

Abstract
Nitrogen scanning in aryl fragments is a valuable aspect of the drug discovery process, but current strategies require time-intensive, parallel, bottom-up synthesis of each pyridyl isomer because of a lack of direct carbon-to-nitrogen (C-to-N) replacement reactions. We report a site-directable aryl C-to-N replacement reaction allowing unified access to various pyridine isomers through a nitrene-internalization process. In a two-step, one-pot procedure, aryl azides are first photochemically converted to 3H-azepines, which then undergo an oxidatively triggered C2-selective cheletropic carbon extrusion through a spirocyclic azanorcaradiene intermediate to afford the pyridine products. Because the ipso carbon of the aryl nitrene is excised from the molecule, the reaction proceeds regioselectively without perturbation of the remainder of the substrate. Applications are demonstrated in the abbreviated synthesis of a pyridyl derivative of estrone, as well as in a prototypical nitrogen scan.
Shape complementarity between a ligand and its target strongly influences binding phenomena in medicinal chemistry. Consequently, isoteric atom replacements that retain the three-dimensional contour of the molecule figure prominently in the discovery process. Within this class, the replacement of an aromatic carbon atom with a nitrogen to afford the corresponding pyridine (or higher azaarene) is privileged for its ability to impart critical drug-like properties through the modulation of physicochemical properties, introduction of hydrogen-bond acceptors, and management of oxidative metabolic liabilities (1–3). This effect has been so frequently observed by medicinal chemists that it has become known colloquially as the “necessary nitrogen effect” (4, 5). With drug discovery still a largely empirical practice, the identification of such necessary nitrogens frequently requires the examination of each isomeric permutation—a practice referred to in discovery chemistry as a “nitrogen scan” (Fig. 1A). Specific nitrogen scans that were involved in the identification of three recently Food and Drug Administration (FDA)–approved pharmaceuticals are shown in Fig. 1B (6–8).
Fig. 1. Background and motivation.
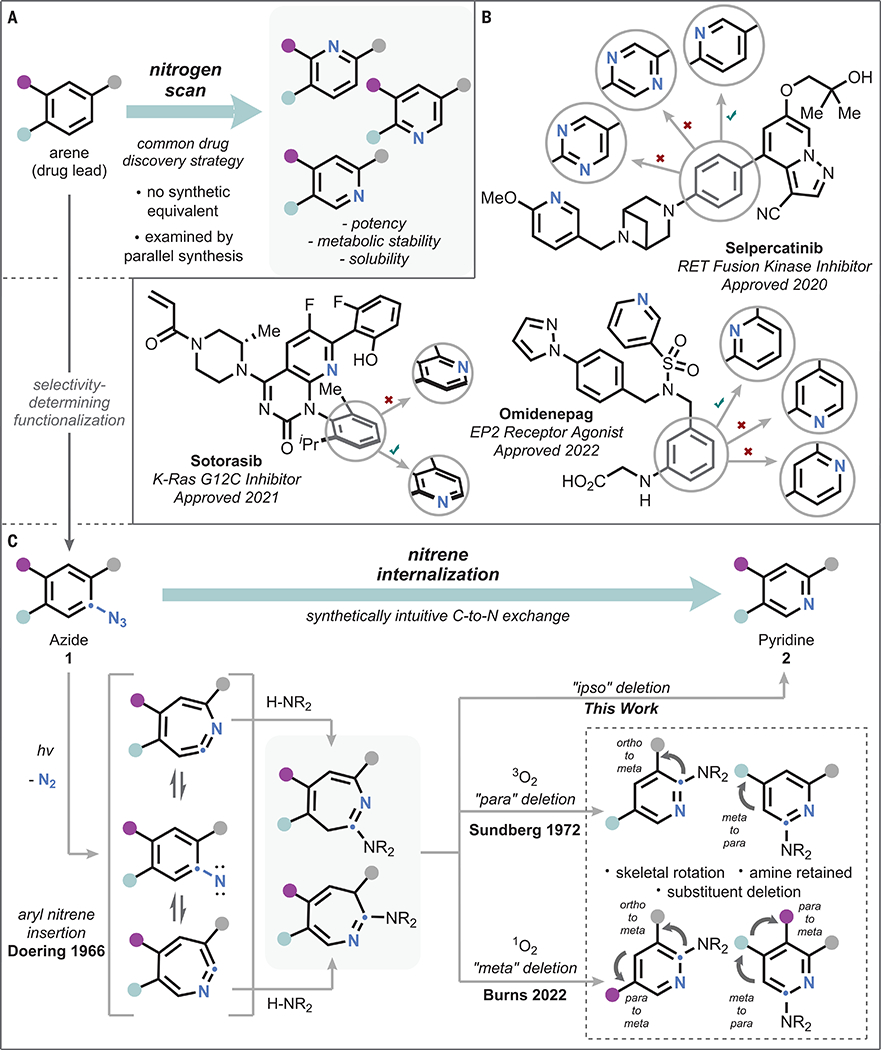
(A) General outline of the nitrogen scan strategy. (B) Examples of nitrogen scans examined in the discovery of recently approved pharmaceuticals. (C) Outline of relevant precedent for formal nitrene internalization, emphasizing drawbacks associated with the prior art. EP2, prostaglandin E2 receptor; RET, rearranged during transfection proto-oncogene; K-Ras G12C, Kirsten rat sarcoma protein with glycine 12 to cysteine mutation; iPr, isopropyl; Me, methyl.
Although a nitrogen scan is conceptually simple, its practice is divorced from this ideal by a conspicuous lack of direct atom-replacement techniques. Conducting a nitrogen scan therefore almost invariably requires bottom-up synthesis of each azine isomer (9). Each of the analogs shown in Fig. 1B followed this template, requiring iterative syntheses to identify the final clinical candidate. This need for synthetic iteration represents a substantial bottle-neck in the discovery of new medicines, and the availability of appropriate heterocycle syntheses can often limit the viability of the strategy altogether.
Accordingly, the development of synthetic methods that can enable the direct interrogation of such C-to-N replacements has attracted substantial interest (10–12). The reverse reaction (pyridine to benzene) has been reported with the use of stoichiometric titanium alkylidynes, and other atom-swapping approaches (for example, O to N) have been demonstrated in aliphatic systems (13–15). We recently reported a C2 selective carbon deletion of quinolines, which when paired with nitrogen insertion into the resulting indole, affords the corresponding replacement product, a cinnoline (16, 17). This sequence, however, is currently limited to specific heterocyclic scaffolds and can only afford a single C-to-N replacement isomer.
In the search for a strategy that would enable arbitrary site-directable C-to-N replacement, we took inspiration from the literature of 2-amino-3H-azepines, synthesis of which from aryl azides (1) and protic nucleophiles dates back to Doering’s seminal 1966 report (Fig. 1C) (18–21). Specifically, Sundberg’s subsequent finding that photolysis in the presence of air leads to a mixture of pyridine products, including those featuring the formal “para”–carbon deletion products, suggested that oxidation of the azepines could serve as a potential path forward (22). In a further development, Burns recently reported that singlet oxygen acts on the 3H-azepines to induce formal “meta”-carbon deletion (23). Both of these approaches are problematic in that the nitrogen insertion and carbon deletions are conducted with differing selectivity, leading to an overall sequence that produces multiple products (in the case of nonsymmetric starting aryl azides), removes distal functional groups, retains the incoming amine nucleophile, and thereby promotes rearrangement of the arene skeleton. Indeed, such rearrangements are the norm in the handful of other ring-contraction reactions of azepines that have been reported (24–34). Whereas in certain instances this may be desirable, such skeletal rotation precludes straightforward structure-activity interrogation and complicates retrosynthetic analysis. We reasoned that selective ipso-carbon deletion of azepines would instead allow direct formation of a single pyridine isomer (2) with neither skeletal nor functional group perturbation. This would also enable nitrogen scanning because the site selectivity of azide installation would, in a predictable and straightforward manner, guide the final site of nitrogen placement. In effect, the net transformation would be internalization of the nitrene nitrogen, replacing the carbon atom to which it was formerly attached. We report the successful realization of such an ipso-selective nitrene-internalization reaction.
Development of an oxidative carbon extrusion
Our design centered on the hypothesis that oxidation of the azepine would lead to an azaheptatriene species (I) from which cheletropic extrusion of the ipso carbon could be achieved through the corresponding azanorcaradiene isomer (II) (Fig. 2A). In this regard, we were guided by precedent in the analogous benzenoid systems; whereas decarbonylation of tropone requires temperatures >400°C, spirocyclic diamido-aminal derivatives have been shown to extrude benzene at or near room temperature (Fig. 2B) (35–37). This difference in reactivity can be attributed to the change in angle strain of the differently hybridized azanorcaradiene intermediates. Accordingly, we envisioned that introduction of a second pendant donor to the amine nucleophile would enable spirocyclization and thereby facilitate carbene elimination through a similar reduction of angle strain. Initial attempts with 1, n-diamines (which would lead to N-heterocyclic carbene leaving groups) provided detectable quantities of pyridine but were consistently plagued by the oxidative sensitivity of the free amine. Consequently, we turned our attention to the aminoalcohol-substituted azepine derivative 3a. On the basis of prior reports of non-destructive oxidation of azepines through the use of N-bromosuccinimide, we initially hypothesized that similar oxidants would facilitate oxidation and spirocycle formation in our system (38). However, treatment of 3a with an organic base and N-bromosuccinimide led to the exclusive formation of the succinimide-trapped oxidation product 4a (Fig. 2C). Having observed the noninnocence of the liberated conjugate anion upon formal Br+ transfer, we hypothesized that further modulation of the oxidant could allow productive reactivity. Upon subsequent examination of N-bromocaprolactam, a less-oxidizing Br+ equivalent, we observed the formation of the desired spirocyclic N,O-ketal 5a (39). Heating of this species to 80°C resulted in the liberation of the corresponding pyridine in moderate yield, whereas thermolysis of 4a led only to nonspecific decomposition. Because further oxidation of 5a begins to compete with 3a as the reaction proceeds, if the oxidation reaction is instead conducted at 80°C with slow addition of the oxidant, much higher yields of the desired pyridine can be obtained. Although the putative carbene by-product (6) could not be detected, we could observe its oxidation product, N-ethyl oxazolidinone, with both nuclear magnetic resonance (NMR) and mass spectrometry. This oxidation can occur either from adventitious dioxygen or the Br+ reagent, with the latter forming the oxazolidinone through hydrolysis upon aqueous workup.
Fig. 2. Design and mechanism.
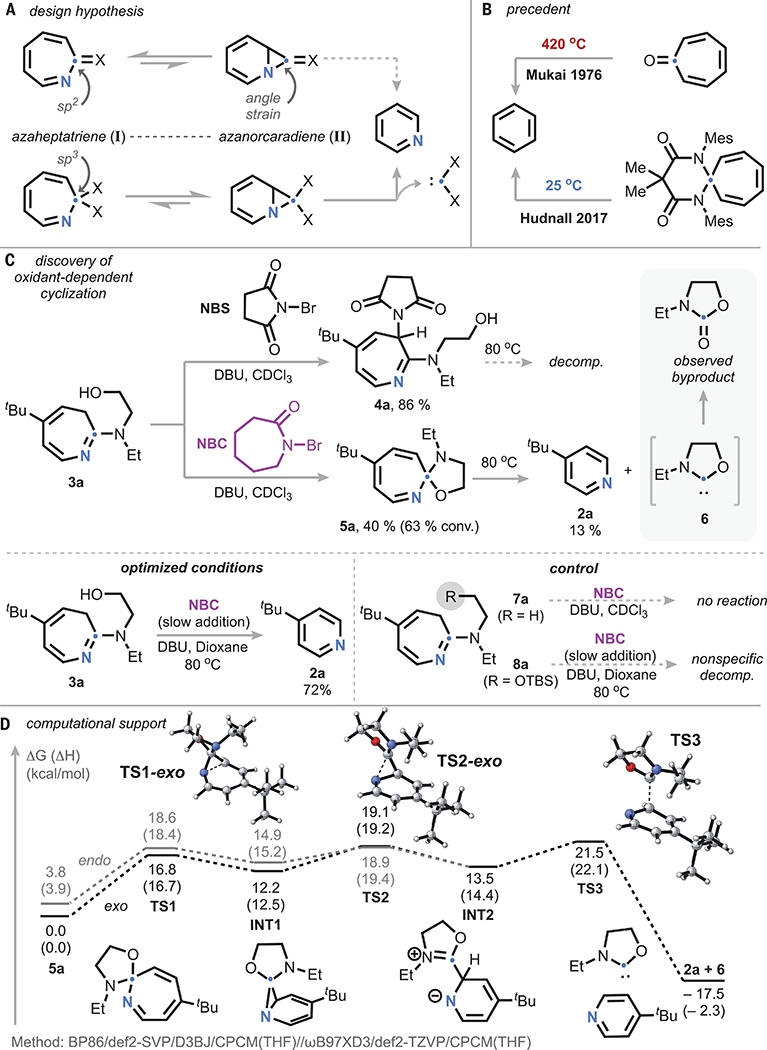
(A) Design hypothesis for ipso-carbon deletion of azepines. (B) Precedent in benzenoid systems, demonstrating hybridization effects. (C) Discovery of oxidant-dependent cyclization and thermolytic extrusion of pyridine, with optimized conditions and control experiments lacking a pendant alcohol. (D) Computed mechanism for carbene extrusion from spirocyclic azahexatriene. NBS, N-bromosuccinimide; NBC, N-bromocaprolactam; decomp., decomposition; conv., conversion; DBU, diazabicycloundecene; OTBS, tert-butyldimethylsilyloxy.
Control experiments with aminoazepines that lacked the pendant alcohol function (7a) or in which the alcohol was silylated (8a) did not afford any detectable pyridine under analogous conditions, supporting the critical role of spirocyclization in the carbon-deletion process. Computational assessment of the formation of 2a from 5a at the BP86/def2-SVP/D3BJ/CPCM(THF)//ωB97XD3/def2-TZVP/CPCM(THF) level of theory revealed an energetic landscape roughly in line with our experimental observations (THF, tetrahydrofuran). The extrusion was predicted to proceed with an overall barrier of 21.5 kcal/mol through a stepwise carbene extrusion by means of a zwitterionic intermediate (INT2) (37). Although various levels of theory disagreed on the rate-determining step, all functionals that we examined afforded an energy surface with three similarly energetic transition states (details provided in supplementary materials). The two diastereomeric pathways were found to be similar in energy, converging at INT2.
Reaction scope and applications
In seeking to translate these observations to a synthetic protocol starting from the aryl azide, we found that the crude photolysate from the initial azepine synthesis could be effectively carried forward to pyridine without intermediate purification, avoiding yield losses associated with purification of the azepine. This two-stage, one-pot transformation was found to be readily applicable to a range of aryl azides (Fig. 3). Various alkyl, aryl, and heteroaryl groups were compatible in a number of different substitution patterns (2a to 2c, 2j, 2k, 2n, 2o, and 2r). Electron-rich arenes (2b, 2j, and 2r) and those bearing both resonance (2d to 2f, 2l, and 2m) and inductive (2h) acceptors were all viable for nitrene internalization. The reaction also tolerates protected amines (2p and 2s), allowing conversion of aminoglutethimide into rogletimide and alcohols (2 g, 2h, and 2i), even those with delicate protecting groups such as silyl ethers. Strained and fused rings were also compatible (2p and 2r).
Fig. 3. Scope and selectivity.
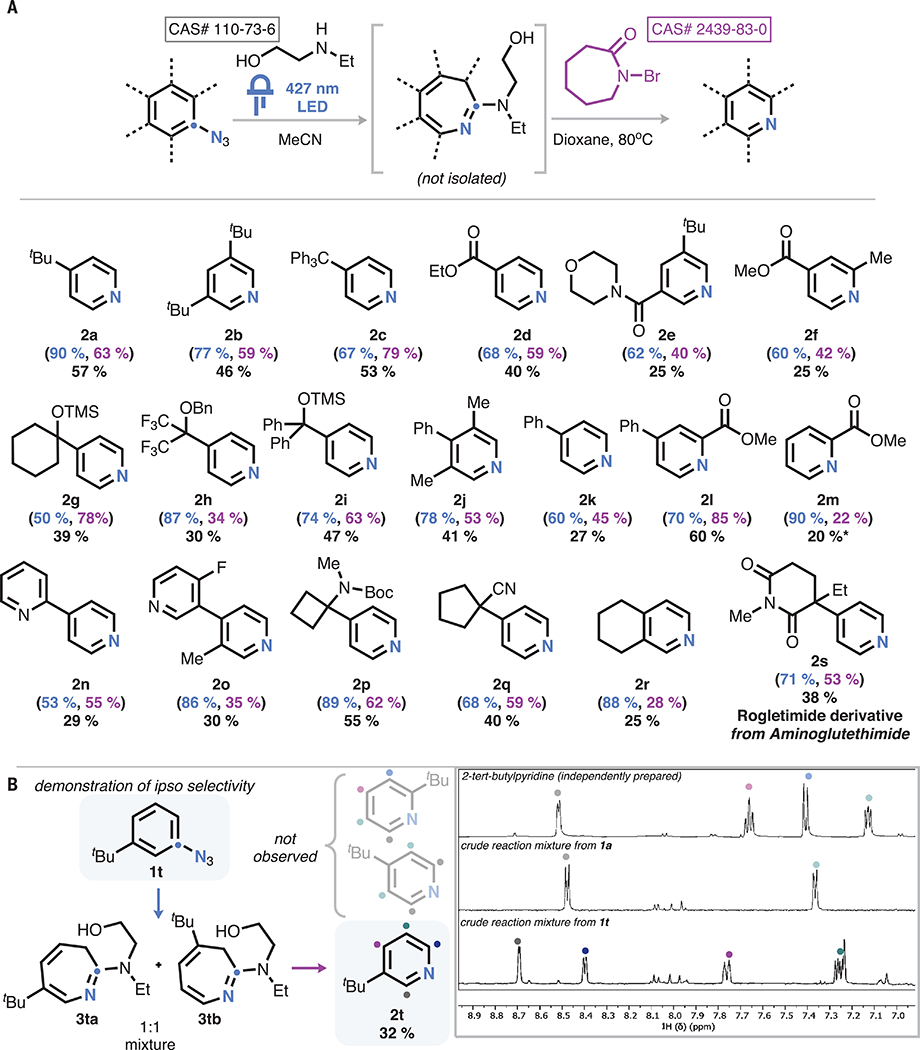
(A) Scope of the reaction. Standard conditions: (i) 0.3 mmol aryl azide, 0.3 mmol ethylaminoethanol, 3 ml MeCN, and 427-nm light-emitting diode (LED); (ii) 0.3 mmol DBU, 3 ml dioxane heated to 80°C, and 0.6 mmol N-bromocaprolactam added dropwise at 80°C. Isolated yield of pyridine from aryl azide (black). Proton NMR (1H-NMR) yield of photolysis (blue) and interpolated yield of pyridine based on azepine yield (purple), shown in parentheses. Asterisk indicates NMR yield. (B) Demonstration that nonsymmetric substrates form only one pyridine isomer.
A critically important feature of our protocol is that in cases in which two different azepine isomers are formed during photolysis, both converge to a common product. Indeed, for substrate 1t, we could observe the formation of both azepine isomers in a 1:1 ratio. Treatment of these azepines, either as a mixture (Fig. 3B) or upon separation (supplementary materials), led to the formation of the ipso-substitution product 2t as the exclusive pyridine product, with no detectable formation of its isomeric pyridines. Although isomers 3ta and 3tb both yielded pyridine, they proceeded with differing yields, suggesting that the specific steric profile of the azepine does influence the oxidation efficiency.
Limitations manifest in both steps of the reaction. For example, functionalities in the ortho position that can competitively react with the photogenerated nitrene (for example, Cadogan cyclization) are not well tolerated during photolysis (supplementary materials) (40, 41). Some substrates also have limitations inherent to their photophysics. For example, 4-azidobiphenyl undergoes facile intersystem crossing relative to ring expansion and therefore generates substantial quantities of triplet nitrene products such as azobiphenyl and 4-phenylaniline (42). Furthermore, resonance donors such as alkoxy and amine substituents in the ortho and para positions of the aryl ring favor the formation of quinone-type resonance forms upon nitrene generation, which we have found not to productively form azepines. The oxidation carries a different set of limitations. Free alcohols and amines do not productively react with N-bromocaprolactam, even in the presence of excess base, necessitating their protection. In some instances, overoxidation of the intermediate azepine leads instead to bromopyridines or bipyridines as prominent by-products. In this regard, we found that substrates bearing sterically demanding groups in the para position tended to avoid these side reactions (compare 2l versus 2m; 2a versus 2t) and as such served as the most general substrate class.
To further demonstrate the applications of this protocol, we examined its use in a more complex setting (Fig. 4A). Estrone can be converted to the corresponding azide in three steps; upon subsequent application of our protocol, the pyridine analog (2u) can be accessed in 10% overall yield from the parent steroid. This compound had previously been synthesized en route to a potential 5α-reductase inhibitor and was prepared from nortestosterone (despite its homology to the far more widely available estrone) in 11 steps and <1% overall yield (43).
Fig. 4. Applications of nitrene internalization.
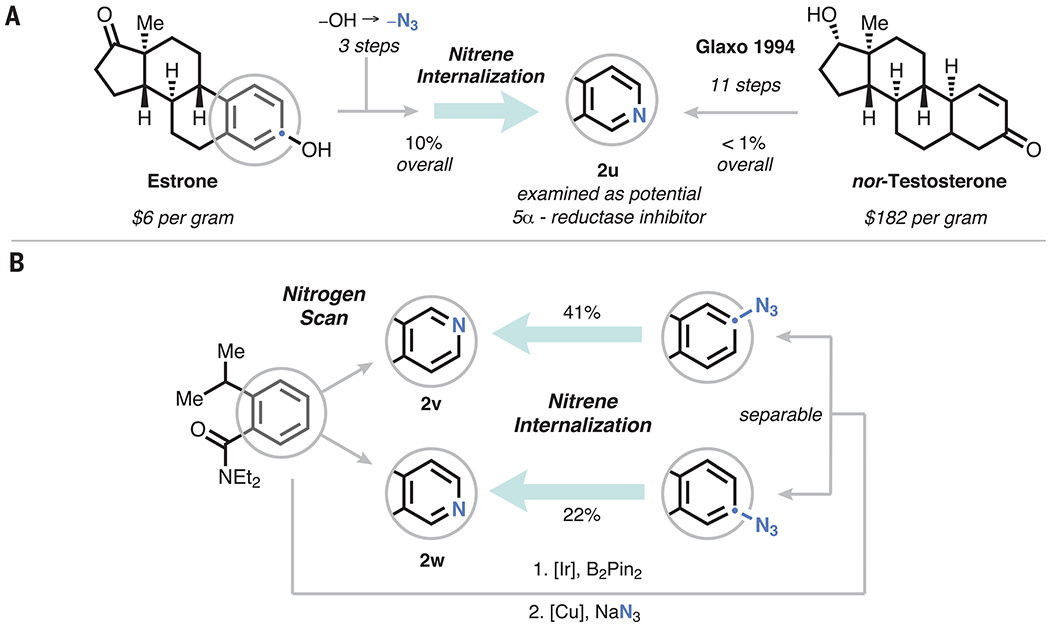
(A) Synthesis of an azasteroid from estrone and comparison with published route from nortestosterone. (B) Demonstration of a prototypical nitrogen scan leveraging an unselective C–H borylation.
Lastly, we conducted a direct nitrogen scan on a simple model arene (Fig. 4B). Application of an unselective iridium-catalyzed C–H borylation of an asymmetrically 1,2-disubstituted benzene followed by Chan-Lam azidation afforded two separable, isomeric aryl azides. Subsequent nitrene internalization of each of these precursors afforded isomeric pyridines 2v and 2w from a single parent without contamination by any other pyridine isomers.
We have developed a transformation that enables site-specific replacement of a carbon atom in an aromatic ring with a nitrogen atom. Because installation of the requisite azide can proceed from a variety of conveniently installed functional groups, and because the reaction promotes deletion of the targeted ipso carbon, site-specific, predictable replacement reactions can be conducted, enabling nitrogen scan operations that serve as a direct chemical analog to the common medicinal chemistry strategy. We anticipate a wide range of exciting applications for this selective benzene-to-pyridine transformation, as well as the underlying spirocyclic azanorcaradiene mechanism that underpins it.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Snyder (UChicago) for helpful discussions. We thank P. Kelly (UChicago) for assistance with separations. The University of Chicago’s Research Computing Center is thanked for computational resources.
Funding:
M.D.L. acknowledges the NIH (R35 GM142768) for research support. J.L.D. thanks the NSF Graduate Research Fellowship (DGE: 2140001).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data are available in the supplementary materials.
REFERENCES AND NOTES
- 1.Hu Y, Stumpfe D, Bajorath J, J. Med. Chem 60, 1238–1246 (2017). [DOI] [PubMed] [Google Scholar]
- 2.St Jean DJ Jr., Fotsch C, J. Med. Chem 55, 6002–6020 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Meanwell NA, J. Med. Chem 54, 2529–2591 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Pennington LD, Moustakas DT, J. Med. Chem 60, 3552–3579 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Pennington LD, Collier PN, Comer E, Med. Chem. Res 32, 1278–1293 (2023). [Google Scholar]
- 6.Lanman BA et al. , J. Med. Chem 63, 52–65 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Andrews SW, Blake JF, Chicarelli MJ, Golos A, Haas J, Jiang Y, Kolakowski GR, Substituted pyrazolo[1,5-A]pyridine compounds as RET kinase inhibitors. US Patent US10023570B2 (2018).
- 8.Iwamura R. et al. , J. Med. Chem 61, 6869–6891 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Joule JA, Mills K, Heterocyclic Chemistry (Wiley-Blackwell, ed. 5, 2010). [Google Scholar]
- 10.Blakemore DC et al. , Nat. Chem 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Campos KR et al. , Science 363, eaat0805 (2019).30655413 [Google Scholar]
- 12.Jurczyk J. et al. , Nat. Synth 1, 352–364 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fout AR, Bailey BC, Tomaszewski J, Mindiola DJ, J. Am. Chem. Soc 129, 12640–12641 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Morofuji T, Inagawa K, Kano N, Org. Lett 23, 6126–6130 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Lyu H, Kevlishvili I, Yu X, Liu P, Dong G, Science 372, 175–182 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Woo J. et al. , Science 376, 527–532 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, Cheng X, Nat. Commun 13, 425 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doering W. von E., Odum RA, Tetrahedron 22, 81–93 (1966). [Google Scholar]
- 19.Gritsan NP, Platz MS, Chem. Rev 106, 3844–3867 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Borden WT et al. Acc. Chem. Res 33, 765–771 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Smalley RK, “Azepines: 3.1. Azepines, Cyclopentazepines and Related Systems” in Hetarenes IV: Six-Membered and Larger Hetero Rings with Maximum Unsaturation, vol. E9d of Methods of Organic Chemistry (Houben-Weyl), Schaumann E, Ed. (Georg Thieme Verlag, 1997), pp. 108–298. [Google Scholar]
- 22.Sundberg RJ, Suter SR, Brenner M, J. Am. Chem. Soc 94, 513–520 (1972). [Google Scholar]
- 23.Patel SC, Burns NZ, J. Am. Chem. Soc 144, 17797–17802 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Streef JW, van der Plas HC, Wei YY, Declercq JP, Van Meerssche M, Heterocycles 26, 685 (1987). [Google Scholar]
- 25.Bátori S, Gompper R, Meier J, Wagner H-U, Tetrahedron 44, 3309–3318 (1988). [Google Scholar]
- 26.Ogata M, Matsumoto H, Kano H, Tetrahedron 25, 5217–5226 (1969). [Google Scholar]
- 27.Anderson M, Johnson AW, J. Chem. Soc. C Org 1966, 1075–1078 (1966). [Google Scholar]
- 28.Satake K. et al. , Chem. Lett 25, 1129–1130 (1996). [Google Scholar]
- 29.Anderson M, Johnson AW, J. Chem. Soc. Resumed 1965, 2411–2422 (1965). [Google Scholar]
- 30.Sundberg RJ, Das BP, Smith RH, J. Am. Chem. Soc 91, 658–668 (1969). [Google Scholar]
- 31.Wentrup C, J. Chem. Soc. D 0, 1386–1387 (1969). [Google Scholar]
- 32.Karimi S. et al. , Tetrahedron 69, 147–151 (2013). [Google Scholar]
- 33.Karimi S. et al. , Tetrahedron Lett. 56, 6886–6889 (2015). [Google Scholar]
- 34.Li G, Lavagnino MN, Ali SZ, Hu S, Radosevich AT, J. Am. Chem. Soc 145, 41–46 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amano A, Mukai T, Nakazawa T, Okayama K, Bull. Chem. Soc. Jpn 49, 1671–1675 (1976). [Google Scholar]
- 36.Perera TA, Reinheimer EW, Hudnall TW, J. Am. Chem. Soc 139, 14807–14814 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Vilsmaier E, Kristen G, Tetzlaff C, J. Org. Chem 53, 1806–1808 (1988). [Google Scholar]
- 38.Cordonier CEJ et al. , J. Org. Chem 70, 3425–3436 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Miles DH, Veguillas M, Toste FD, Chem. Sci 4, 3427–3431 (2013). [Google Scholar]
- 40.Lindley JM, McRobbie IM, Meth-Cohn O, Suschitzky H, J. Chem. Soc., Perkin Trans. 1 1977, 2194–2204 (1977). [Google Scholar]
- 41.Smith PAS, Brown BB, J. Am. Chem. Soc 73, 2435–2437 (1951). [Google Scholar]
- 42.Tsao M-L et al. , J. Am. Chem. Soc 125, 9343–9358 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Haffner C, Tetrahedron Lett. 35, 1349–1352 (1994). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the supplementary materials.