

Abstract
Background
This study aimed to investigate clinicopathological and molecular tumour features associated with intratumoral pks+ Escherichia coli (pks+E.coli+), pks+E.coli- (non-E.coli bacteria harbouring the pks island), Enterotoxigenic Bacteroides fragilis (ETBF) and Fusobacterium nucleatum (F. nucleatum).
Methods
We screened 1697 tumour-derived DNA samples from the Australasian Colorectal Cancer Family Registry, Melbourne Collaborative Cohort Study and the ANGELS study using targeted PCR.
Results
Pks+E.coli+ was associated with male sex (P < 0.01) and APC:c.835-8 A > G somatic mutation (P = 0.03). The association between pks+E.coli+ and APC:c.835-8 A > G was specific to early-onset CRCs (diagnosed<45years, P = 0.02). The APC:c.835-A > G was not associated with pks+E.coli- (P = 0.36). F. nucleatum was associated with DNA mismatch repair deficiency (MMRd), BRAF:c.1799T>A p.V600E mutation, CpG island methylator phenotype, proximal tumour location, and high levels of tumour infiltrating lymphocytes (Ps < 0.01). In the stratified analysis by MMRd subgroups, F. nucleatum was associated with Lynch syndrome, MLH1 methylated and double MMR somatic mutated MMRd subgroups (Ps < 0.01).
Conclusion
Intratumoral pks+E.coli+ but not pks+E.coli- are associated with CRCs harbouring the APC:c.835-8 A > G somatic mutation, suggesting that this mutation is specifically related to DNA damage from colibactin-producing E.coli exposures. F. nucleatum was associated with both hereditary and sporadic MMRd subtypes, suggesting the MMRd tumour microenvironment is important for F. nucleatum colonisation irrespective of its cause.
Subject terms: Oncogenesis, Risk factors, Cancer
Introduction
Colorectal cancer (CRC) was the third most prevalent cancer and the second leading cause of cancer-related death worldwide for both sexes in 2020 [1]. In Australia, it is the second most commonly diagnosed cancer with 1 in 21 (4.8%) and 1 in 30 (3.3%) males and females, respectively, developing CRC by 75 years of age [2]. CRC is a heterogeneous disease with regards to molecular drivers and pathways of tumorigenesis. In 2007, Jass et al. [3] described the molecular heterogeneity of CRC based on the presence or absence of chromosomal instability (CIN), microsatellite instability/mismatch repair deficiency (MSI/MMRd), CpG island methylator phenotype (CIMP), BRAF c.1799T>A p.Val600Glu (BRAF p.V600E), KRAS somatic mutations in codons 12 and 13 and germline pathogenic variants in the DNA MMR genes (Lynch syndrome). This heterogeneity over time has been shown to be associated with differing risk factors, survival, and treatment response. For example, serrated polyps, which are the precursors of the serrated pathway of tumorigenesis, characterised by BRAF p.V600E, CIMP-high and proximal location with or without high levels of MSI, show stronger association with tobacco smoking, alcohol intake and high body mass index (BMI) than conventional adenomas [4].
The etiology of the majority of CRC is multifactorial, involving the interplay between genetic, epigenetic, and environmental/lifestyle factors [5] whilst only ~5% of CRC are caused by germline pathogenic variants in known cancer-predisposition genes [6]. More recently, the gut microbiome is increasingly recognised to play an important role in CRC development [7, 8]. The presence of certain genotoxic gut bacteria is associated with CRC development [9]. Previous studies have shown that pks+ Escherichia coli (pks+ E. coli+), Enterotoxigenic Bacteroides fragilis (ETBF) and Fusobacterium nucleatum (F. nucleatum) are enriched in colonic mucosa of CRC-affected patients compared with healthy individuals [10–12].
E. coli strains from the B2 phylogroup [13] frequently harbour the 54 kb polyketide synthase (pks) island, which encodes enzymes for colibactin biosynthesis [14–16]. Colibactin is a genotoxin that induces DNA damage including inter-strand cross links [17] and double-stranded breaks [14, 18]. Recently, the colibactin-induced DNA damage was found to occur in specific patterns of single base substitution mutations (T > N), frequently in ATN and TTT sequence contexts [19]. This has led to the discovery of tumour mutational signatures associated with colibactin, in particular, the presence of single base substitution (SBS)-88 and short insertions and deletions (indels or ID)-18 signatures [19]. The somatic APC splice mutation (c.835-8 A > G) has been proposed as a biomarker of colibactin-induced DNA damage due to the specific sequence context of ATT > C and this has been observed in multiple adenomas from patients with unexplained colorectal polyposis though the presence of pks + E.coli+ was not measured [20].
ETBF secretes Bacteroides fragilis toxin and can cause symptoms such as inflammatory diarrhoea in humans [21]. ETBF triggers colitis, induces TH17 cell infiltration in murine models and promotes colonic tumorigenesis [22], although the mechanism underlying ETBF-related colorectal tumorigenesis is currently unknown.
F. nucleatum produces F. nucleatum adhesin A, which alters the β-catenin/Wnt signalling pathway and promotes CRC tumour growth [23]. F. nucleatum inhibits T-cell mediated immune responses against tumour cells [24] as well as creates a pro-inflammatory microenvironment that is favourable for colorectal neoplasia progression [25]. F. nucleatum is associated with CRCs that demonstrate MSI-high/MMRd, BRAF p.V600E somatic mutation and CIMP-high, features of the serrated pathways of tumorigenesis [12].
The aim of this study was to identify the intratumoral presence of genotoxic gut bacterial species, namely pks+ in E. coli (pks+ E. coli+), pks+ in non-E. coli (pks+ E. coli−) and ETBF, and F. nucleatum, in population-based CRC tumour samples from the Australasian Colorectal Cancer Family Registry (ACCFR) and Melbourne Collaborative Cohort Study (MCCS), and from the clinic-based CRC tumour samples from the Applying Novel Genomic approaches to Early-onset and suspected Lynch Syndrome colorectal and endometrial cancers (ANGELS) study. The association between the intratumoral bacteria and specific clinicopathological characteristics and molecular features, including the APC c.835-8 A > G somatic mutation were examined.
Methods
Australasian Colorectal Cancer Family Registry (ACCFR)
The ACCFR is the Australasian arm of the International Colon Cancer Family Registry with >42,000 recruited participants [26, 27]. Tumours tested in this study were from the population-based recruitment arm of the ACCFR, recruited from the Victorian Cancer Registry independent of family cancer history and diagnosed with invasive carcinoma of the colon or rectum between 1997 and 2007 during two recruitment phases [26]. Phase I recruitment (1997–2001) involved all CRC patients diagnosed between 18 and 44 years of age and 50% of CRC patients diagnosed between 45 and 59 years of age. Phase II recruitment (2001-2006) involved all CRC patients between 18 and 49 years of age [26]. From the Jeremy Jass Memorial Tissue Bank, a total of 823 primary adenocarcinomas of the colon or rectum during two recruitment phases with tumour tissue collected were available to this study [28]. Cancers were verified using obtainable pathology reports, cancer registry reports, medical records, and/or death certificates [26, 27].
Melbourne Collaborative Cohort Study (MCCS)
The MCCS is a prospective cohort study composed of 41,513 participants - 17,044 males and 24,469 females- recruited between 1990 and 1994 [29, 30], designed to understand the role of diet and lifestyle associated with cancer risk, including CRC [29, 30]. Tumour tissue was collected and molecularly characterised for a total of 858 CRCs, with the diagnosis age ranging from 41 to 86 years [28].
Applying Novel Genomic approaches to Early-onset and suspected Lynch Syndrome colorectal and endometrial cancers (ANGELS)
The ANGELS study recruited patients referred from family cancer clinics across Australia who were: (1) CRC- or endometrial cancer-affected people with an MMRd and/or microsatellite instability high (MSI-H) tumour with a diagnosis of suspected Lynch syndrome (as previously defined [31]), or (2) CRC- or endometrial cancer-affected people with an MMR-proficient (MMRp) and/or microsatellite stable cancer diagnosed <45 years of age. The ANGELS study had collected CRC tumour tissue for 229 participants diagnosed between 2014–2021 [32, 33].
Written informed consent was obtained from all study participants to collect blood and tumour tissue materials. The study protocols were approved by Human Research Ethics Committees at the University of Melbourne (ACCFR and ANGELS) and Cancer Council Victoria (MCCS). Given their rarity across the three study groups, CRCs from germline biallelic MUTYH pathogenic variant carriers (n = 4), constitutional MLH1 epimutation carriers (n = 6) and germline carriers of variant of uncertain significance in the MMR genes (VUS, n = 9) were excluded from this study. The details for samples included in this study are shown in Supplementary Fig. 1. A total of 29 participants had synchronous and metachronous CRCs and individual CRCs were treated independently in the statistical analysis.
Analyses of clinicopathological features and molecular characteristics
CRC tumour tissue was available for 813 and 816 probands for ACCFR and MCCS, respectively, as previously described [28], and 221 probands from the ANGELS study. A standardised pathological review was performed by anatomical pathologist (CR) for all three studies [34]. Tumours from the ileo-cecal junction, cecum, ascending colon, hepatic flexure, and transverse colon were classified as proximal, whereas tumours from the splenic flexure, descending, sigmoid colon and recto-sigmoid junction were grouped as distal. Tumours from the rectum were classified as rectal. Tumour-infiltrating lymphocytes were scored as present when there were ≥ 5 intra-epithelial lymphocytes in at least one high-power field (40×) [35].
Molecular characterisation of each tumour was performed using consistent methodology for the ACCFR and MCCS studies [28]. Tumour MMR status was determined by immunohistochemical (IHC) staining for MLH1, MSH2, MSH6 and PMS2 protein expression on all CRCs and for a subset, MSI status was determined as previously described [36–38]. For the ACCFR and MCCS studies, the primary antibodies used were MLH1 (G168-15, BD PharMingen), MSH2 (G219-1129, BD PharMingen), MSH6 (44, BD Transduction Labs) and PMS2 (A16-4, BD PharMingen). For the ANGELS study, tumour MMR status was determined from MMR IHC testing by clinical diagnostic laboratory or by MMR IHC testing performed internally as previously described [39] and confirmed by whole exome or targeted tumour sequencing using the additive feature combination approach [33]. For the ANGELS study, the primary antibodies used were MLH1 (M1), MSH2 (G219-1129), MSH6 (SP93) and PMS2 (A16-4) supplied from Roche Diagnostics (Basel, Switzerland). Across the three studies, tumours were categorised as: (1) MMRp if they showed retained and normal expression of all four MMR proteins by IHC and were microsatellite stable according to the additive feature combination approach or MSI-PCR method where tested, or (2) MMRd if they demonstrated loss of expression of one or more MMR proteins by IHC and/or were MSI-high according to the additive feature combination approach or MSI-PCR method where tested [28, 33].
MMRd CRCs were further divided into three subgroups: (1) Lynch syndrome - where a germline pathogenic variant in one of the DNA MMR genes (MLH1, MSH2, MSH6 and PMS2) or in EPCAM was identified as previously described [26, 38, 40, 41]; (2) MLH1 methylated CRCs - positive for tumour hypermethylation of the MLH1 gene promoter using the MethyLight assay as previously described for ACCFR and MCCS [28, 40, 42] and using MethyLight and MS-HRM assays as previously described for ANGELS [39] with MLH1 methylation positive tumours showing concomitant loss of MLH1 and PMS2 expression by IHC; (3) Somatic biallelic MMR gene inactivation resulting from two somatic MMR gene mutations (double somatic MMR mutations) determined as described previously from either targeted tumour sequencing assay [39, 43] or from tumour whole exome sequencing [32, 33].
For the ACCFR and MCCS, KRAS codons 12&13 somatic mutations were tested using real-time quantitative PCR (qPCR) with high-resolution melting analysis in the presence of the SYTO9 fluorescent intercalating dye followed by direct Sanger sequencing on cases with differential melting profiles as previously described [34, 44]. BRAF p.V600E somatic mutation was tested using a fluorescent allele-specific polymerase chain reaction assay as previously described [45]. For the ANGELS study, KRAS codons 12&13 and BRAF p.V600E somatic mutations were derived from custom-designed panel sequencing or tested using Sanger or allele-specific PCR as for the ACCFR/MCCS tumours [32, 33, 39]. CIMP-high tumours were defined by tumour hypermethylation at 3 or more of the promoter regions of the 5 tumour suppressor genes: CACNA1G, IGF2, NEUROG1, RUNX3 and SOCS1 using MethyLight [46] for all three studies.
DNA extraction from tumour samples and qPCR assays for detecting pks+E. coli, ETBF and F. nucleatum
The tumour rich regions of the FFPE CRC tumour tissue were macrodissected as previously described for the ACCFR and MCCS studies [28, 43]. For the ANGELS study, the genomic DNA was extracted from FFPE CRC tumour tissues using the QIAamp DNA FFPE Tissue Kit (QIAGEN, Hilden, Germany), and the concentration was assessed using the Qubit fluorometer (Thermo Fisher Scientific, California, USA). The intratumoral presence of pks+ E. coli+, pks+ E. coli−, ETBF, and F. nucleatum was assessed by performing qPCR, which is detailed in Supplementary methods. The pks+ E. coli strains (34351) were kindly provided by Drs Danielle Ingle and Norelle Sherry from the Peter Doherty Institute for Infection and Immunity, collected as part of the Controlling Superbugs study [47] and classed as Extraintestinal Pathogenic Escherichia coli (ExPEC) [48]. The pks- E. coli strains (MG1655) were provided by Dr. Dianna M. Hocking (Department of Microbiology and Immunology, The University of Melbourne). Genomic DNA from pks+ E. coli and pks- E. coli was used as internal controls for qPCR.
Genotyping assay for the APC c.835-8 A > G somatic mutation
A custom-designed TaqMan genotyping assay was used to detect the APC: c.835-8 A > G mutation in tumour DNA (ThermoFisher Scientific, California, USA, Cat# ANGZYCC), which was set up using TaqMan Genotyping Master Mix (ThermoFisher Scientific), and performed on Thermo QuantStudio 7 (ThermoFisher Scientific). The presence of APC: c.835-8 A > G mutation was determined using the QuantStudio Real-time PCR System software (v1.7.2, ThermoFisher Scientific).
Statistical analysis
All statistical tests were performed using R programming software (v4.2.1). Logistic regression was used to assess the association between the presence of intratumoral bacteria and clinicopathological and tumour features. Unless indicated otherwise, all tests were adjusted for sex, age at CRC diagnosis and study. P < 0.05 were considered statistically significant.
Results
Participant and tumour characteristics associated with the intratumoral bacterial presence
In total, 1697 CRC tumours from 1666 individuals from the ACCFR (44.2%), MCCS (43.5%), and ANGELS (12.3%) had results from intratumoral bacteria testing. A description of the participants and their tumour characteristics are provided in Table 1. Of those, 29 (1.7%) participants had a synchronous or metachronous CRC, 26% were diagnosed with CRC before age 45 years (early-onset CRC or EOCRC) and 15.6% were MMRd. Of these MMRd CRCs with an explained etiology, 20.6% were related to Lynch syndrome while the remaining 79.4% were related to somatic MMR inactivation with 47.4% related to tumour MLH1 methylation and 32% related to double somatic MMR mutations.
Table 1.
CRC-affected participants and their tumour characteristics from each of the three studies.
| Characteristics | Total | ACCFR | MCCS | ANGELS |
|---|---|---|---|---|
| Total CRC patients | 1666 | 736 (44.2%) | 725 (43.5%) | 205 (12.3%) |
| Numbers of CRC patients with synchronous and metachronous CRCs | 29 | 5 | 20 | 4 |
| Sex | ||||
| Female | 824 (49.5%) | 356 (48.4%) | 351 (48.4%) | 117 (57.1%) |
| Male | 842 (50.5%) | 380 (51.6%) | 374 (51.6%) | 88 (42.9%) |
| Total CRCsa | 1697 | 742 (43.7%) | 745 (43.9%) | 210 (12.4%) |
| Age at CRC diagnosis | ||||
| Mean ± IQR | 55.7 ± 24.8 | 46.5 ± 11 | 68.5 ± 11.4 | 44.1 ± 19.5 |
| Early-onset CRC (<45 years) | 442 (26%) | 301 (40.6%) | 4 (0.5%) | 137 (65.2%) |
| Late-onset CRC (≥45 years) | 1255 (74%) | 441 (59.4%) | 741 (99.5%) | 73 (34.8%) |
| MMR status | ||||
| MMR-proficient (MMRp) | 1432 (84.4%) | 669 (90.2%) | 645 (86.6%) | 118 (56.2%) |
| MMR-deficient (MMRd) | 265 (15.6%) | 73 (9.8%) | 100 (13.4%) | 92 (43.8%) |
| Anatomical location | ||||
| Proximal | 576 (33.9%) | 201 (27.1%) | 267 (35.8%) | 108 (51.4%) |
| Distal | 467 (27.5%) | 214 (28.8%) | 196 (26.3%) | 57 (27.2%) |
| Rectal | 582 (34.3%) | 288 (38.8%) | 249 (33.4%) | 45 (21.4%) |
| Unknown | 72 (4.3%) | 39 (5.3%) | 33 (4.5%) | 0 (0%) |
| BRAF somatic mutation | ||||
| BRAF V600E | 197 (11.6%) | 53 (7.1%) | 124 (16.7%) | 24 (11.4%) |
| Wildtype | 1420 (83.7%) | 682 (91.9%) | 603 (80.9%) | 149 (71%) |
| Unknown | 80 (4.7%) | 7 (1%) | 18 (2.4%) | 37 (17.6%) |
| KRAS somatic mutation | ||||
| KRAS codon 12&13 somatic mutation | 461 (27.2%) | 211 (28.4%) | 203 (27.3%) | 51 (24.3%) |
| Wildtype | 1088 (64.1%) | 451 (60.8%) | 529 (71%) | 122 (58.1%) |
| Unknown | 148 (8.7%) | 80 (10.8%) | 13 (1.7%) | 37 (17.6%) |
| CIMP | ||||
| CIMP-high | 171 (10.1%) | 35 (4.7%) | 109 (14.6%) | 27 (12.9%) |
| CIMP-negative | 1399 (82.4%) | 613 (82.6%) | 624 (83.8%) | 162 (77.1%) |
| Unknown | 127 (7.5%) | 94 (12.7%) | 12 (1.6%) | 21 (10%) |
| Tumour histological type | ||||
| Adenocarcinoma | 1437 (84.7%) | 654 (88.2%) | 653 (87.6%) | 130 (61.9%) |
| Mucinous | 143 (8.4%) | 64 (8.6%) | 59 (7.9%) | 20 (9.5%) |
| Other type | 22 (1.3%) | 6 (0.8%) | 13 (1.8%) | 3 (1.5%) |
| Unknown | 95 (5.6%) | 18 (2.4%) | 20 (2.7%) | 57 (27.1%) |
| Tumour grade | ||||
| High | 345 (20.3%) | 141 (19%) | 143 (19.2%) | 61 (29%) |
| Low | 1301 (76.7%) | 577 (77.8%) | 581 (78%) | 143 (68.1%) |
| Unknown | 51 (3%) | 24 (3.2%) | 21 (2.8%) | 6 (2.9%) |
| Tumour-infilteration lymphocytes (TILs) | ||||
| Present | 373 (22%) | 147 (19.8%) | 170 (22.8%) | 56 (26.7%) |
| Absent | 1195 (70.4%) | 558 (75.2%) | 538 (72.2%) | 99 (47.1%) |
| Unknown | 129 (7.6%) | 37 (5%) | 37 (5%) | 55 (26.2%) |
| MMRd CRC Subgroupb | ||||
| Lynch syndrome | 47 (20.6%) | 37 (58.8%) | 6 (7.4%) | 4 (4.8%) |
| MLH1 methylation | 108 (47.4%) | 13 (20.6%) | 71 (87.7%) | 24 (28.5%) |
| Double somatic MMR mutationsc | 73 (32%) | 13 (20.6%) | 4 (4.9%) | 56 (66.7%) |
aIncludes synchronous and metachronous CRCs from same individuals.
bMMRd CRCs with unexplained causes are not shown.
cDouble MMR somatic pathogenic mutations identified using NGS-based techniques.
The prevalence of each of the bacteria and their association with participant clinicopathological and tumour characteristics are shown in Table 2. Intratumoral prevalence of pks+ E. coli+, pks+ E. coli−, ETBF and F. nucleatum was 10.3%, 10.4%, 6.1% and 8.9%, respectively. CRCs with intratumoral pks+ E. coli+, were associated with male sex (P < 0.01, odds ratio (OR) = 1.54, 95% confidence interval (CI) = 1.11–2.13) when compared with female sex. CRCs with intratumoral pks+ E. coli− were associated with older age at CRC diagnosis (P < 0.01) and low-grade tumours when compared with high-grade tumours (P = 0.02, ORhigh grade = 0.57, 95% CI = 0.36–0.89).
Table 2.
Intratumoural presence of pks+ E. coli, pks+ E. coli−, ETBF and F nucleatum and the association with tumour characteristics.
| pks+ E.coli+ | pks+ E.coli- | Enterotoxigenic Bacteroides fragilis | Fusobacterium nucleatum | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratioa (95% CI) | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratioa (95% CI) | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratioa (95% CI) | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratioa (95% CI) |
| Total CRCs | 174 (10.3%) | 1339 (78.9%) | 184 (10.8%) | - | - | 176 (10.4%) | 1337 (78.8%) | 184 (10.8%) | - | - | 104 (6.1%) | 1451 (85.5%) | 142 (8.4%) | - | - | 151 (8.9%) | 1440 (84.9%) | 106 (6.2%) | - | - |
| Recruitmentb | 0.4306 | - | 0.2157 | - | 0.5240 | - | 0.0024 | - | ||||||||||||
| Population-based | 152 (10.2%) | 1170 (78.7%) | 165 (11.1%) | - | Ref | 162 (10.9%) | 1160 (78%) | 165 (11.1%) | - | Ref | 93 (6.3%) | 1258 (84.6%) | 136 (9.1%) | - | Ref | 120 (8.1%) | 1266 (85.1%) | 101 (6.8%) | - | Ref |
| Clinic-based | 22 (10.5%) | 169 (80.5%) | 19 (9%) | - | 1.22 (0.73–1.98) | 14 (6.7%) | 177 (84.3%) | 19 (9%) | - | 0.69 (0.37–1.20) | 11 (5.2%) | 193 (91.9%) | 6 (2.9%) | - | 0.80 (0.39–1.51) | 31 (15%) | 174 (83%) | 5 (2%) | - | 2.03 (1.27–3.17) |
| Sex | 0.009701 | - | 0.6114 | - | 0.6181 | - | 0.2619 | - | ||||||||||||
| Female | 67 (8%) | 660 (79%) | 109 (13%) | - | Ref | 86 (10.3%) | 641 (76.7%) | 109 (13%) | - | Ref | 49 (6%) | 723 (86%) | 64 (8%) | - | Ref | 69 (8.3%) | 720 (86.1%) | 47 (5.6%) | - | Ref |
| Male | 107 (12.4%) | 679 (78.9%) | 75 (8.7%) | - | 1.54 (1.11–2.13) | 90 (10.5%) | 696 (80.8%) | 75 (8.7%) | - | 0.92 (0.67–1.27) | 55 (6.4%) | 728 (84.5%) | 78 (9.1%) | - | 1.11 (0.74–1.66) | 82 (9.5%) | 720 (83.6%) | 59 (6.9%) | - | 1.21 (0.87–1.71) |
| Age (mean ± IQR) | 57.8 ± 26.0 | 55.3 ± 24.3 | 58.0 ± 22.1 | 0.27052 | - | 58.8 ± 22.7 | 55.2 ± 25.1 | 58.0 ± 22.1 | 0.0061 | - | 56.7 ± 25.9 | 55.8 ± 25.1 | 55.3 ± 20.2 | 0.8806 | - | 55.8 ± 27.0 | 56.0 ± 25.1 | 54.4 ± 21.4 | 0.7854 | - |
| Age groupc | 0.8462 | - | 0.0533 | - | 0.2520 | 0.4110 | - | |||||||||||||
| LOCRC (≥45) | 132 (10.5%) | 977 (77.9%) | 146 (11.6%) | - | Ref | 144 (11.5%) | 965 (76.9%) | 146 (11.6%) | - | Ref | 75 (6%) | 1071 (85.3%) | 109 (8.7%) | - | Ref | 105 (8.4%) | 1071 (85.3%) | 79 (6.3%) | - | Ref |
| EOCRC (<45) | 42 (9.5%) | 362 (81.9%) | 38 (8.6%) | - | 1.05 (0.67–1.63) | 32 (7.2%) | 372 (84.2%) | 38 (8.6%) | - | 0.63 (0.40–1.00) | 29 (7%) | 380 (86%) | 33 (7%) | - | 1.38 (0.79–2.42) | 46 (10.4%) | 369 (83.5%) | 27 (6.1%) | - | 1.21 (0.77–1.91) |
| CRC site | ||||||||||||||||||||
| Proximal colon | 62 (10.8%) | 457 (79.3%) | 57 (9.9%) | - | Ref | 61 (10.6%) | 458 (79.5%) | 57 (9.9%) | - | Ref | 44 (7.6%) | 485 (84.2%) | 47 (8.2%) | - | Ref | 85 (14.8%) | 457 (79.3%) | 34 (5.9%) | - | Ref |
| Distal colon | 49 (10.5%) | 371 (79.4%) | 47 (10.1%) | 0.9924 | 1.00 (0.66–1.50) | 49 (10.5%) | 371 (79.4%) | 47 (10.1%) | 0.9232 | 1.02 (1.53–0.68) | 24 (5.1%) | 404 (86.5%) | 39 (8.4%) | 0.0862 | 0.63 (0.37–1.06) | 31 (7%) | 406 (87%) | 30 (6%) | 8.22E-05 | 0.42 (0.26–0.64) |
| Rectum | 55 (9.5%) | 460 (79%) | 67 (11.5%) | 0.5165 | 0.88 (0.59–1.30) | 54 (9.3%) | 461 (79.2%) | 67 (11.5%) | 0.5203 | 0.88 (1.31–0.59) | 31 (5%) | 506 (87%) | 45 (8%) | 0.0708 | 0.64 (0.39–1.03) | 34 (6%) | 514 (88%) | 34 (6%) | 3.00E-06 | 0.36 (0.23–0.55) |
| Unknown | 8 (11.1%) | 51 (70.8%) | 13 (18.1%) | - | - | 12 (16.7%) | 47 (65.3%) | 13 (18%) | - | - | 5 (6.9%) | 56 (77.8%) | 11 (15.3%) | - | - | 1 (1.4%) | 63 (87.5%) | 8 (11.1%) | - | - |
| MMR status | 0.5656 | - | 0.1588 | - | 0.0021 | - | 7.33E-12 | - | ||||||||||||
| MMRp | 148 (10.3%) | 1122 (78.4%) | 162 (11.3%) | - | Ref | 155 (10.8%) | 1115 (77.9%) | 162 (11.3%) | - | Ref | 78 (5.5%) | 1232 (86%) | 122 (8.5%) | - | Ref | 94 (6.6%) | 1249 (87.2%) | 89 (6.2%) | - | Ref |
| MMRd | 26 (9.8%) | 217 (81.9%) | 22 (8.3%) | - | 0.87 (0.53–1.37) | 21 (7.9%) | 222 (83.8%) | 22 (8.3%) | - | 0.69 (0.41–1.13) | 26 (9.8%) | 219 (82.6%) | 20 (7.6%) | - | 2.16 (1.30–3.49) | 57 (21.5%) | 191 (72.1%) | 17 (6.4%) | - | 3.90 (2.63–5.75) |
| BRAF somatic mutation | 0.4789 | - | 0.1297 | - | 0.5399 | - | 0.0008 | - | ||||||||||||
| Wildtype | 149 (10.4%) | 1127 (78.6%) | 158 (11%) | - | Ref | 154 (10.7%) | 1122 (78.3%) | 158 (11%) | - | Ref | 91 (6.4%) | 1215 (84.7%) | 128 (8.9%) | - | Ref | 113 (7.9%) | 1225 (85.4%) | 96 (6.7%) | - | Ref |
| V600E | 18 (9%) | 162 (80.6%) | 21 (10.4%) | - | 0.83 (0.48–1.36) | 16 (8%) | 164 (81.6%) | 21 (10.4%) | - | 0.65 (0.36–1.10) | 11 (5.5%) | 179 (89%) | 11 (5.5%) | - | 0.82 (0.40–1.50) | 31 (15.4%) | 162 (80.6%) | 8 (4%) | - | 2.13 (1.35–3.28) |
| Unknown | 7 (11.3%) | 50 (80.6%) | 5 (8.1%) | - | - | 6 (9.7%) | 51 (82.2%) | 5 (8.1%) | - | - | 2 (3.2%) | 57 (91.9%) | 3 (4.9%) | - | - | 7 (11.3%) | 53 (85.5%) | 2 (3.2%) | - | - |
| KRAS somatic mutation | 0.5568 | - | 0.2526 | - | 0.0178 | - | 0.2901 | - | ||||||||||||
| Wildtype | 110 (10%) | 874 (79.3%) | 118 (10.7%) | - | Ref | 113 (10.3%) | 871 (79%) | 118 (10.7%) | - | Ref | 59 (5.4%) | 958 (86.9%) | 85 (7.7%) | - | Ref | 102 (9.3%) | 933 (84.7%) | 67 (6%) | - | Ref |
| codon 12&13 | 50 (10.8%) | 360 (77.4%) | 55 (11.8%) | - | 1.11 (0.77–1.58) | 56 (12%) | 354 (76.2%) | 55 (11.8%) | - | 1.22 (0.86–1.72) | 39 (8.4%) | 384 (82.6%) | 42 (9%) | - | 1.67 (1.09–2.53) | 35 (7.6%) | 401 (86.2%) | 29 (6.2%) | - | 0.80 (0.53–1.19) |
| Unknown | 14 (10.8%) | 105 (80.8%) | 11 (8.5%) | - | - | 7 (5.4%) | 112 (86.1%) | 11 (8.5%) | - | - | 6 (4.6%) | 109 (83.9%) | 15 (11.5%) | - | - | 14 (10.8%) | 106 (81.5%) | 10 (7.7%) | - | - |
| CIMP status | 0.8502 | - | 0.9232 | - | 0.6695 | - | 3.71E-05 | - | ||||||||||||
| CIMP-negative | 149 (11%) | 1098 (78%) | 152 (11%) | - | Ref | 144 (10.3%) | 1103 (78.8%) | 152 (10.9%) | - | Ref | 90 (6.4%) | 1187 (84.9%) | 122 (8.7%) | - | Ref | 112 (8%) | 1198 (85.6%) | 89 (6.4%) | - | Ref |
| CIMP-high | 18 (10.5%) | 135 (79%) | 18 (10.5%) | - | 0.95 (0.54–1.58) | 19 (11.1%) | 134 (78.4%) | 18 (10.5%) | - | 0.97 (0.56–1.61) | 10 (5.8%) | 152 (88.9%) | 9 (5.3%) | - | 0.86 (0.41–1.64) | 31 (18.1%) | 131 (76.6%) | 9 (5.3%) | - | 2.62 (1.64–4.10) |
| Unknown | 7 (5.5%) | 106 (83.5%) | 14 (11%) | - | - | 13 (10%) | 100 (79%) | 14 (11%) | - | - | 4 (3.1%) | 112 (88.2%) | 11 (8.7%) | - | - | 8 (6.3%) | 111 (87.4%) | 8 (6.3%) | - | - |
| CRC tumour grade | 0.2109 | - | 0.0161 | - | 0.2760 | - | 4.79E-05 | - | ||||||||||||
| Low grade | 140 (11%) | 1012 (78%) | 149 (11%) | - | Ref | 148 (11%) | 1004 (77%) | 149 (12%) | - | Ref | 76 (5.8%) | 1119 (86%) | 106 (8.2%) | - | Ref | 97 (7.5%) | 1127 (86.6%) | 77 (5.9%) | - | Ref |
| High grade | 30 (8.7%) | 288 (83.5%) | 27 (7.8%) | - | 0.77 (0.50–1.15) | 24 (7%) | 294 (85.2%) | 27 (7.8%) | - | 0.57 (0.36–0.89) | 25 (7.2%) | 289 (83.8%) | 31 (9%) | - | 1.30 (0.80–2.06) | 51 (15%) | 270 (78%) | 24 (7%) | - | 2.14 (1.48–3.08) |
| Unknown | 4 (7.8%) | 39 (76.5%) | 8 (15.7%) | - | - | 4 (7.8%) | 39 (76.5%) | 8 (15.7%) | - | - | 3 (5.9%) | 43 (84.3%) | 5 (9.8%) | - | - | 3 (5.9%) | 43 (84.3%) | 5 (9.8%) | - | - |
| Tumour histological type | ||||||||||||||||||||
| Adenocarcinoma | 145 (10.1%) | 1133 (78.8%) | 159 (11.1%) | - | Ref | 149 (10%) | 1129 (79%) | 159 (11%) | - | Ref | 85 (5.9%) | 1225 (85.3%) | 127 (8.8%) | - | Ref | 111 (7.7%) | 1229 (85.5%) | 97 (6.8%) | - | Ref |
| Mucinous | 18 (12.6%) | 111 (77.6%) | 14 (9.8%) | 0.4023 | 1.26 (0.72–2.09) | 18 (12.6%) | 111 (77.6%) | 14 (9.8%) | 0.4660 | 1.22 (0.70–2.03) | 14 (9.8%) | 120 (83.9%) | 9 (6.3%) | 0.0851 | 1.69 (0.89–2.98) | 26 (18.2%) | 112 (78.3%) | 5 (3.5%) | 0.0001 | 2.51 (1.54–3.97) |
| Other types | 3 (13.6%) | 19 (86.4%) | 0 (0%) | 0.7714 | 1.20 (0.28–3.61) | 3 (13.6%) | 19 (86.4%) | 0 (0%) | 0.8090 | 1.16 (0.27–3.50) | 2 (9.1%) | 19 (86.4%) | 1 (4.5%) | 0.5889 | 1.50 (0.24–5.31) | 4 (18.2%) | 18 (81.8%) | 0 (0%) | 0.1265 | 2.37 (0.67–6.53) |
| Unknown | 8 (8.4%) | 76 (80%) | 11 (11.6%) | - | - | 6 (6.3%) | 78 (82.1%) | 11 (11.6%) | - | - | 3 (3%) | 87 (92%) | 5 (5%) | - | - | 10 (10.5%) | 81 (85.3%) | 4 (4.2%) | - | - |
aP-values and odd ratios were calculated using multiple logistic regression adjusted for CRC diagnosis age, sex and study cohort. Samples with unknown intratumoural presences and tumour characteristics treated as na and excluded from the statistical testing.
bAdjusted for sex and age only.
cAdjusted for sex and study cohort only.
Statistically significant p < 0.05 values are in bold.
CRCs with intratumoral ETBF were more likely to be MMRd (P < 0.01, OR = 2.16, 95% CI = 1.30–3.49) and have KRAS codons 12&13 somatic mutations (P = 0.02, OR = 1.67, 95% CI = 1.09–2.53) when compared with MMRp and CRCs without KRAS codon 12&13 somatic mutations, respectively. Intratumoral F. nucleatum was associated with proximal tumour location when compared with distal (P < 0.01, ORdistal = 0.42, 95% CI = 0.26–0.64) and rectal locations (P < 0.01, ORrectal = 0.36, 95% CI = 0.23–0.55). CRCs with intratumoral F. nucleatum were also associated with high histology grade (P < 0.01, OR = 2.14, 95% CI = 1.48–3.08) and mucinous adenocarcinoma histologic type (P < 0.01, OR = 2.51, 95% CI = 1.54–3.97). For molecular features, intratumoral F. nucleatum was associated with CRCs with MMRd (P < 0.01, OR = 3.90, 95% CI = 2.63–5.75), BRAF p.V600E somatic mutation (P < 0.01, OR = 2.13, 95% CI = 1.35–3.28), and CIMP-high (P < 0.01, OR = 2.62, 95%CI = 1.64–4.10) when compared with CRCs without these features.
An analysis of the 442 EOCRCs did not show evidence that clinicopathological or tumour characteristics were associated with the presence of pks+ E. coli+ (Supplementary Table 1). In EOCRCs, the presence of ETBF (P < 0.01, OR = 4.17, 95% CI = 1.77–9.67) or F. nucleatum (P < 0.01, OR = 3.36, 95%CI = 1.67–6.65) was both associated with MMRd when compared with MMRp CRCs. F. nucleatum was also associated with the proximal tumour location when compared with distal tumour location (P = 0.01, ORdistal = 0.36, 95% CI = 0.16–7.70) and rectal tumour location (P < 0.01, ORrectal = 0.33, 95% CI = 0.15–7.00) (Supplementary Table 1). ETBF was associated with proximal tumour location when compared with distal location (P = 0.04, ORprox = 2.78/ORdistal = 0.36, 95%CIprox = 0.13–6.25/95%CIdist = 0.16–7.70) but this was not significant when compared with rectal location (P = 0.20, ORprox = 3.03/ORrectal = 0.33, 95%CIprox = 0.14–6.67/95%CIrectal = 0.15–7.00).
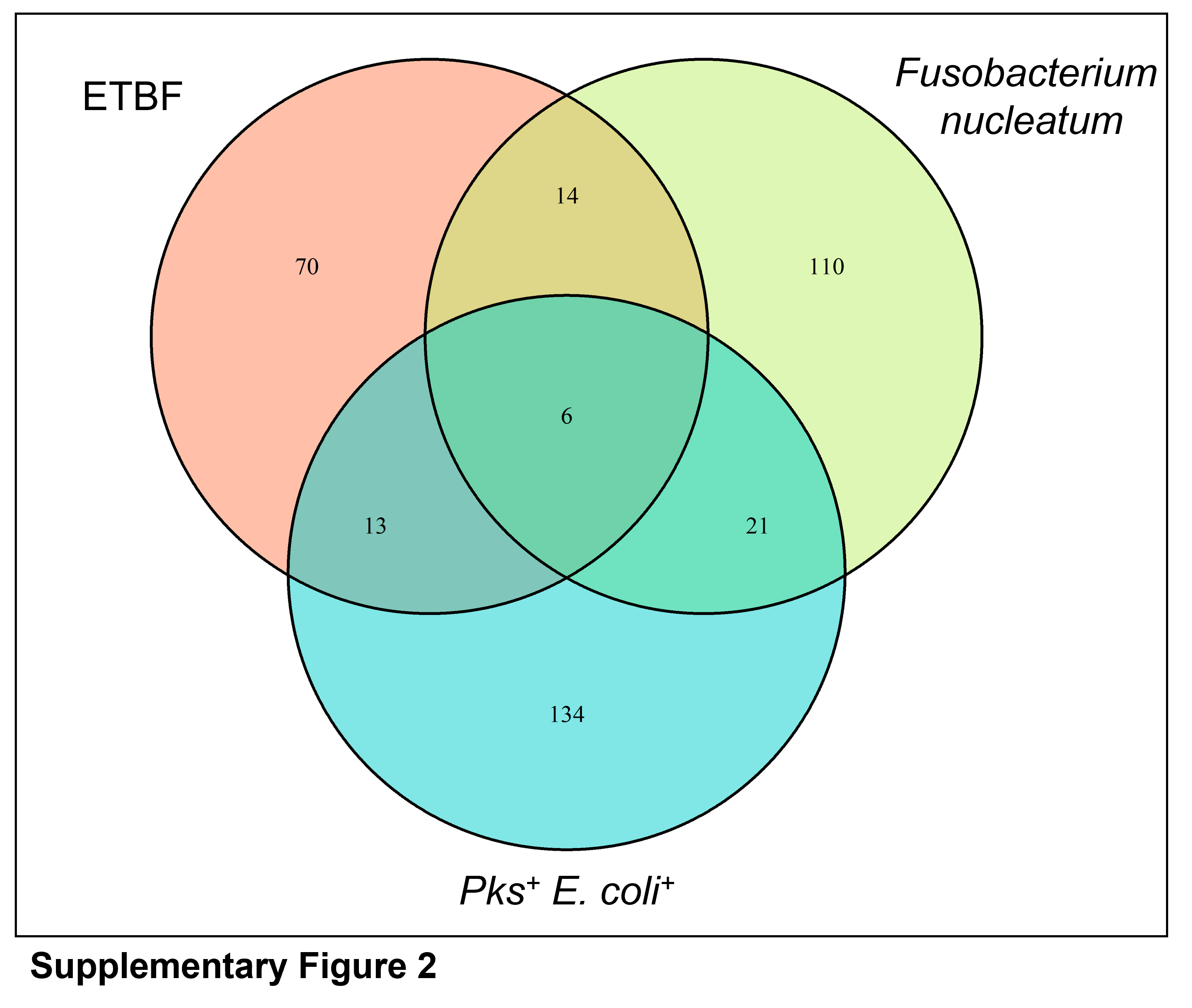
The presence of any two or all three of the bacteria (pks+ E. coli+, ETBF and F. nucleatum) in the same CRC was uncommon, with only 54 (3.2%) tumours having the presence of >1 bacteria (Supplementary Fig. 2). Only 6 (0.4%) tumours were detected with all three bacteria and these CRCs were not associated with any specific clinicopathological characteristics (Supplementary Table 2). CRCs that had both ETBF and F. nucleatum detected were associated with MMRd when compared with CRCs that did not have both ETBF and F. nucleatum detected (P < 0.01, OR = 4.56, 95%CI = 1.38–13.56) (Supplementary Table 2).
The colibactin-associated APC: c.835-8 A > G somatic mutation is associated with pks+ E. coli+
The association between the intratumoral presence of the pks island, with or without E. coli (pks+ E. coli+ and pks+ E. coli−) and the APC: c.835-8 A > G somatic mutation was tested. Due to lower DNA requirement than the intratumoral bacterial screening, 62 additional samples (total n = 1759) were included in the APC: c.835-8 A > G testing. Across all CRCs, 3.3% had the APC: c.835-8 A > G somatic mutation, which was consistent with the frequency observed in both EOCRCs and late-onset CRCs (LOCRCs) (Table 3). The APC: c.835-8 A > G mutation was associated with intratumoral pks+ E. coli+ (P = 0.025, OR = 2.20, 95% CI = 1.05–4.25) but not with other bacteria carrying the pks island (pks+ E. coli−; P = 0.36, OR = 0.61, 95% CI = 0.18–1.54) or with the E. coli bacteria not carrying the pks island (pks- E. coli+; P = 0.16, OR = 1.99, 95% CI = 0.67–4.72) (Table 3). These trends were consistent when tested in the EOCRCs (P = 0.022, OR = 4.26, 95%CI = 1.10–14.02), however, in the LOCRCs, the association between APC: c.835-8 A > G and pks+ E. coli+ was not significant (P = 0.18, OR = 1.78, 95% CI = 0.70–3.94) (Table 3). The APC: c.835-8 A > G mutation was not associated with intratumoral ETBF or F. nucleatum (data not shown). The participant and tumour characteristics associated with the APC: c.835-8 A > G somatic mutation are shown in Supplementary Table 3.
Table 3.
The prevalence of the APC: c.835-8 A > G somatic mutation and its association with the intratumoral presence of pks+ E. coli+, pks+ E. coli−, all pks+ bacteria and pks- E. coli+.
| All CRCs | EOCRCs (CRC < 45 yrs) | LOCRCs (CRC ≥ 45 yrs) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intratumoural bacteria | APC: c.835-8 A > G mutation | APC: c.835-8 A > G wildtype | P valuea | Adjusted Odd Ratio (95% CI)a | APC: c.835-8 A > G mutation | APC: c.835-8 A > G wildtype | P valuea | Adjusted Odd Ratio (95% CI)a | APC: c.835-8 A > G mutation | APC: c.835-8 A > G wildtype | P valuea | Adjusted Odd Ratio (95% CI)a |
| Total CRCs | 58 (3.3%) | 1701 (96.7%) | - | - | 15 (3.3%) | 439 (96.7%) | - | - | 43 (3.3%) | 1262 (96.7%) | - | - |
| pks + E. coli + | 0.0252 | - | 0.0219 | - | 0.1820 | - | ||||||
| Not detected | 41 (3.1%) | 1287 (96.9%) | - | Ref | 9 (2.5%) | 351 (97.5%) | - | Ref | 32 (3.3%) | 936 (96.7%) | - | Ref |
| Detected | 11 (6.3%) | 163 (93.7%) | - | 2.20 (1.05–4.25) | 4 (9.5%) | 38 (90.5%) | - | 4.26 (1.10–14.02) | 7 (5.3%) | 125 (94.7%) | - | 1.78 (0.70–3.94) |
| Unknown | 6 (2.3%) | 251 (97.7%) | - | - | 2 (3.8%) | 50 (96.2%) | - | - | 4 (2%) | 201 (98%) | - | - |
| pks + E. coli− | 0.3570 | - | 0.9931 | - | 0.6098 | - | ||||||
| Not detected | 48 (3.6%) | 1279 (96.4%) | - | Ref | 13 (3.5%) | 357 (96.5%) | - | - | 35 (3.7%) | 922 (96.3%) | - | - |
| Detected | 4 (2.3%) | 171 (97.7%) | - | 0.61 (0.18–1.54) | 0 (0%) | 32 (100%) | - | ntb | 4 (2.8%) | 139 (97.2%) | - | 0.76 (0.22–1.95) |
| Unknown | 6 (2.3%) | 251 (97.7%) | - | - | 2 (3.8%) | 50 (96.2%) | - | - | 4 (2%) | 201 (98%) | - | - |
| pks + (all bacteria) | 0.3775 | - | 0.2537 | - | 0.6323 | - | ||||||
| Not detected | 37 (3.1%) | 1150 (96.9%) | - | Ref | 9 (2.7%) | 327 (97.3%) | - | Ref | 28 (3.3%) | 823 (96.7%) | - | Ref |
| Detected | 15 (4%) | 360 (96%) | - | 1.32 (0.69–2.40) | 4 (5.2%) | 73 (94.8%) | - | 2.03 (0.53–6.54) | 11 (3.7%) | 287 (96.3%) | - | 1.19 (0.56–2.37) |
| Unknown | 6 (3%) | 191 (97%) | - | - | 2 (4.9%) | 39 (95.1%) | - | - | 4 (2.6%) | 152 (97.4%) | - | - |
| pks- E. coli + | 0.1574 | - | 0.8141 | - | 0.0900 | - | ||||||
| Not detected | 47 (3.3%) | 1374 (96.7%) | - | Ref | 12 (3.2%) | 365 (96.8%) | - | Ref | 35 (3.4%) | 1009 (96.6%) | - | Ref |
| Detected | 5 (6.2%) | 76 (93.8%) | - | 1.99 (0.67–4.72) | 1 (4%) | 24 (96%) | - | 1.29 (0.07–7.07) | 4 (7.1%) | 52 (92.9%) | - | 2.56 (0.74–6.82) |
| Unknown | 6 (2.3%) | 251 (97.7%) | - | - | 2 (3.8%) | 50 (96.2%) | - | - | 4 (2%) | 201 (98%) | - | - |
aMultiple logistic regression adjusted for CRC diagnosis age, sex and study cohort. Samples with unknown intratumoural presences and tumour characteristics were excluded in the statistical testing.
bNot tested as deemed non-sensical due to the group containing zero variable.
Statistically significant p < 0.05 values are in bold.
F. nucleatum is associated with both inherited and sporadic subtypes of MMRd CRC
ETBF and F. nucleatum were associated with MMRd CRCs (Table 2). The etiology of MMRd for 228 of these CRCs was known comprising 47 (20.6%) CRCs from people with Lynch syndrome, and 181 (79.4%) related to sporadic causes namely MLH1 promoter methylation (n = 108; 47.4%) and double somatic MMR mutations (n = 73; 32%). We further investigated the association between these bacteria and specific MMRd subgroups. The presence of F. nucleatum was associated with all three MMRd subgroups (Table 4), where the association was strongest for the MLH1 methylated subgroup (P < 0.01, OR = 4.91, 95% CI = 2.84–8.36). No associations were observed between the specific MMRd subgroups and intratumoral pks + E. coli or ETBF, though ETBF was overall associated with MMRd status (Table 4).
Table 4.
Intratumoural presence of pks+ E. coli, pks+ E. coli−, ETBF and F nucleatum and the association with CRC subgroup.
| pks+E.coli+ | pks+E.coli- | Entrotoxigenic Bacteroides fragilis | Fusobacterium nucleatum | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CRC Subgroups | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratio (95% CI) | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratio (95% CI) | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratio (95% CI) | Detected | Not detected | Unknown | P valuea | Adjusted Odd Ratio (95% CI) |
| Total CRCs | 171 (10.3%) | 1311 (79.1%) | 176 (10.6%) | - | - | 173 (10.4%) | 1309 (79%) | 176 (10.6%) | - | - | 95 (5.7%) | 1425 (86%) | 138 (8.3%) | - | - | 146 (8.8%) | 1411 (85.1%) | 101 (6.1%) | - | - |
| Sporadic MMRp CRC | 148 (10.4%) | 1120 (78.3%) | 162 (11.3%) | Ref | Ref | 155 (11%) | 1113 (78%) | 162 (11%) | Ref | Ref | 78 (5.5%) | 1230 (86%) | 122 (8.5%) | Ref | Ref | 94 (6.6%) | 1247 (87.2%) | 89 (6.2%) | Ref | Ref |
| MMRd CRC subgroupsb | ||||||||||||||||||||
| Total MMRd CRCs with known etiology | 23 | 191 | 14 | - | - | 18 | 196 | 14 | - | - | - | 195 | 16 | - | - | 52 | 164 | 12 | - | - |
| Lynch syndrome | 5 (11%) | 41 (87%) | 1 (2%) | 0.8786 | 1.08 (0.36–2.58) | 2 (4.3%) | 44 (93.6%) | 1 (2.1%) | 0.1993 | 0.39 (0.06–1.30) | 3 (6.4%) | 39 (83%) | 5 (10.6%) | 0.6113 | 1.37 (0.32–3.96) | 9 (19.2%) | 33 (70.2%) | 5 (10.6%) | 0.0016 | 3.54 (1.53–7.46) |
| MLH1 methylated | 10 (9%) | 87 (81%) | 11 (10%) | 0.4442 | 0.76 (0.35–1.48) | 8 (7.4%) | 89 (82.4%) | 11 (10.2%) | 0.1102 | 0.53 (0.23–1.09) | 8 (7.4%) | 92 (85.2%) | 8 (7.4%) | 0.5325 | 1.29 (0.54–2.71) | 26 (24%) | 76 (70%) | 6 (6%) | 6.84 × 10-9 | 4.91 (2.84–8.36) |
| Double somatic MMR mutationsc | 8 (11%) | 63 (86.3%) | 2 (2.7%) | 0.7074 | 0.85 (0.34–1.93) | 8 (11%) | 63 (86.3%) | 2 (2.7%) | 0.4687 | 1.39 (0.54–3.28) | 6 (8.2%) | 64 (87.7%) | 3 (4.1%) | 0.2100 | 1.93 (0.64–5.12) | 17 (23.3%) | 55 (75.3%) | 1 (1.4%) | 0.0002 | 3.75 (1.85–7.39) |
aMultiple logistic regression adjusted for CRC diagnosis age, sex and study cohort. Samples with unknown intratumoural presences and tumour characteristics were excluded in the statistical testing.
bExcludes MMRd CRCs with unexplained causes, primary and de novo MLH1 epimutation carriers.
cDouble MMR somatic pathogenic mutations identified using NGS-based techniques.
Statistically significant p < 0.05 values are in bold.
Tumour-infiltrating lymphocytes (TILs) are associated with F. nucleatum but not with pks+ E. coli or ETBF
The intratumoral presence of each of pks+ E. coli+, pks+ E. coli−, ETBF, and F. nucleatum bacteria and the association with mild or marked levels of TILs present (combined as TILs present) within the tumour microenvironment was tested. The presence of TILs was associated with F. nucleatum (P < 0.01, OR = 1.97, 95% CI = 1.35–2.85), but not with pks+ E. coli or ETBF when tested across all CRCs (Table 5). The MMRd status was associated with both F. nucleatum (P < 0.01; Table 2) and TILs (P < 0.01; Table 5), we performed a stratified analysis to test whether the association between TILs and F. nucleatum is independent of MMR status. When the CRCs were stratified by tumour MMR status, the association between TILs and F. nucleatum was no longer present for either MMRp or MMRd CRCs (Table 5). These findings were consistent in both EOCRC and LOCRC (Table 5). The APC: c.835-8 A > G somatic mutation showed an inverse association with the presence of TILs across all CRCs (P < 0.01, ORAPC:c.835-8A>G = 0.19, 95% CI = 0.05–0.53), however, this observation was no longer significant when only MMRp CRCs were included in the analysis (P = 0.056, ORAPC:c.835-8A>G = 0.32, 95% CI = 0.08–0.87) (Supplementary Table 4).
Table 5.
The association between tumour infiltrating lymphocytes (TILs) and intratumoral presences pks+ E. coli, ETBF and F nucleatum.
| Intratumoral bacteria | All CRCs | EOCRCs (CRC < 45 yrs) | LOCRCs (CRC ≥ 45 yrs) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TILs presenta | TILs absent | TILs unknown | P valueb | Adjusted Odd Ratio (95% CI) | TILs presenta | TILs absent | TILs unknown | P valueb | Adjusted Odd Ratio (95% CI) | TILs presenta | TILs absent | TILs unknown | P valueb | Adjusted Odd Ratio (95% CI) | |
| All CRCs | |||||||||||||||
| pks+ E. coli negative | 300 (22.4%) | 934 (69.8%) | 105 (7.8%) | - | Ref | 81 (22.4%) | 235 (64.9%) | 46 (12.7%) | - | Ref | 219 (22.4%) | 699 (71.5%) | 59 (6%) | - | Ref |
| pks+ E. coli+ detected | 34 (19.5%) | 131 (75.3%) | 9 (5.2%) | 0.2216 | 0.78 (0.51-1.15) | 11 (26.2%) | 28 (66.7%) | 3 (7.1%) | 0.8785 | 1.06 (0.48–2.2) | 23 (17.4%) | 103 (78%) | 6 (4.5%) | 0.1002 | 0.66 (0.40–1.06) |
| pks+ E. coli unknown | 39 (21.2%) | 130 (70.6%) | 15 (8.2%) | - | - | 9 (24%) | 24 (63%) | 5 (13%) | - | - | 30 (20.5%) | 106 (72.6%) | 10 (6.8%) | - | - |
| ETBF negative | 315 (21.7%) | 1023 (70.6%) | 113 (7.7%) | - | Ref | 81 (21.3%) | 248 (65.3%) | 51 (13.4%) | - | Ref | 234 (21.8%) | 775 (72.4%) | 62 (5.8%) | - | Ref |
| ETBF positive | 28 (27%) | 67 (64.4%) | 9 (8.6%) | 0.1887 | 1.36 (0.85-2.14) | 11 (37.9%) | 18 (62.1%) | 0 (0%) | 0.1743 | 1.75 (0.76–3.87) | 17 (22.7%) | 49 (65.3%) | 9 (12%) | 0.6984 | 1.12 (0.61–1.97) |
| ETBF unknown | 30 (21.1%) | 105 (74%) | 7 (4.9%) | - | - | 9 (27.3%) | 21 (63.6%) | 3 (9.1%) | - | - | 21 (19.3%) | 84 (77.1%) | 4 (3.7%) | - | - |
| Fn negative | 300 (20.8%) | 1032 (71.7%) | 107 (7.5%) | - | Ref | 76 (20.6%) | 247 (66.9%) | 46 (12.5%) | - | Ref | 224 (20.9%) | 785 (73.3%) | 62 (5.8%) | - | Ref |
| Fn positive | 51 (34%) | 86 (57.3%) | 13 (8.7%) | 0.0004 | 1.97 (1.35–2.85) | 18 (39.1%) | 23 (50%) | 5 (10.9%) | 0.0154 | 2.33 (1.16–4.62) | 33 (31.4%) | 63 (60%) | 9 (8.6%) | 0.0235 | 1.70 (1.06–2.67) |
| Fn unknown | 22 (20.8%) | 77 (72.6%) | 7 (6.6%) | - | - | 7 (25.9%) | 17 (63%) | 3 (11.1%) | - | - | 15 (19%) | 60 (75.9%) | 4 (5.1%) | - | - |
| MMR status | |||||||||||||||
| MMRp CRCs, n (%) | 215 (15.0%) | 1129 (78.8%) | 88 (6.2%) | - | - | 56 (15.4%) | 271 (74.5%) | 37 (10.1%) | - | - | 159 (14.9%) | 858 (80.3%) | 51 (4.8%) | - | - |
| MMRd CRCs, n (%) | 158 (59.8%) | 66 (25%) | 40 (15.2%) | 2 × 10−16 | 12.57 (9.15–17.44) | 45 (57.7%) | 16 (20.5%) | 17 (21.8%) | 1.2 × 10−15 | 13.61 (7.33–26.46) | 113 (60.4%) | 50 (26.7%) | 24 (12.8%) | 2 × 10−16 | 12.20 (8.44–17.84) |
| MMRp CRCs only | |||||||||||||||
| pks+ E. coli negative | 171 (15%) | 881 (79%) | 69 (6%) | - | Ref | 44 (14.8%) | 223 (74.8%) | 31 (10.4%) | - | Ref | 127 (15.4%) | 658 (79.9%) | 39 (4.7%) | - | Ref |
| pks+ E. coli positive | 19 (12.8%) | 123 (83.1%) | 6 (4.1%) | 0.3079 | 0.77(0.45–1.25) | 5 (16.1%) | 24 (77.4%) | 2 (6.5%) | 0.9504 | 0.97 (0.31–2.52) | 14 (12%) | 99 (84.6%) | 4 (3.4%) | 0.1999 | 0.68 (0.36–1.19) |
| pks+ E. coli unknown | 25 (15.4%) | 125 (77.2%) | 12 (7.4%) | - | - | 7 (20%) | 24 (68.6%) | 4 (11.4%) | - | - | 18 (14.2%) | 101 (79.5%) | 8 (6.3%) | - | - |
| ETBF negative | 188 (15.3%) | 968 (78.6%) | 75 (6.1%) | - | Ref | 49 (15.3%) | 236 (73.5%) | 36 (11.2%) | - | Ref | 139 (15.3%) | 732 (80.4%) | 40 (4.4%) | - | Ref |
| ETBF positive | 10 (12.8%) | 61 (78.2%) | 7 (9%) | 0.5959 | 0.83 (0.39–1.58) | 3 (17.6%) | 14 (82.4%) | 0 (0%) | 0.9142 | 1.07 (0.24–3.50) | 7 (11.5%) | 47 (77%) | 7 (11.5%) | 0.4645 | 0.74 (0.30–1.57) |
| ETBF unknown | 17 (13.9%) | 100 (82%) | 5 (4.1%) | - | - | 4 (15.4%) | 21 (80.8%) | 1 (3.8%) | - | - | 13 (13.5%) | 79 (82.3%) | 4 (4.2%) | - | - |
| Fn negative | 189 (15.1%) | 982 (78.7%) | 77 (6.2%) | - | Ref | 48 (15.2%) | 234 (74.3%) | 33 (10.5%) | - | Ref | 141 (15.1%) | 748 (80.1%) | 45 (4.8%) | - | Ref |
| Fn positive | 15 (16%) | 74 (78.7%) | 5 (5.3%) | 0.8544 | 1.06 (0.57–1.83) | 5 (17.9%) | 20 (71.4%) | 3 (10.7%) | 0.6890 | 1.24 (0.39–3.30) | 10 (15.2%) | 54 (81.8%) | 2 (3%) | 0.9470 | 0.98 (0.46–1.89) |
| Fn unknown | 11 (12.4%) | 73 (82%) | 5 (5.6%) | - | - | 3 (14%) | 17 (81%) | 1 (5%) | - | - | 8 (11.8%) | 56 (82.4%) | 4 (5.9%) | - | - |
| MMRd CRCs only | |||||||||||||||
| pks+ E. coli negative | 129 (60%) | 53 (25%) | 34 (15%) | - | Ref | 37 (57.8%) | 12 (18.8%) | 15 (23.4%) | - | Ref | 92 (60.1%) | 41 (26.8%) | 20 (13.1%) | - | Ref |
| pks+ E. coli positive | 15 (57.7%) | 8 (30.8%) | 3 (11.5%) | 0.6230 | 0.79 (0.32–2.10) | 6 (54.5%) | 4 (36.4%) | 1 (9.1%) | 0.3000 | 0.47 (0.11–2.11) | 9 (60%) | 4 (26.7%) | 2 (13.3%) | 0.7740 | 1.21 (0.34–4.97) |
| pks+ E. coli unknown | 14 (64%) | 5 (22%) | 3 (14%) | - | - | 2 (66.7%) | 0 (0%) | 1 (33.3%) | - | - | 12 (63.2%) | 5 (26.3%) | 2 (10.5%) | - | - |
| ETBF negative | 127 (58.3%) | 55 (25.2%) | 36 (16.5%) | - | Ref | 32 (54.2%) | 12 (20.4%) | 15 (25.4%) | - | Ref | 95 (59.4%) | 43 (26.9%) | 22 (13.8%) | - | Ref |
| ETBF positive | 18 (69.2%) | 6 (23.1%) | 2 (7.7%) | 0.7060 | 1.21 (0.46–3.58) | 8 (66.7%) | 4 (33.3%) | 0 (0%) | 0.7410 | 0.79 (0.19–3.57) | 10 (71.4%) | 2 (14.3%) | 2 (14.3%) | 0.4180 | 1.94 (0.46–13.36) |
| ETBF unknown | 13 (65%) | 5 (25%) | 2 (10%) | - | - | 5 (71.4%) | 0 (0%) | 2 (28.6%) | - | - | 8 (61.5%) | 5 (38.5%) | 0 (0%) | - | - |
| Fn negative | 111 (58.1%) | 50 (26.2%) | 30 (15.7%) | - | Ref | 28 (52%) | 13 (24%) | 13 (24%) | - | Ref | 83 (60.6%) | 37 (27%) | 17 (12.4%) | - | Ref |
| Fn positive | 36 (64.3%) | 12 (21.4%) | 8 (14.3%) | 0.4440 | 1.33 (0.65–2.88) | 13 (72.2%) | 3 (16.7%) | 2 (11.1%) | 0.3060 | 2.12 (0.55–10.55) | 23 (59%) | 9 (23.1%) | 7 (17.9%) | 0.7120 | 1.18 (0.50–2.95) |
| Fn unknown | 11 (64.7%) | 4 (23.5%) | 2 (11.8%) | - | - | 4 (66.7%) | 0 (0%) | 2 (33.3%) | - | - | 7 (63.6%) | 4 (36.4%) | 0 (0%) | - | - |
aMarked and mild tumour infiltrating lymphocytes were grouped to TILs present.
bMultiple logistic regression adjusted for CRC diagnosis age, sex and study cohort. Samples with unknown intratumoural presences and tumour characteristics were excluded in the statistical testing.
Statistically significant p < 0.05 values are in bold.
Discussion
In this study of three Australian-based CRC cohorts comprising 1697 CRCs, the intratumoral prevalence of the pks+ E. coli+, ETBF and F. nucleatum genotoxic gut bacteria was 10%, 6% and 9%, respectively. The prevalence of other non-E. coli bacteria harbouring the pks island (pks+ E. coli−) was 10%. An association between the APC: c.835-8 A > G somatic mutation and the intratumoral presence of pks+ E. coli+ was observed, although no association was observed between this somatic mutation and other bacteria harbouring the pks island that produces the genotoxin colibactin, highlighting the specific relationship between this hotspot mutation and pks+ E. coli+ bacteria. The presence of ETBF or F. nucleatum were each associated with MMRd as was the presence of both of these bacteria in the same CRC. The co-occurrence of all three bacteria in the same CRC was uncommon (0.4% of all CRCs), suggesting there is minimal interplay between these bacteria at the time of CRC diagnosis.
Pks+ E. coli
Colibactin-producing pks+ E. coli+ promotes CRC development by causing double-stranded DNA breakage [49, 50] and a specific pattern of mutational signature, namely SBS88 and ID18 [19]. The association between APC: c.835-8 A > G hotspot mutation and SBS88 have been identified in people with unexplained adenomatous polyposis [20], providing a mechanistic link and a potential biomarker for colibactin-induced tumorigenesis. This present study identified a significant association between the APC: c.835-8 A > G mutation and intratumoral pks+ E. coli+ but not with other pks harbouring bacteria (pks+ E. coli−), indicating the specific association of this mutation with pks+ E. coli+.
This study found that the pks+ E. coli+ tumours with the APC: c.835-8 A > G mutation were more prevalent in EOCRCs than LOCRCs (9.5% versus 5.3%), with only the association in EOCRCs showing statistical significance. The reason for this association in the EOCRCs is currently unknown and, if validated, raises interesting questions regarding the mechanism in EOCRC versus LOCRC. It has been hypothesised that early-life exposure to pks+ E. coli+ may influence early-onset tumorigenesis. Pks+ E. coli+ is a common gut bacteria found in ~31% of healthy infants by 1-month post-birth [51]. In addition, Lee-six et al. found that the colibactin-related mutation signature in normal colonic crypts from healthy young individuals and this was most active in younger children before reaching 10 years of age [52]. Therefore, the association between APC: c.835-8 A > G and pks+ E. coli+ in EOCRCs could be related to early-life exposure to the bacteria when our gut microbiome is still undergoing developmental changes [53], posing an especially “sensitive” period to extrinsic influences. Boot et al. argued that in later life when microbiome homoeostasis is established, individuals may be less susceptible to colibactin-related mutagenesis [54]. Studies aimed at prevention of colibactin-related EOCRC may need to focus on detection and eradication of pks+ E. coli+ in children.
In this study, APC: c.835-8 A > G mutation was detected in 3.3% of CRCs, a similar frequency (3.2%) to the previous report [55]. Whilst APC: c.835-8 A > G had a significant association with pks+ E. coli+, only a small subset (6.3%) of CRCs with pks+ E. coli+ had this mutation. As mentioned above, there might be a specific window when colibactin-induced damage is likely to occur, hence not causing the mutation in all CRCs exposed to the bacteria. Alternatively, prolonged exposure to this bacteria may be necessary to result in DNA damage. Studies suggest that up to 31% of healthy infants harbour pks+ E. coli+ by 1-month post-birth [51], though there still is no longitudinal study to investigate how long pks+ E. coli+ persists into the adult life and potentially induces CRC. Our study has focused on intratumoral pks+ E. coli+ at CRC diagnosis/resection and does not exclude prior pks+ E. coli+ infection. It is plausible that the association between APC: c.835-8 A > G and pks+ E. coli+ may be dependent on the duration of the exposure. Terlouw et al. [20], identified the APC: c.835-8 A > G mutation in premalignant adenomas from people with unexplained adenomatous polyposis supporting this mutation and colibactin-induced DNA damage as an early event in tumorigenesis, although further studies are needed to help elucidate this bacteria’s driver role during CRC development.
A recent study by Arima et al. [56] investigated intratumoral pks+ E. coli in 1175 CRCs collected as a part of two large prospective cohort studies. The authors found that intratumoral pks+ E. coli is associated with high western diet score, highlighting the interplay between gut bacterial pathogens and diet in CRC development. Consistent with the results reported by Arima et al. [56], this current study found no association between intratumoral pks+ E. coli+, and age at CRC diagnosis, tumour location, BRAF p.V600E mutation and CIMP-high. However, our study identified a significant association of pks+ E. coli+ with male sex, where this was not detected in Arima et al. Our study measured pks+ E. coli+ using two target genes for the pks island (ClbB) and E. coli (UidA), only capturing the presence of pks+ E. coli+, whereas Arima et al. [56] targeted only the pks island (ClbB) and, therefore, could not differentiate between pks+ in E. coli or pks+ in other bacteria. This may explain the association with the male gender, which was not present in our pks only analysis. In our study, there was no evidence of an association between pks+ E. coli+ and presence of elevated TILs, indicating that pks+ E. coli+ does not cause a highly immunogenic tumour microenvironment (TME), at least at the time of CRC diagnosis/resection.
ETBF
ETBF was significantly enriched in MMRd CRCs when compared with MMRp CRCs but not associated with any specific MMRd subgroups of either hereditary or sporadic etiology. This suggests that ETBF may be associated with the tumour microenvironment related to MMRd, rather than playing a causative role in CRC development. ETBF is infectious bacteria, which cause acute inflammation of the colon and shown to be a risk factor for colitis [57]. ETBF is present in colonic mucosa of people with familial adenomatous polyposis [58] and promotes oncogenic processes in a tumour-prone mice model (ApcMinΔ/+) [59].
Allen et al. reported that ETBF promotes loss of heterozygosity of Apc in the Apc mutant mice, however, the organoids exposed to ETBF showed near identical mutational profiles to unexposed controls, suggesting that ETBF does not cause a unique mutational signature unlike the SBS88 and ID18 signatures associated with pks+ E. coli [60]. This suggests that the carcinogenic mechanism of ETBF may not involve characteristic genomic aberrations and suggests alternative mechanisms including DNA methylation. Maiuri et al. examined genome-wide DNA methylation in mice exposed to ETBF. The authors reported that normal epithelium exposed to ETBF undergoes inflammation-driven tumorigenesis and caused a unique DNA methylation signature [61]. Interestingly, these methylation aberrations were abrogated in mice with dysfunctional Msh2, however, yet still promoting tumorigenesis. This suggests a multi-faceted role of ETBF and warrants further investigation of ETBF as the tumorigenic instigator and the risk mediator via modifying the epigenome.
F. nucleatum
F. nucleatum was associated with tumour characteristics related to MMRd and CRCs of the serrated pathway (e.g., right location, BRAF p.V600E, CIMP-high), consistent with previous reports [62, 63]. In addition, our study identified that F. nucleatum was associated with MMRd related to Lynch syndrome as well as sporadic MMRd CRCs related to MLH1 promoter methylation and double MMR somatic mutations. These findings suggest that F. nucleatum colonisation in the tumour is not related to MMRd etiology. This supports F. nucleatum causing opportunistic infections [64], exploiting the specific tumour microenvironment of MMRd CRCs.
Though F. nucleatum is found to be highly abundant in CRC tumours [64–66], the oncogenic mechanism of F. nucleatum remains to be elucidated, especially lacking are studies investigating their effect on the host genetic and epigenetics. Our study identified an association between TILs and F. nucleatum and this suggests the favourable tumour microenvironment of F. nucleatum. Given this association was no longer significant in the stratified analysis by MMR status, it may suggest that this interrelationship is dependent from the overrepresentation of TILs in MMRd CRCs, rather than suggesting a causative role of F. nucleatum.
Strengths and weaknesses of this study
Our study has several strengths including a large sample size from three CRC cohorts that have extensive molecular characterisation. Our study was the first to screen for intratumoral presence of these three CRC-associated bacteria but also to investigate the association between pks+ E. coli and the APC: c.835-8 A > G somatic mutation that is mechanistically linked to colibactin-related DNA damage. Our study also provided a stratified analysis of clinically relevant MMRd subgroups of both hereditary and sporadic etiologies, providing further insight into the nature of the association between MMRd and these bacteria. Further, by including a large number of EOCRCs, our study has provided separate analysis on EOCRCs, which is an emerging health problem, globally [67]. A further strength of our study was the use of assays that targeted both the pks island and E. coli, ensuring differentiation pks+ E. coli+ from other pks harbouring bacteria. This differentiation highlighted the specific association between pks+ E. coli+ and the APC: c.835-8 A > G somatic mutation.
The limitations of this study include the cross-sectional study design where in our participants, the prior infection of these bacteria could not be examined. Ex vivo or in vitro studies such as organoids co-culture experiments [19, 60] may further elucidate the direct effect these bacteria may have on colonic mucosa. Additionally, bacterial screening on premalignant polyps may help strengthen the early role of these bacteria in driving colorectal neoplasm.
In this study, we utilised FFPE specimens for detecting intratumoral bacteria. Although this biospecimen type is commonly used in such studies [12, 56], a recent meta-analysis indicated that the biospecimen type could influence the detection efficacy and affect the results [68]. Further studies performed on different biospecimen types (e.g., fresh frozen) may be needed to validate the findings from our study. Another limitation includes the lack of TNM stage information.
Conclusion
This study provides novel findings on specific molecular features and pathways of tumorigenesis associated with each genotoxic gut bacterium. The strength of the association between the presence of intratumoral pks+ E. coli+ and APC: c.835-8 A > G somatic mutation is shown for the first time. This has important clinical implication as the APC: c.835-8 A > G somatic mutation may represent a biomarker for colibactin-induced DNA damage in CRC tumours caused by pks+ E. coli+. This finding provides new opportunities for future studies on prevention and treatments of bacterial-driven CRCs.
Supplementary information
{kind=link}
{kind=link}
Acknowledgements
We thank the members of the Colorectal Oncogenomics Group (The University of Melbourne), in particular Emma Barrance. We thank the participants and staff from the ANGELS study, the Colon Cancer Family Registry (CCFR) and the Melbourne Collaborative Cohort Study (MCCS). We especially thank Maggie Angelakos, Samantha Fox, Allyson Templeton for facilitating CCFR resources and supporting this study. BML was supported by a Mid-Career Fellowship from the Victorian Cancer Agency (MRCF-18005). MCS is supported by a Level 3 Investigator Grant from the National Health and Medical Research Council, Australia, GNT2017325). DDB is supported by an NHMRC Investigator grant (GNT1194896) and the University of Melbourne Dame Kate Campbell Fellowship. The E. coli isolate provided was collected as part of the Controlling Superbugs study, funded by the Melbourne Genomics Health Alliance (supported by the State Government of Victoria). The Australasian Colon Cancer Family Registry (ACCFR) is supported in part by funding from the National Cancer Institute (NCI), National Institutes of Health (NIH) (award U01 CA167551). Support for case ascertainment was provided in part from the Victoria Cancer Registry (Australia). The content of this manuscript is solely the responsibility of the authors and does not necessarily reflect the views or policies of the NIH or any of the collaborating centres in the CCFR, nor does mention of trade names, commercial products, or organisations imply endorsement by the US Government, any cancer registry, or the CCFR.
Author contributions
JEJ and DDB conceptualised the study. JEJ, YLC, RW, MC, JC, and SGP performed laboratory experiments. JEJ, YLC, KM, PG, and ALM performed statistical analysis. SJ, ND, JT, DI, NS, AM, BML, RLM, MCS, JLH, AKW, FAM, IMW, and MAJ contributed to the acquisition of study participants and data. DI and NS contributed to the acquisition of materials needed for the laboratory testing. CR oversaw the pathological reviews of tumour specimens. JEJ, YLC and DDB wrote the manuscript. All authors read and provided critical revisions to the manuscript and approved the final manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Data availability
The data and materials presented in this publication were collected according to the guidelines of the Australian Colon Cancer Family Registry, Melbourne Collaborative Cohort Study and the ANGELS studies. The data generated in this study can be accessed via request to the Colon Cancer Family Registry (https://www.coloncfr.org/collaboration) or via contacting the corresponding author (DDB) and study investigators for the MCCS and ANGELS studies.
Competing interests
The authors declare no competing interests.
Ethics approval
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Cancer Council of Victoria (IEC 9001) and University of Melbourne Research Ethics Committees (13094).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jihoon E. Joo, Yen Lin Chu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-023-02554-x.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Australian Institute of Health and Welfare 2017. Cancer in Australia 2017. Cancer series no.101. Cat. no. CAN 100. Canberra: AIHW.
- 3.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–30. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 4.He X, Wu K, Ogino S, Giovannucci EL, Chan AT, Song M. Association between risk factors for colorectal cancer and risk of serrated polyps and conventional adenomas. Gastroenterology. 2018;155:355–73.e318. doi: 10.1053/j.gastro.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12:661–72. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 6.Grady WM. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003;124:1574–94. doi: 10.1016/s0016-5085(03)00376-7. [DOI] [PubMed] [Google Scholar]
- 7.Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66:70–78. doi: 10.1136/gutjnl-2015-309800. [DOI] [PubMed] [Google Scholar]
- 8.Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727. doi: 10.1038/ncomms9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyadorai T, Mariappan V, Vellasamy KM, Wanyiri JW, Roslani AC, Lee GK, et al. Prevalence and association of pks+ Escherichia coli with colorectal cancer in patients at the University Malaya Medical Centre, Malaysia. PLoS One (Artic) 2020;15:1–13. doi: 10.1371/journal.pone.0228217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, He H, Xu H, Li Y, Li Z, Du Y, et al. Association of oncogenic bacteria with colorectal cancer in South China. Oncotarget. 2016;7:80794–802. doi: 10.18632/oncotarget.13094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, et al. Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncol. 2015;1:653–61. doi: 10.1001/jamaoncol.2015.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clermont O, Bonacorsi S, Bingen E. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl Environ Microbiol. 2000;66:4555–8. doi: 10.1128/AEM.66.10.4555-4558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–51. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 15.Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue M, Kim CS, Healy AR, Wernke KM, Wang Z, Frischling MC et al. Structure elucidation of colibactin and its DNA cross-links. Science. 2019;365. 10.1126/science.aax2685. [DOI] [PMC free article] [PubMed]
- 17.Bossuet-Greif N, Vignard J, Taieb F, Mirey G, Dubois D, Petit C et al. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. mBio. 2018;9. 10.1128/mBio.02393-17. [DOI] [PMC free article] [PubMed]
- 18.Iftekhar A, Berger H, Bouznad N, Heuberger J, Boccellato F, Dobrindt U, et al. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat Commun. 2021;12:1003. doi: 10.1038/s41467-021-21162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature. 2020;580:269–73. doi: 10.1038/s41586-020-2080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terlouw D, Suerink M, Boot A, van Wezel T, Nielsen M, Morreau H. Recurrent APC splice variant c.835-8A>G in patients with unexplained colorectal polyposis fulfilling the colibactin mutational signature. Gastroenterology. 2020;159:1612–4.e1615. doi: 10.1053/j.gastro.2020.06.055. [DOI] [PubMed] [Google Scholar]
- 21.Sears CL, Islam S, Saha A, Arjumand M, Alam NH, Faruque AS, et al. Association of enterotoxigenic Bacteroides fragilis infection with inflammatory diarrhea. Clin Infect Dis Off Publ Infect Dis Soc Am. 2008;47:797–803. doi: 10.1086/591130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344–55. doi: 10.1016/j.immuni.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–15. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newcomb PA, Baron J, Cotterchio M, Gallinger S, Grove J, Haile R, et al. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomark Prev. 2007;16:2331–43. doi: 10.1158/1055-9965.Epi-07-0648. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins MA, Win AK, Templeton AS, Angelakos MS, Buchanan DD, Cotterchio M et al. Cohort profile: the colon cancer family registry cohort (CCFRC). Int J Epidemiol. 2018. 10.1093/ije/dyy006. [DOI] [PMC free article] [PubMed]
- 28.Buchanan DD, Clendenning M, Rosty C, Eriksen SV, Walsh MD, Walters RJ, et al. Tumor testing to identify Lynch syndrome in two Australian colorectal cancer cohorts. J Gastroenterol Hepatol. 2017;32:427–38. doi: 10.1111/jgh.13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giles GG, English DR. The Melbourne collaborative cohort study. IARC Sci Publ. 2002;156:69–70. [PubMed] [Google Scholar]
- 30.Milne RL, Fletcher AS, MacInnis RJ, Hodge AM, Hopkins AH, Bassett JK, et al. Cohort profile: the Melbourne collaborative cohort study (Health 2020) Int J Epidemiol. 2017;46:1757–1757i. doi: 10.1093/ije/dyx085. [DOI] [PubMed] [Google Scholar]
- 31.Buchanan DD, Rosty C, Clendenning M, Spurdle AB, Win AK. Clinical problems of colorectal cancer and endometrial cancer cases with unknown cause of tumor mismatch repair deficiency (suspected Lynch syndrome) Appl Clin Genet. 2014;7:183–93. doi: 10.2147/TACG.S48625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georgeson P, Pope BJ, Rosty C, Clendenning M, Mahmood K, Joo JE, et al. Evaluating the utility of tumour mutational signatures for identifying hereditary colorectal cancer and polyposis syndrome carriers. Gut (Artic) 2021;70:2138–49. doi: 10.1136/gutjnl-2019-320462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker R, Georgeson P, Mahmood K, Joo JE, Makalic E, Clendenning M, et al. Evaluating multiple next-generation sequencing–derived tumor features to accurately predict DNA mismatch repair status. J Mol Diagn. 2023;25:94–109. doi: 10.1016/j.jmoldx.2022.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosty C, Young JP, Walsh MD, Clendenning M, Walters RJ, Pearson S, et al. Colorectal carcinomas with KRAS mutation are associated with distinctive morphological and molecular features. Mod Pathol. 2013;26:825–34. doi: 10.1038/modpathol.2012.240. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins MA, Hayashi S, O’Shea AM, Burgart LJ, Smyrk TC, Shimizu D, et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology. 2007;133:48–56. doi: 10.1053/j.gastro.2007.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindor NM, Burgart LJ, Leontovich O, Goldberg RM, Cunningham JM, Sargent DJ, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol. 2002;20:1043–8. doi: 10.1200/jco.2002.20.4.1043. [DOI] [PubMed] [Google Scholar]
- 37.Cicek MS, Lindor NM, Gallinger S, Bapat B, Hopper JL, Jenkins MA, et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J Mol Diagn. 2011;13:271–81. doi: 10.1016/j.jmoldx.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walsh MD, Buchanan DD, Cummings MC, Pearson SA, Arnold ST, Clendenning M, et al. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16:2214–24. doi: 10.1158/1078-0432.CCR-09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker R, Mahmood K, Joo JE, Clendenning M, Georgeson P, Como J, et al. A tumor focused approach to resolving the etiology of DNA mismatch repair deficient tumors classified as suspected Lynch syndrome. J Transl Med. 2023;21:282. doi: 10.1186/s12967-023-04143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poynter JN, Siegmund KD, Weisenberger DJ, Long TI, Thibodeau SN, Lindor N, et al. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol Biomark Prev. 2008;17:3208–15. doi: 10.1158/1055-9965.Epi-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clendenning M, Walsh MD, Gelpi JB, Thibodeau SN, Lindor N, Potter JD, et al. Detection of large scale 3′ deletions in the PMS2 gene amongst Colon-CFR participants: have we been missing anything? Fam Cancer. 2013;12:563–6. doi: 10.1007/s10689-012-9597-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchanan DD, Tan YY, Walsh MD, Clendenning M, Metcalf AM, Ferguson K, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol. 2014;32:90–100. doi: 10.1200/JCO.2013.51.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pope BJ, Clendenning M, Rosty C, Mahmood K, Georgeson P, Joo JE, et al. Germline and tumor sequencing as a diagnostic tool to resolve suspected Lynch syndrome. J Mol Diagn. 2021;23:358–71. doi: 10.1016/j.jmoldx.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosty C, Buchanan DD, Walsh MD, Pearson SA, Pavluk E, Walters RJ, et al. Phenotype and polyp landscape in serrated polyposis syndrome: a series of 100 patients from genetics clinics. Am J Surg Pathol. 2012;36:876–82. doi: 10.1097/PAS.0b013e31824e133f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buchanan DD, Sweet K, Drini M, Jenkins MA, Win AK, English DR, et al. Risk factors for colorectal cancer in patients with multiple serrated polyps: a cross-sectional case series from genetics clinics. PloS One. 2010;5:e11636. doi: 10.1371/journal.pone.0011636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 47.Sherry NL, Lee RS, Gorrie CL, Kwong JC, Stuart RL, Korman TM, et al. Pilot study of a combined genomic and epidemiologic surveillance program for hospital-acquired multidrug-resistant pathogens across multiple hospital networks in Australia. Infect Control Hosp Epidemiol. 2021;42:573–81. doi: 10.1017/ice.2020.1253. [DOI] [PubMed] [Google Scholar]
- 48.Sherry NL, Lee RS, Gorrie CL, Kwong JC, Stuart RL, Korman T et al. Genomic interrogation of the burden and transmission of multidrug-resistant pathogens within and across hospital networks. 2019:764787; 10.1101/764787%JbioRxiv.
- 49.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PloS One. 2013;8:e56964. doi: 10.1371/journal.pone.0056964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 2014;20:859–67. doi: 10.1158/1078-0432.CCR-13-1343. [DOI] [PubMed] [Google Scholar]
- 51.Tsunematsu Y, Hosomi K, Kunisawa J, Sato M, Shibuya N, Saito E, et al. Mother-to-infant transmission of the carcinogenic colibactin-producing bacteria. BMC Microbiol. 2021;21:235. doi: 10.1186/s12866-021-02292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532–7. doi: 10.1038/s41586-019-1672-7. [DOI] [PubMed] [Google Scholar]
- 53.Arrieta MC, Stiemsma LT, Amenyogbe N, Brown EM, Finlay B. The intestinal microbiome in early life: health and disease. Front Immunol. 2014;5:427. doi: 10.3389/fimmu.2014.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boot A, Ng AWT, Chong FT, Ho SC, Yu W, Tan DSW, et al. Characterization of colibactin-associated mutational signature in an Asian oral squamous cell carcinoma and in other mucosal tumor types. Genome Res. 2020;30:803–13. doi: 10.1101/gr.255620.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33:125–136 e123. doi: 10.1016/j.ccell.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arima K, Zhong R, Ugai T, Zhao M, Haruki K, Akimoto N, et al. Western-style diet, pks island-carrying escherichia coli, and colorectal cancer: analyses from two large prospective cohort studies. Gastroenterology. 2022;163:862–74. doi: 10.1053/j.gastro.2022.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rabizadeh S, Rhee KJ, Wu S, Huso D, Gan CM, Golub JE, et al. Enterotoxigenic bacteroides fragilis: a potential instigator of colitis. Inflamm Bowel Dis. 2007;13:1475–83. doi: 10.1002/ibd.20265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359:592–7. doi: 10.1126/science.aah3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung L, Orberg ET, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23:421. doi: 10.1016/j.chom.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen J, Rosendahl Huber A, Pleguezuelos-Manzano C, Puschhof J, Wu S, Wu X, et al. Colon tumors in enterotoxigenic bacteroides fragilis (ETBF)-colonized mice do not display a unique mutational signature but instead possess host-dependent alterations in the APC gene. Microbiol Spectr. 2022;10:e0105522. doi: 10.1128/spectrum.01055-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maiuri AR, Peng M, Podicheti R, Sriramkumar S, Kamplain CM, Rusch DB, et al. Mismatch repair proteins initiate epigenetic alterations during inflammation-driven tumorigenesis. Cancer Res. 2017;77:3467–78. doi: 10.1158/0008-5472.CAN-17-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2016;65:1973–80. doi: 10.1136/gutjnl-2015-310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014;74:1311–8. doi: 10.1158/0008-5472.CAN-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brennan CA, Garrett WS. Fusobacterium nucleatum—symbiont, opportunist and oncobacterium. Nat Rev Microbiol. 2019;17:156–66. doi: 10.1038/s41579-018-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song M, Chan AT, Sun J. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology. 2020;158:322–40. doi: 10.1053/j.gastro.2019.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ugai T, Sasamoto N, Lee HY, Ando M, Song M, Tamimi RM, et al. Is early-onset cancer an emerging global epidemic? Current evidence and future implications. Nat Rev Clin Oncol. 2022;19:656–73. doi: 10.1038/s41571-022-00672-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim Y, Cho NY, Kang GH. Prognostic and clinicopathological significance of Fusobacterium nucleatum in colorectal cancer: a systemic review and meta-analysis. J Pathol Transl Med. 2022;56:144–51. doi: 10.4132/jptm.2022.03.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and materials presented in this publication were collected according to the guidelines of the Australian Colon Cancer Family Registry, Melbourne Collaborative Cohort Study and the ANGELS studies. The data generated in this study can be accessed via request to the Colon Cancer Family Registry (https://www.coloncfr.org/collaboration) or via contacting the corresponding author (DDB) and study investigators for the MCCS and ANGELS studies.