

Abstract
Glioblastoma (GBM) is one of the deadliest types of cancer and highly refractory to chemoradiation and immunotherapy. One of the main reasons for this resistance to therapy lies within the heterogeneity of the tumor and its associated microenvironment. The vast diversity of cell states, composition of cells, and phenotypical characteristics makes it difficult to accurately classify GBM into distinct subtypes and find effective therapies. The advancement of sequencing technologies in recent years has further corroborated the heterogeneity of GBM at the single cell level. Recent studies have only begun to elucidate the different cell states present in GBM and how they correlate with sensitivity to therapy. Furthermore, it has become clear that GBM heterogeneity not only depends on intrinsic factors but also strongly differs between new and recurrent GBM, and treatment naïve and experienced patients. Understanding and connecting the complex cellular network that underlies GBM heterogeneity will be indispensable in finding new ways to tackle this deadly disease. Here, we present an overview of the multiple layers of GBM heterogeneity and discuss novel findings in the age of single cell technologies.
Keywords: Glioblastoma, GBM, myeloid cells, T cells, cancer stem cells, GBM subtypes, heterogeneity, tumor microenvironment
Introduction
Glioblastoma (GBM) is the most common and deadliest malignant brain tumor with a 5-year survival rate of only 7 percent. While overall survival for all malignant brain tumors combined increased significantly in the last decade, rates for GBM remain consistently low [1]. The current standard of care for the treatment of GBM is surgery followed by radiotherapy and chemotherapy [2]. Since the outlook for GBM patients remains grim following standard therapy, a lot of hope has been put on immunotherapeutic approaches to finally overcome this disease. However, despite recent advances in select types of cancer, resistance is commonly observed and, so far, immunotherapy falls flat in the battle against GBM [3]. The inter-patient and intra-tumoral heterogeneity, the different subtypes of GBM, the existence of multiple glioma stem cells, and the heterogeneity of the tumor microenvironment (TME) are all well documented and remain one of the main obstacles in finding an efficient cure (Figure 1) [4–6]. The broad availability of single cell-based technologies allows for the unraveling of heterogeneity at the DNA, RNA, epigenetic, and protein level at an unprecedented resolution (Figure 2). Here, we review recent studies utilizing single cell-based technologies to better understand the causes and consequences of GBM heterogeneity (Table 1).
Figure 1. The heterogenous cell types in GBM.
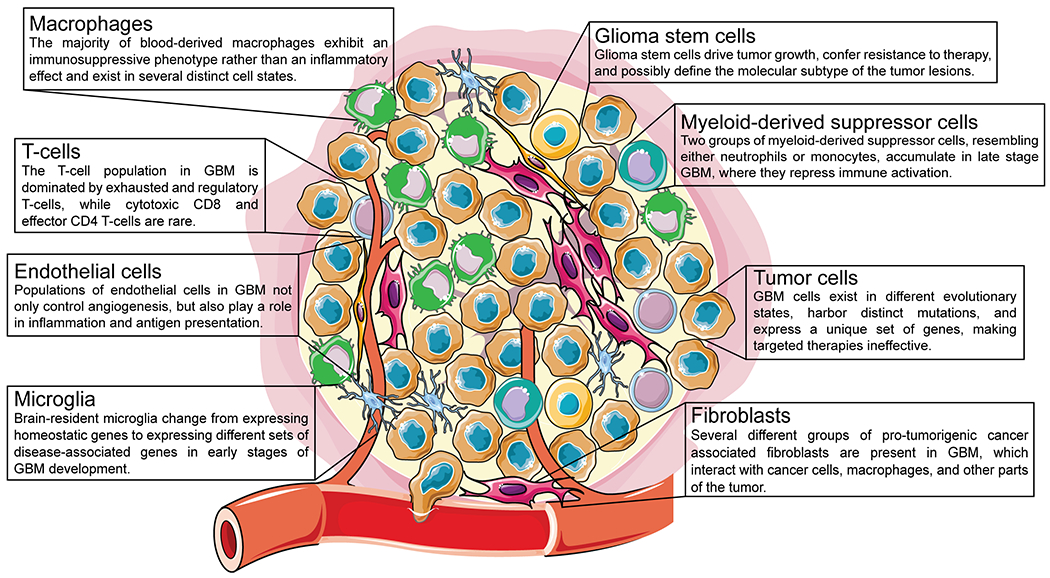
GBM consists of several distinct cell types, including macrophages, T cells, endothelial cells, microglia, glioma stem cells, myeloid-derived suppressor cells, tumor cells, and fibroblasts, all of which display multiple different sub-populations or cell states.
Figure 2. Current single cell-based technologies available to study GBM heterogeneity.
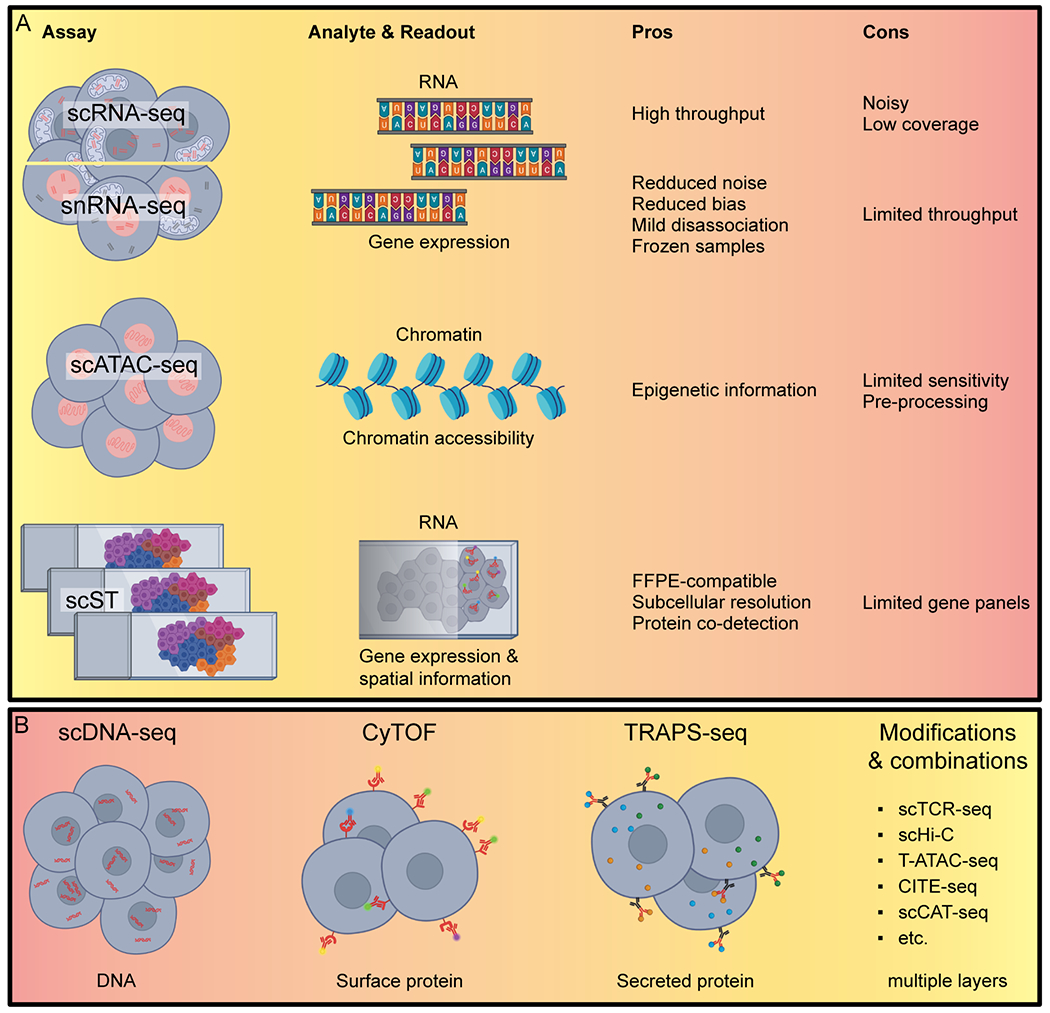
(A) Assays focused on in this review: single cell RNA sequencing (scRNA-seq), single nucleus RNA sequencing (snRNA-seq), single-cell sequencing Assay for Transposase-Accessible Chromatin (scATAC-seq), and single cell Spatial Transcriptomics (scST). (B) Other single cell assays: single cell DNA sequencing (scDNA-seq), Cytometry by Time of Flight (CyTOF), Time-Resolved Assessment of Protein Secretion from single cells by sequencing (TRAPS-seq), single cell T Cell Receptor sequencing (scTCR-seq), Transcript-indexed ATAC-seq (T-ATAC-seq), Cellular Indexing of Transcriptomes and Epitopes (CITE-seq), single cell Cap And Tail sequencing (scCAT-seq) and their read-outs.
Table 1. Single cell studies discussed in this review.
Single cell RNA sequencing (scRNA), single cell whole genome sequencing (scWGS), single-cell sequencing Assay for Transposase-Accessible Chromatin (scATAC), single-cell DNA methylome sequencing (scDNAme), single nucleus RNA sequencing (snRNA), Cytometry by Time of Flight (CyTOF), Cellular Indexing of Transcriptomes and Epitopes (CITE).
| Reference | Assay | Organism | Tissue type | Details | Accession |
|---|---|---|---|---|---|
| Larsson et al [7] | scRNA | human | cell line | U3065MG | E-MTAB-9296, E-MTAB-10615 |
| Abdelfattah et al [12] | scRNA | human | tumor | LGG, pGBM, rGBM | GSE182109 |
| LeBlanc et al [13] | scRNA, scWGS | human | tumor, explants, cell lines | pGBM | GSE173280 |
| Jacob et al [14] | scRNA | human | tumor, organoids | pGBM, rGBM | GSE141946 |
| Patel et al [18] | scRNA | human | CD45− tumor, cell lines | pGBM | GSE57872 |
| Wang et al [19] | scRNA | human | tumor | pGBM, rGBM | EGAS00001001033 |
| Neftel et al [20] | scRNA | human | CD45− tumor | adult, pediatric GBM | GSE131928 |
| Liu et al [21] | scRNA, scATAC | human | tumor | DMG | GSE184357 |
| Couturier et al [23] | scRNA | human | tumor, GSCs, normal | GBM | EGAS00001004422 |
| Yuan et al [26] | scRNA | human | tumor | pGBM, rGBM | GSE103224 |
| Müller et al [27] | scRNA | human | tumor | pGBM | EGAS00001001900 |
| Wu et al [31] | scRNA | human | tumor | multifocal GBM | HRA002913, HRA002914 |
| Yu et al [32] | scRNA | human | tumor | WHO grade II - IV | HRA000179 |
| Darmanis et al [33] | scRNA | human | tumor, peritumor | pGBM | GSE84465 |
| Johnson et al [37] | scRNA, scDNAme | human | tumor | pGBM, rGBM | EGAS00001005300 |
| Bhaduri et al [39] | scRNA, snRNA | human | tumor, PTPRZ1+ tumor, normal | GBM | PRJNA579593, SRP132816 |
| Richards et al [40] | scRNA, snRNA | human | GSCs | pGBM, rGBM | EGAS00001004656 |
| Wang et al [47] | scRNA, snRNA, scATAC | human | tumor | pGBM | EGAS00001003845 |
| Guilhamon et al [48] | scATAC | human | tumor | pGBM | GSE139136 |
| Wang et al [49] | snRNA, scATAC | human | tumor | pGBM, rGBM | EGAS00001004909 |
| Müller et al [54] | scRNA | human | tumor, CD11b+ | pGBM, LGG | EGAS00001002185 |
| Goswami et al [65] | scRNA, CyTOF | human | CD45+ tumor | pGBM | PRJNA588461 |
| Chen et al [66] | scRNA | human | tumor | GBM | GSE141383 |
| Mathewson et al [77] | scRNA | human | T-cells tumor | pGBM, rGBM | GSE163108 |
| Ravi et al [78] | scRNA | human | CD45+ CD3+ tumor | pGBM | DOI 10.17605/OSF.IO/4Q32E |
| Jain et al [86] | scRNA | human | CAFs tumor | pGBM | GSE132825 |
| Xie et al [88] | scRNA | human | CD31+ tumor & peritumor | pGBM | GSE162631 |
| Jessa et al [34] | scRNA | human, mouse | tumor, normal | non-GBM | EGAS00001003368, GSE133531 |
| Pombo Antunes et al [57] | scRNA, CITE | human, mouse | CD45+ tumor | pGBM, rGBM; orthotopic | GSE163120 |
| Zamler et al [61] | scRNA | human, mouse | CD45+ tumor | GEMM, orthotopic | GSE147275 |
| Yeo et al [38] | scRNA | mouse | CD45− & CD45+ tumor | GEMM | GSE196175 |
| Van Hove et al [55] | scRNA | mouse | CD45+ normal | WT, GEMM | GSE128855 |
| Hammond et al [56] | scRNA | mouse | CD45+ CD11b+ Cx3cr1+ normal | Microglia | GSE121654 |
| Yeini et al [60] | scRNA | mouse | CD11b+, CD45+ CD11b− tumor | orthotopic | GSE156663 |
| Rajendran et al [63] | scRNA | mouse | CD45+ tumor | non tumor, LGG, HGG | GSE221440 |
| Sawant et al [79] | scRNA | mouse | Tregs tumor | GEMM | GSE126184 |
Inter-patient heterogeneity
Overall cell identity in GBM is highly dynamic and plastic changes in cell states are patient specific. Moreover, therapeutic intervention results in complex changes in the landscape of GBM, further contributing to its heterogeneity [7]. GBM is not driven by a single driver oncogene and instead can be the result of a plethora of different mutations. The most commonly mutated genes in GBM patients include TP53, EGFR, PDGFR, NF1, IDH1, PTEN, and MDM2 [8, 9]. Another important contributing factor to the development and heterogeneity of GBM is the number and location of copy number variations (CNV) which can vary significantly between GBM patients. The overall impact of CNV in GBM on patient survival, however, remains controversial [10, 11]. Single cell RNA sequencing (scRNA-seq) of a cohort of GBM patients confirmed the heterogeneity of glioma cells states in different patients. Furthermore, the TME also showed diversity between patients regarding specific populations of immune cells. While all patients contributed to all myeloid cell states, there were significant differences in the overall number of T cells and the contribution to different T cell states per patient. CD4 T cells and regulatory T cells (Tregs) were absent or barely detected, and the number of CD8 T cells and Natural Killer (NK) cells varied significantly in a subset of patients. Whole exome sequencing of some of the samples from this cohort revealed both shared mutations, such as loss of chromosome 10/10q and gain of chromosome 7/7q, as well as unique mutational patterns in genes, such as RB1, PRIM1, KLF12, and KDM2B across all patients [12]. Inter-patient heterogeneity makes it difficult to find and establish unified treatment approaches and calls for more individualized treatment plans. Since much of this genetic heterogeneity is retained in explanted organoids, these cultures can be utilized to elucidate the impact of different genetic alterations on the progression of GBM and their role in resistance to therapy [13, 14]. To account for the TME and enable the screening of immunotherapeutic drugs, biopsies can be grown in perfusion reactors, screened with immune checkpoint inhibitors, and monitored by multiplexed microscopy to measure their efficacy at the single cell level [15]. Together, the wide range of inter-patient heterogeneity diminishes hopes for the development of broadly applicable therapies and emphasizes the need for more individualized therapeutic approaches.
GBM subtype heterogeneity
GBM has been divided into different subtypes to better categorize its diverse phenotypes and predict the response to treatment and offer more reliable prognosis [16]. Based on the mutational landscape in combination with transcriptional profiles established from bulk RNA sequencing experiments, GBM has been classified into four separate subtypes (classical, mesenchymal, proneural, and neural). These subtypes are mainly distinguished by the expression levels of EGFR, NF1, and PDGFRA/IDH1 and show different sensitivity to drug treatment [17]. Further sequencing experiments of GBM specimens helped narrow down the previously established profiles into only three main subtypes (proneural, classical, and mesenchymal), by ruling out the neuronal subtype as a normal neuronal lineage contamination. RNA sequencing analysis also revealed that GBM subtypes may intra-convert to a different subtype during tumor progression and recurrence. For example, the mesenchymal subtype is frequently observed in recurrent GBM, while primary tumors from the same patients may display proneural or classical subtypes [18]. However, scRNA-seq experiments revealed that GBM patients harbored cells of all described subtypes and neoplastic cells can exhibit strong differences in their gene expression profiles in regards to the cell cycle, hypoxia, the complement/immune response, and oligodendrocyte function [19]. Subsequent scRNA-seq experiments described four malignant transcriptional states in GBM: oligodendrocyte progenitor-like (OPC), neural-progenitor-like (NPC), astrocyte-like (AC), and mesenchymal-like (MES), with cells progressing through these subtypes during tumor progression [20]. Interestingly, these cell states were also reported in diffuse midline gliomas, further emphasizing their importance [21]. Genomic alterations alone are insufficient to explain the emergence of distinct subtypes of GBM. Accordingly, an unbiased computational approach of biological traits of single cells revealed four new subtypes, distinguished by their neurodevelopmental (neuronal and proliferative/progenitor) or metabolic (mitochondrial and glycolytic/plurimetabolic) traits. These subtypes are also marked by varying degrees of sensitivity to inhibitors, such as inhibitors of oxidative phosphorylation, potentially making this classification system a more valuable tool in choosing the most effective therapeutic approach [22]. Regardless of subtype or origin, the development of GBM in different patients still seems to follow a common developmental progression. Simultaneous sequencing of tumor tissue and normal fetal brain cells revealed remarkable similarities. GBM cells follow a trilinear hierarchy and develop along specific conserved neurodevelopmental gene programs. Moreover, evidence of rapidly dividing progenitor cells supports the notion of a GBM stem cell population [23]. Despite the advancement of our understanding of the different subtypes of GBM, however, cells clearly mapped to one cellular state still exhibit a remarkable plastic potential and their identity can be further influenced by the TME [20].
Intra-tumoral heterogeneity
The intra-tumoral heterogeneity of GBM is well documented and poses a major problem in therapeutically challenging GBM. This is also highlighted by the disappointing results of clinical trials of Rindopepimut, a cancer vaccine targeting the oncogenic mutant of EGFR - EGFRvIII. Despite its power to recognize the mutant EGFR protein and its success in preliminary studies, it fell flat in phase III clinical trials [24]. While the exact reasons for this remain unclear, the heterogeneity of tumor cells might play an important role in this intrinsic resistance to targeted agents. Indeed, patients not only display different subtypes within one tumor but also harbor different copy number alterations in genes such as EGFR, PTEN, and PDGFR, ultimately resulting in several cell lineages coexisting within the same tumor [25]. The identification of transformed cells according to their gene expression is not trivial and, accordingly, not one single marker is sufficient to identify these cells. Analysis of scRNA-seq data revealed that most high grade glioma cells express high levels of SOX2, suggesting that SOX2 expression could be utilized as a universal marker of transformed glioma cells [26]. To study tumor evolution, Mueller et al. established detailed phylogenies of PDGF- and EGF-driven tumors. They mapped out sequentially acquired mutations, associated these mutations with a specific subtype, and finally correlated them to the tumor’s invasive potential [27]. In accordance with these results, a second independent study found that mutations of EGFR accumulate at a later stage, while alterations in PI3KCA occur at an early stage in the developmental process of GBM. This work also confirmed that a favorable response to therapeutic intervention is correlated with the genetic similarity of the tumor [28]. Single cell-derived clones established from the same GBM patient can still exhibit significantly different proliferative and differentiative capabilities as well as responses to drug treatment, highlighting the plasticity of individual cells within the same tumor [29]. This finding leads to the conclusion that multiple biopsies might be necessary to reach a conclusive representation of the tumor. In multifocal GBM, tumor lesions all develop from a common precursor and undergo parallel evolution and, thus, constitute an ideal model to study natural tumor evolution while ruling out any treatment-induced effects [30]. In a recent study, based on scRNA-seq of eight independent multifocal GBM patients, the authors delineate some of the key events in the acquisition of heterogeneity and describe a natural evolution signature (NES) that increases over the course of natural evolution and is defined by the activity of genes, such as HIF1A, FOSL2, and ANXA1 [31]. In an independent cohort, multi-sector biopsies followed by scRNA-seq confirmed that cells from different biopsies of the same patient showed vast differences in gene expression and varying subtype affiliation [32]. Another study, which collected samples from the tumor core and surrounding peripheral tissues that had been infiltrated by tumor cells, further corroborated the different expression profiles of GBM cells within the same patient. Importantly, however, the authors found that the infiltrating GBM cells in all patients shared certain gene expression patterns involved in size regulation, adhesion, developmental processes, and apoptosis. This points at a common strategy of GBM cells, independent of their origin, to infiltrate their surroundings and might represent a potential target for successful therapeutic intervention [33].
Cancer stem cell heterogeneity
While the exact origin of GBM and other tumors of the brain remains unclear, analysis of scRNA-seq data points at specific niches, cellular hierarchies, and transcriptional programs reminiscent of normal development [34]. The presence of cancer stem cells (CSCs) in GBM is largely undisputed, details regarding their evolution and functions, however, remain controversial. Glioblastoma stem cells (GSCs) have been linked to the resistance to chemo- and radiotherapy, and their role in shaping and interacting with the TME has strong implications for the applicability and success of immunotherapy [35]. GSCs are directly associated with the OPC, NPC, AC, and MES cellular states and their respective stem cell markers CD133, CD24, Nestin, and CD44 [36]. The actual number of GSCs and their exact characteristics remain difficult to determine, since classical stem cell markers, such as SOX2, can be widely expressed amongst tumor cells. Johnson et al. proposed three different cell states amongst all glioma cells, two of which relate to a stem cell-like status: 1.) differentiated-like, 2.) stem-like, and 3.) proliferating stem-like tumor cells. Interestingly, they also found that the proportion of SOX2-positive glioma stem-like cells correlates with IDH mutation status. IDH-mutant tumors are enriched for stem-like tumor cells, while IDH wildtype tumors consist mainly of differentiated tumor cells [37]. The presence of these stem-like cells was also confirmed in an independent cohort of GBM patients [38]. Since a single marker is insufficient to distinguish stem-like cells from real GSCs, another study attempted to identify GSCs by looking at the simultaneous expression of stem cell-linked genes, such as PROM1, FUT4, and L1CAM, in addition to SOX2. Within a single tumor there exist several distinguishable subtypes that differ in the expression level of such marker genes, and these heterogeneous populations of GSCs contribute differently to the tumor growth of individual lesions [39]. GSCs are associated with one of two distinct transcriptional programs, related to neuronal development or an inflammatory wound response, respectively. Importantly, matured cancer cells retain the basics of these transcriptional programs throughout their development, making clinical intervention through modulating these transcriptional programs an attractive option [40]. Specific stem cell and subtype-specific stem-like cell markers, however, remain elusive, and some datasets fail to recapitulate the presence of different types of GSCs and stem-like cells [12]. The reasons for this might lie within their scarcity, the mutational landscape, and differences in tissue preparation or other technical limitations.
Epigenetic heterogeneity
Epigenetic gene regulation plays an important role in the development, progression, and prognosis of GBM. Accordingly, GBM can be separated into eight epigenetically distinct classes in regard to the DNA methylation status [41]. Furthermore, epigenetic silencing via promoter methylation of the O6-methylguanine DNA methyltransferase (MGMT) is predictive of a favorable response to therapy and one of the main prognostic markers of survival [42]. The genetic landscape of a tumor cell might determine its affiliation to the different cell states characterized by the status of overall chromatin accessibility [43]. Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) experiments showed that the epigenetic modification of enhancer regions drives the aggressive phenotype of the mesenchymal and classical subtypes, while active enhancers in the proneural subtype regulate genes associated with less detrimental signaling events [44]. Further ChIP-seq analyses revealed that active promoters overlap significantly between GBM patients. Interestingly, however, non-regulatory regions seem to show a higher degree of heterogeneity between GBM patients than promoter regions, emphasizing the importance of delineating the overall chromatin status across GBM patients and individual lesions [45]. Epigenetic markers and chromatin accessibility can reliably discern stem cells from mature cells, both under normal conditions and in cancer [46]. However, the analysis of H3K27 acetylation ChIP-seq data highlights the epigenetic heterogeneity of GSCs, resulting in no clear consensus on the developmental trajectory of GBM. GSCs can be separated into two groups, distinct from neural stem cells, depending on the active enhancer landscapes and associated transcriptional programs, such as MAPK/ERK signaling [47]. Analysis of combined scRNA-seq and single cell assay for transposase-accessible chromatin sequencing (scATAC-seq) suggests that GSCs develop along a single axis, where they gradually transition from a mesenchymal to an intermediary classical and finally to a proneural phenotype [48]. Further scATAC-seq experiments revealed three epigenetically distinct populations of GSCs in a small pool of GBM patients, each featuring a unique set of epigenetically activated transcription factors. The invasive state correlates with lower survival, while the reactive and constructive states exhibit reduced invasive potential and associate with improved prognosis [49]. Another single cell multi-omics study found a significant difference in the chromatin landscape of primary and recurrent tumors, with the latter featuring a shift to the more detrimental mesenchymal subtype and an over-representation of open chromatin peaks belonging to components of the AP-1 signaling complex [50]. Together, the epigenetic landscape of GBM cells reflects their heterogeneity and might hold the key to identifying key signaling events and potential clinical targets.
Tumor microenvironment heterogeneity
The overall microenvironment in GBM can be separated into three distinct regions: the hypoxic tumor niche at the core, the perivascular tumor niche, and the vascular-invasive tumor niche, all exhibiting a unique milieu populated by different cell populations [51]. Since the emergence of immunotherapy as the fourth pillar in cancer treatment and the advancement of high throughput single cell technologies, remarkable progress has been made in delineating which cell populations of the TME are crucial for a positive response to immune checkpoint blockade (ICB). Importantly, these analyses revealed that the main targets of ICB, such as PD-1 and CTLA-4, are only sporadically expressed in GBM, and their expression in the immune compartment is highly heterogenous [19, 33]. This might explain the disappointing results of ICB clinical trials for GBM patients and underlines the need to further categorize the TME and identify the cellular states of the immune cell clusters. The number of different cell populations making up the TME is vast and there is still no clear consensus as to which cell types play a fundamental role in cancer development and resistance to therapy (Table 2). The main components of the CD45+ immune population in GBM are myeloid cells, dendritic cells (DCs), T cells, NK cells, B-cells, neutrophils, and mast cells. Most of these populations harbor several subtypes or states, resulting in a highly diverse and heterogenous environment. The TME of GBM is regarded as one of the most immunosuppressive, which is mainly attributed to the presence of a large number of myeloid cells and Tregs [52, 53]. Myeloid cells, which make up around 45% of the total tumor mass of human GBM, are a highly diverse group of cells in GBM, encompassing microglia, macrophages, monocytes, granulocytes, myeloid-derived suppressor cells (MDSCs), and more [12, 54]. Interestingly, female patients seem to have a higher proportion of myeloid cells than male patients, hinting at potential sex differences in GBM [12]. The sex-specific difference in GBM is also reflected by the distribution of monocytic and granulocytic MDSCs in a GBM animal model [55]. Recent studies have only begun to unravel the multiple layers of interactions between the tumor cells and the different subtypes of myeloid cells and T cells and their impact on therapy.
Table 2. The main cell types of the glioblastoma tumor microenvironment and their cell states.
Microglia-derived tumor associated macrophages (Mg-TAMs), monocyte-derived tumor associated macrophages (Mo-TAMs), myeloid-derived suppressor cells (MDSCs), cancer associated fibroblasts (CAFs), endothelial cells (ECs), high grade glioma-associated microglia (HGG-AM), polymorphonuclear (PMN) MDSCs.
| Mg-TAMs [38, 59, 60] |
Mo-TAMs [12, 59, 63, 64, 66, 68, 80] |
MDSCs [38, 72] |
T-cells [79, 80, 81] |
CAFs [88, 89] |
ECs [90] |
|---|---|---|---|---|---|
| Phagocytic | Phagocytic | Early stage | Effector CD4 | Immature | Quiescent |
| IFN+ | MHCII-high | PMN / granulocytic | Cytotoxic CD8 | Mature | Angiogenic |
| HGG-AM | IFN activated | Monocytic | Exhausted | Steady state-like | Intermediate |
| Proliferative | Proliferating | Regulatory | Mechanoresponsive | Inflammatory | |
| Inflammatory | Cycling | Immunomodulatory | Immune activated | ||
| M2-like | |||||
| SPP1 expressing | |||||
| CD73-high | |||||
| MARCO-high | |||||
| S100A4-high | |||||
| HMOX1 expressing |
Microglia heterogeneity
It can be difficult to clearly distinguish infiltrating bone marrow-derived macrophages (BMDMs) from tissue resident microglia, both of which are present in significant numbers in GBM. Muller et al. utilized scRNA-seq data to elucidate and compile gene signatures that reliably identify either population [56]. Microglia can be identified by the expression of genes, such as TMEM119, P2RY12, and CX3CR1 but can exhibit significant differences in gene expression profiles depending on underlying mutations or progression status [57, 58]. Pombo-Antunes et al. confirmed the presence of both BMDMs and microglia in a cohort of human GBM patients and subsequently labeled them as tissue resident microglia-derived macrophages (Mg-TAMs) and monocyte-derived brain macrophages (Mo-TAMs) (Figure 3). Tumor associated microglia downregulate homeostatic signature genes and start expressing disease-associated markers in GBM patients. Subsets of Mg-TAMs express a phagocytic/lipid signature or an IFN-signaling related gene program [59]. Contrary to the normal brain, microglia in GBM can start to proliferate and adapt a pro-inflammatory phenotype, expressing genes, such as IL1α, IL1β, TNF, and CXCL10, which are gradually replaced by CXCL13 and AP-1 signaling during tumor progression [38]. These proliferating, pro-inflammatory microglia have been termed high-grade glioma-associated microglia (HGG-AM) and can be activated by cancer cells via TGFβ-signaling. Disruption of TGFβ-signaling reduces the number of HGG-AM and inhibits tumor growth [60]. mTORC1 activation in microglia leads to the secretion of anti-inflammatory cytokines, thereby inhibiting peripheral T cell infiltration and promoting tumor growth via mTOR-mediated immunosuppression [61]. Another way tumor cells activate and exploit microglia to facilitate tumor growth involves adhesion molecules, such as P-selectin. Pharmacological inhibition of SELP/PSGL-1 interaction reduces tumor growth and highlights the potential of utilizing microglia as therapeutic targets in the fight against GBM [62].
Figure 3. Origin and cell state of macrophages in GBM.
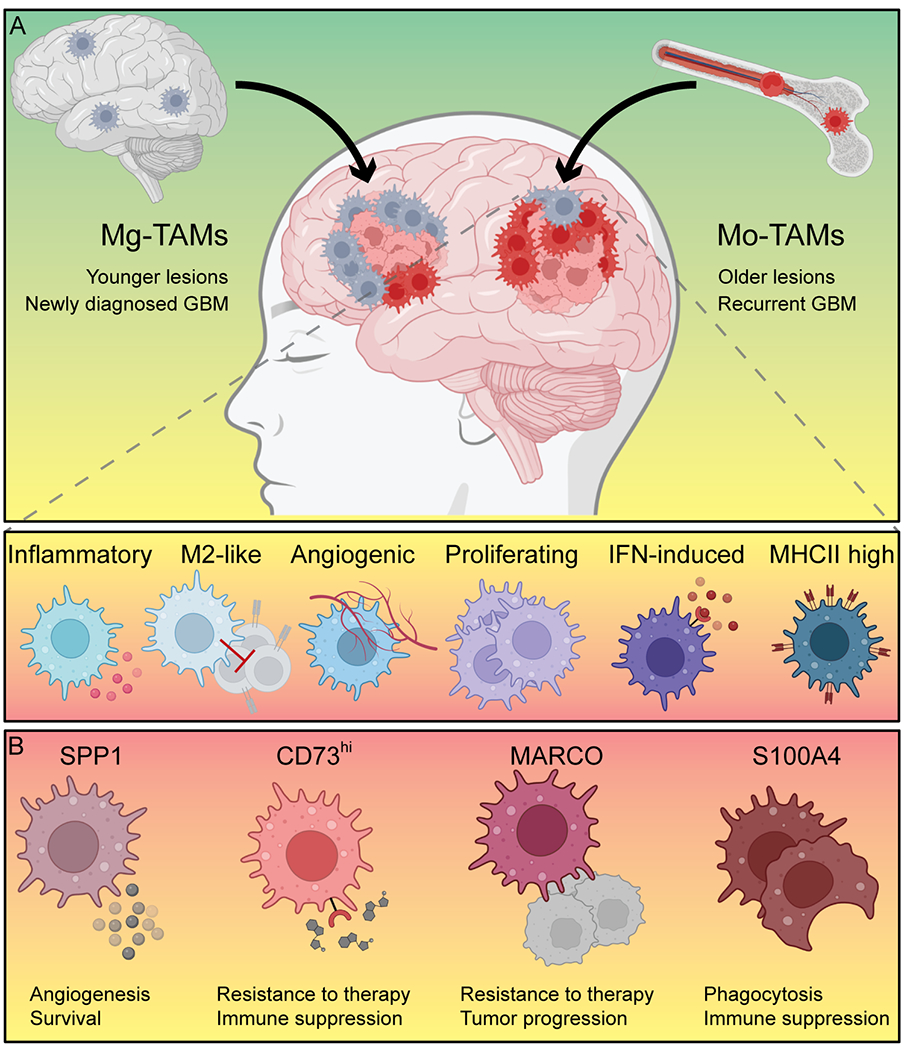
(A) Microglia-derived macrophages (Mg-TAMs) are primarily found in newly diagnosed patients and younger lesions of multifocal GBM, while monocyte-derived brain macrophages (Mo-TAMs) accumulate in recurrent GBM and older lesions. Mo-TAMS can be roughly divided into inflammatory, M2-like, angiogenic, proliferating, interferon (IFN)-induced, and MHCII-high expressing Mo-TAMs. (B) Several novel marker genes, such as SPP1, CD73, MARCO, and S100A4, have been discovered recently that serve distinct functions in the evolution and progression of GBM and might constitute targets for therapeutic intervention.
Macrophage heterogeneity
In vitro, macrophages can be readily divided into non-activated M0, pro-inflammatory M1, and immunosuppressive M2 macrophages. In vivo, however, the divide is far less clear, and tumor-associated macrophages (TAMs) cannot be reliably assigned to one of these subsets, as they often express both M1 and M2 markers simultaneously. Moreover, while some clusters of Mg-TAMs and Mo-TAMs share characteristics, such as an IFN-induced state, phagocytic potential, or proliferative capabilities, they still differ greatly in surface receptor expression and cytokine secretion [59]. Mo-TAMs are highly diverse, and it remains difficult to distinguish the sub-populations that support tumor growth and immune evasion from those that inhibit cancer development. Overall, the populations of blood-derived macrophages include MHCII-high expressing, IFN activated, proliferating, inflammatory, and FN1/ARG1 expressing M2-like macrophages. Additionally, SPP1-expressing macrophages, which have previously been shown to support glioma cell survival and angiogenesis and are a potential target for therapy, have also been identified in a human scRNA-seq dataset (Figure 3) [63, 64]. Macrophages co-evolve with the tumor and adjust their transcriptional program and marker gene expression over time [31, 65]. GBM cells can induce the expression of CD73 by TAMs, turning them into CD8 T cell disrupting agents, and the simultaneous activation of CD73 and CD39 leads to the production of immunosuppressive adenosine [66]. TAMs expressing high levels of CD73 are refractory to anti-PD-1 treatment and can inhibit the effectiveness of immunotherapeutic approaches. The absence of CD73hi BMDMs significantly increases the therapeutic effect of anti CTLA-4 and anti-PD-1 treatment in mice [67]. Another tumor-promoting subset of BMDMs expresses high amounts of the scavenger receptor MARCO. This population is associated with a mesenchymal subtype, poor clinical outcomes, and is negatively correlated with anti-PD-1 treatment [68]. Furthermore, MARCO-expressing macrophages were also identified in small cell lung cancer samples, suggesting that MARCOhi macrophages are a more common occurrence, making them a potentially valuable target for clinical intervention [69]. S100A4 is a protein that has been long associated with metastasis and advocated as a prognostic marker [70]. New evidence suggests that S100A4 is not only expressed by cancer cells but also by pro-tumorigenic myeloid cells, and regulatory and exhausted T cells. Subsequently, S100A4 expression is associated with poor prognosis in GBM patients and its deletion in glioma-associated myeloid cells in a GBM mouse model significantly boosts their phagocytic capabilities to suppress glioma growth (Figure 3) [12].
Myeloid-derived suppressor cell heterogeneity
MDSCs are a heterogeneous group of cells which have been observed and implicated in the pathology of many types of cancer. In humans, they have been divided into three separate groups, distinguished by the expression of several surface markers: Early-stage MDSCs (E-MDSCs) represent a population including myeloid progenitor cells, while polymorphonuclear MDSCs (PMN-MDSCs) or granulocytic MDSCs (G-MDSCs) and monocytic MDSCs (M-MDSCs) more closely resemble neutrophils and monocytes, respectively [71]. Since the identification of MDSCs has, so far, relied primarily on cell surface marker expression, it remains challenging to clearly distinguish this cell type in scRNA-seq experiments. E-MDSCs are attracted by and co-localize with stem-like tumor cells mainly in the pseudopalisading region where they support glioma cell survival. M-MDSCs develop from E-MDSCs and elicit a strong inhibitory effect on T cell proliferation [72]. They are primarily detected in late stage GBM, compared to the normal brain and early tumor lesions, and show increased epigenetic immunoediting, resulting in increased immune evasion [73]. Both E- and M-MDSCs exhibit increased proliferation and an activation of catabolic and anabolic metabolism, distinguishing them from BMDMs. The elevated expression of several related transporter proteins may represent a way to target these cells in a therapeutic setting [72]. PMN-MDSCs also accumulate in GBM tissues, while they are completely absent in the normal brain, further confirming the conclusion of earlier reports that showed the expansion of PMN-MDSCs during GBM progression [38, 74, 75]. Cytometry by time of flight (CyTOF) analysis coupled with scRNA-seq of immune cells infiltrating IDH1 wildtype and mutant mouse tumors revealed that PMN-MDSCs accumulate mostly in IDH1 wildtype tumors, potentially explaining the increased sensitivity to therapy and more favorable prognosis of IDH mutant patients. The mechanism by which IDH1 mutant gliomas restrain the immunosuppressive function of PMN-MDSCs also involves epigenetic reprogramming, further hinting at the importance of epigenetic regulation in the prevalence and activity of all MDSCs [76–78]. Other scRNA-seq studies managed to identify MDSCs as part of the overall macrophage population, however, specific marker gene expression and the distinction of different subtypes of MDSCs remain elusive in this context [12]. Increasing the resolution of scRNA-seq technologies and alternative approaches will lead to better detection and characterization of MDSCs in GBM and allow for the development of therapies targeting this immunosuppressive cell type.
T cell heterogeneity
As the main effector cell type of current cancer immunotherapies, it is crucial to elucidate the different T cell populations and their functions in GBM development. Four main populations of T cells exist in GBM: CD8 T cells, CD4 conventional T cells, CD4 regulatory T cells, and cycling T cells, all of which can be further subdivided into clusters representing different T cells states, including populations expressing cytotoxic, interferon, memory, or stress-related gene signatures [79]. However, expression of PDCD1/PD1 within the T cell population is comparatively low, which might explain the limited effect of therapeutic approaches targeting PD1/PD-L1 in GBM [12]. Cytotoxic CD8 T cells express a robust NK cell signature, suggesting a potentially strong anti-tumor function. However, despite low levels of known inhibitory receptors, such as PDCD1, CTLA4, and LAG3, their actual prowess in killing tumor cells is limited, hinting at the presence of additional receptors. One of these appears to be CD161, which is widely expressed on the surface of cytotoxic CD8 and effector CD4 T cells, but not Tregs. Furthermore, both glioma cells and suppressive myeloid cells robustly express CLEC2D, the main ligand for CD161 [79]. Exhausted T cells and Tregs make up a significant part of the overall T cell population in GBM patients. Tregs can suppress the activity of effector T cells via the secretion of IL10. Recently, the combination of scRNA-seq and spatial transcriptomics revealed a sub-population of HMOX1-expressing macrophages located in close proximity to exhausted T cell populations within the mesenchymal tumor regions. These HMOX1+ macrophages also secrete IL10 and thereby inactivate potentially tumor-suppressive T cells. Inhibition of JAK/STAT signaling can reverse this effect and alleviate T cell exhaustion [80, 81]. GBM tumors are generally considered cold tumors. Being able to therapeutically increase the infiltration of T cells into the brain of GBM patients would greatly increase the chances of successful immunotherapy. Kollis et al. identified several receptors (CCR2, CCR5, CXCR3/4/6, and CD49a/b) in tumor infiltrating T cells, absent in matched peripheral blood samples, that are responsible for T cell infiltration [82]. Whether this finding can be utilized in the clinics to increase T cell infiltration, however, remains to be seen.
Other cell types
B-cells are only found in relatively small numbers in GBM and most analyses of scRNA-seq data do not offer a detailed picture of this cell type [12, 38]. There is some evidence that B-cells may play a bigger role in GBM than anticipated, and B-cell-targeted therapies show promising results in preliminary experiments [83]. However, GBM cells can also promote the conversion of B-cells into immunosuppressive regulatory B-cells, again highlighting a potential heterogeneity that needs to be taken into consideration [84]. In tumors in organs such as the lung, dendritic cells can be divided into several subtypes [85]. While there are different sub-populations present in the normal brain (plasmacytoid DCs and two populations of conventional DCs) [57], the situation in GBM remains unclear. NK cells can also accumulate in GBM tissues and might represent an attractive target for immunotherapy. However, tumor infiltrating NK cells exhibit reduced lytic potential once they come into direct contact with the tumor. Once there, GSCs activate TGF-β signaling in NK cells in an αv integrin–dependent manner, leading to dysfunctional NK cells [86]. The accumulation of fibroblasts has long been associated with most types of cancers, their function in tumor development and their exact cellular identities, however, are strongly context dependent [87]. A new study identified multiple distinct clusters of cancer associated fibroblasts (CAFs), including immature, mature, steady state-like, mechanoresponsive, and immunomodulatory CAFs, in scRNA-seq data, making fibroblasts one of the most diverse cell populations in GBM. CAFs interact with GSCs via PDGF-β, TGF-β, osteopontin, and HGF and can induce an immunosuppressive phenotype in macrophages via the production of the extra domain A fibronectin variant [88]. These data are supported by another report showing the positive effect of fibroblast-derived FN1 on the migration and invasive potential of GBM cells [89]. Overcoming the blood brain barrier (BBB) is one of the main obstacles in the efficient delivery of therapeutic agents to battle GBM. To better understand the endothelial cells (ECs) that make up the BBB, Xie et al. isolated and sequenced single endothelial cells from the tumor and peripheral tissues of human patients and identified five distinct clusters: quiescent, angiogenic, intermediate, inflammatory and antigen presenting, and immune activated ECs. Importantly, while overall gene expression amongst the different clusters was heterogeneous, the expression of BBB-related transporter genes was mostly retained in the tumor samples [90]. Fibroblast activation protein (FAP) is not only highly expressed on tumor cells but also on endothelial cells and pericytes. FAP expression is near ubiquitous in and around blood vessels in GBM tissues, whereas it is mostly absent in the blood vessels of normal tissues, hinting at a potential novel target for therapy [91]. Other cell types present in GBM include mast cells and perivascular cells, their potential heterogeneity and the consequences on the development of GBM, however, remain to be uncovered [38].
Heterogeneity and therapy
The chromatin state and gene expression profile of tumor cells found in recurrent GBM more often resemble the mesenchymal subtype [50]. In the era of ICB, choosing the most promising course of treatment for a patient strongly depends on the state of the TME. However, how therapy reshapes the TME in GBM in regard to changes in the cellular states of single immune cells remains largely unexplored. In cancers, such as non-small cell lung cancer and basal and squamous cell carcinoma, scRNA-seq of tumor samples before and after immune checkpoint therapy, and recurrent tumors revealed stark differences in the immune cell compartment, especially within the T cell and macrophage populations [92, 93]. In GBM, patients receiving anti-PD-1 treatment show an enrichment in PTEN mutations and an associated increase in immunosuppressive gene signatures in non-responders, while responders show elevated alterations associated with the MAPK signaling pathway. Additionally, PTEN wildtype samples exhibit an increase in infiltrating T cells that is not observed in PTEN-mutant samples, which instead exhibit slightly increased levels of macrophage infiltration [94]. Patients receiving anti-PD1 neoadjuvant therapy not only show increased survival rates compared to patients receiving adjuvant therapy following surgery, but also exhibit significant differences regarding the immune cell populations. Neoadjuvant treatment is associated with an increase in T cell and interferon-γ-related gene expression, clonal expansion of T cells, reduced levels of PD-1 on peripheral blood T cells, and a decrease in the number of monocytic cells [95]. Similar results regarding the modulation of immune cells were obtained in a phase II clinical trial with nivolumab, even though no clinical benefit was observed [96]. One study performing scRNA-seq in treatment-naïve and recurrent GBM reported that Mg-TAMs accumulate in newly diagnosed GBM, whereas Mo-TAMs are mainly found in recurrent cancers and highly hypoxic environments. The expression profile of TAMs found in recurrent patient samples shows elevated monocyte chemotaxis, IFN signaling, and phagocytosis, while newly diagnosed samples show increases in gene signatures related to glucocorticoid treatment, a drug often given prior to surgery [59]. This observation seems to be recapitulated in an independent set of GBM patients, however, additional analyses are needed to uncover its implications on the development of GBM and on the choice of therapeutic approach [12]. Together, these data emphasize the value of elucidating how individual therapies reshape the TME. It will be important to build a repository delineating changes at the single cell level in GBM patients undergoing therapy, to better understand and predict the response to treatment, develop novel drugs, and test combinatorial treatments.
Challenges and future direction
While scRNA-seq has drastically improved our understanding of GBM heterogeneity, there remain several limitations that reduce the insight gained by such studies. The initial preparation of single cells can artificially alter cell numbers, depending on the method of choice and the cell/tissue type, and scRNA-seq data is overall noisy, features a low capture efficiency, and suffers from batch effect [97]. Therefore, scRNA analyses rely heavily on bioinformatical pipelines to counteract these restrictions. These pipelines include alignment, quality control, quantification, dimensionality reduction, clustering, expression analysis, and more, all of which introduce a significant amount of uncertainness into the data [98]. Other events that cannot be readily captured by current scRNA-seq technologies include the study of allelic expression, alternative splicing, and other RNA editing mechanisms [97]. Since RNA abundance does not always translate to protein expression and activity, approaches that allow for the simultaneous quantification of cell surface proteins, in addition to transcriptomic data, need to be explored [99]. Tumors are, at least in part, a product of their surroundings, the spatial distribution of tumor cells and the surrounding cells of the TME and tissue organization in the context of gene expression, however, remain difficult to study. Spatial transcriptomics enables the study of cell state distribution and gene expression while maintaining information about the tissue architecture, offering novel insights into the cellular networks in GBM [100]. The emergence of spatial transcriptomics at single cell resolution will no doubt lead to additional discoveries and the development of new therapeutics in the next decade. The option to profile previously preserved patient samples and not rely on fresh tissue unlocks a vast repository and should lead to the fast accumulation of data. Establishing databases will allow for the analysis of the cellular landscape of human patients at a rapid pace and the identification of additional inhibitory receptor/ligand pairs between the tumor core, myeloid population, and effector T cells, ultimately leading to the improvement of immunotherapeutic agents and novel combinatorial treatments. However, many single cell technologies and bioinformatical pipelines to analyze the large amounts of data created are still in their early days, and further improvements and fine-tuning will enable a more complete analysis and comprehensive characterization of GBM tumors. Lastly, the identification of underappreciated cell populations and cell states, that have eluded detection and characterization owing to their comparatively low numbers or ambiguous marker expression, will provide further ways to tackle the difficulties encountered in the battle against GBM.
Conclusion
Since the first histological descriptions our understanding of GBM has steadily increased and has changed our perception of this disease. The genetic landscape alone is insufficient to explain the emergence of heterogeneity, leaving interactions among the tumor cells and the environment as logical explanation. While it is still difficult to reliably classify GBM into distinct subtypes, it seems clear that subtype distribution is directly tied to cancer cell state and TME status. Moreover, immune and cancer stem cell states, invasive potential, and therapy-induced changes are all connected in the context of heterogeneity. With recent advances in single cell technologies, the vast heterogeneity of both the tumor cells and the tumor microenvironment has become apparent. Further increasing their resolution, making them more widely accessible, and adding new functions and pipelines to analyze the data will help answer many of the remaining questions in the fight against GBM. Together, characterizing and understanding the heterogeneity of GBM will be crucial in order to improve treatment options and determine the optimal therapeutic approach for individual patients.
Acknowledgements
This work was in part supported by grants from the National Cancer Institute (1RO1CA231349, 1RO1CA262798) and the Brown Center for Immunotherapy at Indiana University Melvin and Bren Simon Comprehensive Cancer Center. Illustrations were in part generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license and BioRender.com.
Footnotes
The authors declare no potential conflicts of interest.
Competing Interests
The authors declare that no conflicts of interest exist.
Data Availability
N/A
References
- 1.Miller KD, Ostrom QT, Kruchko C, Patil N, Tihan T, Cioffi G et al. Brain and other central nervous system tumor statistics, 2021. CA: A Cancer Journal for Clinicians 2021; 71: 381–406. [DOI] [PubMed] [Google Scholar]
- 2.Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nature Reviews Clinical Oncology 2021; 18: 170–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nature Reviews Clinical Oncology 2018; 15: 422–442. [DOI] [PubMed] [Google Scholar]
- 4.Puchalski RB, Shah N, Miller J, Dalley R, Nomura SR, Yoon JG et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018; 360: 660–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444: 756–760. [DOI] [PubMed] [Google Scholar]
- 6.Lan X, Jörg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017; 549: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsson I, Dalmo E, Elgendy R, Niklasson M, Doroszko M, Segerman A et al. Modeling glioblastoma heterogeneity as a dynamic network of cell states. Molecular Systems Biology 2021; 17: e10105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res 1993; 53: 2736–2739. [PubMed] [Google Scholar]
- 9.Brennan Cameron W, Verhaak Roel GW, McKenna A, Campos B, Noushmehr H, Salama Sofie R et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013; 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchwald ZS, Tian S, Rossi M, Smith GH, Switchenko J, Hauenstein JE et al. Genomic copy number variation correlates with survival outcomes in WHO grade IV glioma. Scientific Reports 2020; 10: 7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Liu Z, Li J, Huang T, Wang Y, Chang L et al. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Scientific Reports 2019; 9: 16058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdelfattah N, Kumar P, Wang C, Leu J-S, Flynn WF, Gao R et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nature Communications 2022; 13: 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeBlanc VG, Trinh DL, Aslanpour S, Hughes M, Livingstone D, Jin D et al. Single-cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter- and intratumor heterogeneity. Cancer Cell 2022; 40: 379–392.e379. [DOI] [PubMed] [Google Scholar]
- 14.Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020; 180: 188–204.e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shekarian T, Zinner CP, Bartoszek EM, Duchemin W, Wachnowicz AT, Hogan S et al. Immunotherapy of glioblastoma explants induces interferon-γ responses and spatial immune cell rearrangements in tumor center, but not periphery. Science Advances 2022; 8: eabn9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9: 157–173. [DOI] [PubMed] [Google Scholar]
- 17.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017; 32: 42–56.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014; 344: 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019; 178: 835–849.e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu I, Jiang L, Samuelsson ER, Marco Salas S, Beck A, Hack OA et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nature Genetics 2022; 54: 1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garofano L, Migliozzi S, Oh YT, D’Angelo F, Najac RD, Ko A et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nature Cancer 2021; 2: 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couturier CP, Ayyadhury S, Le PU, Nadaf J, Monlong J, Riva G et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nature Communications 2020; 11: 3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. The Lancet Oncology 2017; 18: 1373–1385. [DOI] [PubMed] [Google Scholar]
- 25.Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proceedings of the National Academy of Sciences 2013; 110: 4009–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan J, Levitin HM, Frattini V, Bush EC, Boyett DM, Samanamud J et al. Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Medicine 2018; 10: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Müller S, Liu SJ, Di Lullo E, Malatesta M, Pollen AA, Nowakowski TJ et al. Single-cell sequencing maps gene expression to mutational phylogenies in PDGF- and EGF-driven gliomas. Molecular Systems Biology 2016; 12: 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J-K, Wang J, Sa JK, Ladewig E, Lee H-O, Lee I-H et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nature Genetics 2017; 49: 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A 2015; 112: 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abou-El-Ardat K, Seifert M, Becker K, Eisenreich S, Lehmann M, Hackmann K et al. Comprehensive molecular characterization of multifocal glioblastoma proves its monoclonal origin and reveals novel insights into clonal evolution and heterogeneity of glioblastomas. Neuro Oncol 2017; 19: 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu L, Wu W, Zhang J, Zhao Z, Li L, Zhu M et al. Natural Coevolution of Tumor and Immunoenvironment in Glioblastoma. Cancer Discov 2022; 12: 2820–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu K, Hu Y, Wu F, Guo Q, Qian Z, Hu W et al. Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. National Science Review 2020; 7: 1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Reports 2017; 21: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jessa S, Blanchet-Cohen A, Krug B, Vladoiu M, Coutelier M, Faury D et al. Stalled developmental programs at the root of pediatric brain tumors. Nature Genetics 2019; 51: 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev 2015; 29: 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suvà ML, Tirosh I. The Glioma Stem Cell Model in the Era of Single-Cell Genomics. Cancer Cell 2020; 37: 630–636. [DOI] [PubMed] [Google Scholar]
- 37.Johnson KC, Anderson KJ, Courtois ET, Gujar AD, Barthel FP, Varn FS et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nature Genetics 2021; 53: 1456–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeo AT, Rawal S, Delcuze B, Christofides A, Atayde A, Strauss L et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nature Immunology 2022; 23: 971–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhaduri A, Di Lullo E, Jung D, Müller S, Crouch EE, Espinosa CS et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020; 26: 48–63.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richards LM, Whitley OKN, MacLeod G, Cavalli FMG, Coutinho FJ, Jaramillo JE et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nature Cancer 2021; 2: 157–173. [DOI] [PubMed] [Google Scholar]
- 41.Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al. DNA methylation-based classification of central nervous system tumours. Nature 2018; 555: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. New England Journal of Medicine 2005; 352: 997–1003. [DOI] [PubMed] [Google Scholar]
- 43.Nikolic A, Singhal D, Ellestad K, Johnston M, Shen Y, Gillmor A et al. Copy-scAT: Deconvoluting single-cell chromatin accessibility of genetic subclones in cancer. Science Advances 2021; 7: eabg6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall AW, Battenhouse AM, Shivram H, Morris AR, Cowperthwaite MC, Shpak M et al. Bivalent Chromatin Domains in Glioblastoma Reveal a Subtype-Specific Signature of Glioma Stem Cells. Cancer Research 2018; 78: 2463–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stępniak K, Machnicka MA, Mieczkowski J, Macioszek A, Wojtaś B, Gielniewski B et al. Mapping chromatin accessibility and active regulatory elements reveals pathological mechanisms in human gliomas. Nature Communications 2021; 12: 3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wainwright EN, Scaffidi P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017; 3: 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J Exp Med 2019; 216: 1071–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Babikir H, Müller S, Yagnik G, Shamardani K, Catalan F et al. The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation. Cancer Discovery 2019; 9: 1708–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guilhamon P, Chesnelong C, Kushida MM, Nikolic A, Singhal D, MacLeod G et al. Single-cell chromatin accessibility profiling of glioblastoma identifies an invasive cancer stem cell population associated with lower survival. eLife 2021; 10: e64090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Jung J, Babikir H, Shamardani K, Jain S, Feng X et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nature Cancer 2022; 3: 1534–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hambardzumyan D, Bergers G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015; 1: 252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 2008; 216: 15–24. [DOI] [PubMed] [Google Scholar]
- 53.Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol 2012; 14: 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Kleer I, Willems F, Lambrecht B, Goriely S. Ontogeny of Myeloid Cells. Frontiers in Immunology (Review) 2014; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bayik D, Zhou Y, Park C, Hong C, Vail D, Silver DJ et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discovery 2020; 10: 1210–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 2017; 18: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nature Neuroscience 2019; 22: 1021–1035. [DOI] [PubMed] [Google Scholar]
- 58.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019; 50: 253–271.e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nature Neuroscience 2021; 24: 595–610. [DOI] [PubMed] [Google Scholar]
- 60.Liu H, Sun Y, Zhang Q, Jin W, Gordon RE, Zhang Y et al. Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep 2021; 36: 109718. [DOI] [PubMed] [Google Scholar]
- 61.Dumas AA, Pomella N, Rosser G, Guglielmi L, Vinel C, Millner TO et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. The EMBO Journal 2020; 39: e103790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yeini E, Ofek P, Pozzi S, Albeck N, Ben-Shushan D, Tiram G et al. P-selectin axis plays a key role in microglia immunophenotype and glioblastoma progression. Nature Communications 2021; 12: 1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zamler DB, Shingu T, Kahn LM, Huntoon K, Kassab C, Ott M et al. Immune landscape of a genetically engineered murine model of glioma compared with human glioma. JCI Insight 2022; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen P, Zhao D, Li J, Liang X, Li J, Chang A et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019; 35: 868–884.e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rajendran S, Hu Y, Canella A, Peterson C, Gross A, Cam M et al. Single-cell RNA sequencing reveals immunosuppressive myeloid cell diversity during malignant progression in a murine model of glioma. Cell Reports 2023; 42: 112197. [DOI] [PubMed] [Google Scholar]
- 66.Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao C-C et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nature Neuroscience 2019; 22: 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nature Medicine 2020; 26: 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen AX, Gartrell RD, Zhao J, Upadhyayula PS, Zhao W, Yuan J et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Medicine 2021; 13: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chan JM, Quintanal-Villalonga Á, Gao VR, Xie Y, Allaj V, Chaudhary O et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021; 39: 1479–1496.e1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Helfman DM, Kim EJ, Lukanidin E, Grigorian M. The metastasis associated protein S100A4: role in tumour progression and metastasis. British Journal of Cancer 2005; 92: 1955–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nature Immunology 2018; 19: 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jackson C, Cherry C, Bom S, Dykema AG, Thompson E, Zheng M et al. Distinct Myeloid Derived Suppressor Cell Populations Promote Tumor Aggression in Glioblastoma. bioRxiv 2023. [Google Scholar]
- 73.Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021; 184: 2454–2470.e2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HM, ter Laan M et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol 2015; 74: 390–400. [DOI] [PubMed] [Google Scholar]
- 75.Dubinski D, Wölfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol 2016; 18: 807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alghamri MS, McClellan BL, Avvari RP, Thalla R, Carney S, Hartlage CS et al. G-CSF secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Science Advances 2021; 7: eabh3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tran AN, Lai A, Li S, Pope WB, Teixeira S, Harris RJ et al. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol 2014; 16: 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Han S, Liu Y, Cai SJ, Qian M, Ding J, Larion M et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. British Journal of Cancer 2020; 122: 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mathewson ND, Ashenberg O, Tirosh I, Gritsch S, Perez EM, Marx S et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 2021; 184: 1281–1298.e1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ravi VM, Neidert N, Will P, Joseph K, Maier JP, Kückelhaus J et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nature Communications 2022; 13: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nature Immunology 2019; 20: 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kollis PM, Ebert LM, Toubia J, Bastow CR, Ormsby RJ, Poonnoose SI et al. Characterising Distinct Migratory Profiles of Infiltrating T-Cell Subsets in Human Glioblastoma. Frontiers in Immunology (Original Research) 2022; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee-Chang C, Miska J, Hou D, Rashidi A, Zhang P, Burga RA et al. Activation of 4-1BBL+ B cells with CD40 agonism and IFNγ elicits potent immunity against glioblastoma. Journal of Experimental Medicine 2020; 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Han S, Feng S, Ren M, Ma E, Wang X, Xu L et al. Glioma cell-derived placental growth factor induces regulatory B cells. The International Journal of Biochemistry & Cell Biology 2014; 57: 63–68. [DOI] [PubMed] [Google Scholar]
- 85.Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019; 50: 1317–1334.e1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. The Journal of Clinical Investigation 2021; 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kalluri R. The biology and function of fibroblasts in cancer. Nature Reviews Cancer 2016; 16: 582–598. [DOI] [PubMed] [Google Scholar]
- 88.Jain S, Rick JW, Joshi RS, Beniwal A, Spatz J, Gill S et al. Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. The Journal of Clinical Investigation 2023; 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Galbo PM, Liu Y, Peng M, Wei Y, Madsen AT, Graff S et al. Functional Contribution of Cancer-Associated Fibroblasts in Glioblastoma. bioRxiv 2022: 2022.2004.2007.487495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xie Y, He L, Lugano R, Zhang Y, Cao H, He Q et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 2021; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ebert LM, Yu W, Gargett T, Toubia J, Kollis PM, Tea MN et al. Endothelial, pericyte and tumor cell expression in glioblastoma identifies fibroblast activation protein (FAP) as an excellent target for immunotherapy. Clinical & Translational Immunology 2020; 9: e1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020; 182: 1232–1251.e1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nature Medicine 2019; 25: 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nature Medicine 2019; 25: 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nature Medicine 2019; 25: 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nature Medicine 2019; 25: 470–476. [DOI] [PubMed] [Google Scholar]
- 97.Chen G, Ning B, Shi T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Frontiers in Genetics (Review) 2019; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology 2019; 15: e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H et al. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods 2017; 14: 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ravi VM, Will P, Kueckelhaus J, Sun N, Joseph K, Salié H et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 2022; 40: 639–655.e613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
N/A