

Abstract
Immunotherapy has been a paradigm shift in cancer treatment to produce durable responses. Unfortunately, most cancers do not respond to current immunotherapies, and thus, exploring novel mechanisms is crucial. Emerging data now demonstrate that protein modification by the small ubiquitin-like modifiers (SUMO) is a novel target to activate antitumor immunity.
Transcription regulation – the central theme in immune response
Cancer immunotherapy has produced a paradigm shift in generating durable responses in ways not seen with chemotherapy or targeted therapy alone. However, most cancers do not respond to currently available immunotherapies and are still treated with decades old cytotoxic chemo-therapies. This underscores the need to investigate novel mechanisms that inhibit antitumor immune responses.
Immune responses at the molecular level are gene regulation in the expression of cytokines, cell surface receptors, co-stimulatory, or inhibitory proteins, which regulate the activation, expansion, function, and differentiation of immune cells. Post-translational modifications by SUMO play important roles in gene regulation by reversible modification (SUMOylation) of transcription factors and chromatin-modifying proteins. This forum highlights recent advances in our understanding of the role of SUMOylation in antitumor immune responses.
SUMOylation regulates IFN-I response in innate immune cells
Recently, post-translational modifications by SUMO proteins were found to be a central regulator of the expression of interferon (IFN)-β, a type I IFN (IFN-I), and IFNγ-stimulated genes (ISGs) [1,2]. IFN-I inducers first spawned cancer immunotherapy more than 100 years ago when Dr William Coley discovered that bacterial extracts induced spontaneous and durable remission in some sarcoma patients. Now, the molecular mechanism has been elucidated. Bacterial extracts contain molecules that stimulate IFN-I response by binding cell surface pattern recognition receptors (PRRs) that recognize molecules associated with pathogens or tissue damage. The PRR pathways stimulate IFN-I expression by recruiting transcription factors to the promoters of IFN-I (IFN-α and IFN-β) genes. IFN-I activates dendritic cells for antigen presentation, which results in antigen-specific T cell expansion and differentiation. IFN-Is also stimulate innate immune cells [macrophages and natural killer (NK) cells] to enhance their antiviral and anticancer response. SUMOylation is formed by a cascade of reactions catalyzed by the SUMO E1 (a heterodimer of Sae1 and Uba2), E2 (Ubc9, gene name UBE2I), and an E3 (Figure 1). SUMO conjugated on target proteins can be removed by deSUMOylation enzymes. SUMOylation regulates protein functions by altering protein–protein interaction, protein stability, or activity. Deletion of SUMO E2 enzyme (Ubc9), deletion of both SUMO2 and SUMO3, or pharmacologic inhibition of the SUMO E1 enzyme greatly enhances IFN-β and ISG expression and induces dendritic cell maturation, activation, and enhances T cell priming [1–3].
Figure 1. The SUMO conjugation process and approaches available to achieve inhibition.
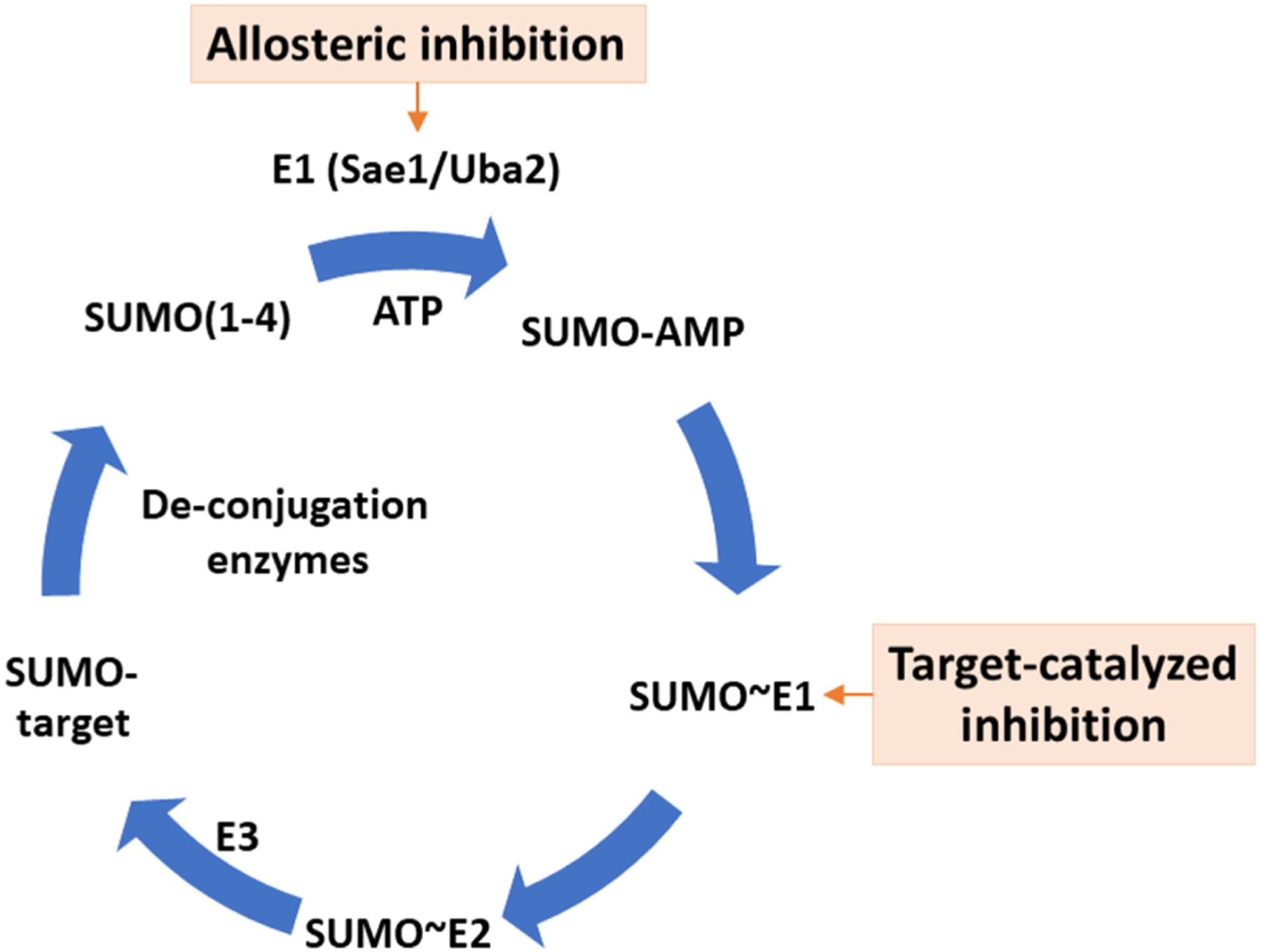
SUMOylation is formed by a cascade of reactions catalyzed by the SUMO E1 (a heterodimer of Sae1 and Uba2), E2 (Ubc9, gene name UBE2I), and an E3 ligase. SUMO modifications on target proteins are removed by deSUMOylation enzymes. Two approaches are available to achieve pharmacologic inhibition of SUMOylation by small molecules. One approach utilizes a target-catalyzed approach, in which binding of the drug to the SUMO E1~SUMO thioester conjugate leads to the formation of a SUMO-AMP mimetic that binds and inhibits SUMO E1. TAK-981 (subasumstat) is based on this mechanism. Another approach uses a covalent allosteric mechanism to target a deep cryptic pocket in the SUMO E1 enzyme, resulting in dramatic conformational changes that destroy the ATP-binding site of the enzyme.
SUMOylation inhibition does not induce IFN-β production through the canonical PRRs and has no effects on the IFNB1 promoter [4]. SUMOylation suppresses the IFNB1 gene enhancer, at least in part through chromatin modification [4]. Consequently, SUMOylation inhibition synergizes with canonical PRR pathway inducers, such as bacterial lipopolysaccharides, in the production of IFN-β and ISGs [1]. Through IFN-I autocrine signaling in macrophages, SUMOylation inhibition increases activities of NK cells and macrophages to eliminate cancer cells [4,5] (Figure 2).
Figure 2. The effects of SUMOylation inhibition on immune cells and on tumor cells are expected to be synergistic in inducing antitumor immunity.
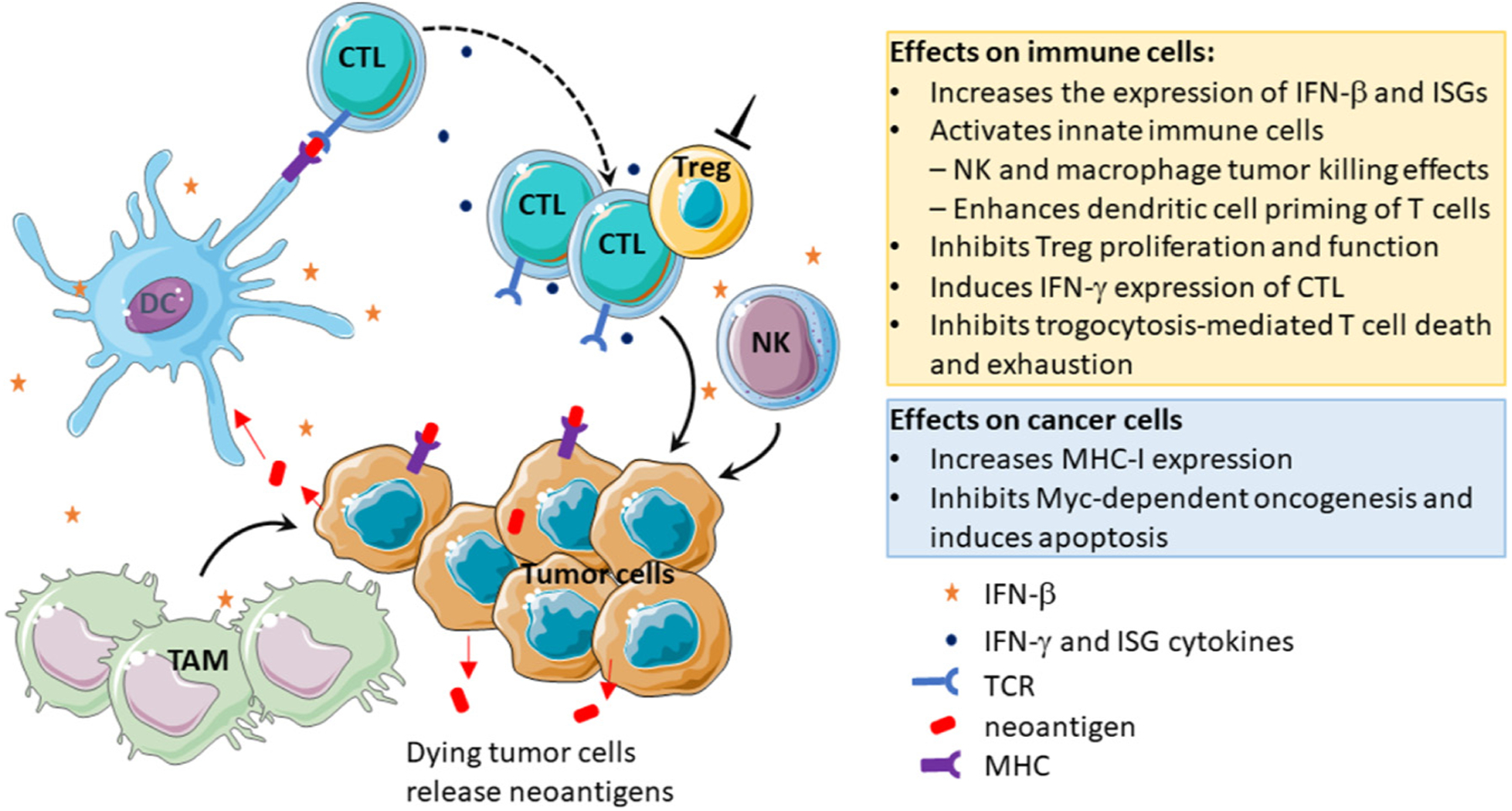
SUMOylation regulates gene expression in various cells through modification of transcription factors and chromatin regulators. SUMOylation inhibition of tumor cells inhibits Myc-dependent oncogenesis and induces apoptosis resulting in the release of neoantigens and could enhance MHC-I expression to enable cytotoxic T lymphocyte (CTL) recognition of neoantigens. SUMOylation inhibition of innate immune cells induces expression of interferon (IFN)-β by these cells that leads to autocrine signaling to activate innate immune cells [natural killer (NK) cells and tumor-associated macrophages (TAMs)] for their tumor elimination and induces dendritic cell (DC) activation and CTL priming. SUMOylation inhibition of CTL promotes the expression of IFN-γ and inhibits trogocytosis-mediated T cell exhaustion and death. Inhibition of SUMOylation in Treg cells inhibits their proliferation and function.
SUMOylation inhibition suppresses Tregs and enhances effector T cell functions through multiple mechanisms
SUMOylation is required for regulatory T cell (Treg) maintenance, proliferation, and suppressive function [6]. Ubc9-deficient Tregs in mice lost Foxp3 expression upon T cell receptor (TCR) stimulation [6]. In primary patient tumor tissues, pharmacological SUMOylation inhibition reduced Tregs [7] (Figure 2).
In contrast to the role of SUMOylation in Treg cells, pharmacological inhibition of SUMOylation directly enhances T cell responses to antigens ex vivo [3] (Figure 2). Pretreatment of mouse T cells with a small molecule SUMO E1 inhibitor, TAK-981 (subasumstat), followed by activation with phytohemagglutinin (PMA)/ionomycin, which bypasses TCRs to activate T cells, increases the percentage of IFNγ or granzyme B-expressing CD8+ T cell populations [3]. TAK-981 also increased the sensitivity of CD8+ T cells ex vivo in a TCR-binding affinity-dependent manner using the ovalbumin antigen (OVA) model [3]. Furthermore, SUMOylation inhibition blocks transfer of tumor-derived factors to T cells, a process known as trogocytosis, using OVA-specific CD8+ T cells and chimericantigen receptor (CAR) T cells [8]. Consequently, SUMOylation inhibition blocks trogocytosis-associated T cell exhaustion and death [8].
Expression of the MHC class I on tumor cells is required for T cell recognition and thus for antitumor T cell-mediated tumor cell killing. It was shown that pharmacologic inhibition of SUMOylation in lymphoma cells increased the expression of MHC-I as a single agent and synergized with IFN-γ to increase the level of MHC-I on tumor cell surfaces [9] (Figure 2).
Pharmacologic inhibition of SUMOylation
Two approaches are available to achieve pharmacologic inhibition of SUMOylation by small molecules. One approach utilizes a target-catalyzed approach, in which the drug binds the SUMO E1~SUMO thioester leading to the formation of a SUMO-AMP mimetic that binds and inhibits SUMO E1 [10] (Figure 1). The first-in-class clinical stage SUMOylation inhibitor, TAK-981 (subasumstat), is based on this mechanism.
Another approach uses a covalent allosteric mechanism [11] (Figure 1). A deep cryptic pocket in the SUMO E1 enzyme enables covalent inhibition by small molecules, resulting in dramatic conformational changes that destroy the ATP-binding site of the enzyme [12]. The duration of action of covalent drugs depends on the turnover rate of the protein targets, which is well suited for targeting SUMO and other Ubl E1 enzymes because their turnover rates are of the order of a day or longer. Both approaches that inhibit the SUMO E1 are applicable or potentially applicable to inhibit other Ubl E1 enzymes [3,11].
Effects of a first-in-class SUMOylation inhibitor in clinical trials
The immune modulatory effects of SUMOylation inhibition observed in preclinical studies have been validated in human clinical trials using TAK-981 [13,14]. Clinical studies of single agent subasumstat (NCT03648372) have shown that it increases chemokine CXCL10, a chemoattractant for T and NK cells, and a known ISG. In addition, subasumstat increased the expression of CD69 in T and NK cells, suggesting their increased activation. Based on the IFN-I and innate immune cell (NK cells and macrophages) activation effects, the current subasumstat Phase 1b/2 trials are in combination with cancer cell targeting anti-bodies, including the combination with an anti-CD38 antibody for treating multiple myeloma (NCT04776018), and with rituximab to treat non-Hodgkin’s lymphoma (NCT04074330). In addition, because TAK-981 activates dendritic cells for cytotoxic T lymphocyte (CTL) priming and suppress Tregs, combination with anti-PD1 (pembrolizumab) is also evaluated in clinical trials in nonsquamous non-small cell lung cancer (NSCLC), cervical cancer, microsatellite stable colorectal cancer (MSS-CRC), and cutaneous melanoma, with additional solid tumor indications planned for Phase 2. Recent meeting presentations have shown some responses in Phase 1 dose escalation in relapsed and refractory patient populations in lymphoma, NSCLC, and in MSS-CRC patients that do not usually respond to immune checkpoint inhibition (ICI) [13,14].
Concluding remarks and future perspectives
Innate immunity is a largely untapped resource for innovation in cancer immune therapies. The human body has two types of immunity – innate and adaptive immunity. Most current FDA-approved cancer immune therapies, such as ICI or CAR-T cell therapies, target adaptive immunity. However, innate immune cells, in particular tumor-associated macrophages (TAMs), often form a major subpopulation in a solid tumor, where as little as 10% of cells are cancer cells. TAM numbers are generally high in tumor types that do not respond to current immune therapies, such as pancreatic ductal adenocarcinoma and MSS-CRC. As summarized in this forum article, recent clinical trials and studies using animal models and human cell lines have collectively established that SUMOylation is a central repressive mechanism for the expression of IFN-β as well as ISGs. Consistently, SUMOylation inhibition activates innate immune cells – especially macrophages and NK cells – for eliminating tumors while having a profound impact on enhancing dendritic cells to induce adaptive antitumor immunity (Figure 2). These findings support the concept that SUMOylation inhibition is an exciting novel mechanism to address innate immunity in developing novel cancer therapeutic strategies.
Despite these exciting recent advances, there are key knowledge gaps and questions. The mechanism by which SUMOylation regulates CTL activation and differentiation remains to be better elucidated. High Myc expression increases cancer cell sensitivity to SUMOylation inhibition [15], suggesting that Myc expression in tumor cells could be a potential biomarker for patient stratification. Future clinical studies are needed to validate whether Myc expression is a biomarker to predict cancer response to SUMOylation inhibition and to develop a precise biomarker strategy. Finally, future studies are needed to increase our knowledge of the biology of SUMOylation inhibition in syngeneic models of various cancer types to understand the complex and tumor type-specific interplay of tumor cells and immune cells to guide the development of combination therapeutic strategies. We expect future breakthroughs in the field, both in expanding scientific knowledge and in developing innovative combination therapeutic strategies.
Acknowledgments
This work was supported by the National Institutes of Health grants R01CA265410, R01 CA212119, and R01CA216987 and Pancreatic Cancer Action Network (Y.C.). We apologize to those authors whose work we could not cite due to space limitations.
Footnotes
Declaration of interests
Y.C. reports board of director, equity ownership, and consulting fees from Suvalent Therapeutics, Inc.
References
- 1.Decque A et al. (2016) Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat. Immunol 17, 140–149 [DOI] [PubMed] [Google Scholar]
- 2.Crowl JT and Stetson DB (2018) SUMO2 and SUMO3 redundantly prevent a noncanonical type I interferon response. Proc. Natl. Acad. Sci. U. S. A 115, 6798–6803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lightcap ES et al. (2021) A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci. Transl. Med 13, eaba7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du L et al. (2023) Mechanism of SUMOylation-mediated regulation of type I IFN expression. J. Mol. Biol 435, 167968. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura A et al. (2022) The SUMOylation inhibitor subasumstat potentiates rituximab activity by IFN1-dependent macrophage and NK cell stimulation. Blood 139, 2770–2781 [DOI] [PubMed] [Google Scholar]
- 6.Ding X et al. (2016) Protein SUMOylation is required for regulatory T cell expansion and function. Cell Rep. 16, 1055–1066 [DOI] [PubMed] [Google Scholar]
- 7.Weitz J et al. (2022) An ex-vivo organotypic culture platform for functional interrogation of human appendiceal cancer reveals a prominent and heterogenous immunological landscape. Clin. Cancer Res 28, 4793–4806 [DOI] [PubMed] [Google Scholar]
- 8.Lu Z et al. (2022) ATF3 and CH25H regulate effector trogocytosis and anti-tumor activities of endogenous and immunotherapeutic cytotoxic T lymphocytes. Cell Metab. 34, 1342–1358.e1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demel UM et al. (2022) Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J. Clin. Invest 132, e152383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langston SP et al. (2021) Discovery of TAK-981, a first-in-class inhibitor of SUMO-activating enzyme for the treatment of cancer. J. Med. Chem 64, 2501–2520 [DOI] [PubMed] [Google Scholar]
- 11.Li YJ et al. (2019) Allosteric inhibition of ubiquitin-like modifications by a class of inhibitor of SUMO-activating enzyme. Cell Chem. Biol 26, 278–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lv Z et al. (2018) Molecular mechanism of a covalent allosteric inhibitor of SUMOE1 activating enzyme. Nat. Commun 9, 5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goel S (2022) A phase 1b, multicenter, dose-escalation study of subasumstat (TAK-981) in combination with pembrolizumab in patients (pts) with advanced solid tumors. In ASCO Annual Meeting, Abstract # 2506 [Google Scholar]
- 14.Dudek AZ et al. (2021) First-in-human phase 1/2 study of the first-in-class SUMO-activating enzyme inhibitor TAK-981 in patients with advanced or metastatic solid tumors or relapsed/refractory lymphoma: phase 1 results. J. Immunother. Cancer 9, A505–A506 [Google Scholar]
- 15.Kessler JD et al. (2012) A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]