

Abstract
The estrogen receptor (ER) is a well-established target for the treatment of breast cancer, with the majority of patients presenting as ER-positive (ER+). Endocrine therapy is a mainstay of breast cancer treatment but the development of resistance mutations in response to aromatase inhibitors, poor pharmacokinetic properties of fulvestrant, agonist activity of tamoxifen, and limited benefit for elacestrant leave unmet needs for patients with or without resistance mutations in ESR1, the gene that encodes the ER protein. Here we describe palazestrant (OP-1250), a novel, orally bioavailable complete ER antagonist and selective ER degrader. OP-1250, like fulvestrant, has no agonist activity on the ER and completely blocks estrogen-induced transcriptional activity. In addition, OP-1250 demonstrates favorable biochemical binding affinity, ER degradation, and antiproliferative activity in ER+ breast cancer models that is comparable or superior to other agents of interest. OP-1250 has superior pharmacokinetic properties relative to fulvestrant, including oral bioavailability and brain penetrance, as well as superior performance in wild-type and ESR1-mutant breast cancer xenograft studies. OP-1250 combines well with cyclin-dependent kinase 4 and 6 inhibitors in xenograft studies of ER+ breast cancer models and effectively shrinks intracranially implanted tumors, resulting in prolonged animal survival. With demonstrated preclinical efficacy exceeding fulvestrant in wild-type models, elacestrant in ESR1-mutant models, and tamoxifen in intracranial xenografts, OP-1250 has the potential to benefit patients with ER+ breast cancer.
Introduction
In 2022, approximately 288,000 U.S. women were diagnosed with breast cancer and approximately 43,000 of them will succumb to their disease (1). Estrogen receptor (ER) α regulates expression of hundreds of target genes, many of which are involved in cell-cycle progression and cellular growth (2, 3), and it is a key driver of approximately 70% of breast cancers (4). Approved treatments for ER-positive (ER+) breast cancer include fulvestrant, tamoxifen, and aromatase inhibitors. Tamoxifen and fulvestrant are ER ligands, displacing estrogens from cognate receptors (5, 6), whereas aromatase inhibitors block the conversion of adrenal androgens to estrogens (7). Fulvestrant, a potent inhibitor and degrader of the ER in vitro, has physiochemical properties that result in poor absorption and pharmacokinetic profile, requiring dosing via intramuscular injection every 28 days (8). Tamoxifen is orally bioavailable but its partial agonist activity limits its effectiveness in breast cancer and may lead to an increase in uterine cancer incidence (9). Aromatase inhibitors are initially effective, but prolonged treatment can lead to therapeutic resistance in the form of mutations that confer constitutive activity of the ER (10).
These mutations of the ER, which confer ligand-independent activity (11) and typically occur in the ligand-binding domain (LBD), are important drivers of tumor progression (12, 13) and have been detected with increasing frequency in recent clinical trials of patients with advanced breast cancer (14, 15); the most often observed are D538G and Y537S (16). Prior approved therapies are ineffective or demonstrate reduced efficacy on ESR1 mutations (10, 17), leaving an unmet need to be addressed by novel agents. The new endocrine agent elacestrant was recently approved specifically for these patients following results of the EMERALD III trial (18).
Binding of estrogens such as 17β-estradiol (E2) stimulates two activation functions, AF1 and AF2, to initiate gene transcription and signaling (19, 20). AF2, located in the C-terminal LBD, undergoes a conformational change upon E2 binding, impacting interactions with regulatory proteins such as NCoR and SRC-3 (21, 22). Many genes regulated by the ER, including key genes involved in cellular proliferation and survival, are also affected by activity of the amino terminal AF1 domain (23, 24). Although selective ER modulators (SERM) such as tamoxifen compete with estrogens in the LBD to block activation of AF2, many SERMs also activate AF1, resulting in agonist activity in some cell types including the uterus (25, 26). The term “complete ER antagonist (CERAN)” refers to ligands such as fulvestrant (5, 27, 28) that completely shut down estrogen signaling and lack any agonist activity on the ER in uterine or breast cells. While CERANs generally result in degradation of the ER and can therefore also be frequently described as selective ER degraders (SERD), we propose that complete antagonist activity is a more reliable metric for activity in ER+ breast cancer than degradation alone (27, 29).
Standard of care for stage IV ER+ breast cancer at first and second line typically includes an endocrine-targeting agent (aromatase inhibitor or fulvestrant) alongside a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor (palbociclib, ribociclib, or abemaciclib; ref. 30). CDK4/6 are key regulators of the G1-to-S-phase cell-cycle transition (31), and expression levels of the D-type cyclins that complex and interact with CDK4/6 are controlled by growth factor signaling, including that of the ER (31). Ensuring the combinability and lack of drug–drug interactions of new ER-targeting ligands with CDK4/6 inhibitors is therefore a key interest (32–34).
Given the limitations of approved therapies, there is an unmet need in metastatic ER+ breast cancer treatment. Here we describe palazestrant (OP-1250), a novel orally bioavailable complete ER antagonist and degrader of both wild-type and mutant ER.
Materials and Methods
Key details of the materials and methods used in this study are described below (see Supplementary Data S1 for additional information).
Reagents and cell culture
MCF7, CAMA-1, Ishikawa (originally ECC-1), and SK-BR-3 cell lines were purchased from ATCC. The ST941C cell line was obtained under license from XenoSTART. Cell lines were authenticated using short tandem repeat DNA profiling and tested for Mycoplasma at Laragen. Cells were cultured for no more than 30 passages following reanimation. E2 (#E8875), 4-hydroxytamoxifen (#H7904), fulvestrant (#I4409), and MG-132 (#M7449) were purchased from Millipore Sigma, and elacestrant (#HY-19822) was bought from MedChemExpress. Vepdegestrant and palazestrant (OP-1250) were synthesized by external vendors from published procedures [patents US 10,899,742 B1; Example 341 (35) and WO 2017/059139; Compound B (36)]. Cell lines were cultured in medium containing 10% FBS (#SH30070.03 Cytiva) or medium supplemented with listed concentration of charcoal/dextran stripped (CDS) FBS (#SH30068.03 Cytiva). Media used were Richter's IMEM (#A1048801 Gibco) supplemented with nonessential amino acids (#11140-050 Gibco) for CAMA-1, Eagle's MEM (#302003 ATCC) supplemented with 0.01 mg/mL human recombinant insulin (#I9278 Sigma) for MCF7, RPMI1640 (#A104910 Gibco) supplemented with 10 mmol/L HEPES (#15630-080 Gibco) and 1 mmol/L sodium pyruvate (#11360-070 Gibco) for Ishikawa, RPMI1640 (#A1049101 Gibco) for ST941C, and McCoy's 5A (#SH30270.01 Cytiva) for SK-BR-3. All cell lines were additionally supplemented with 1% GlutaMax (#35050061 Thermo Fisher Scientific).
LanthaScreen biochemical competitive binding assay
ERα ligand binding was assayed using the LanthaScreen time-resolved fluorescence energy transfer (TR-FRET) ERα competitive binding assay (#A15887 Thermo Fisher Scientific), per manufacturer protocol. Following 2-hour incubation at room temperature, compound binding was measured as a decrease in TR-FRET and normalized to 10 μmol/L E2 and DMSO vehicle.
Alkaline phosphatase assay
Ishikawa cells were plated in medium containing 4.8% CDS FBS. At least 4 hours later, cells were treated with compounds in medium diluted to 2.4% stripped FBS. Cells were incubated for 3 days, medium was removed, and plates were frozen at −80°C. Thawed plates were incubated with a chromogenic substrate of alkaline phosphatase (AP), p-nitrophenyl phosphate (#02212 Thermo Fisher Scientific), for 90 minutes at 37°C, and absorbance read at 405 nm. For antagonist mode assays, cells were cotreated with 500 pmol/L E2. For experiments where Ishikawa cells were transfected, 75 ng indicated ER constructs were introduced using Lipofectamine LTX (#15338 Thermo Fisher Scientific) 4 hours before compound treatment and subsequent AP assay.
Cellular proliferation
Proliferation of breast cancer cells was measured using CyQUANT, a fluorescent DNA-binding dye (#C7026 Invitrogen). Cells were treated with compounds in duplicate in medium containing 4.8% CDS FBS in the presence of 100 pmol/L E2 unless otherwise noted. Treated plates were incubated for 7 days prior to harvest. Following incubation, medium was removed and plates were frozen at –80°C. Thawed plates were prepared and quantified per manufacturer protocol. Fluorescent activity was normalized to the activity of E2 alone, and replicates with values >130% of E2 vehicle were excluded from analysis.
Simple Western assay
Ishikawa, CAMA-1 and MCF7 cell lines were plated in medium containing 4.8% CDS FBS and treated in duplicate with 1 nmol/L E2 or 300 nmol/L ER ligands, in the absence or presence of 10 μmol/L MG-132. Cells were incubated for between 2 and 72 hours then lysed with RIPA buffer (#89900 Thermo Fisher Scientific) and Halt phosphatase and protease inhibitors (#78440 Thermo Fisher Scientific). Protein concentration was quantified using the Pierce bicinchoninic acid Protein Assay Kit (#23225 Thermo Fisher Scientific) per manufacturer protocol. Protein analysis assay was run on Jess platform, as described previously (37), with 12–230 kDa Fluorescence Separation Modules (#SM-FL004-1 Bio-Techne), lysates diluted to a final concentration of 1 mg/mL, and Protein Normalization reagent (#043-824-C Bio-Techne) loading control. ERα primary antibody (#MA139540 Thermo Fisher Scientific) was used with ProteinSimple Anti-Rabbit Detection Module (#DM-001 Bio-Techne). Compass software (Bio-Techne) was used to quantify ERα levels. Percent ERα was calculated by normalizing ERα values to the protein normalization reagent and expressing them as percent of vehicle.
In vitro analysis
Data for LanthaScreen, AP, reporter gene, and cellular proliferation assays were normalized to E2 vehicle control for each assay and expressed as percent total response. IC50 or EC50 values were calculated in Prism software using a four-parameter fit. Emax is the percent effect at the highest drug concentration for each treatment, where 100% represents maximal response and 0% no change from vehicle control.
Generation of ESR1-mutant cell lines
Ishikawa (originally designated as ECC-1) and CAMA-1 cell lines were engineered using CRISPR-Cas9 genome editing technology through the services of Synthego Corporation. Single-guide RNAs (sgRNA) used had at least two mismatches to potential off-target regions. Silent mutations were included in the donor to modify the Protospacer Adjacent Motif (PAM) site for each guide to maximize knock-in efficiency. sgRNA and spCas9 were complexed to form a ribonucleoprotein and delivered via electroporation. Knock-in percent was assessed via PCR amplification and Sanger sequencing, and clones with desired edits selected and expanded. All cells used in this publication were validated to be homozygous mutant at the desired location of ESR1.
mRNA sequencing
RNA was extracted from frozen cells or tumor samples, enriched for mRNA using Oligo d(T) beads and prepared for sequencing using the NEBNext Ultra II RNA Library Prep Kit (#E7770 New England Biolabs). Sequencing was conducted on an Illumina HiSeq instrument, with 20–30 million reads per sample. Gene counts and differential gene expression analyses were carried out using CLC Genomics Workbench (QIAGEN) utilizing cut-off levels of absolute fold change >2 and FDR-adjusted P value ≤0.05 to determine differentially expressed genes.
Precision run-on sequencing
Precision run-on sequencing (PRO-seq) was conducted at Arpeggio Biosciences. Samples were harvested 15 minutes, 1 hour, 6 hours, and 24 hours after dosing in MCF7 and CAMA-1 cell lines. Cells were switched to medium containing 4.8% CDS FBS 48 hours prior to compound treatment. E2 was dosed at 100 pmol/L and other compounds at 316 nmol/L concentration. Run-on and subsequent PRO-seq library preparations were performed on permeabilized cell pellets in a freeze buffer, as described previously (38) but with modifications. Sequencing was performed using the Illumina NextSeq500 (single read, 1×75 bp). Data were analyzed and visualized using Arpeggio Insights portal or R.
Bioinformatic analysis
FASTQ files were processed using CLC Genomics Workbench (QIAGEN). In short, reads were trimmed and filtered with a quality threshold of 0.05. Reads were mapped to the current Human assembly GRCh38 as reference using the mapping algorithm provided by CLC Genomics Workbench. For xenograft samples, reads that mapped exclusively to the murine reference genome GRCm39 were filtered out prior to analysis. log2-fold changes were calculated using the Workbench software, where gene read counts were assumed to follow a negative binomial distribution. Significantly upregulated or downregulated genes were tallied using an absolute fold change >2.0 and FDR-adjusted P value ≤0.05. Pathway analysis was performed using the Ingenuity Pathway Analysis software.
Heat maps were created using log2-fold change values calculated as mentioned above and plotted using R function heat map, which is part of the library non-negative matrix factorization. The log2-fold change values were scaled across columns, that is, on a per-gene basis.
We used R Rtsne library to apply the t-distributed Stochastic Neighbor Embedding (tSNE) dimensionality reduction algorithm to the data after scaling gene log2-fold changes to have a mean of 0 and SD of 1.
To compare the effect of the combination therapy with the two individual monotherapies (1 mg/kg OP-1250 and 25 mg/kg palbociclib, given separately), we looked at the log-fold changes for each of these three conditions for each gene. The effects of the monotherapies were combined according to the Bliss independence model. Using the effect of the combination therapy and the Bliss independence model as an (x, y) coordinate pair, the distance to the y = x diagonal was calculated. A value of 0 indicates that the combination therapy and combined monotherapies had comparable effects. A more negative value indicates that the combination therapy had a stronger repression on gene expression, while a more positive value indicates a stronger enhancement of gene expression.
Uterine wet weight
Ovariectomized BALB/c mice (7–8 weeks old) were purchased from Charles River Laboratories and studies conducted at Bayside BioSciences in accordance with established protocol approved by the vendor's Institutional Animal Care and Use Committees (IACUC). At 7 days after ovariectomy, mice were administered E2 at 0.1 μg/mouse subcutaneously in combination with OP-1250 (0.01-100 mg/kg,), tamoxifen (50 mg/kg), or fulvestrant (125 mg/kg), orally, once daily, for 3 days. The mice were euthanized 24 hours after the last dose, and uterine samples were harvested and weighed.
Mammary fat pad/subcutaneous xenografts
Xenograft studies on OP-1250 as a monotherapy in the ST941 model and combination studies of OP-1250 with CDK4/6 inhibitors were conducted at XenoSTART, in accordance with established protocols, approved by the vendor's IACUC. Female athymic nude mice were implanted with ST941 patient-derived xenograft (PDX) tumor fragments or cell-derived MCF7 subcutaneously. Mice were supplemented with exogenous E2 via drinking water (ad libitum) throughout the duration of the study. Tumor volume was measured twice weekly. Treatment was initiated when tumor volume reached 175–200 mm3 (day 1 of the study). Mice were administered with OP-1250 (0.1, 3, 1, or 10 mg/kg), palbociclib (25 or 75 mg/kg), or ribociclib (25 or 75 mg/kg) as single agents or in combination, orally, once daily for 28 days. Mice were terminated on day 29 for the MCF7 model, or when tumor reached 2,000 mm3 or day 57 (whichever came first) for the ST941 model.
Other xenograft studies were performed at Huntsman Cancer Institute per established protocol, approved by the vendor's IACUC. Female, NOD/SCID mice were implanted with HCI-013 or MCF7 cells in the mammary fat pad and supplemented with E2 pellets. A third cohort of NOD/SCID mice was ovariectomized and implanted with HCI-013EI tumor fragments in the absence of estrogen supplementation. OP-1250 was administered at 0.3, 1, 3, 10, or 30 mg/kg daily, orally for 28 days. Fulvestrant was administered weekly, subcutaneously at 50 mg/kg for the first dose, and 25 mg/kg for subsequent doses. Tumor volume was measured twice weekly, and volumes were calculated as (length × width2)/2 = mm3 and analyzed for tumor growth inhibition. At conclusion of studies, plasma, tumor, and brain samples were collected for bioanalysis.
In all, studies animals were randomly assigned to all groups such that across the group mean tumor volumes were comparable on the day 1 of dosing. The number of animals was the minimum necessary to meet the objectives of the study accounting for interanimal variability. There was no unnecessary duplication of studies regarding species, strain, and test article.
Intracranial xenografts
Intracranial xenograft studies were conducted at Minerva Imaging with established protocol approved by the vendor's IACUC. ST941 cells were injected at a depth of 2–2.5 mm intracranially into female athymic nude mice. E2 was supplemented via drinking water from 2 days prior to inoculation until time of animal inclusion in study (tumor volume 2–5 mm3 based on MRI); it was then withdrawn. Dosing was initiated 2 days after inclusion. Animals were treated for 100 days with fulvestrant (5 mg/animal, once a week), tamoxifen (60 mg/kg, once daily), OP-1250 (10 mg/kg, once daily), or OP-1250 + ribociclib (75 mg/kg, once daily); dosing was then discontinued and animals were monitored for tumor growth and survival. One vehicle cohort was ovariectomized at day of inclusion to discontinue estrogen production. Tumor growth was assessed throughout the study using T2-weighted MRI (intracranial).
Xenograft pharmacokinetic bioanalysis
Plasma samples were taken 24 hours following administration of the last dose in the HCI-013 28-day xenograft study. Brain and tumor samples were collected 24 hours following administration of the last dose. Plasma, brain, and tumor drug levels of OP-1250 were assessed at Quintara Biosciences. Plasma and homogenized brain and tumor were extracted by protein precipitation using acidified acetonitrile containing internal standard. Extracted samples were analyzed by tandem LC/MS-MS in multiple reaction monitoring mode using electrospray ionization in positive mode. Unknown sample concentrations were calculated from prepared standards.
Xenograft tumor IHC and scoring
Tumor samples were collected 8 and 24 hours following administration of the last dose in the ST941 28-day xenograft study. Assessment of Ki67 and ER-alpha levels were assessed at Inotiv HistoTox labs. For IHC staining of Human ERα alpha and Human Ki67 in mouse xenograft tumors, staining was conducted on the Leica Bond RXm platform using standard chromogenic methods. For antigen retrieval, slides were heated in a pH9 Ethylenediaminetetraacetic acid (EDTA)-based buffer for 25 minutes at 94°C, followed by a 45-minute antibody incubation (1:100, Abcam [EPR703(2)] ab79413) and 30-minute antibody incubation (1:500, Abcam [EPR360] ab92742). Antibody binding was detected using an horseradish peroxidase–conjugated secondary polymer, followed by chromogenic visualization with diaminobenzidine. A hematoxylin counterstain was used to visualize nuclei. Slides were scored by a board-certified veterinary anatomic pathologist. Staining intensity in non-necrotic tumors was scored on a scale of 0 to 4 (0, no staining; 1, 25% staining; 2, 25%–50% staining; 3, 50%–75% staining; 4, 75%–100% staining).
Data availability
Sequencing data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO; ref. 39) and are accessible through GEO Series accession number GSE241944 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241944). All other data are available upon request from the corresponding author.
Results
OP-1250 is a complete ER antagonist that completely inhibits ER signaling
We set out to design and synthesize nonsteroidal complete ER antagonists capable of blocking activity of both AF1 and AF2 (Fig. 1A), with favorable drug-like properties. The discovery that the tetrahydro-β-carboline–containing compounds bind avidly to ER (40) led us to explore the functionalization of this ring system to optimize potency and pharmacokinetics while achieving complete ER blockade. Through this effort, we identified palazestrant (OP-1250; Fig. 1B; ref. 36). The properties of OP-1250 were assessed relative to FDA-approved and European Medicines Agency–approved ERα ligands (fulvestrant, tamoxifen, elacestrant, and lasofoxifene) and to ligands currently in clinical trials for advanced breast cancer (imlunestrant and vepdegestrant). Active metabolites of tamoxifen, such as 4-hydroxytamoxifen (4OH-tamoxifen), were used in cell culture experiments rather than tamoxifen citrate.
Figure 1.

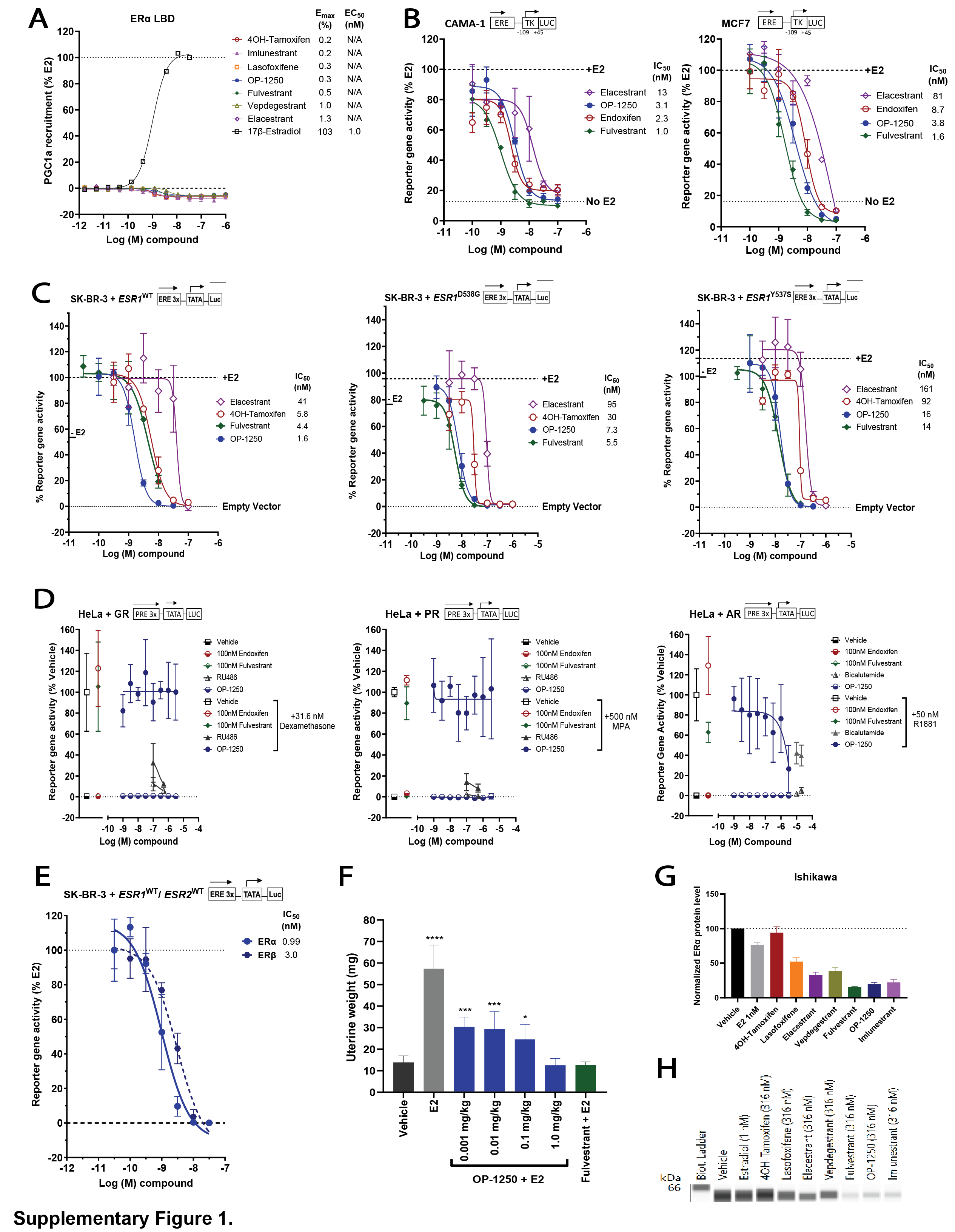
OP-1250 is a complete ER antagonist that completely blocks ER activity. A, Schematic of mechanism of CERAN activity; CERANs bind and inactivate the AF2 domain and additionally induce inactivation of the AF1 domain. B, Chemical structure of OP-1250. C, LanthaScreen competitive binding assay using wild-type ERα LBD comparing OP-1250 and other approved and in-trial comparator molecules. Data are normalized to DMSO vehicle and 10 μmol/L E2-treated wells (100% displacement) and represented as mean and SD of at least three independent experiments. D, UWW assay comparing OP-1250 against fulvestrant and tamoxifen in the presence and absence of E2 supplementation. Mean and SEM from 6 mice per treatment group are plotted with asterisks indicating significance relative to vehicle by one-way analysis of variance. ****, Padjusted value < 0.0001. E, Basal AP activity in Ishikawa cells following transient transfection with vectors encoding ESR1WT, ESR1Y537S, ESR1D538G, AF2 truncated ESR1, AF1 truncated ESR1, and AF1 truncated ESR1Y537S. Data represent activity in estrogen-depleted media. Data are normalized to wild-type receptor and presented as mean and SD of a representative of at least three independent experiments. F, Basal AP activity of Ishikawa cells in estrogen-depleted media following incubation for 72 hours with ERα ligands. Data are normalized to 500 pmol/L E2- and vehicle-treated wells and represented as mean and SD of at least three independent experiments. G, AP activity of Ishikawa cells in estrogen-depleted media following incubation for 72 hours with ER ligands and 500 pmol/L E2. Data are normalized to 500 pmol/L E2 and vehicle-treated wells and represented as mean and SD of at least three independent experiments. N/A = not applicable; WT = wild-type.
In a competitive binding assay using the ERα LBD, OP-1250 demonstrated similar potency to comparator compounds (Fig. 1C), while in a PGC1a coactivator recruitment assay, all compounds demonstrated no recruitment (Supplementary Fig. S1A). In luciferase reporter gene assays of E2-dependent transcriptional activity conducted in ER+ breast cancer cells and ER-negative SK-BR-3 cells transfected with ESR1WT, ESR1Y537S, and ESR1D538G, it was observed that elacestrant, OP-1250, z-endoxifen, and fulvestrant all strongly inhibited transcription in a dose-dependent manner (Supplementary Fig. S1B and S1C). OP-1250 demonstrated no agonist-like cross-reactivity on glucocorticoid, progesterone, or androgen receptors. Inhibition of agonist-induced androgen receptor transcriptional activity was detected only at >1 μmol/L OP-1250 (Supplementary Fig. S1D). OP-1250 inhibition of transfected ESR1WT and ESR2WT displayed approximately 3-fold increased potency on ERα (Supplementary Fig. S1E).
The uterine wet weight (UWW) assay is a well-established model to study ER-mediated agonism (uterine hypertrophy and hyperplasia) and antagonism (26, 41, 42). We compared OP-1250 with fulvestrant and tamoxifen, both in the presence and absence of supplemented E2 (Fig. 1D). E2 and tamoxifen significantly increased UWW, while OP-1250 and fulvestrant did not. OP-1250 and fulvestrant inhibited E2-induced increases in weight, with OP-1250 displaying dose-dependent inhibition as low as 0.01 mg/kg (Supplementary Fig. S1F). At 1 mg/kg, OP-1250 inhibited trophic responses comparable to fulvestrant at 5 mg/mouse or vehicle.
To optimize for compounds with a CERAN profile and compare OP-1250 to comparators of interest, we utilized the AP assay, which monitors uterine agonist activity with higher throughput than the UWW assay and can discriminate between partial and complete antagonists (25, 43, 44). In an AP assay conducted following transfection of Ishikawa cells with AF1 or AF2 domain truncations of ERα, AF2 truncation had little impact on AP activity, while AF1 truncation reduced AP activity to levels comparable to empty vector control (Fig. 1E), suggesting the utility of this assay as a readout of ERα AF1 activity in uterine cells. Interestingly, AP activity of the constitutively active ESR1Y537S mutation also demonstrated AF1 dependence that was unaffected by deletion of AF2.
In the absence of E2 supplementation, 4OH-tamoxifen, lasofoxifene, and vepdegestrant caused dose-dependent increases in AP activity consistent with agonism of AF1 with maximum responses >80% (Fig. 1F). Elacestrant demonstrated partial agonist activity, with maximum response of 24%; imlunestrant, fulvestrant, and OP-1250 did not induce measurable AP activity. Antagonism of AF1-dependent AP activity following supplementation with 500 pmol/L E2 displayed a similar profile; molecules demonstrating full agonist activity in the absence of E2 showed minimal/no inhibition of AP activity with E2. Partial agonist elacestrant did not completely inhibit activity at 1 μmol/L (Emax, 85%), while imlunestrant, fulvestrant, and OP-1250 completely inhibited activity with IC50 values of 32, 9.4, and 6.4 nmol/L, respectively (Fig. 1G). Of note, agonist and antagonist activity in the AP assay was well correlated with available UWW results (Fig. 1D), including published data on elacestrant and lasofoxifene (45, 46). The level of ERα protein in ECC-1 cells was assessed following 72 hours compound treatment (Supplementary Fig. S1G and S1H). Although CERANs reduced levels of ERα in this cell line, a consistent correlation between protein level and AP activity was not observed, particularly regarding agonist activity.
OP-1250 blocks estrogen-induced transcriptional activity and lacks agonist activity on estrogen-induced genes
In a proliferation assay of MCF7 and CAMA-1 ER+ breast cancer cells cultured in E2-depleted media (Fig. 2A), treatment with E2, 4OH-tamoxifen, or lasofoxifene led to increased proliferation in the CAMA-1 cell line (P < 0.0005 at 1 μmol/L dose), which was not observed with CERAN treatment. This suggests that this cell line may be particularly sensitive to agonist activity of SERMs and illustrates the capacity of a SERM to stimulate breast cancer cell proliferation in addition to demonstrating uterine agonist activity.
Figure 2.

OP-1250 effectively blocks estrogen-induced transcriptional activity and lacks agonist activity on breast cancer cells in the absence of estrogen. A, Cellular proliferation assay of MCF7 and CAMA-1 cells treated over 7 days with listed compounds in the absence of E2 supplementation. Proliferation is assessed by CyQUANT reagent and normalized to DMSO vehicle, the value of which is indicated by the horizontal dotted line. Data is represented as mean and SD of at least three independent experiments. ****, Padjusted value < 0.0001 and ***, Padjusted value < 0.001. B, Schematic of PRO-seq treatment strategy and sample collection timepoints for MCF7 and CAMA-1 cells (above) and principal component analysis of DMSO, E2, and E2 + OP-1250 treatments (below), with timepoint indicated by circle size and treatment indicated by color. C and D, Heat maps of genes associated with E2 early gene signature or G2–M gene signature in 24 hours E2 and E2 + OP-1250 PRO-seq samples, with red indicating high expression relative to vehicle and blue indicating low expression relative to vehicle. E, Gene set enrichment analysis of G2–M checkpoint gene set at 24 hours PRO-seq timepoint with genes ranked from high to low expression relative to vehicle (x-axis) and plotted lines tracking running enrichment (y-axis). Ticks below graph represent individual genes. Upward peak toward left side of graph indicates enrichment in activation of gene expression while a downward peak toward right side of graph indicates enrichment in reduction of gene expression. F and G, Heat maps of annotated estrogen response and G2–M gene signatures in CAMA-1 mRNA-seq data. Cells were treated in triplicate with 100 pmol/L E2 or 316 nmol/L antiestrogen in estrogen-depleted media for 24 hours. Red indicates high expression relative to vehicle and blue indicates low expression.
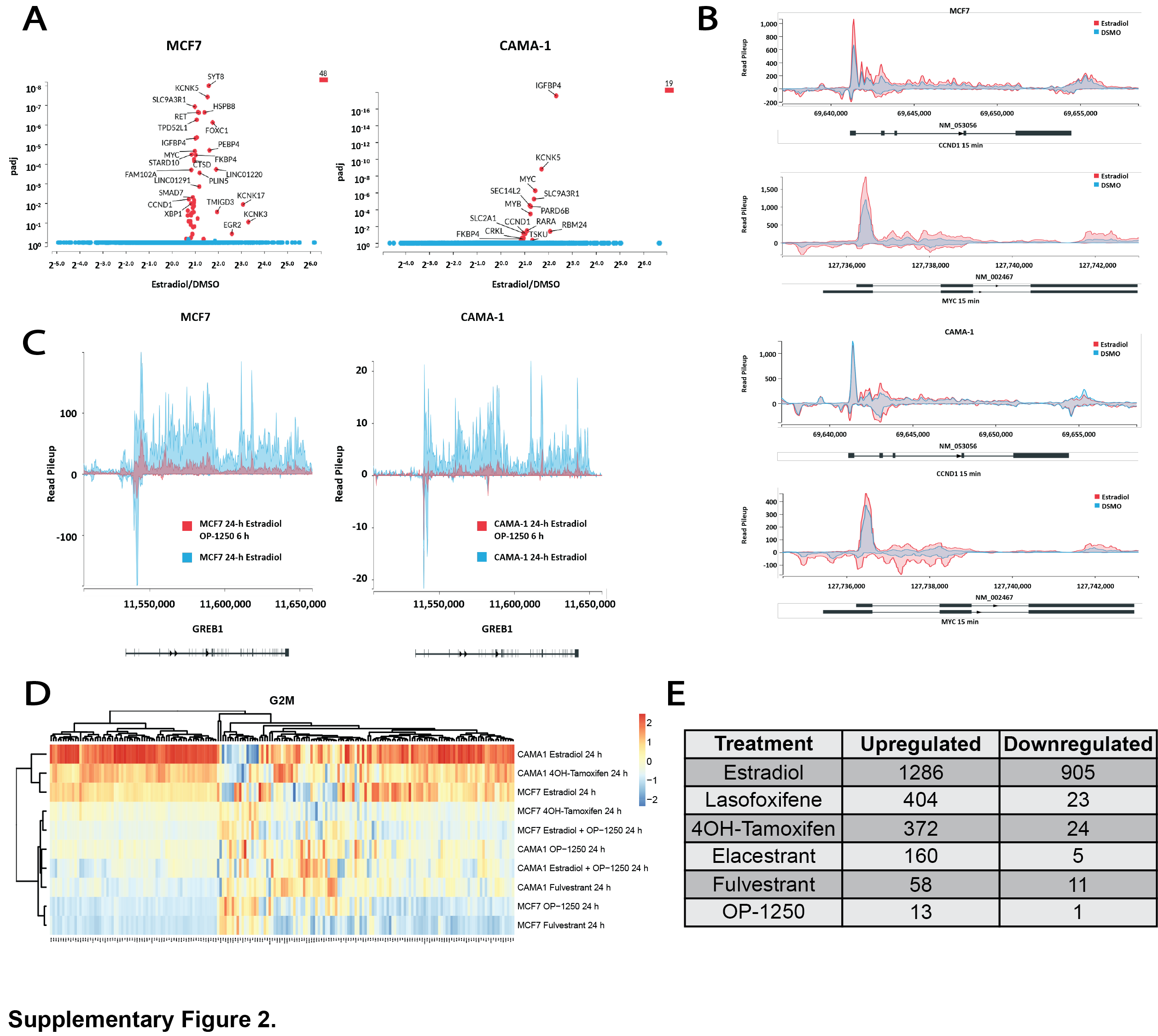
To determine whether OP-1250 can completely reverse transcriptional changes induced by E2 treatment, we conducted PRO-seq with MCF7 and CAMA-1 cells cultured in E2-depleted media prior to stimulation with E2 or treated with OP-1250 after 24-hour E2 treatment. Samples were analyzed after 15 minutes to 24 hours treatment, to track gene expression changes over time. E2 treatment induced gene expression changes within 15 minutes (Supplementary Fig. S2A and S2B), with broader changes observed by 24 hours (Fig. 2B; Supplementary Tables S1 and S2), including activation of known E2 pathway genes (Fig. 2C) and genes associated with cell-cycle progression (Fig. 2D). OP-1250 treatment reversed E2-induced changes in transcriptional activity of known estrogen response genes within 6 hours (Fig. 2C; Supplementary Fig. S2C). Within 24 hours, OP-1250 treatment suppressed genes associated with cell-cycle progression (Fig. 2D; Supplementary Tables S1 and S2) and reversed broader E2-induced changes to the gene expression profile in a principal component analysis (Fig. 2B). In the absence of E2, 4OH-tamoxifen treatment for 24 hours led to enrichment of the G2–M checkpoint gene signature in the CAMA-1 cell line (Fig. 2E and G) and E2-like stimulation of G2–M genes (Supplementary Fig. S2D) not observed with OP-1250 or fulvestrant; this is consistent with observed effects on cellular proliferation (Fig. 2A).
We conducted RNA sequencing (RNA-seq) on CAMA-1 cells cultured in estrogen-depleted media before 24-hour treatment with compound or E2 to further investigate differences of CERANs and SERMs on gene expression in the absence of E2. While E2 treatment resulted in expression changes of genes associated with E2 signaling (Fig. 2E) and cell-cycle progression (Fig. 2F), CERANs OP-1250 and fulvestrant showed an inverse pattern of gene expression on these pathways. Compounds with demonstrated agonist activity in the AP assay (4OH-tamoxifen, lasofoxifene, elacestrant) resulted in more gene expression changes (Supplementary Fig. S2E) and showed gene expression patterns similar to E2 (Fig. 2E and F), albeit with lower fold changes than E2. CERAN treatment led to expression patterns opposite to that of E2 for both E2 and cell-cycle gene signatures, while elacestrant, with partial agonist activity in the AP assay, displayed intermediate effect on gene expression. This analysis demonstrates that compounds with agonist activity in the AP assay can stimulate ER signaling and cellular proliferation in breast cancer cells, while CERANs completely block ER-induced transcriptional activity.
OP-1250 displays in vitro and in vivo efficacy and degrades the ER in breast cancer models
ER degradation is a common metric for identifying candidate antiestrogen compounds, with many clinical candidates promoted as SERDs. In a 24-hour simple Western assay of ERα degradation in the MCF7 cell line (Supplementary Fig. S3A), OP-1250 degrades ERα with comparable potency to fulvestrant (DC50 0.52 nmol/L and 0.56 nmol/L, respectively; Emax 62% and 69%, respectively), while 4OH-tamoxifen stabilizes ERα (Emax –30%) and E2 displays more potent degradation than OP-1250 or fulvestrant (DC50 < 0.1 nmol/L, Emax 49%; Fig. 3A). In a broader comparison of compounds of interest on MCF7 and CAMA-1 cell lines (Fig. 3B; Supplementary Fig. S3A and S3B), cells were treated with 300 nmol/L compound or 1 nmol/L E2 for 24 hours. OP-1250 treatment reduced ERα protein levels by 63% relative to vehicle in MCF7 and 72% in CAMA-1 cells; reductions in levels were comparable to those of previously described SERDs, fulvestrant, vepdegestrant, and imlunestrant, and superior to elacestrant. Of note, treatment with 1 nmol/L E2 resulted in ERα degradation comparable to that of SERDs (65% reduction in MCF7 and 43% in CAMA-1) in both cell lines. 4OH-tamoxifen and lasofoxifene, which demonstrated full agonist activity in the AP assay, did not degrade the ER. In MCF7 cells, degradation of ERα by OP-1250 was observed as early as 2 hours after treatment, levels reduced in a time-dependent manner, and degradation was blocked by treatment with the proteasome inhibitor MG-132 (Supplementary Fig. S3C and S3D).
Figure 3.

OP-1250 displays in vitro and in vivo efficacy and degrades the ER in ER+ breast cancer models. A, Simple Western analysis of ERα protein levels in MCF7 cells after 24 hours treatment with listed compounds. Samples are normalized to untreated control, with mean and SD across three independent treatments shown. B, Simple Western analysis of ERα protein levels in MCF7 or CAMA-1 cells after 24 hours of treatment with 300 nmol/L compound or 1 nmol/L E2. Samples are normalized to untreated control, represented by dotted line, with mean and SD across at least three independent treatments shown. C and D, Cellular proliferation of MCF7 and CAMA-1 cells treated with indicated dose of antiestrogen in the presence of 100 pmol/L E2. Proliferation assessed by CyQUANT reagent is normalized to E2-treated vehicle and shown as mean and SD across at least three experiments. E, Mean and SEM tumor volume over time with listed treatments in the MCF7 breast cancer model, with dotted line representing tumor stasis. Each group represents at least 6 animals. F, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of MCF7 xenograft study, normalized to day 1 tumor volume.
To investigate whether these agents can inhibit ER+ breast cancer growth, compounds were tested in a proliferation assay of MCF7 and CAMA-1 cells in the presence of 100 pmol/L E2 (Fig. 3C and D). In these assays, OP-1250 demonstrated a promising efficacy profile, with IC50 values of 1.4–1.6 nmol/L and favorable Emax values. Importantly, compounds with full agonist activity in the AP assay (4OH-tamoxifen, lasofoxifene, and vepdegestrant) displayed incomplete suppression of cellular proliferation in the CAMA-1 cell line, demonstrated by lower Emax values, while complete ER antagonists were better able to fully shut down estrogen-mediated proliferation. Although elacestrant displayed an unusually high maximum percent inhibition in the CAMA-1 cells, this is likely a nonspecific effect of a 1 μmol/L dose of elacestrant, which is an outlier to the overall dose–response curve. Of note, agonist activity in the AP assay was a better indicator of maximum antiproliferative activity than ERα degradation, with the PROTAC vepdegestrant a notable outlier as it demonstrated superior performance in the degradation assay but poor potency and maximum inhibition of breast cancer cell proliferation.
OP-1250 daily oral doses, 0.3–30 mg/kg, were tested in a MCF7 mammary fat pad xenograft against weekly, subcutaneous fulvestrant (Fig. 3E and F). OP-1250 doses ≥3 mg/kg shrank tumors and outperformed fulvestrant, which did not lead to appreciable tumor shrinkage. A 3 mg/kg dose of OP-1250 led to tumor shrinkage below starting volume in all but one animal, while higher doses led to shrinkage in all animals (Fig. 3F). At the highest tested OP-1250 dose, all tumors displayed >50% reduction in tumor volume.
OP-1250 blocks ER signaling and proliferation in ESR1-mutant breast cancer models
To investigate the activity of compounds of interest against the most prevalent clinical variants of ESR1, we generated Ishikawa and CAMA-1 cell lines with homozygous ESR1Y537S and ESR1D538G mutations using CRISPR/Cas9 knock-in. In AP assays conducted with mutant Ishikawa cells, OP-1250 displayed favorable potency against both mutants, comparable to that of fulvestrant (Fig. 4A and B). Of interest, compounds displaying agonist activity in the wild-type AP assay (Fig. 1E) showed incomplete inhibition of the Y537S receptor but more complete inhibition of the D538G mutant receptor. Similar rank orders of compound potency were observed in cellular proliferation assays of the homozygous mutant CAMA-1 cells (Fig. 4C and D) and with a cell line derivative of the heterozygous ESR1Y537S ST941 PDX model (Fig. 4E; ref. 47), with lasofoxifene and OP-1250 displaying greatest potency. In the ST941C cell line, compounds classified as full agonists in the AP assay displayed incomplete suppression of cellular proliferation as demonstrated by lower Emax values. As in the wild-type setting, there was no reliable correlation between effective degradation of mutant ERα (Supplementary Fig. S3E–S3H) and functional efficacy in blocking cellular proliferation. In both AP and proliferation assays, CERANs showed reduced potency in the mutant cell lines relative to wild-type.
Figure 4.

OP-1250 blocks ER signaling and proliferation in ESR1-mutant breast cancer models. A and B, AP mean and SD of indicated homozygous ESR1-mutant Ishikawa endometrial cells conducted in the presence of 500 pmol/L E2. Data are normalized to 500 pmol/L E2-treated wells and represented as mean and SD of at least three independent experiments. C–E, Cellular proliferation of indicated ESR1-mutant cells treated with antiestrogen in the presence of 100 pmol/L E2. Proliferation assessed by CyQUANT reagent is normalized to E2-treated vehicle and shown as mean and SD across at least three experiments. F, Mean and SEM tumor volume over time with listed treatments in the ESR1Y537S HCI-013 PDX model, with dotted line representing tumor stasis. G, Mean and SEM tumor volume over time with listed treatments in the ESR1Y537S ST941 PDX model, with dotted line representing tumor stasis. H and I, IHC analysis of Ki67 and nuclear ERα in tumor samples, and pharmacokinetic analysis of plasma samples from the ST941 xenograft study collected 24 h after the final dose. Data are shown as the mean values of at least 3 animals. Non-necrotic tumor staining extent was scored as 0 for no staining, 1 for up to 25%, 2 for 25%–50%, 3 for 50%–75%, and 4 for 75%–100%. J, Pharmacokinetic analysis of plasma, tumor, and brain samples from the HCI-013 xenograft study collected 24 hours after final dose. Numbers represent mean values from at least 3 animals.
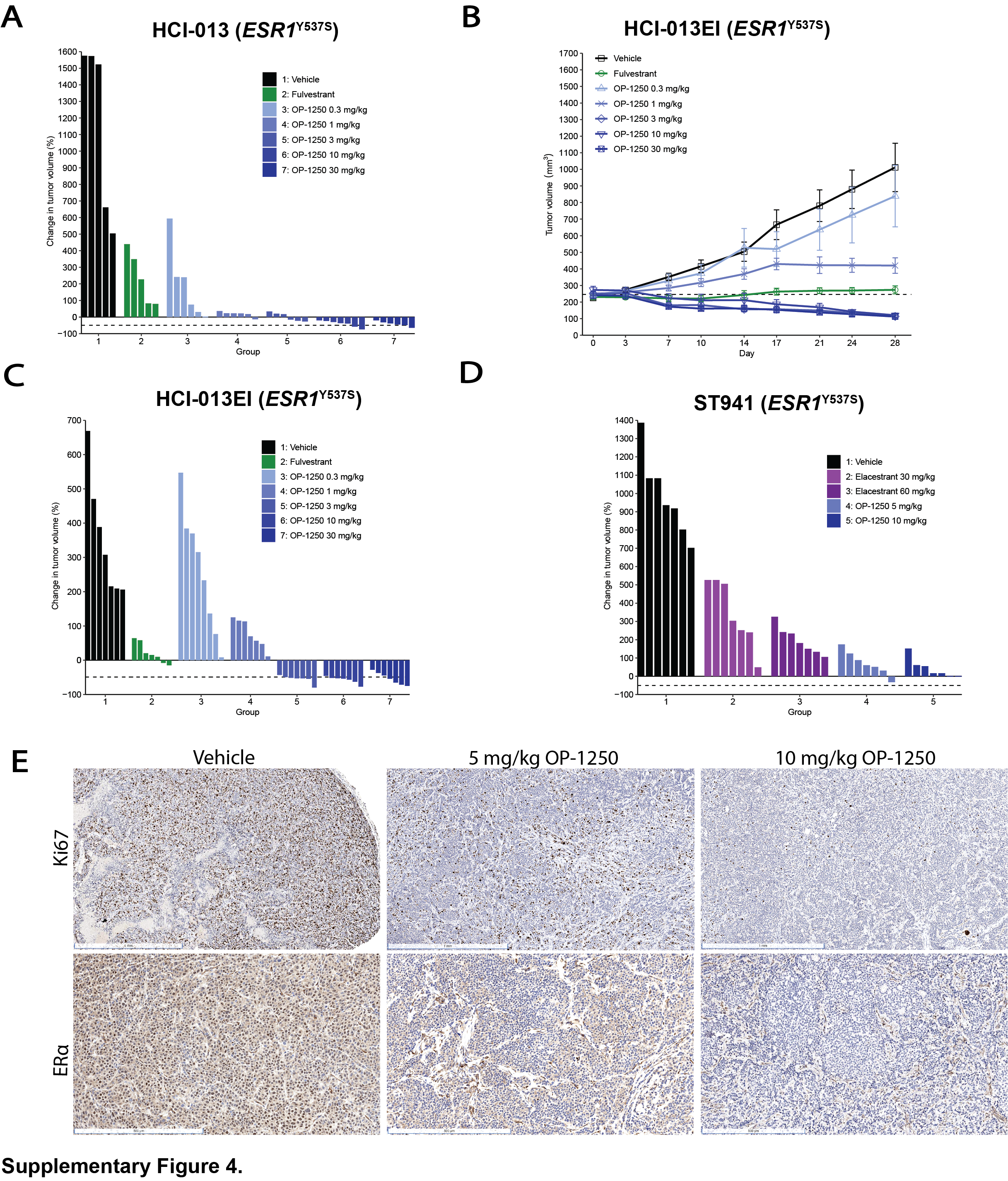
We conducted a mammary fat pad xenograft study using the HCI-013 ESR1Y537S breast cancer PDX model comparing OP-1250, 0.3–30 mg/kg, against fulvestrant (Fig. 4F). In this model, OP-1250 doses of ≥1 mg/kg outperformed fulvestrant, and all treatments led to tumor growth inhibition; 10 and 30 mg/kg doses led to tumor shrinkage below starting volume in all treated animals (Supplementary Fig. S4A). In a similar study conducted in the absence of estrogen with an estrogen-independent version of HCI-013 (HCI-013EI; Supplementary Fig. S4B and S4C), 3 mg/kg OP-1250 performed comparably to 10 and 30 mg/kg, suggesting that potency might be improved in the absence of estrogen.
Because elacestrant is clinically approved specifically for ESR1-mutant breast cancer, we assessed the in vivo performance of OP-1250 relative to elacestrant in this context. We conducted a xenograft study comparing 5 and 10 mg/kg doses of OP-1250 against 30 and 60 mg/kg doses of elacestrant in the ST941 ESR1Y537S breast cancer PDX model (Fig. 4G; Supplementary Fig. S4D). Both OP-1250 doses outperformed elacestrant, with greatest tumor growth inhibition with 10 mg/kg OP-1250. IHC analysis conducted on tumor samples from the ST941 xenograft study showed that OP-1250 reduced Ki67 levels in a dose-dependent manner, in line with observed tumor growth inhibition (Fig. 4H; Supplementary Fig. S4E). At both the 5 and 10 mg/kg doses, nuclear ERα staining was reduced by approximately 75%, compared with the vehicle group, confirming target engagement (Fig. 4I; Supplementary Fig S4E).
Pharmacokinetic analysis was conducted on plasma, tumor, and brain samples from the HCI-013 xenograft study 24 hours following final dose at 3 and 10 mg/kg (Fig. 4J). At this timepoint, OP-1250 demonstrated plasma exposures well above levels needed for complete ER saturation at Ctrough, robust brain penetrance with brain/plasma ratios >1, and tumor accumulation.
OP-1250 improves efficacy of CDK4/6 inhibitors in ER+ xenograft models and enhances inhibition of cell cycle–related gene expression
Because of the clinical relevance of combining endocrine therapy with CDK4/6 inhibitors, we investigated OP-1250 in this setting. In MCF7 cellular proliferation assays of OP-1250 in combination with the three clinically approved CDK4/6 inhibitors, palbociclib, ribociclib, and abemaciclib (Fig. 5A), addition of OP-1250 resulted in a decrease in proliferation relative to CDK4/6 inhibitor alone; the most pronounced effect was observed with palbociclib and ribociclib.
Figure 5.

Addition of OP-1250 improves efficacy of CDK4/6 inhibitors in vitro and in vivo. A, Cellular proliferation assessed by CyQUANT reagent of MCF7 cells treated for 7 days with the CDK4/6 inhibitors palbociclib, ribociclib, or abemaciclib alone or in combination with OP-1250. Dose of indicated CDK4/6 inhibitor is shown on the x-axis, with samples not treated with CDK4/6 inhibitor shown to the left of the axis break. Proliferation is normalized to 100 pmol/L E2 vehicle and shown as mean and SD across three experiments. B and C, Mean and SEM tumor volume over time with monotherapy and combination treatments in the MCF7 breast cancer model, with dotted line representing tumor stasis. D, Heat map showing expression of genes associated with the G2–M gene signature of n = 4 MCF7 xenograft tumors from OP-1250 and palbociclib low dose monotherapy and combination therapy groups. Red corresponds to high expression relative to vehicle and blue corresponds to low expression. E, Differential gene expression comparison between combination therapy and monotherapy MCF7 xenograft samples in hallmark gene sets. Each datapoint represents a gene in the listed gene signature. Vertical axis depicts a datapoint's distance to the y = x diagonal, where 0 represents no change from expected result based on monotherapy values. F and G, Mean and SEM tumor volume over dosing interval with monotherapy and combination treatments in the ESR1Y537S ST941 PDX model, with dotted line representing tumor stasis.
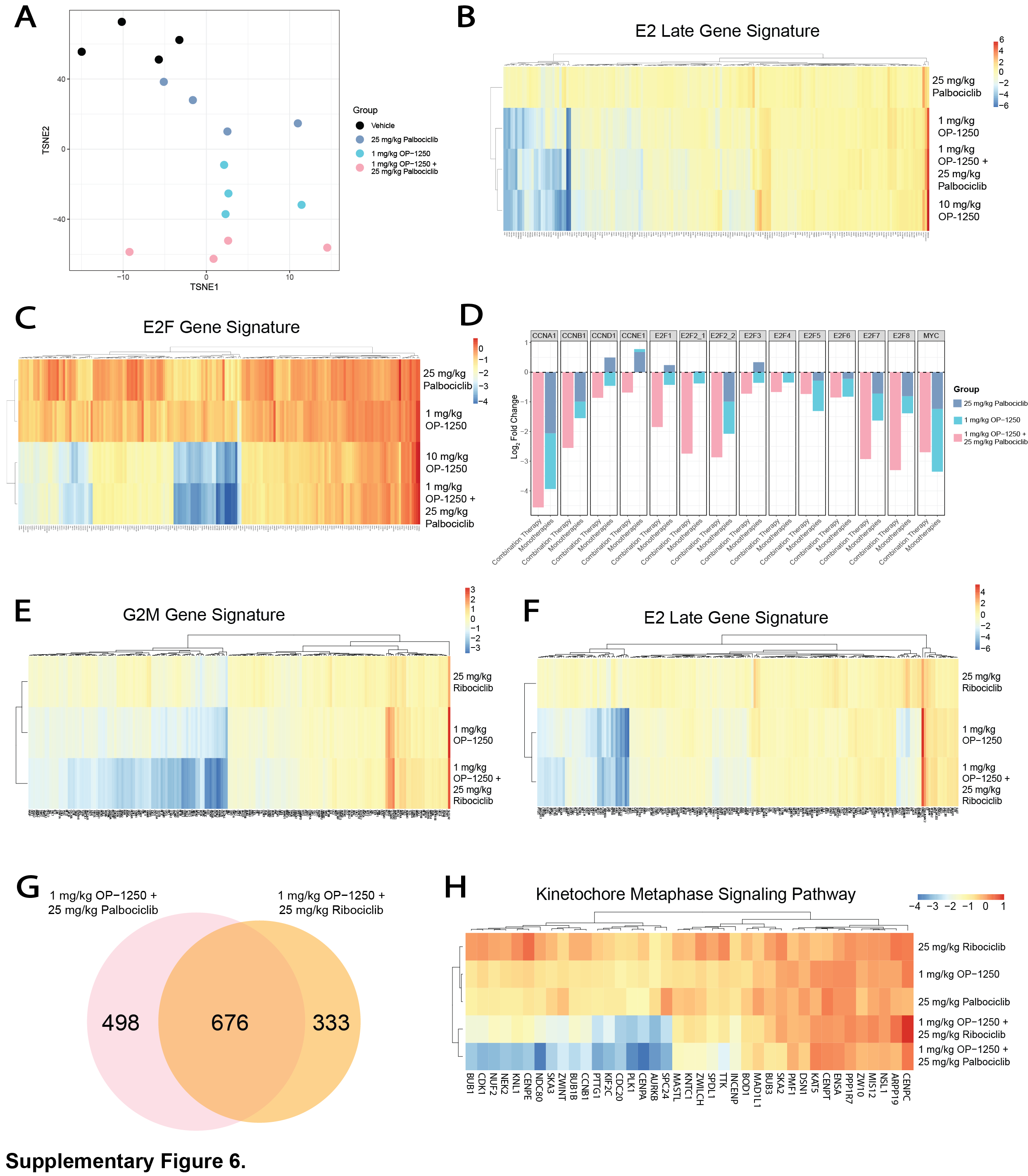
We conducted a xenograft study with the MCF7 cell line combining 1 and 10 mg/kg OP-1250 with 25 and 75 mg/kg palbociclib or ribociclib alongside corresponding monotherapy doses (Fig. 5B and C; Supplementary Fig. S5A and S5B). All doses and combinations resulted in tumor growth inhibition relative to vehicle, with greater inhibition with the higher monotherapy doses. The 10 mg/kg dose of OP-1250, which leads to plasma concentrations similar to those measured in clinical studies, outperformed 75 mg/kg monotherapy doses of both CDK4/6 inhibitors and resulted in tumor shrinkage. Combination treatment resulted in more pronounced and consistent tumor shrinkage across doses, with all but one animal achieving >50% reduction in tumor volume (Supplementary Fig. S5A and S5B) and many in the high-dose combination groups achieving near-complete tumor regression. RNA-seq of tumors from low-dose OP-1250 and palbociclib monotherapy and combination groups revealed that combination treatment enhanced suppression of genes associated with cell-cycle progression, with an observed effect greater than the combination of monotherapies (Fig. 5D and E; Supplementary Fig. S6A–S6D; Supplementary Table S3) and lack of a similar effect in pathways such as ER signaling or apoptosis (Fig. 5E). A similar transcriptional profile was observed for the OP-1250 and ribociclib combination (Supplementary Fig. S6E–S6H), illustrating the profound effect on cell-cycle regulation that can be generated through simultaneous administration of OP-1250 and a CDK4/6 inhibitor.
A similar study was conducted using the ST941 ESR1Y537S PDX model. While this model was more resistant to treatment, with modest or no response to lower monotherapy doses, higher dose combinations resulted in more tumor shrinkage than with monotherapy treatment (Fig. 5F and G; Supplementary Fig. S5C and S5D). Monitoring the number of live animals and tumor volume over 30 days after cessation of dosing revealed that addition of OP-1250 to either CDK4/6 inhibitor extended animal survival (Supplementary Fig. S5E and S5F) and led to a more durable tumor response (Supplementary Fig. S5G and S5H).
OP-1250 inhibits ER+ breast cancer intracranial xenografts and extends survival
In a pharmacokinetic analysis of animals from the HCI-013 mammary fat pad xenograft study, the concentration of OP-1250 in the brain 24 hours following last dose was comparable to or higher than plasma concentration (Fig. 4H), suggesting the compound can effectively penetrate the brain. To further investigate OP-1250 efficacy in a xenograft model of breast cancer brain metastases, mice were implanted intracranially with the ESR1Y537S mutation–containing ST941 breast cancer PDX model. In a preliminary study, 3 and 10 mg/kg doses of OP-1250 were tested alongside and in combination with 75 mg/kg ribociclib (Supplementary Fig. S7A). While ribociclib monotherapy slightly reduced intracranial tumor growth, the addition of ribociclib to 3 mg/kg OP-1250 led to improvement in tumor growth inhibition (Supplementary Fig. S7B and S7C) and prolonged animal survival (Supplementary Fig. S7D). The 10 mg/kg dose of OP-1250 outperformed the 3 mg/kg dose and was chosen for a study comparing OP-1250 against approved antiestrogens of interest, fulvestrant and tamoxifen (Fig. 6A).
Figure 6.

OP-1250 shrinks ER+ breast cancer intracranial xenografts and prolongs animal survival. A, Schematic of intracranial xenograft experiment using ESR1Y537S ST941 PDX model. B, Representative intracranial MRIs from ST941-implanted vehicle and OP-1250-treated mice at indicated timepoints. Arrow indicates implantation site. C, Mean and SEM intracranial tumor volume over time with listed treatments as assessed by MRI in ST941 intracranial xenograft model. D, Percent change in intracranial tumor volume of individual animals in listed treatment groups at day 101 or last recorded timepoint of ST941 intracranial xenograft study, normalized to day 1 tumor volume. E, Kaplan–Meier graphs of animal survival over time in listed treatment groups of mice implanted intracranially with the ST941 PDX model. Dosing was ceased on day 100 of study, as indicated by the shaded area.
A second vehicle-treated group with ovariectomy was included to monitor the effect of estrogen depletion on tumor growth. MRI was used to monitor tumors throughout the study and revealed a notable reduction in tumor size with OP-1250 treatment relative to vehicle animals (Fig. 6B; Supplementary Fig. S7E). Treatment with tamoxifen and OP-1250 inhibited intracranial tumor growth, while fulvestrant treatment and ovariectomy demonstrated minimal impact (Fig. 6C). Treatment with 10 mg/kg OP-1250 resulted in tumor shrinkage >50% in all treated animals with or without ribociclib treatment (Fig. 6D); 4 of 8 animals in the OP-1250 monotherapy group and 6 of 8 in the OP-1250 + ribociclib group lacked detectable tumor at study day 100. Animal survival was monitored throughout the study and for another 50 days following cessation of dosing (Fig. 6E). Tamoxifen treatment prolonged animal survival, with 6 of 8 animals alive at study day 100, but did not result in a durable effect once dosing was terminated. In contrast, OP-1250 treatment (monotherapy or in combination with ribociclib) resulted in all 8 animals alive at study day 100 and 6 alive 50 days after cessation of dosing.
Discussion
Palazestrant (OP-1250) has the potential to be a best-in-class ER antagonist for treatment of ER+ breast cancer due to its oral pharmacokinetic properties and preclinical antitumor efficacy. Unlike the clinically approved SERM tamoxifen, OP-1250 lacks agonist activity in AP assay, UWW assay, breast cancer proliferation assay, and RNA-seq analysis of estrogen signaling and cell-cycle genes in breast cancer cells. OP-1250 completely suppresses estrogen-induced AF1-dependent AP activity and inhibits estrogen-induced increases in UWW as effectively as the CERAN fulvestrant. In a PRO-seq assay of nascent RNA levels in breast cancer cells following estrogen stimulation, OP-1250 completely reverses estrogen's effects on canonical E2 pathway genes within 6 hours and downstream effects on cell-cycle genes within 24 hours. These data characterize a complete antagonist of the ER that blocks both AF1 and AF2 signaling domains and lacks agonist activity in uterine or breast cells. While the mechanism of AF1 inhibition by CERANs such as OP-1250 has not been determined, we hypothesize that this may occur through recruitment of ER corepressor proteins such as NCoR, as suggested previously (22, 48).
OP-1250 degrades ERα similarly to fulvestrant and other comparators of interest, with a DC50 of < 1 nmol/L; thus, it is a member of the SERD class as well as being a CERAN. However, while many groups have focused on degradation of the ER protein as a critical indicator of antitumor efficacy, the potent degradation observed with estradiol treatment raises questions concerning the reliability of ERα degradation as a metric for antagonist activity. Prior studies have also suggested that ER mobility or assays of downstream ER activity, such as gene expression profiling or chromatin accessibility, may correlate better with in vivo efficacy (27).
OP-1250 displays an in vitro profile comparable to fulvestrant, including biochemical binding affinity, ER degradation, and potent activity in wild-type and ESR1-mutant cellular proliferation assays. However, OP-1250 has a superior pharmacokinetic profile and oral bioavailability in mouse xenograft experiments, and significantly outperforms fulvestrant at a 10 mg/kg oral dose, including in PDX, which are characterized by greater heterogeneity and clinical relevance than cell line–derived models (49).
Mutations in ESR1 that confer constitutive activity of the ER are a known resistance mechanism to endocrine therapy (13, 50) and are present more frequently in high-grade (51) and metastatic tumors (16). Therefore, efficacy against ESR1-mutant tumors is critical for antiestrogens in clinical development to treat advanced breast cancer. Although most compounds displayed reduced potency against mutant ESR1, particularly regarding ESR1Y537S, OP-1250 demonstrated competitive in vitro efficacy against the most common clinical variants of ESR1 relative to agents of interest. In PDX models containing the ESR1Y537S mutation, OP-1250 effectively suppressed tumor growth at a 3 mg/kg dose and induced tumor shrinkage at higher doses or in combination with CDK4/6 inhibitors. In a study with the ESR1Y537S ST941 model, OP-1250 outperformed elacestrant, clinically approved for patients with ESR1 mutations, using a fraction of the dose. The superior performance of OP-1250 relative to both fulvestrant and elacestrant in these xenograft studies, and the inability of aromatase inhibitors to inhibit growth of ESR1-mutant tumors indicate that OP-1250 may provide additional benefit beyond currently approved therapies.
Combinability with CDK4/6 inhibitors is another important attribute for ER antagonists and has proven to be challenging for other oral SERDs in clinical trials. Notably, OP-1250 demonstrates combinability with both palbociclib and ribociclib in wild-type and ESR1-mutant preclinical models, establishing potential for combination treatment relevant to both first- and second-line clinical settings.
Even as breast cancer treatments improve, central nervous system metastasis remains important; these metastases occur in approximately 25% of patients with metastatic breast cancer and correlate with poor prognosis (52). In pharmacokinetic analyses of mouse xenograft studies, OP-1250 demonstrates excellent brain penetrance and favorable half-life. In an intracranial xenograft study, treatment with 10 mg/kg OP-1250 resulted in tumor shrinkage and survival of all animals over a 100-day dosing interval, dramatically outperforming fulvestrant and tamoxifen. These properties position OP-1250 to treat a broader patient population than currently available therapies and provide novel benefit to patients with high-risk metastatic breast cancer.
Overall, these data characterize a promising new clinical candidate in the oral SERD/CERAN class with potential for the treatment of ER+ breast cancer brain metastasis. OP-1250 is in clinical trials (NCT04505826, NCT05266105, NCT05508906) for the treatment of ER+ metastatic breast cancer, as monotherapy and in combination with palbociclib, ribociclib, or alpelisib.
Supplementary Material
PROSeq analysis of gene expression changes following treatment of MCF7 and CAMA-1 cells with ER-alpha ligands.
PROSeq gene ontology dataset following treatment of MCF7 and CAMA-1 cells with ER-alpha ligands.
RNASeq dataset of gene pathway changes following treatment with OP-1250, palbociclib and ribociclib in an MCF7 xenograft model.
Supplementary text including supplementary materials and methods and references.
OP-1250 specifically inhibits the ER without cross-reactivity with other hormone receptors. A, LanthaScreen co-activator recruitment assay using wild-type ERα LBD and fluorescein tagged PGC1a comparing OP-1250 and other approved and in-trial comparator molecules. Data are normalized to DMSO vehicle and 10 µM E2-treated wells (100% recruitment) and presented as mean and SD of at least two separate experiments. B, Reporter gene activity assay in CAMA-1 and MCF7 cells transiently transfected with a luciferase reporter gene construct containing an estrogen response element and minimal tk promoter. Cells were cultured in estrogen-depleted media and reporter gene activity was normalized to E2 vehicle. The dotted line represents activity in the absence of estrogen, the hashed line represents activity with E2 supplementation. Data shown represent mean and SD from a representative of at least three independent experiments. C, Reporter gene assay of ER-negative SK-BR-3 cells transiently transfected with wild-type or mutant ESR1 vectors along with a luciferase reporter gene cloned to 3× estrogen response element and TATA minimal promoter. Transcriptional activity was measured as luciferase activity. Cells were cultured in estrogen-depleted media and reporter gene activity was normalized to wild-type E2 vehicle and empty vector + E2. The dotted line represents activity with empty vector control, the hashed line represents activity following supplementation with 31.6 nM E2. Data shown represent mean and SD from a representative of at least three independent experiments. D, Reporter gene activity assay of HeLa cells transiently transfected with glucocorticoid, androgen, or progesterone receptor and a luciferase reporter cloned to 3× PRE and TATA minimal promoter, measuring the impact of OP-1250 on the glucocorticoid receptor in the presence and absence of 31.6 nM dexamethasone, progesterone receptor in the presence and absence of 500 nM MPA (medroxyprogesterone acetate), and androgen receptor in the presence and absence of 50 nM R1881. Data are normalized to the indicated amount of agonist (either MPA, dexamethasone, or R1881). Figures show data as the mean and SD from a representative of at least three independent experiments. E, Reporter gene assay of ER-negative SK-BR-3 cells transiently transfected with ESR1 or ESR2 along with a luciferase reporter gene cloned to 3× estrogen response element and TATA minimal promoter. Transcriptional activity was measured as luciferase activity. Cells were cultured in estrogen-depleted media and reporter gene activity was normalized to E2 vehicle. Data shown represent mean and SD from a representative of at least three independent experiments. F, Uterine wet weight assay following treatment with a dose response of OP-1250 or fulvestrant in the presence of E2 supplementation. Mean and SEM from six mice per treatment group are plotted with asterisks indicating significance relative to vehicle by one-way analysis of variance. * represents adjusted p-value <0.05, *** represents adjusted p-value <0.001, **** represents adjusted p-value <0.0001. G, Simple western analysis of ERα protein levels in Ishikawa cells after 72-h treatment with listed compounds. Samples are normalized to untreated control. Data shown represent mean and SD from a representative of at least three independent experiments. H, Representative blot image from simple western assay shown in panel G after 72-h treatment with 300 nM compound or 1 nM E2 with indicated compound treatments and cell line. AR=androgen receptor; GR=glucocorticoid receptor; PR= progesterone receptor; WT=wild-type.
{kind=link}
Effects of E2 and antiestrogens on transcriptional activity and cellular proliferation in E2-depleted media. A, Volcano plot of genes differentially expressed by 15-min E2 treatment relative to vehicle in PRO-seq experiment. Each dot represents a gene in the annotation set. Adjusted p-value is plotted on the y-axis and fold-change relative to time-matched vehicle on the x-axis. The number in the top right-hand corner represents the number of genes changed, and red dots indicate genes with observed change in expression. B, PRO-seq read distribution at estrogen target genes CCND1 and MYC after 15 min of E2 treatment (red) compared to vehicle control (blue). The x-axis refers to the position along the genome, and the y-axis refers to the number of reads aligning to that position normalized for the depth of each sample. Replicates were averaged. Bars that are less than zero are reverse-strand reads; bars above zero are forward-strand reads. C, PRO-seq read distribution at estrogen target gene GREB1 after 6 h of OP-1250 treatment following E2 pretreatment (red) compared to 24-h E2 treatment (blue). The x-axis refers to the position along the genome, and the y-axis refers to the number of reads aligning to that position normalized for the depth of each sample. Replicates were averaged. Bars that are less than zero are reverse-strand reads; bars above zero are forward-strand reads. D, Heatmap of annotated G2/M checkpoint genes in CAMA-1 and MCF7 cells after 24 h of compound treatment in PRO-seq experiment. Red indicates high expression and blue low expression. Samples and genes with similar expression patterns are grouped together using hierarchical clustering, with sample relationships depicted by dendrograms at the top and left of graph. E, Chart showing number of differentially expressed genes compared to vehicle for each treatment group in CAMA-1 mRNA-seq experiment. Cells were treated with 100 pM E2 or 316 nM antiestrogen in E2-depleted media for 24 h.
{kind=link}
OP-1250 acts as a selective ER degrader (SERD) in wild-type and ESR1 mutant cell lines. A, B, Representative blot images from the simple western assay shown in Figure 3 (see main article) after 24-h treatment with 300 nM compound or 1 nM E2 with indicated compound treatments and cell lines. C, D, Representative blot images from simple western assay of MCF7 cells treated for 2 h, 4 h, or 8 h with the listed concentrations of compounds in the presence or absence of MG-132 and simple western analysis of ERα protein levels after treatment with listed compounds. Samples were normalized to untreated control, with mean and SD across at least three independent experiments shown. E, F, ERα degradation in CAMA-1 cells with the indicated homozygous mutant as assessed by simple western assay after 24-h treatment with 300 nM compound or 1 nM E2. Samples were normalized to untreated control, represented by a dotted line, with mean and SD across at least three independent experiments shown. G, H, Representative blot images from simple western data shown in panels E and F with indicated compound treatments and cell lines.
{kind=link}
OP-1250 demonstrates potent in vivo activity and outperforms competitor compounds in ESR1 mutant xenograft studies. A, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ESR1Y537S HCI-013 PDX study, normalized to day 1 tumor volume. B, Mean and SEM tumor volume over time with listed treatments in the ESR1Y537S HCI-013EI PDX breast cancer model conducted in the absence of estrogen supplementation. The dotted line represents tumor stasis. C, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ESR1Y537S PDX HCI-013EI xenograft study conducted in the absence of estrogen supplementation, normalized to day 1 tumor volume. D, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ESR1Y537S ST941 PDX study, normalized to day 1 tumor volume. E, Representative ST941 PDX study immunostained tumor images of Ki67 and ER⍺ treated with vehicle, OP-1250 at 5 mg/kg, and OP-1250 at 10 mg/kg.
{kind=link}
Addition of OP-1250 to CDK4/6 inhibitors enhances tumor suppression and prolongs animal survival in ER+ breast cancer xenograft models. A, B, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the MCF7 xenograft study, normalized to day 1 tumor volume. C, D, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ST941 xenograft study, normalized to day 1 tumor volume. E, F, Kaplan–Meier graphs of animal survival over time in listed treatment groups of mice implanted with the ESR1Y537S ST941 PDX model. Dosing ended on day 29 of the study, and animals were sacrificed when tumor volume reached 2000 mm3. G, H, Mean and SEM tumor volume over the full study interval with monotherapy and combination treatments in the ESR1Y537S ST941 PDX breast cancer model, with the dotted line representing tumor stasis. Dosing ended on day 29, as indicated by the dashed line.
{kind=link}
Combination of OP-1250 with palbociclib or ribociclib enhances suppression of cell cycle-related gene expression. A, t-SNE plot of MCF7 xenograft samples from vehicle, low-dose OP-1250, and palbociclib monotherapy and combination treatment groups. B, C, Heatmap showing expression of genes associated with the E2 late gene signature and E2F gene signature of n = 3 MCF7 xenograft tumors from OP-1250 and palbociclib low dose monotherapy and combination therapy groups. Red indicates high expression relative to vehicle and blue indicates low expression. D, Comparison of log2-fold change values for 14 cell cycle-related genes between combination therapy (pink) and monotherapies (blue and purple for 1 mg/kg OP-1250 and 25 mg/kg palbociclib, respectively). Stacked bars represent the combined effect of the monotherapies according to the Bliss independence model. E, F, Heatmaps showing expression of genes associated with the G2/M and E2 late gene signatures of n = 3 MCF7 xenograft tumors from OP-1250 and ribociclib low dose monotherapy and combination therapy groups. Red indicates high expression relative to vehicle and blue indicates low expression. G, Venn diagram showing overlap in dysregulated genes relative to vehicle of 1 mg/kg OP-1250 + 25 mg/kg palbociclib and 1 mg/kg OP-1250 + 25 mg/kg ribociclib MCF7 xenograft tumors. H, Heatmap of genes associated with the kinetochore metaphase signaling pathway, the most enriched pathway (p-value 6.15×10-7) in 676 genes changed in both OP-1250 + CDK4/6 inhibitor treatment groups. Genes are color-coded by gene expression relative to vehicle, with red indicating high expression and blue indicating low expression. OP-1250 1 mg/kg treatment groups from two xenograft studies were averaged.
{kind=link}
OP-1250 demonstrates potent activity in an ER+ breast cancer intracranial xenograft and outperforms ribociclib. A, Schematic of intracranial xenograft experiment with data shown in B–D. B, Mean and SEM intracranial tumor volume over time with listed treatments as assessed by MRI in an ESR1Y537S ST941 PDX intracranial xenograft model. C, Percent change in intracranial tumor volume of individual animals in listed treatment groups at day 101 or at the last recorded timepoint in the ST941 intracranial xenograft study, normalized to day 1 tumor volume. D, Kaplan–Meier graphs of animal survival over time in listed treatment groups of mice implanted intracranially with the ST941 PDX xenograft. Dosing ended on day 100 of the study in the OP-1250 10 mg/kg group, as indicated by the shaded area. E, Representative intracranial MRIs from ST941-implanted mice with data shown in Figure 6 (see main article) at indicated timepoints and treatment groups. PO=orally.
{kind=link}
Supplementary Legends
Acknowledgments
PRO-seq was conducted in collaboration with Arpeggio Biosciences. Intracranial xenograft studies were conducted in collaboration with Minerva Imaging. The ST941C cell line was licensed from XenoSTART, and the ST941 PDX model was developed by XenoSTART. The HCI-013 and HCI-013EI PDX models were acquired from the Huntsman Cancer Institute's Preclinical Research Shared Resource (PRR) at the University of Utah (Salt Lake City, Utah). Various xenograft studies mentioned in this publication utilized the PRR. We thank Sean Fanning and Geof Greene for helpful discussions on these topics, and Robert Millikin for his assistance in the initial synthesis of OP-1250.
This article is featured in Selected Articles from This Issue, p. 255
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Authors' Disclosures
A.D. Parisian reports personal fees from Olema Pharmaceuticals outside the submitted work; in addition, A.D. Parisian has a patent for METHODS OF TREATING ESTROGEN RECEPTOR-ASSOCIATED DISEASES pending and licensed to Olema Pharmaceuticals. S.A. Barratt reports personal fees from Olema Pharmaceuticals, Inc outside the submitted work. L. Hodges-Gallagher reports other support from Olema Pharmaceuticals outside the submitted work; in addition, L. Hodges-Gallagher has a patent for PCT/US2020/040863 issued to Olema Pharmaceuticals, a patent for PCT/US2021/021151 issued to Olema Pharmaceuticals, and a patent for PCT/US2022/036351 issued to Olema Pharmaceuticals. F.E. Ortega reports personal fees from Olema Pharmaceuticals outside the submitted work. G. Peña reports other support from Olema Pharmaceuticals outside the submitted work. B. Robello reports personal fees from Olema Pharmaceuticals during the conduct of the study; and B. Robello is currently an employee of Olema Oncology. R. Sun reports a patent for REGIMENS OF ESTROGEN RECEPTOR ANTAGONISTS pending to Olema Pharmaceuticals. G.S. Palanisamy reports personal fees from Olema Oncology during the conduct of the study. D.C. Myles reports a patent for US 10,292,971 issued, a patent for US 10,624,878 issued, a patent for US 11,229,630 issued, a patent for PCT/US2020/040863 pending, a patent for PCT/US2021/021151 pending, and a patent for US 10,292,971 Tetrahydro-1H-pyrido[3,4-b]indole anti-estrogenic drugs US 10,624,878 Tetrahydro-1H-pyrido[3,4-b]indole anti-estrogenic drugs US 11,229,630 Tetrahydro-1H-pyrido[3,4-b]indole anti-estrogenic drugs PCT/US2020/040863 REGIMENS OF ESTROGEN RECEPTOR ANTAGONISTS PCT/US2021/021151 METHODS OF TREATING ESTROGEN RECEPTOR-ASSOCIATED DISEASES PCT/US2022/036351 pending. P.J. Kushner reports grants, personal fees, and other support from Olema Oncology outside the submitted work; in addition, P.J. Kushner holds patents for Tetrahydro-1H-pyrido[3,4-b]indole anti-estrogenic drugs issued and licensed to Olema Oncology, a patent for REGIMENS OF ESTROGEN RECEPTOR ANTAGONISTS pending and licensed to Olema Oncology, and a patent for METHODS OF TREATING ESTROGEN RECEPTOR-ASSOCIATED DISEASES pending and licensed to Olema Oncology. pending, issued, licensed, and with royalties paid from Olema Oncology; and P.J. Kushner is an ex-employee of Olema and holds stock in Olema. C.L. Harmon reports other support from Olema Pharmaceuticals during the conduct of the study; in addition, C.L. Harmon has a patent for Tetrahydro-1H-pyrido[3,4-b]indole anti-estrogenic drugs issued and licensed to Olema Oncology, a patent for REGIMENS OF ESTROGEN RECEPTOR ANTAGONISTS pending and licensed to Olema Oncology, and a patent for METHODS OF TREATING ESTROGEN RECEPTOR-ASSOCIATED DISEASES pending and licensed to Olema Oncology. No disclosures were reported by the other authors.
Authors' Contributions
A.D. Parisian: Formal analysis, investigation, writing–original draft, project administration, writing–review and editing. S.A. Barratt: Supervision, investigation, writing–original draft, writing–review and editing. L. Hodges-Gallagher: Conceptualization, supervision, investigation, writing–review and editing. F.E. Ortega: Software, formal analysis, visualization. G. Peña: Investigation. J. Sapugay: Data curation, project administration. B. Robello: Investigation. R. Sun: Investigation. D. Kulp: Data curation, supervision. G.S. Palanisamy: Supervision, writing–review and editing. D.C. Myles: Conceptualization, supervision, investigation. P.J. Kushner: Conceptualization, supervision, writing–review and editing. C.L. Harmon: Conceptualization, data curation, software, supervision, visualization, writing–review and editing.
References
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 2. Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem 2001;276:36869–72. [DOI] [PubMed] [Google Scholar]
- 3. Yamaga R, Ikeda K, Horie-Inoue K, Ouchi Y, Suzuki Y, Inoue S. RNA sequencing of MCF-7 breast cancer cells identifies novel estrogen-responsive genes with functional estrogen receptor-binding sites in the vicinity of their transcription start sites. Horm Cancer 2013;4:222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LA, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst 2014;106:dju055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer 2004;90:S2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov 2003;2:205–13. [DOI] [PubMed] [Google Scholar]
- 7. Carpenter R, Miller WR. Role of aromatase inhibitors in breast cancer. Br J Cancer 2005;93:S1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Kruchten M, de Vries EG, Glaudemans AW, van Lanschot MC, van Faassen M, Kema IP, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov 2015;5:72–81. [DOI] [PubMed] [Google Scholar]
- 9. Emons G, Mustea A, Tempfer C. Tamoxifen and endometrial cancer: a janus-headed drug. Cancers 2020;12:2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer 2015;15:261–75. [DOI] [PubMed] [Google Scholar]
- 11. Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016;5:e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45:1439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 1997;57:1244–9. [PubMed] [Google Scholar]
- 14. Bardia A, Neven P, Streich G, Montero AJ, Forget F, Mouret-Reynier M-A, et al. Elacestrant, an oral selective estrogen receptor degrader (SERD), vs investigator's choice of endocrine monotherapy for ER+/HER2- advanced/metastatic breast cancer (mBC) following progression on prior endocrine and CDK4/6 inhibitor therapy: results of EMERALD phase 3 trial [abstract]. In: Proceedings of the 2021 San Antonio Breast Cancer Symposium; 2021 Dec 7–10; San Antonio, TX. Philadelphia (PA): AACR; Cancer Res 2022;82(4 Suppl):Abstract nr GS2-02. [Google Scholar]
- 15. Oliveira M, Pominchuck D, Nowecki Z, Hamilton E, Kulyaba Y, Andabekov T, et al. GS3-02 Camizestrant, a next generation oral SERD vs fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: results of the randomized, multi-dose Phase 2 SERENA-2 trial [abstract]. In: Proceedings of the 2022 San Antonio Breast Cancer Symposium; 2022 Dec 6–10; San Antonio, TX. Philadelphia (PA): AACR; Cancer Res 2023;83(5 Suppl):Abstract nr GS3-02. [Google Scholar]
- 16. Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, et al. Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res 2016;22:1130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gelsomino L, Gu G, Rechoum Y, Beyer AR, Pejerrey SM, Tsimelzon A, et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res Treat 2016;157:253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bidard FC, Kaklamani VG, Neven P, Streich G, Montero AJ, Forget F, et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor–positive, human Epidermal growth factor receptor 2–negative advanced breast cancer: results from the randomized phase III EMERALD trial. J Clin Oncol 2022;40:3246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell 1989;59:477–87. [DOI] [PubMed] [Google Scholar]
- 20. Metzger D, Ali S, Bornert JM, Chambon P. Characterization of the amino-terminal transcriptional activation function of the human estrogen receptor in animal and yeast cells. J Biol Chem 1995;270:9535–42. [DOI] [PubMed] [Google Scholar]
- 21. Varlakhanova N, Snyder C, Jose S, Hahm JB, Privalsky ML. Estrogen receptors recruit SMRT and N-CoR corepressors through newly recognized contacts between the corepressor N terminus and the receptor DNA binding domain. Mol Cell Biol 2010;30:1434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Webb P, Nguyen P, Kushner PJ. Differential SERM effects on corepressor binding dictate ERalpha activity in vivo. J Biol Chem 2003;278:6912–20. [DOI] [PubMed] [Google Scholar]
- 23. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol 2000;74:311–7. [DOI] [PubMed] [Google Scholar]
- 24. Webb P, Lopez GN, Uht RM, Kushner PJ. Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol 1995;9:443–56. [DOI] [PubMed] [Google Scholar]
- 25. Sakamoto T, Eguchi H, Omoto Y, Ayabe T, Mori H, Hayashi S, et al. Estrogen receptor-mediated effects of tamoxifen on human endometrial cancer cells. Mol Cell Endocrinol 2002;192:93–104. [DOI] [PubMed] [Google Scholar]
- 26. Abot A, Fontaine C, Raymond-Letron I, Flouriot G, Adlanmerini M, Buscato M, et al. The AF-1 activation function of estrogen receptor α is necessary and sufficient for uterine epithelial cell proliferation in vivo. Endocrinology 2013;154:2222–33. [DOI] [PubMed] [Google Scholar]
- 27. Guan J, Zhou W, Hafner M, Blake RA, Chalouni C, Chen IP, et al. Therapeutic ligands antagonize estrogen receptor function by impairing its mobility. Cell 2019;178:949–63. [DOI] [PubMed] [Google Scholar]
- 28. Ciruelos E, Pascual T, Arroyo Vozmediano ML, Blanco M, Manso L, Parrilla L, et al. The therapeutic role of fulvestrant in the management of patients with hormone receptor-positive breast cancer. Breast 2014;23:201–8. [DOI] [PubMed] [Google Scholar]
- 29. Wardell SE, Marks JR, McDonnell DP. The turnover of estrogen receptor α by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy. Biochem Pharmacol 2011;82:122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gradishar WJ, Moran MS, Abraham J, Aft R, Agnese D, Allison KH, et al. Breast cancer, version 3.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2022;20:691–722. [DOI] [PubMed] [Google Scholar]
- 31. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 2016;13:417–30. [DOI] [PubMed] [Google Scholar]
- 32. Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol 2018;36:2465–72. [DOI] [PubMed] [Google Scholar]
- 33. Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med 2018;379:1926–36. [DOI] [PubMed] [Google Scholar]
- 34. Johnston S, Martin M, Di Leo A, Im SA, Awada A, Forrester T, et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crew AP, Qian Y, Dong H, Wang J. Tetrahydronapthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders. United States patent US 10,899,742 B1; 2021.
- 36. Myles DC, Kushner PJ, Harmon CL. Tetrahydro-1H-pyrido[3,4-b]indole anti-estrogenic drugs. United States patent WO 201705139 A1; 2017.
- 37. Luo X, Archibeque I, Dellamaggiore K, Smither K, Homann O, Lipford JR, et al. Profiling of diverse tumor types establishes the broad utility of VHL-based ProTaCs and triages candidate ubiquitin ligases. iScience 2022;25:103985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 2016;11:1455–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, Andrews DM, et al. Optimization of a novel binding motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. J Med Chem 2015;58:8128–40. [DOI] [PubMed] [Google Scholar]
- 41. Kwekel JC, Forgacs AL, Burgoon LD, Williams KJ, Zacharewski R. Tamoxifen-elicited uterotrophy: cross-species and cross-ligand analysis of the gene expression program. BMC Med Genomics 2009;2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ashby J, Odum J, Foster JR. Activity of raloxifene in immature and ovariectomized rat uterotrophic assays. Regul Toxicol Pharmacol 1997;25:226–31. [DOI] [PubMed] [Google Scholar]
- 43. Littlefield BA, Gurpide E, Markiewicz L, McKinley B, Hochberg RB. A simple and sensitive microtiter plate estrogen bioassay based on stimulation of alkaline phosphatase in Ishikawa cells: estrogenic action of delta 5 adrenal steroids. Endocrinology 1990;127:2757–62. [DOI] [PubMed] [Google Scholar]
- 44. Fanning SW, Hodges-Gallagher L, Myles DC, Sun R, Fowler CE, Plant IN, et al. Specific stereochemistry of OP-1074 disrupts estrogen receptor alpha helix 12 and confers pure antiestrogenic activity. Nat Commun 2018;9:2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer 2015;22:713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peano BJ, Crabtree JS, Komm BS, Winneker RC, Harris HA. Effects of various selective estrogen receptor modulators with or without conjugated estrogens on mouse mammary gland. Endocrinology 2008;150:1897–903. [DOI] [PubMed] [Google Scholar]
- 47. Wick MJ, Diaz A, Thomas M, Moriarty A, Quinn M, Guerra M, et al. Establishment and characterization of ST941/C; an ESR1-mutant ER+ breast cancer cell line and xenograft from a patient with acquired resistance to endocrine therapy [abstract]. In: Proceedings of the 2016 San Antonio Breast Cancer Symposium; 2016 Dec 6–10; San Antonio, TX. Philadelphia (PA): AACR; Cancer Res 2017;77(4 Suppl):Abstract nr P3-04-26. [Google Scholar]
- 48. Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science 2002;295:2465–8. [DOI] [PubMed] [Google Scholar]
- 49. Landis MD, Lehmann BD, Pietenpol JA, Chang JC. Patient-derived breast tumor xenografts facilitating personalized cancer therapy. Breast Cancer Res 2013;15:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013;45:1446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang K, Li L, Franch-Expósito S, Le X, Tang J, Li Q, et al. Integrated multi-omics profiling of high-grade estrogen receptor-positive, HER2-negative breast cancer. Mol Oncol 2022;16:2413–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Darlix A, Louvel G, Fraisse J, Jacot W, Brain E, Debled M, et al. Impact of breast cancer molecular subtypes on the incidence, kinetics and prognosis of central nervous system metastases in a large multicentre real-life cohort. Br J Cancer 2019;121:991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PROSeq analysis of gene expression changes following treatment of MCF7 and CAMA-1 cells with ER-alpha ligands.
PROSeq gene ontology dataset following treatment of MCF7 and CAMA-1 cells with ER-alpha ligands.
RNASeq dataset of gene pathway changes following treatment with OP-1250, palbociclib and ribociclib in an MCF7 xenograft model.
Supplementary text including supplementary materials and methods and references.
OP-1250 specifically inhibits the ER without cross-reactivity with other hormone receptors. A, LanthaScreen co-activator recruitment assay using wild-type ERα LBD and fluorescein tagged PGC1a comparing OP-1250 and other approved and in-trial comparator molecules. Data are normalized to DMSO vehicle and 10 µM E2-treated wells (100% recruitment) and presented as mean and SD of at least two separate experiments. B, Reporter gene activity assay in CAMA-1 and MCF7 cells transiently transfected with a luciferase reporter gene construct containing an estrogen response element and minimal tk promoter. Cells were cultured in estrogen-depleted media and reporter gene activity was normalized to E2 vehicle. The dotted line represents activity in the absence of estrogen, the hashed line represents activity with E2 supplementation. Data shown represent mean and SD from a representative of at least three independent experiments. C, Reporter gene assay of ER-negative SK-BR-3 cells transiently transfected with wild-type or mutant ESR1 vectors along with a luciferase reporter gene cloned to 3× estrogen response element and TATA minimal promoter. Transcriptional activity was measured as luciferase activity. Cells were cultured in estrogen-depleted media and reporter gene activity was normalized to wild-type E2 vehicle and empty vector + E2. The dotted line represents activity with empty vector control, the hashed line represents activity following supplementation with 31.6 nM E2. Data shown represent mean and SD from a representative of at least three independent experiments. D, Reporter gene activity assay of HeLa cells transiently transfected with glucocorticoid, androgen, or progesterone receptor and a luciferase reporter cloned to 3× PRE and TATA minimal promoter, measuring the impact of OP-1250 on the glucocorticoid receptor in the presence and absence of 31.6 nM dexamethasone, progesterone receptor in the presence and absence of 500 nM MPA (medroxyprogesterone acetate), and androgen receptor in the presence and absence of 50 nM R1881. Data are normalized to the indicated amount of agonist (either MPA, dexamethasone, or R1881). Figures show data as the mean and SD from a representative of at least three independent experiments. E, Reporter gene assay of ER-negative SK-BR-3 cells transiently transfected with ESR1 or ESR2 along with a luciferase reporter gene cloned to 3× estrogen response element and TATA minimal promoter. Transcriptional activity was measured as luciferase activity. Cells were cultured in estrogen-depleted media and reporter gene activity was normalized to E2 vehicle. Data shown represent mean and SD from a representative of at least three independent experiments. F, Uterine wet weight assay following treatment with a dose response of OP-1250 or fulvestrant in the presence of E2 supplementation. Mean and SEM from six mice per treatment group are plotted with asterisks indicating significance relative to vehicle by one-way analysis of variance. * represents adjusted p-value <0.05, *** represents adjusted p-value <0.001, **** represents adjusted p-value <0.0001. G, Simple western analysis of ERα protein levels in Ishikawa cells after 72-h treatment with listed compounds. Samples are normalized to untreated control. Data shown represent mean and SD from a representative of at least three independent experiments. H, Representative blot image from simple western assay shown in panel G after 72-h treatment with 300 nM compound or 1 nM E2 with indicated compound treatments and cell line. AR=androgen receptor; GR=glucocorticoid receptor; PR= progesterone receptor; WT=wild-type.
Effects of E2 and antiestrogens on transcriptional activity and cellular proliferation in E2-depleted media. A, Volcano plot of genes differentially expressed by 15-min E2 treatment relative to vehicle in PRO-seq experiment. Each dot represents a gene in the annotation set. Adjusted p-value is plotted on the y-axis and fold-change relative to time-matched vehicle on the x-axis. The number in the top right-hand corner represents the number of genes changed, and red dots indicate genes with observed change in expression. B, PRO-seq read distribution at estrogen target genes CCND1 and MYC after 15 min of E2 treatment (red) compared to vehicle control (blue). The x-axis refers to the position along the genome, and the y-axis refers to the number of reads aligning to that position normalized for the depth of each sample. Replicates were averaged. Bars that are less than zero are reverse-strand reads; bars above zero are forward-strand reads. C, PRO-seq read distribution at estrogen target gene GREB1 after 6 h of OP-1250 treatment following E2 pretreatment (red) compared to 24-h E2 treatment (blue). The x-axis refers to the position along the genome, and the y-axis refers to the number of reads aligning to that position normalized for the depth of each sample. Replicates were averaged. Bars that are less than zero are reverse-strand reads; bars above zero are forward-strand reads. D, Heatmap of annotated G2/M checkpoint genes in CAMA-1 and MCF7 cells after 24 h of compound treatment in PRO-seq experiment. Red indicates high expression and blue low expression. Samples and genes with similar expression patterns are grouped together using hierarchical clustering, with sample relationships depicted by dendrograms at the top and left of graph. E, Chart showing number of differentially expressed genes compared to vehicle for each treatment group in CAMA-1 mRNA-seq experiment. Cells were treated with 100 pM E2 or 316 nM antiestrogen in E2-depleted media for 24 h.
OP-1250 acts as a selective ER degrader (SERD) in wild-type and ESR1 mutant cell lines. A, B, Representative blot images from the simple western assay shown in Figure 3 (see main article) after 24-h treatment with 300 nM compound or 1 nM E2 with indicated compound treatments and cell lines. C, D, Representative blot images from simple western assay of MCF7 cells treated for 2 h, 4 h, or 8 h with the listed concentrations of compounds in the presence or absence of MG-132 and simple western analysis of ERα protein levels after treatment with listed compounds. Samples were normalized to untreated control, with mean and SD across at least three independent experiments shown. E, F, ERα degradation in CAMA-1 cells with the indicated homozygous mutant as assessed by simple western assay after 24-h treatment with 300 nM compound or 1 nM E2. Samples were normalized to untreated control, represented by a dotted line, with mean and SD across at least three independent experiments shown. G, H, Representative blot images from simple western data shown in panels E and F with indicated compound treatments and cell lines.
OP-1250 demonstrates potent in vivo activity and outperforms competitor compounds in ESR1 mutant xenograft studies. A, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ESR1Y537S HCI-013 PDX study, normalized to day 1 tumor volume. B, Mean and SEM tumor volume over time with listed treatments in the ESR1Y537S HCI-013EI PDX breast cancer model conducted in the absence of estrogen supplementation. The dotted line represents tumor stasis. C, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ESR1Y537S PDX HCI-013EI xenograft study conducted in the absence of estrogen supplementation, normalized to day 1 tumor volume. D, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ESR1Y537S ST941 PDX study, normalized to day 1 tumor volume. E, Representative ST941 PDX study immunostained tumor images of Ki67 and ER⍺ treated with vehicle, OP-1250 at 5 mg/kg, and OP-1250 at 10 mg/kg.
Addition of OP-1250 to CDK4/6 inhibitors enhances tumor suppression and prolongs animal survival in ER+ breast cancer xenograft models. A, B, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the MCF7 xenograft study, normalized to day 1 tumor volume. C, D, Percent change in tumor volume of individual animals in listed treatment groups at day 29 of the ST941 xenograft study, normalized to day 1 tumor volume. E, F, Kaplan–Meier graphs of animal survival over time in listed treatment groups of mice implanted with the ESR1Y537S ST941 PDX model. Dosing ended on day 29 of the study, and animals were sacrificed when tumor volume reached 2000 mm3. G, H, Mean and SEM tumor volume over the full study interval with monotherapy and combination treatments in the ESR1Y537S ST941 PDX breast cancer model, with the dotted line representing tumor stasis. Dosing ended on day 29, as indicated by the dashed line.
Combination of OP-1250 with palbociclib or ribociclib enhances suppression of cell cycle-related gene expression. A, t-SNE plot of MCF7 xenograft samples from vehicle, low-dose OP-1250, and palbociclib monotherapy and combination treatment groups. B, C, Heatmap showing expression of genes associated with the E2 late gene signature and E2F gene signature of n = 3 MCF7 xenograft tumors from OP-1250 and palbociclib low dose monotherapy and combination therapy groups. Red indicates high expression relative to vehicle and blue indicates low expression. D, Comparison of log2-fold change values for 14 cell cycle-related genes between combination therapy (pink) and monotherapies (blue and purple for 1 mg/kg OP-1250 and 25 mg/kg palbociclib, respectively). Stacked bars represent the combined effect of the monotherapies according to the Bliss independence model. E, F, Heatmaps showing expression of genes associated with the G2/M and E2 late gene signatures of n = 3 MCF7 xenograft tumors from OP-1250 and ribociclib low dose monotherapy and combination therapy groups. Red indicates high expression relative to vehicle and blue indicates low expression. G, Venn diagram showing overlap in dysregulated genes relative to vehicle of 1 mg/kg OP-1250 + 25 mg/kg palbociclib and 1 mg/kg OP-1250 + 25 mg/kg ribociclib MCF7 xenograft tumors. H, Heatmap of genes associated with the kinetochore metaphase signaling pathway, the most enriched pathway (p-value 6.15×10-7) in 676 genes changed in both OP-1250 + CDK4/6 inhibitor treatment groups. Genes are color-coded by gene expression relative to vehicle, with red indicating high expression and blue indicating low expression. OP-1250 1 mg/kg treatment groups from two xenograft studies were averaged.
OP-1250 demonstrates potent activity in an ER+ breast cancer intracranial xenograft and outperforms ribociclib. A, Schematic of intracranial xenograft experiment with data shown in B–D. B, Mean and SEM intracranial tumor volume over time with listed treatments as assessed by MRI in an ESR1Y537S ST941 PDX intracranial xenograft model. C, Percent change in intracranial tumor volume of individual animals in listed treatment groups at day 101 or at the last recorded timepoint in the ST941 intracranial xenograft study, normalized to day 1 tumor volume. D, Kaplan–Meier graphs of animal survival over time in listed treatment groups of mice implanted intracranially with the ST941 PDX xenograft. Dosing ended on day 100 of the study in the OP-1250 10 mg/kg group, as indicated by the shaded area. E, Representative intracranial MRIs from ST941-implanted mice with data shown in Figure 6 (see main article) at indicated timepoints and treatment groups. PO=orally.
Supplementary Legends
Data Availability Statement
Sequencing data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO; ref. 39) and are accessible through GEO Series accession number GSE241944 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241944). All other data are available upon request from the corresponding author.