

Abstract
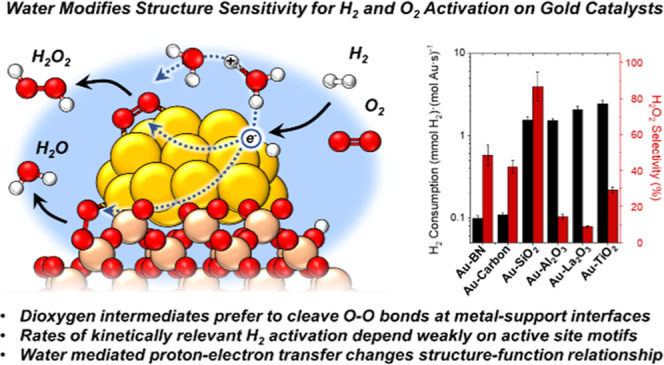
Au nanoparticles catalyze the activation and conversion of small molecules with rates and kinetic barriers that depend on the dimensions of the nanoparticle, composition of the support, and presence of catalytically culpable water molecules that solvate these interfaces. Here, molecular interpretations of steady-state rate measurements, kinetic isotope effects, and structural characterizations reveal how the interface of Au nanoparticles, liquid water, and metal oxide supports mediate the kinetically relevant activation of H2 and sequential reduction of O2-derived intermediates during the formation of H2O2 and H2O. Rates of H2 consumption are 10–100 fold greater on Au nanoparticles supported on metal oxides (e.g., titania) compared to more inert and hydrophobic materials (carbon, boron nitride). Similarly, Au nanoparticles on reducible and Lewis acidic supports (e.g., lanthana) bind dioxygen intermediates more strongly and present lower barriers (<22 kJ mol–1) for O–O bond dissociation than inert interfaces formed with silica (>70 kJ mol–1). Selectivities for H2O2 formation increase significantly as the diameters of the Au nanoparticles increase because differences in nanoparticle size change the relative fractions of exposed sites that exist at Au–support interfaces. In contrast, site-normalized rates and barriers for H2 activation depend weakly on the size of Au nanoparticles and the associated differences in active site motifs. These findings suggest that H2O aids the activation of H2 at sites present across all surface Au atoms when nanoparticles are solvated by water. However, molecular O2 preferentially binds and dissociates at Au–support interfaces, leading to greater structure sensitivity for barriers of O–O dissociation across different support identities and sizes of Au nanoparticles. These insights differ from prior knowledge from studies of gas-phase reactions of H2 and O2 upon Au nanoparticle catalysts within dilute vapor pressures of water (10–4 to 0.1 kPa H2O), in which catalysis occurs at the perimeter of the Au–support interface. In contrast, contacting Au catalysts with liquid water (55.5 M H2O) expands catalysis to all surface Au atoms and enables appreciable H2O2 formation.
Keywords: catalysis, water chemistry, surface science, metals, mechanisms of reactions
1. Introduction
Supported Au nanoparticles catalyze reactions of molecules bound to the sites of the metal, support, and their interfaces,1−4 enabling many industrially relevant reactions (e.g., oxidation of CO5−26 and alcohols,27,28 hydrogenation of heteroatoms,29−33 hydrogenolysis of allylic carbonates,34 Suzuki–Miyaura coupling,35−37 and hydrochlorination of alkynes38). Haruta demonstrated that supported Au nanoparticles show exceptional rates of CO oxidation,5,38 particularly at low temperatures and in the presence of moisture.6−11,13−17,19,27,28 In contrast, unsupported Au powders and Au nanoparticles of similar diameters require much greater temperatures to achieve equivalent rates of CO oxidation,5,39−43 implying Au–support interfaces lower barriers for kinetically relevant oxidation steps. Indeed, decorating Au(111) with metal oxide materials leads to similar conclusions,17,44−47 suggesting that functions at the Au–support interface enable new paths of reacting intermediates sensitive to water molecules.
Generally, the support material can provide acidic or basic functionalities, change the oxidation state of metal atoms, and present oxygen vacancies, among other effects.1,2,48 These interfaces also influence the morphology of metal nanoparticles (e.g., size, orientation, and faceting) and transfer charge between the metal, support, and reactive intermediates.48−53 These effects change reaction barriers and facilitate paths not observed over unsupported metal nanoparticles or extended surfaces.7−12,54 For example, dioxygen binds strongly at the interface of Au nanoparticles and TiO2, which form Au–O–O–Ti species at oxygen vacancy sites, assist O–O dissociation paths, and enable facile oxidation of organic substrates.21−23 Moreover, spectroscopic evidence shows that different Au–support interfaces (e.g., Au–TiO2, Au–Al2O3) stabilize different fractions of oxygen species (e.g., O*, O2*, OH*, and so forth) compared roughened Au foil.55 Thus, the Au–support interface enables the catalysis of critical small molecules (e.g., H2, O2, CO, olefins, and so forth); however, the precise mechanism and site requirements of how these interfaces react with these species remain controversial.1,2,6
Mounting evidence suggests Au catalysts interact with water molecules by facilitating proton–electron transfer (PET) reactions that alter barriers of activating O–O, H–H, and O–H bonds relative to anhydrous conditions.56−58 For example, trace quantities of water vapor (10–4 to 0.1 kPa H2O) promote the activation of dioxygen on supported Au nanoparticles, increasing rates for CO oxidation by 1–2 orders of magnitude.7−12 Indeed, kinetic isotope effect measurements and density functional theory (DFT) calculations suggest that H2O and OH* species cocatalyze the reduction of O2* to OOH*, which transforms CO* to CO2 at Au–support interfaces.7−16 Related PET reactions also occur during reactions of H2 and O2. For example, infrared spectra of H2 dosed over Au–TiO2 shows that electrons transfer to the conduction band of Au–TiO2 and protons “spillover” to the support as hydroxyl groups.29,31,54,59−62 Here, the addition of water simultaneously increases rates of O2 activation6−11,13−17,19,27,28 but inhibits H2 oxidation reactions at the Au–TiO2 interface29,30 by slowing rates of electron transfer. Regardless, these protons and electrons inevitably react with adsorbed O2, forming a litany of species (e.g., O2*, OOH*, O*, OH*) that form either H2O2 or H2O.29,30,59 Thus, modest vapor pressures (10–4 to 0.1 kPa H2O) ubiquitously impact rates of small molecules conversion upon supported Au nanoparticles in the gas phase.7−12 However, past investigations do not explain the emergence of distinct structure–function relationships for thermochemical reactions of H2 and O2 on Au catalysts completely enveloped by liquid water (55.5 M H2O).
Liquid water greatly alters the electronic structure and resulting catalysis of Au surfaces compared to gas-phase conditions. For example, water lowers the work function of Au by ∼2.84–3.21 eV, consistent with an increase in the electrochemical potential of electrons in the metal relative to the vacuum.63 Moreover, the exchange of protons and electrons with reacting species at metal–liquid interfaces leads to the emergence of spontaneous electric fields,64,65 which influence the free energy of binding and reacting polar and charged species at solid–liquid interfaces.66 Such concepts are better understood in purely electrochemical systems. For example, Au catalysts readily convert O2 to H2O2 or H2O under reducing electrochemical potentials within acidic or alkaline electrolyte.67−69 However, the postulation of heterolytic paths in thermal catalysis7,11,27,29,30 and direct comparisons of such systems to electrochemical reactions on Au-based catalysts have garnered considerable attention recently.70−72 For example, Davis and co-workers demonstrated that aqueous-phase OH– and surface-bound OH* cocatalyze alcohol oxidation (e.g., ethanol, glycerol) by forming alkoxide species over Au surfaces while simultaneously transferring protons and electrons to O2.27,28 We have drawn similar conclusions investigating the mechanisms of H2O2 formation from H2 and O2 over Au-based alloys.73,74 However, the role of Au–support interactions at solid–liquid interfaces remains underexplored for heterolytic reactions of H2 and O2 in liquid water.
Here, we describe the role of the Au–support interface during reactions among H2 and O2 over supported Au nanoparticles within liquid water. Kinetic analysis of H2O2 and H2O formation rates combined with isotopic measurements suggest that supported Au nanoparticles share a common PET mechanism involving kinetically relevant H2 activation mediated by H2O molecules. Au nanoparticles supported upon metal oxide materials (e.g., La2O3) show greater rates of H2 consumption than more refractory and hydrophobic classes of materials (e.g., BN, carbon), suggesting that proton acceptors (e.g., M–OH, H2O) assist in the activation of H–H bonds at the metal–liquid–support interface. Still, site-normalized rates and barriers of H2 consumption show a weak dependence on the mean diameter of Au nanoparticles, indicating that H2 can activate across all surface Au atoms with the assistance of liquid water molecules that solvate these interfaces. By comparison, the activation of O–O bonds is far more structure sensitive. Interfaces with reducible or Lewis acidic metal oxides favor the formation of H2O over H2O2, which indicates that oxygen vacancies and moieties that enable direct binding of oxygen between Au and the metal atoms of the support control the free energy of dissociating O–O bonds. Consequently, H2O2 selectivities increase with the size of Au nanoparticles as the fraction of sites at the Au–support interface decreases relative to the total number of metallic surface Au atoms further from these interfaces. Thus, the understanding developed here provides multiple strategies to manipulate the relative rates of H2 and O2 activation, which may guide the design of new Au catalysts and alloys for other thermal and electrochemical reduction and oxidation reactions.
2. Experimental Methods
2.1. Catalyst Preparation
2.1.1. Synthesis of TiO2- and SiO2-Supported Au Catalysts by Strong Electrostatic Adsorption
Catalytic nanoparticles were formed upon anatase TiO2 nanoparticles (US Research Nanomaterials Inc., US3838) and mesoporous SiO2 (Sigma-Aldrich, Davisil 646) by strong electrostatic adsorption (SEA) using techniques reported elsewhere.75−77 Briefly, the TiO2 support was placed within a quartz tube furnace and heated at a rate of 5 K min–1 to a temperature of 873 K and held for 4 h under a flowing mixture (100 cm3 min–1) dry air (21 kPa O2, 80 kPa N2; Airgas, UHP 99.999%) with the intent of removing residual organic ligands present on the material. By comparison, the SiO2 support was used as received. Cationic Au ethylenediamine complexes were synthesized by combining HAuCl4 (15.6 mM; Sigma-Aldrich, >49 wt % Au) and ethylenediamine (113 mM; Sigma-Aldrich, >99%) in 1 L of deionized (DI) water (>17.8 MΩ cm resistivity) with the intent to exchange chloride ligands with ethylenediamine on the Au center. The resulting solution initially forms a dark brown precipitate that evolves into a clear orange solution once the ligand exchange reaches completion. Separately, DI water (2.5 L) and 1 L of concentrated NH4OH (Macron, 28–30% NH4OH) were combined in a beaker. Either SiO2 or TiO2 (85 g) was added to the basic solution with the intent to deprotonate the hydroxylated surface of the support. The suspension was stirred continuously for 10 min, after which the solution of Au–ethylene diamine (1 L) was added while stirring for an additional 10 min. The mixture was then stirred intermittently every 10 min for 1 h and left overnight to provide time for cationic Au species to adsorb to the anionic surface of the support. The solution was then decanted from the solids until a slurry of catalyst remained. Analogous procedures were used to prepare samples with different weight loadings of Au on the support. However, the mass of the Au precursor and support material was adjusted while maintaining the ratio of HAuCl4 to ethylenediamine.
Further treatment of these materials differed with the identity of the support. The Au–SiO2 solids were filtered and washed with DI water (∼40 cm3 g–1) to remove residual ions from the material, and these materials were then vacuum-filtered overnight at ambient temperature. The Au–TiO2 solids were similarly washed (∼40 cm3 g–1) and centrifuged to recover a gel, which was dried under vacuum at ambient temperature (∼298 K) for 1 week. The recovered solids (i.e., Au–SiO2 or Au–TiO2) were then placed within a quartz tube furnace and heated at a rate of 5 K min–1 to a maximum temperature (573–1073 K) and held for 4 h under a flowing mixture (300 cm3 min–1) of He (67 kPa; Airgas, UHP 99.999%) and dry air (7 kPa O2, 26 kPa N2; Airgas, UHP 99.999%). The range of maximum oxidation temperatures was chosen to produce a series of materials with different mean diameters of Au nanoparticles on each support. Au–TiO2 samples prepared at calcination temperatures of 573, 773, 823, 873, 973, and 1073 K are referred to as Au–TiO2–O573, Au–TiO2–O773, Au–TiO2–O823, Au–TiO2–O873, Au–TiO2–O973, and Au–TiO2–O1073, respectively. Similarly, Au–SiO2 samples prepared at calcination temperatures of 573, 673, and 1073 K are referred to as Au–SiO2–O573, Au–SiO2–O673, and Au–SiO2–O1073, respectively.
2.1.2. Synthesis of TiO2-, Al2O3-, SiO2-, and La2O3-Supported Au Catalysts by Deposition Precipitation
Deposition precipitation (DP) was used to create supported Au nanoparticles with smaller mean diameters than those formed by SEA of Au-ethylenediamine (vide supra). Briefly, catalytic nanoparticles were formed upon anatase TiO2 nanoparticles (US Research Nanomaterials Inc., US3838), γ-Al2O3 (Catalox, HP 14/150 Alumina), SiO2 (Sigma-Aldrich, Davisil 646), and La2O3 (Sigma-Aldrich, L4000) by deposition precipitation using techniques reported elsewhere.76,78 The TiO2 support was placed within a quartz tube furnace and heated at a rate of 5 K min–1 to a temperature of 873 K and held for 4 h under a flowing mixture (100 cm3 min–1) dry air (21 kPa O2, 80 kPa N2; Airgas, UHP 99.999%). The Al2O3, SiO2, and La2O3 were used as received.
First, the Al2O3, TiO2, and La2O3 support materials (5 g) were dispersed into 500 mL of DI water and combined with 50 mL of aqueous HAuCl4 (15.3 mM) in DI water. In contrast, the SiO2 support material (5 g) was added to a 500 mL aqueous solution of NH4NO3 (62.5 mM) and HAuCl4 (0.35 mM) in DI water. In either case, the resulting suspension was mixed overnight with the intent to allow Au complexes to fully adsorb to the support. The solution was then titrated dropwise with aqueous NH4OH (175 mM) until reaching a pH of 10 for the purpose of converting adsorbed HAuCl4 into Au(OH)3. The solution was then mixed for an additional 1 h and decanted from the solids until a slurry of catalyst remained. The final Au–TiO2, Au–Al2O3, and Au–La2O3 solids were then washed and centrifuged to extract a paste, which was dried in a convection oven at 323 K overnight in ambient air. The dried solids (Au–TiO2, Au–Al2O3, and Au–La2O3) were then transferred to quartz boats and heated at a rate of 5 K min–1 to 473 K and held for 4 h within a flowing mixture (200 cm3 min–1) of dilute H2 (20 kPa H2, 81 kPa He; both Airgas, UHP 99.999%) within a quartz tube placed in a three-zone furnace.
2.1.3. Synthesis of Carbon and BN-Supported Au Catalysts by Incipient Wetness Impregnation
Incipient wetness impregnation (IWI) was used to create supported Au nanoparticles on carbon and boron nitride (BN) with similarly small Au nanoparticles achieved in Section 2.1.2 (vide supra), because these materials do not readily facilitate precipitation deposition. Catalytic nanoparticles were formed upon nonfunctionalized carbon (Cabot Corporation, Vulcan XC-72) and BN (Sigma-Aldrich, 25475) by incipient wetness impregnation techniques, in which the support materials were used as received.75,79 Here, a 5 mL solution of HAuCl4 (91.4 mM) in DI water was prepared and added dropwise to the support material (3 g) until incipiently wet. The materials were washed with a diluted solution of NH4OH (175 mM) and dried in a convection oven at 323 K overnight in ambient air. The dried solids (i.e., Au-BN and Au-carbon) were then heated at a rate of 5 K min–1 to 473 K and held for 4 h under a flowing mixture within a flowing mixture (200 cm3 min–1) of dilute H2 (20 kPa H2, 81 kPa He; both Airgas, UHP 99.999%) within a quartz tube placed in a three-zone furnace.
2.1.4. Synthesis of Supported Au Catalysts by Deposition of Colloidal Nanoparticles
Colloidal Au nanoparticles were synthesized and then deposited upon anatase TiO2 nanoparticles (US Research Nanomaterials Inc., US3838), γ-Al2O3 (Catalox, HP 14/150 Alumina), and La2O3 (Sigma-Aldrich, L4000) to enable comparisons of supported Au nanoparticles formed by other methods (vide supra). The TiO2 support was placed within a quartz tube furnace and heated at a rate of 5 K min–1 to a temperature of 873 K and held for 4 h under a flowing mixture (100 cm3 min–1) dry air (21 kPa O2, 80 kPa N2; Airgas, UHP 99.999%). Other supports were used as received. First, a solution of colloidal Au nanoparticles was prepared using the Turkevich citrate method.80 Here, a 900 mL solution of trisodium citrate (6.7 mM Na3C6H5O7; Sigma-Aldrich, >99.0%) was prepared with DI H2O in a flask. The solution was heated in an oil bath at ∼398 K to achieve a rolling boil while stirring with a Teflon-coated magnetic stir bar. A 20 mL solution of HAuCl4 (15.2 mM) was prepared in a separate vial of DI H2O and added to the boiling solution. After 30 s of mixing, the corresponding support material (2 g) was added to the solution. The slurry was heated and stirred continuously until the liquid completely evaporated to deposit the colloids onto the support material. The resulting precursor materials were then placed in quartz boats and heated at a rate of 5 K min–1 to 737 K and held for 4 h within a flowing mixture (100 cm3 min–1) of dry air (21 kPa O2, 80 kPa N2; Airgas, UHP 99.999%) in a quartz tube furnace. This treatment was intended to burn off any remaining citrate ligands present on the catalytic material and graft the nanoparticles to the support. Notably, this method deposits a greater quantity of Na and Cl ions onto the supports than the other methods above; however, all materials are washed with over 40 L of DI H2O in situ before catalytic measurements (vide infra).
2.1.5. Synthesis of Alloyed PdAux Nanoparticles by Electroless Depositions of Pd(NO3)2
Pd was deposited onto supported Au nanoparticles by electroless deposition of Pd(NO3)2 (Sigma-Aldrich, ∼40% Pd) to compare rates and selectivities of PdAux alloys to the parent Au nanoparticle catalysts. First, the Au–TiO2 material (2 g) was stirred in DI water (100 cm3) for 10 min in a round-bottom flask. Next, the slurry was blanketed with a flowing mixture of dilute H2 (1 kPa H2, 100 kPa He, 100 cm3 min–1; Airgas, UHP 99.999%) and constantly stirred. Then, an aqueous solution of ∼50 mL Pd(NO3)2 (∼0.05 mM Pd(NO3)2) was added to the vessel in a single injection, and the mixture was stirred for 3 h. The intent of this final step is to allow Pd2+ to reduce by reaction with hydrogen atoms upon the Au nanoparticle surfaces to deposit metallic Pd upon Au nanoparticles and form the alloy. The atomic ratios of Au and Pd were selected to synthesize a material with a bulk composition of PdAu100 on TiO2. Following electroless deposition, the supernatant was decanted, and the remaining slurry was centrifuged before drying it in a convection oven at 323 K overnight. The dried catalyst was then transferred to quartz boats and heated at a rate of 5 K min–1 to 573 K and held for 1 h under a flowing stream (300 cm3 min–1) of dilute air (7 kPa O2, 26 kPa N2, 67 kPa He; Airgas, UHP 99.999%) within a quartz tube located in a three-zone furnace. This calcination was intended to distribute surface Pd atoms homogeneously on the Au, as discussed elsewhere.75
2.2. Characterization of Catalytic Materials
2.2.1. Characterization of Au Nanoparticle Size by TEM, XRD, and UV–Vis Spectroscopy
The numerical average diameter (⟨dTEM,N⟩) of supported Au nanoparticles was calculated from the mean diameter of particle size distributions obtained by bright-field transmission electron microscopy (TEM; JEOL, 2010 LaB6) determined by measurement of at least 100 nanoparticles but more commonly 200–2000 nanoparticles on each material. Each sample was prepared by grinding the catalyst into a fine powder (<200 mesh), which was dispersed in ethanol (Decon Laboratories, >99.9%) and dripped onto a Cu holey-carbon TEM grid (200 mesh, Ted Pella Inc.). The surface area normalized average diameter (⟨dTEM,S⟩) for each catalyst was calculated using eq 1.
| 1 |
where ni is the number of nanoparticles with the diameter di. Figure 1 shows a representative TEM image of 2 nm Au nanoparticles present on Au–TiO2 formed by deposition precipitation and includes an inset histogram of the particle size distribution.
Figure 1.
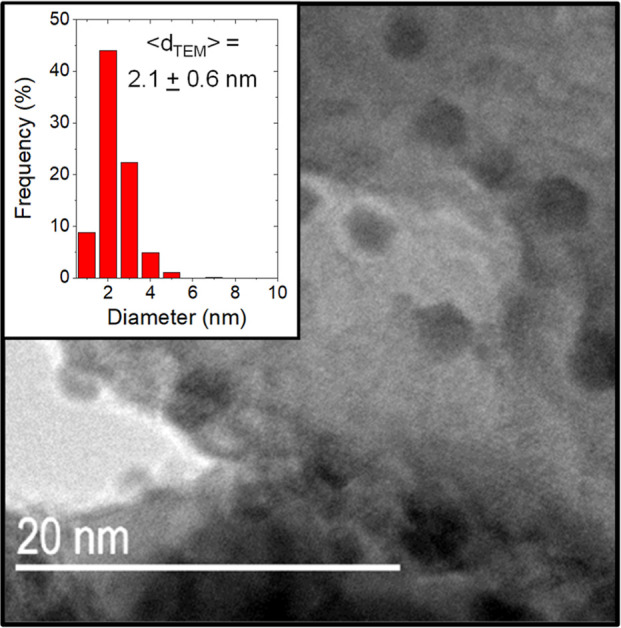
Representative TEM image of ∼3 wt % Au nanoparticles (2–3 nm) supported on calcined anatase TiO2 with an inset histogram of the particle size distribution. Figure S1 shows corresponding images and histograms of particle diameters for the other Au-based catalysts prepared by reductive and oxidative treatments between 473 and 1073 K.
The size of Au nanoparticles was also investigated by diffuse reflectance UV–vis spectroscopy (DRUV–vis) using a diffuse reflection probe (Avantes, solarization-resistant fibers) coupled to a fiber-optic spectrometer (Avantes, AvaFast 2048) with a compact deuterium-halogen light source (Avantes, AvaLight-DHc). Figure S2a,b shows ex situ DRUV–vis spectra of Au–TiO2 and Au–SiO2 materials, respectively, with nanoparticle diameters ranging between 2 and 25 nm. These extinction spectra show features with a peak absorbance wavelength (λmax), related to the surface plasmon resonance frequency of Au nanoparticles, increasing from ∼545 to ∼630 nm for nanoparticle diameters between 2 and 25 nm, respectively (Table 1). Such features are broader on materials treated at the greatest temperatures (>573 K), agreeing with TEM measurements showing increased polydispersity as the particle size increases on these catalysts. In addition, Figure S2c presents photographs that reveal the catalyst color evolves from pink to purple to blue as the values of ⟨dTEM,S⟩ increase from 2 to 25 nm, consistent with shifts in λmax measured by DRUV–vis.
Table 1. Preparation Methods and Characterization of Supported Au Nanoparticles, Including Mean Nanoparticle Diameters from TEM (⟨dTEM,S⟩ and ⟨dTEM,N⟩), XRD (⟨dXRD⟩), and DRUV–Vis Using the Peak Absorbance Wavelength (λmax)a.
| preparation of Au materialb | thermal treatmentc | ⟨dTEM,S⟩ (nm)d | ⟨dTEM,N⟩ (nm)e | ⟨dXRD⟩ (nm)fg | peak absorbance wavelength (nm) |
|---|---|---|---|---|---|
| Au–TiO2–R473 | H2 at 473 K 4 h | 2.1 ± 0.6 | 1.9 ± 0.6 | 6.1 | 545 |
| Au–TiO2–O573 | O2 at 573 K 4 h | 5.0 ± 1.2 | 4.8 ± 1.2 | 9.9 | 565 |
| Au–TiO2–O773 | O2 at 773 K 4 h | 7.4 ± 2.0 | 7.4 ± 2.0 | 9.9 | 563 |
| Au–TiO2–O823 | O2 at 823 K 4 h | 10.6 ± 2.4 | 9.9 ± 2.4 | 10.4 | 575 |
| Au–TiO2–O873 | O2 at 873 K 4 h | 14.5 ± 2.7 | 13.9 ± 2.7 | 12.3 | 592 |
| Au–TiO2–O973 | O2 at 973 K 4 h | 18.5 ± 5.2 | 15.4 ± 5.2 | 17.6 | 610 |
| Au–TiO2–O1073 | O2 at 1073 K 4 h | 24.7 ± 7.5 | 19.1 ± 7.5 | 22.9 | 633 |
| Au–SiO2–R473 | H2 at 473 K 4 h | 2.4 ± 0.7 | 2.1 ± 0.7 | n.d. | 520 |
| Au–SiO2–O573 | O2 at 573 K 4 h | 5.6 ± 2.2 | 3.3 ± 2.2 | 8.7 | 523 |
| Au–SiO2–O673 | O2 at 673 K 4 h | 15.3 ± 3.5 | 13.3 ± 3.5 | 13.5 | 504 |
| Au–SiO2–O1073 | O2 at 1073 K 4 h | 26.0 ± 7.0 | 21.4 ± 7.0 | 22.8 | 504 |
Reported materials were prepared by strong electrostatic adsorption and deposition precipitation methods, followed by different temperature treatments. Further details of characterization are presented in Table S1.
Nomenclature for samples of Au nanoparticles supported on TiO2 or SiO2, prepared by strong electrostatic adsorption and calcined at distinct temperatures.
Thermal treatments were conducted at 20 kPa H2 and 81 kPa He for reductive treatments (R) and at 7 kPa O2, 26 kPa N2, and 67 kPa He for oxidative treatments.
Nominal mean nanoparticle diameters (⟨dTEM,N⟩ = ∑idi/∑ini).
Average crystallite size of Au nanoparticles using the Scherrer equation of Au(111) facets.
Very small Au nanoparticles supported on silica were not detected (n.d.).
X-ray diffraction (XRD) patterns of supported Au nanoparticles were obtained using powder samples within a diffractometer (Bruker D8 Advance) with a Cu Kα radiation source (λ = 0.15418 nm) under ambient conditions.
Measurements were conducted between the range of 2θ = 20–80° with a 0.01° resolution and a scan rate of ∼1.5° min–1. The resulting diffractograms were compared to reference spectra from the powder diffraction file database and analyzed using the Scherrer equation using shape factors for cuboctahedral nanoparticles.81 These diffractograms show an increase in Au(200) crystallinity on materials with increasing calcination temperatures (Figures S3 and S4). Moreover, fits of the Scherrer equation (eq S1.1) on these diffractograms show that the increase in Au crystallite size with oxidation temperature agrees with the increase in nanoparticle diameter shown by TEM and UV–vis.81 These XRD measurements also show that the crystal structure of anatase TiO2 remains mostly unchanged from 573 to 1073 K (Figure S3a); however, greater temperatures can lead to the formation of the rutile phase (Figure S5).
Table 1 reports the average particle size of Au nanoparticles supported on TiO2 and SiO2, as characterized by TEM, XRD, and DRUV–vis spectroscopy. Table S1 reports the metal content of these samples, determined by energy dispersive X-ray fluorescence (EDXRF; Shimadzu, EDX-7000) and inductively coupled plasma optical emission spectroscopy (ICP; PerkinElmer, Optima 2000DV) measurements (Section S1). Figure S1 also reports TEM images and histograms for all other samples of supported Au nanoparticles used in this study.
Table 1 also shows that Au nanoparticles increase in size (⟨dTEM,S⟩ = 2–25 nm) and polydispersity after oxidizing the precursor at greater temperatures (573–1073 K). Samples prepared by deposition precipitation, however, showed smaller and more monodisperse particle size distributions (⟨dTEM,S⟩ = 2–3 nm; Table S1).
2.2.2. Infrared Spectroscopy of CO Adsorbed to Au Surfaces
Infrared spectra of CO adsorbed to Au–TiO2 provide one method to estimate the relative fractions of interfacial Au atoms versus metallic facets of Au that exist far from the interface.21−23 Samples were prepared by grinding catalysts (∼60 mg) into a fine powder and pelletizing the powder into a mesh disc (McMaster-Carr, 40 × 40, 25 mm diameter, stainless steel) with a hydraulic press (Carver, model C). These discs were placed within two stainless steel retaining rings and loaded into a custom transmission IR cell with CaF2 windows, as described previously.75,82,83 The cell was sealed with graphite ferrules (Chromalytic Technology Pty. Ltd.) within a stainless-steel retaining ring on the cell exterior. Here, gaseous CO, He, and O2 were introduced to the system by digital mass flow controllers (Alicat, MC Series) mounted on a gas manifold in the flow path of the cell.
Initially, Au samples were heated from ambient at 5 K min–1 to 573 K under flowing (40 cm3 min–1) mixtures of oxygen (20 kPa O2, 81 kPa He) and held at 573 K for 1 h with the intent to remove water or adventitious carbon and water from the catalyst surface. The samples were cooled overnight to 303 K in pure He (101 kPa He, 40 cm3 min–1) before collecting background spectra. Afterward, spectra were collected until the spectra ceased changing. Then, a stream of dilute carbon monoxide (1 kPa CO, 100 kPa He) was introduced from a calibrated gas mixture (Airgas, 1 vol % CO, balance He), and spectra were collected until the sample reached a steady state. Next, the pressure of CO was decreased by adding pure He (Airgas, 99.999%) to attain a range of lower CO partial pressures (0.005–1 kPa CO), and steady-state spectra were obtained at each condition. Finally, samples were returned to the initial composition of CO (1 kPa) to measure differences in the final and initial steady-state spectra at these conditions.
2.2.3. Point of Zero Charge of Catalytic Support Materials
The points of zero charge (PZC) of catalytic support materials were measured using a pH sensor (Oakton, CON 450) submerged in a slurry of solids (∼500 mg) and DI H2O. A solution of DI H2O was sparged with He (101 kPa He; Airgas 99.999%) to remove dissolved CO2 from the atmosphere. This solution was added to a separate beaker containing a given support material in increments of 2.5 cm3 while measuring the pH. The slurry transitioned from a paste-like consistency and approached a well-dispersed mixture as the total volume of solution (40 cm3) was transferred. The functional dependence of pH with solid mass was extrapolated to the point of incipient wetness using the pore volume of each material (reported by the manufacturer) to find the PZC. Table 2 reports these PZC values on the untreated BN, carbon, SiO2, Al2O3, La2O3, and TiO2, determined by analyzing Figure S6 with techniques reported elsewhere.84,85
Table 2. Points of Zero Charge of Support Materials from the Dependence of pH as a Function of Solid Content within a Slurry of Support and DI H2O.
| material | point of zero charge (pH) |
|---|---|
| TiO2 | 3.5 |
| SiO2 | 5.4 |
| La2O3 | 8.9 |
| Al2O3 | 6.7 |
| carbon | 4.2 |
| BN | 7.4 |
2.3. Steady-State Reaction Rate Measurements
All steady-state rates of H2O2 (rH2O2) and H2O (rH2O) formation were measured in a continuous-flow trickle-bed reactor (TBR; 48 cm length, 1 cm inner diameter) housed within a stainless-steel cooling jacket (Figure S7).75,82 The reactor was loaded with 0.15–2 g of catalyst (pelletized to 80–120 mesh), which was supported by plugs of glass wool (∼10 mg) and borosilicate glass rods (8 mm diameter). These rods were secured between silver-coated fritted VCR gaskets (Swagelok, SS-4-VCR-2-60M), which were also used to seal the reactor. The temperature was controlled across the reactor by flowing aqueous ethylene glycol (50% volume; Fisher Scientific E178, 99.8%) through the cooling jacket from a recirculating temperature bath (Cole-Parmer Polystat). The temperature within the reactor was monitored using a K-type thermocouple attached to a cooling jacket and in contact with the wall surrounding the catalyst bed. H2 and O2 compositions in the reactor were controlled by flowing certified gas mixtures (25% H2/N2, 99.9% D2, 5% O2/N2, Airgas, 99.999%) through digital mass-flow controllers (Bronkhorst, F-211CV). Warning: pressurized mixtures of H2 and O2 are explosive when both components simultaneously exceed a mole fraction of 0.05. Before contacting the catalyst, the gaseous reactant stream was contacted and mixed with flowing DI water (>17.8 MΩ cm) that served as the solvent and was delivered by a high-performance liquid chromatography pump (SSI, LS class). The pressure of the resultant gas–liquid mixture was maintained by a back-pressure regulator (Equilibar, LF) and controlled by an electronic pressure regulator (Equilibar, GP1). The upstream pressure of the reactor was monitored by a digital pressure transducer (Omega, PXM409-USBH).
Sampling and analysis of the liquid and gaseous effluent streams were automated and operated continuously. The reactor effluent entered a gas–liquid separator (GLS) machined from polycarbonate and polyvinyl chloride. The gas stream flowed from the GLS to a gas chromatograph (Agilent, 7890B) equipped with a capillary column (Vici, Molecular Sieve 5 Å, 30 m × 0.53 mm × 20 μm) and a thermal conductivity detector that used Ar gas (Airgas, 99.999%) as a reference. Gas chromatograms of catalytic measurements were compared to reference chromatograms using equivalent reaction conditions within a bypass reactor loaded with an equivalent mass of support material, which did not contain catalytic nanoparticles. Comparisons to these control measurements enabled quantification of the conversion of reagents, and together with known molar flow rates, and the calculation of rates of H2 (−rH2) and O2 (−rO2) consumption for a given catalyst. An electronically actuated valve (ALSCO Inc., LEV025PL) drained the liquid fraction from the GLS at 10 min intervals. The liquid effluent flowed into an electronic two-position valve (Vici Valco, 10 port EPC10W), which injected ∼1 cm3 of the effluent and ∼1 cm3 of a colorimetric titrant (12 mM neocuproine [Sigma-Aldrich, >99%], 8.3 mM CuSO4 [Fisher Scientific, >98.6%], 25/75 (v/v) ethanol/deionized water mixture [Decon Laboratories, >99.9%]) into test tubes held in an automated fraction collector (Biorad, 2110). Each tube was analyzed by a UV–vis spectrophotometer (Spectronic, 20 Genesys) at a wavelength of 454 nm to measure the H2O2 concentration using a corresponding calibration curve to determine rates of H2O2 formation. Measurements were also conducted at high space velocities (35 mL min–1 DI H2O; 110 SCCM gas) and typically low temperatures (278 K) to suppress secondary reactions that decompose H2O2, such that measured rates of H2O2 formation mostly reflect its primary formation pathway. Such conditions were also demonstrated to avoid external mass transfer limitations in prior work.86
Product formation rates were determined by normalizing the measured rates by the total molar metal content of the materials. In comparison, turnover rates were estimated by dividing measured rates by estimates for the number of active sites. Independent calculations for turnover rates considered the number of active sites to equal the number of Au atoms at perimeter sites of Au nanoparticles, the Au atoms at corners of faceted Au nanoparticles, or the total number of Au atoms present at surfaces, based upon the particle diameter histograms measured by TEM (vide supra).87,88 Reported H2O2 selectivities were calculated by dividing the formation rate of H2O2 formation by the rate of H2 consumption to determine the fraction of H2 gas that forms the desired H2O2 product (rH2O2/–rH2). For pressure dependence and activation enthalpy measurements, reported rates were corrected by accounting for the rate of deactivation, which assumes these changes arise from an exponential reduction in the number of active sites over time.89 These corrections were determined by returning to the initial condition and quantifying the extent of deactivation over time. Generally, most materials showed little deactivation except for Au–SiO2 materials, which still showed similar selectivities of H2O2 formation before and after the measurement (vide infra).
All materials are subjected to multiple activation enthalpy measurements (278–308 K) until they reach a consistent initial and final selectivity to H2O2 formation to ensure a consistent catalytic state before measurements. These pretreatments also wash all catalytic materials with over 40 L or DI H2O, ensuring minimal residual ions are present on materials before measurements. Finally, all samples satisfy the Madon-Boudart criterion, enabling physically meaningful comparisons of catalytic rates and stability in the absence of mass transfer constraints (Section S1).90−92
2.4. Rate Measurements Using Semibatch Reactors
Transient concentration profiles were measured in round-bottom three-neck flasks (Wilmad Lab Glass, 100 cm3) in a semibatch configuration (Figure S8).82 The top neck of each flask was connected to a condenser, which was chilled to 273 K with a 20/80 (v/v) mixture of ethylene glycol (>99.8%, Fisher Scientific E178) and DI water that flowed from a refrigerated recirculating bath (Neslab ENDOCAL). The reactant gas mixture (99.999% H2 and 5% O2/N2, Airgas) was supplied to the three-neck flasks using digital mass-flow controllers (Alicat, MC series) connected to custom-built gas dispersion tubes (GDT). Each GDT was placed into 80 cm3 of a solvent such that the GDT frits (40–60 μm) were completely submerged. The solvents used in these measurements include DI water, deuterated water (D2O; Cambridge Isotopes, 99.9 atom % D), methanol (CH3OH; Fisher Chemicals, HPLC grade), acetonitrile (CH3CN; Fisher Chemicals, HPLC grade), and acetone (C3H6O; Fisher Chemicals, HPLC grade). The gas outlet from each flask was vented through a rotameter (Omega, FLDA) that was vented into a fume hood. Comparisons of the inlet and outlet flow rates were ensured to be equal, so no leaks were present. All experiments were conducted at 293 K at ambient pressure, and the solution media was stirred at 1500 rpm with magnetic stir bars to avoid external mass transfer limitations.82 Each solvent solution was saturated with gas for 10 min before the catalyst was injected into the flask (100 mg). Aliquots (∼1 cm3) were extracted every 5–10 min during the first hour of the measurement and then every hour for 3 h. These samples were titrated with a CuSO4-neocuproine solution to determine concentrations of H2O2 (composition described in Section 2.3). The resulting concentration profiles were then fitted to determine apparent rate constants for the formation of H2O2 using methods reported elsewhere.82
3. Results and Discussion
3.1. Effect of Au Nanoparticle Size and Support Identity on Rates and Selectivities toward H2O2 Formation
Scheme 1 shows pathways for reactions involving H2 and O2 that form H2O2 and H2O by primary reactions and additional H2O by secondary decomposition reactions that consume H2O2. Rates of H2 and O2 consumption are immeasurable on support materials without Au nanoparticles, indicating the supports are catalytically inert by themselves. By comparison, Figure S9 shows that a physical mixture of the support material and colloidal Au nanoparticles present reaction rates 100 times lower than similar-sized Au nanoparticles synthesized by DP, SEA, or IWI methods (Section S2). Calcination of colloidal materials at 773 K should oxidatively remove any remaining citrate ligands before catalysis. Consequently, the low reactivity of colloidal materials may result from poor adhesion of Au nanoparticles to the support rather than site-blocking from residual capping agents. Thus, activation of H2 and O2 likely requires Au nanoparticles covalently bound to the support material to form a catalytically active Au-support interface.
Scheme 1. Reactions of H2 and O2 Over Supported Au Nanoparticles Lead to the Primary Formation of (a) H2O2 and (b) H2O, as Well as the Secondary (c) Hydrogenation of H2O2 to H2O and (d) the Disproportionation of H2O2 to H2O and O2.

Figure 2 shows rates of H2 consumption and selectivities of H2O2 formation depend strongly on the mean diameter of Au nanoparticles supported on TiO2 and SiO2 (200 kPa H2, 60 kPa O2, 278 K; catalysts synthesized by SEA and DP methods). Rates of H2 consumption (normalized by total moles of Au content) decrease as the size of Au nanoparticles increases on both materials, which demonstrates that the number of active sites for H2 activation decreased or the average rate of H2 activation per active site is lower for larger Au nanoparticles.13,93 Rates of H2 consumption on TiO2-supported Au nanoparticles are greater than those of SiO2-supported Au nanoparticles with similar diameters, and these disparities increase as the average diameter of the nanoparticles decreases (Figure 2). Comparisons of rates normalized by estimates for the numbers of surface, perimeter, or corner Au atoms present (vide infra) suggest that all surface Au atoms contribute to H2 activation and consumption at these conditions (i.e., samples fully immersed in liquid water). Therefore, all surface Au atoms may catalyze reactions of H2 by water-assisted mechanisms, but chemical functions at the Au–support interface also play a crucial role in activating H–H bonds.
Figure 2.

Steady-state rates of H2 consumption and H2O2 selectivities as a function of the surface-area normalized Au nanoparticle diameter on (a) Au–TiO2 and (b) Au–SiO2 materials (200 kPa H2, 60 kPa O2, 278 K). Dashed lines represent trends and are intended to guide the eyes.
Comparisons between Au nanoparticles of similar mean diameters show that Au–TiO2 gives lower H2O2 selectivities (Figure 2a, ∼30–55%) than Au–SiO2 (Figure 2b, ∼85–95%), indicating the Au–TiO2 interface facilitates the rupture of O–O bonds within surface dioxygen species more readily than sites present on Au–SiO2. Moreover, H2O2 selectivities increase with the mean diameter of Au nanoparticles for both Au–SiO2 and Au–TiO2 catalysts. These trends suggest that catalytic sites at or near the Au–support interface offer greater rates of O–O bond rupture per exposed Au atom than Au atoms far from this interface (e.g., Au terrace sites). Such conclusions concur with past reports that distinct Au–support interfaces strongly affect the adsorption and activation of small molecules (e.g., O2, CO, H2, etc.),7,11,12,29,30 mirroring comparisons between rates of CO oxidation on Au–TiO2 and Au–SiO2.10 Thus, the Au–support interface is critical to O–O bond activation steps.
Figure 3 shows rates of H2 consumption are greater by one to 2 orders of magnitude when 2–3 nm Au nanoparticles are supported on metal oxides (e.g., SiO2, Al2O3, La2O3, TiO2) in comparison to Au upon refractory (e.g., BN, carbon) supports (200 kPa H2, 60 kPa O2, 278 K; catalysts synthesized by DP and IWI methods). These observations suggest oxygen functionalities (or other nucleophilic or Brønsted basic moieties)98−101 near Au nanoparticles may assist in activating H–H bonds,29,30,94−97 while the more refractory and hydrophobic interfaces of carbon and BN weakly activate H2.
Figure 3.
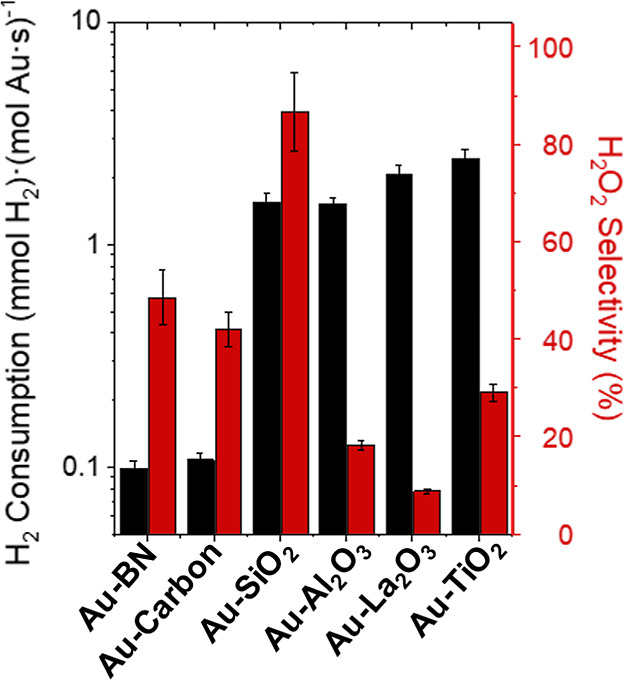
Steady-state rates of H2 consumption (black) and H2O2 selectivities (red) on 2–3 nm Au nanoparticles supported on BN, carbon, SiO2, Al2O3, La2O3, and TiO2 (200 kPa H2, 60 kPa O2, 278 K).
Although PET pathways likely mediate the elementary steps described here, the interfacial pH of the support (related to Brønsted acidity) weakly influences the kinetics of activating O–O bonds. Rates of H2 consumption do not correlate with differences in the point of zero charge (Figure S10a), which indicates the concentration of protons at the surface weakly affects H–H bond activation. Support materials with greater points of zero charge (La2O3 > BN > Al2O3 > SiO2 > carbon > TiO2; Table 2) show somewhat lower selectivities of H2O2 formation (Figure S10b), consistent with greater rates of H2O2 decomposition on Au and PdAux nanoparticles supported on materials with greater isoelectric points.79 Thus, Brønsted acid sites may weakly suppress O–O bond dissociation. However, differences in rates and selectivities of H2O2 formation may depend more on differences in the reducibility, Lewis acidity, or other chemical properties that affect the free energy of binding and activating H2 and O2 at Au–support interfaces.
Together, these observations show the reactions of H2 and O2 depend on multiple factors. First, Au atoms must be present and covalently bound to the support. Second, all surface Au atoms can activate H–H bonds but require assistance from a nearby proton acceptor (e.g., H2O, OH group), which may differ in quantity and proton affinity depending on the identity of the support. Third, Au atoms at the Au–support interface preferentially activate O–O bonds but are sensitive to the chemical functionality of the support (e.g., reducibility, Brønsted basicity, Lewis acidity, and so forth).
3.2. Rate Measurements and Mechanistic Interpretation
Next, we investigated mechanisms for reactions among H2 and O2 on Au nanoparticles on diverse support materials. Steady-state rates of H2O2 (Figure 4) and H2O (Figure 5) formation depend on the pressures of H2 and O2 in ways consistent with reactions involving kinetically relevant H2 activation and subsequent O2 reduction steps over Au nanoparticles supported on TiO2. Figures 4a and 5a show that rates of H2O2 and H2O formation do not depend on the pressure of O2 (10–400 kPa O2, 60 kPa H2, 278 K), which indicates that oxygen-derived intermediates saturate active sites on Au–TiO2. Figures 4b and 5b show H2O2 and H2O formation rates increase linearly with the pressure of H2 (10–400 kPa H2, 60 kPa O2, 278 K), signifying that H2 or H2-derived species participate in a kinetically relevant and mostly irreversible process. These trends hold for Au nanoparticles of all diameters (⟨dTEM,S⟩ = 2–25 nm) on TiO2 and for 2–3 nm Au nanoparticles on all other supports examined (SiO2, Al2O3, La2O3, BN, and carbon; Figure S11), suggesting a consistent mechanism across all supported Au catalysts.
Figure 4.

Steady-state rates of H2O2 formation on 2 nm (red), 5 nm (blue), 7.5 nm (brown), 10 nm (green), 15 nm (orange), 20 nm (purple), and 25 nm (black) Au nanoparticles supported on TiO2 as a function of the pressure of (a) O2 (10–400 kPa H2, 60 kPa O2) and (b) H2 (10–400 kPa O2, 60 kPa H2) at 278 K. Dashed lines fitted to eq 7.
Figure 5.

Steady-state rates of H2O formation on 2 nm (red), 5 nm (blue), 7.5 nm (brown), 10 nm (green), 15 nm (orange), 20 nm (purple), and 25 nm (black) Au nanoparticles supported on TiO2 as functions (a) pressure of O2 (60 kPa H2), and (b) pressure of H2 (60 kPa O2) at 278 K. Dashed lines fitted to eq 8.
Figure S12 shows that H2O2 selectivities do not depend on the pressure of O2 (10–400 kPa O2, 60 kPa H2, 278 K) but increase slightly as H2 pressures increase (10–400 kPa H2, 60 kPa O2, 278 K) on supported Au nanoparticles. Thus, increasing coverages of H2-derived species may stabilize intermediates with O–O bonds (e.g., O2*, OOH*, and H2O2*), as reported on Pd and other noble metals.75,82,98 Product formation rates increase linearly with the pressure of H2 over Au nanoparticles (Figures 4b and 5b) across a broader range of pressures (10–400 kPa H2, 60 kPa O2) than observed for Pd- and Pt-based nanoparticles (10–150 kPa H2, 60 kPa O2).33,75,82,98−100 Thus, the coverage of H2-derived species remains low on Au nanoparticle catalysts compared to Pd- and Pt-based materials.33,34,93,100
Figure 6 shows that formation rates of peroxides (i.e., H2O2, HDO2, and D2O2;Figure 6a) and HD (Figure 6b) increase in proportion with the combined pressure of H2 and D2 (10–200 kPa H2, 10–200 kPa D2, 60 kPa O2, 278 K; equimolar H2 and D2). Moreover, the total formation rates of peroxides exceed HD formation rates by more than 10-fold. Thus, the formation of O2 reduction products likely involves the kinetically relevant activation of H2 and D2 upon sites surrounded by high coverages of O2-derived intermediates. This interpretation also agrees with rates of oxygen reduction that exhibit a normal primary kinetic isotope effect (kH2/kD2 ∼ 1.5 for both peroxide and water formation, Table 3) when D2 replaces H2 as the reductant (200 kPa H2 or D2, 60 kPa O2, 298 K, Figure S13). These observations demonstrate that H2 and D2 activation occurs in a mostly irreversible manner. The H/D atoms produced react immediately to form intermediates that reduce O2-derived surface species on Au nanoparticles at rates 20–30 times faster than rates for their recombination to H2/HD. Furthermore, the similar kinetic isotope effects for primary formation rates of peroxides and water suggest these pathways share common elementary steps or possess similar transition states for their kinetically relevant steps. Consequently, we conclude that hydrogen activation primarily limits the reduction of O2.
Figure 6.

Steady-state formation rates of (a) hydrogen peroxides (i.e., sum of H2O2, HDO2, and D2O2), and (b) HD on 2 nm (red) and 25 nm (black) Au nanoparticles supported on TiO2 as functions of the combined pressure of H2 and D2 (10–200 kPa H2, 10–200 kPa D2, 60 kPa O2; equimolar H2 to D2) at 278 K. Dashed lines intended to guide the eyes.
Table 3. Effects of Isotopic Substitution on Rate Constants of H2O2 and H2O Formation on 2 and 25 nm Au Nanoparticles Supported on TiO2 (200 kPa H2 or 200 kPa D2, 60 kPa O2, 278 K).
| reaction | Au nanoparticles |
|
|---|---|---|
| 2 nm | 25 nm | |
| H2 + O2 → H2O2 | 1.5 ± 0.1 | 1.6 ± 0.1 |
| H2 + 1/2O2 → H2O | 1.5 ± 0.3 | 1.5 ± 0.3 |
Figure 7 shows rates of H2O2 formation over 2 nm Au–TiO2 in protic solvents (water, heavy water, methanol) and aprotic solvents (acetonitrile, acetone) within a semibatch reactor (4.8 kPa H2, 4.8 kPa O2, 298 K). H2O2 forms at rates at least 100-fold greater in protic solvents than aprotic solvents. These findings suggest H2O2 forms on Au nanoparticles by proton transfer reactions, as shown previously for Pd, Pt, and bimetallic nanoparticles.75,82,86,90,101−103 The rates of peroxide formation are marginally greater in H2O versus D2O solvents (rH2O2H2O/rH2O2D2O = 1.1 ± 0.3), indicating that proton transfer steps weakly affect the kinetically relevant step of peroxide formation. These observations agree with analogous isotopic studies of electrochemical oxygen reduction on polycrystalline Au and other noble metals, which suggest electron transfer steps limit oxygen reduction rates.75,104,105 In comparison, the formation of water from the secondary hydrogenation of H2O2 (H2 + H2O2 → 2H2O) exhibits significant differences between rates in water and heavy water (rH2O2H2O/rH2O2D2O = 3.4 ± 0.4; Figure S14), indicating proton transfer to peroxide presents a greater degree of rate control than proton transfer to dioxygen.
Figure 7.

H2O2 formation rates of reactions of H2 and O2 in a semibatch reactor using methanol, water, acetonitrile, and acetone as solvents (4.8 kPa H2, 4.8 kPa O2, 80 cm3 solvent, 100 mg of 2 nm Au–TiO2, 298 K). The dashed line shows the detection limit of the reactor. Figure S14 shows the transient concentration profiles fitted for these data.
These kinetic observations (Figures 4–7 and Table 3) suggest that H2O2 and H2O formation involve kinetically relevant activation of H2 and coupled proton transfer reactions to O2-derived surface intermediates that saturate active sites.
Scheme 2 shows a sequence of elementary steps consistent with the observed dependence of H2O2 and H2O formation rates on the pressures of H2 and O2, isotopic measurements, and the need for a protic solvent to mediate O2 reduction. This scheme invokes sites that bind hydrogen as # and sites that bind oxygen species as *.67−69,75,105−107 This two-site model accounts for the noncompetitive adsorption of H2- and O2-derived species and invokes heterolytic H2 oxidation steps,29,30 yielding analytical rate expressions for H2O2 and H2O formation.29,31,60−62,108
Scheme 2. Proposed Elementary Steps for H2O2 and H2O Formation during Reactions of H2 and O2 on Supported Au Nanoparticles, Which Couple Heterolytic (i.e., Electrochemical) Hydrogen Oxidation and Oxygen Reduction Reactions.
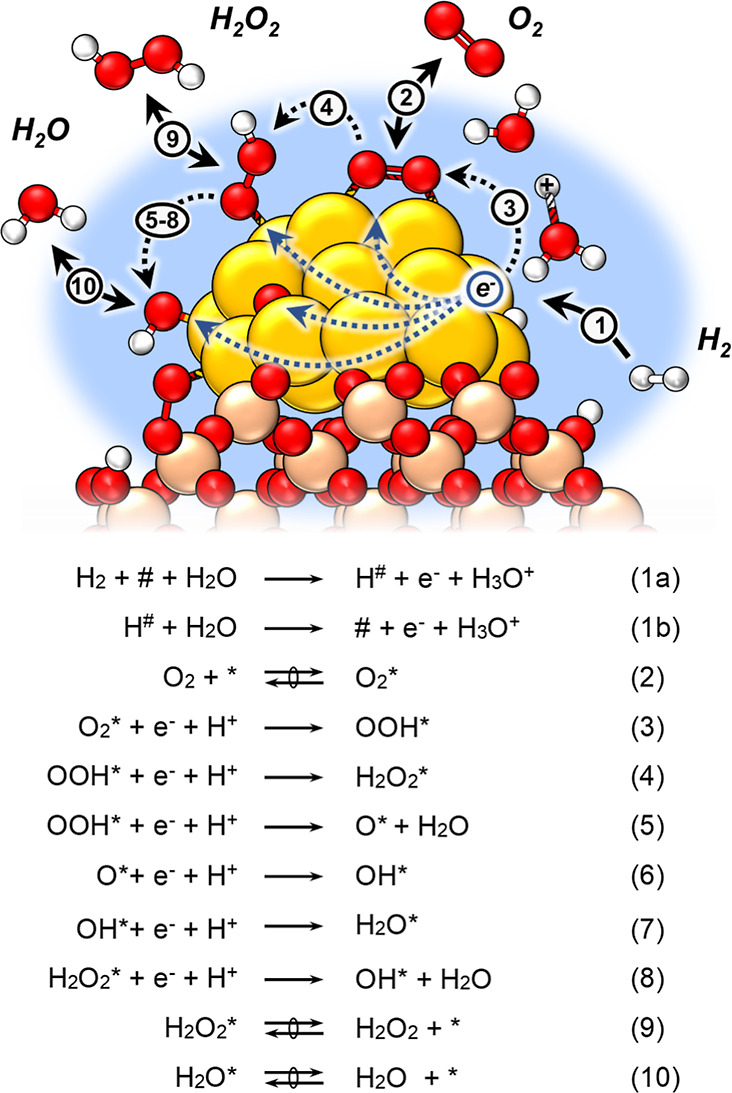
Sites
that bind hydrogen and
oxygen-derived species are denoted as # and *, respectively. The symbols  and
→ indicate that elementary steps are quasi-equilibrated
or mostly irreversible, respectively. Note that this scheme depicts
proton-electron transfer steps mediated by water molecules on Au surface;
however, hydroxyl groups on the support may facilitate equivalent
steps with oxygen species at the Au–support interface.
and
→ indicate that elementary steps are quasi-equilibrated
or mostly irreversible, respectively. Note that this scheme depicts
proton-electron transfer steps mediated by water molecules on Au surface;
however, hydroxyl groups on the support may facilitate equivalent
steps with oxygen species at the Au–support interface.
The kinetically relevant dissociation of H2 (step 1a) proceeds with the assistance of water molecules to generate chemisorbed H# atoms, which then oxidize (step 1b) to form hydronium ions and electrons. These steps occur mostly irreversibly (Figure 6) and resemble the electrochemical Heyrovsky and Volmer reactions.75,109 The resulting protons (H+) may react with water in the solution to form H3O+ or with hydroxyl groups (M–OH) on the support to form M–OH2+.110 Simultaneously, quasi-equilibrated adsorption of O2 (step 2) forms O2*,106 which reduces to form OOH* through PET steps (step 3).29,30,75,82,104 Prior DFT calculations suggest that these O2 reduction steps occur irreversibly since they present low barriers and occur exothermically.29,30 Still, the hydrogen bonding of liquid water stabilizes O–O bonds and lowers barriers of proton transfer steps to O2-derived species bound to metal surfaces relative to the gas phase.82 Subsequently, the resulting OOH* reduces to H2O2* (step 4) or dissociates into H2O and O* (step 5) that reduce further and form OH* (steps 6 and 8).75,111 The dissociation of dioxygen intermediates (steps 5 and 8) is highly exothermic, and prior isotope labeling studies using mixtures of 18O2 and 16O2 suggest that O–O bonds do not reform once cleaved.75,112 This scheme assumes that dioxygen species directly form water upon dissociation (e.g., OOH* + e– + H+ → O* + H2O). Ultimately, the O* and OH* species reduce to water (H2O*) by step 7. Bound H2O2* and H2O* then undergo quasi-equilibrated desorption into the solution (steps 9 and 10) to yield H2O2 and H2O. The crucial implications of this mechanism are described below, and the derivation appears with details from Section S3.
Scheme 2 and the dependence of rates on reactant pressures (Figures 4–6) suggest that kinetically relevant hydrogen consumption (−rH2) limits the total rate of oxygen reduction (−rO2) on all supported Au nanoparticle catalysts examined. Thus, the reaction rate depends mainly on water-assisted activation of H2 (step 1a) during the formation of H2O2 and H2O, as shown by
| 2 |
where kx is the rate constant of step x, and the total rate is a function of the number of unoccupied sites that bind H2 ([#]) and the activities of H2 and H2O ([H2] and [H2O], respectively). Here, the intrinsic rate constants kx reflect the weighted reactivity of all active sites. Note that M–OH functions at the Au–support interface can also activate H–H bonds instead of H2O, as suggested elsewhere.29,30Equation 2 then relates to the rate of electron transfer to O2* species, given by
| 3 |
where −rO2 depends on step 3 and determines the H2O2 and H2O formation rates (rH2O2 and rH2O). Rearrangement of eq 3 yields the product formation rates
| 4 |
and
| 5 |
where eqs 4 and 5 suggest that the average number of electrons transferred during oxygen reduction reflects the ratio of the rates of OOH* hydrogenation (k4) to dissociation (k5), which determines the primary selectivity of H2O2.
Expansion of r3 in terms of its relevant surface intermediates gives
| 6 |
where the rate of oxygen reduction depends on the activity of electrons ([e–]) generated from hydrogen activation and the number of adsorbed dioxygen intermediates ([O2*]). These species then react with protons ([H+]) transferred either from hydronium ions (H3O+) or protonated hydroxyl species on the support (M–OH2+). eq 6, therefore, suggests that Au nanoparticles should show changes in the electrochemical potential of its electrons (related to [e–]) that depend on the rates of kinetically relevant oxygen reduction and hydrogen activation steps, explored in detail elsewhere.70,74,75
Application of the pseudosteady-state hypothesis yields coverages for each reactive species, leading to expressions
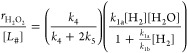 |
7 |
and
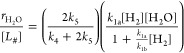 |
8 |
where the product formation rates depend on the total number of sites that bind H2 ([L#]) and multiple rate constants and reactant activities. Specifically, eqs 7 and 8 indicate that H2O2 and H2O turnover rates depend on the rate of hydrogen consumption (shown by eq 2) multiplied by their selectivities. Here, the total H2O2 selectivity reflects the average H2O2 selectivity (k4/(k4 + 2k5)) of all catalytic active sites that perform oxygen reduction.
Equations 7 and 8 predict that H2O2 and H2O turnover rates should increase in proportion with [H2] and approach constant values as the H#-species saturate binding sites at the greatest pressures. This conclusion agrees with Figures 4 and 5 where rates increase linearly with H2 pressures, which suggests H# species exist at low fractional coverages across this range of conditions (10–400 kPa H2, 60 kPa O2, 278 K). The low coverages of H# on Au surfaces agree with the kinetic relevance of H2 dissociation, rapid consumption of H# by heterolytic oxidation, and weak binding of H atoms to supported Au catalysts.33,99,100 Such expressions suggest rates do not depend on the pressure of O2, consistent with Figures 4 and 5 (10–400 kPa O2, 60 kPa H2, 278 K).
Therefore, eqs 7 and 8 take the form
| 9 |
and
| 10 |
where turnover rates depend directly on the pressure of H2 at the conditions used in this study. Indeed, eqs 9 and 10 also explain the similar H2/D2 kinetic isotope effects (Table 3) for H2O2 and H2O formation since the isotopic label should only affect the value of k1a.
This mechanism resembles that proposed by Chandler et al.29,30 We observe a similar importance of chemical functions at the Au–support interface during O–O bond activation, proportional dependencies on H2 pressure (3–20 kPa H2, 10 kPa O2, 333 K), and a strong influence of H2O on catalysis.29,31 However, the condensed water leads to the significant formation of H2O2, suggesting high concentrations of H2O molecules stabilize the formation of dioxygen species on metallic Au far from the Au–support interface and also stabilize O–O bonds better than under dry or humid conditions. By comparison, work by Chandler et al. suggests that H2O2 dissociation is inevitable and is the primary pathway of H2O formation under dry conditions. Furthermore, our findings suggest that water molecules enable the activation of H2 across all surface Au atoms (vide infra). In contrast, prior studies strictly attribute the perimeter of the Au–support interface as the active site for all catalysis of H2 and O2.
3.3. Rate Ratios of H2O2 and H2O Formation Correlate with the Fraction of Perimeter Sites at the Au–Support Interface
Infrared spectra of adsorbed CO were used to probe the quantity and form of site motifs on TiO2-supported Au nanoparticles and how these sites change with the size of Au nanoparticles. Figure 8A shows steady-state infrared spectra of CO on TiO2 and 5 and 25 nm Au nanoparticles supported on TiO2 (0.01 kPa CO, 303 K) obtained following in situ oxidative pretreatments (Section 2.2.2) and saturation at higher pressures of CO (100 kPa). These spectra show vibrational features consistent with CO adsorbed linearly to Au atoms within Au nanoparticles (η1-COAu*; ν(C–O) = 2119 cm–1) and a weak peak corresponding to CO bound to Ti4+ cations of TiO2 (η1-COTi*; ν(C–O) = 2194 cm–1).113
Figure 8.

(a) Ex situ infrared spectra of CO adsorbed upon surfaces of TiO2 and TiO2-supported Au nanoparticles (3 wt % Au, 0.01 kPa CO, 101 kPa He, 303 K) following an oxidative treatment (20 kPa O2, 573 K). (b) Ratios of H2O2 and H2O formation as a function of the ration of surface and perimeter (θt/θp) sites at varying Au nanoparticle diameters (200 kPa H2, 60 kPa O2, 278 K).
Additionally, multiple forms of carbonate species appear (monodentate, η1-CO3*Ti [νs(C–O) = 1300–1420 cm–1]; bidentate, η2-CO3*Ti [νa(C–O) = 1550–1610 cm–1])113−118 along with features that signify the presence of water molecules (δ(H–O–H) = 1600–1630 cm–1; ν(O–H) = 3460–3630 cm–1)113,116 and surface hydroxyl groups on TiO2 (ν(O–H) = 3630–3720 cm–1).113,116 The peak area of linearly adsorbed CO on TiO2 (η1-CO*Ti) is negligible compared to CO bound linearly to Au atoms (η1-CO*Au), which appears prominently upon samples possessing Au nanoparticles (Section S4).
Prior studies demonstrate that carbonate species bind strongly and inhibit CO oxidation catalysis, which has been attributed to competitive adsorption of carbonate at interfacial sites present at the perimeter of Au nanoparticles.119,120 Thus, we hypothesize that the ratio of the peak area of η1-COAu* (ACO–Au) to the area for to η1-CO3*Ti and η2-CO3*Ti features (ACO3) scales in proportion to the ratio of the number of metallic Au sites to the number of sites at the interface between Au and TiO2. This ratio of features is approximately five times greater for 25 nm Au–TiO2 in comparison to 5 nm Au–TiO2, which agrees with expectations that the fraction of exposed Au atoms that bind CO grows in comparison to the fraction sites that reside at the interface between the support and Au nanoparticles as mean nanoparticle diameters increase. This conclusion agrees with geometric arguments for cuboctahedra Au nanoparticles using correlations established by Ribeiro (eq 11)87 and modeled by Van Hardeveld and Hartog,88 as shown below.
| 11 |
This analysis suggests that the fraction of Au atoms that exist at the surface of nanoparticles (θt) increase relative to the fraction of Au atoms that reside at the exposed perimeter of nanoparticles (θp) as the mean diameter increases, which correlates with integrated peak areas of adsorbed CO (ACO–Au/ACO3 ∝ θt/θp) on different-sized nanoparticles (Figures S15 and S16). Moreover, Figure 8b shows that rate ratios of H2O2 and H2O formation increase with this ratio of surface Au atoms to perimeter atoms (θt/θp). This correlation (R2 = 0.991a) agrees with the hypothesis that perimeter sites at the Au–TiO2 interface preferentially form H2O, but the remaining surface Au atoms favor H2O2 formation.
3.4. Activation Barriers for H2O2 and H2O Formation Depend Differently on Active Site Motifs
The interpretation of measured ratios of product formation rates (eqs 9 and 10) depends strongly upon the fraction of Au atoms present at interfacial sites, demonstrating that the position and coordination of Au atoms within active sites impact the rate constants for OOH* conversion to H2O2 (k4) and H2O (k5) differently. These differences suggest that the stabilization of the associated transition states responds strongly to the proximity of the TiO2 surface.
Product formation rates were measured as functions of temperature to quantify the apparent activation enthalpies for H2O2 (ΔH‡H2O2) and H2O (ΔH‡H2O) formation on each catalyst. Measurements were performed at equivalent conditions to facilitate meaningful comparisons across all catalysts (200 kPa H2, 60 kPa O2, 278–308 K), which corresponds to a kinetic regime where rates increase in proportion to [H2] and do not vary with [O2]. Representative time on stream measurement used for calculating activation enthalpies are reported in Figure S17.
Figure 9a,b show that values of ΔHH2O‡ increase with the mean diameters of Au nanoparticles (2–25 nm) on both TiO2 and SiO2, whereas ΔHH2O2‡ varies only slightly. Both Au–SiO2 and Au–TiO2 present relatively low ΔHH2O2‡ that span a narrow range (16–22 kJ mol–1). Values for ΔHH2O‡ reflect intrinsic barriers of cleaving O–O bonds within OOH* species, which occur with greater barriers upon Au–SiO2 (72–85 kJ mol–1) than on Au–TiO2 (22–35 kJ mol–1). These observations agree with the results in Figure 2 and show that large Au nanoparticles and Au nanoparticles on SiO2 more selectively form H2O2 than small nanoparticles or those with a significant fraction of Au atoms present at interfaces with TiO2. The origins of these differences must arise from distinct site requirements for transition states that activate H2 and cleave H–H bonds and those that either reduce OOH* to H2O2 or break O–O bonds in OOH* to form H2O (e.g., steps 1a, 4, and 5).
Figure 9.

Apparent activation enthalpies of H2O2 (ΔHH2O2‡, red ■) and H2O (ΔHH2O‡, ■) formation as functions of mean Au nanoparticle diameter on (a) Au–TiO2, and (b) Au–SiO2 materials (200 kPa H2, 60 kPa O2, 278–308 K). Figures S18 and S19 show corresponding steady-state rate measurements as a function of temperature. Dashed lines intended to guide the eyes.
These distinct site requirements appear most clearly by simultaneously examining activation enthalpies for H2 activation (ΔH‡H2, determined from measurements of −rH2) and the differences between barriers to form H2O2 and H2O (ΔΔH‡ = ΔH‡H2O – ΔH‡H2O2). Applying Eyring theory to eq 2 expands the rate of hydrogen consumption as
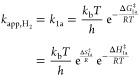 |
12 |
where the apparent rate constant of H2 activation (kapp,H2) depends on the Boltzmann constant (kb), Planck constant (h), and activation Gibbs free energy (ΔG‡1a), entropy (ΔS‡1a), and enthalpy (ΔH‡1a = ΔH‡H2) for H2 consumption. The value of ΔH‡H2 appears directly in expressions for the rates of product formation and the terms ΔH‡H2O2 and ΔH‡H2O (derived in Section S5), because H2 activation represents the kinetically relevant step in the sequence of steps that form both products. While the molecular interpretation of absolute values for ΔH‡H2O2 and ΔH‡H2O remain elusive, analysis of the ratios of H2O2 and H2O formation rates provides molecularly interpretable quantities. Combining these terms with the structures proposed by transition state theory gives the relationship.
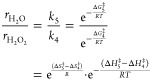 |
13 |
where the differences between the intrinsic activation free energies of steps 4 (ΔG‡4) and 5 (ΔG‡5) represent the differences between apparent barriers of H2O2 and H2O formation (e.g., ΔΔH‡ = ΔH‡H2O – ΔH‡H2O2 = ΔH‡5 – ΔH‡4) and their entropic contributions (ΔS‡5,ΔS‡4). These analyses allow comparisons of ΔH‡H2and ΔΔH‡ across catalysts with different Au nanoparticle diameters and support identities, providing molecular insight into how distinct structural motifs impact the transition states of H–H and O–O bond rupture during the formation of H2O2 or H2O.
Values of ΔH‡H2 increase from 21 to 29 kJ mol–1 on Au–TiO2 as mean Au nanoparticle diameters increase from 2 to 25 nm (Figure S20), while ΔH‡H2 remains equal to ∼35 kJ mol–1 on all Au–SiO2 catalysts. Note that these values are consistent with or slightly lower than barriers of H2 activation reported in the gas phase (∼25 to ∼45 kJ mol–1) on Au–TiO2 materials.30,93 In comparison, ΔΔH‡ values span a much greater range of values and increase from 6 to 14 kJ mol–1 on Au–TiO2 and 59 to 66 kJ mol–1 on Au–SiO2 across the same range of Au nanoparticle diameters (2–25 nm). These observations suggest that intrinsic barriers to activate H–H bonds is less sensitive to its active site than the activation of O–O bonds. Additionally, these trends indicate metallic Au atoms that exist furthest from reducible or Lewis acidic oxide supports present the greatest barriers for activating O–O bonds and slightly higher barriers for activating H2, leading to greater apparent barriers for all pathways on the largest Au nanoparticles and most refractory supports (e.g., SiO2).
Interfaces between Au atoms and TiO2 present significantly lower barriers for dissociating dioxygen surface intermediates and modestly lower barriers for activating H2. Consequently, the smallest Au nanoparticles upon TiO2 give the greatest rates for H2 consumption and H2O formation among all Au–TiO2 and Au–SiO2 catalysts examined. These conclusions agree with computational studies that showed Au–TiO2 interfaces (e.g., Au–TiO2) bind dioxygen more exothermically than metallic Au surfaces (ΔEAu–TiO2ads = −0.82–1.01 eV, ΔEAuads = −0.55 eV) and present lower barriers of dissociating O–O bonds (EA–TiO2a = 0.42–0.6 eV, EAua = 1.59 eV).21−23 The poor activation of O2 on metallic Au surfaces is consistent with its largely occupied d-states that disfavor the binding of most adsorbates.100 However, Yates and Neurock suggested that the Au–TiO2 interface enables the adsorption of O2 in a di-σ configuration.21 Moreover, Au atoms transfer electron density to Ti atoms at these interfaces, strengthening the binding of O2 at these sites.22 These differences in electronic structure may favor the back-donation of electrons from Au to the π* antibonding orbitals of O–O bonds,90 consistent with lower barriers of O–O bond dissociation at Au–support interfaces.
These insights agree with the large barriers for O–O bond dissociation and high H2O2 selectivities (>90%) during electrochemical ORR on polycrystalline Au foils, which do not possess interfacial sites.67,69 Thus, metallic Au and Au–SiO2 interfaces favor transition states of oxygen reduction that stabilize O–O bonds more effectively than Au–TiO2.
We sought to elucidate the active site motifs that favor H2 activation and the formation of H2O2 or H2O by comparing activation barriers and quantifiable structural descriptors for the supported Au catalysts. Estimates for turnover rates for H2 consumption (−rH2 = rH2O + rH2O2) were calculated by normalizing measured rates (Figure 2) by approximated numbers for surface, perimeter, or corner atoms, which were determined from the mean Au nanoparticle diameters and using correlations established by Ribeiro87 and modeled by Van Hardeveld and Hartog88 for a cuboctahedra geometry. Figure 10 shows that H2 consumption turnover rates calculated using the total number of surface Au atoms remain mostly constant across Au nanoparticles of all diameters on SiO2 and vary only slightly among Au–TiO2. In contrast, turnover rates normalized by the number of perimeter or corner sites vary by two to 3 orders of magnitude with changes in the mean diameter of Au nanoparticles.
Figure 10.

Steady-state rates of H2 consumption as a function of the surface-area normalized Au nanoparticle diameter on (a) Au–TiO2 and (b) Au–SiO2 materials (200 kPa H2, 60 kPa O2, 278 K). Rates are normalized by the total number of surface atoms (blue ■), perimeter atoms (●), and corner atoms (red ◆), estimated using correlations reported by Ribeiro and co-workers.87,88
Taken together, the small span of ΔHH2‡ values (21–35 kJ mol–1) across Au–TiO2 and Au–SiO2 catalysts (Figure S20) and H2 consumption rates that vary by a factor of 3 or less (when normalized by estimates for the total number of exposed Au atoms; Figure 10) indicate that H2 activation weakly senses differences between the different forms of active sites formed with Au atoms when the reaction proceeds by heterolytic processes within liquid water. In comparison, the wide span of ΔΔH‡ values (6–66 kJ mol–1) across the same Au–SiO2 and Au–TiO2 materials demonstrate that dissociative reactions of dioxygen surface intermediates (e.g., OOH*) strongly depend on proximity to the Au–support interface (Figure S20). Thus, sites at the Au–support interface preferentially form H2O, while sites far from the interface yield H2O2. These findings expand the understanding of the distinct site requirements of reactions of H2 and O2, which previously attributed all of the catalysis to sites at the perimeter of Au nanoparticles.29,30 Comparisons to prior studies suggest that solvation of Au nanoparticles by liquid water suppresses the structure sensitivity of H2 activation by enabling H2 dissociation across the entire catalyst surface.
The generality of these interpretations was tested by comparing rates and kinetic barriers for H2 consumption, H2O2 formation, and H2O production upon 2–3 nm Au nanoparticles supported on six distinct supports. Figure 11 shows values of ΔHH2‡ and ΔΔH‡ obtained from Au nanoparticles supported on BN, carbon, SiO2, Al2O3, La2O3, and TiO2, all measured within a consistent kinetic regime (vide supra; Figure S11). Moreover, materials with the greatest values of ΔΔH‡ present greater barriers for primary H2O2 decomposition (i.e., ΔH8‡), as determined from reactions among H2O2 and H2 (200 kPa H2, 0.2 mM H2O2, 278–308 K; Figure S22). Thus, catalysts capable of dissociating O–O bonds sourced from O2 are similarly effective at dissociating H2O2.
Figure 11.
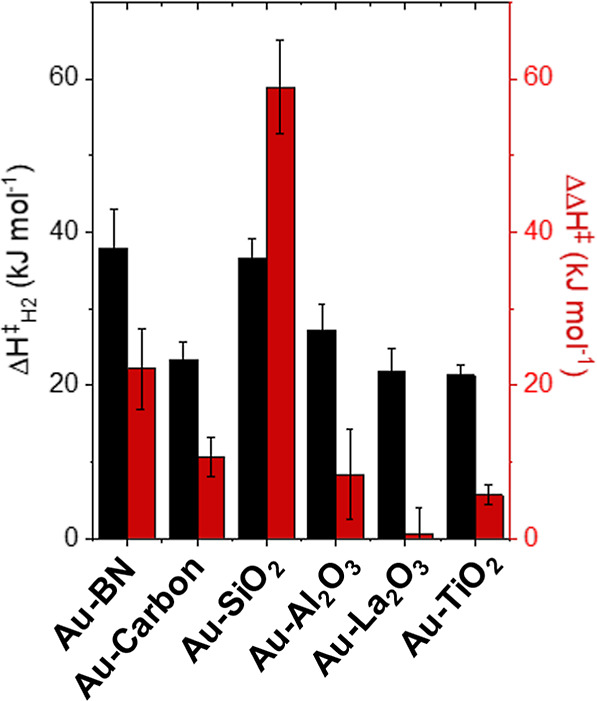
Apparent activation enthalpies of hydrogen consumption (ΔHH2‡; black) and differences between activation enthalpies of H2O2 and H2O formation (ΔΔH‡; red) on 2–3 nm Au nanoparticles supported on BN, carbon, SiO2, Al2O3, La2O3, and TiO2 (200 kPa H2, 60 kPa O2, 278 K). Figure S23 shows associated steady-state rate measurements versus temperature.
Generally, metal oxide supports show lower values of ΔHH2‡ compared to BN, while carbon presents barriers similar to those measured upon metal oxides. We hypothesize that nucleophilic or Brønsted basic functions may aid the activation of H–H bonds by accepting a proton at the Au–support interface, as suggested elsewhere.29,30 Still, the presence of water molecules extends the range of catalysis to all surface Au atoms by facilitating these proton transfer steps (vide supra). Thus, the lower rates of H2 activation on carbon and BN may result from their high level of hydrophobicity, which may lower the concentration of interfacial water molecules that can solvate Au surfaces and disrupt proton transfer steps.
The dissociation of O–O bonds show a much stronger dependence upon the chemical function and identity of the support material. Reducible (e.g., TiO2, La2O3)121 and Lewis acidic (e.g., TiO2, La2O3, Al2O3)122,123 metal oxides show lower values for ΔΔH‡. Thus, we hypothesize that the presence of oxygen vacancies or Lewis acidic metal centers of these Au–support interfaces more readily enable the formation of strongly bound superoxide species (e.g., Au–O–O–M) or related species, which may present lower barriers of O–O dissociation than on more refractory materials (vide supra). By comparison, the Au–SiO2 interface is the most refractory of these catalysts and may bind O2 so weakly such that the majority of H2 and O2 catalysis occurs upon the metallic surfaces of Au nanoparticles rather than the perimeter sites of Au–SiO2 (Figure 9). Finally, the refractory and hydrophobic materials (e.g., BN, Carbon) show intermediate values of ΔΔH‡, which may suggest that the unfavorable interaction of water near these supports may lower the interfacial concentration of water molecules and destabilize O–O bonds relative to hydrophilic and refractory supports like silica. Furthermore, these properties and the identity of the support significantly impact rates and selectivities of H2O2 formation using dilute PdAux nanoparticle catalysts (Figures S24–S28).
4. Conclusions
The interface between Au nanoparticles and supports gives rise to active site motifs that facilitate the reduction and dissociation of dioxygen species; however, the activation of H2 only weakly senses the presence of these interfaces in liquid water. This knowledge expands the understanding from prior work studying reactions of H2 and O2 over supported Au nanoparticles within dry or dilute vapor pressures of water (10–4 to 0.1 kPa H2O). These reports showed that sites at the perimeter of Au–support interfaces catalyze heterolytic reactions that activate H–H and O–O bonds, but water molecules introduce distinct proton-transfer reactions at these interfaces that accelerate O2 activation and inhibit H2 activation. However, we demonstrate that condensed water (55.5 M H2O) further modifies the structure sensitivity of this reaction network by stabilizing O–O bonds, allowing the formation of H2O2, and enabling all surface Au atoms to catalyze heterolytic reactions with H2 at the metal–liquid–support interface. These insights and the accompanying understanding of manipulating relative rates of H2 and O2 activation carry broad significance due to the ubiquity of these reactants (H2, O2, and H2O) in reactions using Au catalysts (e.g., hydrogenation, partial oxidations, electrocatalysis).
Kinetic analyses of H2 and O2 activation combined with isotopic measurements show that Au catalysts follow a similar mechanism. Specifically, proton acceptors (e.g., H2O, M–OH) cocatalyze the kinetically relevant activation of H–H bonds at the interface of Au nanoparticles, liquid water, and the support. Consequently, rates of H2 consumption are 1–2 orders of magnitude greater on Au–MOX interfaces than on refractory and hydrophobic interfaces (e.g., carbon, BN). Yet, comparisons of barriers and rates normalized by the total number of sites at the surface, perimeter, and corner sites of Au nanoparticles show that H–H activation is mostly insensitive to the mean diameter of Au nanoparticles, indicating that all surface atoms may heterolytically activate H–H bonds with liquid water molecules. However, O–O bond dissociation is much more structure-sensitive. More reducible or Lewis acidic metal oxides (e.g., Au–LaO2) favor the dissociation of O–O bonds compared to more inert supports like Au–SiO2, which stabilize O–O bonds. Moreover, increasing the size of Au nanoparticles decreases the fraction of Au atoms at the metal–support interface versus metallic Au atoms far from the support, decreasing the proportion of sites that cleave O–O bonds.
This understanding provides multiple strategies to manipulate the activation of H2- and O2-derived species at metal–liquid–support interfaces. These findings may guide the design of catalytic materials for thermochemical redox reactions within liquid water (e.g., alcohol oxidation, HCOOH decomposition, and so forth). Similarly, these principles provide structure–function relationships to influence heterolytic steps relevant to electrochemical reactions in aqueous electrolyte. Furthermore, future work should further elucidate how the interfaces of water, metal nanoparticles, and metal oxides affect heterolytic paths during the reduction and oxidation of small molecules and organic substrates.
Acknowledgments
We acknowledge the generous funding to support this work provided by the Energy & Biosciences Institute through the EBI-Shell program and support from the National Science Foundation (CBET-1511819 and CCI-1740656). The authors also acknowledge stimulating discussions with Drs. Sander Van Bavel, Andrew Horton, and Sumit Verma of Royal Dutch Shell. J.S.A. was supported by a National Science Foundation Graduate Research Fellowship (DGE-1144245). Portions of this work were carried out in part in the Materials Research Laboratory Central Research Facilities and School of Chemical Sciences Microanalysis Lab at the University of Illinois. The authors declare no competing interests.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c05072.
Additional Characterization of materials by TEM, ICP-OES, EDXRF, and point of zero charge analysis; process flow diagrams of reactors, additional pressure dependence data, data for kinetic isotope measurements, discussion of rate derivations, isotherms from FTIR spectra of adsorbed CO, additional discussion of Eyring theory and relevant Arrhenius plots, and rate data normalized by different site counts (PDF)
Author Present Address
§ Department of Chemical Engineering, California Institute of Technology, 1200 E California Blvd, Pasadena, California 91125, United States
Author Present Address
∥ Amogy, 19 Morris Ave, Brooklyn, New York 11250, United States.
Author Present Address
⊥ Department of Chemical Engineering, Indian Institute of Technology, Palau 382355, Gujarat, India.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Sankar M.; He Q.; Engel R. V.; Sainna M. A.; Logsdail A. J.; Roldan A.; Willock D. J.; Agarwal N.; Kiely C. J.; Hutchings G. J. Role of the Support in Gold-Containing Nanoparticles as Heterogeneous Catalysts. Chem. Rev. 2020, 120 (8), 3890–3938. 10.1021/acs.chemrev.9b00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. Y.; Wang A.; Zhang T.; Mou C. Catalysis by gold: New insights into the support effect. Nano Today 2013, 8 (4), 403–416. 10.1016/j.nantod.2013.07.005. [DOI] [Google Scholar]
- Ciriminna R.; Falletta E.; Della Pina C.; Teles J. H.; Pagliaro M. Industrial Applications of Gold Catalysis. Angew. Chem., Int. Ed. 2016, 55 (46), 14210–14217. 10.1002/anie.201604656. [DOI] [PubMed] [Google Scholar]
- Carabineiro S. A. C. Supported Gold Nanoparticles as Catalysts for the Oxidation of Alcohols and Alkanes. Front. Chem. 2019, 7, 702. 10.3389/fchem.2019.00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruta M.; Kobayashi T.; Sano H.; Yamada N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature far Below 0 °C. Chem. Lett. 1987, 16, 405–408. 10.1246/cl.1987.405. [DOI] [Google Scholar]
- Mullen G. M.; Mullins C. B. Water’s place in Au catalysis. Science 2014, 345 (6204), 1564–1565. 10.1126/science.1259527. [DOI] [PubMed] [Google Scholar]
- Saavedra J.; Doan H. A.; Pursell C. J.; Grabow L. C.; Chandler B. D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345 (6204), 1599–1602. 10.1126/science.1256018. [DOI] [PubMed] [Google Scholar]
- Ojeda M.; Zhan B.; Iglesia E. Mechanistic interpretation of CO oxidation turnover rates on supported Au clusters. J. Catal. 2012, 285 (1), 92–102. 10.1016/j.jcat.2011.09.015. [DOI] [Google Scholar]
- Daté M.; Haruta M. Moisture Effect on CO Oxidation over Au/TiO2 Catalyst. J. Catal. 2001, 201 (2), 221–224. 10.1006/jcat.2001.3254. [DOI] [Google Scholar]
- Daté M.; Okumura M.; Tsubota S.; Haruta M. Vital Role of Moisture in the Catalytic Activity of Supported Gold Nanoparticles. Angew. Chem., Int. Ed. 2004, 43 (16), 2129–2132. 10.1002/anie.200453796. [DOI] [PubMed] [Google Scholar]
- Saavedra J.; Pursell C. J.; Chandler B. D. CO Oxidation Kinetics over Au/TiO2 and Au/Al2O3 Catalysts: Evidence for a Common Water-Assisted Mechanism. J. Am. Chem. Soc. 2018, 140 (10), 3712–3723. 10.1021/jacs.7b12758. [DOI] [PubMed] [Google Scholar]
- Saavedra J.; Whittaker T.; Chen Z.; Pursell C. J.; Rioux R. M.; Chandler B. D. Controlling activity and selectivity using water in the Au-catalysed preferential oxidation of CO in H2. Nat. Chem. 2016, 8, 584–589. 10.1038/nchem.2494. [DOI] [PubMed] [Google Scholar]
- Fujitani T.; Nakamura I.; Takahashi A. H2O Dissociation at the Perimeter Interface between Gold Nanoparticles and TiO2 Is Crucial for Oxidation of CO. ACS Catal. 2020, 10, 2517–2521. 10.1021/acscatal.9b05195. [DOI] [Google Scholar]
- Siemer N.; Munoz-Santiburcio D.; Marx D. Solvation-Enhanced Oxygen Activation at Gold/Titania Nanocatalysts. ACS Catal. 2020, 10, 8530–8534. 10.1021/acscatal.0c01326. [DOI] [Google Scholar]
- Bongiorno A.; Landman U. Water-Enhanced Catalysis of CO Oxidation on Free and Supported Gold Nanoclusters. Phys. Rev. Lett. 2005, 95, 106102. 10.1103/PhysRevLett.95.106102. [DOI] [PubMed] [Google Scholar]
- Costello C. K.; Yang J. H.; Law H. Y.; Wang Y.; Lin J. N.; Marks L. D.; Kung M. C.; Kung H. H. On the potential role of hydroxyl groups in CO oxidation over Au/Al2O3. Appl. Catal., A 2003, 243, 15–24. 10.1016/S0926-860X(02)00533-1. [DOI] [Google Scholar]
- Yan T.; Gong J.; Flaherty D. W.; Mullins C. B. The Effect of Adsorbed Water in CO Oxidation on Au/TiO2 (110). J. Phys. Chem. C 2011, 115 (5), 2057–2065. 10.1021/jp109295u. [DOI] [Google Scholar]
- Mullen G. M.; Gong J.; Yan T.; Pan M.; Mullins C. B. The Effects of Adsorbed Water on Gold Catalysis and Surface Chemistry. Top. Catal. 2013, 56, 1499–1511. 10.1007/s11244-013-0143-x. [DOI] [Google Scholar]
- Kim T. S.; Gong J.; Ojifinni R. A.; White J. M.; Mullins C. B. Water Activated by Atomic Oxygen on Au(111) to Oxidize CO at Low Temperatures. J. Am. Chem. Soc. 2006, 128 (19), 6282–6283. 10.1021/ja058263m. [DOI] [PubMed] [Google Scholar]
- Ojifinni R. A.; Froemming N. S.; Gong J.; Pan M.; Kim T. S.; White J. M.; Henkelman G.; Mullins C. B. Water-Enhanced Low-Temperature CO Oxidation and Isotope Effects on Atomic Oxygen-Covered Au(111). J. Am. Chem. Soc. 2008, 130 (21), 6801–6812. 10.1021/ja800351j. [DOI] [PubMed] [Google Scholar]
- Green I. X.; Tang W.; Neurock M.; Yates J. T. Insights into Catalytic Oxidation at the Au/TiO2 Dual Perimeter Sites. Acc. Chem. Res. 2014, 47 (3), 805–815. 10.1021/ar400196f. [DOI] [PubMed] [Google Scholar]
- Green I. X.; Tang W.; Neurock M.; Yates J. T. Spectroscopic Observation of Dual Catalytic Sites During Oxidation of CO on a Au/TiO2 Catalyst. Science 2011, 333 (6043), 736–739. 10.1126/science.1207272. [DOI] [PubMed] [Google Scholar]
- Green I. X.; Tang W.; McEntee M.; Neurock M.; Yates J. T. Inhibition at Perimeter Sites of Au/TiO2 Oxidation Catalyst by Reactant Oxygen. J. Am. Chem. Soc. 2012, 134 (30), 12717–12723. 10.1021/ja304426b. [DOI] [PubMed] [Google Scholar]
- Ketchie W. C.; Fang Y.; Wong M. S.; Murayama M.; Davis R. J. Influence of gold particle size on the aqueous-phase oxidation of carbon monoxide and glycerol. J. Catal. 2007, 250 (1), 94–101. 10.1016/j.jcat.2007.06.001. [DOI] [Google Scholar]
- Kung H. H.; Kung M. C.; Costello C. K. Supported Au catalysts for low temperature CO oxidation. J. Catal. 2003, 216 (1–2), 425–432. 10.1016/S0021-9517(02)00111-2. [DOI] [Google Scholar]
- Bond G. C.; Thompson D. T. Gold-Catalysed Oxidation of Carbon Monoxide. Gold Bull. 2000, 33 (2), 41–50. 10.1007/BF03216579. [DOI] [Google Scholar]
- Zope B. N.; Hibbitts D. D.; Neurock M.; Davis R. J. Reactivity of the Gold/Water Interface During Selective Oxidation Catalysis. Science 2010, 330 (6000), 74–78. 10.1126/science.1195055. [DOI] [PubMed] [Google Scholar]
- Ide M. S.; Davis R. J. The Important Role of Hydroxyl on Oxidation Catalysis by Gold Nanoparticles. Acc. Chem. Res. 2014, 47 (3), 825–833. 10.1021/ar4001907. [DOI] [PubMed] [Google Scholar]
- Whittaker T.; Kumar K. B. S.; Peterson C.; Pollock M. N.; Grabow L. C.; Chandler B. D. H2 Oxidation over Supported Au Nanoparticle Catalysts: Evidence for Heterolytic H2 Activation at the Metal-Support Interface. J. Am. Chem. Soc. 2018, 140 (48), 16469–16487. 10.1021/jacs.8b04991. [DOI] [PubMed] [Google Scholar]
- Sravan Kumar K. B.; Whittaker T.; Peterson C.; Grabow L. C.; Chandler B. D. Water Poisons H2 Activation at the Au-TiO2 Interface by Slowing Proton and Electron Transfer between Au and Titania. J. Am. Chem. Soc. 2020, 142 (12), 5760–5772. 10.1021/jacs.9b13729. [DOI] [PubMed] [Google Scholar]
- Mahdavi-Shakib A.; Kumar K. B. S.; Whittaker T.; Xie T.; Grabow L. C.; Rioux R. M.; Chandler B. D. Kinetics of H2 Adsorption at the Metal-Support Interface of Au/TiO2 Catalysts Probed by Broad Background IR Absorbance. Angew. Chem., Int. Ed. 2021, 60 (14), 7735–7743. 10.1002/anie.202013359. [DOI] [PubMed] [Google Scholar]
- Mitsudome T.; Kaneda K. Gold nanoparticle catalysts for selective hydrogenations. Green Chem. 2013, 15 (10), 2636–2654. 10.1039/c3gc41360h. [DOI] [Google Scholar]
- Zhao J.; Ge L.; Yuan H.; Liu Y.; Gui Y.; Zhang B.; Zhou L.; Fang S. Heterogeneous gold catalysts for selective hydrogenation: from nanoparticles to atomically precise nanoclusters. Nanoscale 2019, 11 (24), 11429–11436. 10.1039/C9NR03182K. [DOI] [PubMed] [Google Scholar]
- Noujima A.; Mitsudome T.; Mizugaki T.; Jitsukawa K.; Kaneda K. Unique catalysis of gold nanoparticles in the chemoselective hydrogenolysis with H2: cooperative effect between small gold nanoparticles and a basic support. Chem. Commun. 2012, 48 (53), 6723–6725. 10.1039/c2cc32850j. [DOI] [PubMed] [Google Scholar]
- Corma A.; Garcia H. Supported Gold Nanoparticles as Catalysts for Organic Reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. 10.1039/b707314n. [DOI] [PubMed] [Google Scholar]
- Stratakis M.; Garcia H. Catalysis by Supported Gold Nanoparticles: Beyond Aerobic Oxidative Processes. Chem. Rev. 2012, 112, 4469–4506. 10.1021/cr3000785. [DOI] [PubMed] [Google Scholar]
- Takale B. S.; Bao M.; Yamamoto Y. Gold Nanoparticle (AuNPs) and Gold Nanopore (AuNPore) Catalysts in Organic Synthesis. Org. Biomol. Chem. 2014, 12, 2005–2027. 10.1039/c3ob42207k. [DOI] [PubMed] [Google Scholar]
- Nkosi B.; Coville N. J.; Hutchings G. J. Reactivation of a supported gold catalyst for acetylene hydrochlorination. J. Chem. Soc., Chem. Commun. 1988, 71–72. 10.1039/c39880000071. [DOI] [Google Scholar]
- Molina L. M.; Hammer B. Active Role of Oxide Support during Co Oxidation at Au/MgO. Phys. Rev. Lett. 2003, 90 (20), 206102. 10.1103/PhysRevLett.90.206102. [DOI] [PubMed] [Google Scholar]
- Remediakis I.; Lopez N.; Norskov J. K. CO Oxidation on Rutile-Supported Au Nanoparticles. Angew. Chem., Int. Ed. 2005, 44 (12), 1824–1826. 10.1002/anie.200461699. [DOI] [PubMed] [Google Scholar]
- Meier D. C.; Goodman D. W. The Influence of Metal Cluster Size on Adsorption Energies: CO Adsorbed on Au Clusters Supported on TiO2. J. Am. Chem. Soc. 2004, 126 (6), 1892–1899. 10.1021/ja030359y. [DOI] [PubMed] [Google Scholar]
- Valden M.; Lai X.; Goodman D. W. Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties. Science 1998, 281 (5383), 1647–1650. 10.1126/science.281.5383.1647. [DOI] [PubMed] [Google Scholar]
- Iizuka Y.; Tode T.; Takao T.; Yatsu K.; Takeuchi T.; Tsubota S.; Haruta M. A Kinetic and Adsorption Study of CO Oxidation over Unsupported Fine Gold Powder and over Gold Supported on Titanium Dioxide. J. Catal. 1999, 187 (1), 50–58. 10.1006/jcat.1999.2604. [DOI] [Google Scholar]
- Yan T.; Redman D. W.; Yu W.; Flaherty D. W.; Rodriguez J.; Mullins C. B. CO Oxidation on Inverse Fe2O3/Au(111) model catalysts. J. Catal. 2012, 294, 216–222. 10.1016/j.jcat.2012.07.024. [DOI] [Google Scholar]
- Senanayake S.; Stacchiola D.; Evans J.; Estrella M.; Barrio L.; Perez M.; Hrbek J.; Rodriguez J. Probing the reaction intermediates for the water-gas shift over inverse CeOx/Au(111) catalysts. J. Catal. 2010, 271 (2), 392–400. 10.1016/j.jcat.2010.02.024. [DOI] [Google Scholar]
- Rodriguez J.; Hrbek J. Inverse oxide/metal catalysts: A versatile approach for activity tests and mechanistic studies. Surf. Sci. 2010, 604 (3–4), 241–244. 10.1016/j.susc.2009.11.038. [DOI] [Google Scholar]
- Palomino R. M.; Gutierrez R. A.; Liu Z.; Tenney S.; Grinter D. C.; Crumlin E.; Waluyo I.; Ramirez P. J.; Rodriguez J.; Senanayake S. Inverse Catalysts for CO Oxidation: Enhanced Oxide-metal Interactions in MgO/Au(111), CeO2/Au(111), and TiO2/Au(111). ACS Sustain. Chem. Eng. 2017, 5 (11), 10783–10791. 10.1021/acssuschemeng.7b02744. [DOI] [Google Scholar]
- van Deelen T. W.; Hernández Mejía C.; de Jong K. P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955–970. 10.1038/s41929-019-0364-x. [DOI] [Google Scholar]
- Yuan W.; Zhu B.; Fang K.; Li X.; Hansen T. W.; Ou Y.; Yang H.; Wagner J. B.; Gao Y.; Wang Y.; Zhang Z. In situ manipulation of the active Au-TiO2 interface with atomic precision during CO oxidation. Science 2021, 371 (6528), 517–521. 10.1126/science.abe3558. [DOI] [PubMed] [Google Scholar]
- Tang H.; Su Y.; Zhang B.; Lee A. F.; Isaacs M. A.; Wilson K.; Li L.; Ren Y.; Huang J.; Haruta M.; Qiao B.; Liu X.; Jin C.; Su D.; Wang J.; Zhang T. Classical strong metal-support interactions between gold nanoparticles and titanium dioxide. Sci. Adv. 2017, 3 (10), e1700231 10.1126/sciadv.1700231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X.; Huang Y.; Pan X.; Han B.; Su Y.; Jiang Q.; Li M.; Tang H.; Li G.; Qiao B. Size-dependent strong metal-support interaction in TiO2 supported Au nanocatalysts. Nat. Commun. 2020, 11, 5811. 10.1038/s41467-020-19484-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoeva B. G.; Ovoshchnikov D. S.; Golovko V. B. Establishing a Au Nanoparticle Size Effect in the Oxidation of Cyclohexene Using Gradually Changing Au Catalysts. ACS Catal. 2013, 3 (12), 2986–2991. 10.1021/cs400701j. [DOI] [Google Scholar]
- Uchiyama T.; Yoshida H.; Kuwauchi Y.; Ichikawa S.; Shimada S.; Haruta M.; Takeda S. Systematic Morphology Changes of Gold Nanoparticles Supported on CeO2 during CO Oxidation. Angew. Chem., Int. Ed. 2011, 50 (43), 10157–10160. 10.1002/anie.201102487. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Murayama T.; Taketoshi A.; Haruta M. Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes. Chem. Rev. 2020, 120, 464–525. 10.1021/acs.chemrev.9b00551. [DOI] [PubMed] [Google Scholar]
- Sampath A.; Ricciardulli T.; Priyadarshini P.; Ghosh R.; Adams J. S.; Flaherty D. W. Spectroscopic Evidence for the Involvement of Interfacial Sites in O–O Bond Activation over Gold Catalysts. ACS Catal. 2022, 12 (15), 9549–9558. 10.1021/acscatal.2c02076. [DOI] [Google Scholar]
- Huynh M. H. V.; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2007, 107 (11), 5004–5064. 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Stuchebrukhov A. A. Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev. 2010, 110 (12), 6939–6960. 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Soudackov A. V. Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry. J. Phys. Chem. B 2008, 112, 14108–14123. 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green I. X.; Tang w.; Neurock M.; Yates J. T. Low-Temperature Catalytic H2 Oxidation over Au Nanoparticle/TiO2 Dual Perimeter Sites. Angew. Chem., Int. Ed. 2011, 50 (43), 10186–10189. 10.1002/anie.201101612. [DOI] [PubMed] [Google Scholar]
- Panayotov D. A.; Burrows S. P.; Yates J. T.; Morris J. R. Mechanistic Studies of Hydrogen Dissociation and Spillover on Au/TiO2: IR Spectroscopy of Coadsorbed CO and H-Donated Electrons. J. Phys. Chem. C 2011, 115 (45), 22400–22408. 10.1021/jp2065024. [DOI] [Google Scholar]
- Panayotov D. A.; Yates J. T. n-Type doping of TiO2 with atomic hydrogen-observation of the production of conduction band electrons by infrared spectroscopy. Chem. Phys. Lett. 2007, 436 (1–3), 204–208. 10.1016/j.cplett.2007.01.039. [DOI] [Google Scholar]
- Panayotov D. A.; Yates J. T. Spectroscopic Detection of Hydrogen Atom Spillover from Au Nanoparticles Supported on TiO2: Use of Conduction Band Electrons. J. Phys. Chem. C 2007, 111 (7), 2959–2964. 10.1021/jp066686k. [DOI] [Google Scholar]
- Musumeci F.; Pollack G. H. Influence of water on the work function of certain metals. Chem. Phys. Lett. 2012, 536, 65–67. 10.1016/j.cplett.2012.03.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley T. S.; Hulsey M. J.; Westendorff K. S.; Lewis N. B.; Crumlin E. J.; Roman-leshkov Y.; Surendranath Y. Metal nanoparticles supported on a nonconductive oxide undergo pH-dependent spontaneous polarization. Chem. Sci. 2023, 14, 7154–7160. 10.1039/D3SC00884C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley T. S.; Roman-leshkov Y.; Surendranath Y. Spontaneous Electric Fields Play a Key Role in Thermochemical Catalysis at Metal-Liquid Interfaces. ACS Cent. Sci. 2021, 7, 1045–1055. 10.1021/acscentsci.1c00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockris J. O.; Reddy A.; Gamboa-Aldeco M.. Modern Electrochemistry 2A: Fundamentals of Electrodic, 2nd ed.; Springer: Boston, MA, 2000. [Google Scholar]
- Sanchez-Sanchez C. M.; Bard A. J. Hydrogen peroxide production in the oxygen reduction reaction at different electrocatalysts as quantified by scanning electrochemical microscopy. Anal. Chem. 2009, 81 (19), 8094–8100. 10.1021/ac901291v. [DOI] [PubMed] [Google Scholar]
- Zurilla R. W.; Sen R. K.; Yeager E. The Kinetics of the Oxygen Reduction Reaction on Gold in Alkaline Solution. J. Electrochem. Soc. 1978, 125, 1103–1109. 10.1149/1.2131628. [DOI] [Google Scholar]
- Strobl J. R.; Scherson D. On the Mechanism of the Oxygen Reduction Reaction on Au(poly) in Aqueous Alkaline Electrolytes: A Critical Reassessment. J. Phys. Chem. C 2021, 125 (25), 13862–13870. 10.1021/acs.jpcc.1c02683. [DOI] [Google Scholar]
- Ryu J.; Bregante D. T.; Howland W. C.; Bisbey R. P.; Kaminsky C. J.; Surendranath Y. Thermochemical aerobic oxidation catalysis in water can be analysed as two coupled electrochemical half reactions. Nat. Catal. 2021, 4, 742–752. 10.1038/s41929-021-00666-2. [DOI] [Google Scholar]
- Huang X.; Akdim O.; Douthwaite M.; Wang K.; Zhao L.; Lewis R. J.; Pattisson S.; Daniel I. T.; Miedziak P. J.; Shaw G.; Morgan D. J.; Althahban S. M.; Davies T. E.; He Q.; Wang F.; Fu J.; Bethell D.; Mcintosh S.; Kiely C. J.; Hutchings G. J. Au–Pd separation enhances bimetallic catalysis of alcohol oxidation. Nature 2022, 603, 271–275. 10.1038/s41586-022-04397-7. [DOI] [PubMed] [Google Scholar]
- An H.; Sun G.; Hulsey M. J.; Sautet P.; Yan N. Demonstrating the Electron–Proton-Transfer Mechanism of Aqueous Phase 4-Nitrophenol Hydrogenation Using Unbiased Electrochemical Cells. ACS Catal. 2022, 12, 15021–15027. 10.1021/acscatal.2c03133. [DOI] [Google Scholar]
- Adams J. S.; Kromer M.; Rodriguez-Lopez J.; Flaherty D. W. Unifying Concepts in Electro- and Thermocatalysis toward Hydrogen Peroxide Production. J. Am. Chem. Soc. 2021, 143, 7940–7957. 10.1021/jacs.0c13399. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Adams J. S.; Baby A.; Kromer M.; Flaherty D. W.; Rodríguez-López J. Electrochemical Screening of Au/Pt Catalysts for the Thermocatalytic Synthesis of Hydrogen Peroxide Based on Their Oxygen Reduction and Hydrogen Oxidation Activities Probed via Voltammetric Scanning Electrochemical Microscopy. ACS Sustain. Chem. Eng. 2022, 10 (51), 17207–17220. 10.1021/acssuschemeng.2c05120. [DOI] [Google Scholar]
- Ricciardulli T.; Gorthy S.; Adams J. S.; Thompson C.; Karim A. M.; Neurock M.; Flaherty D. W. Effect of Pd Coordination and Isolation on the Catalytic Reduction of O2 to H2O2 over PdAu Bimetallic Nanoparticles. J. Am. Chem. Soc. 2021, 143, 5445–5464. 10.1021/jacs.1c00539. [DOI] [PubMed] [Google Scholar]
- Zanella R. D. L.; Delannoy L.; Louis C. Mechanism of deposition of gold precursors onto TiO2 during the preparation by cation adsorption and deposition-precipitation with NaOH and urea. Appl. Catal., A 2005, 291 (1–2), 62–72. 10.1016/j.apcata.2005.02.045. [DOI] [Google Scholar]
- Lam Y. L.; Boudart M. Preparation of small Au-Pd particles on silica. J. Catal. 1977, 50 (3), 530–540. 10.1016/0021-9517(77)90064-1. [DOI] [Google Scholar]
- Li W.; Comotti M.; Schuth F. Highly reproducible syntheses of active Au/TiO2 catalysts for CO oxidation by deposition-precipitation or impregnation. J. Catal. 2006, 237 (1), 190–196. 10.1016/j.jcat.2005.11.006. [DOI] [Google Scholar]
- Delannoy L.; El Hassan N.; Musi A.; Le To N. N.; Krafft J. M.; Louis C. Preparation of supported gold nanoparticles by a modified incipient wetness impregnation method. J. Phys. Chem. B 2006, 110 (45), 22471–22478. 10.1021/jp062130l. [DOI] [PubMed] [Google Scholar]
- Turkevich J.; Stevenson P. C.; Hillier J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. 10.1039/df9511100055. [DOI] [Google Scholar]
- Patterson A. L. The Scherrer Formula for X-Ray Particle Size Determination. Phys. Rev. 1939, 56 (10), 978–982. 10.1103/PhysRev.56.978. [DOI] [Google Scholar]
- Adams J. S.; Chemburkar A.; Priyadarshini P.; Ricciardulli T.; Lu Y.; Maliekkal V.; Sampath A.; Winikoff S.; Karim A. M.; Neurock M.; Flaherty D. W. Solvent molecules form surface redox mediators in situ and cocatalyze O2 reduction on Pd. Science 2021, 371 (6529), 626–632. 10.1126/science.abc1339. [DOI] [PubMed] [Google Scholar]
- Ricciardulli T.; Adams J. S.; DeRidder M.; van Bavel A. P.; Karim A. M.; Flaherty D. W. H2O-Assisted O2 Reduction by H2 on Pt and PtAu Bimetallic Nanoparticles: Influences of Composition and Reactant Coverages on Kinetic Regimes, Rates, and Selectivities. J. Catal. 2021, 404, 661–678. 10.1016/j.jcat.2021.10.001. [DOI] [Google Scholar]
- Park J.; Regalbuto J. R. A Simple, Accurate Determination of Oxide PZC and the Strong Buffering Effect of Oxide Surfaces at Incipient Wetness. J. Colloid Interface Sci. 1995, 175, 239–252. 10.1006/jcis.1995.1452. [DOI] [Google Scholar]
- Noh J. S.; Schwarz J. A. Estimation of the point of zero charge of simple oxides by mass titration. J. Colloid Interface Sci. 1989, 130, 157–164. 10.1016/0021-9797(89)90086-6. [DOI] [Google Scholar]
- Wilson N. M.; Priyadarshini P.; Kunz S.; Flaherty D. W. Direct synthesis of H2O2 on Pd and AuxPd1 clusters: Understanding the effects of alloying Pd with Au. J. Catal. 2018, 357, 163–175. 10.1016/j.jcat.2017.10.028. [DOI] [Google Scholar]
- Shekhar M.; Wang J.; Lee W.; Williams W. D.; Kim S. M.; Stach E. A.; Miller J. T.; Delgass W. N.; Ribeiro F. H. Size and Support Effects for the Water-Gas Shift Catalysis over Gold Nanoparticles Supported on Model Al2O3 and TiO2. J. Am. Chem. Soc. 2012, 134 (10), 4700–4708. 10.1021/ja210083d. [DOI] [PubMed] [Google Scholar]
- Van Hardeveld R.; Hartog F. The Statistics of Surface Atoms and Surface Sites on Metal Crystals. Surf. Sci. 1969, 15 (2), 189–230. 10.1016/0039-6028(69)90148-4. [DOI] [Google Scholar]
- Domingues L.; Pinheiro C. C.; Oliveira N. M. C.; Vilelas A.; Fernandes J.; Ribeiro F. R. Estimation of catalyst deactivation parameters of ethyl tert-butyl ether (ETBE) reactors based on industrial plant data. Comput. Aided Chem. Eng. 2012, 30, 1002–1006. 10.1016/B978-0-444-59520-1.50059-2. [DOI] [Google Scholar]
- Wilson N. M.; Flaherty D. W. Mechanism for the Direct Synthesis of H2O2 on Pd Clusters: Heterolytic Reaction Pathways at the Liquid-Solid Interface. J. Am. Chem. Soc. 2016, 138 (2), 574–586. 10.1021/jacs.5b10669. [DOI] [PubMed] [Google Scholar]
- Madon R. J.; Boudart M. Experimental criterion for the absence of artifacts in the measurement of rates of heterogeneous catalytic reactions. Ind. Eng. Chem. Fundam. 1982, 21, 438–447. 10.1021/i100008a022. [DOI] [Google Scholar]
- Scott S. L. A Matter of Life(time) and Death. ACS Catal. 2018, 8 (9), 8597–8599. 10.1021/acscatal.8b03199. [DOI] [Google Scholar]
- Fujitani T.; Nakamura I.; Akita T.; Okumura M.; Haruta M. Hydrogen Dissociation by Gold Clusters. Angew. Chem., Int. Ed. 2009, 48 (50), 9515–9518. 10.1002/anie.200905380. [DOI] [PubMed] [Google Scholar]
- Fiorio J. L.; Gonçalves R. V.; Teixeira-Neto E.; Ortuño M. A.; López N.; Rossi L. M. Accessing Frustrated Lewis Pair Chemistry through Robust Gold@N-Doped Carbon for Selective Hydrogenation of Alkynes. ACS Catal. 2018, 8 (4), 3516–3524. 10.1021/acscatal.8b00806. [DOI] [Google Scholar]
- Fiorio J. L.; López N.; Rossi L. M. Gold-Ligand-Catalyzed Selective Hydrogenation of Alkynes into cis-Alkenes via H2 Heterolytic Activation by Frustrated Lewis Pairs. ACS Catal. 2017, 7 (4), 2973–2980. 10.1021/acscatal.6b03441. [DOI] [Google Scholar]
- Cano I.; Chapman A. M.; Urakawa A.; van Leeuwen P. W. N. M. Air-Stable Gold Nanoparticles Ligated by Secondary Phosphine Oxides for the Chemoselective Hydrogenation of Aldehydes: Crucial Role of the Ligand. J. Am. Chem. Soc. 2014, 136 (6), 2520–2528. 10.1021/ja411202h. [DOI] [PubMed] [Google Scholar]
- Almora-Barrios N.; Cano I.; van Leeuwen P. W. N. M.; López N. Concerted Chemoselective Hydrogenation of Acrolein on Secondary Phosphine Oxide Decorated Gold Nanoparticles. ACS Catal. 2017, 7 (6), 3949–3954. 10.1021/acscatal.7b00355. [DOI] [Google Scholar]
- Tu W. F.; Li X. L.; Wang R. Q.; Malhi H. S.; Ran J. Y.; Shi Y. L.; Han Y. F. Catalytic consequences of the identity of surface reactive intermediates during direct hydrogen peroxide formation on Pd particles. J. Catal. 2019, 377, 494–506. 10.1016/j.jcat.2019.07.047. [DOI] [Google Scholar]
- Bond G. C.; Thompson D. T. Catalysis by Gold. Catal. Rev. 1999, 41 (3–4), 319–388. 10.1081/CR-100101171. [DOI] [Google Scholar]
- Hammer B.; Norskov J. K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]
- Wilson N. M.; Schroder J.; Priyadarshini P.; Bregante D. T.; Kunz S.; Flaherty D. W. Direct synthesis of H2O2 on PdZn nanoparticles: The impact of electronic modifications and heterogeneity of active sites. J. Catal. 2018, 368, 261–274. 10.1016/j.jcat.2018.09.020. [DOI] [Google Scholar]
- Agarwal N.; Thomas L.; Nasrallah A.; Sainna M. A.; Freakley S. J.; Edwards J. K.; Catlow R. A.; Hutchings G. J.; Taylor S. H.; Willock D. The direct synthesis of hydrogen peroxide over Au and Pd nanoparticles: A DFT study. Catal. Today 2021, 381, 76–85. 10.1016/j.cattod.2020.09.001. [DOI] [Google Scholar]
- Kim M.; Han S. S. Electrochemically Modeling a Nonelectrochemical System: Hydrogen Peroxide Direct Synthesis on Palladium Catalysts. J. Phys. Chem. Lett. 2021, 12 (19), 4490–4495. 10.1021/acs.jpclett.1c01223. [DOI] [PubMed] [Google Scholar]
- Tse E. C. M.; Varnell J. A.; Hoang T. T. H.; Gewirth A. A. Elucidating proton involvement in the rate-determining step for Pt/Pd-based and non-precious-metal oxygen reduction reaction catalysts using the kinetic isotope effect. J. Phys. Chem. Lett. 2016, 7 (18), 3542–3547. 10.1021/acs.jpclett.6b01235. [DOI] [PubMed] [Google Scholar]
- Mei D.; He Z. D.; Zheng Y. L.; Jiang D. C.; Chen Y. Mechanistic and kinetic implications on the ORR on a Au(100) electrode: pH, temperature and H-D kinetic isotope effects. Phys. Chem. Chem. Phys. 2014, 16 (27), 13762–13773. 10.1039/C4CP00257A. [DOI] [PubMed] [Google Scholar]
- Stiehl J. D.; Kim T. S.; McClure S. M.; Mullins C. B. Evidence for Molecularly Chemisorbed Oxygen on TiO2 Supported Gold Nanoclusters and Au(111). J. Am. Chem. Soc. 2004, 126 (6), 1606–1607. 10.1021/ja039214h. [DOI] [PubMed] [Google Scholar]
- Podlibner B. A.; Nekrasov L. N. Electrokhimiya 1960, 5, 340. [Google Scholar]
- Wan W.; Nie X.; Janik M. J.; Song C.; Guo X. Adsorption, Dissociation, and Spillover of Hydrogen over Au/TiO2 Catalysts: The Effects of Cluster Size and Metal-Support Interaction from DFT. J. Phys. Chem. C 2018, 122 (31), 17895–17916. 10.1021/acs.jpcc.8b05482. [DOI] [Google Scholar]
- Davydova E. S.; Mukerjee S.; Jaouen F.; Dekel D. R. Electrocatalysts for hydrogen oxidation reaction in alkaline electrolytes. ACS Catal. 2018, 8 (7), 6665–6690. 10.1021/acscatal.8b00689. [DOI] [Google Scholar]
- Parks G. A. The isoelectric points of solid oxides, solid hydroxides, and aqueous hydroxo complex systems. Chem. Rev. 1965, 65 (2), 177–198. 10.1021/cr60234a002. [DOI] [Google Scholar]
- Sivadinarayana C.; Choudhary T. V.; Daemen L. L.; Eckert J.; Goodman D. W. The Nature of the Surface Species Formed on Au/TiO2 during the Reaction of H2 and O2: An Inelastic Neutron Scattering Study. J. Am. Chem. Soc. 2004, 126 (1), 38–39. 10.1021/ja0381398. [DOI] [PubMed] [Google Scholar]
- Dissanayake D.; Lunsford J. H. The direct formation of H2O2 from H2 and O2 over colloidal palladium. J. Catal. 2003, 214, 113–120. 10.1016/S0021-9517(02)00171-9. [DOI] [Google Scholar]
- Powell C. D.; Daigh A. W.; Pollock M. N.; Chandler B. D.; Pursell C. J. CO Adsorption on Au/TiO2 Catalysts: Obervations, Quantification, and Explanation of Broad-band Infrared Signal. J. Phys. Chem. C 2017, 121 (44), 24541–24547. 10.1021/acs.jpcc.7b07249. [DOI] [Google Scholar]
- Clark J. C.; Dai S.; Overbury S. H. Operando studies of desorption, reaction and carbonate formation during CO oxidation by Au/TiO2 catalysts. Catal. Today 2007, 126 (1–2), 135–142. 10.1016/j.cattod.2006.10.008. [DOI] [Google Scholar]
- Chang B.; Jang B. W.; Dai S.; Overbury S. H. Transient studies of the mechanisms of CO oxidation over Au/TiO2 using time-resolved FTIR spectroscopy and product analysis. J. Catal. 2005, 236, 392–400. 10.1016/j.jcat.2005.10.006. [DOI] [Google Scholar]
- Litke A.; Su Y.; Tranca I.; Weber T.; Hensen E. J. M.; Hofmann J. P. Role of Adsorbed Water on Charge Carrier Dynamics in Photoexcited TiO2. J. Phys. Chem. C 2017, 121 (13), 7514–7524. 10.1021/acs.jpcc.7b00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mino L.; Spoto G.; Ferrari A. M. CO2 Capture by TiO2 Anatase Surfaces: A Combined DFT and FTIR Study. J. Phys. Chem. C 2014, 118 (43), 25016–25026. 10.1021/jp507443k. [DOI] [Google Scholar]
- Yates D. J. C. Infrared Studies of the Surface Hydroxyl Groups on Titanium Dioxide, and of the Chemisorption of Carbon Monoxide and Carbon Dioxide. J. Phys. Chem. 1961, 65, 746–753. 10.1021/j100823a011. [DOI] [Google Scholar]
- Schumacher B.; Plzak V.; Kinne M.; Behm R. J. Highly active Au/TiO2 catalyst for low-temperature CO oxidation: Preparation, conditioning and stability. Catal. Lett. 2003, 89 (1/2), 109–114. 10.1023/A:1024731812974. [DOI] [Google Scholar]
- Denkwitz Y.; Zhao Z.; Hormann U.; Kaiser U.; Plzak V.; Behm R. J. Stability and deactivation of unconditioned Au/TiO2 catalysts during CO oxidation in a near-stoichiometric and O2-rich reaction atmosphere. J. Catal. 2007, 251, 363–373. 10.1016/j.jcat.2007.07.029. [DOI] [Google Scholar]
- Ruiz Puigdollers A.; Schlexer P.; Tosoni S.; Pacchioni G. Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies. ACS Catal. 2017, 7 (10), 6493–6513. 10.1021/acscatal.7b01913. [DOI] [Google Scholar]
- Zakharova M. V.; Kleitz F.; Fontaine F. Lewis acidity quantification and catalytic activity of Ti, Zr, and Al-supported mesoporous silica. Dalton Trans. 2017, 46 (12), 3864–3876. 10.1039/C7DT00035A. [DOI] [PubMed] [Google Scholar]
- Manoilova O. V.; Podkolzin S. G.; Tope B.; Lercher J.; Stangland E. E.; Goupil J. M.; Weckhuysen B. M. Surface Acidity and Bascisity of La2O4, LaOCl, and LaCl3, Characterized by Ir Spectroscopy, TPD, and DFT Calculations. J. Phys. Chem. B 2004, 108 (40), 15770–15781. 10.1021/jp040311m. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.