC tissues
To verify the biological significance of upregulated genes in 2D HC through the histopathological assessment of CRC tissues, we focused on the overlapping gene signature between the tumor budding gene set, as the tumor budding signature is based on the histological selection of the cell population.16 Among the 29 genes highlighted (Figure S2A), we identified several cytoskeleton-related genes. Notably, KRT80, a key component of cytoplasmic intermediate filaments,29 AHNAK2, linked to actin-dependent cytoskeletal formation,30,31 and AKAP12, a critical regulator of actin cytoskeleton reorganization32 were discerned. We further validated the expression of these three molecules at the protein level through an IHC analysis. Consistent with the gene expression data (Figure 3A), upregulated protein expression of these molecules in 2D HC was well verified in biological triplicates (#1–3) (Figures 3B and 3C). Importantly, an IHC analysis of the three corresponding CRC surgical specimens in sample #1–3 also revealed the higher expression of these cytoskeleton-related molecules in the poorly differentiated cancer cells in the invasive front compared to the differentiated cancer cells in the tumoral center, as shown in the sequential sections including H&E staining (Figures 3D–3F). These results imply that the upregulation of these three molecules in both 2D HC and the tumor invasive front might reflect some commonness between them, although the analyses are too limited to reach a definitive conclusion. Moreover, within the 29 overlapping genes, there might be additional candidate genes warranting further exploration.
Figure 3.

The expression of some 2D HC markers in the invasive front of CRC tissues
(A) A heatmap of Z score-transformed relative expression levels of KRT80, AHNAK2, and AKAP12, normalized by RLE in 9 cultures of 3D MG and 2D HC is shown.
(B) Immunohistochemical images of cytoskeleton-related proteins: KRT80, AHNAK2, and AKAP12 (red) of 3D MG (upper panel) and 2D HC (lower panel) are shown. In MG panels, the region of organoids is indicated with a dashed white line in the left panel. In the right panels, merged images with E-cadherin (gray) are shown. Scale bars, 50 μm.
(C) The protein expression levels of KRT80, AHNAK2, and AKAP12 are verified to be upregulated in 2D HC in each case (#1–3), as indicated by the bar graph. Data are represented as mean ± SD. Statistical significance: ∗p < 0.05, unpaired t-test.
(D) A tiled image of a colorectal cancer surgical specimen is shown. Scale bar, 500 μm. Regions indicated with squares are enlarged in Figure 5E for both the tumoral center and the invasive front.
(E) Differentiated and polarized tumor glands are observed in the tumoral center (top), whereas de-differentiated tumor cells are observed in the invasive front (bottom) after H.E. staining. Immunohistochemical images of KRT80, AHNAK2, and AKAP12 (red) of the tumoral center (upper panel) and the invasive front (lower panel) in the sequential sections are shown. In the H.E. staining map image, the region of the tumoral center is highlighted with a white square, while the invasive front is indicated by a yellow square. In the right panels, merged images with E-cadherin (gray) are shown. Scale bars. 100 μm.
(F) The signal intensity of KRT80, AHNAK2, and AKAP12 in immunofluorescence images are shown by the bar graph in individual cases #1–3. Data are represented as mean ± SD. Statistical significance: ∗p < 0.05, unpaired t-test. See also Figure S2.
We subsequently analyzed the relationship between the gene expression level and the clinicopathological characteristics of CRC. According to the data from the GEPIA website, KRT80 expression was closely associated with the pathological stage in CRC (Figure S2B). AHNAK2 and AKAP12 are not only significantly upregulated in parallel with cancer progression in advanced histopathological stages but also associated with a poor overall survival and disease-free survival rate (Figures S2C and S2D). These results suggest that cell fate change of CRC cells toward a fetal/regenerative/revival state is involved in clinical features, including a prognosis and progression of CRC.
AP-1 and TEAD-mediated transcription and the loss of lineage determination synergize to control the fate of cancer cells
To elucidate the ECM-dependent cell fate conversion in CRC, we investigated the transcriptional regulatory mechanisms in detail. We performed a KEGG pathway analysis of upregulated genes identified by RNA-seq (log2FC ≥ 1, p value < 0.05) in both 3D MG and 2D HC across all nine cases. When compared with normal colon crypts isolated from noncancerous tissues of the surgical specimens of cases #1–3 (Table S2), the Wnt signaling pathway was enriched in both 3D MG and 2D HC (Figure 4A). It was speculated to be consistent with CRC-specific gene mutations such as in APC and β-catenin. The Hippo signaling pathway was also enriched in both 3D MG and 2D HC, however ECM receptor interaction and focal adhesion were characteristically identified in 2D HC (Figure 4A). These observations suggest the potential existence of additional amplification in YAP/TAZ/TEAD-dependent signal cascade in 2D HC, akin to the activation of the cascade via mechanotransduction in the normal colon epithelium. Among the typical YAP target genes,33 six genes (CYR61, CTGF, ANKRD1, F3, LATS2, and TGFB2) showed more than a 2-fold increase of the average TPM value in 2D HC compared to 3D MG (Figure S3A). Moreover, in the analysis of the expression levels of individual genes after subclustering based on their RAS mutational status,34 expression markedly spiked in 2D HC with RAS mutations in cases #1 and #7 (Figure 4B). The expression level of cytoskeleton-related genes such as KRT80, AHNAK2, and AKAP12, which were revealed to be enriched in the invasive front in the current study, showed a similar trend to the representative YAP signature. Notably, a more distinct distribution was observed upon further investigation through subclustering by RAS mutation status (Figure 4C). This observation aligns with published results showing that the RAS oncogenic program modulates cellular mechanosensing by increasing actin stress fibers.35 Considering that the RAS mutational status is also reported to be an important factor in determining the inflammatory state,36 we visualized the expression levels of TNF in RNA sequencing tracks after subclustering by RAS mutational status. The results clearly revealed that TNF is upregulated in both RAS mutation-positive and negative cases, with a notable increase observed in cases #1 and #7 within the RAS mutation-positive group (Figure 4D).
Figure 4.

ECM niche-dependent AP-1 and TEAD-mediated transcription and the loss of lineage determination synergize to control the fate of cancer cells
(A) The top 10 KEGG pathway enriched in upregulated genes in nine cultures (#1–9) of 2D HC (left panel) or 3D MG (right panel) compared to normal colonic epithelial crypts in three samples (#1–3) is shown.
(B) Violin plot analyses comparing the distribution of total transcripts per million (TPM) values of indicated genes (CYR61, CTGF, ANKRD1, F3, LATS2, and TGFB2) in nine cultures (#1–9) of 3D MG and 2D HC and three normal colonic epithelial crypts are shown. Samples are subcategorized by RAS mutation status. WT; wild-type, MT; mutation. The color and shape of each point in the cancer cell lines corresponds to each case.
(C) Violin plot analyses comparing the distribution of total transcripts per million (TPM) values of indicated genes (KRT80, AHNAK2, and AKAP12) in nine cultures of 3D MG and 2D HC and three normal colonic epithelial crypts are shown. Samples are subcategorized by RAS mutation status. WT; wild-type, MT; mutation. The color and shape of each point in cancer cell lines correspond to each case.
(D) A representative RNA sequencing track visualized in Integrative Genomic Viewer (IGV) at TNF loci is shown. Samples are subclustered by RAS mutation status. TNF is generally upregulated in 2D HC compared to 3D MG in both RAS wild-type (WT) and RAS mutant (MT) cases. The data range of the RNA sequencing track was set as 400 and 2000 in RAS WT and RAS MT cases, respectively.
(E) A principal component analysis (PCA) plot from the ATAC sequencing analysis (ATAC-seq) of three 3D MG and 2D HC cultures is shown.
(F) Representative motifs among the top 25 most highly enriched transcriptional factor motifs in either 3D MG (2D HCCLOSE) or 2D HC (2D HCOPEN) are shown. See also Figure S3, Tables S2 and S3.
To dissect the involvement of epigenetic factors, we applied ATAC sequencing analysis (ATAC-seq) in 3D MG and 2D HC in cases #1–3. PCA revealed that the chromatin dynamics of 3D MG and 2D HC were divided into different clusters (Figure 4E). Differentially accessible regions were analyzed using edgeR (log2FC ≥ 1, p value < 0.05) (Table S3). 1464 and 540 enriched accessible peaks were identified in 2D HC (2D HCopen sites) and 3D MG (2D HCclose sites). By integrating ATAC-seq and RNA-seq, TNF, PLAU, F3, MUC16, CCL2, and ATF3 were identified in the overlap between 2D HC up and 2D HCopen genes (Figure S3B). In contrast, several genes among the CBC signature, including OLFM4, SMOC2, BLNK, IRS1, and PHGDH were included in the overlap between 2D HC down and 2D HCclose genes (Figure S3B). To identify the candidate TFs governing cellular reprogramming, we identified enriched motifs in 2D HCopen and 2D HCclose sites. The ATAC-based motif analysis revealed the robust and widespread induction of AP-1 family members such as FRA1, BATF, and ATF3, followed by gradual induction of TEAD family members in the top TFs binding 2D HCopen sites (Figures 4F and S3C). These results are consistent with the previous report on the cooperation between AP-1 and the YAP/TAZ/TEAD complex.37 Moreover, NF-κB-p65, which is known to be activated by TNF and required for the expression of TNF downstream genes,38 was also found in the top TFs (Figure S3B). On the other hand, lineage-determining TFs of colonic epithelium such as CDX2, HNF4A, GATA6 and FOXA214 occupied the top TFs binding 2D HCclose sites (Figures 4F and S3C). Consequently, it is shown that CRC cells with genetic mutations respond to the surrounding microenvironment, including the ECM niche via 1) the activation of AP-1 and TEAD-dependent transcription and 2) the repression of lineage-determining related transcription simultaneously, which would subsequently lead to a distinct YAP state.
TNF serves as a major effector in fetal/regenerative/revival cells which exhibit the potential to convert back again into stem-like CRC cells
We performed a gene network analysis of the 2D HC gene set. The protein-protein interaction network of the 2D HC gene set was constructed by STRING and visualized in Cytoscape. TNF was revealed to be the largest node (degree = 120). Furthermore, IL1B (degree = 95), CCL2 (degree = 60), ICAM1 (degree = 51), and CSF2 (degree = 46), which are all closely related to TNF signaling pathway according to the KEGG-database, were also found in the top 12 largest nodes (Figure 5A). Other hub nodes ranked in the top 12 are FN1 (degree = 105), CXCL8 (degree = 78), COL1A1 (degree = 56), THBS1 (degree = 56), SERPINE1 (degree = 41), CD31 (degree = 42), and MYD88 (degree = 39) (Figure 5A). Given that TNF was identified as a top-ranked pathway in the enrichment analysis of RNA-seq (Figure 2C) and exhibited increased chromatin accessibility in 2D HC in the ATAC-seq (Figure S3B; Table S3), it was assumed to be one of the critical effectors of a fetal/regenerative/revival state. To investigate the effect of TNF on cancer cell phenotype, we applied TNF to 3D MG at a concentration of 50 ng/mL for 10 days and found an increase in representative YAP target genes (F3, CYR61, and CTGF) and a fetal marker gene (TACSTD2) as predicted. However, stem cell markers such as SMOC2 and LGR5 showed a decrease upon exposure to TNF (Figure 5B). Interestingly, when medium supernatant from 2D HC was applied to 3D MG for five days, an increase of YAP target genes (F3, CYR61, and CTGF) and a fetal marker gene (TACSTD2), as well as a decrease of stem cell markers (OLFM4, SMOC2, and LGR5), were induced, suggesting that TNF is secreted from 2D HC cancer cells. This may act on secretary cancer cells themselves or neighboring cancer cells in an autocrine/paracrine manner (Figure 5B). Importantly, the most prominent change in these genes was observed in 2D HC, suggesting that the importance lies not only in TNF, the end product induced by a series of cell fate conversion, but also in the upstream transcriptional dynamics of AP-1 and TEAD involved in mechanotransduction (Figure 5B).
Figure 5.

TNF serves as a major effector in fetal/regenerative/revival cells which exhibit the potential to convert back again into stem-like CRC cells
(A) The protein-protein interaction network of the 2D HC gene set was constructed by STRING and visualized in Cytoscape. Each circle represents a hub node, and the diameter of the circle corresponds with the degree of each node. The largest node, TNF, and the other 11 largest nodes are indicated with gene names.
(B) The quantitative polymerase chain reaction (qPCR) of indicated genes, including typical YAP target genes (F3, CYR61 and CTGF), a fetal marker gene (TACSTD2), and stem cell marker genes (OLFM4, SMOC2 and LGR5) in human colorectal cancer (CRC) organoids cultured under different conditions is shown. The average Ct value of each gene is normalized by the average Ct value of GAPDH, and the normalized value is plotted on the Y axis. The average expression level of seven cultures of 3D MG, 3D MG-TNF, 3D MG-supernatant and 2D HC is presented for each gene. Data are represented as mean ± SD. ∗p < 0.05, paired t-test. 3D MG-TNF; Matrigel CRC organoids were exposed to TNF (50 ng/mL) for 10 days 3D MG-Supernatant; Matrigel CRC organoids were cultured in the 2D HC medium supernatant for 5 days.
(C) The qPCR results for the indicated genes evaluated in (B) are shown. The average expression level of seven cultures of 3D MG, 2D HC and 3D MG from 2D HC is presented for each gene. Data are represented as mean ± SD. ∗p < 0.05, paired t-test. 3D MG from 2D HC; Matrigel CRC organoids which is passaged from 2D HC.
According to the previous reports 5,39 multiple cell types including LGR5high stem-like CRC cells can be reestablished from LGR5low CRC cells. Therefore, the ability of fetal/regenerative/revival cells to produce stem cells is one of the critical matters related to the observed cell fate conversion. We evaluated this issue by transferring 2D HC into Matrigel to assess whether a fetal/regenerative/revival state would be lost or if a stem cell phenotype could arise. In the qPCR analyses, YAP target genes (F3, CYR61, and CTGF) and a fetal marker gene (TACSTD2) were all repressed after replating from 2D HC to 3D MG. In contrast, stem cell markers (OLFM4, SMOC2, and LGR5) all exhibited an increase. These results illustrate that LGR5high stem-like CRC can arise from fetal/regenerative/revival cells, supporting the previously reported fate-shift between LGR5high and LGR5low state (Figure 5C).
Chemoresistance in fetal/regenerative/revival cells can be overcome by inhibiting TEAD/TNF
To explore the relation of cancer cell plasticity toward a fetal/regenerative/revival state and the acquisition of chemoresistance, we investigated whether 2D HC was resistant to chemotherapeutic agents. Therapeutic profiling was performed on seven cultures of 3D MG and their derived 2D HC, and these primary cancer cells were treated by three standard chemotherapeutic agents commonly used in CRC patients: 5-Fluorouracil (5-FU), Oxaliplatin, and Irinotecan (CPT-11),.40 First, 3D MG and 2D HC were treated with 5-FU at various concentrations ranging from 1 to 120 μM for 72 h. At the end of each experiment, cellular viability assay was performed. While 3D MG showed a significant reduction in cellular viability at any concentration, cellular viability remained significantly higher in 2D HC (Figure 6A). The IC50 of 5-FU was significantly higher in 2D HC (IC50 = 91.1 μM) than 3D MG (IC50 = 12.0 μM) (Figure 6A). 2D HC also showed a clear resistance to oxaliplatin and CPT-11 (Figures 6B and 6C). IC50 of oxaliplatin in 2D HC and 3D MG was 45.4 μM and 19.0 μM respectively. IC50 of CPT-11 in 2D HC and 3D MG was 59.1 μM and 15.2 μM respectively (Figures 6B and 6C). The pharmacotyping confirmed that CRC cells in a fetal/regenerative/revival state have a chemoresistance to cytotoxic chemotherapeutic agents in vitro.
Figure 6.

Chemoresistance in fetal/regenerative/revival cells can be overcome by inhibiting TEAD/TNF
(A) Cell proliferation assay after 72 h treatment with 5-Fluorouracil (5-FU) in seven 3D MG and 2D HC cultures are shown (left panel). The median inhibitory concentration (IC50) of 5-FU is shown (right panel). Data are represented as mean ± SD. Statistical significance: ∗p < 0.05, paired t-test.
(B) Cell proliferation assay after 72 h treatment with Oxaliplatin in seven 3D MG and 2D HC cultures is shown (left panel). IC50 of Oxaliplatin is shown (right panel). Data are represented as mean ± SD. Statistical significance: ∗p < 0.05, paired t-test.
(C) Cell proliferation assay after 72 h treatment with CPT-11 in seven 3D MG and 2D HC cultures are shown (left panel). IC50 of CPT-11 is shown (right panel). Data are represented as mean ± SD. Statistical significance: ∗p < 0.05, paired t-test.
(D) Cell proliferation assay after 72 h treatment with the TEAD inhibitor in seven 3D MG and 2D HC cultures is shown (left panel). IC50 of the TEAD inhibitor is shown (middle panel). Cell viability after 72 h treatment with 5-FU at 20 and 40 μM, TEAD inhibitor at 10 μM and a combination of 5-FU at 20 μM plus TEAD inhibitor at 10 μM is shown (right panel). Data are represented as mean ± SD. Statistical significance: ∗p < 0.05, paired t-test.
(E) Cell proliferation assay after 72 h treatment with the TNF inhibitor in seven 3D MG and 2D HC cultures is shown (left panel). IC50 of the TNF inhibitor is shown (middle panel). Cell viability after 72 h treatment with 5-FU at 20 and 40 μM, TNF inhibitor at 7.5 μM and a combination of 5-FU at 20 μM plus TNF inhibitor at 7.5 μM is shown (right panel). Data are represented as mean ± SD. Statistical significance: ∗p < 0.05, paired t-test.
(F) Representative cell-cycle profiling of control and 5-FU treated 3D MG (upper panel) and 2D HC (lower panel) determined by fluorescence image cytometer NucleoCounter NC-250 system is shown. Cell populations in S and G2/M phases are indicated by a square. The >4N cell populations are indicated by a dashed square.
(G) Representative images of cleaved caspase3 staining (red) of 2D HC after 72 h treatment of DMSO (control) (upper panel) or 5-FU at 40 μM (lower panel) are shown. Images are counterstained with DAPI (blue). The adjacent panels present merged images with Phalloidin (gray) for both control and 5-FU. Scale bars, 50 μm.
(H) Structural changes in actin filaments induced by TNF inhibitor administration are depicted by phalloidin staining. Representative images of phalloidin staining (gray) of 2D HC after 72 h treatment of DMSO (control) (upper panel) or TNF inhibitor at 10μΜ are shown. In the adjacent panels, merged images with DAPI (blue) are presented for control and TNF inhibitor. Scale bars, 50 μm. See also Figure S4 and Videos S6 and S7.
Importantly, however, when 3D MG and 2D HC were exposed to a potent TEAD inhibitor (K-975), a small molecule that can inhibit YAP/TAZ-TEAD protein-protein interactions,41 2D HC was efficiently eliminated (Figure 6D). IC50 of the TEAD inhibitor (K-975) in 2D HC and 3D MG was 13.0 μM and 72.1 μM respectively (Figure 6D). Moreover, the combination of the TEAD inhibitor at 10 μM plus 5-FU at 20 μM showed more efficient eradication of 2D HC than both 5-FU monotherapy at 20 and 40 μM and the TEAD inhibitor monotherapy at 10 μM (Figure 6D). Additionally, when 3D MG and 2D HC were exposed to a TNF inhibitor (CAS 1049741-03-8), a small molecule that directly binds to TNF and blocks the formation of trimers,42 the response showed the opposite pattern to that seen in the cell viability assay of standard chemotherapeutic agents, showing the significant sensitivity of 2D HC to a TNF inhibitor (Figure 6E). IC50 of the TNF inhibitor in 2D HC and 3D MG was 4.6 μM and 20.2 μM respectively (Figure 6E). The combination of the TNF inhibitor at 7.5 μM plus 5-FU at 20 μM showed more efficient eradication of 2D HC than both 5-FU monotherapy at 20 and 40 μM and the TNF inhibitor at 7.5 μM (Figure 6E).
To understand the mechanism by which 2D HC exhibits chemoresistance, we turn to cell cycle analysis. In the control state prior to 5-FU treatment, the population of the S phase, which was obviously detected in 3D MG, showed a clear decrease in 2D HC, suggesting that 2D HC has more quiescent characteristics (Figures 6F and S4A). Interestingly, the percentage of >4N cells was statistically higher in 2D HC, which reflects the existence of polyploid cancer cells associated with chromosomal instability (Figures 6F and S4A),43 supporting the presence of multipolar cell division occasionally seen in time-lapse imaging of 2D HC (Video S3). In 3D MG, 5-FU treatment targeting cancer cells in the S phase44 caused a significant reduction of the cell population in the S phase and a significant accumulation of cells in the sub-G1 phase, reflecting the cell-cycle arrest in 3D MG (Figures 6F and S4B). However, 2D HC did not show significant changes in the cell cycle following 5-FU treatment (Figures 6F and S4B), which aligns with the established concepts of a slow-cycling state in drug-persistent tumor cells.45 Cleaved caspase-3 immunostaining revealed that 2D HC did not show apparent changes in cleaved caspase-3 expression after 72 h 5-FU treatment at 40μm M (Figure 6G). The time-lapse imaging, which was performed immediately after application of 5-FU at 40 μM, confirmed that the active cell division was maintained, and the monolayer expansion was observed over time, suggesting that 2D HC acquired chemoresistance to 5-FU (Video S6). However, administration of TNF inhibitor at 10μM induced progressive shrinkage of cancer cells, and time-lapse imaging (Video S7) revealed a complete loss of autonomous cell motility. Phalloidin staining revealed that the formation of actin stress fibers was strongly hindered (Figure 6H).
To evaluate cell death caused by the cytotoxic regent functionally, we performed the replating assay and verified whether 3D MG and 2D HC could be passaged after the treatment of 5-FU and could regrow as 3D MG (Figure S4C). The replating assay revealed that 3D MG could not be passaged following the treatment of 5-FU at 40 μM, suggesting the effective induction of cell death (Figure S4D). However, 2D HC was successfully passaged as spherical growing organoids even after the treatment of 5-FU at the same concentration, indicating that 5-FU did not induce total cell death in 2D HC (Figure S4D). In contrast, the replating assay revealed that 3D MG showed non-affected regrowth followed by passage after the administration of TNF inhibitor at 10 μM, whereas 2D HC did not show re-expansion at all, demonstrating that cell death was truly induced in 2D HC (Figures S4E and S4F).
These results indicate that a TEAD inhibitor or a TNF inhibitor could be effective in treating aggressive, drug-tolerant CRC cells in a fetal/regenerative/revival state within the limited genetic mutation profile analyzed in the current study.
Discussion
In the current study, we revealed that cellular plasticity toward a fetal/regenerative/revival state was induced following activation of YAP transcription via mechanotransduction by using two in-vitro culture systems with different ECM components. Interestingly, 2D HC showed a distinct signature from 3D MG through multiple differences in culture conditions, including the ECM component, physical property (2D or 3D), or the serum in the culture medium. However, 2D HC characteristically showed enrichment of ECM-receptor interaction and focal adhesion according to transcriptome profiling using RNA-seq. This implies that the ECM component plays a pivotal role in determining the cell fate. On the whole, the fetal signature24 as well as the EMT signature were both upregulated in 2D HC, implying that the ECM niche-dependent mechanotransduction synergizes with the genetic mutation-based oncogenic program to promote cell fate conversion and form the landscape of heterogeneity in CRC.
The differences in transcriptome profile between 3D MG and 2D HC reflected the two major characteristics of CRC: CBC signature and RSC signature,7 illustrating the scheme that spatial distribution of the ECM components between the tumoral center and invasive front may influence the cancer phenotype. Expression of cytoskeleton-related genes such as KRT80, AKAP12, and AHNAK2 among upregulated genes in 2D HC were elevated in poorly differentiated CRC cells in the invasive front among limited samples. Additionally, these genes were correlated with the clinical staging and prognosis of CRC. These results suggest that differences in the ECM components of the tumor microenvironment might partially induce morphological or phenotypic heterogeneity. Intriguingly, 2D HC showed the upregulation of mucin secretion-related genes, including MUC16, MUC2, and TFF2. As MUC2 expression was downregulated when normal colonic epithelial cells were cultured in collagen type I,12 the acquisition of mucinous phenotype in CRC cells under collagen-enrich conditions serves as evidence of how the regenerative reaction equipped in the normal colonic epithelium is altered in CRC cells harboring genetic mutations. Given that the expression of MUC16, MUC2, and TFF2 are reported to be associated with advanced tumor stage or poor prognosis,46,47,48 2D HC is a valid tool in the study of CRC of an aggressive phenotype.
The importance of the YAP/TAZ/TEAD complex as a regulator of CRC phenotype has been recently reported.49 In the current study, 2D HC showed a distinct YAP state, especially in the RAS mutation-positive cases. Epigenetic characterization of 2D HC by ATAC-seq revealed that the binding regions of colonic epithelium-specific lineage-determining transcriptional factors such as CDX2, HNF4A, GATA6, and FOXA214 were all closed, whereas AP-1 and TEAD binding regions were open. This illustrates that CRC cells harboring mutations in the Wnt signaling pathway achieve further upregulation of the YAP/TAZ target genes. Direct verification of the interaction between AP-1 and YAP/TAZ/TEAD complex will be essential to identify specific YAP regulatory elements in human CRC. This is particularly important to formulate therapeutic strategies for CRC.
As we presented in this study, 3D MG showed sensitivity to 5-FU, Oxaliplatin, and CPT-11, whereas 2D HC showed a distinct resistance to these cytotoxic agents, illustrating that cancer cells in a fetal/regenerative/revival state display chemoresistance. Importantly, this study identified that a TEAD inhibitor can be effectively used to eradicate these cancer cells, as the phenotype majorly relies on the activated YAP/TAZ-TEAD transcription mechanistically. TNF, which is identified as the largest hub gene and as a major effector in a fetal/regenerative/revival state might also be a promising therapeutic target. In fact, the administration of a TNF inhibitor, a small molecule directly binding to TNF, induced disruption of actin-stress fibers, leading to a progressive shrinkage of cancer cells and complete loss of autonomous cell motility.
It is difficult to determine whether the mechanism of action of TNF is autocrine or paracrine. We hypothesize that both mechanisms are likely to be involved. In any case, an inflammatory network associated with TNF activation seems to play a central role inducing a fetal/regenerative/revival state of CRC cells. The entire network could, therefore, potentially be a druggable target in drug-tolerant persistent CRC cells. In a clinical setting, TNF inhibitors have been the mainstay of treatment for inflammatory bowel disease and autoimmune disease.50 In the oncology field, a TNF inhibitor was also demonstrated to enhance the anti-metastatic effect of cisplatin in a phase I trial in stage IV lung adenocarcinomas.51 Further mechanistic studies are warranted to elucidate how TNF inhibition suppresses the transcriptional dynamics via cooperative molecular function between AP-1 and YAP/TAZ/TEAD complex.
The current study also clearly illustrates the need for further research on mesenchymal cells, including cancer-associated fibroblasts (CAFs), which produce both ECM proteins and cytokines.52 In CRC, trophoblasts were reported to produce hyaluronan, which induces mesenchymal phenotype with a poor prognosis.53 Furthermore, some soluble factors released by fibroblasts were also reported to drive the cell fate conversion toward a fetal/regenerative/revival state in murine CRC models.54 In addition, a previous report showed that a fetal/regenerative/revival state induced by CAFs was associated with chemoresistance.55 We believe that the scheme presented by our study is useful to understand the functions of CAFs in detail.
Limitations of the study
This study’s limitation lies in its focus on a limited number of cases, specifically those involving moderately differentiated CRC samples. However, our culture method could provide a platform for analyzing cellular reprogramming in other colorectal malignancies, such as mucinous adenocarcinoma and neuroendocrine carcinoma. Further analysis in a larger number of cases would be helpful to categorize the typical status of the ECM-mediated mechanotransduction and its resultant reprogramming based on the genetic mutation profile of an individual cancer. Although 3D MG is useful for analyzing the effect of several cytokines, such as the WNT ligand, on cellular plasticity by changing the medium composition, the collaborative application of 3D MG and 2D HC could broaden CRC research and lead to the development of effective therapeutic strategies targeting both genetic mutations and cancer cell plasticity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-E-Cadherin | BD Biosciences | Cat# 610181, RRID:AB_397580 |
| Rabbit anti-Collagen I | Bioss | Cat# bs-10423R |
| Biotinylated hyaluronan binding protein | Hokudo | Cat# BC41 |
| Rabbit anti-Collagen IV | Proteintech | Cat# 19674-1-AP, RRID:AB_2878595 |
| Rabbit anti-Laminin-1+2 | Abcam | Cat# ab7463, RRID:AB_305933 |
| Rabbit anti-KRT80 | Proteintech | Cat# 16835-1-AP, RRID:AB_1851273 |
| Rabbit anti-AHNAK2 | Atlas Antibodies | Cat# HPA004145, RRID:AB_1859546 |
| Rabbit anti-AKAP12 | Proteintech | Cat# 25199-1-AP, RRID:AB_2879954 |
| Rabbit anti-Ki67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| Rabbit anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| Alexa Fluor 555 Goat anti Mouse IgG (H + L) | Thermo Fisher Scientific | Cat# A-21422, RRID:AB_2535844 |
| Alexa Fluor 555 Goat anti Mouse IgG2a (H + L) | Thermo Fisher Scientific | Cat# A-21137, RRID:AB_2535776 |
| Alexa Fluor 488 Donkey anti Rabbit IgG (H + L) | Thermo Fisher Scientific | Cat# R37118, RRID:AB_2556546 |
| Alexa Fluor 555 Donkey Anti-Rat IgG (H + L) | Abcam | Cat# ab150154, RRID:AB_2813834 |
| DyLight 488 Streptavidin | Vector Laboratories | Cat# SA-5488, RRID:AB_2336405 |
| Biological samples | ||
| Surgical specimens of human colorectal cancer | Tokyo Medical and Dental University Hospital | Yoshihito Kano |
| Chemicals, peptides, and recombinant proteins | ||
| Growth factor reduced Matrigel | Corning | Cat# 356231 |
| Cellmatrix Type I-A | Nitta Gelatin | Cat# 631-00651 |
| Cellmatrix Type IV | Nitta Gelatin | Cat# 638-05921 |
| HyStem | Sigma | Cat# HYS020-1KT |
| murine EGF | Peprotech | Cat# 315-09 |
| murine Noggin | R&D systems | Cat# 1967-NG |
| murine R-spondin 1 | R&D systems | Cat# 3474-RS |
| Murine Wnt3a | Abcam | Cat# ab81484 |
| N-2 supplement | Gibco | Cat# 17502-048 |
| B-27 supplement | Gibco | Cat# 17504-044 |
| Nicotinamide | Sigma-Aldrich | Cat# 72340 |
| N-Acetyl-L cysteine | Sigma-Aldrich | Cat# A9165 |
| Gastrin I | Sigma-Aldrich | Cat# G9145 |
| A83-01 | Tocris | Cat# 2939 |
| Prostaglandin E2 | Nacalai Tesque | Cat# 29334-21 |
| Bovine Serum Albumin | Sigma | Cat# A9576 |
| Y-27632 | Tocris | Cat# 1254 |
| TNF-α | Peprotech | Cat# 300-01A |
| 5-Fluorouracil | Selleck Chemicals | Cat# S1209 |
| Oxaliplatin | Selleck Chemicals | Cat# S1224 |
| Irinotecan (CPT-11) | Selleck Chemicals | Cat# S1198 |
| TNF inhibitor | Calbiochem | Cat# 654256 |
| TEAD inhibitor | Selleck Chemicals | Cat# E1329 |
| Critical commercial assays | ||
| DNeasy Blood & Tissue Kits | Qiagen | Cat# 69504 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| SuperScriptⅢ Reverse Transcriptase | Invitrogen | Cat# 18080044 |
| SYBR green master mix | Qiagen | Cat# 20415 |
| Cell Titer 96 Aqueous One Solution Cell Proliferation Assay Kit | Promega | Cat# G3581 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector laboratories | Cat# H-1200-10 |
| Phalloidin FITC Reagent | Abcam | Cat# ab235137 |
| Deposited data | ||
| Bulk RNA-seq of human CRC patient tissue and organoids and cancer sheets | This study | SRA: PRJNA1044535 |
| Bulk ATAC-seq of human CRC organoids and cancer sheets | This study | SRA: PRJNA1044535 Mendeley Data: https://doi.org/10.17632/gbfgn8kfh8.1 |
| Experimental models: Cell lines | ||
| Human CRC Matrigel cancer organoid (3D MG) | This study | N/A |
| Human CRC cancer sheet (2D HC) | This study | N/A |
| L-WRN cells for L-WRN conditioned medium | ATCC | CRL-3276 |
| Oligonucleotides | ||
| Primer: AHNAK2 Fwd: CAGCAGGGGGTAGCTTTCAA |
This study | N/A |
| Primer: AHNAK2 Rev: CTTGCTGTTGCACCAAGTCC |
This study | N/A |
| Primer: KRT80 Fwd: TTCGACCTCGGGCATCTCTA |
This study | N/A |
| Primer: KRT80 Rev: AACTCCATGTCTGTGCGCTT |
This study | N/A |
| Primer: AKAP12 Fwd: AGAAGAGGTTGTGGCCGAAG |
This study | N/A |
| Primer: AKAP12 Rev: CCACCTCCGTTTTCTGCTCT |
This study | N/A |
| Primer: F3 Fwd: AGCTTTTGAGGGGCTGACTT |
This study | N/A |
| Primer: F3 Rev: GGAAGGTGCCCAGAATACCA |
This study | N/A |
| Primer: CYR61 Fwd: CGCCTTGTGAAAGAAACCCG |
This study | N/A |
| Primer: CYR61 Rev: GGTTCGGGGGATTTCTTGGT |
This study | N/A |
| Primer: CTGF Fwd: GGGAAATGCTGCGAGGAGT |
This study | N/A |
| Primer: CTGF Rev: TCCAGTCGGTAAGCCGC |
This study | N/A |
| Primer: TACSTD2 Fwd: CCCCTTTCGGTCCAACAACA |
This study | N/A |
| Primer: TACSTD2 Rev: AACTCCTCTTCTCCTCGGGT |
This study | N/A |
| Primer: OLFM4 Fwd: AGCTGGAGGTGGAGATAAGAAA |
This study | N/A |
| Primer: OLFM4 Rev: CTGAACCACAGACGGTTTGC |
This study | N/A |
| Primer: SMOC2 Fwd: GGCACATCCACAAGGTACGA |
This study | N/A |
| Primer: SMOC2 Rev: GTCCAGAACGCTGGTCAGAA |
This study | N/A |
| Primer: LGR5 Fwd: CCTTCCAACCTCAGCGTCTT |
This study | N/A |
| Primer: LGR5 Rev: AGGGATTGAAGGCTTCGCAA |
This study | N/A |
| Software and algorithms | ||
| RIAS visualization tool | Rhelixa | https://www.rhelixa.com/rias/ |
| R package – ClusterProfiler | Wu T et al.56 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| ShinyGO v.0.77 | Ge SX et al.57 | http://bioinformatics.sdstate.edu/go/ |
| Gene set enrichment analysis tool software (version; 4.3.0) | BROAD INSTITUTE | https://www.gsea-msigdb.org/gsea/index.jsp |
| Cytoscape software Version 3.9.1 | Doncheva NT et al.58 |
http://cytoscape.org RRID:SCR_003032 |
| Fiji | Schneider CA et al.59 |
http://fiji.sc RRID:SCR_002285 |
| Adobe Photoshop (version 24.0.1 x64) | Adobe |
https://www.adobe.com/jp/products/photoshop.html RRID:SCR_014199 |
| GraphPad Prism software (version; 9.3.1) | GraphPad Prism | https://www.graphpad.com/RRID:SCR_002798 |
| NDP.view2 | Hamamatsu | Cat# U12388-01 |
| IGV (version; 2.4.19) | Robinson JT et al.60 | https://software.broadinstitute.org/software/igv/ |
| Other | ||
| Coster 24-well Clear TC-treated Multiple Well Plates | Corning | Cat# 3524 |
| Nunc 48-well Cell-Culture Treated Multidishes | Thermo Scientific Nunc | Cat# 150687 |
| Falcon 96-well Clear Flat Bottom TC-treated Culture Microplate | Corning | Cat# 353072 |
| EZVIEW Glass Bottom Culture Plate LB 24well | Iwaki | Cat# 5826-024 |
| Matsunami 8-well Chamber Plate | Matsunami | Cat# SCS-N28 |
| Matsunami 2-well Chamber Plate | Matsunami | Cat# SCS-N22 |
| Tissue-Tek O.C.T. Compound | Sakura Finetek USA | Cat# 4583 |
| High Profile Microtome Blade | Feather | Cat# FHP-01 |
Resource availability
Lead contact
Further information and requests regarding resources and reagents should be directed to and will be fulfilled by the lead contact, Shiro Yui (yui.arm@tmd.ac.jp).
Materials availability
There are restrictions to the availability of unique materials generated in this study, such as human colorectal cancer (CRC) organoids, due to ethical regulations within Tokyo Medical and Dental University (TMDU).
Data and code availability
-
•
Bulk RNA-seq data and ATAC-seq data which have been deposited into Sequence Read Archive in National Center for Biotechnology Information and Mendeley Data are publicly available. Accession numbers are listed in the key resources table.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Ethics approval and consent for the use of human surgical specimens
Surgical specimens were obtained at Tokyo Medical and Dental University (TMDU) hospital. The Scientific Ethics Committee of TMDU approved the use of this material for research purposes (M2019-285), and informed consent was obtained adequately from all patients.
Study participants
In total, 9 patients of Japanese descent (females: n = 2: males: n = 7, between 31 and 83 years old) volunteered to participate in the current study. The clinicopathological features of the patients are shown in Table 1.
Method details
Primary culture of human colorectal cancer cells
Human CRC cells were harvested from surgical specimens. The 5mm–10 mm pieces of CRC tissue were dissected from a cancerous lesion of the CRC surgical specimen and cut into small pieces. The fragments were washed in 30 mL Phosphate Buffer Saline (PBS; Nacalai Tesque, #14249-24) twice and subsequently in 2% Mucofilin (Eisai) once, followed by 4°C incubations in 15 mM EDTA for 20 min. After being washed in 30 mL PBS, the tissues were incubated in 10 mL collagenase solution (Sigma-Aldrich; 6.25 mg/ml in PBS, #C7657) at 37°C for 20 min. After settling down, the supernatant was transferred into another 50 mL conical tube by passing through a 70 μm pore-size mesh filter (SPL Life Science, #93070). The total volume was adjusted to 10 mL with 0.1% v/v BSA/PBS into a 15 mL conical tube, and cells were pelleted at 500g for 3 min.
When establishing the Matrigel CRC organoids (3D Matrigel; 3D MG), the pellets were suspended in Matrigel (Corning, #356231) and plated onto a 24-well plate (Flat bottom; Corning, #3524) or 48-well plate (Flat bottom; Thermo Scientific Nunc, #150687). Advanced DMEM/F12 (Gibco, #12634-010) with GlutaMAX (Gibco; 1% v/v, #35050-061) and Penicillin/Streptomycin (Nacalai Tesque; 1% v/v, #26253-84) was used as basal medium. The culture medium contains murine EGF (PeproTech; 50 ng/ml, #315-09), L-WRN conditioned medium (20%v/v), Nicotinamide (Sigma-Aldrich; 10 mM, #72340), N-2 (Gibco; 1%v/v, #17502-048)/B-27 supplement (Gibco; 2%v/v, #17504-044), N-Acetyl-L cysteine (Sigma-Aldrich; 1 mM, #A9165), Gastrin I (Sigma-Aldrich; 10 nM, #G9145) and A83-01 (Tocris; 500 nM, #2939).
We also established in vitro culture system of primary CRC cells using Hyaluronan/Collagen gel (HC gel) composed of Cellmatrix Type I-A (collagen type I; 70% v/v, Nitta Gelatin, #631–00651), Cellmatrix Type IV (collagen type IV; 15% v/v, Nitta Gelatin, #638–05921), and HyStem (hyaluronan; 15% v/v, Sigma-Aldrich, #HYS020-1KT), and named it 2D HC. In this case, the pellets were suspended on HC gel and placed on the bottom of a 24-well plate. The culture medium for 2D HC contained the same basal medium above with supplementation of murine EGF (50 ng/ml), murine Noggin (R&D Systems; 100 ng/ml, #1967-NG), mouse R-spondin1 (R&D Systems; 500 ng/ml, #3474-RS), murine Wnt3a (Abcam; 300 ng/ml, #ab81484), Nicotinamide (10 mM), N-2 (1%v/v)/B-27 supplement (2%v/v), Prostaglandin E2 (Nacalai Tesque; 2.5 μM, #29334-21) and Bovine Serum Albumin (BSA; 1%, Sigma-Aldrich, #A9576). The medium was subsequently changed every 2–3 days. Y-27632 (Tocris; 10 μM, #1254) was supplemented for the first 2–3 days after the suspension of the pellets.
2D HC was expanded following the passage of 3D MG on HC gel, which was plated at the bottom of a 24-well plate for RNA and ATAC sequencing analysis, on the bottom of a chamber slide (MATSUNAMI, #SCS-N28, SCS-N22) or a 24-well glass plate (Flat bottom; IWAKI, #5826-024) for immunohistochemical analysis, and on the bottom of a 96-well plate (Flat bottom; Corning, #353072) for cell viability assays using chemotherapeutic agents.
For the replating assays, 2D HC was sequentially passaged into 3D MG. For passaging, a monolayer cancer sheet was isolated from HC gel by collagenase solution (6.25 mg/ml in PBS) and was mechanically dissociated into fragments by pipetting 15 times using P1000 pipettes. These fragments were subsequently cultured in Matrigel under an L-WRN-conditioned medium as described above. Y-27632 was supplemented for the first 2–3 days after passage.
DNA extraction
The pellets of CRC cells were partially cryopreserved in CELLBANKER2 (ZENOGEN PHARMA) for DNA extraction. The total DNA of CRC cells was extracted using the DNeasy Blood & Tissue Kit (Qiagen, #69504) according to the manufacturer’s protocol.
Target enrichment and sequencing
For library construction, the quality of genomic DNA was assessed using the Genomic DNA ScreenTape on a TapeStation 4200 (Agilent Technologies, Santa Clara, CA, USA). The hotspot regions of approximately 2,800 COSMIC mutations from 50 oncogenes and tumor suppressor genes were enriched by multiplex PCR for 10 ng of genomic DNA obtained from each sample, according to the manufacturer’s instructions. The PCR-amplicon-based sequencing libraries were generated using AmpliSeq Library PLUS for Illumina (Illumina). The qualities of the libraries were assessed using High Sensitivity D1000 ScreenTape on a TapeStation 4200. The multiplex barcoded and equally-pooled libraries were sequenced using MiSeq (Illumina) in 151-base-pair (bp) paired-end reads.
Alignment and variant calling were performed using the DNA Amplicon Workflow on an Illumina BaseSpace Sequence Hub (https://jp.illumina.com/products/by-type/informatics-products/basespace-sequence-hub/apps.html). Functional annotations and the possible effects of variants were added using Illumina Annotation Engine on the BaseSpace.
RNA extraction
The total RNA of 3DMG, 2DHC, and normal colonic epithelial crypts were extracted using RNeasy Micro Kit (Qiagen, #74004) according to the manufacturer’s protocol. Normal colonic epithelial crypts were isolated in the same way as described above after being harvested from a non-cancerous tissue of CRC surgical specimen. The isolated crypts were cryopreserved for RNA extraction.
cDNA synthesis and qRT-PCR
The synthesis of cDNA involved using superscript III reverse transcriptase (Invitrogen, #18080044) and random primers with 100-500 ng total RNA. Each reverse-transcription product was subjected to a PCR reaction using SYBR green master mix (QIAGEN, #20415), which was run on StepOnePlus Real-Time PCR system (Applied Biosystems). Each assay was performed in seven cases using specific primer sequences. Primer sequences are listed in the key resources table. The expression level was determined by the ΔCt method, and mRNA levels were normalized to that of GAPDH, shown as the fold-change relative to control.
RNA sequencing analysis
RNA sequencing analysis of 3D MG and 2D HC established from human CRC specimens and normal colonic epithelial crypts was performed by TaKaRa Bio, Inc. (Kusatsu, Japan). Total RNA was reversed transcribed into cDNA with a Clontech SMART-Seq v4 Low Input RNA Kit according to the manufacturer’s instructions. RNA sequencing libraries were constructed from the amplified cDNA using Illumina Nextera XT DNA Library Prep Kits, validated using an Agilent 4200 TapeStation, and sequenced on the Illumina NovaSeq 6000 platform. Gene quantification results were normalized by TPM (transcripts per million). Differentially expressed gene (DEG) analysis was performed with DESeq2 (Version 1.24.0), following normalization of the raw read counts by relative log normalization (RLE). The heatmap of Z score-transformed relative expression levels of particular genes, and a volcano plot of DEGs were generated using the RIAS visualization tool (https://www.rhelixa.com/rias/) with RLE values.
Gene Ontology (GO) enrichment analysis was performed and visualized based on DEGs using the Bioconductor package, clusterProfiler4.0 56 in R software. A KEGG pathway enrichment analysis was performed and visualized based on DEGs using ShinyGO v.0.77 (http://bioinformatics.sdstate.edu/go/).57
A Gene set enrichment analysis (GSEA) was performed using GSEA tool software provided by the Broad Institute (https://www.gsea-msigdb.org/gsea/index.jsp). The hallmark gene set collection included in the Molecular Signatures Database,61 human fetal colon epithelium gene signature,24 and human tumor budding gene signature16 were used for the analysis.
A protein-protein interaction network analysis of differentially expressed genes was constructed by STRING and visualized in Cytoscape software version 3.9.1..58
Database analyses
We investigated the correlation between mRNA expression of KRT80, AHNAK2, and AKAP12 and clinical stages and subsequently analyzed the prognostic value of these genes in CRC patients using the Gene Expression Profiling Interactive Analysis (GEPIA) browser (http://gepia.cancer-pku.cn/).
Time-lapse imaging
The monolayer expansion of 2D HC was recorded by time-lapse imaging every 5 min (BZ-X710, KEYENCE). Just before live imaging, 5-Fluorouracil and a TNF inhibitor were added to the culture medium. Throughout the imaging process, the culture condition was maintained under 5% CO2 at 37°C, in a stage-top incubator (Tokai Hit, Shizuoka, Japan). The images were analyzed using the BZ-X Analyzer 1.4.0.1 (KEYENCE). Serial images of 2D HC were converted to a time-lapse video in chronological order.
Histology and immunohistochemistry
For histology and immunohistochemistry, 3D MG was fixed with 4% Paraformaldehyde (Nacalai Tesque, #09154-85) overnight. Samples were embedded in Tissue-Tek O.C.T. Compound (Sakura) and frozen. 8μm-thick cryosections were prepared by Tissue-Tek Polar Microtome/Cryostat Polar (Sakura). 2D HC was cultured on the chamber plate for immunohistochemistry and was subsequently fixed with 4% Paraformaldehyde overnight. CRC tissues were fixed using 10% formaldehyde, embedded in paraffin, cut into 3μm-thick sections using a microtome, and then fixed on slides. Slides of CRC tissue were deparaffinized and subjected to hematoxylin and eosin (H.E.) staining. A tiled image of H.E. staining was obtained by using NanoZoomer S210 (Hamamatsu Photonics, Hamamatsu, Japan). Immunohistochemical staining of 3D MG, 2D HC and parental CRC tissue was performed with various antibodies, including mouse E-Cadherin (BD Biosciences, #610181), rabbit anti-collagen type I (Bioss, #bs-10423R), biotinylated hyaluronan binding protein (Hokudo, #BC41), rabbit anti-collagen type IV (Proteintech, #19674-1-AP), rabbit anti-laminin-1+2 (Abcam, #ab7463), rabbit anti-KRT80 (Proteintech, #16835-1-AP), rabbit anti-AHNAK2 (Atlas Antibodies, #HPA004145), rabbit anti-AKAP12 (Proteintech, #25199-1-AP), rabbit anti-Ki-67 (Abcam, #ab15580) and Cleaved Caspase-3 (Asp175) antibody (Cell Signaling Technology, #9661). Phalloidin FITC Reagent (Abcam, #ab235137) was used for IHC staining of both MG and HC. Nuclei were counterstained with VECTASHILED mounting medium (Vector, #H-1200). Images were captured with a laser scanning confocal microscope (Leica TCS SP8) and were analyzed in Fiji59 and Adobe Photoshop CS6.
Quantification of immunohistochemical analysis
For the in vitro CRC cells (#1–3) a series of images of 3D MG and 2D HC were acquired under the same confocal microscope settings. They were subsequently converted into a grayscale adapting the same pre-set. The signal intensity of KRT80, AHNAK2, and AKAP12 was determined by measuring around 200 independent points of one organoid in ImageJ using the multi-point tool and taking their average. As for 3D MG, the signal intensity was determined for 3–5 different organoids in the individual cases. As for 2D HC, the signal intensity was determined in 3–5 different colonies of the cancer sheet of each case (#1–3). In this assay, 2D HC samples were processed as 8μm-thick cryosections corresponding to target samples of 3D MG. In addition, background intensity was determined in each image, and this value was subtracted from the actual measured value to determine the correction value. These corrected values were subjected to the statistical analysis described below.
For the parental CRC surgical specimens from #1–3, a series of images of the tumoral center and the invasive front were captured under the same setting of a confocal microscope and were converted into a grayscale adapting the same pre-set. The signal intensity of KRT80, AHNAK2, and AKAP12 was determined by measuring around 200 independent points of the tissues for each image in ImageJ using the multi-point tool and taking their average. The signal intensity was determined in 3–5 different lesions of the tumoral center and the invasive front, respectively. Background intensity was determined in each image, and this value was subtracted from the actual measured value to determine the correction value. These corrected values were subjected to the statistical analysis described below.
ATAC sequencing analysis
A quantity of 5,000 to 10,000 cryopreserved cells were thawed to room temperature, pelleted, and washed with cold PBS. The cell pellets were resuspended and tagmented using the enzyme and buffer provided in the ATAC-Seq Kit (Active Motif, #53150). Tagmented DNA was then purified using the DNA Purification Buffers, amplified with ten cycles of PCR, and subsequently purified by SPRI bead solution. The resulting material was quantified by the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems) and sequenced with PE150 sequencing on the Illumina Novaseq 6000.
The quality of the raw paired-end sequence reads was assessed with FastQC (0.11.7). Illumina Nextera adapter sequences were trimmed by Skewer (0.2.2). The trimmed reads were subsequently aligned to the hg38 version of the human genome using Bowtie2 (2.3.4.2), and non-uniquely mapping fragments were removed by Samtools (1.9). Subsequently, the Bowtie2-resultant. SAM files were converted into binary format (BAM) files with Samtools (1.9). The reads alignment was adjusted by using the ATAC-seq QC R package (1.8.5) and duplicate reads were removed using the MarkDuplicates tool by Picard (2.18.11). Peak calling was performed with MACS2 (2.1.2) with the following setting as default: --shift −75 --extsize 150 --nomodel -q 0.01. The peak regions from each sample were merged with Bedtools (2.27.1). Read summarization was performed using featureCounts (1.6.3) and normalized read counts were obtained by trimmed mean of M values (TMM). Principal component analysis (PCA) of the normalized counts was conducted, and each sample was projected onto the 2D plane of the first and second PCA axes using stats (3.6.1) and gplots (3.0.1.1) R packages. Identification of differentially accessible regions (DARs) was performed by edgeR. For track visualization, normalized bigwig files were uploaded to Integrative Genomics Viewer (IGV).60 Motif enrichment was identified using the hypergeometric test (HOMER).
Cell cycle profiling
Cell-cycle profiling of 3D MG and 2D HC was determined by fluorescence image cytometer NucleoCounter NC-250TM system (M&S TechnoSystems, Inc.) according to the manufacturer’s protocol.
Chemotherapeutic profiling
For the drug sensitivity test, 3D MG was seeded on a 48-well plate, and 2D HC was seeded on a 96-well plate. After confirming that 3D MG formed a budding structure and 2D HC widely spread throughout the well after a few days, chemotherapeutic agents such as 5-Fluorouracil (5-FU) (Selleck Chemicals, #S1209), Oxaliplatin (Selleck Chemicals, #S1224), Irinotecan (CPT-11; Sellek Chemicals, #S1198), a TNF inhibitor (Calbiochem, #654256), and a TEAD inhibitor (Selleck Chemicals, #E1329) were added to the culture medium at various concentrations. The concentrations of 5-FU, Oxaliplatin, and CPT-11 were 1, 10, 20, 40, 80, and 120 μM. The concentrations of the TNF inhibitor were 1, 5, 7.5, 10, 20, and 40 μM. The concentrations of the TEAD inhibitor were 0.1, 0.5, 1, 10, 20, 50, 100, 200, and 400 μM. 3D MG and 2D HC were exposed to the chemotherapeutic agents for 72 h, while DMSO-only treated cells were included as controls. At the end of treatment in each experiment, cellular viability was assessed using Cell Titer 96 Aqueous One Solution Cell Proliferation Assay Kit (Promega, #G3581). The median inhibitory concentration (IC50), which was defined as the concentration producing a 50% decrease in the viability of cells compared with untreated controls, was calculated using GraphPad Prism computer software (version; 9.3.1).
Quantification and statistical analysis
All error bars indicate the SD. Statistical assessment of overlap between different gene sets was done using Fishers’ exact t-test, assuming that the number of total genes is 25,000 in humans in Figure S2A. Student t-tests were performed for determining statistical significance in Figures 3C, 3F, 5B, 5C, 6A–6E, S4A, and S4B, using GraphPad Prism computer software (version; 9.3.1). All p values of <0.05 were considered significantly different.
Acknowledgments
We thank all members of the Department of Gastroenterology and Hepatology, Tokyo Medical and Dental University (TMDU) for their feedback on this study. We also thank Professor Ichiro Sekiya, Dr. Norio Shimizu, Dr. Hisako Katano,and all other members of the Center for Stem Cell and Regenerative Medicine, TMDU, for their feedback on this study. We also thank Professor Yasunari Miyazaki of the Department of Respiratory Medicine for his valuable comments on this study. This research was supported by MEXT/JSPS KAKENHI (20H03657 to S. Yui, 23H02887 to S. Yui, 22H00472 to M.W.), Japan Science and Technology Agency (JST) Forrest Program (JPMJFR2012 to S. Yui), Japan Agency for Medical Research and Development (AMED) (20bm0704029h0003 for S. Yui, 20bm0304001h0008, 20bk0104008h0003 to M.W.), Young Innovative Medical Science Unit (TMDU) to S. Yui, and Naoki Tsuchida Research Grant to S. Yui. S.T. is supported by a BOF-Fundamental Clinical Research mandate (FKO) from KU Leuven. Y.H. is supported by a FWO Junior Postdoctoral fellowship (12D5823N).
Author contributions
Conceptualization, N.O., Y. Kano, S.T., and S. Yui; Methodology, N.O. and S. Yui; Investigation, N.O., S. Kobayashi, S.W., S. Kirino, I.O., H.Y., G.I., and S. Yui; Analysis, N.O., Y.Y., F.D.V.B., Y.H., A.O., P.L., and S. Yui; Resources, S. Yamauchi, Y. Kinugasa, I.O., K.O., R.O., and Y. Kano; Writing the draft, N.O., S.T., and S. Yui; Supervision, M.W.; Funding; M.W. and S. Yui.
Declaration of interests
The authors declare no competing interests.
Published: February 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109247.
Contributor Information
Sabine Tejpar, Email: sabine.tejpar@uzleuven.be.
Shiro Yui, Email: yui.arm@tmd.ac.jp.
Supplemental information
Differentially expressed genes identified by DESeq2, following normalization of the raw read counts by relative log normalization (RLE) are shown.
Sheet1. Gene quantification results normalized by TPM (transcripts per million) are shown. Sheet2: Upregulated genes in 3D MG compared to normal crypt (log2FC ≥ 1, p < 0.05) are identified by differentially expressed gene (DEG) analysis with DESeq2, following normalization of the raw read counts by relative log normalization (RLE). Sheet3: Upregulated genes in 2D HC compared to normal crypt (log2FC ≥ 1, p < 0.05) are identified by DEG analysis with DESeq2, following normalization of the raw counts by RLE.
Sheet1. Normalized read counts obtained by trimmed mean of M values (TMM) are indicated. Sheet2: 2D HCopen sites (log2FC ≥ 1, p < 0.05) among the differentially accessible regions (DARs) identified by edgeR are indicated. Sheet3: 2D HCclose sites (log2FC ≤ −1, p < 0.05) among the DARs identified by edgeR are indicated.
References
- 1.Sánchez Alvarado A., Yamanaka S. Rethinking differentiation: stem cells, regeneration, and plasticity. Cell. 2014;157:110–119. doi: 10.1016/j.cell.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pérez-González A., Bévant K., Blanpain C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer. 2023;4:1063–1082. doi: 10.1038/s43018-023-00595-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brabletz T., Spaderna S., Kolb J., Hlubek F., Faller G., Bruns C.J., Jung A., Nentwich J., Duluc I., Domon-Dell C., et al. Down-regulation of the homeodomain factor Cdx2 in colorectal cancer by collagen type I: an active role for the tumor environment in malignant tumor progression. Cancer Res. 2004;64:6973–6977. doi: 10.1158/0008-5472.CAN-04-1132. [DOI] [PubMed] [Google Scholar]
- 4.Brabletz T., Kalluri R., Nieto M.A., Weinberg R.A. EMT in cancer. Nat. Rev. Cancer. 2018;18:128–134. doi: 10.1038/nrc.2017.118. [DOI] [PubMed] [Google Scholar]
- 5.Fumagalli A., Oost K.C., Kester L., Morgner J., Bornes L., Bruens L., Spaargaren L., Azkanaz M., Schelfhorst T., Beerling E., et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell. 2020;26:569–578.e7. doi: 10.1016/j.stem.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moorman A., Cambuli F., Benitez E., Jiang Q., Xie Y., Mahmoud A., Lumish M., Hartner S., Balkaran S., Bermeo J., et al. Progressive plasticity during colorectal cancer metastasis. bioRxiv. 2023 doi: 10.1101/2023.08.18.553925. Preprint at. [DOI] [Google Scholar]
- 7.Gil Vazquez E., Nasreddin N., Valbuena G.N., Mulholland E.J., Belnoue-Davis H.L., Eggington H.R., Schenck R.O., Wouters V.M., Wirapati P., Gilroy K., et al. Dynamic and adaptive cancer stem cell population admixture in colorectal neoplasia. Cell Stem Cell. 2022;29:1612. doi: 10.1016/j.stem.2022.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin X., Rodriguez F.C., Sufi J., Vlckova P., Claus J., Tape C.J. A single-cell perturbation landscape of colonic stem cell polarisation. bioRxiv. 2023 doi: 10.1101/2023.02.15.528008. Preprint at. [DOI] [Google Scholar]
- 9.Yui S., Azzolin L., Maimets M., Pedersen M.T., Fordham R.P., Hansen S.L., Larsen H.L., Guiu J., Alves M.R.P., Rundsten C.F., et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell. 2018;22:35–49.e7. doi: 10.1016/j.stem.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nusse Y.M., Savage A.K., Marangoni P., Rosendahl-Huber A.K.M., Landman T.A., de Sauvage F.J., Locksley R.M., Klein O.D. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature. 2018;559:109–113. doi: 10.1038/s41586-018-0257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayyaz A., Kumar S., Sangiorgi B., Ghoshal B., Gosio J., Ouladan S., Fink M., Barutcu S., Trcka D., Shen J., et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature. 2019;569:121–125. doi: 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi S., Ogasawara N., Watanabe S., Yoneyama Y., Kirino S., Hiraguri Y., Inoue M., Nagata S., Okamoto-Uchida Y., Kofuji S., et al. Collagen type I-mediated mechanotransduction controls epithelial cell fate conversion during intestinal inflammation. Inflamm. Regen. 2022;42:49. doi: 10.1186/s41232-022-00237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yui S., Nakamura T., Sato T., Nemoto Y., Mizutani T., Zheng X., Ichinose S., Nagaishi T., Okamoto R., Tsuchiya K., et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat. Med. 2012;18:618–623. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 14.Miura S., Suzuki A. Generation of Mouse and Human Organoid-Forming Intestinal Progenitor Cells by Direct Lineage Reprogramming. Cell Stem Cell. 2017;21:456–471.e5. doi: 10.1016/j.stem.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 15.Brabletz T., Jung A., Reu S., Porzner M., Hlubek F., Kunz-Schughart L.A., Knuechel R., Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA. 2001;98:10356–10361. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Smedt L., Palmans S., Andel D., Govaere O., Boeckx B., Smeets D., Galle E., Wouters J., Barras D., Suffiotti M., et al. Expression profiling of budding cells in colorectal cancer reveals an EMT-like phenotype and molecular subtype switching. Br. J. Cancer. 2017;116:58–65. doi: 10.1038/bjc.2016.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolácná L., Bakesová J., Varga F., Kostáková E., Plánka L., Necas A., Lukás D., Amler E., Pelouch V. Biochemical and biophysical aspects of collagen nanostructure in the extracellular matrix. Physiol. Res. 2007;56:S51–S60. doi: 10.33549/physiolres.931302. [DOI] [PubMed] [Google Scholar]
- 18.Ganem N.J., Godinho S.A., Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shankar J., Messenberg A., Chan J., Underhill T.M., Foster L.J., Nabi I.R. Pseudopodial actin dynamics control epithelial-mesenchymal transition in metastatic cancer cells. Cancer Res. 2010;70:3780–3790. doi: 10.1158/0008-5472.CAN-09-4439. [DOI] [PubMed] [Google Scholar]
- 20.Fackler O.T., Grosse R. Cell motility through plasma membrane blebbing. J. Cell Biol. 2008;181:879–884. doi: 10.1083/jcb.200802081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollard T.D., Cooper J.A. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broders-Bondon F., Nguyen Ho-Bouldoires T.H., Fernandez-Sanchez M.E., Farge E. Mechanotransduction in tumor progression: The dark side of the force. J. Cell Biol. 2018;217:1571–1587. doi: 10.1083/jcb.201701039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao S., Yan L., Wang R., Li J., Yong J., Zhou X., Wei Y., Wu X., Wang X., Fan X., et al. Tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. Nat. Cell Biol. 2018;20:721–734. doi: 10.1038/s41556-018-0105-4. [DOI] [PubMed] [Google Scholar]
- 25.Muñoz J., Stange D.E., Schepers A.G., van de Wetering M., Koo B.K., Itzkovitz S., Volckmann R., Kung K.S., Koster J., Radulescu S., et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent '+4' cell markers. EMBO J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mustata R.C., Vasile G., Fernandez-Vallone V., Strollo S., Lefort A., Libert F., Monteyne D., Pérez-Morga D., Vassart G., Garcia M.I. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 2013;5:421–432. doi: 10.1016/j.celrep.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Jang B.G., Kim H.S., Bae J.M., Kim W.H., Kim H.U., Kang G.H. SMOC2, an intestinal stem cell marker, is an independent prognostic marker associated with better survival in colorectal cancers. Sci. Rep. 2020;10:14591. doi: 10.1038/s41598-020-71643-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W., Liu Y., Zhu J., Wright E., Ding I., Rodgers G.P. Reduced hGC-1 protein expression is associated with malignant progression of colon carcinoma. Clin. Cancer Res. 2008;14:1041–1049. doi: 10.1158/1078-0432.CCR-07-4125. [DOI] [PubMed] [Google Scholar]
- 29.Perone Y., Farrugia A.J., Rodríguez-Meira A., Győrffy B., Ion C., Uggetti A., Chronopoulos A., Marrazzo P., Faronato M., Shousha S., et al. SREBP1 drives Keratin-80-dependent cytoskeletal changes and invasive behavior in endocrine-resistant ERalpha breast cancer. Nat. Commun. 2019;10:2115. doi: 10.1038/s41467-019-09676-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benaud C., Gentil B.J., Assard N., Court M., Garin J., Delphin C., Baudier J. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J. Cell Biol. 2004;164:133–144. doi: 10.1083/jcb.200307098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komuro A., Masuda Y., Kobayashi K., Babbitt R., Gunel M., Flavell R.A., Marchesi V.T. The AHNAKs are a class of giant propeller-like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc. Natl. Acad. Sci. USA. 2004;101:4053–4058. doi: 10.1073/pnas.0308619101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akakura S., Gelman I.H. Pivotal Role of AKAP12 in the Regulation of Cellular Adhesion Dynamics: Control of Cytoskeletal Architecture, Cell Migration, and Mitogenic Signaling. J. Signal Transduct. 2012;2012:529179. doi: 10.1155/2012/529179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Xu X., Maglic D., Dill M.T., Mojumdar K., Ng P.K.S., Jeong K.J., Tsang Y.H., Moreno D., Bhavana V.H., et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018;25:1304–1317.e5. doi: 10.1016/j.celrep.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piccolo S., Panciera T., Contessotto P., Cordenonsi M. YAP/TAZ as master regulators in cancer: modulation, function and therapeutic approaches. Nat. Cancer. 2023;4:9–26. doi: 10.1038/s43018-022-00473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panciera T., Citron A., Di Biagio D., Battilana G., Gandin A., Giulitti S., Forcato M., Bicciato S., Panzetta V., Fusco S., et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat. Mater. 2020;19:797–806. doi: 10.1038/s41563-020-0615-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joanito I., Wirapati P., Zhao N., Nawaz Z., Yeo G., Lee F., Eng C.L.P., Macalinao D.C., Kahraman M., Srinivasan H., et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 2022;54:963–975. doi: 10.1038/s41588-022-01100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanconato F., Forcato M., Battilana G., Azzolin L., Quaranta E., Bodega B., Rosato A., Bicciato S., Cordenonsi M., Piccolo S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015;17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Y., Zhou B.P. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer. 2010;102:639–644. doi: 10.1038/sj.bjc.6605530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cañellas-Socias A., Cortina C., Hernando-Momblona X., Palomo-Ponce S., Mulholland E.J., Turon G., Mateo L., Conti S., Roman O., Sevillano M., et al. Metastatic recurrence in colorectal cancer arises from residual EMP1(+) cells. Nature. 2022;611:603–613. doi: 10.1038/s41586-022-05402-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashiguchi Y., Muro K., Saito Y., Ito Y., Ajioka Y., Hamaguchi T., Hasegawa K., Hotta K., Ishida H., Ishiguro M., et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2019 for the treatment of colorectal cancer. Int. J. Clin. Oncol. 2020;25:1–42. doi: 10.1007/s10147-019-01485-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaneda A., Seike T., Danjo T., Nakajima T., Otsubo N., Yamaguchi D., Tsuji Y., Hamaguchi K., Yasunaga M., Nishiya Y., et al. The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am. J. Cancer Res. 2020;10:4399–4415. [PMC free article] [PubMed] [Google Scholar]
- 42.He M.M., Smith A.S., Oslob J.D., Flanagan W.M., Braisted A.C., Whitty A., Cancilla M.T., Wang J., Lugovskoy A.A., Yoburn J.C., et al. Small-molecule inhibition of TNF-alpha. Science. 2005;310:1022–1025. doi: 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- 43.Ganesh K., Wu C., O'Rourke K.P., Szeglin B.C., Zheng Y., Sauvé C.E.G., Adileh M., Wasserman I., Marco M.R., Kim A.S., et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019;25:1607–1614. doi: 10.1038/s41591-019-0584-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Longley D.B., Harkin D.P., Johnston P.G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 45.Rehman S.K., Haynes J., Collignon E., Brown K.R., Wang Y., Nixon A.M.L., Bruce J.P., Wintersinger J.A., Singh Mer A., Lo E.B.L., et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell. 2021;184:226–242.e21. doi: 10.1016/j.cell.2020.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Björkman K., Mustonen H., Kaprio T., Kekki H., Pettersson K., Haglund C., Böckelman C. CA125: A superior prognostic biomarker for colorectal cancer compared to CEA, CA19-9 or CA242. Tumour Biol. 2021;43:57–70. doi: 10.3233/TUB-200069. [DOI] [PubMed] [Google Scholar]
- 47.Börger M.E., Gosens M.J.E.M., Jeuken J.W.M., van Kempen L.C.L.T., van de Velde C.J.H., van Krieken J.H.J.M., Nagtegaal I.D. Signet ring cell differentiation in mucinous colorectal carcinoma. J. Pathol. 2007;212:278–286. doi: 10.1002/path.2181. [DOI] [PubMed] [Google Scholar]
- 48.Emami S., Rodrigues S., Rodrigue C.M., Le Floch N., Rivat C., Attoub S., Bruyneel E., Gespach C. Trefoil factor family (TFF) peptides and cancer progression. Peptides. 2004;25:885–898. doi: 10.1016/j.peptides.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 49.Della Chiara G., Gervasoni F., Fakiola M., Godano C., D'Oria C., Azzolin L., Bonnal R.J.P., Moreni G., Drufuca L., Rossetti G., et al. Epigenomic landscape of human colorectal cancer unveils an aberrant core of pan-cancer enhancers orchestrated by YAP/TAZ. Nat. Commun. 2021;12:2340. doi: 10.1038/s41467-021-22544-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rungoe C., Langholz E., Andersson M., Basit S., Nielsen N.M., Wohlfahrt J., Jess T. Changes in medical treatment and surgery rates in inflammatory bowel disease: a nationwide cohort study 1979-2011. Gut. 2014;63:1607–1616. doi: 10.1136/gutjnl-2013-305607. [DOI] [PubMed] [Google Scholar]
- 51.Paik P.K., Luo J., Ai N., Kim R., Ahn L., Biswas A., Coker C., Ma W., Wong P., Buonocore D.J., et al. Phase I trial of the TNF-alpha inhibitor certolizumab plus chemotherapy in stage IV lung adenocarcinomas. Nat. Commun. 2022;13:6095. doi: 10.1038/s41467-022-33719-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nurmik M., Ullmann P., Rodriguez F., Haan S., Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer. 2020;146:895–905. doi: 10.1002/ijc.32193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez-Ordoñez A., Duran A., Ruiz-Martinez M., Cid-Diaz T., Zhang X., Han Q., Kinoshita H., Muta Y., Linares J.F., Kasashima H., et al. Hyaluronan driven by epithelial aPKC deficiency remodels the microenvironment and creates a vulnerability in mesenchymal colorectal cancer. Cancer Cell. 2023;41:252–271.e9. doi: 10.1016/j.ccell.2022.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roulis M., Kaklamanos A., Schernthanner M., Bielecki P., Zhao J., Kaffe E., Frommelt L.S., Qu R., Knapp M.S., Henriques A., et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature. 2020;580:524–529. doi: 10.1038/s41586-020-2166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramos Zapatero M., Tong A., Opzoomer J.W., O'Sullivan R., Cardoso Rodriguez F., Sufi J., Vlckova P., Nattress C., Qin X., Claus J., et al. Trellis tree-based analysis reveals stromal regulation of patient-derived organoid drug responses. Cell. 2023;186:5606–5619.e24. doi: 10.1016/j.cell.2023.11.005. [DOI] [PubMed] [Google Scholar]
- 56.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ge S.X., Jung D., Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36:2628–2629. doi: 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doncheva N.T., Morris J.H., Gorodkin J., Jensen L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019;18:623–632. doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson J.T., Thorvaldsdottir H., Turner D., Mesirov J.P. igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV) Bioinformatics. 2023;39:btac830. doi: 10.1093/bioinformatics/btac830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentially expressed genes identified by DESeq2, following normalization of the raw read counts by relative log normalization (RLE) are shown.
Sheet1. Gene quantification results normalized by TPM (transcripts per million) are shown. Sheet2: Upregulated genes in 3D MG compared to normal crypt (log2FC ≥ 1, p < 0.05) are identified by differentially expressed gene (DEG) analysis with DESeq2, following normalization of the raw read counts by relative log normalization (RLE). Sheet3: Upregulated genes in 2D HC compared to normal crypt (log2FC ≥ 1, p < 0.05) are identified by DEG analysis with DESeq2, following normalization of the raw counts by RLE.
Sheet1. Normalized read counts obtained by trimmed mean of M values (TMM) are indicated. Sheet2: 2D HCopen sites (log2FC ≥ 1, p < 0.05) among the differentially accessible regions (DARs) identified by edgeR are indicated. Sheet3: 2D HCclose sites (log2FC ≤ −1, p < 0.05) among the DARs identified by edgeR are indicated.
Data Availability Statement
-
•
Bulk RNA-seq data and ATAC-seq data which have been deposited into Sequence Read Archive in National Center for Biotechnology Information and Mendeley Data are publicly available. Accession numbers are listed in the key resources table.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.