

ABSTRACT
Bovine enterovirus (BEV) consisting of enterovirus species E (EV-E) and F (EV-F) is the causative agent associated with respiratory and gastrointestinal diseases in cattle. Here, we reported the characterization, genetic diversity, and recombination of novel BEV strains isolated from the major cattle-raising regions in China during 2012–2018. Twenty-seven BEV strains were successfully isolated and characterized. Molecular characterization demonstrated that the majority of these novel BEV strains (24/27) were EV-E, while only few strains (3/27) were EV-F. Sequence analysis revealed the diversity of the circulating BEV strains such as species and subtypes where different species or subtype coinfections were detected in the same regions and even in the same cattle herds. For the EV-E, two novel subtypes, designated as EV-E6 and EV-E7, were revealed in addition to the currently reported EV-E1–EV-E5. Comparative genomic analysis revealed the intraspecies and interspecies genetic exchanges among BEV isolates. The representative strain HeN-B62 was probably from AN12 (EV-F7) and PS-87-Belfast (EV-F3) strains. The interspecies recombination between EV-E and EV-F was also discovered, where the EV-F7-AN12 might be from EV-E5 and EV-F1, and EV-E5-MexKSU/5 may be recombined from EV-F7 and EV-E1. The aforementioned results revealed the genetic diversity and recombination of novel BEV strains and unveiled the different BEV species or subtype infections in the same cattle herd, which will broaden the understanding of enterovirus genetic diversity, recombination, pathogenesis, and prevention of disease outbreaks.
IMPORTANCE
Bovine enterovirus (BEV) infection is an emerging disease in China that is characterized by digestive, respiratory, and reproductive disorders. In this study, we first reported two novel EV-E subtypes detected in cattle herds in China, unveiled the coinfection of two enterovirus species (EV-E/EV-F) and different subtypes (EV-E2/EV-E7, EV-E1/EV-E7, and EV-E3/EV-E6) in the same cattle herds, and revealed the enterovirus genetic exchange in intraspecies and interspecies recombination. These results provide an important update of enterovirus prevalence and epidemiological aspects and contribute to a better understanding of enterovirus genetic diversity, evolution, and pathogenesis.
KEYWORDS: bovine enterovirus, isolation, recombination, cattle, molecular analysis
INTRODUCTION
The Enterovirus genus of the family Picornaviridae is taxonomically grouped into 12 Enterovirus (A–L) and 3 Rhinovirus (A–C) species (1). These viruses share similar characteristics that are nonenveloped and icosahedral with the positive-stranded RNA genome (2, 3). They infect humans and animals and cause respiratory and gastrointestinal diseases (4–8). Bovine enterovirus (BEV) consists of two enterovirus species, the species E (EV-E) and F (EV-F) (7–9). EV-E and EV-F are separately divided into five subtypes (E1–E5) and seven subtypes (F1–F7) currently, respectively (3). The genome of enterovirus is approximately 7,100 to 7,450 nucleotides long, containing a single open-reading frame (ORF) that encodes a polyprotein. The polyprotein is later cleaved into three precursor proteins (P1, P2, and P3). The P1 protein is proteolytically processed into four structural viral proteins (VP1, VP2, VP3, and VP4), forming the viral capsid. The P2 precursor protein is cleaved to yield three nonstructural proteins (2A, 2B, and 2C). The P3 precursor protein is cleaved to generate four nonstructural proteins (3A, 3B, 3C, and 3D) (2, 3).
Recombination is a pervasive process generating diversity in most viruses, especially in Enterovirus. Recombination between serotypes for EVs was first observed in poliovirus in 1991 (10) and since then in a wide range of human enteroviruses. Among the members of Enterovirus A, the recombination occurred quite commonly in spite of lower recombination rates in EV-A71 in comparison with other subtypes in this species (11–13). Recombination rates in Enterovirus B are high, and a vast number of recombinant forms have been identified in this species (14–16). Recently, Luo et al. reported that BEV strains (GXNN2204 and GXGL2215) were, respectively, obtained from the genomic recombination of EV-E4 with EV-F3, and EV-E2 to EV-E4 (17). Recombination events in EVs are almost exclusively detected at the edges of the structural P1 region and within the nonstructural 5′UTR, P2, or P3 regions (14, 18). Although recombination has been shown to occur across the whole genome, not all recombinants may be viable. Thus, there would be selection of recombinants with breakpoints at particular locations (19). Since recombination is usually demonstrated by showing phylogeny inconsistencies (20, 21), the recombinant forms (RFs) can then be assigned based on clustering of the strains in the nonstructural region (16, 22), and breakpoints can be detected by performing bootscanning (21, 23). According to the criteria by the International Committee on Taxonomy of Viruses, members of species in the genus Enterovirus should have a high degree of amino acid identity (amino acid >70% in the polyprotein and amino acid >60% in P1) (1). In 1999, Oberste et al. found that the VP1 coding region carries major neutralization epitopes among the capsid proteins and is likely the optimal region for virus identification and molecular typing (24). Analysis of the VP1 sequences has been applied to genotype the most enteroviruses such as EV-A71 (11), CVA16 (25), CVA6 (26), and EV-E3 (27). Recently, Zell et al. suggested that enteroviruses were classified based on the amino acid sequence identities of the VP1 capsid protein, of which 70% to 85% were defined as heterologous types and >90% as homologous types (28).
In this study, we have isolated 27 strains of the BEV from cattle herds with diarrhea in various regions in China from 2012 to 2018, identified two novel EV-E subtypes in cattle herds in China, unveiled the coinfection of two enterovirus species and different subtypes in the same cattle herds, and revealed the enterovirus genetic exchange in intraspecies and interspecies recombination. These results provide an important update of the enterovirus prevalence and epidemiological aspects and contribute to a better understanding of enterovirus genetic diversity, evolution, and pathogenesis.
RESULTS
Isolation and characterization of the novel BEV strains
To isolate the potential BEV, the fecal samples collected from cattle herds with diarrhea in Jilin, Henan, and Ningxia provinces during 2012–2018 were processed. After infection, the Vero cells began to show cytopathic effects (CPEs) as early as 12 hours post-infection (hpi) and detached at 24–36 hpi compared to the uninfected cells (Fig. S1A and B). The isolates were further identified by RT-PCR with BEV-specific primers (5′UTR). After sequence analyses, the amplified fragments were turned out to contain the sequences of BEV. Information on these novel bovine virus strains is listed in Table 1. The TCID50 titers for the isolated BEV strains were determined to range from 102/0.1 to 105.5/0.1 mL (Fig. S1C).
TABLE 1.
Molecular typing of enterovirus E and F isolates collected in China
| Isolate | Location | EV type | Years | Isolation source | Host | Available sequence |
|---|---|---|---|---|---|---|
| HeN-A2 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| HeN-A3 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| HeN-B72 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| HeN-B78 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| HeN-B81 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| HeN-B83 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| HeN-B84 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| HeN-C4 | Henan | E | 2018 | Feces | Cattle | Complete genome |
| JL-40 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY1 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY7 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY12 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY29 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY33 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY37 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY40 | Jilin | E | 2018 | Serum | Cattle | Complete genome |
| JL-JY41 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| JL-JY42 | Jilin | E | 2018 | Feces | Cattle | Complete genome |
| NX-FY40 | Ningxia | E | 2018 | Feces | Cow | Complete genome |
| NX-b-DR47 | Ningxia | E | 2018 | Feces | Cattle | Complete genome |
| JL-HY61 | Jilin | E | 2013 | Feces | Cattle | Complete genome |
| JL-HY68 | Jilin | E | 2012 | Feces | Cattle | Complete genome |
| JL-DH13 | Jilin | E | 2012 | Feces | Cattle | Complete genome |
| JL-GZL4 | Jilin | E | 2014 | Feces | Cattle | Complete genome |
| HeN-A12 | Henan | F | 2018 | Feces | Cattle | Complete genome |
| HeN-B62 | Henan | F | 2018 | Feces | Cattle | Complete genome |
| HeN-YR91 | Henan | F | 2018 | Feces | Cattle | Complete genome |
Decoding of the complete genome sequences for novel BEV strains
To decode the complete sequences for the novel BEV strains, a series of primers were designed individually for each strain and were used to amplify the genomic sequences (Table S1). After sequencing and joining the overlapped PCR-amplified fragments, the complete genomes for these viruses were obtained. Analyses showed that the lengths of complete genome sequences for these novel BEV strains varied from 7,433 to 7,470 nucleotides; the lengths of the 3′UTR were relatively diverse, ranging from 93 nucleotides to 136 nucleotides; and the lengths of the 5′UTR ranged from 810 nucleotides to 828 nucleotides with the relative conserved sequence. Additionally, the ORFs of these novel strains were highly conserved, ranging from 6,501 nucleotides to 6,531 nucleotides and encoding a polyprotein of 2,167–2,177 amino acids. Moreover, all strains shared a typical picornavirus genome organization. The newly identified viral strains and their GenBank accession numbers are listed in Table 2.
TABLE 2.
The complete genome sequence of the newly identified virus strains
| Strains | Complete genome (bp) | 5'UTR (bp) | ORF (bp) | 3'UTR (bp) | Accession no. |
|---|---|---|---|---|---|
| HeN-A2 | 7,459 | 810 | 6,531 | 118 | MN598008 |
| HeN-A3 | 7,453 | 814 | 6,525 | 114 | MN598009 |
| HeN-B72 | 7,457 | 815 | 6,525 | 117 | MN598012 |
| HeN-B78 | 7,453 | 812 | 6,525 | 116 | MN598013 |
| HeN-B81 | 7,457 | 815 | 6,525 | 117 | MN598014 |
| HeN-B83 | 7,451 | 812 | 6,525 | 114 | MN598015 |
| HeN-B84 | 7,469 | 819 | 6,525 | 125 | MN598016 |
| HeN-C4 | 7,469 | 821 | 6,525 | 123 | MN598017 |
| JL-40 | 7,434 | 816 | 6,525 | 93 | MN598019 |
| JL-JY1 | 7,453 | 818 | 6,525 | 110 | MN598024 |
| JL-JY7 | 7,455 | 816 | 6,525 | 114 | MN598025 |
| JL-JY12 | 7,444 | 814 | 6,525 | 102 | MN598026 |
| JL-JY29 | 7,450 | 815 | 6,525 | 107 | MN598027 |
| JL-JY33 | 7,454 | 813 | 6,525 | 116 | MN598028 |
| JL-JY37 | 7,465 | 814 | 6,525 | 126 | MN598029 |
| JL-JY40 | 7,457 | 814 | 6,525 | 118 | MN598030 |
| JL-JY41 | 7,461 | 814 | 6,528 | 119 | MN598031 |
| JL-JY42 | 7,467 | 815 | 6,525 | 127 | MN598032 |
| NX-FY40 | 7,470 | 817 | 6,531 | 122 | MN598039 |
| NX-b-DR47 | 7,449 | 810 | 6,525 | 114 | MN598037 |
| JL-HY61 | 7,462 | 816 | 6,531 | 115 | MN598022 |
| JL-HY68 | 7,463 | 828 | 6,531 | 104 | MN598023 |
| JL-DH13 | 7,451 | 817 | 6,531 | 103 | MN598020 |
| JL-GZL4 | 7,447 | 815 | 6,528 | 104 | MN598021 |
| HeN-A12 | 7,433 | 822 | 6,504 | 107 | MN598010 |
| HeN-B62 | 7,458 | 821 | 6,501 | 136 | MN598011 |
| HeN-YR91 | 7,460 | 825 | 6,504 | 131 | MN598018 |
The new BEV strains shared high sequence identity with EV-E and EV-F reference strains
To determine the sequence identity of these novel BEV strains with the reference strains, a comprehensive analysis of the nucleotide and amino acid sequences of VP1, P1, and 2C + 3 CD was performed. As given in Table 3, five strains, namely, HeN-A2, NX-FY40, JL-HY61, JL-HY68, and JL-DH13, shared the nucleotide sequence identity of 80.3%–89.1% and amino acid sequence identity of 96.5%–97.5% with the VP1 of EV-E3 reference strain HY12 (KF748290), indicating that these five isolated strains are evolutionarily closer to EV-E3 strains. In Table 3, JL-JY12, JL-JY29, JL-JY41, and JL-GZL4 strains shared a relatively high nucleotide and amino acid sequence identity with the VP1 of the EV-E1 reference strain VG-5–27 (NC_001859), respectively, varying from 79.8% to 80.9% and 94.3% to 96.1%, indicating that these four isolated strains belong to the EV-E1 strains. HeN-B78, HeN-B84, and HeN-C4 strains from Henan province shared a relatively high nucleotide identity of 73.6% to 75.6% and amino acid sequence identity of 91.4% to 94.3% with the VP1 of EV-E2 reference strain PS 42 (DQ092792), suggesting they are close to EV-E2 strains. The strain HeN-B62 shared the highest nucleotide and amino acid sequence identity with VP1 of EV-F3 strain PS-87-Belfast (DQ092794), 76.9% and 92.7%, respectively. HeN-A12 and HeN-YR91 strains shared a relatively high nucleotide and amino acid sequence identity with VP1 of the EV-F1 strain BEV-261 (DQ092770), varying from 75.5% to 81.3% and 89.8% to 97.5%, respectively, indicating that they were evolutionarily closer to EV-F1 strains. Interestingly, twelve strains, namely, JL-40, JL-JY1, JL-JY7, HeN-B72, HeN-B81, HeN-B83, JL-JY33, JL-JY37, JL-JY40, JL-JY42, HeN-A3, and NX-b-DR47, shared low nucleotide and amino acid identity with all of the EV-E strains, with the highest only 88.6% amino acid identity with EV-E2 strain PS 42 (DQ092792), indicating that those strains likely belong to a new subtype (Table S3). Taken together, the aforementioned results indicate these novel BEV strains are either close to EV-E or EV-F based on their sequence identities.
TABLE 3.
Percent identity of nucleotide and amino acid sequences between isolated strains and enterovirus reference strains
| Strains | Reference enterovirus strains | Identity (%) | Subgenotype | |||||
|---|---|---|---|---|---|---|---|---|
| VP1 | P1 | 2C + 3 CD | ||||||
| Nt | Aa | Nt | Aa | Nt | Aa | |||
| HeN-A2 | HY12 (KF748290) | 80.3 | 96.5 | 81.1 | 96.2 | 82.5 | 96.8 | EV-E3 |
| NX-FY40 | HY12 (KF748290) | 82.6 | 97.5 | 82.8 | 97.4 | 83.3 | 97 | EV-E3 |
| JL-HY61 | HY12 (KF748290) | 89.1 | 97.5 | 88.5 | 98.1 | 84.5 | 96.8 | EV-E3 |
| JL-HY68 | HY12 (KF748290) | 88.8 | 97.5 | 88.7 | 97.9 | 84.3 | 96.7 | EV-E3 |
| JL-DH13 | HY12 (KF748290) | 88.5 | 97.5 | 88.5 | 98.1 | 84.4 | 96.9 | EV-E3 |
| JL-JY12 | VG-5–27 (NC_001859) | 80.9 | 96.1 | 80.4 | 97 | 79.4 | 95.6 | EV-E1 |
| JL-JY29 | VG-5–27 (NC_001859) | 79.8 | 94.3 | 79.1 | 96.4 | 79.6 | 95.7 | EV-E1 |
| JL-JY41 | VG-5–27 (NC_001859) | 80 | 95.4 | 79.3 | 96.5 | 79.2 | 96.1 | EV-E1 |
| JL-GZL4 | VG-5–27 (NC_001859) | 80.7 | 95 | 79.8 | 96.7 | 81 | 97.6 | EV-E1 |
| HeN-B78 | PS 42 (DQ092792) | 73.9 | 91.8 | 76.3 | 93.9 | 80.4 | 96.3 | EV-E2 |
| HeN-B84 | PS 42 (DQ092792) | 73.6 | 91.4 | 76.4 | 94.2 | 79.2 | 95.1 | EV-E2 |
| HeN-C4 | PS 42 (DQ092792) | 75.6 | 94.3 | 76.9 | 95.8 | 81.1 | 96.9 | EV-E2 |
| JL-40 | PS 42 (DQ092792) | 73.3 | 87.9 | 76.2 | 91.9 | 78.9 | 95.1 | EV-E7 |
| JL-JY1 | PS 42 (DQ092792) | 73.3 | 87.9 | 76.4 | 92.1 | 78.7 | 95.1 | EV-E7 |
| JL-JY7 | PS 42 (DQ092792) | 73.3 | 87.9 | 76.3 | 91.7 | 78.9 | 95.1 | EV-E7 |
| HeN-B72 | PS 42 (DQ092792) | 73.1 | 88.6 | 75.7 | 91.8 | 80.2 | 96.5 | EV-E7 |
| HeN-B81 | PS 42 (DQ092792) | 72.9 | 87.1 | 75.5 | 91.4 | 79.5 | 95.1 | EV-E7 |
| HeN-B83 | PS 42 (DQ092792) | 73.1 | 87.5 | 75.6 | 91.4 | 80.4 | 95.6 | EV-E7 |
| JL-JY33 | PS 42 (DQ092792) | 73.3 | 88.2 | 75.7 | 92.3 | 78.8 | 95.7 | EV-E7 |
| JL-JY37 | PS 42 (DQ092792) | 73.1 | 88.2 | 75.4 | 92.1 | 78.7 | 95.7 | EV-E7 |
| JL-JY40 | PS 42 (DQ092792) | 73.1 | 88.2 | 75.4 | 2.1 | 78.6 | 95.3 | EV-E7 |
| JL-JY42 | PS 42 (DQ092792) | 73 | 87.9 | 75.6 | 92 | 78.8 | 95.7 | EV-E7 |
| HeN-A3 | PS 42 (DQ092792) | 72.9 | 85 | 75.6 | 90.3 | 78.1 | 92.8 | EV-E6 |
| NX-b-DR47 | PS 42 (DQ092792) | 72.1 | 85.4 | 75.1 | 90.3 | 77.7 | 92.3 | EV-E6 |
| HeN-B62 | PS-87-Belfast (DQ092794) | 76.9 | 92.7 | 76.9 | 93.8 | 82 | 97.7 | EV-F3 |
| HeN-A12 | BEV-261 (DQ092770) | 81.3 | 97.5 | 81 | 97.5 | 83 | 97.8 | EV-F1 |
| HeN-YR91 | BEV-261 (DQ092770) | 75.5 | 89.8 | 75.5 | 90.5 | 80.9 | 95.1 | EV-F1 |
Phylogenetic analyses clustered the novel BEV strains to EV-E and EV-F species
To further confirm the phylogenetic status of these novel bovine enterovirus strains, phylogenetic analysis was performed based on the amino acid sequences of P1 and 2C + 3 CD using the maximum likelihood method. The representative sequences of all known types of EV-E and EV-F, and the representative sequences of all 12 enterovirus species and 3 rhinovirus within the Enterovirus genus were used as outgroup sequences. The HY12 virus was used as a reference strain of EV-E3 for 2C + 3 CD phylogenetic tree analyses since it is the only EV-E3 virus strain with 2C + 3 CD sequences available. As shown in Fig. 1 and 2, 24 isolated strains were clustered in the same clade as EV-E, and 3 isolated strains, namely, HeN-A12, HeN-B62, and HeN-YR91, belong to the branch of EV-F strains. These results were congruent with the species and subtypes determined based on sequence identity, revealing the inconsistency of phylogenetic clustering between those of P1 and 2C + 3 CD.
Fig 1.

Phylogenetic analyses of P1 on the novel bovine enterovirus strains. The reference sequences include the representative sequences of all known EV-E and EV-F types and the representative sequences of all 12 enterovirus species and 3 rhinovirus species in the Enterovirus genus were used as outgroup sequences. The amino acid sequences of P1 were used to construct the phylogenetic tree using the maximum likelihood method with 1,000 bootstrap replications. Bootstrap values of >50 are shown at the nodes. The scale bar represents 10% nucleotide sequence divergence for maximum likelihood methods. The analysis models for ML methods for the P1 amino acid are LG + G + I. Viruses are marked with symbols as follows: ● refers to the EV-E strains obtained in this study; ▲ refers to the EV-F strains obtained in this study; ◇ stands for the CEV-JL14 strain; ○ refers to the HY12 EV-E strain isolated from cattle; △ stands for the SD-S67 strain (an EV-F) isolated from goats.
Fig 2.

Phylogenetic analyses of 2C + 3 CD on novel bovine enterovirus strains. The reference sequences include the representative sequences of all known EV-E and EV-F types and the representative sequences of all 12 enterovirus species and 3 rhinovirus species in the Enterovirus genus were used as outgroup sequences. The amino acid sequences of 2C + 3 CD were used to construct the phylogenetic tree using the maximum likelihood method with 1000 bootstrap replications. Bootstrap values of >50 are shown at the nodes. The scale bar represents 5% nucleotide sequence divergence for maximum likelihood methods. The analysis models for ML methods for the 2C + 3 CD amino acid is LG + G + I. Viruses are marked with symbols as follows: ● refers to the EV-E strains obtained in this study; ▲ refers to the EV-F strains obtained in this study; ◇ stands for the CEV-JL14 strain; ○ refers to the HY12 EV-E strain isolated from cattle; △ stands for the SD-S67 strain (an EV-F) isolated from goats.
Two novel EV-E subtypes revealed for bovine enterovirus
To determine the subtypes for the novel BEV strains, phylogenetic analyses were further performed for these EV-E and EV-F strains based on the sequence identity of VP1. Twenty-four novel EV-E strains were clustered separately into five subtypes. The strains JL-JY12, JL-JY29, JL-JY41, and JL-GZL4 were clustered to the same subtype as VG-5–27 (EV-E1), sharing 94.3%–96.1% (Table 3) VP1 amino acid sequence identity. The strains HeN-B78, HeN-B84, and HeN-C4 were clustered to the same subtype with PS 42, an EV-E2 since they shared the VP1 amino acid sequence identity of 91.4%–94.3% (Table 3). The strains HeN-A2, NX-FY40, JL-HY61, JL-HY68, and JL-DH13 were clustered to the same type as HY12 (EV-E3), where they shared 96.5%–97.5% VP1 amino acid sequence identity (Table 3). In addition to the aforementioned three subtypes (EV-E1–EV-E3), two novel subtypes excluding EV-E4 and EV-E5 were identified (Fig. 3). The first new subtype contained the strains HeN-A3 and NX-b-DR47. They had the VP1 amino acid sequence identity of 97.5% of each other, with VP1 amino acid sequence identity of 50.7%–85.4% from all other EV-E reference strains (Table S3). These results demonstrated that HeN-A3 and NX-b-DR47 belong to a new subtype, designated as EV-E6. The second novel subtype consisted of JL-40, JL-JY1, JL-JY7, HeN-B72, HeN-B81, HeN-B83, JL-JY33, JL-JY37, JL-JY40, and JL-JY42 strains, with the identity of 96.1%–100% in the VP1 amino acid sequence among them (Table S3). These strains presented 50.4%–88.6% amino acid identity on the VP1 with other EV-E reference strains (Table S3), indicating they are also a new subtype, designated as EV-E7.
Fig 3.

Phylogenetic analyses for BEV subtyping. Phylogenetic analysis was performed to subtype the BEV strain based on the VP1 amino acid sequences. The reference sequences including the representative sequences of all known EV-E and EV-F types and the representative sequences of all 12 enterovirus species and 3 rhinovirus species within the Enterovirus genus were used as outgroup sequences. A phylogenetic tree was generated using the maximum likelihood method with 1000 bootstrap replications. The scale bar represents 10% nucleotide sequence divergence for maximum likelihood methods. The analysis model for ML methods for the VP1 amino acid is LG + G. Viruses are marked with symbols as follows: red characters refer to the EV-E and EV-F strains obtained in this study, and white characters in the EV-E1 subtype refer to the EV-E strains obtained in this study; the green character stands for the strains isolated in the laboratory. The subtypes of the strains are shown in the figure. R-A stands for rhinovirus A, R-B stands for rhinovirus B, R-C stands for rhinovirus C.
Similarly, analysis on the subtypes for EV-F showed that HeN-A12 and HeN-YR91 strains were clustered to the same subtype as BEV-261 (EV-F1), with 89.8%–97.5% amino acid sequence identity of VP1 (Table 3). The strain HeN-B62 was clustered to the subtype as PS-87-Belfast (EV-F3), with 92.7% amino acid sequence identity (Table 3).
Different subtypes and species of enterovirus detected in the same cattle herd
Further analysis on the enterovirus species showed that the HeN-B78, HeN-B84, and HeN-B62 strains from the same cattle herd were clustered to different enterovirus species, where HeN-B78 and HeN-B84 strains were grouped to EV-E and HeN-B62 clustered to EV-F. These results clearly demonstrated the mixed infections of EV-E and EV-F simultaneously in the same cattle herd. Similarly, HeN-A12 and HeN-A3 strains from another cattle herd also belonged to different species, where HeN-A3 was grouped to EV-E and HeN-A12 was grouped to EV-F. These results demonstrated the unusual coinfection of EV-E and EV-F in the cattle herd.
It is interesting to note that different BEV subtype infections were also revealed in the same cattle herds. As shown in Fig. 1 and 3, JL-JY12, JL-JY29, and JL-JY7; HeN-A2 and HeN-A3; HeN-B78, and HeN-B84 and HeN-B81 from the same herds were clustered to different subtypes. These results demonstrated the complexity of BEV infection.
Recombination detected in the novel isolated bovine enterovirus strains
To characterize the genomic features and explore the origin of the isolated strains, recombination analyses were performed using SimPlot software and the RDP4 package with multiple algorithms. The P values by the individual tools implemented in the RDP are shown in Table S4. The complete genome sequences of 24 isolated EV-E and 3 EV-F strains were selected as the query sequences for similarity and bootscan analysis with EV-E and EV-F reference sequences, respectively. As shown in Fig. 4A and B, the HeN-B62 strain resembled the EV-F7-AN12 strain in the sequence before the VP2 junction (nearby 1,160 bp), while it was more similar to the EV-F3-PS-87-Belfast strain than other strains for the fragment region (1,160–3,480 bp). Additionally, HeN-B62 contained an unidentified sequence that was apparently not related to the PS-87-Belfast strain in the fragment region (3,480–7,400 bp). Therefore, two recombination events possibly occurred in the HeN-B62 strain: one occurred between EV-F7-AN12 and EV-F3-PS-87-Belfast strains, with the breakpoint located in the VP2 gene, and the other occurred between EV-F3-PS-87-Belfast and unidentified sequences, with one breakpoint located in the 2A gene (Fig. 4B). The HeN-A12 (EV-F1) strain showed the highest degree of similarity to the EV-F1-SD-S67 strain in the P1 region. However, it contained an unidentified sequence that was apparently not related to the SD-S67 strain in the P2 and P3 regions (Fig. S2A). No apparent recombination was found in the HeN-YR91 strain with EV-F (data not shown). The HeN-A2 strain showed the highest degree of similarity to HY12 (EV-E3) in the P1 region. However, it contained an unidentified sequence that was apparently not related to the HY12 strain in the P2 and P3 regions (Fig. S2B). No apparent recombination events were found in other isolated EV-E strains (data not shown).
Fig 4.
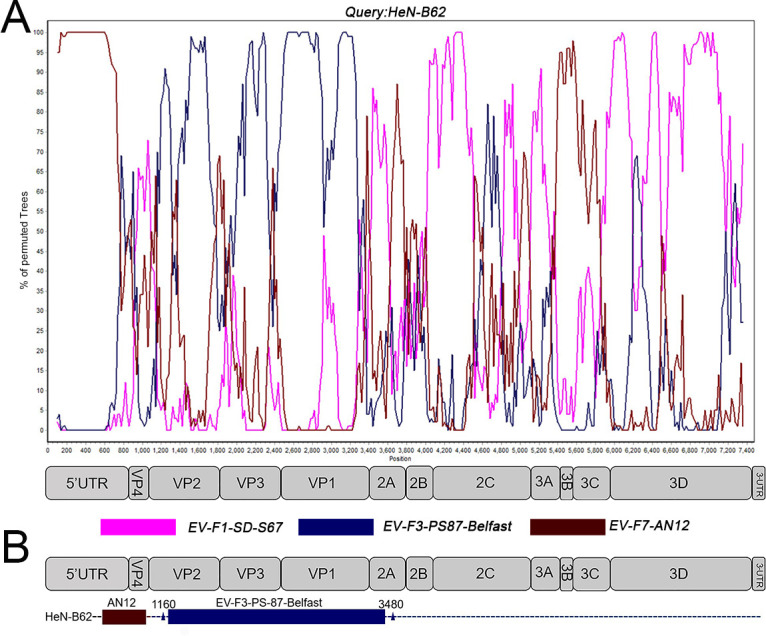
Recombination analyses of the HeN-B62 strain with EV-F types. The genome sequence of HeN-B62 (A) was used as query sequences in the bootscan analysis. The structural map of the bovine enterovirus genome is displayed below each panel. The possible crossover breakpoints were marked with a rectangular box with the enterovirus subtypes and represented by corresponding colors (B). Each point plotted is the percentage identity within a sliding window 200 nt wide centered on the position plotted, with a step size of 20 nt between points.
Interspecies and intraspecies recombination revealed on bovine enterovirus strains
To characterize the genomic features of the predominant EV-E and EV-F strains, the complete genome sequences of EV-E1-EV-E3, and EV-E5; EV-F1-EV-F4, and EV-F7 standard strains; and VG-5–27, PS 42, HY12, MexKSU/5, BEV-261, PS-89, PS-87-Belfast, W1, and AN12 were selected as the query sequences for similarity and bootscan analysis (Fig. 5). As shown in Fig. 5A, the interspecies recombination between EV-E and EV-F was analyzed. Bootscan analyses indicated that EV-F7-AN12 might be a recombinant virus between EV-E5-MexKSU/5 and EV-F1-BEV-261 strains, with one breakpoint located at the VP1/2A junction (3,340 bp). EV-F7-AN12 shared the highest similarity to the EV-E5-MexKSU/5 strain in the sequence region (1,160–3,340 bp), while it had the highest similarity to the EV-F1-BEV-261 strain in the sequence after 4,160 bp. As observed in Fig. 5B, the possible recombination events occurred with at least three recombinant breakpoints on EV-E5-MexKSU/5, corresponding to the VP4/VP2 junction, VP1/2A junction, and 3A/3B junction. The EV-E5-MexKSU/5 strain contained alternative regions between the VP1/2A and 3A/3B junctions that were closely related to EV-E3-HY12 and EV-E2-PS 42 strains. EV-E5-MexKSU/5 shared the highest similarity to the EV-F7-AN12 strain within the sequence region (1,160–3,340 bp), while it had the highest similarity to the EV-E1-VG-5–27 strain in the sequence after 5,140 bp. These results demonstrated the interspecies recombination among bovine enteroviruses.
Fig 5.

Recombination analyses between EV-E and EV-F types. The genome sequences of EV-F7-AN12 (A) and EV-E5-MexKSU/5 (B) were used as query sequences in the bootscan analysis. The structural map of the bovine enterovirus genome was displayed below each panel. The possible crossover breakpoints were marked with a rectangular box with the enterovirus subtypes and represented by corresponding colors. Each point plotted is the percentage identity within a sliding window 200 nt wide centered on the position plotted, with a step size of 20 nt between points.
The intraspecies recombination among different EV-F subtypes was also analyzed. For the EV-F1-BEV-261 strain (Fig. S3A), it had the similar sequence as that of the EV-E5-MexKSU/5 strain before the 5′UTR junction (nearby 700 bp), while it was closer to EV-F2-PS_89 in the fragment region (700–4,100 bp). Additionally, EV-F1-BEV-261 shared the highest similarity to the EV-F7-AN12 strain in the sequence after 4,100 bp. For the EV-F2-PS_89 strain (Fig. S3B), the fragment (1,160–3,920 bp) was more similar to that of EV-F1-BEV-261, and the subsequent sequence (3,920–7,400 bp) shared the highest similarity to the EV-F3-PS-87-Belfast strain. As for EV-F3-PS-87-Belfast (Fig. S3C), the fragment (1,140–3,940 bp) was more similar to that of EV-F4-W1, and the subsequent sequence (3,940–7,400 bp) shared the highest similarity to EV-F2-PS_89 (Fig. S3C). The fragment (1,160–3,840 bp) of EV-F4-W1 was more similar to that of EV-F3-PS-87-Belfast (Fig. S3D).
DISCUSSION
In this study, we have isolated 27 novel bovine enteroviruses from the major cattle-raising regions during 2012–2018 in China, explored the genetic diversity and recombination of the novel BEV strains, unveiled two new subtypes of EV-E designated as EV-E6 and EV-E7, and revealed the different BEV species or subtype infection on the same cattle herds.
BEV is the causative agent associated with an emerging disease in China, which leads to an important economic loss to the cattle industry. Based on virus genetic variability and molecular difference, BEV is divided into two species (genotype or serotype), EV-E and EV-F, where EV-E and EV-F are further divided into five subtypes (subgenotype) and seven subtypes, respectively (3). Although EV-E and EV-F infections were reported in China in the early 2010s (9, 27), they remain largely unknown on many aspects such as the molecular difference, the virus genetic diversity and origin, the geographical distribution, and the transmission pattern. To unveil the underlying aspects of BEV infections, we performed BEV isolation using the representative specimens detected as BEV-positive by ELISA methods (29). Molecular characterization demonstrated that 27 BEV strains were successfully obtained. Sequencing and analyses of these BEV strains revealed that the majority of them are EV-E, suggesting that the predominant BEV infections are caused by EV-E. Interestingly, we found that different subtype strains within EV-E or EV-F were isolated from the same cattle herds; even EV-E (HeN-B78 and HeN-B84) and EV-F (HeN-B62) were both revealed in the same cattle herds. These results clearly demonstrated the coinfection of enterovirus species or subtypes. Since mutation and recombination extensively occurred among different BEV genotypes, the emergence and importation of new genotypes or subtypes of BEV should be continuously monitored. The findings that the same subtypes were revealed in different regions or provinces indicate that BEV may be transmitted between different regions by live cattle distribution. More importantly, we found that some of these newly identified strains are likely the recombinants resulting from either enterovirus intraspecies or interspecies recombination, which enriches the understanding of underlying enterovirus pathogenicity and evolution.
The amino acid sequence identity of VP1 is currently considered the standard for enterovirus classification (30). Recently, according to the suggestion by Zell et al., enteroviruses were classified based on the amino acid sequence identities of the VP1 capsid protein, of which 70% to 85% were defined as heterologous types and >90% as homologous types (28). Based on the criteria for enterovirus demarcation and our analyses on the available EV-E sequences, we proposed that the EV-E strains should be divided into seven subtypes. In addition to the five current reported subtypes EV-E1–EV-E5, obviously two novel subtypes for EV-E strains were discovered. We designated them as EV-E6 and EV-E7. This finding will broaden our current knowledge on enterovirus classification.
Genetic recombination plays important roles in the genetic variation and evolution of viruses. Although EV-A71 was mostly selected for recombination analysis in previous studies on Enterovirus, there were few studies on recombination analysis of bovine enterovirus (31–33). In the present study, the complete genome sequences of the novel BEV strains were used as the query sequences. Comprehensive recombination analyses revealed two recombination events that occurred possibly in the HeN-B62 strain. One recombination event occurred between EV-F7-AN12 and EV-F3-PS-87-Belfast strains, with the breakpoint located in the VP2 gene, and the other occurred between EV-F3-PS-87-Belfast and unidentified sequences, with the breakpoint located in the 2A gene, suggesting an intraspecies recombination. More importantly, interspecies recombination between EV-E and EV-F was found, where the EV-F7-AN12 strain is likely a recombinant from EV-E5 and EV-F1, and EV-E5-MexKSU/5 may be a recombinant from EV-F7 and EV-E1. It was also found that there was intraspecific recombination between the EV-F strains. We found that recombination breakpoints in the Enterovirus genome were frequently located in the nonstructural regions 5′UTR, P2, and P3 but rarely in the structural region P1, which is consistent with previous reports (34). The discovery that the nonstructural regions P2 and P3 act as the hot spots for recombination in BEV may be due to the higher homology of P2 and P3 regions than the structural region shared by BEV strains. Moreover, the inconsistent topology of phylogenetic trees suggested that it was not sufficient to genotype the recombinant viruses by VP1 alone.
The EV-F strains were first isolated in Northeast China in 2011. Since the discovery of the EV-E strain in 2012 in China, many BEV infections were reported. Our analyses on BEV strains demonstrated the EV-E strains as the predominant genotype in China. Since mutation and recombination extensively occurred among different BEV genotypes, the emergence and importation of new genotypes or subtypes of BEV should be continuously monitored (9, 27, 30).
MATERIALS AND METHODS
Sample collection and detection
A total of 2,721 fecal swabs collected from Jilin, Ningxia, and Henan were processed as previously described (27). All the samples were shipped on ice and then stored at −80°C.
Cell culture and virus isolation
Vero cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (HyClone, Beijing, China), 2 µg/mL gentamycin, and 2 mM L-glutamine (Invitrogen). Virus isolation was performed as previously described (27). Briefly, the fecal swabs were diluted at 1:10 (W/V) in a 10 mM phosphate-buffered saline (PBS) (pH 7.4) and clarified by low-speed centrifugation (3,000 × g for 10 min). The supernatants were filtered through 0.45-µm pore membrane filters (JIN TENG, Tianjin, China). Virus isolation was performed on these specimens using Vero cells. The CPE was captured using a Canon digital camera (Canon, Tokyo, Japan). The noninfected cells were used as negative controls.
TCID50 titration
TCID50 titration of the isolated strains was performed using 96-well plates following the procedure previously described (27). Briefly, the viruses were diluted at 10 × serial dilutions and used to infect the cells for each dilution. The cytopathic effects on the Vero cells were observed and counted at 48 hpi. TCID50 was calculated following a standard procedure based on the Reed–Muench method (35).
RNA extraction and PCR
Viral RNA was extracted from the sample using the TRIzol reagent (Invitrogen) as previously described (27). cDNA was synthesized by reverse transcription using the Bio RT-cDNA kit (Invitrogen) following the manufacturer’s instructions. The primers for amplification of the complete genome sequence were designed based on the variation of each strain (Table S1). cDNA ends were determined using the FirstChoice RLM-RACE kit (Thermos Fisher Scientific, Carlsbad, CA). PCR products were subjected to electrophoresis using 1.0% agarose gel and purified for DNA sequencing (Sangon Biotechnology, Shanghai, China). The resulting sequences were analyzed using the Basic Local Alignment Search Tool (BLAST). The complete genome sequences were assembled using Lasergene software (version 7.0, DNASTAR, Madison, WI, USA).
Data collection and nucleotide sequence data sets
Genome sequences for enteroviruses from species E and F were retrieved from the GenBank database (to March 2023) (Table S2). Wrongly annotated sequences and coding sequences (CDSs) containing internal termination codons were discarded. A sequence alignment including the complete genomes of enterovirus strains was generated with Clustal W implemented in BioEdit, version 7.2.3, software (http://www.mbio.ncsu.edu/bioedit/bioedit.html) and split into three partitions corresponding to the VP1, P1, and 2C + 3 CD genomic regions.
Maximum likelihood phylogenetic analysis and recombination analysis
Maximum likelihood trees were generated with MEGA-7 software (36) using the best evolutionary model and 1,000 bootstrap replicates for the estimation of node consistency (37). The merged sequence file (2C + 3 CD) was first aligned by MEGA-7 with the Clustal W method to generate nucleotide alignment. Phylogenetic trees were visualized by using the Interactive Tree of Life version 6 (https://itol.embl.de/).
Recombination analyses were performed using a recombination detection software package (RDP 4). A total of seven methods implemented in RDP4 were applied: RDP, Bootscan, GENECONV, 3Seq, Chimaera, MaxChi, and SiScan. Recombination detected by at least three of the seven methods with a P value cutoff 0.05 was considered true recombination. The similarity plot and bootscan analysis were performed by SimPlot 3.5.1 (JHK University, Baltimore, MD, USA). The pairwise similarity between all sequences in multiple sequence alignment was calculated with a 200-nt window moved along the sequence in 20-nt steps.
ACKNOWLEDGMENTS
This study was supported by the National Key Research & Development Program 2017YFD0500104 and 2016YFD0500904; National Natural Science Foundation of China (31572531).
Contributor Information
Yidi Guo, Email: guoyd@jlu.edu.cn.
Xinping Wang, Email: wangxp88@hotmail.com.
Gabriel I. Parra, US Food and Drug Administration, USA
DATA AVAILABILITY
All nucleotide sequence data are available in GenBank under accession numbers MN598008-MN598032, MN598037, and MN598039.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/spectrum.02800-23.
Cytopathic effects (CPE) and growth characteristics analysis in Vero cells.
Recombination analyses of HeN-A12 and HeN-A2 strains with EV-E and EV-F types.
Recombination analyses between EV-F subtypes.
Supplemental figure legends.
Primer sequences to amplify full-length sequences.
Reference strains for enterovirus.
Percent identities of amino acid sequence in VP1 of bovine enterovirus strains compared with reference strains of EV-E and EV-F.
Algorithms of the RDP4 package used to predict the recombination event.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Zerbini FM, Siddell SG, Lefkowitz EJ, Mushegian AR, Adriaenssens EM, Alfenas-Zerbini P, Dempsey DM, Dutilh BE, García ML, Hendrickson RC, Junglen S, Krupovic M, Kuhn JH, Lambert AJ, Łobocka M, Oksanen HM, Robertson DL, Rubino L, Sabanadzovic S, Simmonds P, Smith DB, Suzuki N, Van Doorslaer K, Vandamme A-M, Varsani A. 2023. Changes to virus taxonomy and the ICTV statutes ratified by the International Committee on Taxonomy of viruses. Arch Virol 168:175. doi: 10.1007/s00705-023-05797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. 2010. Picornaviruses. Curr Top Microbiol Immunol 343:43–89. doi: 10.1007/82_2010_37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zell R, Delwart E, Gorbalenya AE, Hovi T, King AMQ, Knowles NJ, Lindberg AM, Pallansch MA, Palmenberg AC, Reuter G, Simmonds P, Skern T, Stanway G, Yamashita TIctv Report Consortium . 2017. ICTV virus taxonomy profile: picornaviridae. J Gen Virol 98:2421–2422. doi: 10.1099/jgv.0.000911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. 2010. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis 10:778–790. doi: 10.1016/S1473-3099(10)70194-8 [DOI] [PubMed] [Google Scholar]
- 5. Kang HJ, Yoon Y, Lee YP, Kim HJ, Lee DY, Lee JW, Hyeon JY, Yoo JS, Lee S, Kang C, Choi W, Han MG. 2021. A different epidemiology of Enterovirus A and Enterovirus B co-circulating in Korea, 2012-2019. J Pediatric Infect Dis Soc 10:398–407. doi: 10.1093/jpids/piaa111 [DOI] [PubMed] [Google Scholar]
- 6. Sadeuh-Mba SA, Joffret ML, Mazitchi A, Endegue-Zanga MC, Njouom R, Delpeyroux F, Gouandjika-Vasilache I, Bessaud M. 2019. Genetic and phenotypic characterization of recently discovered enterovirus D type 111. PLoS Negl Trop Dis 13:e0007797. doi: 10.1371/journal.pntd.0007797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moll T, Davis AD. 1959. Isolation and characterization of cytopathogenic enteroviruses from cattle with respiratory disease. Am J Vet Res 20:27–32. [PubMed] [Google Scholar]
- 8. Huck RA, Cartwright SF. 1964. Isolation and classification of viruses from cattle during outbreaks of Mucosal or respiratory disease and from herds with reproductive disorders. J Comp Pathol 74:346–365. doi: 10.1016/s0368-1742(64)80041-2 [DOI] [PubMed] [Google Scholar]
- 9. Li YL, Chang JT, Wang Q, Yu L. 2012. Isolation of two Chinese bovine enteroviruses and sequence analysis of their complete genomes. Arch Virol 157:2369–2375. doi: 10.1007/s00705-012-1424-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tatem JM, Weeks-Levy C, Mento SJ, DiMichele SJ, Georgiu A, Waterfield WF, Sheip B, Costalas C, Davies T, Ritchey MB. 1991. Oral poliovirus vaccine in the United States: molecular characterization of Sabin type 3 after replication in the gut of vaccinees. J Med Virol 35:101–109. doi: 10.1002/jmv.1890350206 [DOI] [PubMed] [Google Scholar]
- 11. McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Koike S, Kroes ACM, Lukashev A, Perera D, Roivainen M, Susi P, Trallero G, Evans DJ, Simmonds P. 2012. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J Virol 86:2676–2685. doi: 10.1128/JVI.06065-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lukashev AN, Shumilina Ey, Belalov IS, Ivanova OE, Eremeeva TP, Reznik VI, Trotsenko OE, Drexler JF, Drosten C. 2014. Recombination strategies and evolutionary dynamics of the human enterovirus A global gene pool. J Gen Virol 95:868–873. doi: 10.1099/vir.0.060004-0 [DOI] [PubMed] [Google Scholar]
- 13. van der Sanden S, van Eek J, Martin DP, van der Avoort H, Vennema H, Koopmans M. 2011. Detection of recombination breakpoints in the genomes of human enterovirus 71 strains isolated in the Netherlands in epidemic and non-epidemic years. Infect Genet Evol 11:886–894. doi: 10.1016/j.meegid.2011.02.011 [DOI] [PubMed] [Google Scholar]
- 14. Simmonds P, Welch J. 2006. Frequency and dynamics of recombination within different species of human enteroviruses. J Virol 80:483–493. doi: 10.1128/JVI.80.1.483-493.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Kroes ACM, Lukashev A, Muir P, Odoom J, Roivainen M, Susi P, Trallero G, Evans DJ, Simmonds P. 2010. Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J Virol 84:9292–9300. doi: 10.1128/JVI.00783-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cabrerizo M, Trallero G, Simmonds P. 2014. Recombination and evolutionary dynamics of human echovirus 6. J Med Virol 86:857–864. doi: 10.1002/jmv.23741 [DOI] [PubMed] [Google Scholar]
- 17. Luo Y, Liu H, Zou Y, Qiao C, Su Y, Zhu X, Zhang G, Huang W, Qin Y, Pan Y, Huang W. 2023. Molecular epidemiology of bovine enteroviruses and genome characterization of two novel bovine enterovirus strains in Guangxi, China. Microbiol Spectr 11:e0378522. doi: 10.1128/spectrum.03785-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kyriakopoulou Z, Pliaka V, Amoutzias GD, Markoulatos P. 2015. Recombination among human non-polio enteroviruses: implications for epidemiology and evolution. Virus Genes 50:177–188. doi: 10.1007/s11262-014-1152-y [DOI] [PubMed] [Google Scholar]
- 19. Runckel C, Westesson O, Andino R, DeRisi JL. 2013. Identification and manipulation of the molecular determinants influencing Poliovirus recombination. PLoS Pathog 9:e1003164. doi: 10.1371/journal.ppat.1003164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smura T, Blomqvist S, Vuorinen T, Ivanova O, Samoilovich E, Al-Hello H, Savolainen-Kopra C, Hovi T, Roivainen M. 2014. Recombination in the evolution of enterovirus C species sub-group that contains types CVA-21. PLoS One 9:e94579. doi: 10.1371/journal.pone.0094579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bessaud M, Joffret ML, Holmblat B, Razafindratsimandresy R, Delpeyroux F. 2011. Genetic relationship between cocirculating human enteroviruses species C. PLoS One 6:e24823. doi: 10.1371/journal.pone.0024823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McWilliam Leitch EC, Bendig J, Cabrerizo M, Cardosa J, Hyypiä T, Ivanova OE, Kelly A, Kroes ACM, Lukashev A, MacAdam A, McMinn P, Roivainen M, Trallero G, Evans DJ, Simmonds P. 2009. Transmission networks and population turnover of echovirus 30. J Virol 83:2109–2118. doi: 10.1128/JVI.02109-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lukashev AN, Lashkevich VA, Ivanova OE, Koroleva GA, Hinkkanen AE, Ilonen J. 2005. Recombination in circulating human enterovirus B: independent evolution of structural and non-structural genome regions. J Gen Virol 86:3281–3290. doi: 10.1099/vir.0.81264-0 [DOI] [PubMed] [Google Scholar]
- 24. Oberste MS, Maher K, Kilpatrick DR, Flemister MR, Brown BA, Pallansch MA. 1999. Typing of human enteroviruses by partial sequencing of VP1. J Clin Microbiol 37:1288–1293. doi: 10.1128/JCM.37.5.1288-1293.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang DY, Chen H, Yan DM, Zhang Y, An JJ, Ma JT, Zhu SL, Ma XM, An HQ, Xu WB. 2010. Genetic characterization of coxsackievirus A16 isolated in Ningxia Hui municipality in 2008. Zhonghua Liu Xing Bing Xue Za Zhi 31:904–908. [PubMed] [Google Scholar]
- 26. He YQ, Chen L, Xu WB, Yang H, Wang HZ, Zong WP, Xian HX, Chen HL, Yao XJ, Hu ZL, Luo M, Zhang HL, Ma HW, Cheng JQ, Feng QJ, Zhao DJ. 2013. Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus A6- and coxsackievirus A10-associated hand, foot. J Clin Microbiol 51:3560–3566. doi: 10.1128/JCM.01231-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu L, Xing Z, Gai X, Li S, San Z, Wang X, Rong L. 2014. Identification of a novel enterovirus E isolates HY12 from cattle with severe respiratory and enteric diseases. PLoS ONE 9:e97730. doi: 10.1371/journal.pone.0097730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zell R, Krumbholz A, Dauber M, Hoey E, Wutzler P. 2006. Molecular-based reclassification of the bovine enteroviruses. J Gen Virol 87:375–385. doi: 10.1099/vir.0.81298-0 [DOI] [PubMed] [Google Scholar]
- 29. Li X, Hu J, Lin Q, Wang X, Zheng B, Zhang Z, Hu L, Wang X. 2019. Epidemiological investigation on enterovirus infections in cattle/sheep/goat herds in Shandong province. Chin J Vet Sci 39:223–227. [Google Scholar]
- 30. Earle JA, Skuce RA, Fleming CS, Hoey EM, Martin SJ. 1988. The complete nucleotide sequence of a bovine enterovirus. J Gen Virol 69 ( Pt 2):253–263. doi: 10.1099/0022-1317-69-2-253 [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Wang J, Guo W, Wang H, Zhu S, Wang D, Bai R, Li X, Yan D, Wang H, Zhang Y, Zhu Z, Tan X, An H, Xu A, Xu W, Jin D-Y. 2011. Emergence and transmission pathways of rapidly evolving evolutionary branch C4A strains of human enterovirus 71 in the Central plain of China. PLoS ONE 6:e27895. doi: 10.1371/journal.pone.0027895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Tan X, Cui A, Mao N, Xu S, Zhu Z, Zhou J, Shi J, Zhao Y, Wang X, Huang X, Zhu S, Zhang Y, Tang W, Ling H, Xu W, Jin D-Y. 2013. Complete genome analysis of the C4 subgenotype strains of enterovirus 71: predominant recombination C4 viruses persistently circulating in China for 14 years. PLoS ONE 8:e56341. doi: 10.1371/journal.pone.0056341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X, Zhu C, Bao W, Zhao K, Niu J, Yu X-F, Zhang W, Pyrc K. 2012. Characterization of full-length enterovirus 71 strains from severe and mild disease patients in northeastern China. PLoS ONE 7:e32405. doi: 10.1371/journal.pone.0032405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yip CCY, Lau SKP, Zhou B, Zhang M-X, Tsoi H-W, Chan K-H, Chen X-C, Woo PCY, Yuen K-Y. 2010. “Emergence of enterovirus 71 "double-recombinant" strains belonging to a novel genotype D originating from Southern China: first evidence for combination of Intratypic and Intertypic recombination events in EV71”. Arch Virol 155:1413–1424. doi: 10.1007/s00705-010-0722-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Epidemiol 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408 [DOI] [Google Scholar]
- 36. Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cytopathic effects (CPE) and growth characteristics analysis in Vero cells.
Recombination analyses of HeN-A12 and HeN-A2 strains with EV-E and EV-F types.
Recombination analyses between EV-F subtypes.
Supplemental figure legends.
Primer sequences to amplify full-length sequences.
Reference strains for enterovirus.
Percent identities of amino acid sequence in VP1 of bovine enterovirus strains compared with reference strains of EV-E and EV-F.
Algorithms of the RDP4 package used to predict the recombination event.
Data Availability Statement
All nucleotide sequence data are available in GenBank under accession numbers MN598008-MN598032, MN598037, and MN598039.