

Abstract

Trans-disubstituted porphyrins are highly valuable intermediates across diverse fields, but they pose a significant synthesis challenge in some cases due to scrambling and formation of complex mixtures. Conditions that minimize scrambling also lower yields, but steric hindrance around the meso-aryl substituent can effectively suppress scrambling altogether. Here we report a straightforward approach to valuable trans-A2B2 porphyrin intermediates that are otherwise very difficult to obtain, through use of removable blocking bromide substituents.
The synthetic chemistry of porphyrins was first revolutionized by Adler and Longo’s simple procedure that permitted easy access to meso-aryl porphyrins in a single step from pyrrole and aromatic aldehydes by refluxing in propionic acid open to air.1 The general methodology was subsequently refined and expanded by Lindsey and co-workers who, among many other developments, introduced higher yielding protocols that employed milder acidic conditions in organic solvents to allow the incorporation of more sensitive substrates (Scheme 1).2
Scheme 1. Direct Synthesis of meso-Aryl Porphyrins.

Unsymmetrically substituted derivatives can be accessed through a mixed condensation of pyrrole with two different aldehydes. As expected, reactions of this type produce a complex mixture with low yields of the individual products isolated after challenging separations. The reactions can be useful for synthesis of A3B type porphyrins, and we have exploited this in our own work for building symmetrical diporphyrins as precursors to multidecker systems,3 and unsymmetrical chromophore dyads.4 The strategy is rarely useful for A2B2 derivatives where both AABB (cis) and ABAB (trans) isomers are formed. The trans isomers are highly valued intermediates and have been widely employed across diverse fields including supra-/supermolecule construction and catalysis.5−7 A rational approach to the synthesis of trans-ABAB porphyrins exists6 whereby a preformed dipyrromethane is condensed with a different aldehyde. The synthesis, which follows from MacDonald’s original use of a dipyrromethane dialdehyde + dipyrromethane,8 is widely employed and is highly successful in selected cases. However, a major problem that is inevitably encountered in these syntheses is that of scrambling, whereby acidolysis of dipyrromethane and/or higher oligomers (essentially reverse condensation) leads to a set of products identical to that expected from a simple mixed condensation with two aldehydes. The reaction has been carefully and systematically investigated, and conditions have been developed to minimize scrambling. Typically, conditions that minimize scrambling have a negative impact on yield, but a key observation is that significant steric hindrance around the meso-aryl substituent can effectively suppress scrambling altogether (Scheme 2).6 In many cases this is a benefit because the same bulky substituents aid the porphyrin solubility (useful in supra- and supermolecule construction and characterization) and can affect the environment above and below the porphyrin plane around the axial position of any incorporated metal ion, a feature that can be exploited in catalysis.9
Scheme 2. Efficient Rational Synthesis of transA2B2meso-Aryl Porphyrins Using a Dipyrromethane Bearing a Sterically Hindered Aryl (e.g., Mesityl, Top) and Inefficient Synthesis Due to Scrambling When Unhindered Aryl Substituents Are Employed (Bottom)6b.

In our ongoing work on heteroleptic triple-decker porphyrin–phthalocyanine complexes3 we required efficient syntheses of differentially substituted trans ABAB-meso-aryl porphyrins suitable for further elaboration at either of the 5,15-positions only, or separately at the 5,15- and then 10,20-positions. The planned chemistry is one example where the use of sterically hindered aryl substituents cannot be used, because the hindrance required for efficient trans-porphyrin synthesis prevents the subsequent face-to-face assembly of multidecker complexes. Here even fluorine substituents on the 2,6-positions prevent face-to-face assembly and essentially only hydrogen can be accommodated. However, porphyrins bearing only remote functionality (3- and 4-positions) are valuable for elaboration in many other areas also, for example to build oligomers and polymers, and for attachment to complementary organic and inorganic species and surfaces.10 We particularly targeted trans-porphyrins bearing opposite pairs of hydroxyl and/or methoxy groups, knowing the latter can be selectively hydrolyzed to reveal phenolic residues following alkylation of the first pair of phenols and therefore provide valuable versatility for further stepwise elaboration. trans-Bis(4-methoxyphenyl)porphyrin 2 can be prepared using the dipyrromethane route, but the outcome is similar to the standard mixed porphyrin synthesis from aldehyde precursors. The reactions yielded the full mixture of scrambled products from which the dimethoxy isomeric mixture can be isolated in low (5–12%) yield (Scheme 3). The isomers cannot be separated, but NMR analysis of the mixture shows that the ratio of isomers is between 2:1 and 1:1 (Supporting Information). Hydrolysis allows careful separation of the isomeric diols and reveals the major isomer to be cis (5,10-). The presence of the activating methoxy substituent no doubt accelerates acidolysis. The reason for domination of the cis-isomer over trans is less clear, but the result is consistent with reported direct synthesis of di(4-hydroxyphenyl)porphyrins where the cis-isomer is also formed preferentially.11 The differentially substituted trans di(4-methoxyphenyl)-di(3-hydroxyphenyl)porphyrin 4 is unknown, and there is no obvious direct synthesis possible. Our brief investigation of mixed cyclizations confirmed that separation of the porphyrin mixture, and particularly the isomers, would be impractical.
Scheme 3. Attempted Synthesis of trans-Dimethoxyphenyl Porphyrin 2 Is Inefficient Due to Scrambling and Favors cis-Isomer Production (Top); Lightly Functionalized Opposite-Opposite trans-Substituted Porphyrins Like the Target 4 (Bottom) Are Not Accessible.

While multistep synthesis via cross-coupling strategies is possible,12 we reasoned that the most pragmatic solution to overcoming the scrambling issue would be to employ steric blocking groups that could later be removed after guiding efficient dipyromethane + aldehyde porphyrin synthesis without scrambling. The success of the sequence would rely on the ready availability of suitable precursors, so that the overall effectiveness of the sequence outweighed the inherent low atom economy of protecting group strategies. A survey of suitably designed benzaldehyde derivatives highlighted the potential of 3-hydroxy-2,4,6-tribromobenzaldehyde 5 which is readily available both commercially and from bromination of 3-hydroxybenzaldehyde.13 We recognized that aldehyde 5 could act as a precursor for both the complex, differentially substituted porphyrin 4, and the simple (but difficult to access) trans-dimethoxyporphyrin 2 via common intermediate trans-porphyrin 7. The sequence is shown in Scheme 4 along with the simple statistical porphyrin synthesis that was employed to generate a suitable model porphyrin 9 for the initial evaluation of deprotection (reduction) conditions.
Scheme 4. Synthesis of Lightly Substituted Trans Porphyrins Employing Removable Bromide Substituents To Prevent Scrambling.
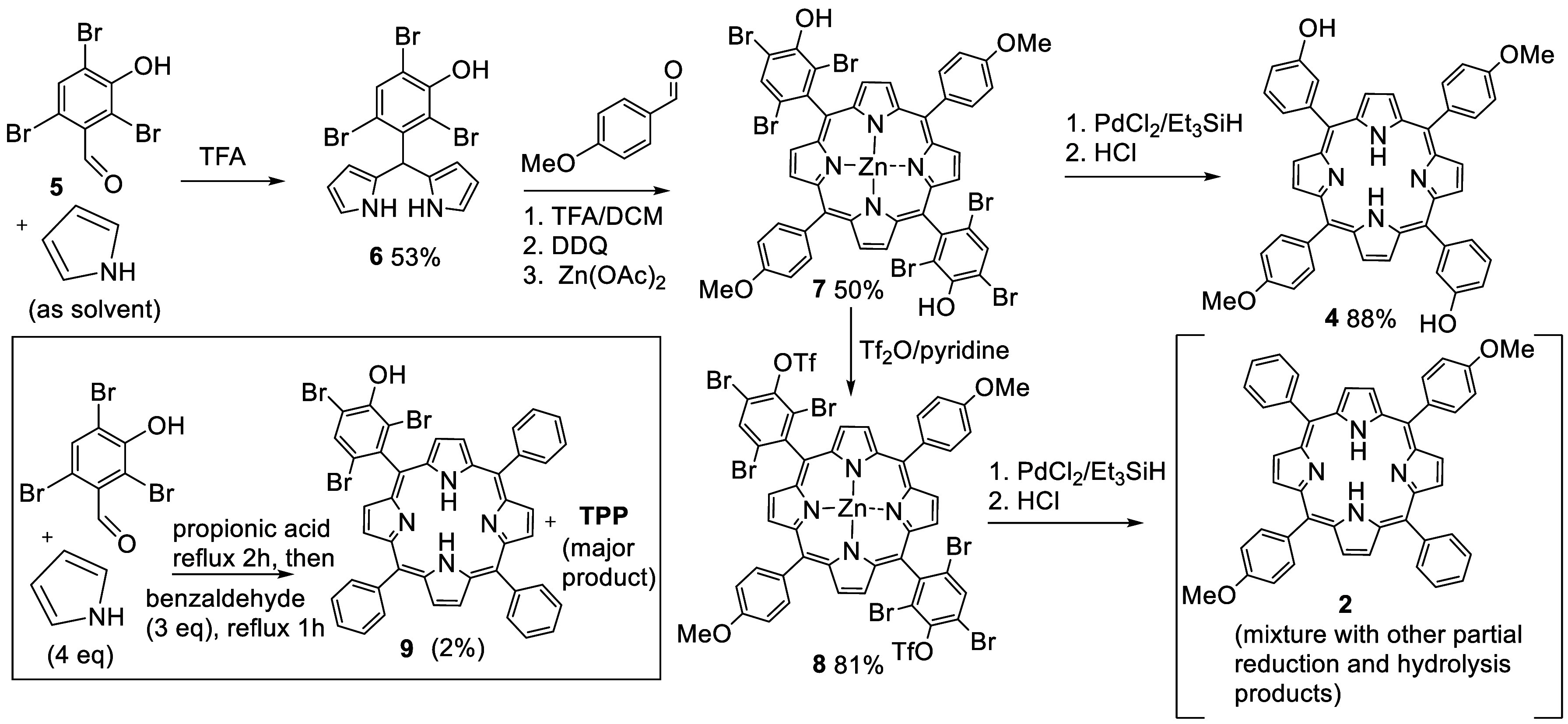
Unsurprisingly the hindered aldehyde 5 proved to be less reactive than benzaldehyde itself, resulting in a 2% yield of the 3:1 porphyrin 9 alongside tetraphenylporphyrin (TPP) as the major porphyrin product. Nevertheless, sufficient porphyrin 9 was isolated to allow investigation of known reduction conditions employing triethylsilane and palladium chloride catalyst (Scheme 5).14 Porphyrin 9 and PdCl2 (5 mol %) were heated in triethylsilane at 120 °C in a sealed tube, and the reaction was monitored periodically by analysis of aliquots by MALDI-MS. Reduction proceeded slowly, and it was clear that palladium porphyrin derivatives were also formed in the process. While this is not surprising, and the palladium can be easily removed in the acidic workup, the side reaction effectively removes the palladium catalyst from the system and slows the rate. Nevertheless, mono-(3-hydroxyphenyl)porphyrin 10 was isolated after workup (HCl) in 62% yield.
Scheme 5. Reductive Debromination of Porphyrin 9 Using Triethyl Silane and Palladium Chloride.

The main synthesis of trans-porphyrin targets began with straightforward synthesis of dipyrromethane 6 from the reaction of aldehyde 5 with excess pyrrole (used as reactant and solvent). As expected, dipyrromethane 6 proved to be relatively stable and could be stored for several weeks as a crystalline solid in the dark at 0–5 °C without any noticeable degradation. Pleasingly the synthesis of the corresponding trans-porphyrin was also smoothly achieved using 4-methoxybenzaldehyde following conditions developed by Lindsey,2 and it is worth noting this electron-rich, unhindered aldehyde represents one of the most challenging reactants in terms of suppressing scrambling during porphyrin synthesis. However, based on the previous observations during reductive debromination of the model porphyrin 9, we decided to insert zinc at the end of the reaction in order to prevent palladium sequestration during subsequent reduction. Dipyrromethane 6 and 4-methoxybenzaldehyde were therefore reacted together in DCM (0.85 mM) with TFA catalyst at 0 °C. At the end of the reaction, DDQ was added followed (after 1 h) by addition of Zn(OAc)2. Trans porphyrin 7 was isolated as the only observed porphyrin product in a 50% yield. Porphyrin 7 exists as an equilibrated mixture of atropisomers. They appear as two distinct spots by tlc but are essentially identical in 1H NMR spectroscopy. Atropisomer interconversion occurs in minutes at room temperature (tlc).
Reductive debromination of zinc porphyrin 7 was achieved smoothly by using triethylsilane and PdCl2 at 120 °C for 3–5 days. The crude reaction mixture was treated with HCl to remove zinc, neutralized, and separated to give the target differentially substituted trans porphyrin 4 in 88% yield. Alternative reduction conditions using formate and palladium, successfully employed by us in other projects for reduction of Ar–X,15 gave very slow reduction, an observation that likely reflects the effect of the electron-donating hydroxyl group in retarding palladium insertion (oxidative addition) into the Ar–Br bonds.
Conversion of intermediate 7 into trans di(methoxyphenyl) porphyrin 2 required reductive removal of both the bromide and hydroxyl substituents and was achieved by first converting the free phenols to triflates using triflic anhydride. Reduction of triflate 8 was attempted by using both PdCl2/Et3SiH and Pd/formate conditions. In each case the reduction of both triflate and bromide could be achieved, but the reactions were slow and impractical. Each reaction was monitored periodically by MALDI-MS. Under PdCl2/Et3SiH conditions, the MALDI-MS spectra clearly demonstrated initial preferential (faster) removal of bromides but very sluggish aryl triflate reduction (incomplete after 7 days at 120 °C). Reductions using Pd/formate conditions were also inefficient and slow, with evidence for competing triflate hydrolysis at long reaction times. Rather than pursue investigation of alternative conditions to achieve this, more simple, target, we elected to instead use the developed blocking strategy with an alternative bromoaryl aldehyde. Fortunately, 2,6-dibromobenzaldehyde is available. Indeed, it has been previously employed for synthesis of trans porphyrins, although not with challenging electron-rich partners.9 The convenient synthesis of trans di(methoxyphenyl) porphyrin 2 is shown in Scheme 6.
Scheme 6. Convenient Synthesis of trans Dimethoxyporphyrin 2 (Inset Shows the Single Mass Observed in MALDI-MS of a Sample before Addition of Zinc Acetate), and the X-ray Crystal Structure for Porphyrin 13 (Shown as Elipsoids at 65% Probability, H-Atoms and Molecule of Chloroform Removed for Clarity).

Dipyrromethane 11 was synthesized as reported,9 and reaction first with 4-methoxybenzaldehyde, then DDQ, and then zinc acetate gave the corresponding trans Zn-porphyrin 13 with no evidence for other scrambled products; Scheme 6 (inset) shows the MALDI-MS taken for an aliquot of the reaction mixture, before addition of zinc acetate, with essentially a single signal (cluster at m/z 990.62) corresponding to porphyrin 12. The zinc porphyrin 13 showed a strong tendency toward crystallization that complicated chromatographic purification. It was found to be more convenient to first isolate the metal-free porphyrin 12 (57%) and then insert zinc in a separate step (94%). Crystals of porphyrin 13 suitable for X-ray diffraction were obtained (CCDC 2313839), and the structure is also shown in Scheme 6. The space-filling representations clearly illustrate the effective steric blocking of meso-sites provided by the o-bromines. Reduction also proceeded smoothly using PdCl2/Et3SIH and gave the desired porphyrin 2 in 74% yield after workup (HCl) and straightforward isolation. Direct reductive debromination of metal-free trans porphyrin 12 was also investigated by using palladium on carbon. Pleasingly the reduction works well, again employing triethylsilane, giving porphyrin 2 in 60% yield.
In conclusion, we have developed a straightforward approach to valuable trans-A2B2 porphyrin intermediates that are otherwise very difficult to obtain by direct methods. Access to such porphyrins, which bear remote functionality but lack excessive steric blocking on the porphyrin core, opens the potential for wide application, particularly in super/supramolecule construction and surface grafting.
Acknowledgments
Financial support is gratefully acknowledged from Northern Border University (MA) and Almajmaah University (SA).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.3c04215.
Experimental and characterization data for synthesized compounds plus crystallography details for porphyrin 13 (PDF)
Author Contributions
M.A. and S.A. performed the experimental synthetic work with equal contribution. I.C. and D.L.H. performed X-ray crystallographic analysis. A.C. conceived and led the research and prepared the manuscript draft.
The authors declare no competing financial interest.
Supplementary Material
References
- Adler A. D.; Longo F. R.; Finarelli J. D.; Goldmacher J.; Assour J.; Korsakoff L. A simplified synthesis of meso-tetraphenylporphin. J. Org. Chem. 1967, 32, 476. 10.1021/jo01288a053. [DOI] [Google Scholar]
- Lindsey J. S.; Schreiman I. C.; Hsu H. C.; Kearney P. C.; Marguerettaz A. M. Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987, 52, 827–836. 10.1021/jo00381a022. [DOI] [Google Scholar]
- González-Lucas D.; Soobrattee S. C.; Hughes D. L.; Tizzard G. J.; Coles S. J.; Cammidge A. N. Straightforward and controlled synthesis of porphyrin-phthalocyanine-porphyrin heteroleptic triple-decker assemblies. Chem.—Eur. J. 2020, 26, 10724–10728. 10.1002/chem.202002500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan G.; Cammidge A. N.; Jones G. A.; Heisler I. A.; González-Lucas D.; Remiro-Buenamañana S.; Meech S. R. Electronic Energy Transfer in a subphthalocyanine-Zn porphyrin dimer studied by linear and nonlinear ultrafast spectroscopy. J. Phys. Chem. 2019, 123, 5724–5733. 10.1021/acs.jpca.9b04398. [DOI] [PubMed] [Google Scholar]
- Marschner S. M.; Haldar R.; Fuhr O.; Wöll W.; Bräse S. Modular synthesis of trans-A2B2-porphyrins with terminal esters: systematically extending the scope of linear linkers for porphyrin-based MOFs. Chem.—Eur. J. 2021, 27, 1390–1401. and references therein 10.1002/chem.202003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Littler B. J.; Ciringh Y.; Lindsey J. S. Investigation of conditions giving minimal scrambling in the synthesis of trans-porphyrins from dipyrromethanes and aldehydes. J. Org. Chem. 1999, 64, 2864–2872. and references therein 10.1021/jo982452o. [DOI] [PubMed] [Google Scholar]; b Geier G. R. III; Littler B. J.; Lindsey J. S. Investigation of porphyrin-forming reactions. Part 3. The origin of scrambling in dipyromethane + aldehyde condensations yielding trans-A2B2-tetraarylporphyrins. J. Chem. Soc., Perkin Trans. 2001, 2, 701–711. 10.1039/b009098k. [DOI] [Google Scholar]
- Zwick P.; Dulić D.; van der Zant H. S. J.; Mayor M. Poprhyrins as building blocks for single-molecule devices. Nanoscale 2021, 13, 15500–15525. 10.1039/D1NR04523G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Arsenault G. P.; Bullock E.; MacDonald S. F. Pyrromethanes and porphyrins therefrom. J. Am. Chem. Soc. 1960, 82, 4384–4389. 10.1021/ja01501a066. [DOI] [Google Scholar]; b Lash T. D. What’s in a name? The MacDonald condensation. J. Porphyrins Phthalocyanines 2016, 20, 855–888. 10.1142/S1088424616300147. [DOI] [Google Scholar]
- Lu H.; Li C.; Jiang H.; Lizardi C. L.; Zhang X. P. Chemoselective amination of propargylic C(sp3)-H bonds by cobalt(II)-based metalloradical catalysis. Angew. Chem.Int. Ed. 2014, 53, 7028–7032. 10.1002/anie.201400557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Walter M. G.; Rudine A. B.; Wamser C. C. Porphyrins and phthalocyanines in solar photovoltaic cells. J. Porphyrins Phthalocyanines 2010, 14, 759–792. 10.1142/S1088424610002689. [DOI] [Google Scholar]; b Day N. U.; Wamser C. C.; Walter M. G. Porphyrin polymers and organic frameworks. Polym. Int. 2015, 64, 833–857. 10.1002/pi.4908. [DOI] [Google Scholar]
- Esquivel Guzmán J. A.; Zhang H.; Rivera E.; Lavertu M.; Zhu X.-X. Porphyrin-based polyesters synthesized by enzymatic catalysis. ACS Appl. Polym. Mater. 2021, 3, 3659–3665. 10.1021/acsapm.1c00547. [DOI] [Google Scholar]
- Shi B.; Boyle R. W. Synthesis of unsymmetrically substituted meso-phenylporphyrins by Suzuki cross coupling reactions. J. Chem. Soc., Perkin Trans. 2002, 1, 1397–1400. 10.1039/b201622b. [DOI] [Google Scholar]
- Osuna M. R.; Aguirre G.; Somanathan R.; Molins E. Asymmetric synthesis of amathamides A and B: novel alkaloids isolated from Amathia wilsoni. Tetrahedron Asymm. 2002, 13, 2261–2266. 10.1016/S0957-4166(02)00586-4. [DOI] [Google Scholar]
- Villemin D.; Nechab B. Rapid and efficient palladium catalysed reduction of aryl halides by triethylsilane under microwave irradiation. J. Chem. Res. (S) 2000, 2000, 432–434. 10.3184/030823400103167976. [DOI] [Google Scholar]
- Cammidge A. N.; Crépy K. C. Application of the Suzuki reaction as the key step in the synthesis of a novel atropisomeric biphenyl derivative for use as a liquid crystal dopant. J. Org. Chem. 2003, 68, 6832–6835. 10.1021/jo034652s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.