

Abstract
Adeno-associated virus (AAV) vectors are used for correcting multiple genetic disorders. Although the goal is to achieve lifelong correction with a single vector administration, the ability to redose would enable the extension of therapy in cases in which initial gene transfer is insufficient to achieve a lasting cure, episomal vector forms are lost in growing organs of pediatric patients, or transgene expression is diminished over time. However, AAV typically induces potent and long-lasting neutralizing antibodies (NAbs) against capsid that prevents re-administration. To prevent NAb formation in hepatic AAV8 gene transfer, we developed a transient B cell-targeting protocol using a combination of monoclonal Ab therapy against CD20 (for B cell depletion) and BAFF (to slow B cell repopulation). Initiation of immunosuppression before (rather than at the time of) vector administration and prolonged anti-BAFF treatment prevented immune responses against the transgene product and abrogated prolonged IgM formation. As a result, vector re-administration after immune reconstitution was highly effective. Interestingly, re-administration before the immune system had fully recovered achieved further elevated levels of transgene expression. Finally, this immunosuppression protocol reduced Ig-mediated AAV uptake by immune cell types with implications to reduce the risk of immunotoxicities in human gene therapy with AAV.
Keywords: gene therapy, AAV, immune suppression, redosing, re-administration, capsid, transgene, CD20, BAFF
Graphical abstract
Rana and colleagues identify a role for the cytokine B cell-activating factor BAFF in promoting adaptive immune responses to AAV gene therapy. Combination therapy with anti-CD20 and extended use of anti-BAFF enables redosing of systemic AAV gene therapy and can improve transgene expression levels.
Introduction
Adeno associated virus (AAV) vectors are widely used in clinical gene therapy because of their versatility in transferring therapeutic genes in vivo to various organs and cell types, in part owing to the large number of viral capsids with varying tropism that have been isolated, generated through capsid library screens, or otherwise engineered.1,2 AAV-based gene therapy products have received regulatory approval for the treatment of Leber congenital amaurosis, spinal muscular atrophy, hemophilia A and B, Duchenne muscular dystrophy (DMD), and aromatic l-amino acid decarboxylase deficiency.3,4,5,6,7 However, there have also been several hurdles to success in AAV gene therapy, including loss of transgene expression and immune responses to both the encoded protein and vector capsid, which need to be addressed to further improve outcomes.
In some cases, gradual loss of transgene expression may be due to persistence of the AAV genome, mostly as episomes in transduced cells, affecting the stability of gene transfer if the transduced target cells are dividing. This limits the ability to achieve long-term therapy by gene transfer to pediatric patients, depending on the target cell type. For instance, it is known from preclinical studies that hepatic AAV gene transfer is not stable upon transduction of young animals.8,9,10 Although turnover of muscle fibers is normally rare even in a growing individual, these cells regenerate in patients with DMD. To prevent this, a minimum threshold level of modified dystrophin expression in muscles is required for disease prevention. The degeneration-regeneration process may not be entirely halted if dystrophin replacement therapy, using microdystrophin transgenes, is not robust enough, which will lead to the gradual elimination of episomal transgene and hence expression. Following hepatic gene therapy in adult patients with hemophilia A, a gradual loss of factor VIII expression was observed, likely reflecting properties of this particular transgene product that may lead to translational and/or transcriptional silencing.11,12,13 These considerations and outcomes support the development of protocols for re-administration of AAV vectors to achieve lifelong therapy.
The most critical challenge in vector re-administration is managing the host immune responses. The initial vector administration results in the formation of high-titer neutralizing antibodies (NAbs) against the viral capsid that persist for many years, thereby preventing gene transfer upon vector re-administration.14,15,16 Moreover, cross-reactivity of these Abs among AAV serotypes/capsids complicates the use of alternative capsids for redosing, which would also require the development of two gene therapy products. Hence, a transient immune suppression (IS) protocol that effectively prevents NAb formation during initial vector administration is desirable.
Previously, we have successfully achieved AAV8 vector re-administration to the livers of hemophilia A mice that were treated with the mammalian target of rapamycin (mTOR) inhibitor rapamycin for 8 weeks, starting at the time of initial vector delivery.11 However, rapamycin has been found to be less effective in nonhuman primates (NHPs), prompting efforts to improve its ability to block antibody (Ab) formation against AAV.17,18 The coadministration of synthetic vaccine particles encapsulating rapamycin (SVP[Rapa]) with initial vector dose in both mice and NHPs reduced both B and T cell responses and enabled re-administration.19 Alternatively, rapamycin can be combined with other drugs such as B cell-depleting monoclonal Ab (mAb) against CD20.20,21 Here, we report a novel IS protocol, based on combined depletion of B cells and their survival factor BAFF (B cell activating factor). Our strategy shows high efficacy for liver-directed re-administration of AAV vector in a murine model with several superior characteristics to overcome the limitations of other drug combinations.
Results
Concurrent initiation of IS fails to achieve re-administration and exacerbates transgene-specific immune responses
To enable AAV8 redosing for liver gene therapy, we used single-drug or combination IS regimens using α-CD20, rapamycin, or α-BAFF, as described in Figure 1A. IS was initiated at the time of vector administration. We gauged the efficacy of these treatments in preventing immune responses to the transgene product and AAV8 capsid. AAV8-OVA (ovalbumin) was used for primary dosing and AAV8-hFIX (human factor IX) was used for re-administration after an interval of 8 weeks. Circulating levels of transgene products, OVA and hFIX, were used as indicators of successful gene delivery following each AAV8 dosing.
Figure 1.

Effect of concurrent IS treatment on transgene and AAV8-specific immune responses
(A) Schematic outline of IS treatments and AAV8 dosing. (B) CD8+ T cell responses to the OVA transgene quantified using MHC-I H2-Kb-SIINFEKL tetramer for various IS treatments (n = 5–8/group). Red dots indicate (Tet+) animals and blue dots indicate (Tet−) animals. Frequency of Tet+ animals in each group is indicated above each bar. (C) Circulating plasma levels of OVA at 4 weeks post-AAV8-OVA administration. (D) Representative images of OVA staining in liver tissue from mice with or without CD8+ T cell response to OVA (20× magnification). (E) Longitudinal analysis of AAV8 capsid-specific IgM Ab levels in plasma samples post primary and -secondary AAV8 administrations. The dotted line represents baseline IgM levels averaged from 8 pre-bled animals. (F) AAV8 capsid-specific IgG2c Ab levels in plasma samples. The dotted line represents baseline IgG2c levels averaged from 8 pre-bled animals. (G) Circulating human FIX levels at 4 weeks post-AAV8-hFIX administration. Statistical significance was calculated by one-way ANOVA (Dunnett’s multiple comparisons) for (B), (C), and (G), and two-way ANOVA for (E) and (F). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
AAV8-OVA has previously been shown to have a dose-dependent effect on the induction of transgene-specific CD8+ T cell responses.22 Vector doses of ≥1010 vector genome (vg)/mouse resulted in expression levels sufficient to induce immune tolerance to the transgene product, whereas lower doses trigger CD8+ T cell responses.22,23 For this IS treatment regimen, a mildly immunogenic AAV8-OVA dose of 2 × 109 vg/mouse was administered to C57BL/6 mice. Frequencies of OVA-specific CD8+ T cells (Tet+, tetramer positive) among peripheral blood mononuclear cells were determined at 4 weeks postadministration. Surprisingly, IS treatments involving B cell depletion with α-CD20 and/or α-BAFF increased the incidences of Tet+ responses from 12% in the control group (no IS) to 37% in α-CD20, 75% in α-BAFF, 25% in α-CD20+Rapa and 50% in α-CD20+α-BAFF groups (Figure 1B). Detectable levels of circulating OVA in plasma samples correlated with the absence of Tet+ cells (Tet−) (Figure 1C). Furthermore, immunohistochemistry confirmed the loss of OVA expression in the livers of Tet+ animals, and Tet− animals showed hepatic OVA expression (Figure 1D). To confirm these findings, we compared 3 different AAV8-OVA doses: 1 × 109, 2 × 109, and 5 × 109 vg/mouse. At the mildly immunogenic doses of 2 × 109 and 5 × 109 vg/mouse, we again observed that treatment with α-CD20 and α-BAFF increased the incidences of Tet+ responses from 12% to 20% in the no-IS group to 50% and 70%, respectively, in the treated groups (Figure S1A). There was no difference in Tet+ responses at the immunogenic dose of 1 × 109 vg/mouse (75 vs. 87%). We also tested OVA-specific Abs in plasma at 4 weeks. Only the α-BAFF group had elevated levels of α-OVA immunoglobulin G1 (IgG1) and IgG2c (Figures S1C and S1D).
Next, we quantified AAV8 capsid-specific IgM and IgG2c Ab responses. A single α-CD20 treatment depleted >99% of circulating B cells by day 2 (Figure S1B). In the absence of IS, AAV8-OVA administration resulted in development of both IgM and IgG2c Abs to the AAV8 capsid (Figures 1E and 1F). Except for α-BAFF, IS treatment failed to attenuate IgM responses, and α-CD20 (1.36 ± 0.32 μg/mL), Rapa (1.150 ± 0.11 μg/mL) and α-CD20+Rapa (1.36 ± 0.19 μg/mL) groups had significantly higher IgM levels compared to the no-IS control group (0.53 ± 0.10 μg/mL) at 4 weeks (Figure 1E). However, IgG2c Abs against AAV8 were detected only in Rapa (0.39 ± 0.22 μg/mL) and α-BAFF (0.56 ± 0.28 μg/mL) IS groups and were comparable with the no-IS group (0.58 ± 0.28 μg/mL) (Figure 1F). For vector re-administration, all of the animals, as well as naive C57BL/6 mice, were injected with 1 × 1011 vg/mouse of AAV8-hFIX. The analysis of AAV8-specific Abs post-re-administration showed comparable IgMs among all of the experimental groups, irrespective of the initial IS treatment (Figure 1E). Circulating hFIX expression was observed only in previously naive animals. All of the animals that had received initial AAV8-OVA gene transfer failed to express hFIX or had comparatively low levels (Figures 1G and S1E). In the α-CD20+α-BAFF group, re-administration of AAV8 in half of the animals resulted in a 5-fold lower hFIX expression (1.01 ± 0.17 μg/mL) compared to previously naive control animals (5.12 ± 0.46 μg/mL).
Overall, these results demonstrated that concurrent IS treatment exacerbated transgene-specific T cell responses (except for the Rapa group), and largely failed to mitigate Ab responses against AAV8 capsid, resulting in failed re-administration.
Enhanced plasma BAFF levels following IS treatment correlates with increased IgM formation against capsid
Even though concurrent IS treatments were able to prevent IgG formation against the AAV8 capsid, most treatments were not efficient in preventing IgM responses. The resulting low to undetectable levels of hFIX suggest that IgM Abs were neutralizing, hence preventing re-administration of AAV8.
To confirm the neutralizing property of IgMs, we performed in vitro NAb assays using serum samples from the 4-week time point and AAV8-expressing renilla luciferase (RLuc). Serum samples from uninjected C57BL/6 mice served as the negative control. Serum samples from control animals (no IS), served as positive controls for NAbs (half-maximal inhibitory concentration [IC50] = 67.59). We examined only those serum samples that had detectable levels of IgM but lacked IgG2c against AAV8 capsid (IC50 = 5.23 for α-CD20, 16.76 for Rapa, 12.23 for α-CD20+Rapa, and 4.05 for α-CD20+α-BAFF group). Our results confirmed that serum IgM to capsid can neutralize AAV8 (Figures 2A and 2B).
Figure 2.

AAV8 neutralizing IgM Ab titers and correlation with plasma BAFF levels
(A) Comparison of IgM and IgG2c Ab titers and IC50 NAb dilution factors for representative samples from control (no-IS) and various IS treatment groups (n = 3–5/group) at 4 weeks. (B) Representative IC50 dose-response curves showing percentage of transduction at various plasma dilutions. (C) Two-axis combined bar graph correlating circulating BAFF levels (left y axis) and IgM levels (right y axis) for naive, no-IS, and various IS groups. Significantly elevated BAFF levels are indicated. Statistical significance was calculated by 1-way ANOVA (Dunnett’s multiple comparisons) for (C). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
Next, we sought to identify factors that could promote the development of prolonged IgM formation. One candidate is BAFF, which was found to be significantly increased upon α-CD20 (38.3 ± 7.93 ng/mL) and α-CD20+Rapa (51.14 ± 2.65 ng/mL) treatments compared to the no-IS group (7.48 ± 0.91 ng/mL) or naive animals (1.95 ± 0.21 ng/mL) (Figure 2C). We recently reported sustained, elevated levels of BAFF in hemophilia A patients with Abs against coagulation factor VIII (FVIII) that failed to achieve tolerance to FVIII following α-CD20 treatment.24 Hence, we assessed the correlation between plasma BAFF levels and IgM development in representative samples from Figure 1E. IgM titers against AAV8 were higher for those treatments (α-CD20 and α-CD20+Rapa) that resulted in increased levels of circulating BAFF (Figure 2C). BAFF levels were undetectable after α-CD20+α-BAFF treatment (4-week time point). These results suggest that IS treatments that elevate BAFF predispose to prolonged neutralizing IgM formation.
Initiation of immune suppression before AAV administration abrogates NAb development
Because the concurrent IS treatment regimen was unable to block both T cell and Ab responses, we assessed the effect of initiating IS treatments before AAV8-OVA dosing as described in Figure 3A. Transgene-specific CD8+ T cell responses were reduced upon pre-α-CD20 (12%) and pre-α-CD20+Rapa (12%) treatment compared to the no-IS control group (50%) and completely absent in the pre-α-CD20+α-BAFF (0%) treatment group (Figure 3B). Average plasma levels of OVA were higher in pre-α-CD20+Rapa (124.25 ± 97.420 ng/mL)- and pre-α-CD20+α-BAFF (131.59 ± 34.84 ng/mL)-treated groups compared to the no-IS control group (60.08 ± 73.35 ng/mL), with significant differences in the pre-α-CD20 (164.30 ± 98.30 ng/mL) group (Figure 3C). Prior IS treatments also prevented Ab responses to OVA protein, indicating effective prevention of B cell responses against the transgene product (Figures S2A and 2B).
Figure 3.

IS before low-dose AAV administration abrogates NAb development
(A) Schematic representation of IS pretreatment regimens and AAV8 dosing. (B) CD8+ T cell response to the OVA transgene at 4 weeks post-AAV8-OVA administration (n = 7–8/group). (C) Circulating plasma levels of OVA at 4 weeks post-AAV8-OVA administration. (D) Longitudinal analysis of AAV8 capsid-specific IgM Ab levels in plasma samples post primary and -secondary AAV8 administrations. (E) AAV8 capsid-specific IgG2c Ab levels in plasma samples. (F) Circulating hFIX levels at 4 weeks post-re-administration. (G) Representative images of liver tissue for control (no-IS) and various IS treatment groups stained for hFIX (in red, 40× magnification) expression at 12 weeks post-re-administration. Statistical significance was calculated by one-way ANOVA (Dunnett’s multiple comparisons) for (B), (C), and (F), and two-way ANOVA for (D) and (E). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001; ns, not significant.
Next, we assessed the ability of IS pretreatments to prevent Ab formation against AAV8. Serum samples collected at 4 and 8 weeks post-AAV8-OVA administration showed no detectable IgM or IgG2c Abs against AAV8 in pre-α-CD20 and pre-α-CD20+α-BAFF groups, whereas 25% of the animals in the pre-α-CD20+Rapa group developed low levels of IgM (0.5–1 μg/mL) (Figures 3D, 3E, S2C, and S2D). To evaluate the re-administration efficacy in these groups, all of the experimental groups and naive control animals received 1 × 1011 vg/mouse of AAV8-hFIX. Analysis of plasma samples 4 weeks post-re-administration (week 12) revealed that pre-α-CD20+Rapa IS resulted in significantly higher levels of IgM against capsid (1.96 ± 0.42 μg/mL) compared to the no-IS group (1.23 ± 0.62 μg/mL), whereas the pre-α-CD20 and pre-α-CD20+α-BAFF groups had lower levels (0.66 ± 0.25 μg/mL and 1.15 ± 0.09 μg/mL, respectively) (Figure 3D). IgG2c was undetectable in pre-α-CD20- and pre-α-CD20+α-BAFF-treated groups, whereas 30% of animals in pre-α-CD20+Rapa developed IgG2c (Figure 3E). The ability to re-administer with AAV8 was assessed by evaluating the expression of hFIX from the second vector, resulting in average levels of 5.09 ± 0.57 μg/mL in pre-α-CD20, 5.15 ± 1.08 μg/mL in pre-α-CD20+Rapa, and 5.85 ± 1.01 μg/mL in pre-α-CD20+α-BAFF groups, which are comparable to previously naive control animals that received only AAV8-hFIX (4.59 ± 0.51 μg/mL; Figure 3F). In the pre-α-CD20+Rapa group, two animals did not show any detectable hFIX in plasma samples, which correlated with the formation of AAV8-specific IgM (Figure S2C). hFIX immunostaining confirmed robust expression of the re-administered vector in the pre-IS-treated groups (Figure 3G). As expected, re-administration was unsuccessful in mice that initially received AAV8-OVA without IS (Figures 3F and 3G).
These results demonstrated that initiating IS before, rather than at the time of initial vector delivery, prevents both transgene and AAV-specific Ab responses, thereby enabling re-administration. Importantly, this approach also prevented antitransgene immune responses.
Initial high vector dose reduces re-administration efficacy following IS pretreatment
Our results have shown the effectiveness of IS treatment before AAV dosing. However, the initial dose used for these experiments was comparatively lower than the typical clinical doses. To determine whether these IS treatments are equally efficient when using a higher dose, we administered an initial dose of 1 × 1011 vg/mouse AAV8-OVA (∼5 × 1012 vg/kg) to C57BL/6 animals with the dosing regimen represented in Figure 4A. We again found a reduced T cell response against OVA in IS-pretreated mice (Figure 4B). Circulating OVA levels were significantly higher in pre-α-BAFF (263.479 ± 30.527 ng/mL) and pre-α-CD20+Rapa groups (217.151 ± 29.739 ng/mL) compared to the no-IS group (101.742 ± 28.570 ng/mL), whereas other treatment groups showed no significant differences (Figure 4C). These results confirmed that prior IS treatments were successful in preventing cellular responses against the transgene product even at high vector doses. We also observed the prevention of Ab formation against OVA protein in treated animals (Figures S3A and S3B).
Figure 4.

Initial high vector dose reduces the re-administration efficacy of IS pretreatments
(A) Schematic outline of IS pretreatment regimens and AAV8 dosing. (B) CD8+ T cell response to the OVA transgene at 4 weeks post-AAV8-OVA administration (n = 5–10/group). (C) Circulating plasma levels of OVA at 4 weeks post-AAV8-OVA administration. AAV8 capsid-specific IgM (D) and IgG2c (E) Ab levels in plasma samples collected at different time points before re-administration. AAV8 capsid-specific IgM (F) and IgG2c (G) Ab levels in plasma samples collected post-re-administration. (H) Circulating human FIX levels at 4 weeks post-re-administration with AAV8-hFIX. Statistical significance was calculated by one-way ANOVA (Dunnett’s multiple comparisons) for (B), (C), and (H), and two-way ANOVA for (D)–(G). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗∗p ≤ 0.0001.
Next, we tested the efficacy of these treatments in preventing Ab responses against the AAV8 capsid. The analysis of plasma samples collected at 4 and 8 weeks post-high-dose AAV8-OVA administration showed IgM formation in 55% of animals in pre-α-CD20, 20% in pre-α-CD20+Rapa and pre-α-CD20+α-BAFF, and 100% of the animals in the pre-α-BAFF treatment group (Figures 4D and S3C). AAV-specific IgG2c Abs were not detectable until 8 weeks post-AAV8-OVA dosing in treated groups, except for pre-α-BAFF treatment (here, 80% of animals developed low levels of IgG2c Abs; Figures 4E and S3D). To test for the ability to re-administer, all of the animals received 1 × 1011 vg/mouse AAV8-hFIX at 8 weeks postinitial dosing. Following secondary dosing, all of the mice developed both IgM and IgG2c Abs to AAV (Figures 4F and 4G). This contrasts with experiments described above with a lower initial vector dose, in which only IgM but not IgG2c development was observed in pre-IS-treated animals (compare to Figures 3D and 3E).
Circulating hFIX levels were significantly lower in pre-α-CD20 (5.00 ± 1.73 μg/mL)-treated groups compared to the control group receiving AAV8-FIX only (9.72 ± 0.41 μg/mL) (Figure 4H) and were undetectable in no-IS and pre-α-BAFF-treated groups. Circulating hFIX levels were reduced by only ∼25% in pre-α-CD20+Rapa (7.65 ± 1.09 μg/mL) and pre-α-CD20+α-BAFF (7.02 ± 1.61 μg/mL) compared to only the AAV8-FIX control group (not statistically significant; Figures 4H and S3E). These differences in the immune responses compared to results with a lower initial vector dose demonstrate that not only the timing of IS treatments but also the initial vector dose play an important role in determining the efficacy and durability of these treatments.
Extending the α-BAFF treatment prevents Ab responses and enhances re-administration efficacy
We have found substantial increases in systemic BAFF levels following α-CD20 treatment, which is favorable to Ab formation. Measurements of BAFF levels at 4- and 8-week time points following different IS treatments in the high-dose vector groups showed that pre-α-CD20 (32.42 ± 2.28 and 17.57 ± 3.83 ng/mL) and pre-α-CD20+Rapa (55.16 ± 1.60 and 43.24 ± 10.58 ng/mL) treatments resulted in significantly higher BAFF levels compared to the no-IS group (4.120 ± 0.45 and 5.57 ± 0.58 ng/mL) (Figure 5A). However, in the pre-α-CD20+α-BAFF group, BAFF levels were very low at 4 weeks (0.33 ± 0.04 ng/mL) but significantly increased at 8 weeks (21.84 ± 2.49 ng/mL) (Figure 5A). Because we observed a correlation between increased plasma BAFF levels and Ab formation, we hypothesized that prolonged depletion of BAFF may improve the efficacy of α-CD20 and α-BAFF combination therapy. For this purpose, animals received two more maintenance doses of α-BAFF postvector dosing, as described in Figure 5B.
Figure 5.

Extending α-BAFF treatment improves the re-administration efficacy
(A) Longitudinal quantification of plasma BAFF levels for control (no-IS) and various IS treatment groups (n = 5–10/group). (B) Schematic representation for timing and dosing of α-CD20 and extended α-BAFF treatments and AAV8. AAV8 capsid-specific IgM (C) and IgG2c (D) Ab levels in plasma samples collected at different time points post primary and -secondary AAV8 administration. (E) Circulating hFIX levels at 4 weeks post-re-administration with AAV8-hFIX. Statistical significance was calculated by one-way ANOVA (Dunnett’s multiple comparisons) for (E), and two-way ANOVA for (A), (C), and (D). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
This modified regimen completely prevented IgM and IgG2c formation against AAV8 capsid (Figures 5C and 5D) and also reduced CD8+ T cell responses against the transgene product, resulting in sustained OVA expression (Figures S4A–S4C). The secondary dosing of AAV8-hFIX at 8 weeks showed the development of a de novo IgM response against AAV8 capsid, but no detectable levels of IgG2c (Figures 5C and 5D). Interestingly, not only was re-administration 100% successful in the pre-α-CD20+Ext (extended) α-BAFF treatment group but also the resulting hFIX expression levels were 2.5-fold higher (25.80 ± 1.37 μg/mL) compared to AAV8 FIX controls (10.00 ± 0.96 μg/mL) (Figure 5E). Furthermore, the treated animals maintained the higher FIX expression levels 4 months post-AAV8-hFIX administration compared to the control groups (Figure S4D). On independently repeating this experiment, re-administration was 100% successful, and we again found significantly increased (albeit more modest) expression of FIX in pre-α-CD20+Ext α-BAFF-treated animals (1.4-fold higher levels; Figure S4E). However, this did not correlate with increased liver transduction (Figure S4F). Overall, these results demonstrated that maintaining lower BAFF levels along with B cell depletion prevents immune responses during initial vector dosing, therefore allowing for efficient re-administration.
Combination treatment with α-CD20 and extended α-BAFF allows for efficient re-administration following immune cell repopulation
In the experiments outlined above, re-administration was performed at a time point when the immune compartments affected by the IS protocols were not completely repopulated with immune cells. Therefore, we decided to evaluate re-administration at a later time point, when the B cell population was completely reconstituted (Figure S5A), as outlined in Figure 6A. All of the experimental groups showed similar levels of transgene expression from the first vector administration, 1 × 1011 vg AAV8-OVA (Figure 6B). We again did not observe CD8+ T cell responses against OVA with this dosing regimen (data not shown). Analysis of AAV8-specific Ab responses showed an increase in IgM responses over time in pre-α-CD20 and pre-α-CD20+Rapa groups, whereas IgG2c formation was not detected (Figures 6C and 6D). Moreover, pre-α-CD20+α-BAFF treatment prevented Ab responses to AAV8 (Figures 6C and 6D). In the pre-α-CD20 treatment group, the increase in IgM was observed up to 12 weeks (Figure 6C). However, in the pre-α-CD20+Rapa group, IgM titers continued to increase over the entire time course, with 60% of animals forming IgM against capsid. Average titers were ∼8-fold higher than for the no-IS group, in which only a single animal still had detectable IgM at the time point of vector re-administration (Figure 6C).
Figure 6.

Combination treatment with α-CD20 and extended α-BAFF allows re-administration following immune compartment repopulation
(A) Schematic outline of IS pretreatment regimens and AAV8 dosing. (B) Circulating plasma levels of OVA at 4 weeks post-AAV8-OVA administration (n = 5–10/group). AAV8 capsid-specific IgM (C) and IgG2c (D) Ab levels in plasma samples collected at different time points prior to AAV8-hFIX re-administration. AAV8 capsid-specific IgM (E) and IgG2c (F) Ab levels in plasma samples collected at different time points post-AAV8-hFIX re-administration. (G) Circulating hFIX levels at 4 weeks post-AAV8-hFIX re-administration. Statistical significance was calculated by one-way ANOVA (Dunnett’s multiple comparisons) for (B) and (G), and two-way ANOVA for (C)–(F). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
Before re-administration, we analyzed immune cell populations (at 12 and 14 weeks postinitial dosing). We found that B cell frequencies approached normal by week 14 (Figures S5A and S5B). Similarly, T cell, dendritic cell (DC), neutrophil, and monocyte frequencies were comparable to non-IS mice by week 14 (Figures S5B–S5G). Based on these observations, we decided to re-administer 1 × 1011 vg/mouse AAV8-hFIX at the 15-week time point. Since no IS was used during re-administration, this second vector dosing triggered a de novo immune response in the pre-α-CD20+Ext α-BAFF group (Figures 6E and 6F). AAV8-specific Abs (IgM and IgG2c) initially showed no significant differences among experimental groups following re-administration. However, control animals without initial IS eventually developed IgG2c titers that were significantly higher than for the other groups (Figures 6E and 6F). Levels of transgene expression from the second vector administration (hFIX) were similar for pre-α-CD20+Ext α-BAFF group (16.05 ± 0.47 μg/mL, with successful re-administration in 100% of animals) compared to control mice that received only the AAV-FIX vector (19.36 ± 2.71 μg/mL) (Figure 6G). Subsequent expression of hFIX in pre-α-CD20+Ext α-BAFF was stable over time (Figure S5I). In contrast, re-administration was much less efficient in the other IS groups, which achieved hFIX expression in only 38% of mice in pre-α-CD20 (average 2.56 ± 1.31 μg/mL) and 66% in pre-α-CD20+Rapa (average 5.30 ± 1.57 μg/mL) groups (Figure 6G), with substantially lower levels. This correlated with IgM levels before to re-administration (Figure S5H). In summary, these results demonstrate that combination treatment with α-CD20 and α-BAFF allows for efficient re-administration of vector at a time point when the immune system has recovered from immunodepleting treatments.
Presence of Abs against capsid alters AAV8 uptake by immune cells
We hypothesized that preexisting Abs against capsid would alter the uptake of viral particles by immune cells. To study this in detail, control mice (no-IS) or mice pretreated with α-CD20+Ext α-BAFF protocol were injected initially with 1 × 1011 vg/mouse AAV8-OVA followed by re-administration with Atto-590 labeled AAV8 (2 × 1011 vg/mouse) 12 weeks later. Another control group was naive mice receiving the second vector only. Spleens and liver tissue were harvested 2 h later to identify Atto-590+ immune cell types.
We first confirmed in vitro in 2V6.11 cells that Atto-590 labeling does not affect cellular uptake (Figure S6). Next, among splenocytes, we analyzed B cells and other immune cells for AAV8 uptake (Figure S7A). Frequencies of B cell subsets were similar between groups, indicating complete B cell repopulation (Figures S7B and S7C). We did not find evidence for AAV8 uptake by B cells, with the exception of transitional B cells, ∼5% of which were Atto-590+ in the no-IS group (while IS treated and naive animals showed no uptake; Figure S7D). Importantly, we saw a complete absence of AAV-specific plasma cells in the bone marrow and spleen by ELISpot even following B cell repopulation (Figure S7E).
In contrast, substantial uptake of AAV particles was detected in splenic neutrophils (23.61 ± 3.5%) and marginal zone macrophages (MZMs; 11.72 ± 2.4%) of mice in the no-IS group, which was much reduced in IS-treated and naive mice (Figure 7A). A small proportion of monocytes (1.75 ± 0.2%) and metallophilic macrophages (4.54 ± 1.4%) were also Atto-590+ in the no-IS group. These results illustrate that the presence of Abs against capsid enhances uptake by myeloid cells. Conventional DCs in all three experimental groups took up AAV particles at low frequencies (0.5%–1% Atto-590+).
Figure 7.

B cell-depleting IS therapy mitigates AAV8 uptake
Control mice (no-IS) or mice pretreated with α-CD20+Ext α-BAFF initially received 1 × 1011 vg/mouse AAV8-OVA, followed by re-administration with AAV8-Atto-590 12 weeks later. Naive mice received AAV8-Atto-590 only (n = 4–6/group). (A) Proportion of neutrophils, monocytes, MZMs, metallophilic macrophages (MMM) and DCs from splenocytes that take up AAV8-Atto-590. (B) Proportion of neutrophils, monocytes, Kupffer cells, and DCs from the liver that take up AAV8-Atto-59. Statistical significance was calculated by 1-way ANOVA (Dunnett’s multiple comparisons). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
Next, we assessed AAV uptake by hepatic immune cells. Similar to spleen, the majority of Atto-590+ cells in the no-IS group were neutrophils and Kupffer cells/resident macrophages (Figure 7B). As we observed in the spleen, there was a reduction in the uptake of AAV8 in these cells from animals lacking anti-capsid Abs. Therefore, preexisting Abs enhance the uptake of AAV particles by splenic and hepatic neutrophils and macrophages, which is prevented by IS (combination treatment with α-CD20 and α-BAFF) at the time of first exposure to AAV capsid, creating an environment during re-administration that mimics vector administration in naive animals.
Discussion
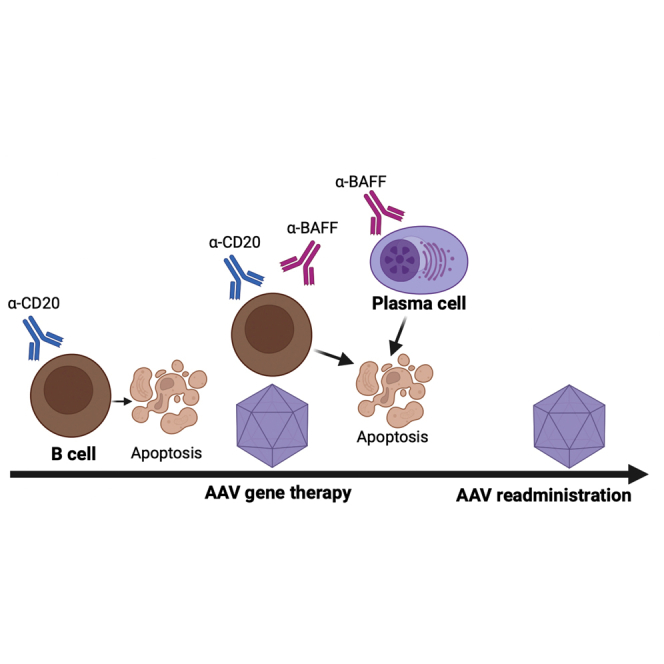
AAV re-administration to achieve desired therapeutic protein levels is challenging due to the development of immune responses against viral capsid and the encoded transgene product during the initial vector administration. In this study, we have used an IS regimen that greatly targets the B cell compartment of the immune system to prevent adaptive immune responses during initial vector administration. This combination therapy uses α-CD20 to deplete the majority of the B cells and α-BAFF to deplete plasma cells/plasmablasts by dissipation of their survival factor.24 We have systematically adapted our combination drug regimen and schedule to completely inhibit immune responses during initial vector dosing and allow for vector re-administration without compromising therapeutic transgene expression levels (Figure 8).
Figure 8.

Proposed mechanism of AAV-specific immune responses in untreated and α-CD20 and α-BAFF-treated animals
AAV8 administration in naive animals without any IS (no-IS) results in AAV uptake, presentation, T cell help, and activation of adaptive immune responses, resulting in the formation of high-titer, affinity-matured AAV capsid-specific Abs, which can prevent vector redosing. IS pretreatment with α-CD20+Ext α-BAFF during initial AAV administration prevents the selection of high-affinity B cell clones and further deprives B cells and plasma cells of the B cell survival cytokine BAFF, thereby enabling vector redosing. The IS effect is transient, and the repopulated B cells initiate a de novo adaptive immune response to the re-administered AAV vector.
Several IS drug treatments have been used in clinical trials to preserve transgene expression following initial dosing such as corticosteroids, rapamycin, mycophenolate mofetil, rituximab, calcineurin inhibitors, eculizumab.25 However, achieving re-administration using combinations of these immunosuppressants has shown mixed responses. In preclinical studies, the use of tolerogenic nanoparticles encoding rapamycin has shown success in redosing with the same AAV serotype and improved transgene expression following both doses compared to control groups.19,26,27 However, this formulation is not yet clinically approved. A combination of rapamycin and ibrutinib has been reported to block IgG formation against capsid.18 Use of abatacept (CTLA4-Ig) with a low initial dose (5 × 1011 vg/kg) permitted re-administration with a subsequent higher dose of 4 × 1012 vg/kg.28 However, re-administration was performed 21 days following the initial low vector dose; hence, the efficacy of the treatment is not known following clinically relevant vector dosing and at a later time point.
A combination of anti-CD20 and rapamycin has shown promising results in preventing both capsid and transgene-specific immune responses in preclinical models and initial human experience (this study was registered at Clinicaltrials.gov: NCT00976352).20,29,30 This combination is being tested for re-administration of AAV9 in gene therapy for Pompe disease and has also shown some positive outcomes for immune tolerance induction to FVIII protein in hemophilia A patients.31 Nonetheless, rapamycin has also been shown to inhibit B cell class switching, producing a skewed Ab pattern of IgG and IgM specificities.32 In our study, we observed a prolonged IgM response following rapamycin treatment, with the ability to neutralize AAV. Moreover, we find that α-CD20 treatment alone or in combination with rapamycin leads to an increase in circulating BAFF levels, which increases the incidence of IgM development, thereby reducing treatment efficacy. Several studies have shown increased BAFF levels in autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and progressive systemic sclerosis33,34,35,36—in some cases correlating with autoantibody development.37 Some studies suggested that increased BAFF levels following α-CD20 treatment may be responsible for relapses due to the development of delayed autoreactive B cells.38,39 Therefore, monitoring plasma BAFF levels and targeting BAFF expression is a potential approach to improve outcomes for α-CD20-mediated B cell depletion. Importantly, the combination of α-CD20 and α-BAFF does not affect the survival of non-B lineage cells, as our findings (Figure S5) and that of others show.40,41
Our study also demonstrated that the timing of IS affects the efficacy of the treatment. Concurrent B cell targeting IS treatments unexpectedly enhanced the cellular immune response against the transgene product. This is probably due to inflammation induced by natural killer cell activation and Ab-dependent cellular cytotoxicity caused by B cell depletion,42 although further studies are required to confirm this. Concurrent rapamycin treatment blocked B cell class switching and enhanced IgM production against AAV capsid. On the contrary, IS before vector administration prevented skewing of the immune response, resulting in reduced capsid and transgene-specific immune responses. A previous study has similarly reported the effect of timing of IS using α-thymocyte globulin (ATG) in a NHP model.43
Our results show that combining α-CD20 with α-BAFF prevents not only IgG but also IgM formation against AAV capsid. Furthermore, extending the duration of α-BAFF treatment prevented de novo Ab formation. Importantly, both of these Abs are approved for clinical trials and can also be evaluated in NHPs, facilitating the translation of the approach. In high-dose systemic delivery of AAV for neuromuscular disorders, complement activation has been observed in multiple patients, resulting in various immunotoxicities.44,45 Therefore, our protocol may be useful not only to enable vector re-administration but also to prevent complement activation, which has been found to largely be Abdependent in response to AAV.44,46,47,48 Similarly, Salabarria et al. demonstrated in study participants receiving systemic high-dose AAV9 gene therapy that α-CD20 and rapamycin IS therapy was able to prevent Ab-mediated induction of the classical pathway and amplification via the alternative pathway of complement activation.30 The α-CD20/α-BAFF protocol may also allow spreading the vector dose over time to reduce potential toxicities such as liver toxicity associated with a single high dose. It is intriguing that vector re-administration after α-CD20/α-BAFF treatment at a time point when the immune system was not yet fully reconstituted modestly enhanced efficacy compared to naive controls. Although the underlying mechanism remains to be uncovered, this phenomenon could be exploited to achieve therapy at reduced vector doses. However, the effectiveness of this protocol may still be limited by vector dose (>1014 vg/kg in current trials for neuromuscular disorders, whereas the dose used here was ∼4 × 1012 vg/kg). In the absence of NAb formation, AAV particles capable of triggering B cell activation may persist longer, so that adjustments to the protocol may be needed for higher vector doses. At the same time, more recently developed capsids may direct the efficacy of systemic AAV delivery at much reduced vector doses.2,49
In conclusion, initiating B cell depletion combined with depletion of the B cell survival factor BAFF before vector administration represents a promising approach to prevent NAb upon systemic AAV delivery, thereby enabling effective vector re-administration. A short maintenance course of α-BAFF is required after B cell-depleting Ab is discontinued to prevent not only IgG but also IgM formation. Such an optimized B cell-targeted protocol also reduces the risk of immune responses against the transgene product.
Materials and methods
Mice
Male 6- to 8-week-old C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Animals were housed at Indiana University, Indianapolis, and treated under Institutional Animal Care and Use Committee-approved protocol 21017.
AAV8 viral vectors
AAV-EF1α-OVA, AAV-ApoE/hAAT-hFIX, and scAAV-RLuc vectors were used for this study. Vector genomes were packaged into AAV8 capsids by triple transfection of HEK293 cells, and viral particles were purified by iodixanol gradient as previously described.50 Vector titers were determined by real-time qPCR.51
Cellular uptake studies were performed with Atto-590-labeled AAV8. Briefly, AAV8 was column purified as previously described52 and reacted with Atto-590 NHS ester in PBS buffer pH 8, with a molar ratio of 10,000 molecules/vg. The reaction was protected from light and incubated overnight at 4°C with gentle shaking. The labeled virus was filtered and purified by extensive buffer exchange (∼10 rounds) with 1× PBS buffer pH 7.2, using ultracentrifugal filter 100K molecular weight cutoff (Sigma-Aldrich, St. Louis, MO). Chemically labeled AAV8-Atto-590 was titrated using real-time qPCR.
All of the vectors were free of endotoxins and administered intravenously (i.v.). The doses and timing of administration are described in the results.
Reagents
α-mCD20 IgG2a subtype (clone 18B12) was purified from transfected HEK293 cells (ATUM, Newark, CA). Rapamycin was purchased from LC Laboratories (Woburn, MA). α-mBAFF IgG1 subtype (clone Sandy-2) was purchased from AdipoGen Life Sciences (San Diego, CA). Atto-590 (catalog no. 70425) was purchased from Sigma-Aldrich. Ponasterone A, an analog of insect steroid hormone ecdysone, was purchased from Invitrogen (Carlsbad, CA).
IS treatments
For B cell depletion, mice were treated i.v. with 2 doses of 200 μg (∼8 mg/kg) α-CD20, spaced 3 weeks apart. For T cell suppression, 100 μg (∼4 mg/kg) of rapamycin was administrated by oral gavage 3×/week for 5 weeks. To deplete mature B cells, 2 doses of 60 μg (∼2.4 mg/kg) α-BAFF were administered by intraperitoneal injection at an interval of 10 days. For extended α-BAFF treatments, 2 maintenance doses of 40 μg (∼1.6 mg/kg) were administered at an interval of 2 weeks. Schematics representing the timing of administration and type of treatment are indicated in the figures.
Detection of transgene and AAV8-specific Abs
Antibody assays were performed using ELISA and in vitro neutralization assays. α-AAV8-specific IgG1, IgG2c, and IgM levels were deermined by ELISA. Briefly, microtiter plates were coated overnight with 5 × 1010 vg/mL AAV8. Plasma samples were used at 1:40 or 1:60 dilutions with serial dilutions of purified murine IgG1, IgG2c, or IgM as standards. For detection, horseradish peroxidase (HRP)-conjugated α-IgG1/IgG2c/IgM Abs (Southern Biotech, Birmingham, AL) were used. For transgene specific Abs, microtiter plates were coated with either 10 μg/mL OVA or 0.5 μg/μL hFIX and assayed for IgG1 and IgG2c Abs as described above.
To determine the level of NAbs to AAV8, plasma samples were added to 2V6.11 cells, as described earlier.53 The 2V6.11 cells indelibly express human adenovirus E4 protein under control of the ecdysone promoter.54 Cells were seeded (20,000/well) in optical polymer base 96-well tissue culture plates and incubated for 24 h at 37°C in DMEM media containing 10% fetal bovine serum (FBS) and 1 μg/mL ponasterone A. The next day, before the neutralization assay, plasma samples were heat inactivated at 50°C for 30 min. Plasma serial dilutions were prepared in serum-free media ranging from 1:5 to 1:1,280, mixed with AAV8-RLuc (5 × 103 MOI) and incubated for 1 h at 37°C. Culture media was removed from the 2V6.11 cells and a mixture of plasma:AAV8-RLuc was added to the cells. Cells were incubated at 37°C for 2 h, followed by the addition of DMEM containing 5% FBS and 1 μg/mL ponasterone A. Cells were cultured for 24 h at 37°C before the Luc assay. Renilla-Glo luciferase reagent (Promega, Madison, WI) was mixed with the buffer and added to the cells. The plate was incubated in the dark for 10 min at room temperature for complete cell lysis, and relative luminescence units (RLUs) were measured using Synergy LX reader (Biotek, Winooski, VT). Each sample was assessed in triplicate, and the results were normalized to a 100% control (preincubation of vector without serum). A 0% control (cells not transduced with virus) was also included in each plate for the background analysis. Transduction efficiency was calculated according to the formula [mean RLU-sample/mean RLU-100% reference value] × 100.
Detection of transgene products
Plasma levels of OVA and FIX transgene products were measured by ELISA. For OVA detection, microtiter plates were coated overnight with affinity-purified rabbit OVA Ab (1:80,000 dilution; Sigma catalog no. C-6534) and mouse plasma samples were applied at 1:40 or 1:60 dilution. For detection, HRP-conjugated rabbit OVA Ab (1:10,000 dilution; Rockland Immunochemicals, Pottstown, PA, catalog no. 200-4333) was used. Serial dilutions of endotoxin-free OVA protein from InvivoGen (San Diego, CA) were used to generate a standard curve.
For determining hFIX plasma concentrations, serial dilutions of control human plasma, Trinicheck level 1 (Diagnostica Stago, Parsippany, NJ) was used to form standards. Briefly, microtiter plates were coated with affinity-purified rabbit α-hFIX (1:850 dilution; Sigma anti-hFIX clone HIX-1 catalog no. F2645) overnight and blocked with 6% nonfat dry milk in 1× PBS/0.05% Tween 20. Plasma samples were applied at a 1:60 dilution and incubated overnight. hFIX level was detected with HRP-conjugated goat anti-hFIX (1:2,333 dilution, GAFIX-APHRP, Affinity Biologicals, Ancaster, ON, Canada).
Immunohistochemistry
Liver tissues were snap-frozen in liquid nitrogen in optimal cutting temperature embedding media (Fisher Scientific, Waltham, MA). Frozen liver tissues were sectioned (∼10 μm), mounted on polylysine-coated slides, and immunohistochemistry performed, as previously described.55
In brief, cryosections were fixed in acetone at room temperature, blocked with 5% donkey serum (Sigma-Aldrich). Goat anti-hFIX (1:400 dilution; Affinity Biologicals) or rabbit α-OVA (1:200 dilution; Abcam, Boston, MA) in 2% donkey serum was added for 90 min. After washing, tissue sections were incubated with secondary Ab Alexa Fluor 568 donkey α-goat IgG for hFIX (1:100 dilution; Invitrogen) or Alexa Fluor 647 donkey α-rabbit (1:200 dilution; Invitrogen). Sections were mounted with ProLong Diamond Antifade Mountant (Life Technologies, Carlsbad, CA) containing DAPI. Fluorescence microscopy was performed on a Zeiss Axio Observer microscope (Carl Zeiss, White Plains, NY).
Flow cytometry analysis for AAV8-Atto-590 uptake
To understand the uptake of AAV8 vector by different immune cell types, we used Atto-590-labeled AAV8. For this, three study groups of C57BL/6 animals were analyzed: naive, preimmunized with AAV8-OVA, and pre-α-CD20+Ext α-BAFF-treated preimmunized animals. These groups were i.v. injected with 2 × 1011 vg/mouse of AAV8-Atto-590, and 2 h later, spleen and liver tissue were harvested. Spleen tissue was homogenized and passed through a 70-μm cell strainer (BD Falcon, San Jose, CA) to obtain a single-cell suspension. Liver tissue was homogenized and treated with 0.5 mg/mL collagenase for 10 min at 37°C. Following incubation, liver cell suspensions were washed with PBS containing 4% FBS and 0.5 mM EDTA and centrifuged at 50 × g for 3 min to sediment hepatocytes. Nonparenchymal cells (NPCs) were collected and washed again to remove any residual hepatocytes.
To assess AAV8-Atto590 uptake, splenocytes and NPCs were incubated with mouse anti-CD16/32 (BioLegend, San Diego, CA) to block Fc receptors and surface stained with fluorochrome-conjugated mAbs for 20 min at 4°C, followed by PBS washes. To identify the myeloid cell population, cells were stained with PDCA-PERCP efluor710 (eBio927, Invitrogen), CD11b-BV605 (M1/70, BioLegend), CD11c-BV421 (N418, BioLegend), Ly6c-APC/Fire 750 (HK1.4, BioLegend), Ly6G-AF647 (1A8, BioLegend), F4/80-BV786 (BM8, BioLegend), and CD45-AF488 (30-F11, BioLegend) for liver or TIM4-AF488 (RMT4-54, Invitrogen) for spleen. For analysis of B cell compartments, splenocytes were stained with CD19-APC efluor780 (eBio1D3, Invitrogen), TACI-BV421 (8F10, BD Biosciences, Franklin Lakes, NJ), CD138-BV711 (281-2, BioLegend), CD21-AF647 (7E9, BioLegend), and CD23-PECy7 (B3B4, BioLegend). Flow cytometry data were collected using BD LSR Fortessa and analyzed with FCS Express 7 (De Novo Software Solutions, Pasadena, CA).
ELISpot assay
The frequency of AAV8-specific Ab secreting cellswas quantified by a B cell ELISpot assay, as described previously.56 Briefly, red blood cells from spleen or bone marrow single-cell suspensions were lysed (eBioscience, San Diego, CA) and double filtered through 70-μm cell strainers. Cells (2 × 106) were seeded in duplicate in RPMI 1640 plus 10% FBS onto B cell ELISpot-specific plates (Millipore, Burlington, MA) precoated with AAV8 (5 × 1010 vg/mL). After overnight incubation at 37°C in 5% CO2, cells were removed by washing in PBS plus 0.5% Tween 20. Goat α-mIgG2c-HRP (Southern Biotech) was used for detection, followed by the addition of AEC substrate (BD Biosciences) for spot development. Plates were analyzed using the ImmunoSpot system (Cellular Technology, Cleveland, OH).
Data and code availability
The datasets presented in this study will be made available upon request to the corresponding author. All of the Ab clones used in this study have been described.
Acknowledgments
This work was supported by NIH grants R01HL131093, to C.T. and R.W.H.; by P01HL160472, to R.W.H.; by R01 AI51390, Indiana Collaborative Initiative for Talent Enrichment funds (provided by Lilly Endowment), and the Riley Children's Foundation, to R.W.H.; and by R01 AI177600, to D.D. and R.W.H., and R21 HL170146 to M.B. The authors thank the members of the Indiana University Melvin and Bren Simon Cancer Center Flow Cytometry Resource Facility for their outstanding technical support. The Indiana University Melvin and Bren Simon Comprehensive Cancer Center Flow Cytometry Resource Facility (FCRF) is funded in part by NIH, National Cancer Institute grant P30CA082709, and National Institute of Diabetes and Digestive and Kidney Diseases grant U54DK106846 (Cooperative Center of Excellence in Hematology). The FCRF is further supported in part by NIH instrumentation grant 1S10D012270.
Author contributions
J.R., S.R.P.K., M.M.-M., K.Y., and A.K.L. performed the experiments; J.R., R.W.H., S.R.P.K., D.M.M., and M.B. designed the experiments; J.R., R.W.H., D.M.M., D.D., C.T., B.J.B., M.C., and M.B. interpreted the data and wrote the manuscript; M.B., R.W.H., and C.T. conceptualized the study; R.W.H. and M.B. supervised the study; All of the authors read and approved the final manuscript.
Declaration of interests
R.W.H. serves on the scientific advisory boards and committees of Regeneron Pharmaceuticals, Pfizer, Biomarin, Spark Therapeutics, Hoffman-La Roche, and Prevail Therapeutics. D.D. serves on the scientific advisory boards of Solid Biosciences and Sardocor Corporation. M.B. serves on the steering committee of Pfizer.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101216.
Supplemental information
References
- 1.Becker J., Fakhiri J., Grimm D. Fantastic AAV Gene Therapy Vectors and How to Find Them-Random Diversification, Rational Design and Machine Learning. Pathogens. 2022;11:756. doi: 10.3390/pathogens11070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rana J., Marsic D., Zou C., Muñoz-Melero M., Li X., Kondratov O., Li N., de Jong Y.P., Zolotukhin S., Biswas M. Characterization of a Bioengineered AAV3B Capsid Variant with Enhanced Hepatocyte Tropism and Immune Evasion. Hum. Gene Ther. 2023;34:289–302. doi: 10.1089/hum.2022.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendell J.R., Al-Zaidy S.A., Rodino-Klapac L.R., Goodspeed K., Gray S.J., Kay C.N., Boye S.L., Boye S.E., George L.A., Salabarria S., et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021;29:464–488. doi: 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pierce G.F., Herzog R.W. Two gene therapies for hemophilia available: Now what? Mol. Ther. 2023;31:919–920. doi: 10.1016/j.ymthe.2023.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herzog R.W., VandenDriessche T., Ozelo M.C. First hemophilia B gene therapy approved: More than two decades in the making. Mol. Ther. 2023;31:1–2. doi: 10.1016/j.ymthe.2022.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.VandenDriessche T., Pipe S.W., Pierce G.F., Kaczmarek R. First conditional marketing authorization approval in the European Union for hemophilia "A" gene therapy. Mol. Ther. 2022;30:3335–3336. doi: 10.1016/j.ymthe.2022.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herzog R.W., Bricker-Anthony C. ASGCT 2023-Gene therapy is becoming medicine. Mol. Ther. 2023;31:1859. doi: 10.1016/j.ymthe.2023.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han S.O., Li S., McCall A., Arnson B., Everitt J.I., Zhang H., Young S.P., ElMallah M.K., Koeberl D.D. Comparisons of Infant and Adult Mice Reveal Age Effects for Liver Depot Gene Therapy in Pompe Disease. Mol. Ther. Methods Clin. Dev. 2020;17:133–142. doi: 10.1016/j.omtm.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kok C.Y., Cunningham S.C., Carpenter K.H., Dane A.P., Siew S.M., Logan G.J., Kuchel P.W., Alexander I.E. Adeno-associated virus-mediated rescue of neonatal lethality in argininosuccinate synthetase-deficient mice. Mol. Ther. 2013;21:1823–1831. doi: 10.1038/mt.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L., Wang H., Bell P., McMenamin D., Wilson J.M. Hepatic gene transfer in neonatal mice by adeno-associated virus serotype 8 vector. Hum. Gene Ther. 2012;23:533–539. doi: 10.1089/hum.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butterfield J.S.S., Yamada K., Bertolini T.B., Syed F., Kumar S.R.P., Li X., Arisa S., Piñeros A.R., Tapia A., Rogers C.A., et al. IL-15 blockade and rapamycin rescue multifactorial loss of factor VIII from AAV-transduced hepatocytes in hemophilia A mice. Mol. Ther. 2022;30:3552–3569. doi: 10.1016/j.ymthe.2022.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George L.A. Hemophilia A Gene Therapy - Some Answers, More Questions. N. Engl. J. Med. 2023;388:761–763. doi: 10.1056/NEJMe2212347. [DOI] [PubMed] [Google Scholar]
- 13.Handyside B., Ismail A.M., Zhang L., Yates B., Xie L., Sihn C.R., Murphy R., Bouwman T., Kim C.K., De Angelis R., et al. Vector genome loss and epigenetic modifications mediate decline in transgene expression of AAV5 vectors produced in mammalian and insect cells. Mol. Ther. 2022;30:3570–3586. doi: 10.1016/j.ymthe.2022.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shirley J.L., de Jong Y.P., Terhorst C., Herzog R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020;28:709–722. doi: 10.1016/j.ymthe.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verdera H.C., Kuranda K., Mingozzi F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulz M., Levy D.I., Petropoulos C.J., Bashirians G., Winburn I., Mahn M., Somanathan S., Cheng S.H., Byrne B.J. Binding and neutralizing anti-AAV antibodies: Detection and implications for rAAV-mediated gene therapy. Mol. Ther. 2023;31:616–630. doi: 10.1016/j.ymthe.2023.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mingozzi F., Hasbrouck N.C., Basner-Tschakarjan E., Edmonson S.A., Hui D.J., Sabatino D.E., Zhou S., Wright J.F., Jiang H., Pierce G.F., et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. 2007;110:2334–2341. doi: 10.1182/blood-2007-03-080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiang Z., Kuranda K., Quinn W., Chekaoui A., Ambrose R., Hasanpourghai M., Novikov M., Newman D., Cole C., Zhou X., et al. The Effect of Rapamycin and Ibrutinib on Antibody Responses to Adeno-Associated Virus Vector-Mediated Gene Transfer. Hum. Gene Ther. 2022;33:614–624. doi: 10.1089/hum.2021.258. [DOI] [PubMed] [Google Scholar]
- 19.Meliani A., Boisgerault F., Hardet R., Marmier S., Collaud F., Ronzitti G., Leborgne C., Costa Verdera H., Simon Sola M., Charles S., et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018;9:4098. doi: 10.1038/s41467-018-06621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corti M., Cleaver B., Clément N., Conlon T.J., Faris K.J., Wang G., Benson J., Tarantal A.F., Fuller D., Herzog R.W., Byrne B.J. Evaluation of Readministration of a Recombinant Adeno-Associated Virus Vector Expressing Acid Alpha-Glucosidase in Pompe Disease: Preclinical to Clinical Planning. Hum. Gene Ther. Clin. Dev. 2015;26:185–193. doi: 10.1089/humc.2015.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biswas M., Palaschak B., Kumar S.R.P., Rana J., Markusic D.M. B Cell Depletion Eliminates FVIII Memory B Cells and Enhances AAV8-coF8 Immune Tolerance Induction When Combined With Rapamycin. Front. Immunol. 2020;11:1293. doi: 10.3389/fimmu.2020.01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S.R.P., Hoffman B.E., Terhorst C., de Jong Y.P., Herzog R.W. The Balance between CD8(+) T Cell-Mediated Clearance of AAV-Encoded Antigen in the Liver and Tolerance Is Dependent on the Vector Dose. Mol. Ther. 2017;25:880–891. doi: 10.1016/j.ymthe.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S.R.P., Biswas M., Cao D., Arisa S., Muñoz-Melero M., Lam A.K., Piñeros A.R., Kapur R., Kaisho T., Kaufman R.J., et al. TLR9-independent CD8(+) T cell responses in hepatic AAV gene transfer through IL-1R1-MyD88 signaling. Mol. Ther. 2024;32:325–339. doi: 10.1016/j.ymthe.2023.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doshi B.S., Rana J., Castaman G., Shaheen M.A., Kaczmarek R., Butterfield J.S., Meeks S.L., Leissinger C., Biswas M., Arruda V.R. B cell-activating factor modulates the factor VIII immune response in hemophilia A. J. Clin. Invest. 2021;131 doi: 10.1172/JCI142906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prasad S., Dimmock D.P., Greenberg B., Walia J.S., Sadhu C., Tavakkoli F., Lipshutz G.S. Immune Responses and Immunosuppressive Strategies for Adeno-Associated Virus-Based Gene Therapy for Treatment of Central Nervous System Disorders: Current Knowledge and Approaches. Hum. Gene Ther. 2022;33:1228–1245. doi: 10.1089/hum.2022.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ilyinskii P.O., Michaud A.M., Rizzo G.L., Roy C.J., Leung S.S., Elkins S.L., Capela T., Chowdhury A., Li L., Chandler R.J., et al. ImmTOR nanoparticles enhance AAV transgene expression after initial and repeat dosing in a mouse model of methylmalonic acidemia. Mol. Ther. Methods Clin. Dev. 2021;22:279–292. doi: 10.1016/j.omtm.2021.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ilyinskii P.O., Michaud A.M., Roy C.J., Rizzo G.L., Elkins S.L., Capela T., Chowdhury A.C., Leung S.S., Kishimoto T.K. Enhancement of liver-directed transgene expression at initial and repeat doses of AAV vectors admixed with ImmTOR nanoparticles. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abd0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frentsch M., Japp A.S., Dingeldey M., Matzmohr N., Thiel A., Scheiflinger F., Reipert B.M., de la Rosa M. Blockade of the costimulatory CD28-B7 family signal axis enables repeated application of AAV8 gene vectors. J. Thromb. Haemost. 2020;18:1075–1080. doi: 10.1111/jth.14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corti M., Elder M., Falk D., Lawson L., Smith B., Nayak S., Conlon T., Clément N., Erger K., Lavassani E., et al. B-Cell Depletion is Protective Against Anti-AAV Capsid Immune Response: A Human Subject Case Study. Mol. Ther. Methods Clin. Dev. 2014;1 doi: 10.1038/mtm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salabarria S.M., Corti M., Coleman K.E., Wichman M.B., Berthy J.A., D'Souza P., Tifft C.J., Herzog R.W., Elder M.E., Shoemaker L.R., et al. Thrombotic microangiopathy following systemic AAV administration is dependent on anti-capsid antibodies. J. Clin. Invest. 2024;134 doi: 10.1172/JCI173510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doshi B.S., Raffini L.J., George L.A. Combined anti-CD20 and mTOR inhibition with factor VIII for immune tolerance induction in hemophilia A patients with refractory inhibitors. J. Thromb. Haemost. 2020;18:848–852. doi: 10.1111/jth.14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keating R., Hertz T., Wehenkel M., Harris T.L., Edwards B.A., McClaren J.L., Brown S.A., Surman S., Wilson Z.S., Bradley P., et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat. Immunol. 2013;14:1266–1276. doi: 10.1038/ni.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groom J., Kalled S.L., Cutler A.H., Olson C., Woodcock S.A., Schneider P., Tschopp J., Cachero T.G., Batten M., Wheway J., et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren's syndrome. J. Clin. Invest. 2002;109:59–68. doi: 10.1172/JCI14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohata J., Zvaifler N.J., Nishio M., Boyle D.L., Kalled S.L., Carson D.A., Kipps T.J. Fibroblast-like synoviocytes of mesenchymal origin express functional B cell-activating factor of the TNF family in response to proinflammatory cytokines. J. Immunol. 2005;174:864–870. doi: 10.4049/jimmunol.174.2.864. [DOI] [PubMed] [Google Scholar]
- 35.Stohl W., Metyas S., Tan S.M., Cheema G.S., Oamar B., Xu D., Roschke V., Wu Y., Baker K.P., Hilbert D.M. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum. 2003;48:3475–3486. doi: 10.1002/art.11354. [DOI] [PubMed] [Google Scholar]
- 36.Tan S.M., Xu D., Roschke V., Perry J.W., Arkfeld D.G., Ehresmann G.R., Migone T.S., Hilbert D.M., Stohl W. Local production of B lymphocyte stimulator protein and APRIL in arthritic joints of patients with inflammatory arthritis. Arthritis Rheum. 2003;48:982–992. doi: 10.1002/art.10860. [DOI] [PubMed] [Google Scholar]
- 37.Mariette X., Roux S., Zhang J., Bengoufa D., Lavie F., Zhou T., Kimberly R. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren's syndrome. Ann. Rheum. Dis. 2003;62:168–171. doi: 10.1136/ard.62.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thien M., Phan T.G., Gardam S., Amesbury M., Basten A., Mackay F., Brink R. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–798. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Lesley R., Xu Y., Kalled S.L., Hess D.M., Schwab S.R., Shu H.B., Cyster J.G. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 40.Ng L.G., Sutherland A.P.R., Newton R., Qian F., Cachero T.G., Scott M.L., Thompson J.S., Wheway J., Chtanova T., Groom J., et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J. Immunol. 2004;173:807–817. doi: 10.4049/jimmunol.173.2.807. [DOI] [PubMed] [Google Scholar]
- 41.Chang S.K., Mihalcik S.A., Jelinek D.F. B lymphocyte stimulator regulates adaptive immune responses by directly promoting dendritic cell maturation. J. Immunol. 2008;180:7394–7403. doi: 10.4049/jimmunol.180.11.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiner G.J. Rituximab: mechanism of action. Semin. Hematol. 2010;47:115–123. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samelson-Jones B.J., Finn J.D., Favaro P., Wright J.F., Arruda V.R. Timing of Intensive Immunosuppression Impacts Risk of Transgene Antibodies after AAV Gene Therapy in Nonhuman Primates. Mol. Ther. Methods Clin. Dev. 2020;17:1129–1138. doi: 10.1016/j.omtm.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lek A., Atas E., Hesterlee S.E., Byrne B.J., Bönnemann C.G. Meeting Report: 2022 Muscular Dystrophy Association Summit on 'Safety and Challenges in Gene Transfer Therapy'. J. Neuromuscul. Dis. 2023;10:327–336. doi: 10.3233/JND-221639. [DOI] [PubMed] [Google Scholar]
- 45.Duan D. Lethal immunotoxicity in high-dose systemic AAV therapy. Mol. Ther. 2023;31:3123–3126. doi: 10.1016/j.ymthe.2023.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Byrne B.J., Corti M., Muntoni F. Considerations for Systemic Use of Gene Therapy. Mol. Ther. 2021;29:422–423. doi: 10.1016/j.ymthe.2021.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.West C., Federspiel J.D., Rogers K., Khatri A., Rao-Dayton S., Ocana M.F., Lim S., D'Antona A.M., Casinghino S., Somanathan S. Complement Activation by Adeno-Associated Virus-Neutralizing Antibody Complexes. Hum. Gene Ther. 2023;34:554–566. doi: 10.1089/hum.2023.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emami M.R., Espinoza A., Young C.S., Ma F., Farahat P.K., Felgner P.L., Chamberlain J.S., Xu X., Pyle A.D., Pellegrini M., et al. Innate and adaptive AAV-mediated immune responses in a mouse model of Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2023;30:90–102. doi: 10.1016/j.omtm.2023.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biswas M., Marsic D., Li N., Zou C., Gonzalez-Aseguinolaza G., Zolotukhin I., Kumar S.R.P., Rana J., Butterfield J.S.S., Kondratov O., et al. Engineering and In Vitro Selection of a Novel AAV3B Variant with High Hepatocyte Tropism and Reduced Seroreactivity. Mol. Ther. Methods Clin. Dev. 2020;19:347–361. doi: 10.1016/j.omtm.2020.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooper M., Nayak S., Hoffman B.E., Terhorst C., Cao O., Herzog R.W. Improved induction of immune tolerance to factor IX by hepatic AAV-8 gene transfer. Hum. Gene Ther. 2009;20:767–776. doi: 10.1089/hum.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y.L., Wagner K., Robinson N., Sabatino D., Margaritis P., Xiao W., Herzog R.W. Optimized production of high-titer recombinant adeno-associated virus in roller bottles. Biotechniques. 2003;34:184–189. doi: 10.2144/03341dd07. [DOI] [PubMed] [Google Scholar]
- 52.Lam A.K., Zhang J., Frabutt D., Mulcrone P.L., Li L., Zeng L., Herzog R.W., Xiao W. Fast and high-throughput LC-MS characterization, and peptide mapping of engineered AAV capsids using LC-MS/MS. Mol. Ther. Methods Clin. Dev. 2022;27:185–194. doi: 10.1016/j.omtm.2022.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meliani A., Leborgne C., Triffault S., Jeanson-Leh L., Veron P., Mingozzi F. Determination of anti-adeno-associated virus vector neutralizing antibody titer with an in vitro reporter system. Hum. Gene Ther. Methods. 2015;26:45–53. doi: 10.1089/hgtb.2015.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mohammadi E.S., Ketner E.A., Johns D.C., Ketner G. Expression of the adenovirus E4 34k oncoprotein inhibits repair of double strand breaks in the cellular genome of a 293-based inducible cell line. Nucleic Acids Res. 2004;32:2652–2659. doi: 10.1093/nar/gkh593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rogers G.L., Hoffman B.E. Optimal Immunofluorescent Staining for Human Factor IX and Infiltrating T Cells following Gene Therapy for Hemophilia B. J. Genet. Syndr. Gene Ther. 2012;S1 doi: 10.4172/2157-7412.s1-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X., Moghimi B., Zolotukhin I., Morel L.M., Cao O., Herzog R.W. Immune tolerance induction to factor IX through B cell gene transfer: TLR9 signaling delineates between tolerogenic and immunogenic B cells. Mol. Ther. 2014;22:1139–1150. doi: 10.1038/mt.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study will be made available upon request to the corresponding author. All of the Ab clones used in this study have been described.