

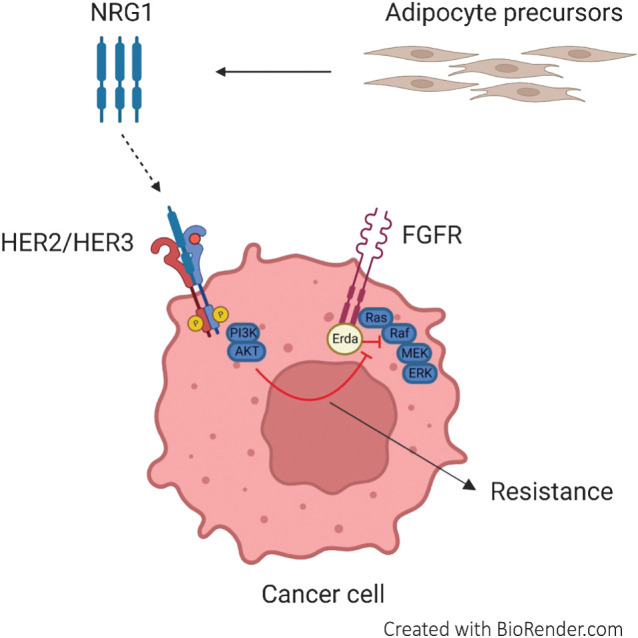
Acquired resistance to FGFR inhibition can be rapidly promoted by paracrine activation of the NRG1/HER3 axis mediated by adipocyte precursors and can be overcome by the combination of pertuzumab and erdafitinib treatment.
Abstract
Aberrations of the fibroblast growth factor receptor (FGFR) family members are frequently observed in metastatic urothelial cancer (mUC), and blocking the FGF/FGFR signaling axis is used as a targeted therapeutic strategy for treating patients. Erdafitinib is a pan-FGFR inhibitor, which has recently been approved by the FDA for mUC with FGFR2/3 alterations. Although mUC patients show initial response to erdafitinib, acquired resistance rapidly develops. Here, we found that adipocyte precursors promoted resistance to erdafitinib in FGFR-dependent bladder and lung cancer in a paracrine manner. Moreover, neuregulin 1 (NRG1) secreted from adipocyte precursors was a mediator of erdafitinib resistance by activating human epidermal growth factor receptor 3 (ERBB3; also known as HER3) signaling, and knockdown of NRG1 in adipocyte precursors abrogated the conferred paracrine resistance. NRG1 expression was significantly downregulated in terminally differentiated adipocytes compared with their progenitors. Pharmacologic inhibition of the NRG1/HER3 axis using pertuzumab reversed erdafitinib resistance in tumor cells in vitro and prolonged survival of mice bearing bladder cancer xenografts in vivo. Remarkably, data from single-cell RNA sequencing revealed that NRG1 was enriched in platelet-derived growth factor receptor-A (PDGFRA) expressing inflammatory cancer-associated fibroblasts, which is also expressed on adipocyte precursors. Together, this work reveals a paracrine mechanism of anti-FGFR resistance in bladder cancer, and potentially other cancers, that is amenable to inhibition using available targeted therapies.
Significance:
Acquired resistance to FGFR inhibition can be rapidly promoted by paracrine activation of the NRG1/HER3 axis mediated by adipocyte precursors and can be overcome by the combination of pertuzumab and erdafitinib treatment.
Graphical Abstract
Introduction
For decades, platinum-based combination chemotherapy has been the standard therapy for metastatic urothelial cancer (mUC). However, this treatment is generally not curative and has a limited impact on patient survival. Although the advent of immunotherapy and antibody–drug conjugates has broadened the therapeutic armamentarium for mUC, only a minority of patients respond (1, 2). The fibroblast growth factor receptor (FGFR) family are receptor tyrosine kinases (RTK), which regulate various cellular functions including cell proliferation, survival, differentiation, and migration. Activation of the members of the FGFR family (FGFR1–4) leads to activation of downstream signaling pathways such as Ras/Raf-MEK-MAPKs and PI3K/AKT (3). Genomic aberrations of FGFR family members are frequently observed in various cancers, most commonly in UC (32% FGFR aberrant; refs. 4–6). Therefore, blocking the FGF/FGFR signaling axis has been developed as a targeted therapeutic strategy in various tumor types including mUC. In 2019, the FDA granted accelerated approval to erdafitinib, a pan-FGFR inhibitor, for patients with FGFR2/3-driven urothelial cancer, based on the BLC2001 study (7, 8). Erdafitinib is a small molecule inhibitor that binds to FGFR and inhibits FGFR autophosphorylation and the resulting downstream signaling (9). Preliminary results from the phase III THOR clinical trial (NCT03390504) suggest significantly improved overall survival and progression-free survival of erdafitinib-treated patients compared with chemotherapy-treated patients (10). Although erdafitinib shows a considerable objective response rate of 40%, the response is not durable in most patients, indicating rapid development of resistance. To date, several efforts have been made to understand the resistance mechanisms to FGFR inhibition, which have primarily focused on tumor cell-intrinsic events (11–14). However, extrinsic factors driven by the tumor microenvironment (TME) play a key role in the development of RTK inhibitors resistance by activating alternative growth-promoting pathways. We hypothesize that the rapid development of erdafitinib resistance is triggered by a prompt protumoral response of the TME. One of the principal components of the TME is the mesenchymal stromal cells (MSC), which are multipotent cells that can differentiate into cancer-associated fibroblasts (CAF), osteocytes, chondrocytes, and adipocytes (15–17). The correlation between obesity and increased cancer progression has been established in multiple cancer types (18). There is increasing evidence suggesting the protumoral role of adipose-derived stem cells (ADSC) or adipocyte precursors in cancer (19). ADSCs are MSCs of adipose tissue and comprise inflammatory, myofibroblastic, and pro-adipogenic subpopulations (19). ADSCs can also promote tumor growth by remodeling the extracellular matrix, promoting angiogenesis, contributing to the recruitment of immunosuppressive cells, and inducing epithelial–mesenchymal transition through paracrine signaling (16, 20–25). Growth arrest and a variety of hormones, for example dexamethasone and insulin, are established means of inducing adipogenic differentiation of adipocyte precursors in vitro (26). Despite the abundant presence of tumor-infiltrating adipose tissue in bladder tumors (27, 28), the influence of ADSCs within the TME on the development of erdafitinib resistance has not been explicitly investigated in urothelial cancer. In this study, we sought to investigate whether adipocyte precursors can induce erdafitinib resistance in bladder cancer cell lines, and whether these cells can be specifically targeted by rational combination therapies.
Materials and Methods
Cell culture
All cells were cultured in a humidified 37°C incubator with 5% CO2. RT4, T24, TCCSUP, and 3T3-L1 cells were purchased from ATCC. MB49 cell line was purchased from Addexbio, RT112 cell line was purchased from DSMZ (German Collection of Microorganisms and Cell Cultures), and human adipose-derived stem cells (hADSC) were purchased from Lonza. LK2 lung cancer cells were a gift from Dr. Hanibal Bohnenberger (University Hospital Göttingen). RT4, RT112, T24, TCCSUP, and LK2 cell lines were cultured in RPMI1640 supplemented with 10% FCS, 1% penicillin/streptomycin, and 1% l-glutamine. MB49 cells and 3T3-L1 preadipocytes were cultured in DMEM supplemented with 10% FCS, 1% penicillin/streptomycin (complete DMEM). hADSCs were cultured in hADSCs medium (Lonza) supplemented with 10% FCS, 1% penicillin/streptomycin, and 1% l-glutamine. RT4, RT112, TCCSUP, and T24 are human bladder cancer cell lines. MB49 is a murine bladder cancer cell line. 3T3-L1 are preadipocytes derived from the mouse embryo, which can differentiate into adipocytes under certain conditions (described separately in Materials and Methods). Cells were split twice per week, and regularly checked for Mycoplasma contamination by MycoBlue Mycoplasma Detector (NeoBiotech). All cell culture reagents were obtained from Gibco (Thermo Fisher Scientific) unless otherwise specified.
Collection of conditioned media from 3T3-L1 and hADSCs
The 3T3-L1 cells and hADSCs were seeded in a T75 flask and incubated until 70% to 80% confluent (3–4 days). Conditioned media was collected from 3T3-L1 cells and hADSCs, filtered using 0.22 μm filters, aliquoted, and stored at −20°C.
Crystal violet proliferation assay
Depending on the cell line, 2,500 to 5,000 cells were seeded per well in a 96-well plate. After overnight incubation, cells were treated and incubated for 7 days. All conditions were run in triplicate wells. Treatments were refreshed on day 4. For staining, cells were fixed with 37% paraformaldehyde per well for 10 minutes, then washed with distilled water, and stained 0.05% crystal violet for 30 minutes. Cells were washed twice with distilled water and dried. 0.1% acetic acid was added per well to solubilize the dye. Finally, the absorbance was measured at a wavelength of 570 nm. The mean values of the triplicate wells were divided by a day 0 control. Relative optical density was normalized with respect to the vehicle control.
Concentration of conditioned media
Conditioned media was filtered through Pierce Protein Concentrators PES, 3K MWCO by centrifugation at 2,600 × g, at 4°C, for 1.5 hours. Flow through was collected and tested as the protein-depleted fraction.
Pertuzumab combination treatment with erdafitinib
Cells were pretreated with 10 μg/mL pertuzumab (Perjeta; Roche) in DMEM 1 hour before treatment with erdafitinib (Selleck Chemicals). Erdafitinib treatment was done in media control or conditioned media, with/without pertuzumab. Treatments were refreshed on day 4. Cells were stained with crystal violet on day 7 as described above.
Recombinant NRG1 treatment
A total of 50 ng/mL recombinant Human Heregulin-β1 (Biolegend) in complete DMEM was used to treat the cells. Treatments were refreshed on day 4 by a full media change. Cells were stained with crystal violet on day 7 as described earlier.
Isolation and enrichment of primary murine preadipocytes
Inguinal white adipose tissue was surgically removed from mice, then minced and digested with collagenase II in 0.5% BSA in PBS at 37°C with agitation. The digestion was quenched by adding AT buffer (0.5% BSA in PBS). Dissociated cells were filtered through a 100 μmol/L filter and centrifuged at 500 × g for 10 minutes. The supernatant containing mature adipocytes was aspirated, and the pellet, consisting of the stromal vascular fraction, was resuspended in red blood cell lysis buffer for 2 minutes at room temperature. The reaction was stopped by adding AT buffer and centrifugation at 500 × g for 10 minutes. Cells were washed in 2 mL magnetic-activated cell sorting (MACS) buffer (0.5% BSA and 2 mmol/L EDTA in PBS) and labeled with biotin-conjugated antibodies against lineage markers of endothelial cells (anti-CD31; clone MEC13.3; #102503), immune cells (anti-CD45; clone 30-F11; #103103), and erythrocytes (anti-TER119; clone TER-119; #116203). Cells were then incubated with Streptavidin MicroBeads (Miltenyi, #130–048–101) and subjected to MACS. The lineage depleted cells were harvested and maintained in DMEM/F12, supplemented with 1% GlutaMAX-I, 1% penicillin–streptomycin (all Gibco/Life Technologies), 10% FCS (Biochrom), 33 mmol/L biotin (Sigma), and 17 mmol/L D-pantothenate (Sigma) at 37°C with 5% CO2. All antibodies were purchased from BioLegend.
Isolation and enrichment of human CAFs
Urothelial carcinoma tumors from four patients were obtained from the University Hospital Bonn under the ethical approval number 363/20. Tumors were minced and digested as described above for the murine adipose tissue. Cells were washed in 2 mL MACS buffer (0.5% BSA and 2 mmol/L EDTA in PBS) and labeled with biotin-conjugated antibodies against lineage markers of endothelial cells (anti-CD31; clone AC128; #130–119–893), immune cells (anti-CD45; clone 5B1; #130–113–116), and epithelial cells [anti-EpCAM (CD326); clone REA764; #130–110–997]. Cells were then incubated with Streptavidin MicroBeads (Miltenyi, #130–048–101) and subjected to MACS. The lineage depleted cells were harvested and maintained in DMEM/F12, supplemented with 1% GlutaMAX-I, 1% penicillin–streptomycin (all Gibco/Life Technologies), 10% FCS (Biochrom), 33 mmol/L biotin (Sigma), and 17 mmol/L D-pantothenate (Sigma) at 37°C with 5% CO2. Conditioned media (CM) was collected when cells reached confluence of approximately 70%. All antibodies were purchased from BioLegend.
Adipogenic differentiation of primary murine preadipocytes
A total of 200,000 3T3-L1 cells were seeded in complete DMEM per well of a six-well plate and incubated for 2 days to allow them to grow to 100% confluence. Differentiation was induced by 5 μg/mL insulin (Sigma), 1 μmol/L dexamethasone (Sigma), 100 μmol/L 3-isobutyl-1-methylxanthine (IBMX; Sigma), and 1 μmol/L Rosiglitazone (Sigma) in complete DMEM. Following 2 days of induction, adipocytes were maintained in complete DMEM supplemented with 1 μg/mL insulin for five more days, with complete media change every other day.
Adipogenic differentiation of 3T3-L1 cells
A total of 200,000 cells were seeded per well of a six-well plate and incubated for 2 days to allow them to grow to 100% confluence. Differentiation was induced by 0.4 μg/mL insulin, 0.1 μmol/L dexamethasone, and 20 μmol/L 3-isobutyl-1-methylxanthine in complete DMEM. Following 2 days of induction, adipocytes were maintained in complete DMEM supplemented with 1 μg/mL insulin for 6 more days, with media change every other day. Conditioned media was harvested from adipocytes 7 days post-differentiation induction, filtered using 0.22 μm filters, aliquoted, and stored at −20°C.
Western blot analysis
A total of 800,000 cells were seeded per well of a 6-well plate. Following overnight incubation, cells were treated with the indicated treatments and incubated overnight. Cells were washed and lysed using RIPA buffer (Cell Signaling Technology; #9806) freshly supplemented with phenylmethylsulfonyl fluoride (PMSF; Carl Roth; #329–98–6). Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific; #23225) according to the manufacturer's instructions. Proteins were separated using SDS-PAGE, transferred to nitrocellulose membranes, then blotted with polyclonal anti-ERK1/2 (#9102), anti-pERK1/2 (clone 197G2; #4377), anti-AKT (clone 40D4; #2920), anti-pAKT (clone D9E; #4060), anti-FGFR3 (Santa Cruz Biotechnology; clone B-9; #sc-13121), anti-HER3 (clone D22C5; #12708), anti-pHER3 (clone 21D3; #4791), anti-β-actin (Sigma-Aldrich; clone AC-74; #A2228), polyclonal anti-Heregulin (#2573), polyclonal anti-EGFR (Santa Cruz Biotechnology; #sc-03), anti-pEGFR (clone 1H12, #2236), anti- HER2 (D8F12, #4290), anti-pHER2 (clone 21D3, #2247). All Western blot antibodies were purchased from Cell Signaling Technology unless otherwise specified.
RNA extraction and qRT-PCR
Total RNA extraction was done using TRizol RNA isolation reagent (Invitrogen). RNA concentration was measured using the Thermo Scientific NanoDrop 2000/2000c. Reverse transcription was performed using Prime Script RT Reagent Kit (Takara; #RR064A). RT-PCR mix was prepared using TB Green Premix Ex Taq I (Takara; #RR82WR), and quantified with the QuantStudio 5 RT-PCR System (Applied Biosystems). Primers used: human NRG1 forward AGAGCCTGTTAAGAAACTCGC, human NRG1 reverse GTCCACTTCCAATCTGTTAGCA, human FGFR1 forward AAACCGTATGCCCGTAGCTC, human FGFR1 reverse AGGTGGCATAACGGACCTTG, human FGFR2 forward CCTGCGGAGACAGGTAACAG, human FGFR2 reverse TGCCCAGTGTCAGCTTATCT, human FGFR3 forward CCCAAATGGGAGCTGTCTCG, human FGFR3 reverse CCCGGTCCTTGTCAATGCC, human GAPDH forward CTCTGCTCCTCCTGTTCGAC, human GAPDH reverse ACGACCAAATCCGTTGACTC, murine Nrg1 forward TTCCCATTCTGGCTTGTCTAGT, murine Nrg1 reverse CCAGGGTCAAGGTGGGTAG, murine Fgfr1 forward ACTCTGCGCTGGTTGAAAAAT, murine Ffgfr1 reverse GGTGGCATAGCGAACCTTGTA, murine Fgfr2 forward GCTATAAGGTACGAAACCAGCAC, murine Fgfr2 reverse GGTTGATGGACCCGTATTCATTC, murine Fgfr3 forward GCCTGCGTGCTAGTGTTCT, murine Fgfr3 reverse CCTGTACCATCCTTAGCCCAG, murine Cd36 forward GCAGGTCTATCTACGCTGTGTT, murine Cd36 reverse GCAAAGGCATTGGCTGGAAG murine Lpl forward CATCAACTGGATGGAGGAGGAG, murine Lpl reverse GTCAGACTTCCTGCTACGCC, murine Glut4 forward CATGTCTCGAAGTAGTGTGCAG, murine Glut4 reverse TGACAGTGACAGCCACAATGATG, murine Lep forward CCAGAAAGTCCAGGATGACACC, murine Lep reverse GGCGGATACCGACTGCGT, murine Gapdh forward AGGTCGGTGTGAACGGATTTG, murine Gapdh reverse TGTAGACCATGTAGTTGAGGT. All procedures were performed according to the manufacturer's protocol.
Lentiviral transduction
RT4 cells stably overexpressing NRG1 were generated by lentiviral transduction. Lentivirus was generated in HEK 293T cells through cotransfection of VSV-G, Gag Pol, and pLV[Exp]-Bsd-hPGK>hNRG1 [Vector Builder (NM_001322205.1)] using jetPRIME (Polyplus). RT4 NRG1 cells were selected with 5 μg/mL Blasticidin (Gibco) for 1 week.
3T3-L1 cells and hADSCs stably overexpressing shRNA targeting mNrg1/hNRG1, respectively, were generated by lentiviral transduction. Lentivirus was generated in HEK 293T cells through cotransfection of VSV-G, Gag Pol, and pLV[shRNA]-Puro-U6>mNrg1[shRNA#1] [Vector Builder (VB230706–1085msf)]/ pLV[shRNA]-Puro-U6>hNRG1[shRNA#2] [Vector Builder (VB230706–1109zqs)] using jetPRIME (Polyplus). 3T3-L1 mNrg1 shRNA cells and hADSCs hNRG1 shRNA cells were selected with 1.6 μg/mL Puromycin (Gibco) for 3 days. Control 3T3-L1 and hADSCs cell lines were transduced with pLV[shRNA]-Puro-U6>Scramble_shRNA#1 [Vector Builder (VB010000–0005 mme)] and selected as described above.
Phospho-RTK array
A total of 800,000 cells were seeded per well of a 6-well plate. Following overnight incubation, cells were treated with the indicated treatments and incubated overnight. Proteome Profiler Human Phospho-RTK Array Kit (R&D Systems; #ARY001B) was used to perform the array. Mean pixel density was quantified using ImageJ, and the average of duplicate dots per receptor were plotted.
In vivo experiments
All procedures involving animals were conducted in compliance with the 3R principle and the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines, after approval by the Italian Ministry of Health (auth. no. 608/2022-PR). In this study, a xenograft model was set up by subcutaneously injecting RT4-WT or RT4-NRG1 cells (2 × 106 cell/200 μL/mouse) in the flank of CB17-SCID mice (8 ± 3 week of age; Charles River Laboratories). Tumor mass was measured twice a week, and tumor volume was calculated according to the following formula:
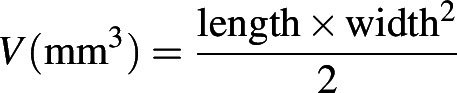 |
Treatment was initiated once the tumor mass was palpable. Mice inoculated with RT4 WT cells were randomized into two experimental groups: control and erdafitinib (5 mg/kg bw by daily gavage). Mice inoculated with RT4-NRG1 cells were randomized into four experimental groups, that is, control, erdafitinib (5 mg/kg bw by daily gavage), pertuzumab (5 mg/kg bw, EV three times a week), combination (erdafitinib 5 mg/kg bw daily gavage + pertuzumab 5 mg/kg bw three times a week). Mice were sacrificed once tumor measured with caliper reached the volume of about 1,500 mm2, when a sudden increase of tumor growth was observed, or if the two previous conditions were not reached, at day 37 (final experimental endpoint). Signs of distress, for example, unusual behaviors, excessive weight loss, and hunched posture, were also monitored during treatment. At sacrifice, the tumor was isolated, washed in saline solution, and fixed in a 4% formalin solution. After 24 hours, the tissues were embedded in paraffin blocks for further IHC analysis.
Ki67 IHC
Two to three micrometers of sections were prepared from formalin-fixed, paraffin-embedded tissues. Antigen retrieval was done by microwaving for 10 minutes at 600 W in boiling 10 mmol/L citrate buffer, pH 6.0. 1:200 dilution of mouse anti-Ki67 antibody (Zytomed; MSK018–05) was used for staining. Staining pattern was nuclear. Quantification was done using QuPath Version 0.3.2.
Single-cell RNA sequencing analysis
Single-cell RNA sequencing (scRNA-seq) data of eight bladder cancer samples and three para tumor samples was downloaded from Chen and colleagues (29), using their interactive Shiny R interface. Analysis and dataset processing was performed using Seurat version 4.1.1 running on a mac OS version 12.4 (Monterey). Analysis was performed using standard Seurat dataset processing pipeline. Data were visualized using the Nebulosa (version 1.4.0) and scCustomize (version 0.7.0) packages. The color-blind friendly, perceptually uniform and ordered “batlow” color pallet was used via the R package scico (version 1.3.0). The publication ready figure was arranged and formatted using Adobe Illustrator version 27. Data availability: Publicly available scRNA-seq data were obtained from Chen and colleagues (29). Code availability: Code to reproduce scRNA-seq data can be found at https://github.com/BaldLab.
The Cancer Genome Atlas data analysis
Log2-transformed RSD (RNA-Seq by Expectation Maximization) RNA-sequencing data (RNA-Seq v2) of PDGFRA, MMP3, DPP4, and NRG1 generated by The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/) were downloaded from the UCSC Xena browser (http://xena.ucsc.edu) for n = 408 urothelial bladder carcinoma (BLCA). Correlation analysis were based on the median expression of NRG1 among the samples.
NRG1 IHC
Two to three micrometers of sections were prepared from formalin-fixed, paraffin-embedded tissues. The sections were mounted on adhesion microscope slides (TOMO). Dewaxing (EZ Prep #950–102), heat pretreatment (Ultra CC1 buffer at pH 8; #950–224), and further steps were performed in the Ventana BenchMark Ultra (Roche) according to the manufacturer's instructions. All reagents were purchased from Ventana Medical Systems unless otherwise specified. Staining was done using 1:100 dilution of the monoclonal anti-NRG1 antibody (Thermo Fisher Scientific; clone 7D5; #MA5–12986). OptiView DAB Detection Kit (#760–700) was used for detection. A cutoff H-score of 150 was applied for NRG1 cytoplasmic intensity on tumor cells, whereas cytoplasmic staining intensity was classified as low and high on stromal NRG1 intensity.
Statistical analysis
GraphPad Prism 8 was used for statistical analysis. For comparisons between two independent variables, unpaired t test was performed. For comparisons among more than two independent variable, two-way ANOVA was performed. At least three biological replicates of all in vitro experiments were performed. Log-rank (Mantel-0-Cox) test was used for statistical analysis of survival data. In all figures, mean ± SD was reported. 4 to 9 mice were treated for in vivo experiments. Not significant (n.s.), P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****).
Data availability statement
The scRNA-seq data analyzed in this paper are available from the Sequence Read Archive (SRA) at PRJNA662018 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA662018). The RNA-seq data generated by the TCGA for urothelial BLCA that was analyzed in this study are available in the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/). All other raw data generated in this study are available upon request from the corresponding author.
Results
Adipocyte precursors promote resistance to erdafitinib in bladder cancer cell lines
To test the susceptibility to growth inhibition by erdafitinib treatment, several bladder cancer cell lines (human RT4, RT112, TCCSUP, T24, and the murine MB49) were screened for their FGFR2 and FGFR3 mRNA expression. qRT-PCR analysis showed that RT4 and RT112 cell lines express high levels of FGFR3 and moderate levels of FGFR2, whereas all other tested cell lines express scant levels of FGFR2/3 (Supplementary Fig. S1A). Because RT4 and RT112 cell lines are known to be FGFR3-dependent (30–32), FGFR3 expression was also analyzed at the protein level. Western blot analysis confirmed the expression of FGFR3 in RT4 and RT112 cell lines (Fig. 1A), which was detected as multiple bands indicative of FGFR3–Transforming Acidic Coiled-Coil Containing Protein 3 (TACC3) fusions at different breakpoints (33, 34). FGFR3–TACC3 fusions lead to constitutive phosphorylation of the FGFR3 tyrosine kinase residues, promoting aberrant FGFR3 activation and consequent growth induction (35). FGFR3–TACC3 fusions have been clinically associated with a greater sensitivity to FGFR3 inhibitors (36, 37). Proliferation analysis demonstrated that MB49, TCCSUP, and T24 are not susceptible to erdafitinib treatment, whereas RT4 and RT112 respond with an IC50 of 10 nmol/L (Fig. 1B), as reported previously (14). To assess an on-target effect at the molecular level, RT4 and RT112 cells were treated with vehicle, 10 or 100 nmol/L erdafitinib. As expected, Western blot analysis showed a dose-dependent inactivation of ERK1/2 in RT4 and RT112 cells (Fig. 1C), whereas AKT was only inactivated in RT112 (Fig. 1C; ref. 9). On the basis of the recent reports describing the protumoral role of adipocyte precursors in several cancers (16, 20–23), we aimed to investigate the potential paracrine effect of adipocyte precursors on erdafitinib susceptibility (Fig 1D). CM of adipocyte precursor cell lines, 3T3-L1 cells and hADSCs, conferred significant resistance against erdafitinib in RT4 and RT112 cells, shown by unrepressed proliferation (Fig. 1E). To note, CM of 3T3-L1 cells collected without serum conferred equivalent resistance to that collected with serum, which indicates that the resistance factor is not stimulated by serum (Supplementary Fig. S1B). Importantly, 3T3-L1 cells were refractory to erdafitinib treatment, even at a concentration of 1,000 nmol/L (Supplementary Fig. S1C). At the molecular level, AKT pathway was activated in RT4 and RT112 cells grown in CM of 3T3-L1 cells compared with media control under erdafitinib treatment (Fig. 1F). Of note, deactivation of ERK1/2 in response to erdafitinib treatment was still observed when RT112 cells were treated in 3T3-L1 CM (Fig. 1F), suggesting that AKT activation may serve as a compensatory growth-promoting pathway in the case of ERK1/2 inhibition. We postulated that the resistance-promoting effect of adipocyte precursors’ CM on cancer cells during anti-FGFR treatment could occur through three possible mechanisms: metabolites, vesicles that carry genetic material, or proteins such as growth factors secreted by adipocyte precursor cells. To test this hypothesis, conditioned media was heat-inactivated at 95°C for 10 minutes. Indeed, heat inactivation abolished the ability of adipocyte precursors’ CM to rescue proliferation during erdafitinib treatment, suggesting that the soluble factors responsible for conferring resistance are proteins (Fig. 1G). To confirm this, CM from 3T3-L1 cells was filtered through a protein-binding column and the flowthrough (protein-depleted fraction) was collected. The flowthrough did not confer any resistance to erdafitinib, confirming that the factor responsible for the observed resistance is a protein (Supplementary Fig. S1D). Taken together, these data show that adipocyte precursors can confer anti-FGFR3 resistance in bladder cancer via soluble proteins in a paracrine manner.
Figure 1.

Adipocyte precursors promote resistance against erdafitinib in bladder cancer cell lines. A, Western blot analysis of FGFR3 in four bladder cancer cell lines: RT4, RT112, TSCCUP, and T24. β-Actin served as a loading control. B, Proliferation analysis of five bladder cancer cell lines RT4, RT112, TSCCUP, T24, and MB49 treated with vehicle (Veh) or erdafitinib (1, 10, 100, 1,000 nmol/L). Crystal violet staining was done on day 7. Three biological replicates were performed. Data are represented as mean ± SD. C, Western blot analysis of pAKT and pERK1/2 in RT4 and RT112 cells treated with DMSO (Veh) or erdafitinib (10 and 100 nmol/L) for 16 hours. β-Actin served as a loading control. D, Schematic diagram of the proliferation assay performed to investigate the effect of CM of different stromal cells on erdafitinib response. E, Proliferation analysis of RT4 and RT112 cells treated with 10 nmol/L erdafitinib in media control (ctrl) or CM of 3T3-L1 cells or hADSC. Crystal violet staining was performed on day 7. Data are normalized to cells treated with vehicle. Four biological replicates were performed. Data are represented as mean ± SD. F, Western blot analysis of pAKT and pERK1/2 in RT4 and RT112 cells treated with 10 nmol/L erdafitinib in media control or CM of 3T3-L1 cells. β-Actin served as a loading control. G, Proliferation analysis of RT4 cells and RT112 cells treated with 10 nmol/L erdafitinib in CM of 3T3-L1 cells or heat-inactivated (HI) CM of 3T3-L1. Data were normalized to cells treated with vehicle. Crystal violet staining was performed on day 7. Three biological replicates were performed. Data are represented as mean ± SD. Two-way ANOVA was used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
NRG1 is secreted from adipocyte precursors and induces erdafitinib resistance in bladder cancer cells by activating HER3 signaling
To screen for the mediator responsible for the observed erdafitinib resistance, we performed a phosphorylated-receptor tyrosine kinase (phospho-RTK) array analysis on RT4 cells cultured in conditioned media of 3T3-L1 cells or media control. The phospho-RTK array revealed phosphorylated human epidermal growth factor receptor 3 (pHER3) as the only activated RTK in the conditioned media treatment compared to the media control (Fig. 2A). In parallel, Western blot analysis confirmed the activation of pHER3 in RT4 and RT112 cells when cultured with 3T3-L1 cells and hADSCs CM (Fig. 2B; Supplementary Fig. S2A). Because the phospho-RTK array was conducted on human RT4 cells stimulated with murine 3T3-L1 CM (Fig. 2A), we sought to investigate potential species-related differences in modulating RTKs. To address this, an additional phospho-RTK was performed on RT4 cells treated with CM of hADSCs or media control. In line with the previous results (Fig. 2A), this phospho-RTK revealed that pHER3 is the only upregulated kinase in the CM treatment compared to media control (Supplementary Fig. S2B), suggesting no bias related to species-mismatch. Because neuregulin 1 (NRG1) is the best characterized and principal ligand of HER3 (38), we investigated whether recombinant NRG1 (rNRG1) can recapitulate the resistance conferred by the CM of adipocyte precursors. Notably, rNRG1 induced significant resistance against erdafitinib in RT4 cells (Fig. 2C). To confirm this phenotype in an additional model, we stably overexpressed NRG1 in RT4 cells (RT4 NRG1), where HER3 activation was shown (Fig. 2D; Supplementary Fig. S2C). In line with our previous results, RT4 NRG1 cells are resistant against erdafitinib compared with the wild-type cells (Fig. 2D). Further, qRT-PCR analysis using NRG1/Nrg1-specific primers showed that 3T3-L1 and hADSCs express high levels of Nrg1, whereas RT4 and RT112 cells barely express NRG1 (Fig. 2E). To gain further insights on the dynamics of NRG1 expression upon FGFR3 inhibition, we analyzed the protein levels of NRG1 in bladder cancer cell lines and adipocyte precursors treated with erdafitinib or vehicle control. Consistent with the NRG1/Nrg1 mRNA expression (Fig. 2E), NRG1 protein levels were highest in both adipocyte precursor cell lines compared with the bladder cancer cell lines (Fig. 2F). Although the NRG1/NRG1 mRNA and protein levels remained unchanged upon erdafitinib treatment in adipocyte precursors, they slightly increased upon erdafitinib treatment in RT4 cells (Fig. 2F; Supplementary Fig. S2D) as reported previously (11, 12, 14). Nevertheless, the mRNA and protein levels of NRG1/NRG1 in erdafitinib-treated RT4 cells remain negligible compared with those in adipocyte precursors, supporting a paracrine mechanism of resistance to anti-FGFR inhibition, mediated by NRG1. Further Western blot analysis of pHER3 demonstrated clear upregulation of pHER3 in RT4 and RT112 cells cultured in 3T3-L1 CM, with and without erdafitinib treatment, compared with media control (Fig. 2G). Although pHER3 was upregulated upon erdafitinib treatment in media control in RT4 cells, pHER3 expression upon erdafitinib treatment in CM was higher (Fig. 2G). To investigate whether NRG1 is the principal mediator of resistance in the CM of adipocyte precursors, Nrg1/NRG1 shRNA was stably expressed by means of lentiviral transduction in 3T3-L1 cells and hADSCs, respectively. Western blot analysis of NRG1 revealed a robust knockdown of NRG1 in 3T3-L1 Nrg1 shRNA cells (Fig. 2H), and a complete knockdown of NRG1 in hADSCs NRG1 shRNA (Supplementary Fig. S2E). Unlike CM of 3T3-L1 cells and hADSCs expressing control shRNA, CM of Nrg1/NRG1 shRNA cells failed to confer comparable resistance to erdafitinib in RT4 and RT112 cells (Fig. 2H; Supplementary Fig. S2E). Of note, CM of 3T3-L1 Nrg1 shRNA cells conferred slight resistance in RT4 cells, which could be attributed due to the incomplete knockdown of Nrg1 in 3T3-L1 cells (Fig. 2H), and to the intrinsic upregulation of NRG1 in RT4 cells upon erdafitinib treatment (Fig. 2F and G). To verify the effect of Nrg1/NRG1 knockdown on downstream signaling, further Western blot analysis of RT4 and RT112 cells treated with CM of 3T3-L1 cells and hADSCs expressing control or Nrg1/NRG1 shRNA was performed. This Western blot revealed a downregulation of pHER3 and pAKT upon treatment with CM of Nrg1/NRG1 shRNA cells compared with CM of control shRNA cells (Fig. 2I; Supplementary Fig. S2F). Moreover, to confirm the observed HER3 and AKT regulation by CM of 3T3-L1 cells in species-matched cells, murine MB49 cells were treated with media control, or CM of 3T3-L1 cells expressing control or Nrg1 shRNA. Western blot analysis of these cells showed upregulation of pHER3 and pAKT upon treatment with CM of 3T3-L1 control shRNA cells compared with media control, and downregulation of pHER3 and pAKT upon treatment with CM of 3T3-L1 Nrg1 shRNA cells compared with CM of 3T3-L1 control shRNA cells (Supplementary Fig. S2G). This corroborates the previously observed regulation of pHER3 and pAKT upon CM treatment in human RT4 and RT112 cells, and implies the lack of disparity due to species-mismatch. Because HER3 lacks kinase activity, it is known to heterodimerize with HER2 or EGFR upon ligand binding (39). Therefore, we analyzed pHER2 and pEGFR upon conditioning RT4 and RT112 cells with 3T3-L1 CM by Western blot analysis (Supplementary Fig. S2H). This analysis showed downregulation of pHER2 in CM treated cells compared with media control treated cells, and a lack of pEGFR expression in both conditions, suggesting the dimerization of HER3 with HER2 upon HER3 activation by NRG1. To sum, these results suggest that NRG1 is secreted into the CM of 3T3-L1 cells and hADSCs, and mediates resistance against erdafitinib by activating the HER3/HER2 pathway.
Figure 2.

NRG1 is secreted from preadipocytes and induces erdafitinib resistance in RT4 and RT112 bladder cancer cells by activating HER3. A, Left, RTK array on RT4 cells cultured in media control (ctrl) or CM of 3T3-L1 cells for 16 hours. Right, quantification of mean pixel density of the RTK array shown on the left. Data are represented as mean of the duplicate dots per kinase. B, Western blot analysis of pHER3 in RT4 and RT112 cells cultured in media control or CM of 3T3-L1 cells for 16 hours. β-Actin served as a loading control. C, Proliferation analysis of RT4 cells treated with 10 nmol/L erdafitinib in DMEM, with or without 50 ng/mL recombinant NRG1. Crystal violet staining was performed on day 7. Data were normalized to cells treated with vehicle. Data are represented as mean ± SD. Three biological replicates were performed. Unpaired t test was used for statistical analysis. D, Left, Western blot analysis of baseline NRG1 and pHER3 in RT4 WT cells and NRG1-overexpressing RT4 (RT4 NRG1) cells. β-Actin served as a loading control. Right, proliferation analysis of RT4 WT cells and RT4 NRG1 cells treated with 10 nmol/L erdafitinib in DMEM. Crystal violet staining was performed on day 7. Data are normalized to cells treated with vehicle. Data are represented as mean ± SD. Three biological replicates were performed. Unpaired t test was used for statistical analysis. E, qRT-PCR analysis of baseline NRG1 expression in RT4, RT112, 3T3-L1, and hADSC cells. Two to three biological replicates were performed. Expression levels are normalized to GAPDH. Data are represented as mean ± SD. F, Western blot analysis of NRG1 in RT4, RT112, 3T3-L1, and hADSC cells treated for 16 hours in 10 nmol/L erdafitinib or vehicle control. β-Actin served as a loading control. G, Western blot analysis of pHER3 in RT4 and RT112 cells treated for 16 hours with 10 nmol/L erdafitinib or vehicle control in media control or 3T3-L1 CM. β-Actin served as a loading control. H, Left, Western blot analysis of NRG1 in 3T3-L1 cells transduced with lentivirus encoding control shRNA or Nrg1 shRNA. β-Actin served as a loading control. Right, proliferation analysis of RT4 and RT112 cells treated with 10 nmol/L erdafitinib in media control or CM of 3T3-L1 ctrl/Nrg1 shRNA cells. Crystal violet staining was performed on day 7. Data were normalized to cells treated with vehicle. Three biological replicates were performed. Data are represented as mean ± SD. Two-way ANOVA was used for statistical analysis. I, Western blot analysis of pHER3 and pAKT in RT4 and RT112 cells treated with CM of 3T3-L1 ctrl/Nrg1 shRNA cells for 16 hours. β-Actin served as a loading control. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
NRG1-mediated resistance against erdafitinib is restricted to preadipocytes
The 3T3-L1 cell line is widely used in adiposity research and is an adipocyte precursor cell line that can, under specific conditions, differentiate into adipocytes (26). To test whether NRG1 secretion is restricted to preadipocytes, we applied a differentiation protocol to generate adipocytes from 3T3-L1 cells (26). The production of lipid droplets (Fig. 3A), and the upregulation of the main adipocytic markers such as cluster of differentiation 36 (Cd36), Leptin (Lep), solute carrier family 2 (facilitated glucose transporter), member 4 (Glut4), and lipoprotein lipase (Lpl; refs. 40–43) confirmed the differentiation process (Supplementary Fig. S3). Remarkably, CM from differentiated adipocytes (7 days post-differentiation) failed to rescue the growth inhibition in RT4 cells treated with erdafitinib, compared with the media control (Fig. 3B), implying that the resistance phenotype is restricted to adipocyte precursor-derived NRG1. Importantly, Nrg1 mRNA levels were greatly reduced in terminally differentiated adipocytes when compared with 3T3-L1 progenitor cells (Fig. 3C). In line with the mRNA levels of Nrg1, Western blot analysis of NRG1 revealed gradual downregulation of protein expression during adipogenic differentiation (Fig. 3D). Primary preadipocytes from murine white adipose tissue were isolated to validate the dynamic expression of Nrg1 (Fig. 3E). Indeed, Nrg1 mRNA expression was significantly downregulated after adipogenic differentiation of primary preadipocytes (Fig. 3F), confirming the dynamic expression observed upon differentiation of 3T3-L1 cells (Fig. 3C and D). Moreover, 3T3-L1-derived adipocytes’ CM failed to activate HER3 and AKT in RT4 and RT112 cells (Fig. 3G). Taken together, these results highlight the restriction of NRG1 expression in preadipocytes, and the impact of this dynamic expression on resistance.
Figure 3.

NRG1-mediated resistance against erdafitinib is restricted to preadipocytes. A, Representative images showing the adipogenic differentiation of 3T3-L1 cells (preadipocytes) to differentiated adipocytes. The differentiation process was carried out for 7 days. Arrows, formation of lipid droplets in adipocytes. B, Proliferation analysis of RT4 cells treated with 10 nmol/L erdafitinib in media control (ctrl) or CM of adipocytes collected after 7 days of differentiation. Crystal violet staining was performed on day 7. Data were normalized to cells treated with vehicle. Three biological replicates were performed. Data are represented as mean ± SD. C, qRT-PCR analysis of baseline NRG1 expression in 3T3-L1 cells and 3T3-L1-derived adipocytes. Three biological replicates were performed. Expression levels are normalized to GAPDH. Data are represented as mean ± SD. D, Western blot analysis of NRG1 in 3T3-L1 under adipogenic differentiation. β-Actin served as a loading control. E, Schematic illustration of the process of isolating primary preadipocytes from mice. F, qRT-PCR analysis of baseline NRG1 expression in primary preadipocytes and primary preadipocyte-derived adipocytes. Three biological replicates were performed. Expression levels are normalized to GAPDH. Data are represented as mean ± SD. G, Western blot analysis of pHER3 and pAKT in RT4 and RT112 cells treated with CM of 3T3-L1 cells or 3T3-L1-derived adipocytes, or the respective media control. β-Actin served as a loading control. Unpaired t test was used for statistical analysis. ***, P < 0.001; ****, P < 0.0001. (E, Created with BioRender.com.)
Pertuzumab reverses NRG1-mediated resistance against erdafitinib
Having established the paracrine NRG1/HER3 signaling as a driver of rapid resistance to FGFR inhibition, we next explored the inhibitory effect of this signaling pathway in vitro and in vivo. We first treated RT4 and RT112 cell lines cultured in adipocyte precursors CM with erdafitinib alone or in combination with a HER2/HER3 dimerization-inhibitory antibody, pertuzumab (44, 45). A negligible growth inhibition on proliferation was observed in RT4 and RT112 cells cultured in adipocytes precursors’ CM and treated with pertuzumab as single agent (Fig. 4A). However, cotreatment with erdafitinib and pertuzumab significantly reversed the resistance mediated by adipocyte precursors’ CM (Fig. 4A). To investigate whether pertuzumab leads to reversal of resistance or resensitization, RT4 and RT112 cells were temporally treated with erdafitinib in media ctrl or 3T3-L1 CM for 3 days, and pertuzumab was added (or not added) on the third day of treatment. This proliferation analysis revealed a marked growth arrest in the cells that were treated with the combination treatment of erdafitinib and pertuzumab in 3T3-L1 CM, compared with those that were only treated with erdafitinib in 3T3-L1 CM. These results imply that pertuzumab leads to the reversal of resistance to erdafitinib conferred by 3T3-L1 CM, rather than resensitization (Supplementary Fig. S4A). Accordingly, combination treatment of erdafitinib and pertuzumab abolished activation of AKT and HER3 in cells cultured in CM of 3T3-L1 (Fig. 4B). We then evaluated the antitumoral effect of the combined treatment using a xenograft mouse model of RT4 and RT4 NRG1 cells (Fig. 4C). In line with the in vitro results, erdafitinib treatment demonstrated potent and sustained antitumor activity, indicated by prolonged survival of RT4 tumor-harboring mice and diminished tumoral Ki-67 expression (Fig. 4D and E). Thereafter, we carried out an in vivo approach to assess the combinational effect of both erdafitinib and pertuzumab on RT4 NRG1 xenografts. Interestingly, erdafitinib induced only insignificant prolongation of survival and Ki-67 reduction in RT4 NRG1 xenografts, confirming that NRG1/HER3 signaling confers resistance to anti-FGFR3 treatment in vivo (Fig. 4F and G; Supplementary Fig. S4B). Similar to erdafitinib treatment as a single agent, pertuzumab monotreatment showed minor antitumoral effect on RT4 NRG1 xenografts (Fig. 4F and G; Supplementary Fig. S4B). Importantly, RT4 NRG1 xenografts treated with combination of erdafitinib and pertuzumab showed increased overall survival of mice as indicated by Kaplan–Meier cumulative survival curve (Fig. 4F), and reduced Ki-67 staining, and tumor volume (Fig. 4G; Supplementary Fig. S4B). These data demonstrate that NRG1 mediates resistance to erdafitinib in vitro and in vivo, and can potentially be exploited for therapeutic targeting using clinically approved inhibitors of the NRG1/HER3 signaling axis, such as pertuzumab.
Figure 4.

Pertuzumab reverses NRG1-mediated resistance against erdafitinib. A, Proliferation analysis of RT4 and RT112 treated with 10 μg/mL pertuzumab (Pertuz) and vehicle control (Veh), 10 nmol/L erdafitinib, or erdafitinib and pertuzumab. Cells were treated in media control (ctrl) or CM of 3T3-L1 cells or hADSCs. Crystal violet staining was done on day 7. Data are normalized to cells treated with vehicle. Three biological replicates were performed. Data are represented as mean ± SD. Two-way ANOVA was used for statistical analysis. B, Western blot analysis of pHER3, pAKT, pERK1/2 in RT4 and RT112 treated with 10 μg/mL pertuzumab and vehicle control (Veh), 10 nmol/L erdafitinib, or erdafitinib and pertuzumab in 3T3-L1 CM. β-Actin served as a loading control. C, Schematic diagram of in vivo model performed by injecting RT4 or RT4 NRG1 cells in CB17-SCID mice. D, Kaplan–Meier survival curve of RT4-tumor bearing CB17-SCID mice treated with vehicle or 5 mg/kg erdafitinib. Four mice were tested per group in D and E. Log-rank (Mantel–Cox) test was used for statistical analysis. E, Left, IHC staining of Ki67 on RT4 xenografts treated as in D. Right, quantification of IHC staining of Ki67. Unpaired t test was used for statistical analysis. F, Kaplan–Meier survival curve of RT4 NRG1 tumor-bearing CB17-SCID mice treated with vehicle, 5 mg/kg erdafitinib, 5 mg/kg pertuzumab, or combinational treatment. The number of mice tested per group was: 9 mice in the erdafitinib and the combination treatment groups, 10 mice in the vehicle and pertuzumab treatment groups. Log-rank (Mantel–Cox) test was used for statistical analysis. G, Left, IHC staining of Ki67 on RT4 NRG1 xenografts treated as in F. Right, quantification of IHC staining of Ki67. The number of xenografts (each from a different mouse) analyzed per group is 8 in the vehicle group, 7 in the erdafitinib group, 6 in the pertuzumab group, and 5 in the combination treatment group. One-way ANOVA was used for statistical analysis. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant. (C, Created with BioRender.com.)
NRG1 mediates resistance against erdafitinib in FGFR1-dependent lung carcinoma
Because FGFRs are altered in other tumor types like lung cancer, we investigated whether the observed NRG1-driven resistance mechanism can be of clinical relevance in other tumor entities. Here we utilized the non–small cell lung carcinoma (NSCLC) cell line, LK2, that is FGFR1-driven (46). First, we confirmed the overexpression of FGFR1 mRNA in this cell line (Supplementary Fig. S5). Proliferation analysis revealed significant growth inhibition by erdafitinib (Fig. 5A), and Western blot analysis showed inactivation of ERK1/2 in LK2 cells (Fig. 5B). Similar to bladder cancer cell lines, CM of 3T3-L1 cells and hADSCs, conferred significant resistance against erdafitinib-mediated growth inhibition in LK2 cell line (Fig. 5C). At the molecular level, the HER3/AKT axis was activated in LK2 cells grown in CM of 3T3-L1 cells compared with media control with or without erdafitinib treatment (Fig 5D and E). Of note, similar to RT112 cells, deactivation of ERK1/2 in response to erdafitinib treatment was still observed in LK2 cells treated in CM of 3T3-L1 cells (Fig. 5D). Similarly, HER3 and AKT were also upregulated upon treatment of LK2 cells in hADSCs CM compared with media control (Fig. 5F). We next tested the inhibitory effect of erdafitinib in combination with pertuzumab in LK2 cell line. Although a minor proliferation inhibition effect was observed in LK2 cells cultured in adipocytes precursors’ CM and treated with erdafitinib or pertuzumab as single agents, combinational treatment of both drugs significantly reversed the resistance phenotype mediated by adipocyte precursors’ CM (Fig. 5G). In line with our results in bladder cancer cell lines, cotreatment of erdafitinib and pertuzumab abolished the activation of HER3 and AKT in LK2 cells cultured in 3T3-L1 CM (Fig 5H). Together, our results demonstrate that the activation of NRG1/HER3 axis induces erdafitinib resistance in FGFR1-dependent NSCLC, which may imply potential clinical relevance in other FGFR-driven tumor entities.
Figure 5.

NRG1 mediates resistance against erdafitinib in lung carcinoma cell line LK2. A, Proliferation analysis of LK2 cells treated with vehicle (Veh) or erdafitinib (1, 10, 30, 100, and 1,000 nmol/L). Crystal violet staining was done on day 7. Three biological replicates were performed. Data are represented as mean ± SD. One-way ANOVA was used for statistical analysis. B, Western blot analysis of pAKT and pERK12/ in LK2 cells treated with vehicle control, 10 or 100 nmol/L Erda for 16 hours. β-Actin served as a loading control. C, Proliferation analysis of LK2 cells treated with 30 nmol/L erdafitinib in media control (ctrl) or CM of 3T3-L1 cells or hADSCs. Crystal violet staining was done on day 7. Data are normalized to cells treated with vehicle. Three biological replicates were performed. Data are represented as mean ± SD. One-way ANOVA was used for statistical analysis. D, Western blot analysis of pAKT and pERK1/2 in LK2 cells treated with vehicle control, or 100 nmol/L Erda in media control or CM of 3T3-L1 cells for 16 hours. β-Actin served as a loading control. E, Western blot analysis of pHER3 in LK2 cells treated with vehicle control, or 100 nmol/L Erda in media control or CM of 3T3-L1 cells for 16 hours. β-Actin served as a loading control. F, Western blot analysis of pHER3 and pAKT in LK2 cells treated with media control or CM of hADSCs for 16 hours. β-Actin served as a loading control. G, Proliferation analysis of LK2 cells treated with 10 μg/mL pertuzumab (Pertuz) and vehicle control (Veh), 10 nmol/L Erda, or Erda and pertuzumab. Cells were treated in media control or CM of 3T3-L1 cells or hADSCs. Crystal violet staining was done on day 7. Data were normalized to cells treated with vehicle. Three biological replicates were performed. Data are represented as mean ± SD. Two-way ANOVA was used for statistical analysis. H, Western blot analysis of pHER3, pAKT, and pERK1/2 in LK2 cells treated with vehicle, 10 μg/mL pertuzumab, 100 nmol/L Erda, or Erda and pertuzumab. Cells were treated in CM of 3T3-L1 cells. β-Actin served as a loading control. **, P < 0.01; ***, P < 0.001; n.s., not significant.
NRG1 expression correlates with preadipocytic markers and is predominantly expressed in inflammatory CAFs in human bladder carcinoma
The platelet-derived growth factor receptor-A (PDGFRA) is one of the most established markers of mesenchymal cells, and is differentially expressed on adipocyte precursors but not on mature adipocytes (47, 48). To assess whether NRG1 expression correlates with PDGFRA expression in urothelial cancer, we analyzed RNA-seq data from TCGA BLCA cohort. This analysis revealed that NRG1 expression is significantly higher in patients that have high PDGFRA expression (PDHFRA-High) compared with those with low expression (PDGFRA-Low; Fig. 6A). Interestingly, higher PDGFRA expression correlated with significantly lower survival (Fig. 6A). To assess the clinical relevance of NRG1 in urothelial cancer, we performed IHC staining of NRG1 on a cohort of 154 paraffin-embedded human muscle-invasive urothelial bladder cancer (MIBC) samples (49, 50). We found that both tumors and stroma showed expression of NRG1 (Supplementary Fig. S6A). Interestingly, stromal NRG1 expression levels increased with tumor stage (Supplementary Fig. S6B; Table 1). To gain further insights into the clinical relevance of NRG1-expressing mesenchymal cells in urothelial cancer, we accessed a publicly available scRNA-seq data set of urothelial bladder (29). As evident in the tSNE overview of sequenced cells, the authors of the dataset have identified two CAF subsets; myo-CAFs (mCAF) and inflammatory CAFs (iCAF; Fig. 6B). NRG1 was predominantly expressed within the iCAF subset (Fig. 6C). Importantly, the expression pattern of NRG1 clearly correlated with the expression of PDGFRA, a marker of iCAFs (29) and adipocyte precursors (Fig. 6D; refs. 47, 48). NRG1 expression also correlated with preadipocyte markers, MMP3 and DPP4 (Supplementary Figs. S6C and S6D), which are known to be differentially expressed in preadipocytes but not in differentiated adipocytes (51–53). In line with this, NRG1 expression significantly correlated with the expression of MMP3 and DPP4 in the TCGA BLCA cohort (Supplementary Fig. S6E). To add more rigor to our findings, we utilized an additional scRNA-seq study that described four subclusters (C1–C4) of CAFs in bladder cancer (54). In this study, subcluster three (C3) showed high expression levels of NRG1, MMP3, and PDGFRA. Interestingly, the gene signature of C3 was clearly enriched in the iCAF population presented in Chen and colleagues (Supplementary Fig. S6F). In addition, Luo and colleagues (55) revealed that “adipogenic CAFs” (CAFadi) were delineated in the same activation trajectory as iCAFs, which was identified as CAFstate3. CAFadi were described to be expressing PDGFRA, TWIST2, TCF21, CFD, and CREB3L1, which were also enriched in the NRG1-expressing iCAF population presented in Chen and colleagues (Supplementary Fig. S6G; ref. 29). Moreover, the expression of several ADSC markers described in Zhu and colleagues (53), was also enriched in the iCAF population and most of them correlated with NRG1 expression (Table 2). To investigate the effect of primary CAFs derived from urothelial bladder, we isolated CAFs by depleting the dissociated UC tumors of CD31, CD45, and EpCAM-expressing cells by MACS (Fig. 6E). CM from urothelial bladder-derived CAFs conferred significant resistance to erdafitinib treatment in RT4 and RT112 cells (Fig. 6F). Taken together, these results highlight the clinical relevance of CAFs in mediating erdafitinib resistance in bladder tumors.
Figure 6.

NRG1 expression correlates with preadipocytic markers and is predominantly expressed in inflammatory CAFs in human bladder carcinoma. A, Data obtained from the TCGA BLCA data set, N = 426 samples. Left, expression levels of NRG1 in bladder cancer stratified based on PDGFRA expression. The low and high cutoffs were determined based on the median expression levels of PDGFRA among the samples. Ten samples did not have the expression level provided and therefore could not be included. Two-tailed unpaired t test was used for statistical analysis. Right, Kaplan–Meier survival analysis of patients with bladder cancer stratified based on the quartile expression of PDGFRA (PDGFRA-High, upper quartile; PDGFRA-Low, lower quartile). Statistical test: log-rank (Mantel–Cox) test. B, tSNE plot of single cells from bladder cancer and paratumor mucosa samples taken from Chen and collegues (29). Plot is colored by major cell types of the tumor microenvironment in bladder cancer. mCAF, myo-cancer–associated fibroblast. C, Imputed gene expression of NRG1 displayed as a function of expression density (left) or in a violin plot (right). D, Imputed gene expression of PDGFRA displayed as a function of expression density (left) or in a violin plot (right). E, Schematic diagram of isolation of CAFs from human bladder cancer (BCa) by MACS. Bladder cancer cell lines were treated with erdafitinib in CM from these CAFs. F, Proliferation analysis of RT4 and RT112 cells treated with 10 nmol/L erdafitinib in media control (ctrl) or CM of CAFs. Crystal violet staining was done on day 7. Data are plotted in a scatter bar graph (left) and in an aligned graph showing matched values of the CM from the same tumor (right). Data were normalized to cells treated with vehicle. Four biological replicates were performed. Data are represented as mean ± SD. Two-way ANOVA was used for statistical analysis. *, P < 0.05; ****, P < 0.0001; n.s., not significant. (E, Created with BioRender.com.)
Table 1.
NRG1 expression in different MIBC stages.
| pT2 | pT3 | pT4 | Total | |
|---|---|---|---|---|
| NRG1 TC high/stroma high | 5 | 23 | 7 | 35 |
| NRG1 TC high/stroma low | 14 | 12 | 5 | 31 |
| NRG1 TC low/stroma high | 5 | 21 | 16 | 42 |
| NRG1 TC low/stroma low | 20 | 18 | 8 | 46 |
| Total: | 44 | 74 | 36 | 154 |
Table 2.
Enrichment of ADSCs' markers in iCAFs.
| iCAF (Chen et al., 2022) | NRG1 overlap | |
|---|---|---|
| DCN | Yes | Yes |
| LUM | Yes | Yes |
| APOD | Yes | Partially |
| CFD | Yes | Yes |
| MGP | Yes | No |
| SERPINF1 | Yes | Yes |
| DPT | Yes | No |
| COL1A2 | Yes | Yes |
| COL6A3 | Yes | Yes |
| CXCL12 | Yes | Partially |
| SRPX | Yes | Yes |
| MMP2 | Yes | Yes |
| CCDC80 | Yes | Partially |
Discussion
It is well established that resistance to RTK inhibitors commonly occurs through feedback activation of additional signaling pathways. Indeed, cancer cell-intrinsic activation of HER2/HER3 (11, 12, 14) and EGFR (13) has been previously reported as resistance determinants to FGFR inhibitors; BGJ398 (14), AZD4547 (12), and PD173074 (13). These reports have documented autocrine resistance mechanisms that develop in response to FGFR inhibition and do not consider the contribution of the TME. In the last decade, research has shed light on the association of the TME in cancer progression and therapy resistance (56–58). In this study, we investigated the effect of the crosstalk between mesenchymal cells and FGFR-driven UC and NSCLC cells in the response to erdafitinib. In summary, our results demonstrated that the CM of adipocyte precursors (3T3-L1 cells and hADSCs) confer resistance to erdafitinib in three FGFR-dependent cell lines (RT4, RT112, and LK2 cells). NRG1 secreted from adipocyte precursor cell lines was identified as a mediator of paracrine resistance against erdafitinib by activating the HER3 pathway. Interestingly, the pharmacologic blockade of the NRG1/HER3 axis using pertuzumab resensitized cancer cells to erdafitinib in vitro and in vivo. Importantly, primary preadipocytes isolated form bladder cancer tumors recapitulated the resistance to erdafitinib conferred by the adipocyte precursor cell lines. To our knowledge, the role of adipocyte precursors in treatment resistance in urothelial bladder is largely unknown and only few reports suggest their involvement in chemotherapy resistance in ovarian (23), breast (22), and pancreatic (16) cancers. Here, our study reveals the role of adipocyte precursors in promoting erdafitinib treatment resistance in urothelial bladder. The dynamics of NRG1 expression in adipocyte precursors was also demonstrated in our study. Our results showed that the expression of NRG1 is downregulated in terminally differentiated adipocytes at both RNA and protein levels compared with their progenitors. In line with this, we observed that CM of differentiated adipocytes failed to confer resistance to erdafitinib. In contrast, one report showed that CM of 3T3-L1 adipocytes and not that of parental preadipocytes conferred resistance to lapatinib in HER2+ breast cancer cells (59). However, this study did not identify the specific factor(s) responsible for this phenotype, nor did it reveal the modulation of downstream signaling proteins in response to the adipocytes’ CM treatment. This impedes the constructive comparison of results. Nevertheless, we believe that the discrepancy in results could be related to the type of drug and cancer investigated (context-dependent).
Interestingly, secreted NRG1 has been reported to be implicated in paracrine anti-androgen resistance in prostate cancer (60, 61). Although Gil and colleagues (60) revealed that NRG1 derived from murine bone marrow–derived macrophages and myeloid-derived suppressor cells promotes prostate cancer growth, Zhang and colleagues (61) showed that CAFs secrete NRG1, which drives resistance against anti-androgen treatment in prostate cancer. Thus, our work corroborates the findings of these studies in urothelial cancer, albeit involving a specific subtype of fibroblasts, adipocyte precursors, and a novel drug, erdafitinib. FGFR3 alterations are present in 5% to 20% of muscle-invasive bladder cancer cases, and are particularly prevalent in the luminal-papillary molecular subtype (40% FGFR3-mutated; refs. 5, 6, 62). However, our data suggest that stroma-rich tumors, which mainly display luminal tumor cell differentiation (63), are less likely to respond to erdafitinib as a single-agent therapy. Importantly, HER3 is also enriched in the luminal subtype of bladder cancer (64, 65). Therefore, the enrichment of NRG1-secreting stroma in the TME could be a negative predictive biomarker for erdafitinib single-agent therapy. These tumors may be the prime candidates for our proposed combination therapy of erdafitinib and pertuzumab (66). Moreover, because obesity results in a higher frequency of ADSCs, and an altered ADSC biology towards increased protumorigenic signaling (67, 68), our proposed combination therapy could be even more relevant in obese patients. Further investigations through biomarker-driven clinical trials is necessary to confirm these hypotheses.
Our results demonstrated that the NRG1/HER3 axis also induces erdafitinib resistance to the NSCLC cell line, LK2, that is FGFR1-driven (46). These indicate that our identified TME-driven resistance mechanism can be of clinical relevance in other tumor entities, where FGFR1–14 inhibitors are used/in clinical investigation, for example, cholangiocarcinoma (NCT04083976; ref. 69).
In conclusion, this study provides preclinical evidence confirming the efficacy of cotargeting the FGFR1–4 and NRG1/HER3 pathways to overcome resistance to erdafitinib in FGFR-dependent tumors.
Supplementary Material
Supplementary Figure1
Supplementary Figure2
Supplementary Figure3
Supplementary Figure4
Supplementary Figure5
Supplementary Figure6
Acknowledgments
S. Hosni was supported by the Deutsche Forschungsgemeinschaft (DFG; SPP2084)-AL 2692/1. V. Kilian was funded by the Sci-Med stipend (application no.: 2021–4-12). Marina Bertlich was funded by DFG (DFG SPP 2048). Research in the lab of D. Wachten was funded by DFG: SFB 1454 – Project-ID 432325352, TRR333/1 – Project-ID 450149205, FOR5547 – Project-ID 503306912, WA 3382/8–1 – Project-ID 513767027, under Germany's Excellence Strategy – EXC2151 – Project-ID 390873048. K. Sieckmann was supported by a fellowship from the DFG. T. Bald is supported by the DFG under Germany's Excellence Strategy – EXC2151–390873048. M. Hölzel was supported by the DFG under Germany's Excellence Strategy–EXC2151–390873048. Research in the lab of A. Alajati was funded by DFG (SPP2084)-AL 2692/1.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
S. Hosni reports grants from Deutsche Forschungsgemeinschaft (DFG; German research society) during the conduct of the study. V. Kilian reports grants from BONFOR Förderungsprogramm during the conduct of the study. N. Klümper reports personal fees from Astellas, Novartis, Ipsen, Photocure, MSD, and Eisai outside the submitted work. K. Sieckmann reports personal fees from Studienstiftung des Deutschen Volkes during the conduct of the study. O. Hahn reports grants from Janssen-Cilag and DFG, and personal fees and nonfinancial support from Bristol-Myers Squibb, AstraZeneca, Bayer, Medac, Astellas, and Merck Sharp & Dohme outside the submitted work. M. Bertlich reports grants from DFG during the conduct of the study and grants from Wolfgang.Dieckmann-Stiftung outside the submitted work. M. Eckstein reports grants and personal fees from AstraZeneca, Janssen, Owkin, and Gilead; personal fees from MSD and Diaceutics outside the submitted work. M. Hölzel reports grants from DFG during the conduct of the study; grants and personal fees from TME Pharma AG; and personal fees from Novartis and BMS outside the submitted work. D. Wachten reports grants from DFG during the conduct of the study. A. Alajati reports grants from DFG during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
S. Hosni: Conceptualization, validation, investigation, writing–original draft, writing–review and editing. V. Kilian: Data curation. N. Klümper: Conceptualization. D. Gabbia: Data curation, formal analysis. K. Sieckmann: Investigation. D. Corvino: Formal analysis. A. Winkler: Methodology. M. Saponaro: Formal analysis, validation, investigation. K. Wörsdörfer: Investigation. D. Schmidt: Methodology. O. Hahn: Investigation. I. Zanotto: Methodology. M. Bertlich: Methodology. M. Toma: Investigation. T. Bald: Supervision. M. Eckstein: Methodology. M. Hölzel: Supervision. M. Geyer: Supervision. M. Ritter: Funding acquisition. D. Wachten: Supervision. S. De Martin: Validation. A. Alajati: Conceptualization, supervision, funding acquisition, writing–original draft, writing–review and editing.
References
- 1. Sharma P, Callahan MK, Bono P, Kim J, Spiliopoulou P, Calvo E, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol 2016;17:1590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Powles T, Rosenberg JE, Sonpavde GP, Loriot Y, Durán I, Lee JL, et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N Engl J Med 2021;384:1125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krook MA, Reeser JW, Ernst G, Barker H, Wilberding M, Li G, et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer 2021;124:880–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 2016;22:259–67. [DOI] [PubMed] [Google Scholar]
- 5. Kamoun A, de Reyniès A, Allory Y, Sjödahl G, Robertson AG, Seiler R, et al. A consensus molecular classification of muscle-invasive bladder cancer. Eur Urol 2020;77:420–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Rhijn BWG, Mertens LS, Mayr R, Bostrom PJ, Real FX, Zwarthoff EC, et al. FGFR3 mutation status and FGFR3 expression in a large bladder cancer cohort treated by radical cystectomy: implications for anti-FGFR3 treatment?†. Eur Urol 2020;78:682–7. [DOI] [PubMed] [Google Scholar]
- 7. Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med 2019;381:338–48. [DOI] [PubMed] [Google Scholar]
- 8. Zheng X, Wang H, Deng J, Yao M, Zou X, Zhang F, et al. Safety and efficacy of the pan-FGFR inhibitor erdafitinib in advanced urothelial carcinoma and other solid tumors: a systematic review and meta-analysis. Front Oncol 2023;12:907377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perera TPS, Jovcheva E, Mevellec L, Vialard J, De Lange D, Verhulst T, et al. Discovery and pharmacological characterization of JNJ-42756493 (Erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol Cancer Ther 2017;16:1010–20. [DOI] [PubMed] [Google Scholar]
- 10. Loriot Y, Matsubara N, Park SH, Huddart RA, Burgess EF, Houede N, et al. Phase 3 THOR study: Results of erdafitinib (erda) versus chemotherapy (chemo) in patients (pts) with advanced or metastatic urothelial cancer (mUC) with select fibroblast growth factor receptor alterations (FGFRalt). J Clin Oncol 2023;41(17_suppl):LBA4619. [Google Scholar]
- 11. Wang J, Mikse O, Liao RG, Li Y, Tan L, Janne PA, et al. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene 2014;34:2167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang L, Šuštić T, Leite de Oliveira R, Lieftink C, Halonen P, van de Ven M, et al. A functional genetic screen identifies the phosphoinositide 3-kinase pathway as a determinant of resistance to fibroblast growth factor receptor inhibitors in FGFR mutant urothelial cell carcinoma. Eur Urol 2017;71:858–62. [DOI] [PubMed] [Google Scholar]
- 13. Herrera-Abreu MT, Pearson A, Campbell J, Shnyder SD, Knowles MA, Ashworth A, et al. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3 -mutant cancer. Cancer Discov 2013;3:1058–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weickhardt AJ, Lau DK, Hodgson-Garms M, Lavis A, Jenkins LJ, Vukelic N, et al. Dual targeting of FGFR3 and ERBB3 enhances the efficacy of FGFR inhibitors in FGFR3 fusion-driven bladder cancer. BMC Cancer 2022;22:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol OncolJ Hematol Oncol 2019;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miyazaki Y, Oda T, Inagaki Y, Kushige H, Saito Y, Mori N, et al. Adipose-derived mesenchymal stem cells differentiate into heterogeneous cancer-associated fibroblasts in a stroma-rich xenograft model. Sci Rep 2021;11:4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atiya H, Frisbie L, Pressimone C, Coffman L. Mesenchymal stem cells in the tumor microenvironment. Adv Exp Med Biol 2020;1234:31–42. [DOI] [PubMed] [Google Scholar]
- 18. Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol 2019;15:139–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saha A, Kolonin MG, DiGiovanni J. Obesity and prostate cancer - microenvironmental roles of adipose tissue. Nat Rev Urol 2023;20:579–96. [DOI] [PubMed] [Google Scholar]
- 20. Maj M, Kokocha A, Bajek A, Drewa T. The interplay between adipose-derived stem cells and bladder cancer cells. Sci Rep 2018;8:15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fajka-Boja R, Szebeni GJ, Hunyadi-Gulyás É, Puskás LG, Katona RL. Polyploid adipose stem cells shift the balance of IGF1/IGFBP2 to promote the growth of breast cancer. Front Oncol 2020;10:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu Y, Yang Y, Liu Y, Hao Y, Zhang Y, Hu Y, et al. Upregulation of PAG1/Cbp contributes to adipose-derived mesenchymal stem cells promoted tumor progression and chemoresistance in breast cancer. Biochem Biophys Res Commun 2017;494:719–27. [DOI] [PubMed] [Google Scholar]
- 23.Koren Carmi Y, Khamaisi H, Adawi R, Noyman E, Gopas J, Mahajna J. Secreted soluble factors from tumor-activated mesenchymal stromal cells confer platinum chemoresistance to ovarian cancer cells. Int J Mol Sci 2023;24:7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang T, Tseng C, Zhang Y, Sirin O, Corn PG, Li-Ning-Tapia EM, et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat Commun 2016;7:11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Daquinag A, Traktuev DO, Amaya-Manzanares F, Simmons PJ, March KL, et al. White adipose tissue cells are recruited by experimental tumors and promote cancer progression in mouse models. Cancer Res 2009;69:5259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zebisch K, Voigt V, Wabitsch M, Brandsch M. Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal Biochem 2012;425:88–90. [DOI] [PubMed] [Google Scholar]
- 27. Cheng L, Montironi R, Davidson DD, Lopez-Beltran A. Staging and reporting of urothelial carcinoma of the urinary bladder. Mod Pathol Off J U S Can Acad Pathol Inc 2009;22Suppl 2:S70–95. [DOI] [PubMed] [Google Scholar]
- 28. Philip AT, Amin MB, Tamboli P, Lee TJ, Hill CE, Ro JY. Intravesical adipose tissue: a quantitative study of its presence and location with implications for therapy and prognosis. Am J Surg Pathol 2000;24:1286–90. [DOI] [PubMed] [Google Scholar]
- 29. Chen Z, Zhou L, Liu L, Hou Y, Xiong M, Yang Y, et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat Commun 2020;11:5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lamont FR, Tomlinson DC, Cooper PA, Shnyder SD, Chester JD, Knowles MA. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br J Cancer 2010;104:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomlinson DC, Hurst CD, Knowles MA. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene 2007;26:5889–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Earl J, Rico D, Carrillo-de-Santa-Pau E, Rodríguez-Santiago B, Méndez-Pertuz M, Auer H, et al. The UBC-40 urothelial bladder cancer cell line index: a genomic resource for functional studies. BMC Genomics [Electronic Resource] 2015;16:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet 2012;22:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahe M, Dufour F, Neyret-Kahn H, Moreno-Vega A, Beraud C, Shi M, et al. An FGFR 3/MYC positive feedback loop provides new opportunities for targeted therapies in bladder cancers. EMBO Mol Med 2018;10:e8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Costa R, Carneiro BA, Taxter T, Tavora FA, Kalyan A, Pai SA, et al. FGFR3-TACC3 fusion in solid tumors: mini review. Oncotarget 2016;7:55924–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nassar AH, Lundgren K, Pomerantz M, Van Allen E, Harshman L, Choudhury AD, et al. Enrichment of FGFR3-TACC3 fusions in patients with bladder cancer who are young, asian, or have never smoked. JCO Precis Oncol 2018:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan–fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 2015;33:3401–8. [DOI] [PubMed] [Google Scholar]
- 38. Ieguchi K, Fujita M, Ma Z, Davari P, Taniguchi Y, Sekiguchi K, et al. Direct binding of the EGF-like domain of neuregulin-1 to integrins (αvβ3 and α6β4) is involved in neuregulin-1/ErbB signaling. J Biol Chem 2010;285:31388–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lyu H, Han A, Polsdofer E, Liu S, Liu B. Understanding the biology of HER3 receptor as a therapeutic target in human cancer. Acta Pharm Sin B 2018;8:503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Christiaens V, Van Hul M, Lijnen HR, Scroyen I. CD36 promotes adipocyte differentiation and adipogenesis. Biochim Biophys Acta BBA - Gen Subj 2012;1820:949–56. [DOI] [PubMed] [Google Scholar]
- 41. Bengestrate L, Virtue S, Campbell M, Vidal-Puig A, Hadaschik D, Hahn P, et al. Genome-wide profiling of MicroRNAs in adipose mesenchymal stem cell differentiation and mouse models of obesity. Navarro A., editor. PLoS One 2011;6:e21305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dalen KT, Ulven SM, Bamberg K, Gustafsson JÅ, Nebb HI. Expression of the insulin-responsive glucose transporter GLUT4 in adipocytes is dependent on liver X receptor α. J Biol Chem 2003;278:48283–91. [DOI] [PubMed] [Google Scholar]
- 43. Gonzales AM, Orlando RA. Role of adipocyte-derived lipoprotein lipase in adipocyte hypertrophy. Nutr Metab 2007;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2002;2:127–37. [DOI] [PubMed] [Google Scholar]
- 45. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004;5:317–28. [DOI] [PubMed] [Google Scholar]
- 46. Hibi M, Kaneda H, Tanizaki J, Sakai K, Togashi Y, Terashima M, et al. FGFR gene alterations in lung squamous cell carcinoma are potential targets for the multikinase inhibitor nintedanib. Cancer Sci 2016;107:1667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berry R, Jeffery E, Rodeheffer MS. Weighing in on adipocyte precursors. Cell Metab 2014;19:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee YH, Petkova AP, Mottillo EP, GJG. In Vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab 2012;15:480–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eckstein M, Strissel P, Strick R, Weyerer V, Wirtz R, Pfannstiel C, et al. Cytotoxic T-cell-related gene expression signature predicts improved survival in muscle-invasive urothelial bladder cancer patients after radical cystectomy and adjuvant chemotherapy. J Immunother Cancer 2020;8:e000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saponaro M, Flottmann S, Eckstein M, Hommerding O, Klümper N, Corvino D, et al. CDCP1 expression is frequently increased in aggressive urothelial carcinoma and promotes urothelial tumor progression. Sci Rep 2023;13:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alexander CM, Selvarajan S, Mudgett J, Werb Z. Stromelysin-1 regulates adipogenesis during mammary gland involution. J Cell Biol 2001;152:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chowdhury HH, Velebit J, Radić N, Frančič V, Kreft M, Zorec R. Hypoxia alters the expression of dipeptidyl peptidase 4 and induces developmental remodeling of human preadipocytes. J Diabetes Res 2016;2016:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu K, Cai L, Cui C, de Los Toyos JR, Anastassiou D. Single-cell analysis reveals the pan-cancer invasiveness-associated transition of adipose-derived stromal cells into COL11A1-expressing cancer-associated fibroblasts. PLoS Comput Biol 2021;17:e1009228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma Z, Li X, Mao Y, Wei C, Huang Z, Li G, et al. Interferon-dependent SLC14A1+ cancer-associated fibroblasts promote cancer stemness via WNT5A in bladder cancer. Cancer Cell 2022;40:1550–65. [DOI] [PubMed] [Google Scholar]
- 55. Luo H, Xia X, Huang LB, An H, Cao M, Kim GD, et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat Commun 2022;13:6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mink SR, Vashistha S, Zhang W, Hodge A, Agus DB, Jain A. Cancer-associated fibroblasts derived from EGFR-TKI–resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol Cancer Res 2010;8:809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res 2009;15:6630–8. [DOI] [PubMed] [Google Scholar]
- 58. Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther 2021;6:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Geneste A, Duong MN, Molina L, Conilh L, Beaumel S, Cleret A, et al. Adipocyte-conditioned medium induces resistance of breast cancer cells to lapatinib. BMC Pharmacol Toxicol 2020;21:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gil V, Miranda S, Riisnaes R, Gurel B, D'Ambrosio M, Vasciaveo A, et al. HER3 is an actionable target in advanced prostate cancer. Cancer Res 2021;81:6207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Z, Karthaus WR, Lee YS, Gao VR, Wu C, Russo JW, et al. Tumor microenvironment-derived NRG1 promotes antiandrogen resistance in prostate cancer. Cancer Cell 2020;38:279–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cox A, Klümper N, Stein J, Sikic D, Breyer J, Bolenz C, et al. Molecular urothelial tumor cell subtypes remain stable during metastatic evolution. Eur Urol 2023;S0302283823026982. [DOI] [PubMed] [Google Scholar]
- 64. Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014;25:152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Czerniak B, Dinney C, McConkey D. Origins of bladder cancer. Annu Rev Pathol Mech Dis 2016;11:149–74. [DOI] [PubMed] [Google Scholar]
- 66. Guardia C, Bianchini G, Arpí-LLucià O, Menendez S, Casadevall D, Galbardi B, et al. Preclinical and clinical characterization of fibroblast-derived neuregulin-1 on trastuzumab and pertuzumab activity in HER2-positive breast cancer. Clin Cancer Res 2021;27:5096–108. [DOI] [PubMed] [Google Scholar]
- 67. Bunnell BA, Martin EC, Matossian MD, Brock CK, Nguyen K, Collins-Burow B, et al. The effect of obesity on adipose-derived stromal cells and adipose tissue and their impact on cancer. Cancer Metastasis Rev 2022;41:549–73. [DOI] [PubMed] [Google Scholar]
- 68. Strong AL, Pei DT, Hurst CG, Gimble JM, Burow ME, Bunnell BA. Obesity enhances the conversion of adipose-derived stromal/stem cells into carcinoma-associated fibroblast leading to cancer cell proliferation and progression to an invasive phenotype. Stem Cells Int 2017;2017:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2 -rearranged intrahepatic cholangiocarcinoma. N Engl J Med 2023;388:228–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure1
Supplementary Figure2
Supplementary Figure3
Supplementary Figure4
Supplementary Figure5
Supplementary Figure6
Data Availability Statement
The scRNA-seq data analyzed in this paper are available from the Sequence Read Archive (SRA) at PRJNA662018 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA662018). The RNA-seq data generated by the TCGA for urothelial BLCA that was analyzed in this study are available in the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/). All other raw data generated in this study are available upon request from the corresponding author.