

Abstract
Nucleosides are ubiquitous to life and are required for the synthesis of DNA, RNA, and other molecules crucial for cell survival. Despite the notoriously difficult organic synthesis of nucleosides, 2′-deoxynucleoside analogues can interfere with natural DNA replication and repair and are successfully employed as anticancer, antiviral, and antimicrobial compounds. Nucleoside 2′-deoxyribosyltransferase (dNDT) enzymes catalyze transglycosylation via a covalent 2′-deoxyribosylated enzyme intermediate with retention of configuration, having applications in the biocatalytic synthesis of 2′-deoxynucleoside analogues in a single step. Here, we characterize the structure and function of a thermophilic dNDT, the protein from Chroococcidiopsis thermalis (CtNDT). We combined enzyme kinetics with structural and biophysical studies to dissect mechanistic features in the reaction coordinate, leading to product formation. Bell-shaped pH-rate profiles demonstrate activity in a broad pH range of 5.5–9.5, with two very distinct pKa values. A pronounced viscosity effect on the turnover rate indicates a diffusional step, likely product (nucleobase1) release, to be rate-limiting. Temperature studies revealed an extremely curved profile, suggesting a large negative activation heat capacity. We trapped a 2′-fluoro-2′-deoxyarabinosyl-enzyme intermediate by mass spectrometry and determined high-resolution structures of the protein in its unliganded, substrate-bound, ribosylated, 2′-difluoro-2′-deoxyribosylated, and in complex with probable transition-state analogues. We reveal key features underlying (2′-deoxy)ribonucleoside selection, as CtNDT can also use ribonucleosides as substrates, albeit with a lower efficiency. Ribonucleosides are the building blocks of RNA and other key intracellular metabolites participating in energy and metabolism, expanding the scope of use of CtNDT in biocatalysis.
Keywords: deoxyribonucleoside transferase, nucleosides, biocatalysis, protein engineering, thermophilic
Introduction
Nucleosides are the building blocks of DNA and RNA and are key molecules for metabolism and energetics. Nucleoside analogues have been extensively used to interfere with biological processes governing nucleotide incorporation into DNA and RNA, successfully targeting viral and cellular replication1 and yielding a plethora of drugs against cancer, herpes simplex, HIV, and hepatitis C.2 However, overcoming synthetic challenges, specifically creating concise and adaptable synthetic pathways, remains a challenge.3 Enzymes present a promising strategy to generate nucleosides in a stereospecific manner, with fewer steps and the potential to utilize a single catalyst for the production of multiple products.4−11 Recent work employing solely biocatalysis to produce the nucleoside drugs Islatravir4 and Molnupiravir,5 as well as the key modified RNA precursor pseudouridine,6 has further highlighted the power of enzymes in the production of a series of therapeutically relevant nucleosides.
Nucleoside 2′-deoxyribosyltransferases (EC 2.4.2.6, dNDTs) participate in nucleoside recycling in organisms that lack purine and pyrimidine nucleoside phosphorylases.12 In these organisms, dNDTs play a crucial role in scavenging deoxyribonucleosides for DNA synthesis. The reaction catalyzed by dNDT proceeds via a ping-pong mechanism, with the formation of a 2′-deoxyribosyl-enzyme intermediate covalently linked to an active-site glutamate (Scheme 1).13 Transglycosylation occurs in a stereospecific manner, preserving the anomeric carbon in the β configuration in the newly formed 2′-deoxyribonucleoside. Enzymes from this class have strict specificity for 2′-deoxyribonucleosides14,15 and have been employed as biocatalysts to synthesize a large number of 2′-deoxyribonucleoside analogues with potential biomedical applications.7−11 dNDTs have also been investigated as model systems to understand glycosyl transfer and principles of catalysis in sugar-transfer reactions.16−18 Prior quantum mechanics–molecular mechanics (QM/MM) free-energy landscape calculations with a dNDT enzyme supported an oxocarbenium ion intermediate in the reaction,19 but no empirical evidence was available shedding light on catalysis and the transition state for this reaction.
Scheme 1. Top: Ping-Pong Reaction Catalyzed by dNDT Enzymes. Bottom: Steps Included in Kinetic Parameters under Evaluation for CtNDT.

A full turnover cycle includes nucleobase transfer to generate a novel 2′-deoxyribonucleoside product. Diffusional steps are shown with orange arrows. Under steady-state initial rate conditions, dashed arrows depicting k–3 and k–6 are ≅0.
The thermal adaptation and plasticity present in thermostable enzymes are key to molecular adaptations to heat and other environmental stressors.20 Despite a preliminary characterization of the first thermostable dNDT enzyme, details on the kinetics, structure, and substrate selection by this thermostable enzyme are lacking. Preliminary work proposed the dNDT from Chroococcidiopsis thermalis PCC 7203 (CtNDT) as a promising tool in biocatalysis to produce novel 2′-deoxynucleosides.21Chroococcidiopsis spp. occur in a wide range of extreme temperature and salinity environments, being highly desiccation resistant.22 Thermostability is a desirable feature in biocatalysis,23 given that many chemical transformations will require high temperatures, contamination is less widespread, and substrate solubility is increased at high temperatures.24
Here, we dissected the mechanism, rate-limiting steps, and kinetic and structural determinants of CtNDT’s catalyzed reaction. We obtained snapshots of different steps in the reaction coordinate using X-ray crystallography, including a ribosyl–CtNDT covalent intermediate and the tight-binding interaction to a potential transition-state analogue, further characterized by isothermal titration calorimetry. Steady-state and pre-steady-state kinetics of CtNDT and mutants uncovered specific interactions and rate-limiting steps in nucleoside transfer, highlighting a significant contribution of negative activation heat capacity for turnover and key amino-acid residues mediating substrate selection, transition-state stabilization, and catalysis. This work sets the stage for the engineering of CtNDT toward the synthesis of novel (2′-deoxy)nucleosides of pharmaceutical interest.
Materials and General Methods
The codon-optimized synthetic gene was ordered from Integrated DNA Technologies (IDT). General chemicals and reagents were from Fluorochem, Merck, and Fisher Scientific. Data were fitted using GraphPad Prism. Pre-steady-state data were fitted using KinTek Global Explorer.25 Additional methods for protein production, enzymatic assays, mass spectrometry, high-performance liquid chromatography (HPLC) conditions, and differential scanning fluorimetry (DSF) experiments are available in the Supporting Information.
Cloning, Expression, and Purification of CtNDT and Its Mutants
The synthetic gene encoding theC. thermalis NDT (Uniprot code: K9TVX3) was ordered as a Gblock (Integrated DNA Technologies) and cloned into a pJ411 expression plasmid with a cleavable 6-histidine tag at the N-terminus by Gibson assembly. Construct design was carried out using NEBuilder (New England Biolabs), following the cloning protocol suggested to amplify the plasmid pJ411 backbone as well as the GBlock to generate an overlap of 20bp between sequences. Following cloning, the gene sequence was confirmed by sequencing (Eurofins). pJ411::CtNDT was transformed into Escherichia coli BL21 (DE3) cells for overexpression in LB medium with 50 μg mL–1 kanamycin at 37 °C with shaking at 180 rpm until cells reached OD600 = 0.8. Protein expression was induced by addition of 0.5 mM IPTG overnight at 16 °C while being shaken at 180 rpm. The cells were harvested by centrifugation at 12,000g for 20 min, resuspended in wash buffer (50 mM MES, 250 mM NaCl, 30 mM imidazole, pH 6.5), and lysed using a cell homogenizer (Constant Systems). Following centrifugation for 30 min at 51,000g at 4 °C, the supernatant was filtered with a 0.8 mm filter to remove particulates and loaded onto a 5 mL HisTrap column pre-equilibrated with wash buffer. The column was washed with 10 column volumes of wash buffer, and CtNDT was eluted with 50 mM MES, 250 mM NaCl, and 500 mM imidazole, pH 6.5. Fractions containing CtNDT were pooled and dialyzed with 2 mg mL–1 tobacco etch virus (TEV) protease (prepared in-house)26 in buffer (50 mM MES, 250 mM NaCl, pH 6.5) overnight at 4 °C. The dialyzed mixture was loaded onto a 5 mL HisTrap column, and the flow-through fractions were collected and analyzed by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Fractions containing purified CtNDT were pulled, flash-frozen, and stored at −80 °C (Figure S1). The mutants CtNDTD62N, CtNDTE88Q, CtNDTE88A, and CtNDTM120C were expressed and purified using the same method. Protein concentration was determined by the extinction coefficient at 280 nm calculated using the ExPASy ProtParam tool.27 Intact protein mass spectra for all proteins produced are shown in Figure S2.
Standard Activity Assay for CtNDT
(2′-Deoxy)nucleoside Substrates
A standard assay contained CtNDT (between 1 nM and 1 μM), a 2′-deoxynucleoside (10–5000 μM), and a nucleobase (10–2.5 mM) in a mixed buffer solution (a final volume of 50 μL and the final concentration of the mixed buffer was 30 mM for each individual buffer component (CHES, MES, and HEPES, pH 8.5) incubated at 45 °C.
Ribonucleoside Substrates
A standard assay contained CtNDT (1 μM), 10 μM–1000 mM ribonucleoside (Ado, Gua, or Ino), and a nucleobase (10 mM Hyp or Ade) in a mixed buffer solution (50 μL and the final concentration of the mixed buffer was 30 mM for each individual buffer component (CHES, MES, and HEPES), pH 7) incubated at 65 °C.
For all substrates tested, 50 μL reaction mixture was quenched with 200 μL of 10 M urea at 5, 10, and 15 min, followed by centrifugation at 24,000g for 10 min. Half of this volume (100 μL) was placed into a 96-well round-bottom microplate (Agilent Technologies), and 10 μL was injected onto the HPLC column (HSS T3, Waters employing a Shimadzu Prominence HPLC and the method described in “HPLC Conditions for Reaction Monitoring” in the Supporting Information). All experiments were carried out in duplicate. Product formation was quantified using calibration curves with reference standard compounds (Figure S5) for each 2′-deoxyribonucleoside or ribonucleoside product and conversion of the integrated area values into concentration. Data were fitted using the Michaelis–Menten equation in GraphPad Prism, and the kinetic parameters were plotted for substrates or enzyme variants. Individual concentrations of enzymes and substrates used for different experiments are described in the Supporting Information. Retention times for reference standard compounds were obtained by running each standard (see the table in Figure S5).
Enzyme Assays of CtNDT with Varying pH and Temperature Values
Assays contained CtNDT (5 nM), 10–1000 μM dGuo, and 10 mM Ade in a mixed buffer solution (50 μL total; 30 mM CHES, MES, and HEPES each) in a range of pH values from 5 to 9.5 at 0.5 pH unit intervals. Prior to varying temperature and/or pH, protein stability in the conditions required was verified. For that, the protein was preincubated for 15 min in test assay buffer under the conditions to be tested (varying pH or varying temperature), after which the protein was diluted in the activity assay (at 45 °C pH 8.5), and activity was monitored. This activity test was compared to the protein preincubated under the standard conditions, and only pH and temperature values in which activity was unchanged from control were used. For temperature studies, reactions were carried out using CHES, MES, and HEPES (30 mM each; pH 8.5) and incubated from 35 to 65 °C at 5 °C intervals. To obtain reaction rates, 50 μL reaction mixture was quenched with 200 μL of 10 M urea at 5, 10, and 15 min, followed by centrifugation at 24,000g for 10 min. Analysis proceeded by HPLC as described above.
Fast Kinetics
Assays of CtNDT under pre-steady-state conditions were carried out with 2′-dAdo or immucillin-H (ImmH), monitoring tryptophan fluorescence emission with an excitation wavelength of 280 nm and an emission wavelength above 320 nm (using a cutoff filter) in an Applied Photophysics SX-20 stopped-flow spectrophotometer containing a 5 μL mixing cell (0.5 cm path length, and 0.9 ms dead time) at 25 °C. The assay buffer contained 50 mM MES and 250 mM NaCl, pH 6.5. One syringe contained 1.5 μM CtNDT (or mutants), and the other contained 0–200 μM 2′-dAdo. The reaction was triggered by mixing 55 μL from each syringe by the stopped-flow instrument with an applied pressure hold. The fluorescence decrease was measured for 1 s with 4000 data points collected per trace. At least 3 traces were averaged per condition and used for data fitting. Data were analyzed using KinTek Global Explorer 10.25,28,29 As controls, readings with the substrate or enzyme in buffer were performed in the concentration range utilized for each experiment. The maximum concentration used of ligands was 100 μM to avoid inner filter effects, in agreement with prior experiments.30
Crystallization of CtNDT
Recombinant CtNDT was purified and concentrated to ∼10 mg mL–1 for use in crystallization experiments. Proteins were crystallized using the sitting-drop vapor diffusion technique (using a 1:1 ratio of protein to reservoir). Crystals formed after 1 week of incubation at 20 °C. For ligand cocrystallization experiments, the enzyme was incubated with 1 mM ligand (Ado for the ribosylated structure, Clofarabine, DADmeH, or ImmH) at 4 °C for 20 min prior to screening. Crystals were cryoprotected in mother liquor supplemented with 20% ethylene glycol and flash-cooled prior to data collection at Diamond Light Source (beamline I03 or I04). Crystallization conditions are given in Table S6.
Data Collection, Structure Determination, Model Building, Refinement, and Validation
Following diffraction data collection, data reduction and processing were carried out with the automated processing pipeline at Diamond with XIA2 or DIALS. The structure of wild-type CtNDT was solved by molecular replacement with PHASER.31 Search components comprised an ensemble of other NDTs (6EVS, 6QAI, 2F62 and 3EHD) and further modified using sculptor. Wild-type CtNDT was used as a search model for complex structures. Buccaneer32 was used to trace chains and build the initial models. Refinement was carried out with Phenix.refine,31 and manual model building with Coot.33 The model was validated using Coot and Molprobity34 online. Figures were made using ChimeraX35 and Pymol (Schrodinger, version 2.5.0). Crystallographic data are shown in Table S7. All coordinates and structure factor files have been deposited in the Protein Data Bank (PDB accession codes 8PQP, 8PQQ, 8PQR, 8PQS, 8PQT, 8QC0, 8RH3).
Equations for Data Fitting
pH data were plotted with pH and the log kinetic parameter. Data for pH-rate profiles were fitted to eq 1, where C is the pH-independent value of kcat; pKa1 and pKa2 are the dissociation constants for ionizable groups.
 |
1 |
The entire range of temperatures showing a nonlinear temperature dependence was fitted to an Eyring equation accounting for heat capacity (C‡p) (eq 2),36−38 where R is the gas constant; T is the temperature (K); T0 is a reference temperature value (45 °C or 318 K was used); k is the kinetic rate constant or parameter; ΔH‡ is the enthalpy of activation; ΔC‡p is the activation heat capacity; kB is the Boltzmann constant; h is Planck’s constant; and ΔS‡ is the entropy of activation.
| 2 |
Fitted values for kinetic rate constants were converted into energy barriers using eq 3
| 3 |
where ΔG‡i is the Gibbs free energy of activation for step i, R is the gas constant, T is the temperature (K), and k is the rate constant for step i.
For all experiments performed in duplicate, values are reported as the mean and standard error of the mean. As a note of caution, these error values are not strictly speaking a true estimate of error (which would require data at least in triplicate) but an indication of experimental variability. Whenever possible, individual replicate values are shown.
Results and Discussion
pH Dependence of the Reaction
For CtNDT, kcat/KM-nucleoside1 encompasses all steps from free enzyme and free substrate up to and including the first irreversible step (Scheme 1). For the first half-reaction, this is the release of the first nucleobase product (nucleobase1), while for the second half-reaction, this is the release of the newly formed nucleoside (nucleoside2). kcat includes all steps after nucleoside1 binding, up to and including regeneration of the free enzyme. In CtNDT’s case, our HPLC assays monitored the formation of a new nucleoside product (nucleoside2), and therefore, kcat includes the release of nucleobase1 and nucleoside2 from the enzyme (Scheme 1). Protein production, purity, and molecular weight determination are available in Figure S1.
pH-rate profiles for CtNDT were bell-shaped for kcat (pKa1 = 5.8 ± 0.1, pKa2 = 9.7 ± 0.2) and for kcat/KM-2′dGuo (pKa1 = 6.1 ± 0.3, pKa2 = 9.0 ± 0.2) with a single group in each limb giving rise to the pH dependence in both cases (Figure 1a).
Figure 1.

Effect of pH, temperature, and viscosity on the CtNDT-catalyzed reaction. (a) pH dependence of the CtNDT-catalyzed reaction. The line is a fit to eq 1, yielding acid dissociation constants for kcat (pKa1 = 5.8 ± 0.1, pKa2 = 9.7 ± 0.2, R2 = 0.8) and for kcat/KM-2′dGuo (pKa1 = 6.1 ± 0.3, pKa2 = 9.0 ± 0.2; R2 = 0.6). Figure S4D depicts residual plots for fits. (b) Temperature dependence of kcat; data were fitted to eq 2, yielding a ΔC‡p of −1.8 ± 0.3 kcal mol–1 K–1, and at 45 °C, a ΔH‡ of 28.9 ± 2.2 kcal mol–1 and a ΔS‡ of 0.032 ± 0.006 kcal mol–1. R2 = 0.95. Figure S4E depicts residual plots for fit. (c) Solvent viscosity effect on kcat using glycerol as a microviscogen at two different temperatures. The dotted line is shown for reference as a reaction completely limited by a diffusional step, with slope = 1. Here, the slope at 35 °C equals 1.0 ± 0.2, and the slope at 55 °C equals 0.9 ± 0.1. A control experiment was performed with PEG 8000, and no effect was observed (Figure S4C). Data for (a–c) were obtained using 2′dGuo and Ade as substrates. For parts (a, b), duplicates are shown; for part (c), data were collected in triplicate and are shown as mean plus standard error of the mean.
pKa1: The pKa1 for kcat/KM-2′dGuo and kcat profiles are likely reporting on the protonation state of E88, which must be deprotonated for catalysis to occur,39 in agreement with Lactobacillus leishmanii (LlNDT).40
pKa2: The pKa2 of ∼9.0 could be reporting on the pKa of D111 (discussed below), which could be donating a proton to the nucleobase leaving group, facilitating departure, a step included in both kcat/KM-2′dGuo and kcat. The environment surrounding D111 could determine its pKa2; however, no other negatively charged residues in the vicinity explain this large shift from the typical pKa of an aspartate residue.41
Alternatively, neighboring groups have been proposed to facilitate substrate binding and/or catalysis.40,42 Schramm proposes that a drastic increase in the pKa from around 2 to 9 in the case of protonation of N7 of inosine and purine nucleobases more generally occurs upon C1′ bond cleavage during the reaction catalyzed by purine nucleoside phosphorylase (PNP).27 Since there are similarities between PNP- and CtNDT-catalyzed reactions, an analogous pKa shift may occur here, enabling nucleobase leaving group stabilization by D111. The pKa for the N7 of guanosine in solution is between 3 and 4.43 Y7 is in close proximity to catalytic E88, and its deprotonation would decrease its nucleophilicity, impacting turnover.26
A comparison of the residues in the vicinity of the active site does not provide any obvious rationale for the shifted pKa values when comparing LlNDT and CtNDT (Figure S14), but the stickiness of substrates44 and rate-limiting product release45 can lead to outward shifted pKa values when comparing for kcat in comparison to kcat/KM-2′dGuo.
Temperature-Rate Profiles and Negative Heat Capacity
Figure 1b depicts the temperature dependence of kcat. The clearly curved temperature dependence profile was the best fit of the data to eq 2, yielding a ΔC‡p of −1.8 ± 0.3 kcal mol–1 K–1, and at 45 °C, a ΔH‡ of 28.9 ± 2.2 kcal mol–1 and a ΔS‡ of 0.032 ± 0.006 kcal mol–1.
Careful consideration was taken to exclude factors other than activation heat capacity, which could lead to nonlinear dependence. CtNDT was not denatured in the range evaluated here (between 35 and 65 °C), as the melting temperature for CtNDT was determined to be 72 °C under the conditions employed (pH 7.0) using differential scanning fluorimetry (Figure S3).
Changes in the rate-limiting steps across the temperature range under evaluation can lead to curved profiles. We carried out viscosity studies to determine whether the rate-limiting step was changing in the temperature range in which the curvature is observed.37 Using different concentrations of glycerol and determining kcat at 35 and 55 °C revealed a large and equal viscosity effect on the two temperatures investigated, with a slope of 1 within the experimental error (Figures 1c and S4). Furthermore, in the viscosity studies, we ensured experimentally, by varying substrates near the saturation range, that the enzyme was saturated with both substrates, and the lack of response in rate with increasing temperature was not due to sub-Vmax rates.
Such a high viscosity effect points toward a diffusional step, likely nucleobase1 release (further discussed below under “transient kinetics”), as a limiting step for turnover at both temperatures,37,46,47 excluding changes in the rate-limiting step between 35 and 55 °C as a reason for deviations from classic Arrhenius behavior. We performed a viscosity experiment with 5% PEG 8000,48 to probe whether a “medium” or crowding effect was taking place. No viscosity effect was observed with PEG (Figure S4C), further corroborating the importance of diffusion steps for kcat.
Prior work on CtNDT indicated a nonlinear dependence of rate as a function of temperature.21 These studies did not evaluate enzyme stability, change of rate-limiting steps, and kinetic parameters in the temperature range under investigation, and therefore, conclusions on the origin of nonlinear curves could not be drawn. Enzymes that display classic Arrhenius behavior have rates of reaction that increase exponentially as a function of temperature until denaturation starts to occur.49 The change in activation heat capacity ( ΔC‡p) associated with enzyme catalysis50 has been invoked to explain deviations from classic Arrhenius behavior.
A negative ΔC‡p suggests a decrease in the number of vibrational modes as the enzyme–substrate complex approaches the transition state.36,50 As illustrated in Scheme 1, kcat contains physical steps (substrate binding and product release, as well as potential conformational changes not shown in the figure for simplicity) and chemical steps,38 all of which can contribute to the effect observed on ΔC‡p. Because the viscosity effect is close to 1 in the temperature range under study, the transition state in this case is not the chemical TS but that for the diffusional release of the product.
Another possibility has been put forward to account for nonclassic Arrhenius behavior in the reaction catalyzed by an α-amylase from a psychrophilic organism, invoking the presence of different conformational states in the ES complex, giving rise to changes of opposite magnitudes to ΔH‡ and −TΔS‡.51,52 In this particular system, ΔG‡ had sizable contributions from ΔH‡ and −TΔS‡ at different temperatures, whereas in the CtNDT-catalyzed reaction, at 45 °C, −TΔS‡ is negligible, while ΔH‡ is 28.9 kcal mol–1. and therefore, it is unlikely that changes in the magnitude of thermodynamic parameters could lead to the curvature observed. Therefore, after ruling out denaturation, subsaturation, and changes in the rate-limiting step, a negative ΔC‡p remains the most likely model to account for nonclassic Arrhenius behavior in the reaction catalyzed by CtNDT.
The microorganism producing CtNDT, C. thermalis, is adapted for growth in desertic arid environments with high optimal temperatures.22 The optimal temperature of enzymes often correlates with environmental temperatures surrounding their organisms of origin,53 and heat capacity has been suggested to influence transition-state stabilization at high temperatures, as well as protein adaptability54 and evolvability.55 Negative ΔC‡p can reflect a higher preorganization state of CtNDT as it approaches the reaction transition state, providing advantages to catalysis at high temperatures.
Substrate Specificity of CtNDT and Mutants
dNDT enzymes have been previously determined to catalyze sugar-transfer reactions in a ping-pong mechanism, forming a covalent 2′-deoxyribosylated intermediate, followed by nucleobase transfer preserving stereochemistry on the anomeric carbon.39,40 To determine substrate specificity, we compared a series of purine and pyrimidine substrates, obtaining kinetic parameters for both nucleoside donor and nucleobase acceptor substrates (Figure 2a). Our results show that CtNDT prefers purines to pyrimidines, both in terms of nucleoside sugar donors and nucleobase acceptors (Figure 2b and Table S2). For the first half-reaction, 2′-deoxyadenosine (2′-dAdo) is the preferred sugar donor (kcat/KM = 50.5 ± 12.2 mM–1 s–1), and adenine (Ade) is the preferred sugar acceptor (kcat/KM = 63.3 ± 12.1 mM–1 s–1) for the second half-reaction. This substrate preference pattern is in agreement with a prior study on CtNDT.21 For purines, the higher substrate specificity is mostly KM-driven, while kcat values for different purine substrates are similar within the experimental error (Table S2).
Figure 2.

Substrate preference by CtNDT. (a) General scheme for the reaction catalyzed by CtNDT. To obtain kcat/KM-nucleoside, nucleoside substrates were varied and nucleobase was present at a saturating concentration, and to obtain kcat/KM-nucleobase, a 2′-deoxyribonucleoside substrate was present at a saturating concentration. Transglycosylation was monitored, and formation of nucleoside2 was quantified as a function of time. (b) Initial velocity studies were performed, and data were fitted to a Michaelis–Menten equation to yield kinetic parameters KM (purple), kcat (blue), and kcat/KM (green) when varying (2′-deoxy)ribonucleoside (left panel) or nucleobase (middle panel). The right panel shows kinetic parameters for 2′-dAdo (varying) and Gua (fixed) for different enzyme variants as a fraction of kinetic parameters for the wild-type enzyme. Colored boxes depict 2′-deoxypurine (orange box), 2′-deoxypyrimidine (light purple box), and ribonucleoside (dark purple box) substrates. All experiments were carried out in triplicate, and data are reported as the mean and standard error of the mean. All data are shown in Figure S5. Here, the “kinetic parameter” stands for the apparent steady-state constant obtained after the Michaelis–Menten fitting (Figure S5) when using different substrates and enzyme variants.
We tested the ribonucleosides Ado, Guo, and Ino as substrates for transglycosylation and obtained KM values of comparable magnitude to the ones obtained with 2′-deoxyribonucleosides (KM-Ado = 0.25 ± 0.05 mM, KM-Guo = 1.1 ± 0.3 mM, KM-Ino = 0.9 ± 0.2 mM). However, an over 100-fold difference in kcat between 2′-dGuo and Guo (6.9 ± 0.7 s–1 at 45 °C for 2′-dGuo and 0.06 ± 0.01 s–1 at 65 °C for Guo) indicates that nucleobase transfer is unfavorable with ribonucleoside substrates. Prior work on dNDT from Trypanosoma brucei(56) demonstrated that it could use ribonucleosides as substrates for transglycosylation, but kinetic parameters for ribonucleoside substrates were not determined for this wild-type enzyme. We further discuss the acceptance of ribonucleoside substrates below, as we solved the structure of a ribosylated CtNDT intermediate, providing crucial insights into substrate selection.
Based on prior reports and on our own structural data for CtNDT (see below), we produced mutants CtNDTD62N, CtNDTE88Q, CtNDTE88A, and CtNDTM120C to determine the importance of each in the catalytic mechanism. The identity of each mutant was further verified by intact protein mass spectrometry (Figure S2). By homology, E88 acts as a nucleophile, forming a glycosidic bond with the donor substrate. Our data show that CtNDTE88A is catalytic inactive, as expected, but CtNDTE88Q retains some catalytic activity, albeit the catalytic efficiency in comparison to the wild type is reduced by almost 50-fold. Any residual activity is unexpected given the crucial role proposed for E88 and might indicate that another residue in the active site vicinity can play a compensatory role in lieu of E88. This residue could be D62, as it is within hydrogen-bonding distance from the C1 of the sugar and E88 (kinetic parameters for mutants are given in Table S4and a discussion on the structure is given below). Alternatively, glutamine deamidation could occur, leading to the reversion of part of the protein into the active form with E88. To investigate this possibility, we conducted experiments incubating CtNDTE88Q with clofarabine, as detailed below. No covalent modifications were observed. If glutamine deamidation had taken place, it would have resulted in modifications in the enzyme fraction undergoing deamidation. Additionally, we performed a multiple turnover experiment to quantify the product formed after one turnover with CtNDTE88Q. After a 60 s reaction (the expected time for one turnover with CtNDTE88Q), 10 μM of the product was formed, in excellent alignment with the concentration of active sites for CtNDTE88Q. We calculated that 0.25% deamidation would be below our limit of detection by mass spectrometry, and the same amount of the product would be observed in a single turnover experiment. Therefore, while we cannot completely rule out deamidation, we have no evidence to support it.
Computational studies proposed the formation of an oxocarbenium ion in the transition state for the reaction, and D62 is equivalent to a residue proposed to interact with the anomeric carbon in this putative oxocarbenium ion transition state.16CtNDTD62N, however, retained ∼50% catalytic efficiency in comparison to WT, and it is unlikely to play such a crucial role in transition-state stabilization.
Prior work in a pyrimidine-preferring dNDT observed some large effects in the substitution of a methionine residue (M125A) by an alanine, also in proximity to the catalytic glutamate.40 This mutation increased KM for substrates, so we carried out substitution on the equivalent position of CtNDTM120C, aimed instead at decreasing KM for substrates. This mutation had no effect on KM but led to a mild ∼30% reduction in catalytic efficiency, demonstrating that M120 might play an indirect role in substrate positioning, as it does not interact with any substrate/analogues according to structural data discussed below.
Transient Kinetics
To probe individual rate constants for the mechanism depicted in Scheme 1, we performed fast kinetic experiments monitoring substrate binding and chemistry in the first half-reaction with 2′-dAdo as a substrate. We used the inactive mutant CtNDTE88A to isolate the substrate binding step, the less active mutants CtNDTE88Q and CtNDTD62N to determine the involvement of each of these residues in binding and/or 2′-deoxyribosylation, and a wild-type enzyme to obtain boundaries for the magnitude of the rate constant for the 2′-deoxyribosylation reaction (Figure 3a). As previously reported for another dNDT enzyme,30 there is a decrease in protein fluorescence upon substrate binding and/or 2′-deoxyribosylation (Figure 3b–d). Experiments were performed at 25 °C due to the presence of transients for binding and ribosylation that were too fast to capture at higher temperatures. After numerical integration of the data using KinTek Global Explorer25,28,29 as described in Supporting Information, values for individual rate constants were obtained (Table S5 and Figures S6–S9).
Figure 3.

Pre-steady-state kinetics of the first half-reaction. (a) Steps and rate constants obtained with different CtNDT mutants. Brackets indicate the number of transients observed with each enzyme variant, using 2′-dAdo for binding or 2′-deoxyribosylation. (b–d) Raw data after mixing 0.75 mM CtNDT. (c) Left for wild type and right for CtNDTD62N. (d) CtNDTE88A with increasing concentrations of 2′-dAdo as indicated. With CtNDTD62N, two transients are observed, where the initial decay is followed by a slow fluorescence increase. (b) Comparison between wild-type CtNDT and CtNDTD62N showed no increase in fluorescence in the wild-type protein. (e) Rate constants obtained after fitted values (Table S5) were converted into ΔG (kcal mol–1) using eq 3, and relative barriers for each step comparing mutants are shown.
The association rate constant for 2′-deoxyadenosine binding (obtained with CtNDTE88A) was 0.76 ± 0.01 μM–1 s–1 with a dissociation rate constant of 39.3 ± 0.7 s–1. kcat/KM-2′Ado-25°C = 0.016 μM–1 s–1 (determined with the saturating concentration of nucleobase) sets a lower boundary for this bimolecular association step. Rates for 2′-deoxyribosylation, on the other hand, were very fast (532 s–1 for CtNDT WT) and therefore did not limit kcat-25°C (3.2 ± 0.2 s–1).
In our pre-steady-state experiments using mutants CtNDTD62N, two transients with opposite amplitudes were observed, while in experiments performed with the wild-type enzyme using identical conditions, only a single transient showing fluorescence decay was observed. The second transient showed a hyperbolic dependence on ligand concentration and therefore can be attributed to an unimolecular transformation. Although this unimolecular transformation could be a conformational change in the enzyme, independent of chemistry, it could also be due to 2′-deoxyribosyl-enzyme hydrolysis (energy barrier depicted in Figure 3e). Figure S10 depicts the extent of nucleoside substrate hydrolysis by the wild type, CtNDTD62N, and CtNDTE88Q. More futile cycles of nucleoside hydrolysis that do not result in transglycosylation take place with CtNDTD62N and CtNDTE88Q than with the wild-type enzyme. Stopped-flow data were used with enzyme variants CtNDTE88A to estimate the rates of 2′-deoxyadenosine binding and release, wild-type CtNDT to obtain the rate of 2′-deoxyribosylation, and CtNDTD62N to obtain the rate of 2′-deoxyribosyl hydrolysis. The rate of 2′-deoxyribosylation was not significantly altered in E88Q-CtNDT compared with the WT enzyme, in agreement with covalent intermediate formation not being rate-limiting for kcat.
The reaction catalyzed by CtNDT is fully reversible, and both substrates and products are equivalent, as depicted in Scheme S1. Therefore, our pre-steady-state data make clear that 2′-deoxynucleoside release is not rate-limiting, as binding experiments showed association and dissociation rate constants for 2′-deoxyadenosine 100 times faster than kcat. Because identical kcat values were determined for 2′-deoxyadenosine and 2′-deoxyguanosine when Gua and Ade were employed as nucleobases, respectively (Table S2, steady-state kinetics of CtNDT), steady-state turnover is likely to be limited by the same step when these different 2′-deoxynucleosides are used as substrates. Taking together the large viscosity effect on kcat as expected for a completely diffusion-controlled reaction and the fast dissociation rate constant for 2′-deoxyadenosine (approximately 10x faster than kcat), we propose that a diffusional step, i.e., nucleobase1 departure, limits turnover in the CtNDT-catalyzed reaction.
Binding of Potential Transition-State Analogues
Schramm and co-workers developed tight-binding transition-state analogues for the reaction catalyzed by human purine nucleoside phosphorylase (PNP) and revolutionized the design of specific and potent inhibitors by studying enzymatic transition states.57 Such analogues have exquisite potency as drugs and also serve as important nonhydrolyzable probes to study principles of catalysis in enzymes that catalyze nucleoside transfer reactions. We compared two potential transition-state analogues for transglycosylation, immucillin-H (ImmH) and DADme–immucillin-H (DADmeH), as the distance between the bond being broken/formed in the nucleobase and sugar varies (Figure 4a).58 For ImmH, ΔH was −5.25 ± 0.07 kcal mol–1, −TΔS was −0.36 ± 0.14 kcal mol–1, and KD was 78.2 ± 6.6 μM, while for DADmeH, ΔH was −11.43 ± 0.29 kcal mol–1, −TΔS was 0.38 ± 0.73 kcal mol–1, and KD was 9 ± 5 nM (Table S6). ΔG was −5.60 ± 0.08 for ImmH and −11.05 ± 0.42 kcal mol–1 for DADmeH; therefore, binding is determined by an enthalpic contribution (Figure 4b).
Figure 4.

Equilibrium binding of potential transition-state analogues. Data for the equilibrium binding of analogues ImmH and DADmeH measured using isothermal titration calorimetry (ITC). (a) Wild-type CtNDT was present in the cell while each analogue was titrated. Raw data and fitted traces to a one-binding-site model are shown. Representative traces are shown here, and all data are available in Figures S11 and S12. (b) Fitted results for thermodynamic parameters ΔH, ΔG, and −TΔS. The average for duplicate experiments and error spread are shown.
Enzymes evolved to bind tightly to their cognate transition state and therefore to stable compounds that most closely mimic the charge distribution and geometry of that transition state.59 Based on Scheme 1 and considering 2′-deoxyribonucleside1 as a substrate, releasing nucleobase1 and generating a 2′-deoxyribosylated enzyme, ImmH would mimic an early transition state in which the distance between ribose C1′ and the nucleobase is ∼1.5 Å, while DADmeH would mimic a late transition state, since the presence of a methylene bridge between ribitol N1′ (which replaces C1′ in DADmeH) keeps the nucleobase at a distance of ∼3.0 Å from these groups. In the second half-reaction, the “early” and “late” definitions would have the opposite meaning, as the final product would be 2′-deoxyribonucleside2 following C–N bond formation.
Prior detailed work on the thermodynamic analysis of a series of transition-state analogues of human PNP revealed that tighter-binding TS analogues had an enthalpically driven binding process with little entropic contribution.60 This is in agreement with our results, as most of the ΔG for binding is derived from ΔH in both analogues studied, more pronounced for the tighter-binding late-transition-state analogue DADmeH (Figure 4b).
Furthermore, we performed kinetic binding experiments to probe whether ImmH and DADmeH binding was taking place as a single step process. Although no signal change was observed with DADmeH, ImmH binding led to a small fluorescence decrease in a single transient (Figure S13).
Overall Structure of CtNDT
The overall structure of CtNDT is similar to that of T. brucei nucleoside 2′deoxyribosyltransferase (TbNDT),61 as structural superposition reveals an RMSD of 0.859 Å. Size exclusion data reveal CtNDT to be a tetramer in solution formed by a dimer of dimers (Figure S1), which is also corroborated by the analysis of protein interfaces with PISA.62 The best studied dNDT in terms of kinetics, the protein from L. leishmanii (LlNDT),63 is a hexamer (a trimer of dimers), and the structural alignment of each dimer of LlNDT shows an RMSD of 1.093 Å when compared to CtNDT. As with other NDT enzymes, the active site of CtNDT is formed by residues from two monomers, and therefore, it is only fully formed in the dimer/tetramer. Figure S14 shows a comparison between the structures of CtNDT and LlNDT, depicting conserved residues that play similar roles in both proteins.
Snapshots of the Reaction Coordinate of CtNDT
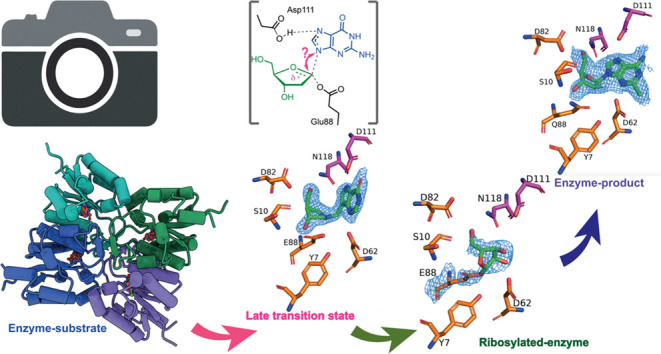
To further understand how CtNDT binds 2′-deoxyribonucleosides and ribonucleosides and catalyzes sugar transfer, we cocrystallized CtNDT in complex with adenosine (to yield a ribosylated enzyme), clofarabine (2-chloro-9-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-9H-purin-6-amine, a substrate/product analogue), and the transition-state analogues ImmH and DADmeH (Figure 5). Specific interactions with each ligand shed light on ligand binding and transition-state stabilization.
Figure 5.

Steps in the reaction coordinate of CtNDT. (a) CtNDTE88Q bound to clofarabine, depicting interactions of an enzyme–substrate or enzyme–product complex depending on the substrates employed. (b) CtNDTD62N bound to immucillin-H and (c) CtNDT bound to DADme–immucillin-H, respectively, designed as “early” or “late” nucleoside-like transition-state analogues for nucleoside phosphorolysis catalyzed by PNP. (d) Ribosylated CtNDT, depicting 5′-OH in close proximity to D82. An alternate conformation depicting 5′-OH facing S10 is depicted in Figure S15. Omit maps (2Fo-Fc) are depicted in Figures S16 and 6.
Enzyme–substrate complex (or enzyme–product complex, depending on the reaction direction): the complex CtNDTE88Q-clofarabine shows the 3′-OH of the arabinofuranosyl sugar within the hydrogen-bonding distance (3.3 Å) of S10. E88Q is 3.2 Å from C1′ in the sugar, 2.8 Å from Y7, and 3.3 Å from D62. E88Q and D62 are 3.3 Å apart, and therefore, Y7 can interact with either or both residues depending on the state in the reaction coordinate to ensure optimal positioning. Similar interaction patterns between the catalytic glutamate and the conserved tyrosine residue were observed in the dNDT from Lactobacillus helveticus (pdb 1S2G).13 Interactions with the nucleobase are mediated by hydrogen-bonding interactions with D111 and D62, and the 5′-OH is within the hydrogen-bonding distance of N118 (2.9 Å).
Possible Transition States for the First Half-Reaction
Structures were obtained with the two potential transition-state analogues ImmH and DADmeH. The structure of bound ImmH largely resembles that of the clofarabine-bound complex, in agreement with the relatively weak binding of ImmH to CtNDT, and it is therefore substrate-like. In the structure of bound DADmeH, however, E88 is 2.5 Å from N1′ in the sugar (analogous to C1′ in the substrate), 2.9 Å from Y7, and 3.5 Å from D62. D62 and Y7 are 3.4 Å apart. Stabilizing the nascent positive charge in the leaving group is D111, 2.5 Å away from Nitrogen 7 on DADmeH and 3.2 Å away from D62. The distance between D111 and N7 from DADmeH is shorter than the sum of the van der Waals radii for O and N, indicative of a very strong hydrogen bond between the two groups.64,65 Therefore, in the complex with DADmeH, E88 is closer to the bond that needs to be broken, D111 is closer to the nucleobase leaving group, and D62 is farther from interactions with the nucleobase when compared to an enzyme–substrate complex and also farther from the 3′-OH in comparison to the 2′-deoxyribosyl intermediate. This, coupled with the nM binding affinity of DADmeH for CtNDT, points toward a late transition state for the first half-reaction in which the bond order between sugar and nucleobase is negligible. The proximity of E88 to N1′ might suggest significant nucleophile participation at the TS. Both ImmH and DADmeH were developed as mimics of the transition state of the reaction catalyzed by purine nucleoside phosphorylase (PNP).66 Human and bovine enzymes share 87% sequence identity, but the reactions catalyzed proceed via different transition states. While the reaction catalyzed by bovine PNP (bPNP) proceeds via an early transition state with a relatively close 1.8 Å distance between the leaving-group nitrogen and the anomeric carbon, human PNP (hPNP) ribosidic bond cleavage occurs via a fully dissociated purine leaving group with a fully developed oxocarbenium ion, with a distance of 3.0 Å between the two groups.67
Covalent Enzyme–Ribosyl Intermediate
After nucleobase1 departure, the enzyme is 2′-deoxyribosylated until hydrolysis or the second half-reaction occurs. Although dNDT enzymes are selective for 2′-deoxyribonucleosides, molecular detail underlying this preference is lacking. We therefore determined the structure of a ribosylated enzyme intermediate by cocrystallization of wild-type CtNDT in the presence of Ado. Residues D82 and N118 are in close proximity to the 5′-OH of the intermediate (3.3 and 3.5 Å, respectively). D62 is in very close proximity (2.6 Å) from 2′-OH and to 3′-OH (3.2 Å), and the proximity to 2′-OH (absent in 2′-deoxyribonucleosides) can lead to less optimal positioning of this intermediate for nucleophilic attack by the incoming nucleobase 2.
An important observation is the positioning of Y7, likely polarizing the –COO– side chain of E88. Y7 does not directly interact with ligands, nor is it at a close enough distance to clash with a potential 2′-OH in the ribonucleoside ligand. One possibility is that Y7 restricts the movement of catalytic E88, but this optimal positioning for 2′-deoxyribonucleosides results in clashes between D62 and the 2′-OH of ribonucleoside substrates, leading to a reduced catalytic efficiency with these substrates.
The electron density surrounding the ribosylated intermediate is poorly defined, which could indicate that the intermediate is sampling multiple conformations (Figure S14 depicts one of such conformations in which the 5′-OH is turned away from D82) or that there is partial density for adenosine, which is not defined well enough to be accurately modeled. We therefore confirmed the presence of covalent adduct formation via an orthogonal method. Prior work with the LhNDT13 trapped a 2′-deoxy-2′-fluoro-ribosyl intermediate upon incubation with a 2′-deoxy-2′-fluoro-β-d-arabinofuranosyladenine substrate, and it was hypothesized that 2′-fluoro-containing substrates undergo slower hydrolysis in comparison to compounds lacking fluorine on position 2′. We report a structure of a 2-difluoro-2-deoxy-ribosylated CtNDT after cocrystallization with Gemcitabine for comparison with the ribosylated enzyme. Figure S17 shows a comparison between ribosylated and 2-difluoro-2-deoxy-ribosylated CtNDT. The main difference is in the conformation sampled by the 3′-OH of the 2-difluoro-2-deoxy-ribosylated intermediate, positioned closer to S10 than to D62. After the ligand was added to the protein, refinement (with Phenix Refine) produced three monomers with a covalently modified E88 and one with a covalent modification on D62. We do not have additional biochemical evidence for the formation of a covalent intermediate with D62, and because this could be an artifact, we have not discussed this in detail.
To further confirm that the ribosylated intermediate was trapped in solution as well as in crystallo, wild-type CtNDT was incubated with the substrate clofarabine. After incubation, the intact mass of wild-type CtNDT increased by 135 Da, in agreement with what would be expected if a 2′-deoxy-2′-fluoro-ribosyl intermediate was present (Figure S18a–c).
This is the first structural characterization of a dNDT enzyme covalently modified by a ribonucleoside, enabling further engineering efforts to improve catalytic efficiency toward ribonucleoside analogues (Figure 6).
Figure 6.

Snapshots of the reaction coordinate for the first half-reaction catalyzed by CtNDT. Proposed mechanism for the CtNDT-catalyzed reaction, combining data from protein mass spectrometry and X-ray crystallography. Electron density for ligands is depicted as −2Fo-Fc at 2σ contour (maps prepared using likelihood-weighted laps on Phenix, and image prepared using Pymol). Distances shown for ImmH and DADmeH are from ref (58).
Conclusions and Implications for dNDT Enzymes
Our work provides a detailed mechanistic understanding of a thermophilic nucleoside transferase enzyme. Combining enzymology and biophysical techniques, we present a unique insight into the reaction coordinate for (2′-deoxy)nucleoside transfer, including interactions important for substrate selection and catalysis. Our data support a late transition state for the first half-reaction and determine that nucleobase1 release is rate-limiting during steady-state turnover.
In recent years, several have hypothesized that nonzero activation heat capacity might explain the presence of curved Arrhenius plots in enzyme catalysis.50 This model has been proposed to account for temperature optima in enzymes that are not associated with the initiation of protein unfolding.38 In the CtNDT-catalyzed reaction, the significant contribution of negative heat capacity to a reaction is limited by a diffusional step, highlighting the importance of determining rate-limiting steps when evaluating the contribution of heat capacity to enzyme-catalyzed reactions, as well as considering changes in rate-limiting steps in the temperature range under investigation.37
CtNDT demonstrated promiscuity in terms of substrate scope and reactions catalyzed—(2′-deoxy)ribosylated hydrolysis or (2′-deoxy)ribosyl transfer. Enzyme promiscuity has been considered an ancestral trait, often lost in modern enzymes under prolonged selection for efficient substrate turnover.68,69 This knowledge has been applied to generate enzymes with expanded function,70 as well as in ancestral enzyme reconstruction efforts. Our work is a crucial step to understand dNDT enzymes and informs future enzyme evolution55 and engineering71 efforts on this enzyme class aimed at producing novel nucleosides.
Acknowledgments
The authors thank Professor Vern Schramm for his kind donation of DADmeH and the mass spectrometry facility in St Andrews for support in mass spectrometry. P.T. is funded by IBioIC (IBioIC 2020-2-1), and C.M.C. is funded by the Wellcome Trust (217078/Z/19/Z). C.M.C. and D.H. are funded by research grants from NuCana plc.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c06260.
Additional methods for protein production, enzymatic assays, mass spectrometry, HPLC conditions, and DSF experiments; raw data for assays; melting temperature determination using differential scanning fluorimetry; hydrolysis of nucleoside substrates uncoupled from base transfer; primers for cloning and mutagenesis; pre-steady-state parameters globally fitted using KinTek Global Explorer; and crystallization conditions and crystallization table for pdb entries 8PQP, 8PQQ, 8PQR, 8PQS, 8PQT, 8QC0, and 8RH3 (PDF)
Author Contributions
§ A.L.D. and D.J.H. are part-time employed by NuCana plc. C.J.H. performed experiments on crystallography and molecular modeling, A.L.D. performed mass spectrometry experiments and aided in data analysis, P.T. designed and performed all kinetics and ITC experiments. C.M.C. and P.T. wrote the manuscript. C.M.C. and D.J.H. planned the project, and R.G.S. participated in data analysis and interpretation. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- De Clercq E. A cutting-edge view on the current state of antiviral drug development. Med. Res. Rev. 2013, 33 (6), 1249–1277. 10.1002/med.21281. [DOI] [PubMed] [Google Scholar]
- Jordheim L. P.; Durantel D.; Zoulim F.; Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery 2013, 12 (6), 447–464. 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- Kaspar F.; Stone M. R. L.; Neubauer P.; Kurreck A. Route efficiency assessment and review of the synthesis of β-nucleosides via N-glycosylation of nucleobases. Green Chem. 2021, 23 (1), 37–50. 10.1039/D0GC02665D. [DOI] [Google Scholar]
- Huffman M. A.; Fryszkowska A.; Alvizo O.; Borra-Garske M.; Campos K. R.; Canada K. A.; Devine P. N.; Duan D.; Forstater J. H.; Grosser S. T.; et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366 (6470), 1255–1259. 10.1126/science.aay8484. [DOI] [PubMed] [Google Scholar]
- McIntosh J. A.; Benkovics T.; Silverman S. M.; Huffman M. A.; Kong J.; Maligres P. E.; Itoh T.; Yang H.; Verma D.; Pan W.; et al. Engineered Ribosyl-1-Kinase Enables Concise Synthesis of Molnupiravir, an Antiviral for COVID-19. ACS Cent. Sci. 2021, 7 (12), 1980–1985. 10.1021/acscentsci.1c00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer M.; Ribar A.; Nidetzky B. A selective and atom-economic rearrangement of uridine by cascade biocatalysis for production of pseudouridine. Nat. Commun. 2023, 14 (1), 2261 10.1038/s41467-023-37942-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco J.; Acosta J.; Fernandez-Lucas J. New trends in the biocatalytic production of nucleosidic active pharmaceutical ingredients using 2’-deoxyribosyltransferases. Biotechnol. Adv. 2021, 51, 107701 10.1016/j.biotechadv.2021.107701. [DOI] [PubMed] [Google Scholar]
- Rinaldi F.; Fernandez-Lucas J.; de la Fuente D.; Zheng C.; Bavaro T.; Peters B.; Massolini G.; Annunziata F.; Conti P.; de la Mata I.; et al. Immobilized enzyme reactors based on nucleoside phosphorylases and 2’-deoxyribosyltransferase for the in-flow synthesis of pharmaceutically relevant nucleoside analogues. Bioresour. Technol. 2020, 307, 123258 10.1016/j.biortech.2020.123258. [DOI] [PubMed] [Google Scholar]
- Vichier-Guerre S.; Ku T. C.; Pochet S.; Seley-Radtke K. L. An Expedient Synthesis of Flexible Nucleosides through Enzymatic Glycosylation of Proximal and Distal Fleximer Bases. Chembiochem 2020, 21 (10), 1412–1417. 10.1002/cbic.201900714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo N.; Sanchez-Murcia P. A.; Gago F.; Cejudo-Sanches J.; Galmes M. A.; Fernandez-Lucas J.; Mancheno J. M. 2’-Deoxyribosyltransferase from Leishmania mexicana, an efficient biocatalyst for one-pot, one-step synthesis of nucleosides from poorly soluble purine bases. Appl. Microbiol. Biotechnol. 2017, 101 (19), 7187–7200. 10.1007/s00253-017-8450-y. [DOI] [PubMed] [Google Scholar]
- Kaminski P. A.; Labesse G. Phosphodeoxyribosyltransferases, designed enzymes for deoxyribonucleotides synthesis. J. Biol. Chem. 2013, 288 (9), 6534–6541. 10.1074/jbc.M112.446492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnutt W. S. The enzymically catalysed transfer of the deoxyribosyl group from one purine or pyrimidine to another. Biochem. J. 1952, 50 (3), 384–397. 10.1042/bj0500384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R.; Kaminski P. A.; Ealick S. E. Structures of purine 2’-deoxyribosyltransferase, substrate complexes, and the ribosylated enzyme intermediate at 2.0 A resolution. Biochemistry 2004, 43 (9), 2384–2393. 10.1021/bi035723k. [DOI] [PubMed] [Google Scholar]
- Del Arco J.; Mills A.; Gago F.; Fernandez-Lucas J. Structure-Guided Tuning of a Selectivity Switch towards Ribonucleosides in Trypanosoma brucei Purine Nucleoside 2’-Deoxyribosyltransferase. Chembiochem 2019, 20 (24), 2996–3000. 10.1002/cbic.201900397. [DOI] [PubMed] [Google Scholar]
- Acosta J.; Del Arco J.; Pisabarro V.; Gago F.; Fernandez-Lucas J. N-Ribosyltransferase From Archaeoglobus veneficus: A Novel Halotolerant and Thermostable Biocatalyst for the Synthesis of Purine Ribonucleoside Analogs. Front. Bioeng. Biotechnol. 2020, 8, 593 10.3389/fbioe.2020.00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco J.; Perona A.; Gonzalez L.; Fernandez-Lucas J.; Gago F.; Sanchez-Murcia P. A. Reaction mechanism of nucleoside 2’-deoxyribosyltransferases: free-energy landscape supports an oxocarbenium ion as the reaction intermediate. Org. Biomol. Chem. 2019, 17 (34), 7891–7899. 10.1039/C9OB01315F. [DOI] [PubMed] [Google Scholar]
- Kaminski P. A.; Dacher P.; Dugue L.; Pochet S. In vivo reshaping the catalytic site of nucleoside 2’-deoxyribosyltransferase for dideoxy- and didehydronucleosides via a single amino acid substitution. J. Biol. Chem. 2008, 283 (29), 20053–20059. 10.1074/jbc.M802706200. [DOI] [PubMed] [Google Scholar]
- Ye W.; Paul D.; Gao L.; Seckute J.; Sangaiah R.; Jayaraj K.; Zhang Z.; Kaminski P. A.; Ealick S. E.; Gold A.; Ball L. M. Ethenoguanines undergo glycosylation by nucleoside 2’-deoxyribosyltransferases at non-natural sites. PLoS One 2014, 9 (12), e115082 10.1371/journal.pone.0115082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco J.; Perona A.; González L.; Fernández-Lucas J.; Gago F.; Sánchez-Murcia P. A. Reaction mechanism of nucleoside 2′-deoxyribosyltransferases: free-energy landscape supports an oxocarbenium ion as the reaction intermediate. Org. Biomol. Chem. 2019, 17 (34), 7891–7899. 10.1039/C9OB01315F. [DOI] [PubMed] [Google Scholar]
- Marasco R.; Fusi M.; Coscolin C.; Barozzi A.; Almendral D.; Bargiela R.; Nutschel C. G. N.; Pfleger C.; Dittrich J.; Gohlke H.; et al. Enzyme adaptation to habitat thermal legacy shapes the thermal plasticity of marine microbiomes. Nat. Commun. 2023, 14 (1), 1045 10.1038/s41467-023-36610-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco J.; Sánchez-Murcia P. A.; Mancheño J. M.; Gago F.; Fernández-Lucas J. Characterization of an atypical, thermostable, organic solvent- and acid-tolerant 2’-deoxyribosyltransferase from Chroococcidiopsis thermalis. Appl. Microbiol. Biotechnol. 2018, 102 (16), 6947–6957. 10.1007/s00253-018-9134-y. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Gan F.; Shen G.; Bryant D. A. RfpA, RfpB, and RfpC are the Master Control Elements of Far-Red Light Photoacclimation (FaRLiP). Front. Microbiol. 2015, 6, 1303 10.3389/fmicb.2015.01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E. L.; Finnigan W.; France S. P.; Green A. P.; Hayes M. A.; Hepworth L. J.; Lovelock S. L.; Niikura H.; Osuna S.; Romero E.; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1 (1), 46 10.1038/s43586-021-00044-z. [DOI] [Google Scholar]
- Dumorne K.; Cordova D. C.; Astorga-Elo M.; Renganathan P. Extremozymes: A Potential Source for Industrial Applications. J. Microbiol. Biotechnol. 2017, 27 (4), 649–659. 10.4014/jmb.1611.11006. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.Fitting Enzyme Kinetic Data with KinTek Global Kinetic Explorer. In Methods in Enzymology; Elsevier, 2009; Vol. 467, pp 601–626. [DOI] [PubMed] [Google Scholar]
- Oke M.; Carter L. G.; Johnson K. A.; Liu H.; McMahon S. A.; Yan X.; Kerou M.; Weikart N. D.; Kadi N.; Sheikh M. A.; et al. The Scottish Structural Proteomics Facility: targets, methods and outputs. J. Struct Funct. Genomics 2010, 11 (2), 167–180. 10.1007/s10969-010-9090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud S.; Wilkins M. R.; Appel R. D.; Bairoch A.. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker J. M., Ed.; Humana Press, 2005; pp 571–607. [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 2009, 387 (1), 30–41. 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 2009, 387 (1), 20–29. 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Porter D. J.; Short S. A. Nucleoside 2-deoxyribosyltransferase. Pre-steady-state kinetic analysis of native enzyme and mutant enzyme with an alanyl residue replacing Glu-98. J. Biol. Chem. 1995, 270 (26), 15557–15562. 10.1074/jbc.270.26.15557. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 1002–1011. 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (Pt 4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. J.; Headd J. J.; Moriarty N. W.; Prisant M. G.; Videau L. L.; Deis L. N.; Verma V.; Keedy D. A.; Hintze B. J.; Chen V. B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27 (1), 293–315. 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Meng E. C.; Couch G. S.; Croll T. I.; Morris J. H.; Ferrin T. E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30 (1), 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcus V. L.; Prentice E. J.; Hobbs J. K.; Mulholland A. J.; Van der Kamp M. W.; Pudney C. R.; Parker E. J.; Schipper L. A. On the Temperature Dependence of Enzyme-Catalyzed Rates. Biochemistry 2016, 55 (12), 1681–1688. 10.1021/acs.biochem.5b01094. [DOI] [PubMed] [Google Scholar]
- Machado T. F. G.; Gloster T. M.; da Silva R. G. Linear Eyring Plots Conceal a Change in the Rate-Limiting Step in an Enzyme Reaction. Biochemistry 2018, 57 (49), 6757–6761. 10.1021/acs.biochem.8b01099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kamp M. W.; Prentice E. J.; Kraakman K. L.; Connolly M.; Mulholland A. J.; Arcus V. L. Dynamical origins of heat capacity changes in enzyme-catalysed reactions. Nat. Commun. 2018, 9 (1), 1177 10.1038/s41467-018-03597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D. J. T.; Merrill B. M.; Short S. A. Identification of the Active Site Nucleophile in Nucleoside 2-Deoxyribosyltransferase as Glutamic Acid 98 (*). J. Biol. Chem. 1995, 270 (26), 15551–15556. 10.1074/jbc.270.26.15551. [DOI] [PubMed] [Google Scholar]
- Short S. A.; Armstrong S. R.; Ealick S. E.; Porter D. J. T. Active Site Amino Acids That Participate in the Catalytic Mechanism of Nucleoside 2′-Deoxyribosyltransferase (*). J. Biol. Chem. 1996, 271 (9), 4978–4987. 10.1074/jbc.271.9.4978. [DOI] [PubMed] [Google Scholar]
- Khavrutskii I. V.; Compton J. R.; Jurkouich K. M.; Legler P. M. Paired Carboxylic Acids in Enzymes and Their Role in Selective Substrate Binding, Catalysis, and Unusually Shifted pK(a) Values. Biochemistry 2019, 58 (52), 5351–5365. 10.1021/acs.biochem.9b00429. [DOI] [PubMed] [Google Scholar]
- Schramm V. L. Development of transition state analogues of purine nucleoside phosphorylase as anti-T-cell agents. Biochim. Biophys. Acta, Mol. Basis Dis. 2002, 1587 (2–3), 107–117. 10.1016/S0925-4439(02)00073-X. [DOI] [PubMed] [Google Scholar]
- Jang Y. H.; Goddard W. A.; Noyes K. T.; Sowers L. C.; Hwang S.; Chung D. S. pKa Values of Guanine in Water: Density Functional Theory Calculations Combined with Poisson–Boltzmann Continuum–Solvation Model. J. Phys. Chem. B 2003, 107 (1), 344–357. 10.1021/jp020774x. [DOI] [Google Scholar]
- Shen R.; Crean R. M.; Johnson S. J.; Kamerlin S. C. L.; Hengge A. C. Single Residue on the WPD-Loop Affects the pH Dependency of Catalysis in Protein Tyrosine Phosphatases. JACS Au 2021, 1 (5), 646–659. 10.1021/jacsau.1c00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland W. W.The Use of pH Studies to Determine Chemical Mechanisms of Enzyme-Catalyzed Reactions. In Methods in Enzymology; Elsevier, 1982; Vol. 87, pp 390–405. [DOI] [PubMed] [Google Scholar]
- Adams J. A.; Taylor S. S. Energetic limits of phosphotransfer in the catalytic subunit of cAMP-dependent protein kinase as measured by viscosity experiments. Biochemistry 1992, 31 (36), 8516–8522. 10.1021/bi00151a019. [DOI] [PubMed] [Google Scholar]
- Fisher G.; Pecaver E.; Read B. J.; Leese S. K.; Laing E.; Dickson A. L.; Czekster C. M.; da Silva R. G. Catalytic Cycle of the Bifunctional Enzyme Phosphoribosyl-ATP Pyrophosphohydrolase/Phosphoribosyl-AMP Cyclohydrolase. ACS Catal. 2023, 13 (11), 7669–7679. 10.1021/acscatal.3c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadda G.; Sobrado P. Kinetic Solvent Viscosity Effects as Probes for Studying the Mechanisms of Enzyme Action. Biochemistry 2018, 57 (25), 3445–3453. 10.1021/acs.biochem.8b00232. [DOI] [PubMed] [Google Scholar]
- Wolfenden R.; Snider M. J. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001, 34 (12), 938–945. 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- Hobbs J. K.; Jiao W.; Easter A. D.; Parker E. J.; Schipper L. A.; Arcus V. L. Change in heat capacity for enzyme catalysis determines temperature dependence of enzyme catalyzed rates. ACS Chem. Biol. 2013, 8 (11), 2388–2393. 10.1021/cb4005029. [DOI] [PubMed] [Google Scholar]
- Åqvist J.; Socan J.; Purg M. Hidden Conformational States and Strange Temperature Optima in Enzyme Catalysis. Biochemistry 2020, 59 (40), 3844–3855. 10.1021/acs.biochem.0c00705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sočan J.; Purg M.; Aqvist J. Computer simulations explain the anomalous temperature optimum in a cold-adapted enzyme. Nat. Commun. 2020, 11 (1), 2644 10.1038/s41467-020-16341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs J. K.; Shepherd C.; Saul D. J.; Demetras N. J.; Haaning S.; Monk C. R.; Daniel R. M.; Arcus V. L. On the origin and evolution of thermophily: reconstruction of functional precambrian enzymes from ancestors of Bacillus. Mol. Biol. Evol. 2012, 29 (2), 825–835. 10.1093/molbev/msr253. [DOI] [PubMed] [Google Scholar]
- Nguyen V.; Wilson C.; Hoemberger M.; Stiller J. B.; Agafonov R. V.; Kutter S.; English J.; Theobald D. L.; Kern D. Evolutionary drivers of thermoadaptation in enzyme catalysis. Science 2017, 355 (6322), 289–294. 10.1126/science.aah3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunzel H. A.; Anderson J. L. R.; Hilvert D.; Arcus V. L.; van der Kamp M. W.; Mulholland A. J. Evolution of dynamical networks enhances catalysis in a designer enzyme. Nat. Chem. 2021, 13 (10), 1017–1022. 10.1038/s41557-021-00763-6. [DOI] [PubMed] [Google Scholar]
- Pérez E.; Sánchez-Murcia P. A.; Jordaan J.; Blanco M. D.; Mancheño J. M.; Gago F.; Fernández-Lucas J. Enzymatic Synthesis of Therapeutic Nucleosides using a Highly Versatile Purine Nucleoside 2’-DeoxyribosylTransferase from Trypanosoma brucei. ChemCatChem 2018, 10 (19), 4406–4416. 10.1002/cctc.201800775. [DOI] [Google Scholar]
- Miles R. W.; Tyler P. C.; Furneaux R. H.; Bagdassarian C. K.; Schramm V. L. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry 1998, 37 (24), 8615–8621. 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]
- Vetticatt M. J.; Itin B.; Evans G. B.; Schramm V. L. Distortional binding of transition state analogs to human purine nucleoside phosphorylase probed by magic angle spinning solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (40), 15991–15996. 10.1073/pnas.1313657110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm V. L. Transition States and transition state analogue interactions with enzymes. Acc. Chem. Res. 2015, 48 (4), 1032–1039. 10.1021/acs.accounts.5b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A. A.; Mason J. M.; Clinch K.; Tyler P. C.; Evans G. B.; Schramm V. L. Altered enthalpy-entropy compensation in picomolar transition state analogues of human purine nucleoside phosphorylase. Biochemistry 2009, 48 (23), 5226–5238. 10.1021/bi9005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch J.; Robien M. A.; Mehlin C.; Boni E.; Riechers A.; Buckner F. S.; Van Voorhis W. C.; Myler P. J.; Worthey E. A.; DeTitta G.; et al. Using fragment cocktail crystallography to assist inhibitor design of Trypanosoma brucei nucleoside 2-deoxyribosyltransferase. J. Med. Chem. 2006, 49 (20), 5939–5946. 10.1021/jm060429m. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372 (3), 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Armstrong S. R.; Cook W. J.; Short S. A.; Ealick S. E. Crystal structures of nucleoside 2-deoxyribosyltransferase in native and ligand-bound forms reveal architecture of the active site. Structure 1996, 4 (1), 97–107. 10.1016/S0969-2126(96)00013-5. [DOI] [PubMed] [Google Scholar]
- Bondi A. van der Waals Volumes and Radii. J. Phys. Chem. A 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- Zhou S.; Wang L. Unraveling the structural and chemical features of biological short hydrogen bonds. Chem. Sci. 2019, 10 (33), 7734–7745. 10.1039/C9SC01496A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm V. L. Enzymatic transition state theory and transition state analogue design. J. Biol. Chem. 2007, 282 (39), 28297–28300. 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- Lewandowicz A.; Schramm V. L. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry 2004, 43 (6), 1458–1468. 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Jensen R. A. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 1976, 30, 409–425. 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Toscano M. D.; Woycechowsky K. J.; Hilvert D. Minimalist active-site redesign: teaching old enzymes new tricks. Angew. Chem., Int. Ed. 2007, 46 (18), 3212–3236. 10.1002/anie.200604205. [DOI] [PubMed] [Google Scholar]
- van der Ent F.; Skagseth S.; Lund B. A.; Socan J.; Griese J. J.; Brandsdal B. O.; Aqvist J. Computational design of the temperature optimum of an enzyme reaction. Sci. Adv. 2023, 9 (26), eadi0963 10.1126/sciadv.adi0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.