

Abstract
Background
RARS2-related mitochondrial disorder is an autosomal recessive mitochondrial encephalopathy caused by biallelic pathogenic variants in the gene encoding the mitochondrial arginyl-transfer RNA synthetase 2 (RARS2, MIM *611524, NM_020320.5). RARS2 catalyzes the transfer of L-arginine to its cognate tRNA during the translation of mitochondrially-encoded proteins. The classical presentation of RARS2-related mitochondrial disorder includes pontocerebellar hypoplasia (PCH), progressive microcephaly, profound developmental delay, feeding difficulties, and hypotonia. Most patients also develop severe epilepsy by three months of age, which consists of focal or generalized seizures that frequently become pharmacoresistant and lead to developmental and epileptic encephalopathy (DEE).
Case presentation
Here, we describe a six-year-old boy with developmental delay, hypotonia, and failure to thrive who developed an early-onset DEE consistent with Lennox-Gastaut Syndrome (LGS), which has not previously been observed in this disorder. He had dysmorphic features including bilateral macrotia, overriding second toes, a depressed nasal bridge, retrognathia, and downslanting palpebral fissures, and he did not demonstrate progressive microcephaly. Whole genome sequencing identified two variants in RARS2, c.36 + 1G > T, a previously unpublished variant that is predicted to affect splicing and is, therefore, likely pathogenic and c.419 T > G (p.Phe140Cys), a known pathogenic variant. He exhibited significant, progressive generalized brain atrophy and ex vacuo dilation of the supratentorial ventricular system on brain MRI and did not demonstrate PCH. Treatment with a ketogenic diet (KD) reduced seizure frequency and enabled him to make developmental progress. Plasma untargeted metabolomics analysis showed increased levels of lysophospholipid and sphingomyelin-related metabolites.
Conclusions
Our work expands the clinical spectrum of RARS2-related mitochondrial disorder, demonstrating that patients can present with dysmorphic features and an absence of progressive microcephaly, which can help guide the diagnosis of this condition. Our case highlights the importance of appropriate seizure phenotyping in this condition and indicates that patients can develop LGS, for which a KD may be a viable therapeutic option. Our work further suggests that analytes of phospholipid metabolism may serve as biomarkers of mitochondrial dysfunction.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12883-024-03571-w.
Keywords: RARS2, Mitochondrial disease, Dysmorphic features, Lennox-Gastaut Syndrome, Untargeted metabolomics analysis
Introduction
RARS2 (MIM*611524, NM_020320.5) is a nuclear gene that encodes the mitochondrial arginyl-transfer RNA synthetase 2, an aminoacyl-tRNA synthetase (aaRS), which charges human mitochondrial tRNA with arginine during the translation of mitochondrial proteins required for ATP production. Biallelic pathogenic variants in RARS2 cause an autosomal recessive mitochondrial encephalopathy known as RARS2-related mitochondrial disorder [1]. Approximately seventy patients with this condition have been reported in the literature [2–6]. The classic presentation of RARS2-related mitochondrial disorder includes pontocerebellar hypoplasia (PCH), epilepsy, profound developmental delay, feeding difficulties, progressive microcephaly, and hypotonia. Various metabolic derangements have also been observed, including metabolic acidosis, hypoglycemia, hyperammonemia, abnormalities in acylcarnitine profile and urine organic acid analysis, and lactic acidosis in blood, cerebrospinal fluid (CSF), and urine [3–5].
Most patients with RARS2-related mitochondrial disorder also develop severe epilepsy, experiencing their first seizure by three months of age [6]. A wide variety of focal, multifocal, or generalized seizure types can be observed in RARS2-related mitochondrial disorder [6]. The most common of these are myoclonic, focal clonic, or generalized tonic–clonic seizures. However, patients may experience several other seizure types, including focal with impaired awareness, atonic, tonic, or absence seizures, and they may develop episodes of convulsive or non-convulsive status epilepticus [6]. Epilepsy in RARS2-related mitochondrial disorder often becomes pharmacoresistant and evolves to developmental and epileptic encephalopathy (DEE) [1–6].
We describe a six-year-old boy with RARS2-related mitochondrial disorder who developed an early-onset DEE consistent with Lennox-Gastaut syndrome (LGS), which has not been reported before. He presented with progressive, generalized brain atrophy and lacked PCH on brain MRI. Furthermore, he exhibited several dysmorphic features on exam that have not been previously observed. A ketogenic diet (KD) reduced seizure frequency and allowed him to progress developmentally. Untargeted metabolomics analysis demonstrated elevations in compounds involved in lysophospholipid and sphingomyelin metabolism. Our report expands the clinical spectrum of RARS2-related disorder and suggests that patients may develop LGS, for which a KD may be beneficial. In addition, our patient’s metabolomics analysis may shed light on potential biomarkers associated with mitochondrial disorders.
Case presentation
The patient was born uneventfully at thirty-nine weeks gestation. At one month of age, he developed myoclonia of his right eyelid. The myoclonia was found to represent focal motor epilepsy and improved with phenobarbital. At four months of age, he developed infantile spasms and experienced a developmental plateau. A brain MRI at seven months of age was notable for large subarachnoid spaces, ventriculomegaly, and brain atrophy. Family history was negative for epilepsy or intellectual disability. However, by nine months of age, he showed minimal alertness and could not babble, orient to sounds, reach for toys, pull to stand, or interact with others. At twelve months of age, he had limited social interactions, demonstrated hypotonia and poor truncal stability, lacked head control, and was unable to stand but could move his extremities against gravity. His head circumference (HC), measured as the occipitofrontal circumference (OFC), was at the 87th percentile (WHO Boys HC 0–5 years) at twelve months of age. Brain MRI was notable for large subarachnoid spaces, ventriculomegaly, generalized brain atrophy, and a minimal reduction in brainstem size, but it demonstrated a normal cerebellar volume (Fig. 1 Top Left, Top Right).
Fig. 1.
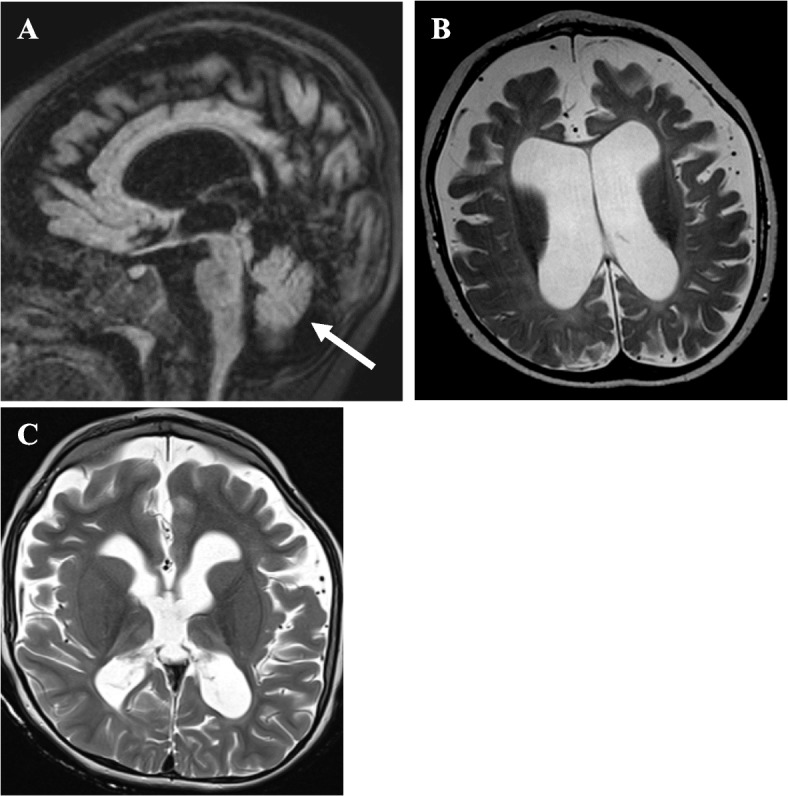
Brain MRIs of a patient with RARS2-related Mitochondrial Disorder. (Top Left) Sagittal FLAIR image of brain MRI at twelve months of age showing large subarachnoid spaces, ventriculomegaly, and generalized brain atrophy. There is a minimal reduction in brainstem size, and the volume of the cerebellum is preserved (white arrow). (Top Right) Axial T2W image at twelve months of age demonstrating ex vacuo enlargement of the ventricles and widening of the subarachnoid spaces. (Bottom Left) Axial T2W image at four years and six months of age demonstrating new T2 hyperintense signals in the bilateral thalami in addition to generalized volume loss, thinning of the corpus callosum, ex vacuo enlargement of the ventricles, and widening of the subarachnoid spaces
A 24-h EEG demonstrated a modified hypsarrhythmic pattern with multifocal spikes greatest posteriorly, which, together with his clinical course, indicated West Syndrome (Supplementary Table 1). Spasms persisted despite the use of ACTH, prednisolone, and vigabatrin; thus, treatment with zonisamide and clobazam was initiated. Screening for a KD was performed and demonstrated a normal serum lactate at 0.7 mmol/L (reference: 0.2–1.7 mmol/L) but revealed carnitine deficiency with a free carnitine of 15 µmol/L (reference: 25–54 µmol/L). KD was deferred in favor of optimizing anti-seizure medications (ASMs). However, carnitine supplementation was initiated with a dose of 100 mg/kg/day. He experienced improvements in tone and began to kick his legs and move his arms, enabling him to participate in physical and occupational therapies. He developed significant oral phase dysphagia related to his underlying mitochondrial disorder that led to poor weight gain. Subsequently, at twenty-six months and one week of age, he was diagnosed with failure to thrive (FTT) with a weight of 10.7 kg (3rd percentile, CDC Boys 2–20). His height was 89.6 cm (64th percentile, CDC Boys 2–20), and his HC was 47 cm (22nd percentile, WHO Boys HC 0–5 years).
By three years and six months of age, his spasms occurred in clusters approximately twice per day. The addition of cannabidiol to his existing ASM regimen reduced the frequency of his spasms to sporadic events. However, he developed new episodes of shaking and teeth clenching, which were found to be caused by multifocal myoclonic seizures. Myoclonic seizures occurred approximately three times a month but were concerning for evolution of his West Syndrome to LGS. As such, rufinamide was added to his ASM regimen. He continued to show some developmental gains. He could recognize when others were talking to him, turn his head, and show interest in others. He continued to have difficulty gaining weight (12.7 kg at four years of age, < 5th percentile, z-score -2.17). Thus, a gastrostomy tube was placed with subsequent improvement in weight gain.
He was admitted to the intensive care unit (ICU) at four years and six months of age in refractory status epilepticus triggered by a rhinoviral infection. Evaluation by the Genetics consult service demonstrated downslanting palpebral fissures, bilateral macrotia, a depressed nasal bridge, retrognathia, and bilateral overriding second toes. A 24-h EEG showed numerous tonic and myoclonic seizures, frequent generalized spike-and-wave discharges at 1.5–2.5 Hz, and generalized paroxysmal fast activity while asleep, which together indicated LGS. A brain MRI was notable for progressive, generalized volume loss of the brain and signal abnormality in bilateral cerebral hemispheres (Fig. 1 Bottom Left). A next-generation sequencing panel for 302 epilepsy genes was obtained but was non-diagnostic. Given the patient’s critical condition and the high risk of clinical decompensation and death, rapid trio whole genome sequencing (WGS) was requested to analyze variants in coding and non-coding regions as well as pathogenic variants and single deletions in mitochondrial DNA (mtDNA). The test was performed by Baylor Genetics (BG). WGS did not detect any pathogenic copy number variants (CNVs) but returned positive for two single nucleotide variants (SNVs) in RARS2, c.419 T > G (p.Phe140Cys) and c.36 + 1G > T. The first variant, c.419 T > G (p.Phe140Cys), is a previously reported pathogenic missense variant inherited from the patient’s father, and c.36 + 1G > T is an unpublished variant inherited from his mother. The c.419 T > G (p.Phe140Cys) variant (ClinVar Variation ID: 215055, Accession SCV000992715.2) has been observed in gnomAD with a frequency of 0.010%. It has an inconclusive theoretical prediction score (CADD: 29.500) but affects a highly conserved residue. The c.36 + 1G > T variant is expected to disrupt a canonical splicing donor site in intron 1, which may lead to splicing defects such as exon skipping or intron retention and, thereby, disrupt normal protein function (SpliceAI: 0.970). It is, therefore, predicted to be deleterious (CADD: 33) and is reported as likely pathogenic (ClinVar Variation ID: 2442235, Accession SCV003835680.1). The level of growth differentiation factor 15 (GDF15) was elevated at 933 pg/mL (reference ≤ 750 pg/mL). Plasma untargeted metabolomics analysis was performed by BG on an ultrahigh-performance liquid chromatography-tandem mass spectrometry platform, as previously reported [7]. The test revealed elevations of carnitine metabolites due to carnitine supplementation and increased levels of analytes in the lysophospholipid and sphingomyelin subpathways (Table 1).
Table 1.
Metabolomic perturbations in complex lipid pathways in a patient with RARS2-related mitochondrial disorder
| BIOCHEMICAL | Z-score | CATEGORY | HMDB_ID |
|---|---|---|---|
| 1-docosapentaenoyl-GPC (22:5n3) | 3.6 | Lysophospholipid | HMDB0010403 |
| 1-eicosapentaenoyl-GPC (20:5) | 3.2 | Lysophospholipid | HMDB0010397 |
| 1-stearoyl-GPI (18:0) | 2.0 | Lysophospholipid | HMDB0240261 |
| 1-dihomo-linolenoyl-GPC (20:3n3 or 6) | 2.0 | Lysophospholipid | HMDB0010394 |
| 1-docosahexaenoyl-GPC (22:6) | 2.3 | Lysophospholipid | HMDB0010404 |
| 1-stearoyl-2-oleoyl-GPC (18:0/18:1) | 2.0 | Phosphatidylcholine | HMDB0008038 |
| 1-oleoyl-2-dihomo-linolenoyl-GPC (18:1/20:3) | 1.9 | Phosphatidylcholine | HMDB0008113 |
| 1-stearoyl-2-docosapentaenoyl-GPC (18:0/22:5n3) | 3.9 | Phosphatidylcholine | HMDB0008056 |
| 1-palmitoyl-2-eicosapentaenoyl-GPC (16:0/20:5) | 3.3 | Phosphatidylcholine | HMDB0007984 |
| 1-stearoyl-2-dihomo-linolenoyl-GPC (18:0/20:3n3 or 6) | 3.1 | Phosphatidylcholine | HMDB0008047 |
| phosphatidylcholine (18:0/20:5, 16:0/22:5n6) | 2.8 | Phosphatidylcholine | HMDB0008050, HMDB0007989 |
| 1-stearoyl-2-docosahexaenoyl-GPC (18:0/22:6) | 2.2 | Phosphatidylcholine | HMDB0008057 |
| 1-palmitoyl-2-docosahexaenoyl-GPC (16:0/22:6) | 2.2 | Phosphatidylcholine | HMDB0007991 |
| 1-stearoyl-2-docosahexaenoyl-GPE (18:0/22:6) | 2.6 | Phosphatidylethanolamine | HMDB0009012 |
| 1-palmitoyl-2-docosahexaenoyl-GPE (16:0/22:6) | 2.2 | Phosphatidylethanolamine | HMDB0008946 |
| N-stearoyl-sphingosine (d18:1/18:0) | 2.4 | Ceramides | HMDB0004950 |
| palmitoyl dihydrosphingomyelin (d18:0/16:0) | 2.1 | Dihydrosphingomyelins | HMDB0010168 |
| sphingomyelin (d18:0/18:0, d19:0/17:0) | 2.0 | Dihydrosphingomyelins | HMDB0012087 |
| stearoyl sphingomyelin (d18:1/18:0) | 2.7 | Sphingomyelins | HMDB0001348 |
| palmitoyl sphingomyelin (d18:1/16:0) | 2.4 | Sphingomyelins | HMDB0010169 |
| sphingomyelin (d18:1/24:1, d18:2/24:0) | 2.2 | Sphingomyelins | HMDB0012107 |
| behenoyl sphingomyelin (d18:1/22:0) | 2.2 | Sphingomyelins | HMDB0012103 |
| lignoceroyl sphingomyelin (d18:1/24:0) | 2.1 | Sphingomyelins | HMDB0011697 |
The genomic findings, in conjunction with his clinical picture and elevated levels of GDF15, were thought to confirm the diagnosis of RARS2-related mitochondrial disorder. Given the diagnosis of a mitochondrial disorder, ubiquinol supplementation was initiated with a dose of 8 mg/kg/day, and zonisamide was discontinued due to a lack of efficacy. Given his failure of several ASMs and the refractory nature of his epilepsy, a KD was implemented at four years and nine months of age. A KD reduced spasm frequency to rare events and markedly increased his alertness and awareness. He showed improvements in his gross motor skills and gained the ability to stand, which enabled him to better engage in therapies. HC at four years and nine months was 49.4 cm (20th percentile, WHO Boys HC 0–5). Repeat EEG, after initiation of a KD, showed only one generalized myoclonic-tonic seizure and one cluster of epileptic spasms. He has rare myoclonic-tonic seizures and continues to make developmental gains.
Discussion and conclusions
RARS2 is an aminoacyl tRNA synthetase that charges cognate tRNAs with L-arginine during mitochondrial protein translation [1]. The pathomechanism of disease in RARS2-related mitochondrial disorder is caused by loss of function biallelic variants in RARS2 that lead to reduced RARS2 expression and enzymatic activity [8, 9]. The classic findings of PCH, progressive microcephaly, developmental delay, and severe epilepsy occur because tissues with high metabolic demands, including the brain, lose the ability to effectively generate adenosine triphosphate (ATP) via oxidative phosphorylation [1, 10]. As such, patients with RARS2-related mitochondrial disorder frequently demonstrate structural abnormalities on brain MRI (Supplementary Table 1). Forty-eight percent of patients have been reported to have PCH [11], while 71% demonstrate supratentorial defects such as cerebral atrophy, thinning of the corpus callosum, or ventriculomegaly [3]. Cerebral atrophy leads to loss of cerebral white matter, and this finding is observed in several patients with RARS2-related mitochondrial disorder (Supplementary Table 1). The exact consequence of white matter loss in RARS2-related mitochondrial disorder is unclear. However, a selective loss of white matter tracts is observed in patients with aaRS disorders more generally [12]. Moreover, in other forms of leukoencephalopathy, the degree of white matter involvement correlates with phenotypic severity. The degree of white matter loss in RARS2-related mitochondrial disorder may, therefore, correspond with the level of encephalopathy or epilepsy [13, 14]. Cerebral atrophy may also reduce the amount of cortical gray matter, which can be a late finding in RARS2-related mitochondrial disorder (Supplementary Table 1). Gray matter abnormalities frequently cause seizures. Thus, cortical volume loss with or without cerebral atrophy may contribute to epilepsy in RARS2-related mitochondrial disorder. Our patient’s brain MRI at four years and six months of age was notable for significantly progressive, generalized volume loss of the brain. It also demonstrated ex vacuo dilatation of the supratentorial ventricular system, which is likely secondary to the global brain atrophy and a thinned corpus callosum, and it did not show PCH. Generalized atrophy and the absence of PCH have been described previously, so our patient’s MRI findings are consistent with those observed in other patients and likely contributed to his neurologic presentation.
Over fifty RARS2 variants have been reported (Supplementary Table 1). Approximately 60% are missense variants and 20% are splice-site variants [3]. The remainder is a combination of small deletions or insertions, nonsense pathogenic variants, translation initiation codon variants, and frameshift pathogenic variants [2–4, 15]. Rapid WGS was performed in our patient given his critical status and high risk of deterioration and death and revealed two variants in RARS2, c.419 T > G, (p.Phe140Cys) and c.36 + 1G > T. c.419 T > G. The first variant (p.Phe140Cys) is a previously published pathogenic missense variant, and c.36 + 1G > T is an unpublished variant that is predicted to disrupt a splicing donor site in intron 1 and is, thereby, predicted to be deleterious. Both variants likely reduce RARS2 expression or activity, thereby leading to disease via a loss-of-function mechanism. Our report, therefore, expands the spectrum of known pathogenic variants in this condition.
Our patient experienced severe epilepsy, first developing focal motor seizures and subsequently West Syndrome, which ultimately evolved into LGS. The diagnosis of LGS was based on his tonic and myoclonic seizures and an EEG, which demonstrated a diffuse slow spike and wave pattern and generalized paroxysms of fast activity in sleep. Our report is the first to document LGS in RARS2-related mitochondrial disorder. However, given the prevalence and severity of epilepsy in this condition, other unreported patients with RARS2-related mitochondrial disorder may have LGS. Given the diagnosis of LGS and the pharmacoresistance of his epilepsy, a KD was implemented at four years and nine months of age, which dramatically reduced his seizure frequency, improved his alertness, and facilitated his acquisition of developmental milestones. The degree of seizure control experienced by our patient has not been seen in other patients with RARS2-related mitochondrial disorder treated with a KD (Supplementary Table 1). Valles-Ibanez et al. report a girl who developed a progressive movement disorder by nine months of age that subsequently progressed to a myoclonic DEE. Despite having tried over ten ASMs in addition to a KD, this patient suffers from daily atypical absence, myoclonic, and atonic seizures [6]. Ngoh et al. describe two male siblings with RARS2-related mitochondrial disorder who developed West syndrome and refractory epilepsy requiring several ASMs [16]. Treatment with a KD led to a modest reduction in seizure frequency in one sibling, while the other experiences daily seizures despite multiple ASMs and a KD. A female patient reported by Nishri et al. developed a severe DEE with focal, multifocal, and myoclonic seizures, which, too, was not well-controlled with multiple ASMs in addition to a KD [17]. More studies are needed to determine the exact efficacy of a KD in RARS2-related mitochondrial disorder. However, several clinical trials have demonstrated its benefit in managing epilepsy for other mitochondrial disorders [18–21]. In a group of fourteen patients with mitochondrial disorders treated with a KD, 10/14 patients demonstrated a greater than 50% reduction in seizure frequency, and 7/14 achieved complete seizure freedom [18]. Another study of twenty patients with LGS and mitochondrial complex I deficiency who received either a KD or Modified Atkins Diet (MAD) reported that seven patients experienced at least a 50% reduction in seizure frequency after two years of treatment [19]. A systematic review by Zweers et al. identified eight patients with defects in other genes associated with mitochondrial disease, including POLG, SLC25A12, MT-TL1, and MTO1, of whom seven experienced control of therapy-refractory epilepsy with a KD [20]. Ketosis achieved through such dietary therapies (DT) has been shown to reduce oxidative damage in the brain, decrease levels of mitochondrial reactive oxygen species, and increase the amount of glutathione peroxidase in hippocampal neurons [21]. Thereby, DT may exert neuroprotective and anticonvulsant effects by improving mitochondrial function, and their benefits may extend beyond epilepsy. Indeed, one patient with lysyl tRNA synthetase 2 (KARS2)–related mitochondrial disorder showed an improvement in psychomotor regression on a KD [22]. As our patient experienced fewer seizures with a KD and improved seizure control enabled him to make developmental gains, a KD may have a role in treating both epilepsy and developmental delay in RARS2-related mitochondrial disorder. The experience with our patient also emphasizes the need for careful seizure phenotyping in RARS2-related mitochondrial disorder, as a diagnosis of LGS may open new treatment options for patients, including a KD.
Metabolic abnormalities have been frequently reported in RARS2-related mitochondrial disorder, and the most common of these is lactic acidosis in serum and CSF [3]. Lactic acidosis has been observed in approximately 40% of patients with RARS2-related mitochondrial disorder and is likely a result of impaired mitochondrial respiration and oxidative ATP production [3, 12, 23]. Interestingly, we did not detect lactic acidosis in our patient. Multiple patients with RARS2-related mitochondrial disorder without lactic acidosis have been reported, so this feature may be variable [3]. However, untargeted plasma metabolomics was analyzed in our patient. This study showed several abnormalities, including mild to moderately increased levels of lysophospholipids, phosphatidylcholines, and sphingomyelins (and their precursors–ceramide and dihydrosphingomyelins). Untargeted plasma metabolomics analyses have not been previously reported in RARS2-related mitochondrial disorder or in other aaRS disorders. Thus, it is difficult to determine the exact cause and prevalence of these abnormalities. One possible explanation, however, given the degree of generalized atrophy and volume loss on brain MRI, is that the elevations in serum of the lysophospholipid compounds may reflect a peripheral process that mirrors the loss of myelin in the cerebral white matter of the central nervous system (CNS). Although the precise etiology of these perturbations is not clearly understood, a previous publication on another mitochondrial syndrome, sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD) (MIM #616084), caused by biallelic pathogenic variants in TRNT1 also demonstrated abnormalities in sphingolipid and lysophospholipid metabolism in untargeted metabolomics analysis of plasma [24]. Furthermore, when the plasma metabolomics profiles of twenty-five patients with mitochondrial disorders were interrogated using high-throughput targeted semiquantitative analysis, elevations in the phospholipid ethanolamine were observed in patients with progressive external ophthalmoplegia (PEO) [25]. As lysophospholipid and sphingomyelin are precursors of phospholipid metabolites, our patient’s untargeted metabolomics analysis and those of prior publications suggest a common pathway that may be affected in several mitochondrial disorders. However, the reliability of these changes in lysophospholipid compounds needs to be validated in a larger cohort of patients with RARS2-related mitochondrial disorder and other mitochondrial disorders.
Dysmorphology in RARS2-related mitochondrial disorder has been observed in less than 20% of patients [3]. Multiple patients have been described with bitemporal narrowing, edematous hands and feet, full cheeks, and a high-arched or narrow palate [9, 26]. Such features are likely to be consequences of hypotonia, severe developmental delay, and FTT. Other common dysmorphic features include adducted thumbs, high nasal bridges, prominent ears, a low philtrum, and trichiasis [3]. Our patient had bilateral macrotia, and this finding has been noted in previous reports. However, he demonstrated other features that have not been previously reported including a depressed nasal bridge, downslanting palpebral fissures, retrognathia, and bilateral overriding second toes (Supplementary Table 1). Although dysmorphisms are not cardinal features and do not follow a specific pattern in this mitochondrial syndrome, it is worth noting that they could be part of the spectrum. Progressive microcephaly is also commonly observed in RARS2-related mitochondrial disorder. Our patient demonstrated a normal HC of 49.4 cm at four years and nine months (20th percentile, WHO Boys HC 0–5) (Supplementary Table 1). Therefore, this feature may not be present in all patients with this condition.
Collectively, our case demonstrates several unique features of RARS2-related mitochondrial disorder, including dysmorphic features, the absence of progressive microcephaly, the presence of a unique metabolomics signature, and the development of LGS, for which a KD may prove beneficial. Our report expands the known phenotypes of RARS2-related mitochondrial disorder and should help broaden the clinical spectrum of the disease. The use of a KD should be considered to help manage epilepsy in patients with this disorder. Moreover, elevations in analytes of phospholipid metabolism are observed in plasma untargeted metabolomics analysis performed in this patient. Further metabolomics analyses in additional patients with mitochondrial disease are needed to determine whether this could be a relevant metabolomics signature in this patient population.
Supplementary Information
Acknowledgements
We thank Dr. Gary Clark of the Baylor College of Medicine’s Division of Pediatric Neurology and Developmental Neuroscience for supporting the publication of this study.
Abbreviations
- RNA
Ribonucleic acid
- tRNA
Transfer RNA
- RARS2
Arginyl-tRNA synthetase 2
- PCH
Pontocerebellar hypoplasia
- DEE
Developmental and epileptic encephalopathy
- LGS
Lennox-Gastaut Syndrome
- Phe
Phenylalanine
- Cys
Cysteine
- KD
Ketogenic diet
- MRI
Magnetic resonance imaging
- HC
Head circumference
- ACTH
Adrenocorticotropic Hormone
- FTT
Failure to thrive
- CDC
Centers for Disease Control and Prevention
- WHO
World Health Organization
- ASM
Anti-seizure medication
- EEG
Electroencephalogram
- BG
Baylor Genetics
- GDF15
Growth differentiation factor 15
- ATP
Adenosine triphosphate
- aaRS
Aminoacyl tRNA synthetase
- CADD
Combined annotation dependent depletion
- gnomAD
Genome Aggregation Database
- MAD
Modified Atkins Diet
- POLG
DNA polymerase gamma, catalytic subunit
- SLC25A12
Solute carrier family 25 member 12
- MT-TL1
Mitochondrially encoded tRNA leucine 1
- MTO1
Mitochondrial tRNA translation optimization 1
- DT
Dietary therapies
- KARS2
Lysyl-tRNA synthetase 2
- SIFD
Sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay
- TRNT1
tRNA nucleotidyl transferase 1
- PEO
Progressive external ophthalmoplegia
- mtDNA
mitochondrial DNA
- ICU
Intensive care unit
- CNV
Copy number variant
- SNV
Single nucleotide variant
- WGS
Whole genome sequencing
Authors’ contributions
K.M., F.S., Y.G., M.R.B., L.T.E. provided the case. A.S.W., K.M., L.T.E., and F.S. wrote and edited the manuscript. A.S.W collected data from the selected articles and performed the literature search. S.F.K. provided MRI images for Fig. 1 and feedback on the neuroradiologic findings. C.G. and S.E. analyzed metabolomics data, and C.G. prepared Table 1. F.S., K.M., and L.T.E. provided critical feedback and overall guidance. All authors have read and approved the final manuscript to be published.
Funding
The authors received no specific funding for this work.
Availability of data and materials
All data and materials supporting our findings are contained within the manuscript and in the additional data files (Additional File 1 FINAL.docx and Fig. 1 FINAL.docx). Additional File 1 FINAL.docx consists of a data table entitled “Supplementary Table 1: Genotype and phenotype comparison of patients with RARS2-related Mitochondrial Disorder”. Additional File 1 FINAL is a data table describing the various clinical and neuroradiologic findings of all published patients with RARS2-related Mitochondrial Disorder. The document format is.docx. The variants reported in the patient’s whole genome sequencing have been deposited in ClinVar by Baylor Genetics: RARS2 c.36 + 1G > T, Variation ID 2442235, Accession SCV003835680.1; RARS2 c.419 T > G, (p.Phe140Cys), Variation ID 215055, Accession SCV000992715.2.
Declarations
Ethics approval and consent to participate
According to the Baylor College of Medicine Internal Review Board (IRB) guidelines, only studies with n ≥ 3 patients constitute research and require an IRB-approved protocol and consent form. The patient's mother signed a consent form authorized by Texas Children's Hospital to approve publication of her child's case.
Consent for publication
Written informed consent was obtained from the parent of the participant for publication of identifying information/images in an online open-access publication. A copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Joseph JT, Innes AM, Smith AC, Vanstone MR, Schwartzentruber JA, Bulman DE, Majewski J, Daza RA, Hevner RF, Michaud J, Boycott KM; FORGE Canada Consortium. Neuropathologic features of pontocerebellar hypoplasia type 6. J Neuropathol Exp Neurol. 2014;73(11):1009–25. 10.1097/NEN.0000000000000123. [DOI] [PubMed]
- 2.Sevinç S, İnci A, Ezgü FS, Eminoğlu FT. A Patient with a Novel RARS2 Variant Exhibiting Liver Involvement as a New Clinical Feature and Review of the Literature. Mol Syndromol. 2022;13(3):226–234. 10.1159/000519604. [DOI] [PMC free article] [PubMed]
- 3.Zhang Y, Yu Y, Zhao X, Xu Y, Chen L, Li N, Yao R, Wang J, Yu T. Novel RARS2 Variants: Updating the Diagnosis and Pathogenesis of Pontocerebellar Hypoplasia Type 6. Pediatr Neurol. 2022;131:30–41. doi: 10.1016/j.pediatrneurol.2022.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Glamuzina E, Brown R, Hogarth K, Saunders D, Russell-Eggitt I, Pitt M, de Sousa C, Rahman S, Brown G, Grunewald S. Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J Inherit Metab Dis. 2012;35(3):459–467. doi: 10.1007/s10545-011-9413-6. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Wu BB, Wang HJ, Zhou SZ, Cheng GQ, Zhou YF. A term neonate with early myoclonic encephalopathy caused by RARS2 gene variants: a case report. Transl Pediatr. 2020;9(5):707–712. doi: 10.21037/tp-20-110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Valles-Ibáñez G, Hildebrand MS, Bahlo M, King C, Coleman M, Green TE, Goldsmith J, Davis S, Gill D, Mandelstam S, Scheffer IE, Sadleir LG. Infantile-onset myoclonic developmental and epileptic encephalopathy: A new RARS2 phenotype. Epilepsia Open. 2022;7(1):170–180. 10.1002/epi4.12553. [DOI] [PMC free article] [PubMed]
- 7.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81(16):6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 8.Kastrissianakis K, Anand G, Quaghebeur G, Price S, Prabhakar P, Marinova J, Brown G, McShane T. Subdural effusions and lack of early pontocerebellar hypoplasia in siblings with RARS2 mutations. Arch Dis Child. 2013;98(12):1004–1007. doi: 10.1136/archdischild-2013-304308. [DOI] [PubMed] [Google Scholar]
- 9.Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, Saada A, Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81(4):857–62. 10.1086/521227. [DOI] [PMC free article] [PubMed]
- 10.Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases. In: Wong LJC, editor. Mitochondrial disorders caused by nuclear genes. New York: Springer; 2013. pp. 263–276. [Google Scholar]
- 11.Nevanlinna V, Konovalova S, Ceulemans B, Muona M, Laari A, Hilander T, Gorski K, Valanne L, Anttonen AK, Tyynismaa H, Courage C, Lehesjoki AE. A patient with pontocerebellar hypoplasia type 6: Novel RARS2 mutations, comparison to previously published patients and clinical distinction from PEHO syndrome. Eur J Med Genet. 2020;63(3):103766. 10.1016/j.ejmg.2019.103766. [DOI] [PubMed]
- 12.Fine AS, Nemeth CL, Kaufman ML, Fatemi A. Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination. J Neurodev Disord. 2019;11(1):29. doi: 10.1186/s11689-019-9292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Berge L, Hamilton EM, Linnankivi T, Uziel G, Steenweg ME, Isohanni P, Wolf NI, Krägeloh-Mann I, Brautaset NJ, Andrews PI, de Jong BA, al Ghamdi M, van Wieringen WN, Tannous BA, Hulleman E, Würdinger T, van Berkel CG, Polder E, Abbink TE, Struys EA, Scheper GC, van der Knaap MS; LBSL Research Group. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: clinical and genetic characterization and target for therapy. Brain. 2014;137(Pt 4):1019–29. 10.1093/brain/awu026. [DOI] [PubMed]
- 14.van der Knaap MS, van der Voorn P, Barkhof F, Van Coster R, Krägeloh-Mann I, Feigenbaum A, Blaser S, Vles JS, Rieckmann P, Pouwels PJ. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann Neurol. 2003;53(2):252–258. doi: 10.1002/ana.10456. [DOI] [PubMed] [Google Scholar]
- 15.Bendeck JL, Villamizar I, Prieto C, Celis LG. Mutación heterocigota, autosómica recesiva del gen RARS2 en una paciente colombiana de padres no consanguíneos [Autosomal recessive heterocygote mutation of the RARS2 gene in a Colombian patient with non- consanguineous parents]. Arch Argent Pediatr. 2022;120(1): e39-e48. Spanish. 10.5546/aap.2022.e39. [DOI] [PubMed]
- 16.Ngoh A, Bras J, Guerreiro R, Meyer E, McTague A, Dawson E, Mankad K, Gunny R, Clayton P, Mills PB, Thornton R, Lai M, Forsyth R, Kurian MA. RARS2 mutations in a sibship with infantile spasms. Epilepsia. 2016;57(5): e97-e102. 10.1111/epi.13358. [DOI] [PMC free article] [PubMed]
- 17.Nishri D, Goldberg-Stern H, Noyman I, Blumkin L, Kivity S, Saitsu H, Nakashima M, Matsumoto N, Leshinsky-Silver E, Lerman-Sagie T, Lev D. RARS2 mutations cause early onset epileptic encephalopathy without ponto-cerebellar hypoplasia. Eur J Paediatr Neurol. 2016;20(3):412–417. doi: 10.1016/j.ejpn.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Kang HC, Lee YM, Kim HD, Lee JS, Slama A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia. 2007;48(1):82–88. doi: 10.1111/j.1528-1167.2006.00906.x. [DOI] [PubMed] [Google Scholar]
- 19.Na JH, Kim HD, Lee YM. Effective and safe diet therapies for Lennox-Gastaut syndrome with mitochondrial dysfunction. Ther Adv Neurol Disord. 2020;6(13):1756286419897813. doi: 10.1177/1756286419897813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zweers H, van Wegberg AMJ, Janssen MCH, Wortmann SB. Ketogenic diet for mitochondrial disease: a systematic review on efficacy and safety. Orphanet J Rare Dis. 2021;16(1):295. doi: 10.1186/s13023-021-01927-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paoli A, Bianco A, Damiani E, Bosco G. Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed Res Int. 2014;2014:474296. 10.1155/2014/474296. [DOI] [PMC free article] [PubMed]
- 22.Murofushi Y, Hayakawa I, Abe Y, Ohto T, Murayama K, Suzuki H, Takenouchi T, Kosaki K, Kubota M. Ketogenic Diet for KARS-Related Mitochondrial Dysfunction and Progressive Leukodystrophy. Neuropediatrics. 2022;53(1):65–68. doi: 10.1055/s-0041-1732446. [DOI] [PubMed] [Google Scholar]
- 23.Steenweg ME, Ghezzi D, Haack T, Abbink TE, Martinelli D, van Berkel CG, Bley A, Diogo L, Grillo E, Te Water Naudé J, Strom TM, Bertini E, Prokisch H, van der Knaap MS, Zeviani M. Leukoencephalopathy with thalamus and brainstem involvement and high lactate 'LTBL' caused by EARS2 mutations. Brain. 2012;135(Pt 5):1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- 24.Odom J, Amin H, Gijavanekar C, Elsea SH, Kralik S, Chinen J, Lin Y, Yates AMM, Mizerik E, Potocki L, Scaglia F. A phenotypic expansion of TRNT1 associated sideroblastic anemia with immunodeficiency, fevers, and developmental delay. Am J Med Genet A. 2022;188(1):259–268. doi: 10.1002/ajmg.a.62482. [DOI] [PubMed] [Google Scholar]
- 25.Buzkova J, Nikkanen J, Ahola S, Hakonen AH, Sevastianova K, Hovinen T, Yki-Järvinen H, Pietiläinen KH, Lönnqvist T, Velagapudi V, Carroll CJ, Suomalainen A. Metabolomes of mitochondrial diseases and inclusion body myositis patients: treatment targets and biomarkers. EMBO Mol Med. 2018;10(12):e9091. doi: 10.15252/emmm.201809091.PMID:30373890;PMCID:PMC6284386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rankin J, Brown R, Dobyns WB, Harington J, Patel J, Quinn M, et al. Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am J Med Genet Part A. 2010;152(8):2079–2084. doi: 10.1002/ajmg.a.33531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials supporting our findings are contained within the manuscript and in the additional data files (Additional File 1 FINAL.docx and Fig. 1 FINAL.docx). Additional File 1 FINAL.docx consists of a data table entitled “Supplementary Table 1: Genotype and phenotype comparison of patients with RARS2-related Mitochondrial Disorder”. Additional File 1 FINAL is a data table describing the various clinical and neuroradiologic findings of all published patients with RARS2-related Mitochondrial Disorder. The document format is.docx. The variants reported in the patient’s whole genome sequencing have been deposited in ClinVar by Baylor Genetics: RARS2 c.36 + 1G > T, Variation ID 2442235, Accession SCV003835680.1; RARS2 c.419 T > G, (p.Phe140Cys), Variation ID 215055, Accession SCV000992715.2.