

Key Points
-
•
In a mouse model of oligoclonal MPN, IL-1β favors disease initiation by promoting early expansion of a subclinical JAK2-V617F clone.
-
•
Anti–IL-1β antibody treatment during the early expansion phase of the JAK2-mutant clone reduced the frequency of MPN disease initiation.
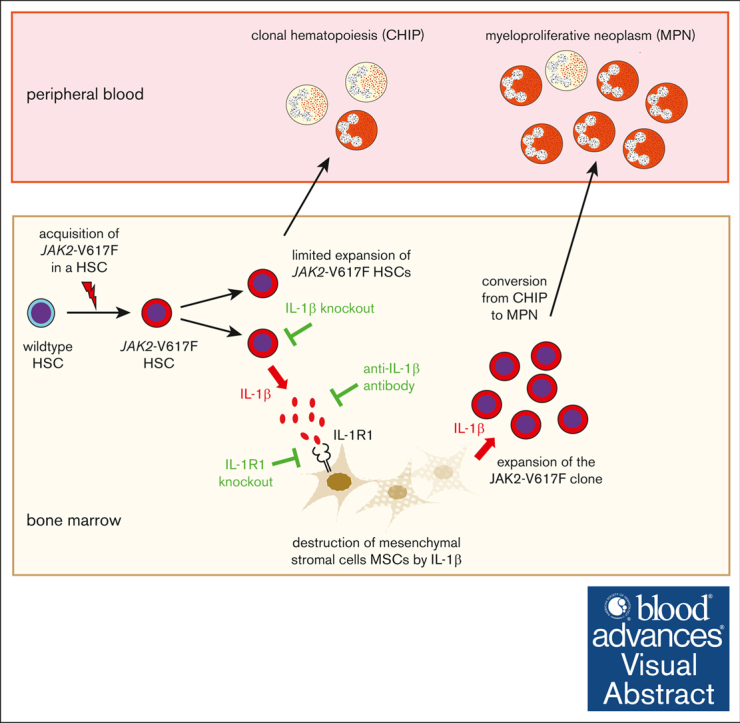
Visual Abstract
Abstract
JAK2-V617F is the most frequent somatic mutation causing myeloproliferative neoplasm (MPN). JAK2-V617F can be found in healthy individuals with clonal hematopoiesis of indeterminate potential (CHIP) with a frequency much higher than the prevalence of MPNs. The factors controlling the conversion of JAK2-V617F CHIP to MPN are largely unknown. We hypothesized that interleukin-1β (IL-1β)–mediated inflammation can favor this progression. We established an experimental system using bone marrow (BM) transplantations from JAK2-V617F and GFP transgenic (VF;GFP) mice that were further crossed with IL-1β−/− or IL-1R1−/− mice. To study the role of IL-1β and its receptor on monoclonal evolution of MPN, we performed competitive BM transplantations at high dilutions with only 1 to 3 hematopoietic stem cells (HSCs) per recipient. Loss of IL-1β in JAK2-mutant HSCs reduced engraftment, restricted clonal expansion, lowered the total numbers of functional HSCs, and decreased the rate of conversion to MPN. Loss of IL-1R1 in the recipients also lowered the conversion to MPN but did not reduce the frequency of engraftment of JAK2-mutant HSCs. Wild-type (WT) recipients transplanted with VF;GFP BM that developed MPNs had elevated IL-1β levels and reduced frequencies of mesenchymal stromal cells (MSCs). Interestingly, frequencies of MSCs were also reduced in recipients that did not develop MPNs, had only marginally elevated IL-1β levels, and displayed low GFP-chimerism resembling CHIP. Anti–IL-1β antibody preserved high frequencies of MSCs in VF;GFP recipients and reduced the rate of engraftment and the conversion to MPN. Our results identify IL-1β as a potential therapeutic target for preventing the transition from JAK2-V617F CHIP to MPNs.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal disorders of the hematopoietic stem cell (HSC) in most cases caused by activating mutations in JAK2, CALR, or MPL that increase the proliferation of erythroid, megakaryocytic, and myeloid lineages.1, 2, 3, 4, 5, 6, 7 MPNs can be subclassified as polycythemia vera, essential thrombocythemia, and primary myelofibrosis.8,9 JAK2-V617F is the most frequent driver gene mutation in patients with MPN, but JAK2-V617F can also be detected as clonal hematopoiesis of indeterminate potential (CHIP) in healthy individuals, with a frequency much higher than the incidence of MPN.10, 11, 12, 13 Thus, the acquisition of JAK2-V617F is not the rate-limiting step for developing MPN, and there are factors that can suppress or promote the expansion of the JAK2 mutated clone and thereby control the transition from CHIP to MPN.
Inflammation is a hallmark of advanced MPN.14 Chronic immune stimulation in patients with a history of infectious or autoimmune diseases was associated with increased risk of myeloid malignancies including MPN.15, 16, 17 Therefore, we hypothesized that inflammation might be one of the factors promoting the transition from JAK2-V617F–positive CHIP to MPN. Interleukin-1β (IL-1β), a pleiotropic cytokine with diverse innate and adaptive immune functions, is a master regulator of inflammatory state18,19 and has been implicated in promoting several hematological malignancies.20,21 Previous studies have shown that IL-1β is favoring progression to myelofibrosis at advanced stages of MPN.22,23
Here, we examined the role of IL-1β in the very early stages of MPN initiation by JAK2-V617F using competitive bone marrow (BM) transplantations at high dilutions (1:100), with very few (1-3) long-term HSCs (LT-HSCs). The initiation of MPN from a single mutant HSCs is fundamentally different from the progression of chronic phase MPN to myelofibrosis, which is mediated by profibrotic cytokines such as transforming growth factor β and is linked to augmented megakaryopoiesis and the emergence of myofibroblasts and monocyte-derived profibrotic fibrocytes.
We found that IL-1β secreted by JAK2-mutant hematopoietic cells promotes the early expansion of the JAK2-V617F clone and requires the presence of the IL-1 receptor type 1 (IL-1R1) on the responding BM stromal cells. Genetic ablation of IL-1β or IL-1R1 and treatment with a neutralizing IL-1β antibody reduces MPN disease initiation.
Methods
Mice
We used conditional JAK2-V617F transgenic mice,24 SclCreER mice,25 UBC-GFP mice,26 IL-1β–knockout mice,27 and IL-1R1–knockout mice.28 SclCreER;V617F (VF) mice were characterized previously.29 CreER was activated by intraperitoneal injections of 2 mg tamoxifen (Sigma Aldrich) for 5 consecutive days. All mice were of pure C57BL/6N background and kept under specific pathogen-free conditions with free access to food and water in accordance with Swiss federal regulations. All animal experiments were approved by the Cantonal Veterinary Office of Basel-Stadt, Switzerland.
Blood samples and clinical data of patients with MPN were collected at the University Hospital Basel, Switzerland. Blood samples from healthy controls were obtained from the local blood donation center (Stiftung Blutspendezentrum SRK beider Basel). The study was approved by the local ethics committee (Ethik Kommission Beider Basel). Written informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
BM transplantations and drug treatment
BM from transgenic mice was harvested 6 to 8 weeks after tamoxifen induction, mixed with wild-type BM competitor cells, and injected into the tail vein of lethally irradiated recipient mice (12 Gy). Blood was drawn from the tail vein every 4 to 6 weeks. Green fluorescent protein (GFP)-chimerism was assessed by flow cytometry, and complete blood counts were performed on Advia120 Hematology Analyzer with Multispecies software version 5.9.0-MS (Bayer). Aspirin (Sigma) was added to drinking water (150 μg/mL) and bottles were changed every 3 to 4 days. Mouse immunoglobulin G2a (IgG2a) anti-mouse IL-1β antibody (01BSUR; Novartis Pharma AG, Basel, Switzerland),30, 31, 32 or mouse IgG2a isotype were injected intraperitoneally once per week (10 mg/kg every week, intraperitoneal).
Results
Loss of IL-1β from JAK2-mutant hematopoietic cells reduces the rate of MPN disease initiation
To test the hypothesis that IL-1β favors MPN disease initiation by promoting the early expansion of the JAK2-V617F clone, we used competitive BM transplantations at limiting dilutions that mimic the monoclonal origin of MPN.33 As BM donors, we used triple-transgenic SclCreER;JAK2-V617F;UBC-GFP (VF;GFP) mice that allow tracking of the transplanted BM cells by detection of GFP. VF;GFP mice were crossed with IL-1β−/− mice to obtain VF;IL-1β−/−;GFP mice. Expression of the VF transgene was induced with tamoxifen. After 6 to 8 weeks, 2 × 104 unfractionated BM cells containing only 1 to 3 JAK2-mutant LT-HSCs were mixed with a 100-fold excess of IL-1β−/− BM competitor cells and transplanted into recipient mice (Figure 1A).33 Within 36 weeks, ∼60% of these recipients developed MPN phenotype characterized by elevated hemoglobin and/or platelet counts and splenomegaly (Figure 1B). Mice with MPN phenotype also showed increased IL-1β levels in the plasma and BM, whereas mice without MPN phenotype displayed very low IL-1β levels (Figure 1C). Very similar results were obtained in experiments when WT BM competitor cells were used instead of IL-1β−/− competitors (supplemental Figure 1), indicating that WT competitor cells did not contribute to the increased IL-1β production.
Figure 1.


Loss of IL-1β from JAK2-mutant hematopoietic cells reduces MPN disease initiation. (A) Schematic drawing of the experimental setup for competitive transplantation at 1:100 dilution. BM from VF;GFP or VF;IL-1β−/−;GFP donor mice was mixed with a 100-fold excess of BM competitor cells from an IL-1β−/− donor. (B) Time course of blood counts from individual mice that received BM from VF;GFP (upper panel) or VF;IL-1β−/−;GFP donors (lower panel). (C) IL-1β protein levels in plasma and BM lavage (1 femur and 1 tibia) of mice with or without MPN phenotype is shown (right panel). Nonparametric Mann-Whitney 2-tailed t test was performed for statistical comparisons. (D) Time courses of GFP chimerism in the peripheral blood are shown. Multiple t tests were performed for statistical analyses. (E) Bar graphs show the percentages of mice that showed engraftment defined as GFP chimerism of >1% at 18 weeks after transplantation and the percentages of mice that developed MPN phenotype (elevated hemoglobin and/or platelet counts). P values in lower panel were computed using Fisher exact test. (F) Time course of mean blood counts and GFP chimerism in the peripheral blood of WT mice transplanted with BM from VF;GFP or VF;IL-1β−/−;GFP and IL-1β−/− competitor cells that developed MPN phenotype during 36-weeks follow-up. Multiple t tests were performed for statistical analyses. (G) GFP chimerism in HSPCs at 36 weeks after transplantation in the BM (left) and spleen (right) of WT mice transplanted with BM from VF;GFP or VF;IL-1β−/−;GFP and IL-1β−/− competitor cells that developed MPN phenotype. Multiple t tests were performed for statistical analyses. (H) Representative images of reticulin fibrosis staining in the BM (left panel) and hematoxylin and eosin staining in the spleen (right panel) of mice showed MPN phenotype at 36 weeks after transplantation. Histological grade of reticulin fibrosis in BM is shown in a bar graph (right). (I) Levels of inflammatory cytokines in BM lavage (1 femur and 1 tibia) and plasma of mice that displayed MPN phenotype at 36 weeks after transplantation. (J) Levels of IL-1 cytokines in the BM. Gray shaded area represents normal range. All data are presented as mean ± standard error of the mean (SEM); ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; and ∗∗∗∗P < .0001. See also supplemental Figures 1 and 2.
Transplantation of VF;IL-1β−/−;GFP BM with IL-1β−/− competitor cells at a 1:100 ratio produced considerably fewer recipients with MPN phenotype than mice that received VF;GFP BM, and only 1 mouse showed thrombocytosis (Figure 1B, lower panel). Recipients with MPN phenotype did not show increased IL-1β levels, suggesting that excess IL-1β was produced by the JAK2-V617F expressing cells (Figure 1C). Recipients transplanted with VF;GFP BM showed higher GFP chimerism (Figure 1D), higher percentage of engraftment (90% vs 66%) defined as GFP chimerism of >1% in peripheral blood Gr1+ granulocytes, and a higher percentage of MPN initiation (48% vs 12%) than mice transplanted with VF;IL-1β−/−;GFP BM (Figure 1E). We have previously shown that VF and VF;IL-1β−/− mice have comparable frequencies of HSCs in the BM.22
When only recipients with MPN phenotype were compared, blood counts and peripheral blood GFP chimerism were similar, except for slightly lower platelet count and GFP chimerism in platelets of VF;IL-1β−/−;GFP recipients (Figure 1F). Furthermore, mice transplanted with VF;IL-1β−/−;GFP BM that developed MPN phenotype showed lower GFP chimerism in hematopoietic stem and progenitor cells (HSPCs; Figure 1G), less reticulin fibrosis and osteosclerosis in the BM, and partial restoration of splenic architecture (Figure 1H). Inflammatory cytokines were largely unchanged, except for a trend toward lower IL-6 levels (Figure 1I). Loss of IL-1β in VF donor cells resulted in reduced levels of IL-1α in BM of mice that developed MPN (Figure 1J). Interestingly, loss of IL-1β in VF donor cells also increased the levels of IL-1R antagonist (IL-1Ra). As a result, the ratio of IL-1Ra to IL-1α increased in VF;IL-1β−/− recipients with MPN phenotype, correlating with the less severe MPN phenotype and less myelofibrosis than recipients of VF BM.
Finally, BM transplantations into IL-1β−/− recipients using IL-1β−/− competitor cells demonstrated that VF BM cells alone were the source for the increased IL-1β levels in the plasma and BM (supplemental Figure 2A, upper panel). Unexpectedly, IL-1β−/− recipients transplanted with VF;IL-1β−/− BM showed higher frequencies of engraftment and MPN disease initiation than expected and almost equal to IL-1β−/− recipients transplanted with VF BM (supplemental Figure 2A, middle panel). GFP chimerism was reduced when all recipient mice were considered (supplemental Figure 2A, lower panel), but when only mice that developed MPN were compared, very few alterations were noted (supplemental Figure 2B). At 36 weeks, IL-1β−/− recipients transplanted with VF;IL-1β−/− BM showed a trend toward reduced reticulin fibrosis, reduced GFP chimerism in HSPCs, and partial restoration of splenic architecture (supplemental Figure 2C-D), and a trend toward reduction of IL-6 and KC/GRO levels (supplemental Figure 2E). The reason why engraftment and MPN disease initiation increased when IL-1β was absent in both donors and recipients is currently unknown. No increase in IL-1Ra or in the ratio between IL-1α and IL-1Ra was found in IL-1β−/− recipients transplanted with VF;IL-1β−/− BM (supplemental Figure 2F), suggesting that lack of upregulation of IL-1RA might contribute to the more efficient MPN disease initiation. Complete loss of IL-1β partially prevented this outcome, possibly because of diminished anti–IL-1 inflammatory responses in the BM.
Overall, our data show that loss of IL-1β in hematopoietic cells reduced MPN initiation, suggesting that IL-1β produced by the JAK2-mutant cells is promoting early expansion of the JAK2-V617F clone and conversion to MPN.
JAK2-V617F mutant HSCs need IL-1β for optimal expansion of the HSC population with long-term repopulation capacity
To examine the effects of IL-1β loss on mutant HSC function, we performed secondary transplantations into WT recipients (Figure 2). As donors, we selected 3 recipients of VF;GFP BM and 3 recipients of VF;IL-1β−/−;GFP BM that displayed MPN phenotype (supplemental Figure 3). BM cells were pooled and used for noncompetitive (Figure 2A-C) and competitive secondary transplantations (Figure 2D-F). Secondary recipients of VF;IL-1β−/−;GFP BM showed significantly reduced blood counts and lower GFP chimerism in the peripheral blood, BM, and spleen than recipients of VF;GFP BM (Figure 2B-C,E-F; supplemental Figure 4A-B). By comparing the frequencies of engraftment (defined as >1% in Gr1+ granulocytes at 18 weeks) between the competitive and noncompetitive secondary transplantations, and using the "extreme limiting dilution analysis" algorithm,34 we estimated that the numbers of functional HSCs were approximately fivefold reduced in donor mice that initially received VF;IL-1β−/− BM compared with in donors initially transplanted with VF BM (Figure 2G). Overall, the HSC pool in primary recipient mice transplanted with 1 to 3 HSCs from VF donors expanded to a total of ∼300 functional HSCs after 36 weeks, whereas primary recipients transplanted with 1 to 3 HSCs from VF;IL-1β−/− donors had only ∼50 functional HSCs. These results indicate that JAK2-V617F mutant HSCs require IL-1β for optimal expansion of the HSC population with long-term repopulation capacity.
Figure 2.

JAK2-V617F HSCs need IL-1β for long-term stem cell function. (A) Schematic drawing of noncompetitive (1:0) transplantations. Primary recipients of VF;GFP or VF;IL-1β−/−;GFP BM were sacrificed at 36 weeks after transplantation and their BM cells (2 × 106) were transplanted into WT recipients (n = 18 per group). (B) The time course of blood counts from mice that received BM from VF;GFP (solid symbols) or VF;IL-1β−/−;GFP donors (open symbols), and their GFP chimerism in the peripheral blood (lower panel) are shown. (C) Analysis of GFP chimerism in HSPCs from the BM and spleen is shown. Multiple t tests were performed for statistical analyses. (D-F) Schematic drawing of the identical aforementioned experiment, in which BM competitor cells from a WT mouse was mixed to a 1:1 ratio for the transplantations into secondary WT recipients. Annotations as in panels B and C. Multiple t tests were performed for statistical analyses. (G) Bar graphs show the percentages of mice that showed engraftment defined as GFP chimerism of >1% at 18 weeks after noncompetitive (left) and competitive 1:1 transplantation (right). Middle panel shows the log of nonengrafted mice (GFP chimerism in Gr1 of <1% at 18 weeks) vs the number of donor BM cells transplanted in each group. Estimated frequency of functional stem cells in the BM was calculated using extreme limiting dilution analysis.34 Solid dark blue line represents the estimated frequency of HSCs in mice transplanted with BM from VF;GFP donors. Solid light blue line represents the estimated frequency of HSCs in mice transplanted with BM from VF;IL-1β−/−;GFP donors. Dotted lines show 95% confidence interval. Gray shaded areas represent the normal range. All data are presented as mean ± SEM; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; and ∗∗∗∗P < .0001. See also supplemental Figures 3 and 4.
MPN initiation by JAK2-V617F is favored by signaling through the IL-1R1 in both hematopoietic and nonhematopoietic cells
Next, we examined the role of IL-1R1 in MPN disease initiation. VF;GFP mice were crossed with IL-1R1−/− mice to obtain VF;IL-1R1−/−;GFP mice that were used as donors for competitive BM transplantations into WT recipients (Figure 3A-E), or IL-1R1−/− recipients (Figure 3F-J). GFP-chimerism in peripheral blood was higher in mice transplanted with VF;GFP BM than in recipients of VF;IL-1R1−/−;GFP BM, when all mice that showed engraftment were considered (Figure 3D). Transplantations into WT recipients resulted in high frequency of engraftment, but fewer recipients transplanted with VF;IL-1R1−/−;GFP BM developed MPN phenotype (27%) than recipients of VF;GFP BM (55%) (Figure 3E). GFP chimerism was similar when only mice with MPN phenotype were compared (supplemental Figure 5A), but loss of IL-1R1 from JAK2-mutant cells resulted in reduced GFP chimerism in HSPCs in the BM and spleen (supplemental Figure 5B). Recipients of VF;IL-1R1−/−;GFP BM displayed low IL-1β levels in the plasma and BM, and no differences were noted between mice with or without MPN phenotype (Figure 3C). Nevertheless, IL-1β levels were higher in WT recipients transplanted with VF;IL-1R1−/− BM than in those transplanted with VF;IL-1β−/− BM (supplemental Figure 5C). Although VF;IL-1β−/− cells are unable to produce IL-1β, cells from VF;IL-1R1−/− mice can produce IL-1β, although at lower levels, because of loss of the positive IL-1β-feedback mediated by the IL-1R1.18,19,35,36 Loss of IL-1R1 in JAK2-mutant cells also reduced reticulin fibrosis and osteosclerosis in the BM and partially corrected the splenic architecture in mice that developed MPN phenotype (supplemental Figure 5D). Cytokine levels did not change, except for decreased IL-1β in plasma (supplemental Figure 5E).
Figure 3.

JAK2-mutant cells need IL-1R1 expression on both hematopoietic and nonhematopoietic cells for optimal MPN initiation. (A) Schematic drawing of the experimental setup for competitive transplantation at 1:100 dilution. BM from VF;GFP or VF;IL-1R1−/−;GFP donor mice was mixed with a 100-fold excess of BM competitor cells from an IL-1R1−/− donor. (B) Time course of blood counts from individual mice that received BM from VF;GFP (upper panel) or VF;IL-1R1−/−;GFP donors (lower panel). (C) IL-1β protein levels in plasma and BM lavage (1 femur and 1 tibia) of mice with or without MPN phenotype is shown. Nonparametric Mann-Whitney 2-tailed t test was performed for statistical comparisons. (D) GFP chimerism in the peripheral blood. Multiple t tests were performed for statistical analyses. (E) Bar graphs show the percentages of mice that showed engraftment defined as GFP chimerism of >1% at 18 weeks after transplantation and the percentages of mice that developed MPN phenotype (elevated hemoglobin and/or platelet counts). P values in lower panel were computed using Fisher exact test. (F-J) Schematic drawing of the identical aforementioned experiment, in which IL-1R1−/− mice were used as the recipients instead of WT mice. Annotations as in panels B through E. Gray shaded area represents normal range. All data are presented as mean ± SEM. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; and ∗∗∗∗P < .0001. See also supplemental Figures 5 and 6.
Transplantations into recipients deficient for IL-1R1−/− also resulted in high frequency of engraftment (Figure 3F-J), but MPN disease initiation in IL-1R1−/− deficient recipients transplanted with VF;GFP BM was reduced to 29% (Figure 3J), compared with 55% in WT recipients (Figure 3E). In mice that developed MPN phenotype, blood counts did not change, but GFP chimerism in the peripheral blood was slightly reduced with complete loss of IL-1R1 (supplemental Figure 6A), whereas GFP chimerism remained unchanged in HSPCs (supplemental Figure 6B). Compared with WT recipients, IL-1R1−/− recipients transplanted with VF;GFP showed reduced grade of reticulin fibrosis in the BM, normalization of splenic architecture, (supplemental Figure 6C), and reduced levels of inflammatory cytokines (supplemental Figure 6D). However, loss of IL-1R1 in both donor and recipient did not affect reticulin fibrosis or the levels of inflammatory cytokines (supplemental Figure 6C-D).
Taken together, these results show that increased expression of IL-1β by the JAK2-mutant hematopoietic cells and the presence of IL-1R1 on both mutant hematopoietic cells and WT nonhematopoietic cells are required for efficient MPN disease initiation and progression.
Pharmacological inhibition of inflammation can reduce MPN disease initiation in a mouse model of JAK2-V617F driven clonal hematopoiesis
Because reducing proinflammatory signaling by IL-1β knockout in VF donor cells or deletion of IL-1R1 in the recipient cells both reduced MPN disease initiation in competitive transplantations at high dilution, we examined whether the same effect can be observed with pharmacologic inhibition of inflammation. We first tested the effects of aspirin, which was added to the drinking water on day 1 after transplantation (supplemental Figure 7). We observed a slight reduction in the frequencies of engraftment (96% in the controls vs 70% in the aspirin-treated mice), but there were no differences in the blood counts or frequencies of MPN disease initiation (supplemental Figure 7).
Next, we examined the effects of a neutralizing anti–IL-1β antibody in mice transplanted with BM from a VF;GFP donor (Figure 4A). Treatment with anti–IL-1β antibody or isotype started on day 1 after transplantation and was continued for 18 weeks. Anti–IL-1β antibody reduced the percentage of mice with engraftment, defined as >1% GFP at 18 weeks and fewer mice developed MPN phenotype than mice treated with isotype control (Figure 4B). GFP chimerism was already higher at 6 weeks and further increased in VF mice that developed MPN phenotype under isotype treatment than in VF mice that engrafted but failed to develop MPN (Figure 4C). In contrast, no differences in GFP chimerism were observed after 6 weeks under anti–IL-1β antibody treatment, but GFP chimerism decreased between week 6 and 18 under anti–IL-1β antibody treatment in VF mice that engrafted but failed to develop MPN (Figure 4D). HSPCs from mice with MPN phenotype displayed high GFP chimerism that did not change by anti–IL-1β antibody treatment, whereas HSPCs from mice with engraftment >1% but without MPN phenotype, resembling a state of CHIP, showed low GFP chimerism and, in some cases, a decrease in chimerism was noted upon anti–IL-1β treatment (Figure 4E). Control mice transplanted with BM from WT:GFP donor showed no differences in phenotype or percentages of engraftment between the anti–IL-1β antibody and isotype treatment groups (supplemental Figure 8). Interestingly, anti–IL-1β antibody treatment did not decrease the GFP chimerism in HSPCs from the BM or spleen compared with isotype control (supplemental Figure 8D).
Figure 4.

Anti–IL-1β antibody reduces MPN disease initiation in a mouse model of JAK2-V617F–driven clonal hematopoiesis. (A) Schematic drawing of the experimental setup for competitive transplantation at 1:100 dilution. BM cells from a VF;GFP donor mouse were mixed with a 100-fold excess of WT BM competitor cells and transplanted into lethally irradiated WT recipient mice. Transplanted mice were randomized into 2 treatment arms (n = 36 per group) and treated with anti–IL-1β antibody or isotype control for 18 weeks, starting 1 day after transplantation (Tx). (B) The percentage of mice that showed engraftment defined as GFP chimerism of >1% at 18 weeks after transplantation and the percentages of mice that developed MPN phenotype (elevated hemoglobin and/or platelet counts) are indicated. P value was computed using Fisher exact test. (C-D) The upper panel shows the time course of blood counts from all individual mice for the 2 treatment arms, as indicated. The lower panel shows the mean GFP chimerism in the peripheral blood for mice with MPN phenotype (MPN) and without MPN phenotype (no MPN) that engrafted, defined as GFP chimerism of >1% at 18 weeks. Erythrocytes (Ter119), platelets (CD61), and granulocytes (Gr1) cells are shown separately. Multiple t tests were performed for statistical analyses. (E) GFP chimerism in HSPCs after 18 weeks of treatment. Multiple t tests were performed for statistical analyses. All data are presented as mean ± SEM; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; and ∗∗∗∗P < .0001. See also supplemental Figures 7 and 8.
We confirmed that in our transplantations with VF;GFP BM at 1:100, mice that developed MPN phenotype displayed strongly elevated IL-1β levels in the BM and plasma compared with in mice transplanted with WT;GFP BM, whereas VF;GFP transplanted mice without MPN phenotype had marginally elevated IL-1β levels only in the BM (Figure 5A). Anti–IL-1β antibody treatment massively reduced IL-1β levels in all treated mice. Primitive BM mesenchymal stromal cells (MSCs), sympathetic nerve fibers, and Schwann cells were reduced at 18 weeks in the BM from isotype treated VF;GFP mice compared with WT;GFP mice. Interestingly, this decrease was also observed in VF;GFP transplanted mice without MPN phenotype that display low GFP chimerism and resemble a state of CHIP (Figure 5C-E). Thus, the damage to the niche cells does not appear to require strongly elevated IL-1β levels that are found in mice that developed MPN phenotype. Anti–IL-1β antibody largely normalized the frequencies of primitive MSCs and the densities of sympathetic nerve fibers and Schwann cells in all treated mice (Figure 5C-E).
Figure 5.

Inhibition of IL-1β preserves the HSC niche in the BM. (A) IL-1β protein concentration in BM lavage (1 femur and 1 tibia) and (B) plasma of WT mice (from supplemental Figure 8), and JAK2-V617F mice (from Fig. 4) with or without MPN phenotype. Nonparametric Mann-Whitney 2-tailed t test was performed for statistical comparisons. The lower limit of detection was 0.11 pg/ml. (C) Frequency of primitive stromal cells (CD45−CD31−Ter119−Sca1−PDGFRα+) in the skull of mice transplanted with BM from WT;GFP or VF;GFP donor mouse after 18 weeks of treatment with isotype or anti–IL-1β antibody (from Figure 4). Two-tailed unpaired t tests were performed for statistical comparisons. (D) Bar graphs show the quantification of tyrosine hydroxylase (TH) area and (E) of glial fibrillary acidic protein (GFAP) area in the BM. One-way analysis of variance with uncorrected Fisher least significant difference test was performed for statistical comparisons. All data are presented as mean ± SEM; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; and ∗∗∗∗P < .0001.
Myelomonocytic cells and megakaryocytes are main sources of IL-1β production in the BM of VF mice
To analyze the distribution of IL-1β protein in BM sections, we used in situ BM 3D fluorescence imaging with proximity ligation assay (PLA) that can visualize single IL-1β molecules.37, 38, 39 WT mice showed low densities of IL-1β PLA signals, whereas VF mice that developed MPN phenotype displayed the highest densities of PLA signals (Figure 6; supplemental Figure 9A). IL-1β PLA signals were enriched in close proximity with CD11b+ myeloid cells in mice of all genotypes, and these gradients were reduced in VF mice that were treated with anti–IL-1β antibody.
Figure 6.

In situ BM 3-dimensional fluorescence imaging with PLA for IL-1β single-molecule visualization. Representative images show the distribution of the IL-1β PLA signals relative to CD11b+ myeloid cells and CD41+ megakaryocytes in BM sections of VF and WT mice. The 3-dimensional spatial densities of PLA signals relative to the distances of the closest CD11b+ myeloid cells, CD41+ megakaryocytes or GFP+JAK2-mutant cells are shown in the graphs next to the pictures. See also supplemental Figure 9.
In VF mice, the IL-1β PLA signals were also enriched in the vicinity of CD41+ megakaryocytes and these signals were strongly reduced in mice treated with anti–IL-1β antibody (Figure 6). These results show that JAK2-V617F upregulates IL-1β expression not only in myeloid cells but also in megakaryocytes and suggest that the treatment with anti–IL-1β antibody interrupts the feedback activation of IL-1β. Consistent with the IL-1β PLA data, expression of IL-1β messenger RNA was also increased in sorted monocytic and megakaryocytic cells from the BM of VF mice compared with that from WT mice (supplemental Figure 9B-C).
IL-1β enhances inflammatory cytokine and chemokine responsiveness in JAK2-mutant HSCs
To investigate the cell autonomous mechanisms of IL-1β in JAK2-mutant HSCs and MPN disease initiation, we performed messenger RNA sequencing of JAK2-mutant LT-HSCs (Lin−Sca1+cKit+CD48–CD150+). BM cells from VF, VF;IL-1β−/−, VF;IL-1R1−/−, or WT donors that have not been induced with tamoxifen before euthanasia were transplanted into lethally irradiated recipient mice (Figure 7A). These recipient mice received tamoxifen injections starting 16 weeks after transplantation and were euthanized 2 or 4 weeks after tamoxifen induction when they already displayed mild MPN phenotype (Figure 7B).
Figure 7.

IL-1β enhances inflammatory cytokine and chemokine responsiveness in JAK2-mutant HSCs. (A) Schematic drawing of the experimental setup for competitive transplantation. BM cells from VF;GFP, VF;IL-1β−/−;GFP, VF;IL-1R1-/;GFP, or WT;GFP donor mice that were not induced by tamoxifen were mixed with competitor BM cells from a WT donor in a 1:1 ratio. Recipient mice were induced with tamoxifen 16 weeks after transplantation to induce JAK2-V617F expression. Mice from each genotype (n = 3-4 per group) were euthanized 2 or 4 weeks after tamoxifen induction and LT-HSCs (Lin−Sca1+cKit+CD48–CD150+) were purified by fluorescence-activated cell sorter for RNA sequencing. (B) The time course of blood counts of recipient mice after tamoxifen induction. (C) Human JAK2-V617F messenger RNA gene expression in LT-HSCs (normalized log2 counts per million [CPM]) at 2 and 4 weeks after tamoxifen induction. Multiple t tests were performed for statistical analyses. (D) Pathway analysis 4 weeks after tamoxifen induction performed with WebGestalt online tool40, using Gene Ontology as the functional database. Minimum and maximum number of genes for a category was set to 15 and 500, respectively. TOP method (TOP means the categories will be first ranked based on the false discovery rate [FDR] and then the top N most significant categories will be selected) was used to identify significance levels of the enriched categories, and number of permutations was 1000. Bars representing significant pathways (FDR of <0.05) are colored according to their normalized enrichment score and the FDR as indicated. The names of the pathways upregulated in VF vs all 3 other genotypes are colored in red or orange according to their FDRs. (E) Expression of genes from the top 3 pathways identified by Gene Ontology in panel D to be commonly upregulated in LT-HSCs from VF mice vs mice from all 3 other genotypes. All data are presented as mean ± SEM; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; and ∗∗∗∗P < .0001. See also supplemental Figure 10.
LT-HSCs from mice transplanted with BM from VF;IL-1R1−/− mice displayed significant increase in the expression of JAK2-V617F compared with recipients transplanted with BM from VF mice, whereas no differences were observed between VF and VF;IL-1β−/− mice (Figure 7C). To identify significantly enriched biological pathways in the RNA sequencing data sets, we used the WebGestalt online tool,40 and used the weighted set cover method of WebGestalt to identify the least redundant and most representative gene sets from the data set (Figure 7D; supplemental Figure 10).
At 2 weeks after tamoxifen induction, we observed no significant enrichment (false discovery rate < 0.05) of any biological pathways (supplemental Figure 10). At 4 weeks after tamoxifen, we identified several significantly enriched Gene Ontology pathways (false discovery rate < 0.05) between VF vs WT, VF vs VF;IL-1β−/−, and VF vs VF;IL-1R1−/−genotypes (Figure 7D). Interestingly, in all 3 comparisons, LT-HSCs from VF mice displayed significantly higher response to interferon signaling than LT-HSCs from WT, VF;IL-1β−/−, or VF;IL-1R1−/− mice. Furthermore, heat map analysis showed increased expression of several genes including IL-1β, Tnf, Cxcl4, Cxcl3, Cxcl10, Ccl3, and Ccl5 from the top 3 Gene Ontology pathways (response to interferon-β, response to interferon-γ, and cytokine-mediated signaling pathway) that are commonly upregulated in VF LT-HSCs compared with LT-HSCs from the other 3 genotypes (Figure 7E). Similarly, the loss of IL-1β or IL-1R1 in JAK2-V617F-expressing LT-HSCs resulted in a reduction In the expression of the same genes (Figure 7E). Overall, our results suggest that loss of IL-1β or IL-1R1 in LT-HSCs expressing JAK2-V617F reverse the inflammatory signature to make them in this respect more WT-like.
IL1B polymorphisms are associated with increased serum IL-1β levels in patients with MPN
Because IL-1β serum levels were increased in patients with MPN and correlated with the JAK2-V617F VAF,22 we examined whether genetic polymorphisms located in the IL1B gene correlated with higher IL-1β serum levels in patients with MPN (supplemental Figure 11). Several functionally relevant polymorphisms in the IL1B gene have been reported.41,42 We selected 2 IL1B gene polymorphisms that were associated with increased production of IL-1β in nonhematological cancers,43, 44, 45 to compare their frequencies in patients with MPN and normal controls (supplemental Figure 11A). We found that the frequencies of the homozygous AA genotype of rs16944 (G511>A) located in the promoter region of IL1B and of the homozygous GG genotype of rs1143627 (A31>G) located closer to the IL1B coding regions were almost threefold higher in patients with JAK2-V617F–positive MPN than in normal controls. Furthermore, patients with MPN with AA and GG genotypes displayed higher IL-1β serum levels than patients with MPN with GG/GA or AA/AG genotypes (supplemental Figure 11B). Although validation in a larger cohort of patients with MPN is needed, our data support the model that presence of IL-1β favors the expansion of the JAK2-mutant clone.
Discussion
In this study, we investigated whether inflammation mediated by IL-1β promotes the early expansion of the JAK2-V617F mutant clone and can thereby favor the conversion from a CHIP-like state to MPN. This process is fundamentally different from the progression from chronic phase MPN to myelofibrosis, which we studied previously.22 Competitive BM transplantations at high dilutions allowed us to examine oligoclonal or monoclonal MPN disease initiation starting from 1 to 3 LT-HSCs per transplanted mouse.33 Genetic ablation of IL-1β from JAK2-mutant donor HSCs significantly reduced the frequency of engraftment and lowered the frequency of conversion from CHIP to MPN among mice that engrafted (Figure 1), and these HSCs were also less functional in secondary transplantations (Figure 2), consistent with a role of IL-1β for optimal HSC expansion and long-term repopulation capacity. Although the complete genetic loss of IL-1β in donor and recipients eliminated the differences in engraftment and MPN disease initiation compared to VF (supplemental Figure 2), treatment with anti–IL-1β antibody decreased engraftment and MPN disease initiation (Figure 4). Our data establish IL-1β as an important trigger for the conversion from a CHIP-like state to MPN in carriers of JAK2-V617F.
The IL-1R1–knockout data are in-line with the IL-1β–knockout results. Because the IL-1R1 is required for the positive autocrine feedback loop that augments IL-1β production,18,19,35,36 loss of IL-1R1 in VF;IL-1R1−/− hematopoietic cells prevented the massive overproduction of IL-1β observed in VF hematopoietic cells. IL-1β produced by the VF;IL-1R1−/− hematopoietic cells was still adequate to assure a high percentage of engraftment, but the levels were not sufficient to reach the same high frequency of conversion from CHIP to MPN observed with VF hematopoietic cells (Figure 3A). Loss of IL-1R1 in the recipients did not reduce the frequency of engraftment but lowered the conversion to MPN (Figure 3B). Thus, IL-1β promotes engraftment in a cell autonomous fashion, also in secondary transplantations, and is not entirely dependent on the presence of the IL-1R1, whereas the conversion to MPN required the presence of the IL-1R1 in the BM niche. Consistent with the genetic deletion of IL-1β, treatment with anti–IL-1β antibody reduced both the frequency of engraftment and the conversion to MPN (Figure 4B).
We have previously shown that in the VF mouse model, IL-1β mediates loss of sympathetic nerve fibers and the Schwann cells that surround them, resulting in a reduction of nestin-positive BM MSCs,21 and these changes were also associated with progression to myelofibrosis.22 The reduction of BM MSCs was also observed in isotype-treated mice transplanted with VF;GFP BM that did not develop MPN phenotype and displayed low GFP chimerism resembling a state of CHIP (Figure 5C-E). These results suggest that reducing the frequency of MSCs alone is not sufficient to assure conversion to MPN. Conversely, preserving high frequencies of MSCs by anti–IL-1β antibody treatment did reduce both the rate of engraftment and conversion to MPN. The mechanism of why the presence of MSCs interferes with the expansion of the JAK2-V617F clone is currently unknown and will be the subject of future studies.
Recent studies have reported associations between inflammatory cytokines and the expansion of CHIP clones carrying mutations other than JAK2-V617F. Tumor necrosis factor α promoted the expansion of hematopoietic cells with mutations in Dnmt3a or Asxl1,46,47 and IL-1α and IL-1β mediated expansion of hematopoietic clones carrying Tet2 mutations.48 For example, inhibiting inflammation, by aspirin, could decrease the probability of MPN disease initiation from JAK2-mutant HSCs. Although aspirin monotherapy slightly reduced the frequency of engraftment of VF BM, it did not decrease the frequency of MPN initiation (supplemental Figure 7). However, aspirin in combination with anti-PD1 showed therapeutic benefit in a Braf-V600E melanoma model.49 JAK2-V617F clones induce strong IL-1–mediated responses, as seen by the relatively high levels of IL-1β and IL-6 in plasma as compared with other cytokines such as interferon-γ or KC/GRO (Figure 1E). Genetic deletion of IL-1β or IL-1R1 in LT-HSCs expressing JAK2-V617F made them more WT-like in respect to VF-induced responsiveness to interferon and cytokine signaling (Figure 7D-E). The anti–IL-1β antibody canakinumab is currently being tested in a clinical trial for clonal cytopenias of unknown significance.50
Our results provide evidence that IL-1β production by JAK2-mutant hematopoietic cells generates an inflammatory environment in the BM that promotes JAK2-V617F clonal expansion and conversion to MPN. Inhibition of IL-1β by anti–IL-1β antibody may be beneficial in individuals with JAK2-V617F CHIP who are at increased risk of progression to MPN.
Conflict-of-interest disclosure: R.C.S. is a scientific adviser for, scientific advisory board member of, and has equity in Ajax Therapeutics; and has consulted for, and/or received honoraria from, Novartis, Bristol Myers Squibb/Celgene, AOP, GlaxoSmithKline, Baxalta, and Pfizer. N.H. owns stocks in Cantargia. C.J.F. is a full-time employee of Novartis Pharma AG. The anti–IL-1β antibody studies were carried out in the laboratory of R.C.S. with the antibody provided by Novartis. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Marc Donath for helpful discussions and members of our laboratory for critical reading of the manuscript. The authors thank Christian Beisel, Elodie Burcklen, and Philippe Demougin of the Genomics core facility for their support. They also thank the Bioinformatics core facility, Animal core facility, and Flow cytometry core facility of the Department of Biomedicine for excellent technical support. Bioinformatic calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing center at the University of Basel.
This work was supported by grants from the Swiss National Science Foundation (310030_185297 /1, and 310030_185297 /2) and Swiss Cancer Research (KFS-4462-02-2018); the Stiftung für Hämatologische Forschung (R.C.S.); core support grants from Medical Research Council (MRC) to the Cambridge Stem Cell Institute, National Health Service Blood and Transplant (United Kingdom); European Union’s Horizon 2020 research (ERC-2014-CoG-648765); MRC-AMED grant MR/V005421/1; and a Programme Foundation Award (C61367/A26670) from Cancer Research UK (S.M.-F.).
Authorship
Contribution: S.R. designed and performed the research, analyzed data, and wrote the manuscript; Y. Zhang and E.G. performed research and analyzed data; N.H., Q.K., C.B.S., M.U., H.H.-S., and Y. Zhu performed research; D.L.P. and J.R. analyzed sequencing data; M.S.B. analyzed clinical data; S.D. performed and analyzed histopathology of mouse tissues; C.J.F., T.S., and S.M.-F. designed research and analyzed data; R.C.S. designed the research, analyzed the data, and wrote the manuscript.
Footnotes
Presented as an oral presentation at the 61st Annual Meeting of the American Society of Hematology, Orlando, FL, 7 December 2019.
The RNA sequencing data set used in this study is available at GEO database with accession number GSE250507. Additional methods are described in the supplemental Material. Requests for original data, resources, and reagents should be addressed to the corresponding author, Radek C. Skoda (radek.skoda@unibas.ch).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 2.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 3.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 4.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 5.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 6.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7) doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112(6):2190–2198. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667–679. doi: 10.1182/blood-2016-10-695940. [DOI] [PubMed] [Google Scholar]
- 10.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordua S, Kjaer L, Skov V, Pallisgaard N, Hasselbalch HC, Ellervik C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood. 2019;134(5):469–479. doi: 10.1182/blood.2019001113. [DOI] [PubMed] [Google Scholar]
- 14.Kleppe M, Koche R, Zou L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018;33(4):785–787. doi: 10.1016/j.ccell.2018.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson LA, Pfeiffer RM, Landgren O, Gadalla S, Berndt SI, Engels EA. Risks of myeloid malignancies in patients with autoimmune conditions. Br J Cancer. 2009;100(5):822–828. doi: 10.1038/sj.bjc.6604935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kristinsson SY, Landgren O, Samuelsson J, Bjorkholm M, Goldin LR. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica. 2010;95(7):1216–1220. doi: 10.3324/haematol.2009.020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kristinsson SY, Bjorkholm M, Hultcrantz M, Derolf AR, Landgren O, Goldin LR. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J Clin Oncol. 2011;29(21):2897–2903. doi: 10.1200/JCO.2011.34.8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39(6):1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–27. doi: 10.1111/imr.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Mooij CEM, Netea MG, van der Velden W, Blijlevens NMA. Targeting the interleukin-1 pathway in patients with hematological disorders. Blood. 2017;129(24):3155–3164. doi: 10.1182/blood-2016-12-754994. [DOI] [PubMed] [Google Scholar]
- 21.Arranz L, Sanchez-Aguilera A, Martin-Perez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512(7512):78–81. doi: 10.1038/nature13383. [DOI] [PubMed] [Google Scholar]
- 22.Rai S, Grockowiak E, Hansen N, et al. Inhibition of interleukin-1beta reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat Commun. 2022;13(1):5346. doi: 10.1038/s41467-022-32927-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahman MF, Yang Y, Le BT, et al. Interleukin-1 contributes to clonal expansion and progression of JAK2V617F-induced myeloproliferative neoplasms. Nat Commun. 2022;13(1) doi: 10.1038/s41467-022-32928-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111(8):3931–3940. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 25.Gothert JR, Gustin SE, Hall MA, et al. In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood. 2005;105(7):2724–2732. doi: 10.1182/blood-2004-08-3037. [DOI] [PubMed] [Google Scholar]
- 26.Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cell Immunol. 2001;214(2):110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- 27.Horai R, Asano M, Sudo K, et al. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med. 1998;187(9):1463–1475. doi: 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glaccum MB, Stocking KL, Charrier K, et al. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol. 1997;159(7):3364–3371. [PubMed] [Google Scholar]
- 29.Kubovcakova L, Lundberg P, Grisouard J, et al. Differential effects of hydroxyurea and INC424 on mutant allele burden and myeloproliferative phenotype in a JAK2-V617F polycythemia vera mouse model. Blood. 2013;121(7):1188–1199. doi: 10.1182/blood-2012-03-415646. [DOI] [PubMed] [Google Scholar]
- 30.Osborn O, Brownell SE, Sanchez-Alavez M, Salomon D, Gram H, Bartfai T. Treatment with an interleukin 1 beta antibody improves glycemic control in diet-induced obesity. Cytokine. 2008;44(1):141–148. doi: 10.1016/j.cyto.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez D, Baylis RA, Durgin BG, et al. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med. 2018;24(9):1418–1429. doi: 10.1038/s41591-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potus F, Pauciulo MW, Cook EK, et al. Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation. 2020;141(24):1986–2000. doi: 10.1161/CIRCULATIONAHA.119.044320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lundberg P, Takizawa H, Kubovcakova L, et al. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J Exp Med. 2014;211(11):2213–2230. doi: 10.1084/jem.20131371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1-2):70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 36.Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010;3(105):cm1. doi: 10.1126/scisignal.3105cm1. [DOI] [PubMed] [Google Scholar]
- 37.Coutu DL, Kokkaliaris KD, Kunz L, Schroeder T. Multicolor quantitative confocal imaging cytometry. Nat Methods. 2018;15(1):39–46. doi: 10.1038/nmeth.4503. [DOI] [PubMed] [Google Scholar]
- 38.Kunz L, Schroeder T. A 3D tissue-wide digital imaging pipeline for quantitation of secreted molecules shows absence of CXCL12 gradients in bone marrow. Cell Stem Cell. 2019;25(6):846–854.e4. doi: 10.1016/j.stem.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Hausmann A, Felmy B, Kunz L, et al. Intercrypt sentinel macrophages tune antibacterial NF-kappaB responses in gut epithelial cells via TNF. J Exp Med. 2021;218(11) doi: 10.1084/jem.20210862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404(6776):398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 42.Khazim K, Azulay EE, Kristal B, Cohen I. Interleukin 1 gene polymorphism and susceptibility to disease. Immunol Rev. 2018;281(1):40–56. doi: 10.1111/imr.12620. [DOI] [PubMed] [Google Scholar]
- 43.Hwang IR, Kodama T, Kikuchi S, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology. 2002;123(6):1793–1803. doi: 10.1053/gast.2002.37043. [DOI] [PubMed] [Google Scholar]
- 44.Hall SK, Perregaux DG, Gabel CA, et al. Correlation of polymorphic variation in the promoter region of the interleukin-1 beta gene with secretion of interleukin-1 beta protein. Arthritis Rheum. 2004;50(6):1976–1983. doi: 10.1002/art.20310. [DOI] [PubMed] [Google Scholar]
- 45.Chen H, Wilkins LM, Aziz N, et al. Single nucleotide polymorphisms in the human interleukin-1B gene affect transcription according to haplotype context. Hum Mol Genet. 2006;15(4):519–529. doi: 10.1093/hmg/ddi469. [DOI] [PubMed] [Google Scholar]
- 46.SanMiguel JM, Eudy E, Loberg MA, et al. Distinct tumor necrosis factor alpha receptors dictate stem cell fitness versus lineage output in Dnmt3a-mutant clonal hematopoiesis. Cancer Discov. 2022;12(12):2763–2773. doi: 10.1158/2159-8290.CD-22-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avagyan S, Henninger JE, Mannherz WP, et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science. 2021;374(6568):768–772. doi: 10.1126/science.aba9304. [DOI] [PubMed] [Google Scholar]
- 48.Caiado F, Kovtonyuk LV, Gonullu NG, Fullin J, Boettcher S, Manz MG. Aging drives Tet2+/- clonal hematopoiesis via IL-1 signaling. Blood. 2023;141(8):886–903. doi: 10.1182/blood.2022016835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zelenay S, van der Veen AG, Bottcher JP, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162(6):1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Canakinumab for the Prevention of Progression to Cancer in Patients With Clonal Cytopenias of Unknown Significance, IMPACT Study. ClinicalTrials.gov identifier: NCT05641831. https://clinicaltrials.gov/ct2/show/NCT05641831 Updated 24 March 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.