

Abstract
Previously, we reported that immunoproteasome (iP)-targeting linear peptide epoxyketones improve cognitive function in mouse models of Alzheimer’s disease (AD) in a manner independent of amyloid β. However, these compounds' clinical prospect for AD is limited due to potential issues, such as poor brain penetration and metabolic instability. Here, we report the development of iP-selective macrocyclic peptide epoxyketones prepared by a ring-closing metathesis reaction between two terminal alkenes attached at the P2 and P3/P4 positions of linear counterparts. We show that a lead macrocyclic compound DB-60 (20) effectively inhibits the catalytic activity of iP in ABCB1-overexpressing cells (IC50: 105 nM) and has metabolic stability superior to its linear counterpart. DB-60 (20) also lowered the serum levels of IL-1α and ameliorated cognitive deficits in Tg2576 mice. The results collectively suggest that macrocyclic peptide epoxyketones have improved CNS drug properties than their linear counterparts and offer promising potential as an AD drug candidate.
Keywords: Proteasome inhibitor, Immunoproteasome, neuroinflammation, LMP2, macrocyclic peptide epoxyketone, Alzheimer’s disease
Graphical Abstract
INTRODUCTION
Over the past decades, almost all drugs developed based on their ability to intervene with the amyloid β (Aβ) pathway have failed in clinical trials for Alzheimer’s disease (AD).1, 2 As a result, much attention has been shifted to developing anti-tau drugs, but no approved anti-tau therapies available so far.3, 4 Strategies of targeting neuroinflammation (regarded as a significant contributing factor to the progression of AD) have also drawn considerable attention.5, 6 Such strategies are further corroborated by recent findings that inflammatory cytokines released by activated microglia can induce astrocytes-mediated7 or tau-mediated neurotoxicity.8 Nevertheless, multiple clinical trials of FDA-approved anti-inflammatory agents targeting cyclooxygenases (COXs) or tumor necrosis factor (TNF)-α signaling have yielded no effective AD therapies.9-12 Thus, identifying new therapeutic agents not reliant on conventional AD targets has become increasingly crucial in AD drug discovery efforts. However, the lack of promising novel drug targets is a major obstacle in making real progress toward developing effective AD drugs.
In mammalian cells, there exist two main types of 20S proteasomes: the constitutive proteasome (cP) and immunoproteasome (iP) which is constitutively expressed in immune cells. The multi-protease complex 20S cP has three catalytic subunits (β1, β2, β5) on inner two β-rings displaying three unique substrate preferences: referred to as caspase-like (C-L), trypsin-like (T-L), and chymotrypsin-like (CT-L) activities, respectively. The cP subunits β1, β2, and β5, which are also commonly referred to as Y, Z, and X, respectively, are replaced by three immunosubunits β1i, β2i, and β5i, commonly referred to as LMP2 (low molecular mass polypeptide-2), MECL-1 (multicatalytic endopeptidase complex subunit-1), and LMP7 (low molecular mass polypeptide-7), respectively, to form the iP. Functionally, the iP has been shown to modulate inflammatory responses.13-15 As such, iP-targeting inhibitors have been investigated in preclinical and clinical trials as potential anti-inflammatory agents. For example, KZR-616, an iP-selective linear peptide epoxyketone (targeting both LMP7 and LMP2, Figure 1a), is currently being evaluated in clinical trials for systemic lupus erythematosus.16 Studies also revealed that the iP is upregulated in the brain of AD patients compared with healthy controls17-19 and that iP expression is closely linked to the activated microglia, which are widely considered as a primary culprit behind AD progression,17, 20, 21 suggesting a potential connection between the iP and AD.
FIGURE 1.

Previously reported linear peptide epoxyketones targeting the immunoproteasome (iP) catalytic subunits LMP7 (a) and LMP2 (b).
Recently, we reported that iP-targeting linear peptide epoxyketones (e.g., DB-310, Figure 1b) have a previously unrecognized effect of alleviating cognitive deficits in mouse models of AD in a manner independent of Aβ and tau aggregation.22, 23 We also showed that the iP inhibitors lower the levels of serum interleukin-1α (IL-1α) and protect the retinal pigment epithelium (RPE) layer from the structural destruction caused by Aβ-triggered inflammation in Tg2576 mice, potentially representing a new class of AD drugs that aim at a previously untapped pathway in AD drug discovery efforts. Despite promising efficacy, however, these linear peptide epoxyketones' clinical prospect seems limited at this time due to issues commonly associated with linear peptides, such as poor brain penetration and metabolic instability (mainly attributable to transporter-mediated efflux and enzymatic metabolism).23, 26-30 Yet, the peptide epoxyketone family (‘short peptides with C-terminal α′,β′-epoxyketone warhead’) provide an attractive drug development platform for related diseases due to pharmacological advantages conferred by their proven target specificity for the proteasomes and long-term safety in the clinic.31
Macrocycles, prepared via constraining the freely rotatable peptide bond, are often introduced to improve linear peptides' physicochemical (drug-like) properties.32, 33 This strategy has been used to boost the potency and chemical stability of linear peptides and improve metabolic stability, penetration across the blood-brain barrier (BBB), and membrane permeability.34-37 Typically, for short linear peptides like the iP inhibitor DB-310 (Figure 1b), a macrocycle can be introduced by a ring-closing reaction between the (n) and (n+2) residues,38 forming a β-turn mimic macrocyclic peptide.
Here, we report the development of iP-targeting macrocyclic peptide epoxyketones prepared via a ring-closing metathesis reaction between two terminal alkenes attached at the P2 and P3/P4 residues of their linear counterparts in the presence of the second-generation Grubbs catalyst. We show that a lead macrocyclic peptide epoxyketone DB-60 (20) potently inhibits the catalytic activity of the iP catalytic subunit LMP2 with an IC50 of 105 nM in cells overexpressing ABCB1 (a major efflux transporter at the BBB), suggesting minimal interactions between DB-60 (20) and ABCB1. DB-60 (20) also showed no interaction with ABCG2, another major efflux transporter that can limit the BBB penetration. Furthermore, DB-60 (20) displayed metabolic stability superior to its linear counterpart. When injected into Tg2576 mice, DB-60 (20) lowered the serum levels of IL-1α and improved cognitive function while showing no gross adverse effects. The results suggest that iP-targeting macrocyclic peptide epoxyketones may offer a new AD drug class that warrants further investigation for clinical potential.
RESULTS AND DISCUSSION
Structure-guided design of macrocyclic peptide epoxyketones.
To assess the feasibility of macrocycle formation, we first built an LMP2 (PDB ID: 3UNF) molecular model complexed with DB-310, previously developed by us as an LMP2-selective linear peptide epoxyketone.23 As shown in Figure 2, both P2-phenylalanine (blue circle) and P4-glycine/N-cap-pyrazine (pink circle) residues of DB-310 are predicted to extend to the surface of the LMP2 catalytic cavity, potentially providing structural flexibility necessary to form a macrocyclic ring between the P2 and P3/P4 residues without disrupting its interactions with LMP2. Based on the molecular analysis, the P3-proline is expected to play a crucial role as an anchor residue by allowing P2 and P3/P4 residues to be positioned for macrocyclization (Figure 2b).
FIGURE 2.

The structure-aided design strategy for macrocyclic peptide epoxyketones. (a) Predicted docking model of DB-310 bound to LMP2 (PDB ID: 3UNF) from the mouse 20S immunoproteasome. (b) A proposed macrocyclization strategy between the P2 and P3/P4 residues.
Chemistry.
Our synthetic approach relied on a convergent strategy that separately synthesizes ά,β´-epoxy amino acid right-hand fragments and macrocyclic peptide left-hand fragments before coupling via a conventional amide bond formation reaction. The right-hand epoxyketone fragments of macrocyclic compounds were prepared from Boc-protected respective amino acids (1) using a previously reported procedure to yield Boc-deprotected 4a-4c (Scheme 1a).23 For the macrocyclic left-hand fragments (Scheme 1b), dipeptide intermediates 6a and 6b were first prepared from serine or tyrosine benzyl ester (5) using conventional amide coupling reactions. O-alkylation of 6a in the presence of potassium carbonate furnished the terminal alkene intermediate 7 in good yield. On the other hand, O-alkylated terminal alkene 10 was prepared from 6b using allyl methyl carbonate and a palladium catalyst, whereas 13 was prepared by adding Boc-Gly to the N-terminus of 10. Di-ene intermediates (8, 11, and 14) were prepared from their respective monoalkene precursors (7, 10, and 13, respectively) via conventional amide coupling reactions.
SCHEME 1. Synthetic strategy for macrocyclic analogs of DB-310.

Reagents and Conditions: a. (a) HN(CH3)OCH3, HOBt, EDCI.HCl, DIPEA, DCM, rt, 12 h; (b) Isopropenylmagnesium bromide, THF, −78 °C, 12 h; (c) i. Benzonitrile, H2O2, DIPEA, methanol, 0 °C to rt, 2 h. ii. TFA, DCM, rt, 1 h, then evaporated and vacuum-dried. b. (a) HBTU, HOBt, DIPEA, DCM, rt, 18 h; (b) Potassium carbonate, 4-Bromo-1-butane, DMF, rt, 6 h; (c) Allyl methyl carbonate, Pd(PPh3)4, THF, 60 °C, 3 h; (d) i. TFA, DCM, rt, 1 h, then evaporated and dried. ii. N-Boc glycine (for compound 13) or alkenyl carboxylic acid, HBTU, HOBt, DIPEA, DCM, rt, 18 h; (e) i. Grubb’s second-generation catalyst, toluene, 90 °C, 1 h, purified by flash column chromatography. ii. H2, Pd/C, methanol, rt, 1 h; (f) Amine deprotected epoxy ketone (4a, 4b or 4c), HBTU, HOBt, DIPEA, DCM, rt, 18 h.
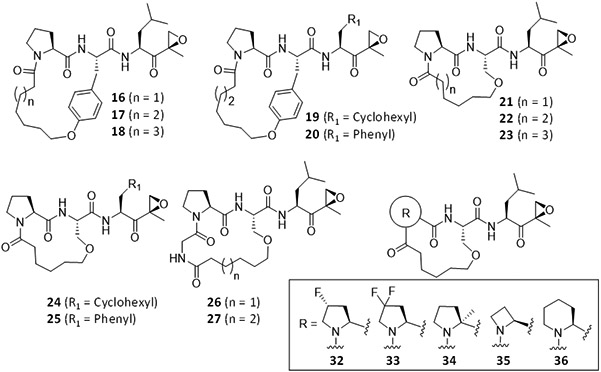
A ring-closing metathesis reaction in the presence of Grubb's second-generation catalyst provided alkene-bridged macrocyclic analogs. Reduction of the cycloalkenes and deprotection of the benzyl ester was simultaneously performed via hydrogenation to afford the macrocyclic left-hand fragments (9, 12, and 15), which were then coupled to the right-hand epoxyketone fragments (4a-4c) to provide the final products (16-27) in moderate to good yields.
Compound 21 (n = 1), chosen based on the initial assessment of its inhibitory potency, selectivity toward LMP2, and low molecular weight, was further modified to provide additional macrocyclic peptide epoxyketones, in which its P3-proline anchor was substituted by proline mimics having different ring size, fluorinated or methylated proline (Scheme 2). Due to the common constrained macrocyclic structures, we expected that these additional analogs would have similar molecular properties as 21.
SCHEME 2. Synthesis of macrocyclic analogs of DB-310 with P3-proline substitutions.

Reagents and Conditions: (a) Allyl methyl carbonate, Pd(PPh3)4, THF, 60 °C, 3 h; (b) TFA, DCM, rt, 1 h then evaporated and dried, N-Boc proline derivative, HBTU, HOBt, DIPEA, DCM, rt, 18 h; (c) 1. TFA, DCM, rt, 1 h then evaporated and dried; 2. alkenyl carboxylic acid, HBTU, HOBt, DIPEA, DCM, rt, 18 h; (d) 1. Grubb’s second-generation catalyst, toluene, 90 °C, 1 h, purified by flash column chromatography; 2. H2, Pd/C, methanol, rt, 1 h; (e) Amine deprotected epoxyketone (4a), HBTU, HOBt, DIPEA, DCM, rt, 18 h.
SAR analysis of macrocyclic peptide epoxyketones.
Seventeen macrocyclic analogs of DB-310 were successfully synthesized and tested for their in vitro activity using purified proteasomes and subunit-selective fluorogenic substrates (Table 1). Most of the macrocyclic compounds inhibited the iP (primarily LMP2) with high selectivity and good potency. Notably, the tetrapeptide backbone-based macrocyclic compounds (26 and 27, resulted from a macrocyclization between P2-serine and P4-glycine) potently inhibited LMP2, with an IC50 value comparable to the previously optimized LMP2-selective linear peptide epoxyketone DB-310: 65.6-69.4 nM for 26 and 27, and 70.8 nM for DB-310. While the tripeptide-based products with P2-tyrosine (16-20) were overall effective against the iP (for LMP2, IC50 = 129-420 nM), 17-19 containing a longer linker or cyclohexyl residue at the P1 exhibited additional inhibitory activity against the constitutive proteasome (cP) subunits X and Y. The tripeptide-based macrocyclic analogs with P2-serine (21-25) also provided good IC50 values against LMP2 (158-563 nM) but with higher selectivity for the iP (specifically LMP2) than the P2-tyrosine-containing ones. Similarly, the P1-leucine substitution with cyclohexyl or phenyl residues (17 vs. 19 and 20; 21 vs. 24 and 25) yielded only mild variations in the proteasome inhibition profile. On the other hand, the P3-proline substitution of 21 with 4-(cis)-fluoroproline (32), 2-methylproline (34) or cyclohexane (36) resulted in a substantial decrease in the LMP2 inhibitory potency, while 33 (with P3-4,4-difluoroproline) and 35 (with P3-azetidine) displayed LMP2 inhibitory potency similar to 21. Collectively, we chose the most potent LMP2 inhibitors (26 and 27), two P2-tyrosine-derived macrocyclic compounds (17 and 20, lacking X inhibition), and a P2-serine-derived one (21, having the smallest MW) for further testing.
TABLE 1.
In vitro proteasome inhibition profiles by macrocyclic peptide epoxyketones. a
| ||||
|---|---|---|---|---|
| IC50 (nM) | ||||
| LMP2 | Y | LMP7 | X | |
| YU102 | 105.2 ± 6.2 | 206.7 ± 5.5 | >10,000 | >10,000 |
| KZR-504 | 157.9 ± 8.5 | 4,763 ± 54 | >10,000 | >10,000 |
| DB-310 | 70.8 ± 1.7 | 589.9 ± 4.7 | >10,000 | >10,000 |
| 16 | 422.8 ± 68.0 | ND | >10,000 | >10,000 |
| 17 | 129.3 ± 21.8 | 715.1 ± 88.0 | 1,516 ± 337 | 4,766 ± 549 |
| 18 | 414.5 ± 30.1 | 414.5 ± 62.7 | 341.8 ± 50.9 | 4,998 ± 704 |
| 19 | 167.7 ± 39.4 | ND | 2,686 ± 489 | 4,998 ± 704 |
| 20 | 184.1 ± 19.0 | 8,385 ± 847 | 399.3 ± 27.6 | 4,640 ± 781 |
| 21 | 412.8 ± 151.1 | 1,503 ± 398 | >10,000 | 1,081 ± 223 |
| 22 | 563.4 ± 87.0 | ND | >10,000 | 4,640 ± 781 |
| 23 | 158.1 ± 30.4 | ND | >10,000 | >10,000 |
| 24 | 522.6 ± 67.5 | ND | 5,773 ± 1007 | >10,000 |
| 25 | 251.1 ± 35.8 | ND | >10,000 | >10,000 |
| 26 | 69.4 ± 12.8 | ND | >10,000 | >10,000 |
| 27 | 65.6 ± 14.5 | ND | >10,000 | >10,000 |
| 32 | 2,131 ± 383 | >10,000 | >10,000 | >10,000 |
| 33 | 237.7 ± 24.4 | ND | >10,000 | >10,000 |
| 34 | 4,583 ± 601 | ND | >10,000 | >10,000 |
| 35 | 261.5 ± 53.2 | ND | >10,000 | >10,000 |
| 36 | 1,245 ± 200 | ND | >10,000 | >10,000 |
The activity of individual proteasome subunits was measured using purified human 20S proteasomes and the respective fluorogenic substrates, Ac-PAL-AMC (for LMP2), Ac-nLPnLD-AMC (for Y), Ac-ANW-AMC (for LMP7), and Ac-WLA-AMC (for X). Data were obtained based on the results of 3 replicates per compound. N.D. denotes “not determined.”
Macrocyclic compounds are more resistant to the ABCB1-mediated efflux than linear counterparts.
To assess the potential ability to cross the BBB, we evaluated the interaction of the representative macrocyclic inhibitors (17, 20, 21, 26, and 27) with ABCB1 by comparing their LMP2 inhibitory potency in the cell line model overexpressing ABCB1 (RPMI-8226/ABCB1, previously established by us30) to that in the parental RPMI-8226 cells. Linear peptide epoxyketones, including carfilzomib (a known substrate of ABCB130, 39, 40), were used for comparison. As expected, carfilzomib required higher concentrations to inhibit the proteasome chymotrypsin-like (CT-L) activity in RPMI-8226/ABCB1 cells than in parental RPMI-8226 cells (~39-fold difference in IC50 values, Figure 3a). The potency of two linear LMP2 inhibitors was also affected by ABCB1 (6.9-fold for YU102 and 7.3-fold KZR-504), but to a lesser extent than carfilzomib. The fold differences in the LMP2 inhibitory potency of the macrocyclic compounds were notably less than the linear counterparts: 3.3-, 4.1-, and 4.7-fold for 20, 21, and 17, respectively. On the other hand, the tetrapeptide-based macrocyclic compounds 26 and 27 displayed no LMP2 inhibition in either cell line (likely attributable in part to low membrane permeability), despite their potent LMP2 inhibitory activity in purified human 20S proteasomes (Table 1). The LMP7 inhibition in RPMI-8226/ABCB1 and RPMI-8226 parental cells by compound 20 showed a similar pattern (a ~2-fold decrease, Supple. Table S1) as the LMP2 inhibition by 20, confirming a moderate interaction between 20 and ABCB1. Similar results were obtained when 20 was incubated with another ABCB1-overexpressing cell line (NCI/ADR-RES in the presence and absence of ABCB1 inhibitor, verapamil (Supple. Table S2). These results cautiously support that the tripeptide-based macrocyclic peptide epoxyketones (17, 20, and 21) are affected by the ABCB1-mediated efflux to a much lesser extent than linear counterparts.
FIGURE 3.

(a) LMP2 inhibitory activity of DB-60 (20) and other compounds in ABCB1-overexpressing RPMI-8226 cells. ABCB1-overexpressing RPMI-8226 cells or the parental RPMI-8226 cells were incubated with varying concentrations of respective compounds (n=3 replicates per concentration). After 72 hours, cell lyses were prepared to measure proteasome catalytic subunit activities (LMP2 and LMP7). Curve fitting analysis was performed to calculate IC50 values reported as the mean ± SD. *Due to carfilzomib's cytotoxic effect (a dual inhibitor of cP and iP), cells were incubated for 3 hours, followed by the measurement of chymotrypsin-like (CT-L) activity. (b) Lack of interactions between DB-60 (20) and ABCG2. The inhibitory interactions with ABCG2 were assessed by measuring the changes in the cellular accumulation of pheophorbide A (PhA, a fluorescent ABCG2 substrate) using MDCKII cells stably expressing ABCG2 (established in our previous study41) and parental cells. The known ABCG2 inhibitor Ko143 was used as a positive control. After cells were preincubated in the complete medium containing PhA (1 μM) with Ko143 (0.2 μM) or DB-60 (20) (2 or 5 μM) at 37°C for 30 min, cells were then washed with ice-cold medium and incubated again with Ko143 or DB-60 (20) for 45 min at 37°C. Subsequently, the fluorescent signal was measured via flow cytometry using the excitation and emission wavelengths at 635 and 670 nm, respectively. Colored lines represent the following groups: grey, PhA only; orange, PhA in the presence of 20 (2 μM); red, PhA in the presence of 20 (5 μM); purple, PhA in the presence of Ko143 (0.2 μM) (n=2 per group).
For compound 20 (dubbed "DB-60") (least affected by the ABCB1-mediated efflux in cell line studies), we further examined for its potential interaction with ABCG2, another major efflux transporter that can limit the BBB penetration.42 We used the MDCKII cells stably expressing ABCG2 to test whether DB-60 (20) can inhibit the efflux of the fluorescent probe substrate pheophorbide A (PhA). The known ABCG2 inhibitor Ko143 (0.2 μM) markedly increased cellular accumulation of PhA as expected (purple-colored distribution showing a rightward shift, left panel, Figure 3b). Under the same conditions, DB-60 (20) (2 or 5 μM) showed no inhibition of the ABCG2-mediated efflux of PhA (leading to no red-colored shift, left panel, Figure 3b), suggesting the lack of interactions between DB-60 (20) and ABCG2. The extent of LMP2 inhibition by DB-60 (20) was similar between MDCKII/ABCG2 and parental MDCKII cells (Supple. Table S3). Based on these results, we then selected DB-60 (20) as a lead macrocyclic LMP2 inhibitor for further study.
Metabolic stability and microglial LMP2 inhibition efficiency of DB-60 (20).
To assess whether macrocyclic peptide epoxyketones are more resistant to enzymatic metabolism than linear peptide epoxyketones, we examined the metabolic stability of DB-60 (20) using whole blood and liver homogenates harvested from ICR mice (male, 7-week-old). The linear counterpart YU102 (containing the same P3-proline as DB-60) was included for comparison. As shown in Figure 4a, DB-60 (20) displayed greater metabolic stability than YU102. In particular, DB-60 (20) stability in liver homogenates, rich in metabolizing enzymes, including epoxide hydrolases and peptidases (major contributors to the metabolism of linear peptide epoxyketones), was superior to YU102. DB-60 (20) was also more effective in suppressing the catalytic activity of LMP2 in inflammatory microglial cells than the linear compounds (YU102, KZR-504) (left panel, Figure 4b). Besides, the treatment of DB-60 (20) decreased the release of IL-1α by LPS-activated microglial BV-2 cells (right panel, Figure 4b), further verifying that iP-targeting macrocyclic peptide epoxyketones retain the inflammation suppression activity of linear counterparts mediated by iP inhibition.23 Encouraged by these results, we investigated the brain permeability of DB-60 (20) via single intravenous dosing (10 mg/kg) to the ICR mice (Supple. Figure S1). When we analyzed brain tissue homogenates at 1-hour post-dosing, the brain homogenates contained a detectable but low level of compound DB-60 (20) (3.50 ± 1.46 pmole/g tissue). When the same brain homogenates were analyzed for LMP2 activity (Supple. Figure S1), however, the results showed no appreciable inhibition of the LMP2 activity (Supple. Figure S1).
FIGURE 4.

(a) In vitro metabolic stability of DB-60 (20) in the whole blood or liver homogenates from mice. The whole blood and the liver were collected from ICR mice and incubated with the test compounds for the indicated time. The remaining drug levels were quantified (details provided in the methods). P values were calculated using Student’s t-test (GraphPad Prism, v.8.4.1). (b) Effects of DB-60 (20) on LMP2 inhibition and IL-1α release in microglial BV-2 cells stimulated with lipopolysaccharide (LPS) (1 μg/mL). Error bars indicate standard deviation derived from three technical replicates.
DB-60 (20) ameliorates cognitive impairment in Tg2576 mice.
Despite modest brain permeability shown in the single intravenous dosing study, we examined whether macrocyclic iP inhibitors are efficacious in a mouse model of AD. Specifically, DB-60 (20) (10 mg/kg) was injected to Tg2576 mice (9-month old) twice weekly for 3 weeks, followed by the assessment of cognitive function via the Morris water maze test for 5 trial days, a single probe trial on day 6, and passive avoidance test on days 7 and 8.22 Similar to the results obtained from its linear counterparts,22, 23 mice treated with DB-60 (20) improved the cognitive function of Tg2576 mice. Specifically, Tg2576 mice treated with DB-60 (20) were more efficient at locating the platform in a pool of water during training days 4 and 5, thus escaping the water with lower latency and shorter distance compared to vehicle-treated mice (Figure 5a).
FIGURE 5.

(a) DB-60 (20) improves cognitive function in Tg2576 mice. The Morris water maze test was performed to measure cognitive function: escape distance (upper) and escape latency time (lower). Statistical analyses were performed using two-way ANOVA. *Difference in days 4-5 between control and DB-60 (20)-treated mice was statistically significant (p-value < 0.05, n=12 for control and n=5 for DB-60 (20)-treated mice). (b) The probe trial (on day 6, upper panel) and passive avoidance test (on days 7-8, lower panel) were performed following the Morris water maze test on days 1-5. The student’s t-test was used for statistical analyses of probe trials and passive avoidance. Differences in time spent in target quadrant or step through latency between control and DB-60 (20)-treated mice were statistically significant (p-value < 0.05, n=5). (c) Serum samples collected from mice treated with vehicle or DB-60 (20) were used to quantify IL-1α levels via ELISA (p-value <0.05, n=3).
The probe trial results showed that the percentages of time spent in the target quadrant (the quadrant with the hidden platform) for DB-60 (20)-treated Tg2576 mice were higher than control mice (upper panel, Figure 5b), demonstrating improved spatial learning and memory in drug-treated Tg2576 mice. Similarly, mice treated with DB-60 (20) performed better in a fear-aggravated test (passive avoidance) than control mice (lower panel, Figure 5b). The serum levels of IL-1α were lower in Tg2576 mice receiving DB-60 (20) treatment than the control mice receiving the vehicle only (Figure 5c), demonstrating an impact of DB-60 (20)-mediated iP inhibition in inflammatory cytokine production. Taken together, we observed that the multiple dosing of DB-60 (20) improved cognitive function in AD mice. As no appreciable brain LMP2 inhibition was observed innon-diseased mice that received a single dosing of DB-60 (20) (Supple. Figure S1), we hypothesize that the efficacy of DB-60 (20) in mouse AD models may be due to the gradual accumulation of LMP2:DB-60 (20) covalent adduct in the brain resulting from repeated administration.
DB-60 (20) protects mouse retinal pigment epithelial (RPE) from inflammation-triggered structural destruction.
As visual evidence for the potential involvement of the iP in neuroinflammatory responses, we previously reported that iP-targeting linear peptide epoxyketones inhibit RPE (retinal pigment epithelium) degeneration caused by Aβ-triggered inflammation in Tg2576 mice.22, 23 We examined whether the macrocyclic DB-60 (20) can also suppress inflammation-triggered RPE degeneration by assessing the RPE samples from Tg2576 mice that completed the Morris water maze and probe/passive avoidance tests. When immunostained using an anti-β-catenin antibody, RPE's orderly standard structure was prominent in age-matching, non-transgenic ICR mice (upper panel, Figure 6a). In contrast, the orderly structure of RPE was severely damaged in Tg2576 mice treated with vehicle only (upper panel, Figure 6b), as reported previously by us and others.22, 23, 43
FIGURE 6.

Protection of Tg2576 mice from retinal pigment epithelium (RPE) destruction by DB-60 (20). Upon completing behavior tests for Tg2576 mice treated with DB-60 (20), RPE samples were collected and immunostained using an anti-β-catenin antibody for visualization. A parallel experiment was also performed using age-matching non-transgenic ICR mice. The images are representative of 3 independent replicates.
On the other hand, RPE collected from Tg2576 mice treated with DB-60 (20) displayed the orderly structure (lower panel, Figure 6a), demonstrating the ability of DB-60 (20) to block RPE destruction caused by Aβ-triggered inflammation. RPE's structural integrity in non-transgenic ICR mice was not affected by DB-60 (20) treatment (lower panel, Figure 6a). No sign of overt toxicity was observed in Tg2576 or non-transgenic ICR mice treated with DB-60 (20) during the test period, which is consistent with our previous results obtained from iP-targeting linear peptide epoxyketones.22, 23
CONCLUSIONS
With the failure of nearly all clinical trials for Aβ or tau-targeting AD drugs in the pipeline to date, identifying a new class of drug candidates has become imperative to bring about effective AD therapies. A major obstacle has been a lack of promising new drug targets unrelated to the events leading to the accumulation of the Aβ and tau protein. We previously reported that linear peptide epoxyketones inhibiting the iP, a previously untapped target in AD drug discovery efforts, have promising anti-AD efficacy in mouse models, potentially offering a new class of AD drugs.22, 23 However, inherent issues associated with the linear peptides prohibit further clinical development. Therefore, in the current study, we aimed to improve the iP-targeting peptide epoxyketones' CNS druggability to be further investigated as potential AD drug candidates in pre-clinical and clinical studies.
One common strategy to improve linear peptide-based drug candidates' CNS druggability is to form a macrocycle by constraining the freely rotatable linear peptide backbone. To this end, we were able to synthesize 17 macrocyclic peptides retaining epoxyketone pharmacophore. These macrocyclic peptide epoxyketones are highly selective to the iP, unlike the previously reported macrocyclic analogs of the anti-cancer agent oprozomib, which target both the cP and iP.44 Besides, these compounds are structurally unique, having a proline or proline mimic at the P3 position as a handle for P2-P3 or P2-P4 macrocyclization. In particular, our lead macrocyclic iP inhibitor, DB-60 (20), showed significantly improved metabolic stability and decreased interaction with the two major drug transporters in the BBB, ABCB1 and ABCG2. Further, DB-60 (20) effectively reduced serum IL-1α and enhanced the cognitive function of Tg2576 mice. Our results support that iP-targeting macrocyclic peptide epoxyketones have promising potential for further pre-clinical and clinical investigations as AD drugs, not reliant on the strategies of targeting Aβ or tau protein accumulation.
In addition to its role in cognitive deficits, accumulating evidence support that Aβ-triggered inflammation plays a crucial role in RPE degeneration in APP (human amyloid precursor protein) transgenic mouse models including Tg2576.24, 25, 45-47 The pathological role of Aβ in the eye has been further supported by the detection of Aβ in RPE and drusen (extracellular waste deposits that accumulate near the RPE region) obtained from patients with age-related macular degeneration (AMD).43, 48-50 This has led researchers to suggest a common neuroinflammation-associated pathogenic mechanism between AMD and AD. Accordingly, the idea of using the retina structure as a potential diagnostic tool in AD has been proposed lately.51, 52 In the current study, the iP-selective inhibitor DB-60 (20) completely protected RPE from an inflammation-triggered structural abnormality in Tg2576 mice, supporting that the iP is potentially linked to the RPE degeneration and AD. Taken together, iP-targeting macrocyclic peptide epoxyketones may also have the potential for AMD treatment.
EXPERIMENTAL SECTION
Chemistry.
All the reagents and solvents used in experiments were purchased commercially and used without additional purification. A pre-coated TLC plate was used to monitor the reaction progress. The different spots were detected by UV or/and staining solutions such as phosphomolybdic acid hydrate (PMA), iodine, and potassium permanganate. All the reactions were performed under inert conditions unless otherwise mentioned. Purification of intermediates and final compounds were performed in flash column chromatography on 60 Å silica gel, 230–400 mesh (Merck) unless otherwise reported. 400 MHz Varian spectrometer was used to record 1H and 13C NMR spectra. Chemical shifts were reported in parts per million (ppm) and coupling constants (J) in Hz with tetramethylsilane as a reference standard. Proton NMR multiplicities were reported using different abbreviations such as singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). LC/MS of final compounds were recorded in an Agilent Technologies 6120 Quadrupole using 0.1% formic acid solution of A: H2O and B: acetonitrile (CH3CN) as eluent. Agilent Eclipse XBD-C18 column with 5-μm particle size, 4.6 mm diameter, and 150 mm length was used in the system with solvent gradient B/A from 5:95 to 90:10 over 18 min and then 100:0 over 7 min. The flow rate of eluent was 0.4 mL/min. The same solvent gradient, column, and flow rate were used to determine the purity of all final compounds in an Agilent Technologies 1200 series equipped with a UV detector at 254 nm. The purity of most of the final compounds was determined to be ≥ 95%. KZR-504 and YU102 used in experiments were synthesized using the previously reported method.
General procedure for Boc deprotection reaction.
Excess of TFA was added to the stirred solution of Boc protected intermediate in DCM at room temperature. The reaction mixture was further stirred at the same temperature for 2 hours. The reaction solution was concentrated under reduced pressure, and the resulting residue was dried under a strong vacuum. Dry TFA salt was used in the next step without further purification.
General procedure for the synthesis of macrocyclic carboxylic acids.
The second-generation Grubbs catalyst (10 mol%) was added at room temperature to the stirred solution of diolefin in toluene. The reaction temperature was increased gradually to 100 °C, and the reaction solution was stirred for 1 hour at the same temperature. After cooling, the solvent was removed under reduced pressure, and the resulting residue was purified by flash chromatography using 50% ethyl acetate in hexane. The above-purified product (a mixture of geometric isomers) was dissolved in methanol, activated palladium in charcoal (10 mol%) was added, and the reaction mixture was stirred under a hydrogen gas environment at room temperature for 2 hours. The reaction solution was then filtered through celite, and the filtrate was concentrated to yield the corresponding macrocyclic carboxylic acid, which was used in the next step without further purification.
(S)-tert-Butyl 1-(methoxy(methyl)amino)-4-methyl-1-oxopentan-2-ylcarbamate (2a) (General procedure for the preparation of Weinreb amide 2a-2c). To the DCM solution of Boc-leucine (2 g, 8.64 mmol), EDCI.HCl (2.48 g, 12.96 mmol), HOBt (1.17 g, 8.64 mmol), N,O-dimethylhydroxylamine hydrochloride (0.84 g, 8.64 mmol) and DIPEA (3.77 mL, 21.6 mmol) were added. The reaction solution was stirred at same temperature overnight. Solvent was removed under reduced pressure and residue was purified by flash column chromatography using 20% ethyl acetate in hexane to yield pure product as sticky colorless solid (2.1 g, 89%). 1H NMR (400 MHz, CDCl3) δ 5.05 (d, J = 9.5 Hz, 1H), 4.70 (s, 1H), 3.76 (s, 3H), 3.17 (s, 3H), 1.69 (dt, J = 6.7, 13.5 Hz, 1H), 1.46-1.37 (m, 11H), 0.92 (dd, J = 6.6, 14.0 Hz, 6H).
(S)-tert-Butyl 3-cyclohexyl-1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate (2b). 2b was prepared from 1b using same general procedure for the preparation of Weinreb amide as colorless oil (90%). 1H NMR (400 MHz, CDCl3) δ 5.00 (d, J = 9.5 Hz, 1H), 4.71 (s, 1H), 3.75 (s, 3H), 3.16 (s, 3H), 1.87 (d, J = 13.0 Hz, 1H), 1.72 – 1.53 (m, 4H), 1.40 (s, 12H), 1.17 (m, 3H), 1.01 – 0.78 (m, 2H).
(S)-tert-Butyl 1-(methoxy(methyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (2c). 2c was prepared from 1c using same general procedure for the preparation of Weinreb amide as colorless thick oil (86%). 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.2 Hz, 2H), 7.24 – 7.19 (m, 1H), 7.15 (d, J = 7.4 Hz, 2H), 5.13 (d, J = 9.0 Hz, 1H), 4.93 (s, 1H), 3.64 (s, 3H), 3.15 (s, 3H), 3.03 (dd, J = 6.1, 13.6 Hz, 1H), 2.86 (t, J = 10.5 Hz, 1H), 1.37 (s, 9H).
(S)-tert-Butyl 2,6-dimethyl-3-oxohept-1-en-4-ylcarbamate (3a) (General procedure for the preparation of enone 3a-3c). 0.5 M isopropenylmagnesium bromide solution in THF (58 mL, 29.15 mmol) was added dropwise to the stirred solution of 2a (2 g, 7.28 mmol) in THF at −78 °C, and the reaction mixture was stirred at the same temperature overnight. The reaction solution was warmed gradually to room temperature, and the reaction was quenched with a saturated solution of ammonium chloride. The reaction product was extracted with ethyl acetate 3 times. The combined organic layer was dried in sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography using 5% ethyl acetate in hexane to yield the pure product as a colorless oil (1.2 g, 64%). 1H NMR (400 MHz, CDCl3) δ 6.06 (s, 1H), 5.86 (s, 1H), 5.20 – 4.96 (m, 2H), 1.83 (s, 3H), 1.81 – 1.64 (m, 1H), 1.41 (s, 9H), 1.36 – 1.18 (m, 2H), 0.98 (d, J = 6.5 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H).
(S)-tert-Butyl 1-cyclohexyl-4-methyl-3-oxopent-4-en-2-ylcarbamate (3b). 3b was prepared from 2b using same general procedure for the preparation of enone as white solid (76%). 1H NMR (400 MHz, CDCl3) δ 6.04 (s, 1H), 5.84 (s, 1H), 5.07 (d, J = 6.7 Hz, 2H), 1.87 (s, 3H), 1.75 – 1.46 (m, 6H), 1.41 (s, 10H), 1.32 – 1.05 (m, 4H), 1.00 – 0.78 (m, 2H).
(S)-tert-Butyl 4-methyl-3-oxo-1-phenylpent-4-en-2-ylcarbamate (3c). 3c was prepared from 2c using same general procedure for the preparation of enone as white solid (79%). 1H NMR (400 MHz, CDCl3) δ 7.30 – 7.12 (m, 3H), 7.05 (d, J = 7.2 Hz, 2H), 5.99 (s, 1H), 5.83 (p, J = 1.5 Hz, 1H), 5.25 (d, J = 8.7 Hz, 2H), 3.19 – 3.01 (m, 1H), 2.97 – 2.80 (m, 1H), 1.84 (s, 3H), 1.39 (s, 9H).
tert-Butyl (S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-ylcarbamate (4a) (General procedure for the preparation of epoxide 4a-4c). DIPEA (6.83 mL, 39.14 mmol) was added dropwise in the stirred solution of 3a (1 g, 3.91 mmol), hydrogen peroxide (6.64 mL, 97.75 mmol), and benzonitrile (4.0 mL, 39.14 mmol) in methanol at 0 °C. The reaction mixture was stirred at the same temperature for 3 hours. Water was added to the solution, and methanol was removed under reduced pressure. The aqueous layer was extracted with ethyl acetate 3 times. The collected organic layer was dried with sodium sulfate, filtered, and concentrated. The resulting crude product was purified by flash column chromatography using 5% ethyl acetate in hexane to yield the pure product as a sticky white solid (0.6 g, 57%).1H NMR (400 MHz, CDCl3) δ 4.82 (d, J = 8.9 Hz, 1H), 4.35 – 4.24 (m, 1H), 3.27 (d, J = 5.0 Hz, 1H), 2.87 (d, J = 5.0 Hz, 1H), 1.78 – 1.64 (m, 1H), 1.50 (s, 4H), 1.39 (s, 9H), 1.15 (ddd, J = 4.3, 10.5, 14.3 Hz, 1H), 0.93 (dd, J = 6.6, 13.0 Hz, 6H).
tert-Butyl (S)-3-cyclohexyl-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-ylcarbamate (4b). 4b was prepared from 3b using same general procedure for the preparation of epoxide sticky white solid (41%). 1H NMR (400 MHz, CDCl3) δ 4.79 (d, J = 8.8 Hz, 1H), 4.32 (td, J = 3.0, 8.6, 9.8 Hz, 1H), 3.26 (d, J = 5.1 Hz, 1H), 2.86 (d, J = 5.0 Hz, 1H), 1.93 – 1.79 (m, 1H), 1.74 – 1.51 (m, 5H), 1.49 (s, 3H), 1.39 (s, 9H), 1.28 – 1.05 (m, 5H), 1.01 – 0.84 (m, 2H).
tert-Butyl (S)-1-((R)-2-methyloxiran-2-yl)-1-oxo-3-phenylpropan-2-ylcarbamate (4c). 4c was prepared from 3c using same general procedure for the preparation of epoxide white solid (32%), 1H NMR (400 MHz, CDCl3) δ 7.33 – 7.19 (m, 3H), 7.19 – 7.09 (m, 2H), 4.98 – 4.86 (m, 1H), 4.62 – 4.49 (m, 1H), 3.27 (d, J = 5.0 Hz, 1H), 3.09 (dd, J = 5.0, 13.9 Hz, 1H), 2.88 (d, J = 5.0 Hz, 1H), 2.72 (dd, J = 7.8, 13.9 Hz, 1H), 1.48 (s, 3H), 1.35 (s, 9H).
(S)-tert-Butyl 2-((S)-1-(benzyloxy)-3-(4-hydroxyphenyl)-1-oxopropan-2-ylcarbamoyl)pyrrolidine-1-carboxylate (6a). General amide coupling reaction was used to prepare 6a from tyrosine benzyl ester and Boc-proline as colorless oil (88%). 1H NMR (400 MHz, CDCl3) δ 7.83 (s, 1H), 7.41 – 7.27 (m, 5H), 6.89 (d, J = 8.1 Hz, 2H), 6.76 – 6.67 (m, 2H), 5.12 (d, J = 5.9 Hz, 2H), 4.80 – 4.76 (s, 1H), 4.63 – 4.52 (m, 1H), 3.42 – 3.22 (m, 2H), 3.08 (dd, J = 5.9, 14.0 Hz, 1H), 2.95 (dd, J = 6.4, 14.0 Hz, 1H), 2.02 – 1.95 (m, 2H), 1.76 – 1.70 (m, 2H), 1.41 (s, 9H).
(S)-tert-Butyl 2-((S)-1-(benzyloxy)-3-hydroxy-1-oxopropan-2-ylcarbamoyl)pyrrolidine-1-carboxylate (6b) (General procedure for amide coupling reaction). To the stirred solution of serine benzyl ester hydrochloride (1 g, 4.31 mmol) DCM, Boc-proline (0.93 g, 4.31 mmol), HATU (3.26 g, 8.62 mmol), HOBt (1.16 g, 8.62 mmol), and DIPEA (3.0 mL, 17.42 mmol), were added respectively at room temperature. The reaction solution was stirred at the same temperature overnight. The reaction mixture was then washed with water, and the organic solution was collected. It was dried over sodium sulfate, filtered, and concentrated. The crude product was purified through flash column chromatography using 50% ethyl acetate in hexane to yield the pure product as a colorless oil (1.37 g, 81%). 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 2.6 Hz, 5H), 7.17 – 7.02 (m, 1H), 5.20 (s, 2H), 4.62 (s, 1H), 4.16 (s, 1H), 4.11 – 3.80 (m, 2H), 3.52 – 3.31 (m, 2H), 2.25 – 2.09 (m, 1H), 2.09 – 1.95 (m, 2H), 1.83 (d, J = 5.7 Hz, 2H), 1.42 (s, 9H).
(S)-tert-Butyl 2-((S)-1-(benzyloxy)-3-(4-(but-3-enyloxy)phenyl)-1-oxopropan-2-ylcarbamoyl)pyrrolidine-1-carboxylate (7). The DMF solution of 6a (0.5 g, 1.06 mmol), potassium carbonate (0.44 g, 3.18 mmol) and 4-bromo-1-butene (0.22 mL, 2.13 mmol) was stirred at room temperature for 6 h. Water was added to the reaction mixture and the product was extracted with ethyl acetate. Combined solution of ethyl acetate was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude product was purified through flash column chromatography using 50% ethyl acetate in hexane to yield pure product as colorless oil (0.45 g, 81%). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.18 (m, 5H), 6.96 – 6.81 (m, 2H), 6.71 (dq, J = 2.9, 8.9 Hz, 2H), 6.47 (s, 1H), 5.95 – 5.71 (m, 1H), 5.20 – 5.00 (m, 4H), 4.81 (s, 1H), 4.21 (d, J = 31.8 Hz, 1H), 3.93 (tq, J = 2.9, 5.5 Hz, 2H), 3.48 – 3.19 (m, 2H), 3.07 (ddq, J = 2.9, 5.5, 14.4 Hz, 1H), 2.96 (ddq, J = 2.8, 6.0, 14.1 Hz, 1H), 2.58 – 2.43 (m, 2H), 1.92 (m, 4H), 1.50 – 1.25 (s, 9H).
(S)-Benzyl 3-(4-(but-3-enyloxy)phenyl)-2-((S)-1-pent-4-enoylpyrrolidine-2-carboxamido)propanoate (8a). Boc deprotected 7 was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to prepare 8a as colorless oil (63%). 1H NMR (400 MHz, CDCl3) 7.47 – 7.21 (m, 5H), 6.91 (dd, J = 6.8, 8.8 Hz, 2H), 6.79 – 6.62 (m, 2H), 6.08 – 5.91 (m, 1H), 5.84 – 5.63 (m, 1H), 5.37 (dq, J = 1.6, 17.3 Hz, 1H), 5.25 (dq, J = 1.6, 10.5 Hz, 1H), 5.19 – 4.88 (m, 4H), 4.75 (td, J = 5.9, 7.2 Hz, 1H), 4.54 (dd, J = 1.9, 8.1 Hz, 1H), 4.45 (tt, J = 1.5, 5.3 Hz, 2H), 3.35 (ddt, J = 4.5, 9.2, 13.2 Hz, 2H), 3.07 (dd, J = 5.8, 14.0 Hz, 1H), 2.98 – 2.89 (m, 1H), 2.36 – 2.26 (m, 1H), 2.20 (ddd, J = 6.8, 8.4, 10.8 Hz, 1H), 2.16 – 2.04 (m, 2H), 2.04 – 1.86 (m, 3H), 1.81 – 1.55 (m, 3H).
(S)-Benzyl 3-(4-(but-3-enyloxy)phenyl)-2-((S)-1-hex-5-enoylpyrrolidine-2-carboxamido)propanoate (8b). Boc deprotected 7 was coupled with 5-hexenoic acid using general procedure for amide coupling reaction to prepare 8b as colorless oil (69%). 1H NMR (400 MHz, CDCl3) δ 7.50 – 7.25 (m, 5H), 6.92 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.6 Hz, 2H), 5.96 – 5.62 (m, 2H), 5.19 – 5.06 (m, 4H), 5.06 – 4.92 (m, 2H), 4.75 (ddd, J = 5.8, 6.9, 7.6 Hz, 1H), 4.54 (dd, J = 1.9, 8.1 Hz, 1H), 3.93 (t, J = 6.7 Hz, 2H), 3.44 – 3.27 (m, 2H), 3.07 (dd, J = 5.8, 14.0 Hz, 1H), 2.93 (dd, J = 6.9, 14.1 Hz, 1H), 2.50 (qt, J = 1.4, 6.7 Hz, 2H), 2.37 – 2.14 (m, 3H), 2.14 – 2.02 (m, 2H), 2.02 – 1.84 (m, 2H), 1.84 – 1.58 (m, 3H).
(S)-Benzyl 3-(4-(but-3-enyloxy)phenyl)-2-((S)-1-hept-6-enoylpyrrolidine-2-carboxamido)propanoate (8c). Boc deprotected 7 was coupled with 6-heptenoic acid using general procedure for amide coupling reaction to prepare 8c as colorless oil (70%). 1H NMR (400 MHz, CDCl3) δ 7.44 (dd, J = 2.3, 7.7 Hz, 1H), 7.37 – 7.14 (m, 5H), 6.89 (tt, J = 4.3, 8.2 Hz, 2H), 6.69 (dd, J = 2.5, 8.7 Hz, 2H), 5.81 (ddddd, J = 3.4, 6.6, 10.4, 14.1, 33.6 Hz, 2H), 5.21 – 5.01 (m, 4H), 5.01 – 4.86 (m, 2H), 4.74 (dd, J = 2.2, 7.2 Hz, 1H), 4.59 – 4.43 (m, 1H), 3.91 (td, J = 2.4, 6.7 Hz, 2H), 3.46 – 3.23 (m, 2H), 3.05 (ddd, J = 2.4, 5.9, 14.0 Hz, 1H), 2.92 (ddd, J = 2.4, 6.9, 14.1 Hz, 1H), 2.57 – 2.40 (m, 2H), 2.35 – 1.82 (m, 7H), 1.74 (dq, J = 3.5, 10.7 Hz, 1H), 1.66 – 1.49 (m, 2H), 1.41 (qd, J = 2.3, 7.8 Hz, 2H).
(S)-tert-Butyl 2-((S)-3-(allyloxy)-1-(benzyloxy)-1-oxopropan-2-ylcarbamoyl)pyrrolidine-1-carboxylate (10). To the solution of 6b (1 g, 2.54 mmol) in THF, Pd(PPh3)4 (0.15 g, 0.12 mmol), and allyl methyl carbonate (0.43 mL, 3.82 mmol) were added under the anhydrous condition at room temperature. The reaction mixture was stirred at 60 °C for 2 hours and then quenched with water. The crude product was extracted with ethyl acetate three times. The combined organic layer was dried under sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified through flash column chromatography using 30% ethyl acetate in hexane as a yellowish oil (94%). 1H NMR (400 MHz, CDCl3) δ 7.30 (d, J = 3.5 Hz, 5H), 6.83 (s, 1H), 5.71 (d, J = 16.8 Hz, 1H), 5.31 – 5.03 (m, 4H), 4.73 (s, 1H), 4.26 (d, J = 27.6 Hz, 1H), 3.98 – 3.73 (m, 3H), 3.61 (d, J = 9.1 Hz, 1H), 3.43 (d, J = 8.2 Hz, 2H), 2.10 (s, 2H), 1.84 (q, J = 6.3, 6.8 Hz, 2H), 1.42 (s, 9H).
(S)-Benzyl 3-(allyloxy)-2-((S)-1-pent-4-enoylpyrrolidine-2-carboxamido)propanoate (11a). Boc deprotected 10 was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to yield 11a as colorless oil (46%). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.25 (m, 5H), 5.94 – 5.64 (m, 2H), 5.28 – 5.07 (m, 4H), 5.07 – 4.87 (m, 2H), 4.68 (dt, J = 3.5, 7.5 Hz, 1H), 4.54 (dd, J = 2.6, 8.1 Hz, 1H), 3.97 – 3.78 (m, 3H), 3.69 – 3.48 (m, 2H), 3.41 (td, J = 6.6, 9.3 Hz, 1H), 2.47 – 2.31 (m, 4H), 2.31 – 2.13 (m, 1H), 2.13 – 1.97 (m, 1H), 1.97 – 1.80 (m, 2H).
(S)-Benzyl 3-(allyloxy)-2-((S)-1-hex-5-enoylpyrrolidine-2-carboxamido)propanoate (11b). Boc deprotected 10 was coupled with 5-hexenoic acid using general procedure for amide coupling reaction to yield 11b as colorless oil (54%). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.22 (m, 5H), 5.83 – 5.63 (m, 2H), 5.27 – 5.05 (m, 4H), 5.05 – 4.85 (m, 2H), 4.67 (dt, J = 3.5, 8.1 Hz, 1H), 4.53 (dd, J = 2.5, 8.3 Hz, 1H), 3.96 – 3.78 (m, 3H), 3.65 – 3.47 (m, 2H), 3.39 (td, J = 6.7, 9.3 Hz, 1H), 2.35 – 2.14 (m, 3H), 2.14 – 1.97 (m, 3H), 1.89 (dddd, J = 2.7, 5.6, 10.3, 11.8 Hz, 2H), 1.81 – 1.61 (m, 2H).
(S)-Benzyl 3-(allyloxy)-2-((S)-1-hept-6-enoylpyrrolidine-2-carboxamido)propanoate (11c). Boc deprotected 10 was coupled with 6-heptenoic acid using general procedure for amide coupling reaction to provide 11c as colorless oil (64%). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.18 (m, 5H), 5.73 (dtd, J = 3.4, 7.0, 17.2 Hz, 2H), 5.28 – 5.02 (m, 4H), 5.02 – 4.81 (m, 2H), 4.65 (dd, J = 3.6, 7.8 Hz, 1H), 4.52 (dd, J = 4.3, 7.2 Hz, 1H), 3.96 – 3.74 (m, 3H), 3.68 – 3.44 (m, 2H), 3.38 (dt, J = 3.1, 6.3 Hz, 1H), 2.37 – 2.11 (m, 3H), 2.11 – 1.95 (m, 3H), 1.89 (dt, J = 5.6, 16.5 Hz, 2H), 1.61 (tt, J = 5.5, 10.8 Hz, 2H), 1.39 (td, J = 2.9, 7.7 Hz, 2H).
(S)-Benzyl 3-(allyloxy)-2-((S)-1-(2-(tert-butoxycarbonylamino)acetyl)pyrrolidine-2-carboxamido)propanoate (13). Boc deprotected 10 was coupled with Boc-glycine using general procedure for amide coupling reaction to provide 13 as colorless oil (57%). 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.27 (m, 5H), 7.15 (d, J = 8.2 Hz, 1H), 5.84 – 5.66 (m, 1H), 5.41 (s, 1H), 5.29 – 5.04 (m, 4H), 4.69 (dt, J = 3.3, 8.1 Hz, 1H), 4.53 (dd, J = 2.7, 8.1 Hz, 1H), 4.03 – 3.78 (m, 5H), 3.63 (dd, J = 3.4, 9.6 Hz, 1H), 3.52 (ddd, J = 3.2, 8.1, 9.9 Hz, 1H), 3.38 (td, J = 6.7, 9.2 Hz, 1H), 2.31 – 2.17 (m, 1H), 2.17 – 2.02 (m, 1H), 1.95 (tdd, J = 4.1, 7.4, 9.6 Hz, 2H), 1.42 (s, 9H).
(S)-Benzyl 3-(allyloxy)-2-((S)-1-(2-pent-4-enamidoacetyl)pyrrolidine-2-carboxamido)propanoate (14a). Boc deprotected 13 was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to prepare 14a as colorless oil (78%). 1H NMR (400 MHz, CDCl3) δ 7.32 (m, 5H), 7.03 (d, J = 8.2 Hz, 1H), 6.47 (s, 1H), 5.77 (dddd, J = 4.9, 10.6, 16.1, 17.9 Hz, 2H), 5.33 – 5.18 (m, 2H), 5.17 – 4.91 (m, 4H), 4.71 (dt, J = 3.3, 8.1 Hz, 1H), 4.52 (dd, J = 3.0, 8.1 Hz, 1H), 4.09 (dd, J = 4.7, 17.8 Hz, 1H), 4.02 – 3.83 (m, 4H), 3.64 (dd, J = 3.3, 9.5 Hz, 1H), 3.55 (ddd, J = 3.2, 7.9, 10.7 Hz, 1H), 3.42 (dd, J = 6.8, 9.3 Hz, 1H), 2.45 – 2.18 (m, 5H), 2.18 – 2.03 (m, 1H), 1.98 (dtd, J = 4.0, 7.3, 13.4 Hz, 2H).
(S)-Benzyl 3-(allyloxy)-2-((S)-1-(2-hex-5-enamidoacetyl)pyrrolidine-2-carboxamido)propanoate (14b). Boc deprotected 13 was coupled with 5-hexenoic acid using general procedure for amide coupling reaction to prepare 14b as colorless oil (68%). 1H NMR (400 MHz, CDCl3) δ 7.40 – 7.26 (m, 5H), 7.07 (d, J = 8.2 Hz, 1H), 6.53 (t, J = 4.2 Hz, 1H), 5.84 – 5.64 (m, 2H), 5.32 – 5.06 (m, 4H), 5.06 – 4.90 (m, 2H), 4.71 (dt, J = 3.3, 8.2 Hz, 1H), 4.51 (dd, J = 3.0, 8.0 Hz, 1H), 4.10 (dd, J = 4.6, 17.8 Hz, 1H), 4.03 – 3.82 (m, 4H), 3.63 (dd, J = 3.3, 9.5 Hz, 1H), 3.55 (ddd, J = 3.4, 7.9, 10.6 Hz, 1H), 3.41 (dt, J = 7.6, 9.9 Hz, 1H), 2.31 (t, J = 7.5 Hz, 1H), 2.27 – 2.15 (m, 3H), 2.15 – 1.90 (m, 4H), 1.79 – 1.62 (m, 2H).
(12S,4S)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-2,14-dioxo-7-oxa-3-aza-1(2,1)-pyrrolidina-6(1,4)-benzenacyclotetradecaphane-4-carboxamide (16). Macrocyclic carboxylic acid intermediate 9a was prepared from 8a using general procedure for synthesis of macrocyclic carboxylic acid. 9a was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 16 as white solid (41%). 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.3 Hz, 1H), 7.01 – 6.89 (m, 2H), 6.86 – 6.76 (m, 2H), 5.85 (d, J = 9.3 Hz, 1H), 4.69 – 4.57 (m, 2H), 4.44 (td, J = 3.6, 9.2 Hz, 2H), 4.06 (td, J = 2.4, 11.7 Hz, 1H), 3.55 – 3.39 (m, 3H), 3.38 (d, J = 5.0 Hz, 1H), 2.88 – 2.81 (m, 1H), 2.64 (dd, J = 6.3, 13.8 Hz, 1H), 2.22 (ddd, J = 3.4, 6.8, 13.7 Hz, 1H), 2.18 – 2.07 (m, 1H), 2.07 – 1.78 (m, 6H), 1.66 – 1.48 (m, 2H), 1.48 – 1.29 (m, 7H), 1.29 – 1.17 (m, 2H), 1.11 (d, J = 3.3 Hz, 1H), 0.97 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H).13C NMR (101 MHz, CDCl3) δ 207.42, 173.50, 171.00, 169.98, 156.20, 130.52, 127.24, 115.85, 66.97, 61.00, 59.19, 52.31, 52.29, 50.24, 47.59, 38.64, 35.41, 33.43, 28.92, 26.38, 24.91, 24.61, 23.63, 23.13, 21.28, 20.65, 16.75. LCMS (ES+) m/z calcd for C30H44N3O6 [M+H]+ 542.3, found: 542.3; Purity: ≥ 96% and retention time is 19.07 min by HPLC analysis.
(12S,4S)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-2,15-dioxo-7-oxa-3-aza-1(2,1)-pyrrolidina-6(1,4)-benzenacyclopentadecaphane-4-carboxamide (17). Macrocyclic carboxylic acid intermediate 9b was prepared from 8b using general procedure for synthesis of macrocyclic carboxylic acid. 9b was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 17 as white solid (48%). 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.3 Hz, 1H), 6.98 – 6.90 (m, 2H), 6.86 – 6.74 (m, 2H), 6.16 (d, J = 9.0 Hz, 1H), 4.73 – 4.54 (m, 2H), 4.46 (dd, J = 3.1, 8.5 Hz, 1H), 4.11 (t, J = 5.4 Hz, 2H), 3.56 – 3.38 (m, 3H), 3.37 (d, J = 5.2 Hz, 1H), 2.85 (d, J = 5.1 Hz, 1H), 2.67 (dd, J = 5.5, 13.9 Hz, 1H), 2.27 (ddd, J = 2.6, 5.6, 12.0 Hz, 1H), 2.15 – 1.90 (m, 5H), 1.71 (dt, J = 6.8, 10.3 Hz, 3H), 1.50 – 1.25 (m, 10H), 0.94 (d, J = 6.1 Hz, 3H), 0.86 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 207.66, 173.74, 170.95, 170.42, 158.22, 130.52, 130.33, 127.20, 114.82, 66.64, 61.02, 59.15, 52.79, 52.33, 50.16, 47.84, 38.96, 35.46, 34.61, 28.70, 27.83, 27.74, 26.28, 24.97, 24.57, 23.58, 23.24, 22.29, 20.87, 16.74. HRMS m/z calcd for C31H46N3O6 [M+H]+ 556.3342, found: 556.3393. LCMS (ES+), found: 556.3; Purity: ≥ 96% and retention time is 19.20 min by HPLC analysis.
(12S,4S)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-2,16-dioxo-7-oxa-3-aza-1(2,1)-pyrrolidina-6(1,4)-benzenacyclohexadecaphane-4-carboxamide (18). Macrocyclic carboxylic acid intermediate 9c was prepared from 8c using general procedure for synthesis of macrocyclic carboxylic acid. 9c was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 18 as white solid (33%). 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.2 Hz, 1H), 6.98 – 6.88 (m, 2H), 6.83 – 6.71 (m, 2H), 6.16 (d, J = 9.4 Hz, 1H), 4.65 (ddd, J = 3.3, 5.5, 9.1 Hz, 1H), 4.61 – 4.51 (m, 1H), 4.47 (dd, J = 3.2, 8.8 Hz, 1H), 4.02 (dt, J = 4.1, 8.5 Hz, 1H), 3.92 (td, J = 2.8, 10.0 Hz, 1H), 3.52 (dd, J = 3.6, 6.9 Hz, 1H), 3.50 – 3.40 (m, 2H), 3.38 (d, J = 5.1 Hz, 1H), 2.84 (d, J = 5.1 Hz, 1H), 2.64 (dd, J = 5.6, 13.9 Hz, 1H), 2.29 (ddt, J = 3.7, 6.3, 12.7 Hz, 1H), 2.17 – 1.88 (m, 6H), 1.84 – 1.58 (m, 3H), 1.46 (d, J = 2.7 Hz, 3H), 1.43 – 1.13 (m, 11H), 0.93 (d, J = 5.8 Hz, 3H), 0.84 (d, J = 5.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 207.56, 173.67, 170.82, 170.17, 158.54, 130.36, 127.04, 114.16, 65.39, 61.15, 59.18, 52.72, 52.33, 50.16, 47.76, 38.74, 35.49, 34.96, 28.79, 27.54, 27.44, 26.28, 24.90, 24.37, 23.76, 23.67, 22.39, 22.07, 20.91, 16.75. HRMS m/z calcd for C32H48N3O6 [M+H]+ 570.3498, found: 570.3534. LCMS (ES+), found: 570.3; Purity: ≥ 96% and retention time is 21.22 min by HPLC analysis.
(12S,4S)-N-((S)-3-Cyclohexyl-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-2,15-dioxo-7-oxa-3-aza-1(2,1)-pyrrolidina-6(1,4)-benzenacyclopentadecaphane-4-carboxamide (19). Macrocyclic carboxylic acid intermediate 9b was prepared from 8b using general procedure for synthesis of macrocyclic carboxylic acid. 9b was coupled with right hand Boc deprotected epoxyketone intermediate 4b using general procedure for amide coupling reaction to prepare 19 as white solid (42%). 1H NMR (400 MHz, CDCl3) δ 7.00 (d, J = 8.2 Hz, 1H), 6.97 – 6.91 (m, 2H), 6.86 – 6.75 (m, 2H), 6.23 (d, J = 8.9 Hz, 1H), 4.69 – 4.59 (m, 2H), 4.46 (dd, J = 3.2, 8.4 Hz, 1H), 4.17 – 4.08 (m, 2H), 3.49 – 3.36 (m, 3H), 3.34 (d, J = 5.0 Hz, 1H), 2.83 (d, J = 5.0 Hz, 1H), 2.70 (dd, J = 5.7, 14.1 Hz, 1H), 2.34 – 2.25 (m, 1H), 2.11 – 1.93 (m, 5H), 1.91 – 1.81 (m, 2H), 1.81 – 1.70 (m, 2H), 1.70 – 1.52 (m, 4H), 1.51 – 1.39 (m, 5H), 1.39 – 1.27 (m, 5H), 1.27 – 1.07 (m, 6H), 0.98 – 0.84 (m, 2H). 13C NMR (101 MHz, cdcl3) δ 207.73, 173.64, 171.09, 170.53, 158.11, 130.35, 127.26, 114.83, 66.34, 60.83, 59.12, 52.75, 52.32, 49.46, 47.78, 37.70, 35.30, 34.57, 33.85, 33.71, 31.57, 28.50, 27.85, 27.59, 26.43, 26.14, 26.08, 25.95, 24.97, 22.91, 22.21, 16.78. HRMS m/z calcd for C34H50N3O6 [M+H]+ 596.3655, found: 596.3534. LCMS (ES+) m/z calcd for C34H50N3O6 [M+H]+ 596.3, found: 596.3; Purity: ≥ 96% and retention time is 21.49 min by HPLC analysis.
(12S,4S)-N-((S)-1-((R)-2-Methyloxiran-2-yl)-1-oxo-3-phenylpropan-2-yl)-2,15-dioxo-7-oxa-3-aza-1(2,1)-pyrrolidina-6(1,4)-benzenacyclopentadecaphane-4-carboxamide (20). Macrocyclic carboxylic acid intermediate 9b was prepared from 8b using general procedure for synthesis of macrocyclic carboxylic acid. 9b was coupled with right hand Boc deprotected epoxyketone intermediate 4c using general procedure for amide coupling reaction to prepare 20 as white solid (38%). 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.16 (m, 5H), 7.08 (d, J = 8.7 Hz, 1H), 6.90 – 6.81 (m, 2H), 6.74 – 6.64 (m, 2H), 6.48 (d, J = 8.0 Hz, 1H), 4.94 (ddd, J = 5.0, 7.4, 8.4 Hz, 1H), 4.59 (dt, J = 5.5, 8.4 Hz, 1H), 4.50 – 4.41 (m, 1H), 4.19 – 4.02 (m, 2H), 3.37 (ddt, J = 3.5, 5.7, 7.8 Hz, 3H), 3.18 (dd, J = 5.8, 14.5 Hz, 1H), 3.11 – 3.00 (m, 1H), 2.84 (dd, J = 2.0, 5.0 Hz, 1H), 2.76 (ddd, J = 2.6, 8.0, 14.3 Hz, 2H), 2.31 – 2.23 (m, 1H), 2.08 – 1.88 (m, 5H), 1.88 – 1.70 (m, 2H), 1.64 (dt, J = 3.4, 6.9 Hz, 1H), 1.52 – 1.38 (m, 4H), 1.38 – 1.22 (m, 4H), 1.21 – 1.07 (m, 2H). 13C NMR (101 MHz, cdcl3) δ 207.37, 173.63, 171.19, 170.96, 157.80, 136.46, 130.10, 129.16, 128.38, 127.15, 126.72, 114.90, 66.35, 60.35, 59.19, 52.98, 52.29, 52.02, 47.65, 36.80, 35.26, 34.51, 28.04, 27.72, 27.62, 26.11, 24.99, 23.07, 22.32, 16.41. HRMS (FAB) m/z calcd for for C34H44N3O6 [M+H]+ 590.3185, found: 590.3214. LCMS (ES+), found: 590.3; Purity: ≥ 96% and retention time is 18.53 min by HPLC analysis
(3S,15aS)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,11-dioxotetradecahydropyrrolo[2,1-f][1]oxa[4,7]diazacyclotridecine-3-carboxamide (21). Macrocyclic carboxylic acid intermediate 12a was prepared from 11a using general procedure for synthesis of macrocyclic carboxylic acid. 12a was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 21 as white solid (60%). 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 7.9 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 4.82 (dd, J = 1.9, 8.1 Hz, 1H), 4.57 (ddd, J = 3.3, 8.2, 10.2 Hz, 1H), 4.39 (ddd, J = 3.7, 6.0, 7.9 Hz, 1H), 3.71 (dd, J = 6.0, 9.9 Hz, 1H), 3.58 (dt, J = 4.3, 10.9 Hz, 2H), 3.53 – 3.34 (m, 3H), 3.27 (d, J = 5.0 Hz, 1H), 2.83 (d, J = 5.0 Hz, 1H), 2.57 – 2.42 (m, 2H), 2.19 (ddd, J = 2.6, 8.2, 12.9 Hz, 1H), 2.08 – 1.94 (m, 2H), 1.94 – 1.73 (m, 3H), 1.73 – 1.61 (m, 2H), 1.61 – 1.43 (m, 6H), 1.43 – 1.28 (m, 2H), 0.91 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 207.80, 174.95, 171.38, 169.69, 69.53, 67.96, 59.50, 58.99, 53.81, 52.33, 50.12, 47.88, 40.04, 34.21, 27.51, 26.50, 25.14, 24.97, 23.85, 23.24, 21.88, 21.38, 16.71. LCMS (ES+) m/z calcd for C23H38N3O6 [M+H]+ 452.2, found: 452.2; Purity: ≥ 96% and retention time is 15.21 min by HPLC analysis.
(3S,16aS)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,12-dioxotetradecahydro-6H-pyrrolo[2,1-f][1]oxa[4,7]diazacyclotetradecine-3-carboxamide (22). Macrocyclic carboxylic acid intermediate 12b was prepared from 11b using general procedure for synthesis of macrocyclic carboxylic acid. 12b was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 22 as white solid (35%). 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.3 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 4.63 (ddt, J = 3.7, 9.0, 22.3 Hz, 2H), 4.30 (dd, J = 3.3, 8.3 Hz, 1H), 3.87 (dd, J = 2.4, 9.7 Hz, 1H), 3.76 – 3.61 (m, 1H), 3.61 – 3.43 (m, 3H), 3.41 – 3.31 (m, 1H), 3.29 (d, J = 5.2 Hz, 1H), 2.82 (d, J = 5.1 Hz, 1H), 2.62 – 2.47 (m, 1H), 2.38 – 2.19 (m, 2H), 2.13 (dd, J = 7.8, 13.1 Hz, 1H), 2.05 – 1.81 (m, 4H), 1.79 – 1.65 (m, 1H), 1.65 – 1.31 (m, 11H), 0.91 (t, J = 7.1 Hz, 6H).13C NMR (101 MHz, CDCl3) δ 207.41, 174.37, 170.61, 170.15, 72.15, 69.60, 60.93, 59.04, 53.67, 52.27, 50.27, 48.16, 39.54, 33.81, 28.42, 27.89, 27.64, 27.27, 25.01, 24.93, 24.69, 23.35, 21.10, 16.76. LCMS (ES+) m/z calcd for C24H40N3O6 [M+H]+ 466.2, found: 466.2; Purity: ≥ 96% and retention time is 19.13 min by HPLC analysis.
(3S,17aS)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,13-dioxohexadecahydropyrrolo[2,1-f][1]oxa[4,7]diazacyclopentadecine-3-carboxamide (23). Macrocyclic carboxylic acid intermediate 12c was prepared from 11c using general procedure for synthesis of macrocyclic carboxylic acid. 12c was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 23 as white solid (20%). 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 9.0 Hz, 1H), 4.73 – 4.57 (m, 2H), 4.40 (ddd, J = 2.2, 3.2, 8.9 Hz, 1H), 3.86 (dd, J = 2.2, 9.1 Hz, 1H), 3.67 (ddd, J = 4.2, 7.2, 9.9 Hz, 1H), 3.57 – 3.42 (m, 3H), 3.42 – 3.34 (m, 1H), 3.33 – 3.24 (m, 1H), 2.83 (d, J = 5.0 Hz, 1H), 2.50 – 2.26 (m, 3H), 2.20 – 2.05 (m, 1H), 2.05 – 1.86 (m, 3H), 1.84 – 1.68 (m, 2H), 1.64 – 1.41 (m, 8H), 1.41 – 1.17 (m, 5H), 0.93 (dd, J = 4.7, 6.6 Hz, 6H).13C NMR (101 MHz, CDCl3) δ 207.63, 174.61, 170.89, 170.07, 72.34, 69.94, 61.32, 59.04, 54.10, 52.29, 50.13, 48.19, 39.66, 33.61, 29.16, 28.62, 28.43, 28.12, 28.08, 24.85, 24.83, 23.43, 23.10, 21.04, 16.76. LCMS (ES+) m/z calcd for C25H42N3O6 [M+H]+ 480.3, found: 480.3; Purity: ≥ 96% and retention time is 14.82 min by HPLC analysis.
(3S,15aS)-N-((S)-3-Cyclohexyl-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-1,11-dioxotetradecahydropyrrolo[2,1-f][1]oxa[4,7]diazacyclotridecine-3-carboxamide (24). Macrocyclic carboxylic acid intermediate 12a was prepared from 11a using general procedure for synthesis of macrocyclic carboxylic acid. 12a was coupled with right hand Boc deprotected epoxyketone intermediate 4b using general procedure for amide coupling reaction to prepare 24 as white solid (33%). 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 7.9 Hz, 1H), 7.02 (d, J = 8.0 Hz, 1H), 4.82 (dd, J = 1.9, 8.2 Hz, 1H), 4.62 – 4.54 (m, 1H), 4.38 (ddd, J = 3.7, 5.9, 7.9 Hz, 1H), 3.75 – 3.70 (m, 1H), 3.59 (dd, J = 3.9, 10.1 Hz, 2H), 3.53 – 3.37 (m, 3H), 3.29 (d, J = 5.0 Hz, 1H), 2.83 (d, J = 5.0 Hz, 1H), 2.55 – 2.44 (m, 2H), 2.19 (ddd, J = 2.5, 8.2, 12.9 Hz, 1H), 2.01 (tdd, J = 2.8, 4.6, 8.4 Hz, 2H), 1.92 – 1.75 (m, 4H), 1.70 – 1.51 (m, 7H), 1.47 (s, 3H), 1.43 – 1.29 (m, 3H), 1.20 – 1.07 (m, 3H), 0.99 – 0.81 (m, 3H); 13C NMR (101 MHz, cdcl3) δ 207.90, 174.93, 171.31, 169.75, 69.42, 68.07, 59.57, 59.05, 53.70, 52.35, 49.52, 47.90, 38.31, 34.34, 34.25, 33.87, 31.87, 27.60, 26.57, 26.38, 26.33, 26.04, 24.98, 24.01, 22.07, 16.73. LCMS (ES+) m/z calcd for C26H42N3O6 [M+H]+ 492.3, found: 492.3; Purity: ≥ 96% and retention time is 17.81 min by HPLC analysis.
(3S,15aS)-N-((S)-1-((R)-2-Methyloxiran-2-yl)-1-oxo-3-phenylpropan-2-yl)-1,11-dioxotetradecahydropyrrolo[2,1-f][1]oxa[4,7]diazacyclotridecine-3-carboxamide (25). Macrocyclic carboxylic acid intermediate 12a was prepared from 11a using general procedure for synthesis of macrocyclic carboxylic acid. 12a was coupled with right hand Boc deprotected epoxyketone intermediate 4c using general procedure for amide coupling reaction to prepare 25 as white solid (31%). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.7 Hz, 1H), 7.30 – 7.22 (m, 2H), 7.22 – 7.09 (m, 4H), 4.80 (td, J = 4.9, 8.2 Hz, 1H), 4.72 (dd, J = 1.7, 8.1 Hz, 1H), 4.38 (td, J = 3.8, 7.3 Hz, 1H), 3.62 (dd, J = 3.8, 10.1 Hz, 1H), 3.59 – 3.49 (m, 2H), 3.49 – 3.42 (m, 1H), 3.35 (ddt, J = 4.0, 8.7, 11.7 Hz, 2H), 3.30 (d, J = 4.9 Hz, 1H), 3.11 (dd, J = 4.9, 14.0 Hz, 1H), 2.85 (d, J = 4.9 Hz, 1H), 2.82 – 2.73 (m, 1H), 2.53 – 2.41 (m, 2H), 2.20 (ddd, J = 2.7, 7.1, 12.9 Hz, 1H), 2.00 (ddt, J = 3.5, 8.4, 12.2 Hz, 2H), 1.86 – 1.75 (m, 2H), 1.55 (tt, J = 5.7, 13.1 Hz, 3H), 1.45 (s, 3H), 1.42 – 1.30 (m, 2H); 13C NMR (101 MHz, cdcl3) δ 207.05, 175.03, 171.64, 169.73, 135.97, 129.34, 128.35, 126.83, 69.05, 67.89, 59.20, 53.41, 52.39, 52.29, 47.85, 37.04, 34.08, 27.50, 26.13, 24.94, 23.87, 21.92, 16.48. LCMS (ES+) m/z calcd for C26H36N3O6 [M+H]+ 486.2, found: 486.2; Purity: ≥ 96% and retention time is 15.50 min by HPLC analysis.
(3S,18aS)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,11,14-trioxohexadecahydro-6H-pyrrolo[2,1-f][1]oxa[4,7,10]triazacyclohexadecine-3-carboxamide (26). Macrocyclic carboxylic acid intermediate 15a was prepared from 14a using general procedure for synthesis of macrocyclic carboxylic acid. 15a was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 26 as white solid (47%). 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.3 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.32 (d, J = 8.1 Hz, 1H), 4.76 – 4.66 (m, 2H), 4.62 (ddd, J = 3.3, 8.4, 9.9 Hz, 1H), 4.32 (dt, J = 3.5, 7.2 Hz, 1H), 3.72 (td, J = 3.6, 8.1, 8.9 Hz, 2H), 3.53 (dd, J = 3.1, 9.6 Hz, 1H), 3.49 – 3.31 (m, 4H), 3.27 (d, J = 4.9 Hz, 1H), 2.84 (d, J = 5.2 Hz, 1H), 2.56 – 2.43 (m, 1H), 2.43 – 2.32 (m, 1H), 2.12 (ddd, J = 2.9, 10.7, 13.7 Hz, 1H), 2.07 – 1.87 (m, 3H), 1.83 – 1.62 (m, 2H), 1.62 – 1.40 (m, 8H), 1.34 (ddd, J = 4.0, 9.3, 18.0 Hz, 2H), 0.92 (dd, J = 1.8, 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 208.16, 173.95, 170.41, 169.99, 169.43, 70.54, 69.06, 60.60, 58.93, 54.70, 52.33, 50.00, 47.01, 41.46, 40.18, 36.47, 29.05, 27.17, 25.24, 25.03, 24.73, 23.39, 21.30, 16.67. LCMS (ES+) m/z calcd for C25H41N4O7 [M+H]+ 509.2, found: 509.3; Purity: ≥ 96% and retention time is 14.36 min by HPLC analysis.
(3S,19aS)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,12,15-trioxooctadecahydropyrrolo[2,1-f][1]oxa[4,7,10]triazacycloheptadecine-3-carboxamide (27). Macrocyclic carboxylic acid intermediate 15b was prepared from 14b using general procedure for synthesis of macrocyclic carboxylic acid. 15b was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 27 as white solid (36%). 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.47 (d, J = 7.1 Hz, 1H), 4.70 (d, J = 6.3 Hz, 1H), 4.67 – 4.55 (m, 2H), 4.38 (dd, J = 2.9, 8.5 Hz, 1H), 3.81 (dd, J = 2.8, 9.4 Hz, 1H), 3.70 – 3.57 (m, 2H), 3.47 – 3.37 (m, 2H), 3.37 – 3.31 (m, 2H), 3.28 (d, J = 5.0 Hz, 1H), 2.85 (d, J = 5.0 Hz, 1H), 2.47 (dd, J = 3.0, 5.9 Hz, 1H), 2.41 – 2.28 (m, 1H), 2.27 – 2.16 (m, 1H), 2.00 (dd, J = 4.6, 7.5 Hz, 3H), 1.87 (s, 2H), 1.72 (dt, J = 3.0, 8.7 Hz, 1H), 1.54 – 1.27 (m, 11H), 0.94 (t, J = 7.1 Hz, 6H).13C NMR (101 MHz, CDCl3) δ 208.21, 173.43, 170.30, 169.76, 169.43, 71.27, 69.95, 60.96, 58.95, 54.44, 52.33, 49.97, 46.82, 41.58, 40.13, 36.24, 30.21, 29.23, 27.49, 25.39, 25.00, 24.66, 24.63, 23.58, 21.31, 16.67. LCMS (ES+) m/z calcd for C26H43N4O7 [M+H]+ 523.3, found: 523.3; Purity: ≥ 96% and retention time is 14.75 min by HPLC analysis.
Benzyl O-allyl-N-(tert-butoxycarbonyl)-L-serinate (28). Intermediate 28 was prepared from Boc-serine benzyl ester and allyl methyl carbonate using same procedure used for preparation of intermediate 10 as colorless oil (78%). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 3.1 Hz, 5H), 5.84 – 5.67 (m, 1H), 5.39 (d, J = 8.8 Hz, 1H), 5.31 – 5.19 (m, 2H), 5.13 (ddd, J = 3.6, 7.9, 12.4 Hz, 2H), 4.45 (dt, J = 3.2, 9.0 Hz, 1H), 3.96 – 3.80 (m, 3H), 3.63 (dd, J = 3.3, 9.4 Hz, 1H), 1.43 (s, 9H).
tert-Butyl (2S,4S)-2-(((S)-3-(allyloxy)-1-(benzyloxy)-1-oxopropan-2-yl)carbamoyl)-4-fluoropyrrolidine-1-carboxylate (29a). Boc deprotected 28 prepared from intermediate 28 using general procedure for Boc deprotection reaction was coupled with N-Boc-cis-4-fluoro-proline using general procedure for amide coupling reaction to prepare 29a as colorless oil (93%). 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.16 (m, 5H), 7.02 (s, 1H), 5.71 (ddt, J = 5.6, 10.4, 17.2 Hz, 1H), 5.34 – 4.97 (m, 4H), 4.75 (dd, J = 3.5, 7.5 Hz, 1H), 4.42 (s, 1H), 3.98 – 3.79 (m, 2H), 3.79 – 3.41 (m, 3H), 2.64 (s, 1H), 2.26 (d, J = 43.7 Hz, 1H), 1.43 (s, 9H).
tert-Butyl (S)-2-(((S)-3-(allyloxy)-1-(benzyloxy)-1-oxopropan-2-yl)carbamoyl)-4,4-difluoropyrrolidine-1-carboxylate (29b). Boc deprotected 28 prepared from intermediate 28 using general procedure for Boc deprotection reaction was coupled with N-Boc-4,4-difluoro-proline using general procedure for amide coupling reaction to prepare 29b as colorless oil (87%). 1H NMR (400 MHz, CDCl3) δ 7.47 – 7.23 (m, 5H), 6.91 (s, 1H), 5.72 (tt, J = 5.3, 10.8 Hz, 1H), 5.34 – 5.01 (m, 4H), 4.73 (d, J = 8.1 Hz, 1H), 4.49 (s, 1H), 4.04 – 3.75 (m, 4H), 3.65 (dt, J = 10.7, 22.5 Hz, 2H), 2.69 (d, J = 60.0 Hz, 2H), 1.44 (s, 9H).
tert-Butyl (S)-2-(((S)-3-(allyloxy)-1-(benzyloxy)-1-oxopropan-2-yl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (29c). Boc deprotected 28 prepared from intermediate 28 using general procedure for Boc deprotection reaction was coupled with N-Boc-α-methyl-proline using general procedure for amide coupling reaction to prepare 29c as colorless oil (65%). 1H NMR (400 MHz, CDCl3) δ 7.31 (s, 5H), 5.72 (tt, J = 5.4, 10.7 Hz, 1H), 5.29 – 5.04 (m, 4H), 4.75 – 4.63 (m, 1H), 3.98 – 3.78 (m, 3H), 3.63 (dd, J = 3.3, 9.5 Hz, 1H), 3.53 (s, 2H), 1.76 (m, 4H), 1.57 (d, J = 16.0 Hz, 3H), 1.50 – 1.32 (s, 9H).
tert-Butyl (S)-2-(((S)-3-(allyloxy)-1-(benzyloxy)-1-oxopropan-2-yl)carbamoyl)azetidine-1-carboxylate (29d). Boc deprotected 28 prepared from intermediate 28 using general procedure for Boc deprotection reaction was coupled with N-Boc-azetidine-2-carboxylic acid using general procedure for amide coupling reaction to prepare 29d as colorless oil (92%). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.17 (m, 5H), 5.74 (ddt, J = 5.6, 10.5, 17.3 Hz, 1H), 5.28 – 5.03 (m, 4H), 4.75 (dt, J = 3.3, 8.3 Hz, 1H), 4.63 (t, J = 8.1 Hz, 1H), 3.95 – 3.83 (m, 4H), 3.77 (td, J = 5.8, 8.3 Hz, 1H), 3.65 (dd, J = 3.3, 9.6 Hz, 1H), 2.38 (d, J = 20.2 Hz, 2H), 1.42 (s, 9H).
tert-Butyl (S)-2-(((S)-3-(allyloxy)-1-(benzyloxy)-1-oxopropan-2-yl)carbamoyl)piperidine-1-carboxylate (29e). Boc deprotected 28 prepared from intermediate 28 using general procedure for Boc deprotection reaction was coupled with N-Boc-2-piperidinecarboxylic acid using general procedure for amide coupling reaction to prepare 29e as colorless oil (81%). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.23 (m, 5H), 6.80 (d, J = 26.1 Hz, 1H), 5.72 (ddt, J = 5.6, 10.9, 17.3 Hz, 1H), 5.32 – 5.00 (m, 4H), 4.74 (d, J = 8.1 Hz, 2H), 3.98 – 3.79 (m, 4H), 3.70 – 3.59 (m, 2H), 2.93 – 2.65 (m, 1H), 2.24 (d, J = 12.1 Hz, 1H), 1.44 (m, 13H).
Benzyl O-allyl-N-((2S,4S)-4-fluoro-1-(pent-4-enoyl)pyrrolidine-2-carbonyl)-L-serinate (30a). Boc deprotected 29a prepared from intermediate 29a using general procedure for Boc deprotection reaction was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to prepare 30a as colorless oil (83%). 1H NMR (400 MHz, CDCl3) δ 7.32 (m, 5H), 6.93 (d, J = 8.8 Hz, 1H), 5.91 – 5.64 (m, 2H), 5.36 – 4.91 (m, 6H), 4.75 (m, 1H), 4.47 (d, J = 9.9 Hz, 1H), 3.97 – 3.64 (m, 4H), 3.61 (dd, J = 3.4, 9.6 Hz, 1H), 3.51 (dd, J = 3.2, 9.5 Hz, 1H), 2.88 – 2.73 (m, 1H), 2.66 (t, J = 16.1 Hz, 1H), 2.57 – 2.07 (m, 5H).
Benzyl O-allyl-N-((S)-4,4-difluoro-1-(pent-4-enoyl)pyrrolidine-2-carbonyl)-L-serinate (30b). Boc deprotected 29b prepared from intermediate 29b using general procedure for Boc deprotection reaction was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to prepare 30b as colorless oil (97%). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.28 (m, 5H), 7.23 (d, J = 8.6 Hz, 1H), 5.91 – 5.67 (m, 2H), 5.32 – 4.93 (m, 6H), 4.81 – 4.66 (m, 2H), 3.99 – 3.74 (m, 5H), 3.63 (dd, J = 3.3, 9.6 Hz, 1H), 2.87 – 2.68 (m, 1H), 2.64 – 2.44 (m, 1H), 2.44 – 2.26 (m, 4H).
Benzyl O-allyl-N-((S)-2-methyl-1-(pent-4-enoyl)pyrrolidine-2-carbonyl)-L-serinate (30c). Boc deprotected 29c prepared from intermediate 29c using general procedure for Boc deprotection reaction was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to prepare 30c as colorless oil (96%). 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 7.7 Hz, 1H), 7.36 – 7.22 (m, 5H), 5.92 – 5.65 (m, 2H), 5.28 – 4.87 (m, 6H), 4.76 – 4.58 (m, 1H), 3.96 – 3.78 (m, 3H), 3.65 (ddd, J = 0.8, 3.5, 9.6 Hz, 1H), 3.61 – 3.44 (m, 2H), 2.53 – 2.40 (m, 1H), 2.34 (s, 4H), 1.96 – 1.78 (m, 2H), 1.69 (ddd, J = 6.6, 8.7, 12.5 Hz, 1H), 1.63 (d, J = 0.8 Hz, 3H).
Benzyl O-allyl-N-((S)-1-(pent-4-enoyl)azetidine-2-carbonyl)-L-serinate (30d). Boc deprotected 29d prepared from intermediate 29d using general procedure for Boc deprotection reaction was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to prepare 30d as colorless oil (78%). 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 7.8 Hz, 1H), 7.39 – 7.24 (m, 5H), 5.78 (dtd, J = 7.0, 10.5, 16.0 Hz, 2H), 5.30 – 4.91 (m, 6H), 4.87 (dd, J = 6.2, 9.4 Hz, 1H), 4.71 (dt, J = 3.9, 7.8 Hz, 1H), 4.10 – 3.98 (m, 2H), 3.92 (ddt, J = 1.4, 2.4, 4.0 Hz, 2H), 3.84 (dd, J = 4.0, 9.7 Hz, 1H), 3.65 (dd, J = 3.7, 9.6 Hz, 1H), 2.61 (ddd, J = 2.1, 5.7, 12.0 Hz, 1H), 2.39 (dq, J = 6.9, 22.3 Hz, 3H), 2.18 (dd, J = 5.8, 8.0 Hz, 2H).
Benzyl O-allyl-N-((S)-1-(pent-4-enoyl)piperidine-2-carbonyl)-L-serinate (30e). Boc deprotected 29e prepared from intermediate 29e using general procedure for Boc deprotection reaction was coupled with 4-pentenoic acid using general procedure for amide coupling reaction to prepare 30e as colorless oil (97%). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.25 (m, 5H), 6.74 (d, J = 7.9 Hz, 1H), 5.91 – 5.66 (m, 2H), 5.30 – 4.88 (m, 7H), 4.70 (dt, J = 3.4, 7.5 Hz, 1H), 3.94 – 3.79 (m, 3H), 3.77 – 3.69 (m, 1H), 3.65 (dd, J = 3.4, 9.6 Hz, 1H), 3.19 (td, J = 2.6, 13.1 Hz, 1H), 2.50 – 2.31 (m, 4H), 2.19 (dt, J = 2.5, 12.7 Hz, 1H), 1.71 – 1.48 (m, 4H), 1.48 – 1.32 (m, 1H).
(3S,14S,15aS)-14-Fluoro-N-((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,11-dioxotetradecahydropyrrolo[2,1-f][1]oxa[4,7]diazacyclotridecine-3-carboxamide (32). Macrocyclic carboxylic acid intermediate 31b was prepared from 30b using general procedure for synthesis of macrocyclic carboxylic acid. 31b was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 33 as white solid (43%). 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 8.4 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 4.90 (d, J = 10.0 Hz, 1H), 4.61 (ddd, J = 3.8, 8.5, 9.6 Hz, 1H), 4.27 (ddd, J = 2.6, 3.6, 8.0 Hz, 1H), 4.03 (dd, J = 2.6, 9.7 Hz, 1H), 3.90 (ddd, J = 1.9, 12.4, 22.9 Hz, 1H), 3.82 – 3.69 (m, 1H), 3.53 (dt, J = 4.6, 9.1 Hz, 1H), 3.38 – 3.31 (m, 2H), 3.28 (d, J = 5.0 Hz, 1H), 2.92 – 2.85 (m, 1H), 2.83 (d, J = 4.9 Hz, 1H), 2.55 (ddd, J = 2.0, 8.0, 12.8 Hz, 1H), 2.30 – 2.15 (m, 2H), 1.92 – 1.77 (m, 3H), 1.71 – 1.58 (m, 3H), 1.54 – 1.38 (m, 7H), 0.91 (dd, J = 1.8, 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 207.52, 174.59, 169.68, 169.63, 93.17, 91.40, 69.60, 68.25, 58.98, 58.94, 54.64, 54.40, 53.87, 52.30, 50.14, 40.05, 34.69, 34.52, 34.30, 27.52, 25.00, 24.09, 23.26, 22.09, 21.33, 16.73. LCMS (ES+) m/z calcd for C23H37FN3O6 [M+H]+ 470.2, found: 470.2; Purity: ≥ 96% and retention time is 15.30 min by HPLC analysis.
(3S,15aS)-14,14-Difluoro-N-((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,11-dioxotetradecahydropyrrolo[2,1-f][1]oxa[4,7]diazacyclotridecine-3-carboxamide (33). Macrocyclic carboxylic acid intermediate 31a was prepared from 30a using general procedure for synthesis of macrocyclic carboxylic acid. 31a was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 32 as white solid (42%). 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.1 Hz, 1H), 6.83 (d, J = 8.3 Hz, 1H), 5.07 (dd, J = 3.7, 9.9 Hz, 1H), 4.58 (ddd, J = 3.2, 8.3, 10.2 Hz, 1H), 4.42 (ddd, J = 3.6, 5.3, 8.5 Hz, 1H), 4.00 – 3.81 (m, 2H), 3.80 – 3.75 (m, 1H), 3.54 (dd, J = 3.7, 9.8 Hz, 1H), 3.46 (dt, J = 3.9, 6.4 Hz, 1H), 3.40 – 3.34 (m, 1H), 3.26 (d, J = 5.0 Hz, 1H), 3.18 – 3.04 (m, 1H), 2.84 (d, J = 5.0 Hz, 1H), 2.56 – 2.40 (m, 2H), 2.26 (ddd, J = 2.6, 8.9, 13.0 Hz, 1H), 1.85 – 1.76 (m, 1H), 1.69 – 1.59 (m, 2H), 1.58 – 1.49 (m, 4H), 1.48 (s, 3H), 1.34 (ddd, J = 4.3, 10.3, 13.9 Hz, 2H), 0.91 (dd, J = 2.5, 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 207.83, 174.62, 169.20, 168.90, 69.55, 67.97, 58.99, 53.95, 52.33, 50.22, 40.11, 34.75, 34.21, 27.31, 25.14, 23.67, 23.25, 21.98, 21.36, 16.70. LCMS (ES+) m/z calcd for C23H36F2N3O6 [M+H]+ 488.2, found: 488.2; Purity: ≥ 96% and retention time is 16.15 min by HPLC analysis.
(3S,15aS)-15a-Methyl-N-((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,11-dioxotetradecahydropyrrolo[2,1-f][1]oxa[4,7]diazacyclotridecine-3-carboxamide (34). Macrocyclic carboxylic acid intermediate 31c was prepared from 30c using general procedure for synthesis of macrocyclic carboxylic acid. 31c was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 34 as white solid (32%). 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 8.6 Hz, 1H), 7.30 (d, J = 7.8 Hz, 1H), 4.66 (td, J = 6.6, 8.5 Hz, 1H), 4.23 (dt, J = 3.6, 7.7 Hz, 1H), 3.97 (dd, J = 3.1, 10.1 Hz, 1H), 3.74 – 3.61 (m, 2H), 3.53 – 3.45 (m, 2H), 3.36 (dt, J = 5.6, 8.8 Hz, 1H), 3.28 (d, J = 4.9 Hz, 1H), 2.83 (d, J = 4.9 Hz, 1H), 2.55 (ddd, J = 1.9, 9.1, 11.3 Hz, 1H), 2.49 – 2.42 (m, 1H), 2.21 – 2.11 (m, 1H), 1.98 – 1.83 (m, 4H), 1.80 (s, 4H), 1.71 – 1.62 (m, 2H), 1.53 (t, J = 5.4 Hz, 2H), 1.47 (d, J = 3.4 Hz, 5H), 1.39 (dd, J = 4.4, 14.2 Hz, 1H), 0.91 (dd, J = 6.6, 9.5 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 207.36, 174.21, 173.20, 170.07, 69.42, 68.95, 68.31, 58.90, 53.98, 52.27, 49.82, 49.75, 40.20, 39.00, 35.35, 27.74, 24.98, 24.40, 23.30, 23.17, 22.52, 22.40, 21.31, 16.76. LCMS (ES+) m/z calcd for C24H40N3O6 [M+H]+ 466.2, found: 466.2; Purity: ≥ 96% and retention time is 16.98 min by HPLC analysis.
(10S,13S)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-2,12-dioxo-8-oxa-1,11-diazabicyclo[11.2.0]pentadecane-10-carboxamide (35). Macrocyclic carboxylic acid intermediate 31d was prepared from 30d using general procedure for synthesis of macrocyclic carboxylic acid. 31d was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 35 as white solid (39%). 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 7.5 Hz, 1H), 6.98 (d, J = 8.1 Hz, 1H), 4.98 (dd, J = 5.4, 9.6 Hz, 1H), 4.55 (ddd, J = 3.3, 8.0, 10.2 Hz, 1H), 4.46 (td, J = 3.7, 7.9 Hz, 1H), 4.11 (td, J = 6.5, 8.6 Hz, 2H), 3.75 (dd, J = 3.7, 9.8 Hz, 1H), 3.57 (dd, J = 8.1, 9.8 Hz, 1H), 3.42 (ddd, J = 4.1, 9.0, 10.4 Hz, 2H), 3.27 (d, J = 5.0 Hz, 1H), 2.84 (d, J = 5.0 Hz, 1H), 2.68 – 2.47 (m, 2H), 2.14 (ddd, J = 2.4, 11.0, 13.2 Hz, 1H), 2.02 (ddd, J = 3.1, 6.6, 12.9 Hz, 1H), 1.78 – 1.68 (m, 2H), 1.66 – 1.56 (m, 2H), 1.56 – 1.45 (m, 5H), 1.34 (dddd, J = 3.4, 10.1, 14.1, 28.3 Hz, 3H), 0.94 – 0.83 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 207.98, 175.42, 172.10, 169.44, 69.56, 67.83, 61.28, 59.01, 53.94, 52.36, 50.09, 48.65, 40.08, 31.02, 27.76, 25.13, 23.26, 23.19, 21.37, 21.30, 18.24, 16.70. LCMS (ES+) m/z calcd for C22H36N3O6 [M+H]+ 438.2, found: 438.2; Purity: ≥ 96% and retention time is 14.55 min by HPLC analysis.
(3S,16aS)-N-((S)-4-Methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-1,11-dioxotetradecahydro-2H-pyrido[2,1-f][1]oxa[4,7]diazacyclotridecine-3-carboxamide (36). Macrocyclic carboxylic acid intermediate 31e was prepared from 30e using general procedure for synthesis of macrocyclic carboxylic acid. 31e was coupled with right hand Boc deprotected epoxyketone intermediate 4a using general procedure for amide coupling reaction to prepare 36 as white solid (28%). 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 8.2 Hz, 1H), 5.42 – 5.35 (m, 1H), 4.54 (ddd, J = 3.3, 8.1, 10.1 Hz, 1H), 4.46 (dt, J = 3.8, 7.9 Hz, 1H), 3.97 (dd, J = 3.6, 9.3 Hz, 1H), 3.81 (d, J = 13.4 Hz, 1H), 3.55 – 3.47 (m, 1H), 3.35 – 3.23 (m, 3H), 2.97 – 2.89 (m, 1H), 2.83 (d, J = 4.9 Hz, 1H), 2.77 (s, 1H), 2.34 (dt, J = 2.6, 14.7 Hz, 1H), 2.19 (ddd, J = 2.2, 10.2, 12.7 Hz, 1H), 1.77 – 1.57 (m, 7H), 1.56 – 1.36 (m, 9H), 1.30 (ddd, J = 4.5, 10.2, 13.7 Hz, 1H), 0.90 (dd, J = 1.8, 6.6 Hz, 6H).13C NMR (101 MHz, CDCl3) δ 207.88, 173.51, 171.11, 169.52, 69.63, 67.76, 59.01, 53.07, 52.34, 51.43, 50.33, 44.73, 40.03, 38.56, 33.18, 27.11, 25.57, 25.22, 24.07, 23.22, 22.50, 22.12, 21.49, 20.36, 16.70. LCMS (ES+) m/z calcd for C24H40N3O6 [M+H]+ 466.2, found: 466.3; Purity: ≥ 96% and retention time is 16.31 min by HPLC analysis.
Biology.
All animal study protocols were approved by the Animal Care and Use Committee (IACUC) of Chungbuk National University (Approval No: CBNUA-144-1001-01) and Seoul National University (SNU-200217-2). In addition, all cell culture experiments were performed following the guidelines and regulations of the University of Kentucky Biosafety Committee (Approval No: B17-3000-M).
Cell culture.
BV-2 murine microglial cell line was a kind gift of Dr. Won-Gon Kim (Korea Research Institute of Bioscience & Biotechnology), and RPMI8226 human multiple myeloma cell line was obtained from the ATCC (American Type Culture Collection). BV-2 cells and RPMI 8226 human multiple myeloma cells were cultured as reported previously.22 RPMI 8226 cells with acquired resistance to carfilzomib (RPMI 8226/ABCB1) previously established by us were used and maintained in 80 nM carfilzomib.30 To assess the in cellulo activity of macrocyclic proteasome inhibitors, RPMI 8226/ABCB1 cells were kept in a drug-free medium for two weeks after drug withdrawal.
Animals.
9-month-old Tg2576 mice were purchased from the Division of Laboratory Animal Resources (Korea FDA, Osong, South Korea). They were quarantined and acclimatized for a month before use. Animals were kept in groups of three per cage and allowed free access to water and food, and maintained on a 12-h light/dark cycle.
Proteasome activity assay.
The in vitro assay of 20S proteasome activities was performed as previously described.22 Briefly, for proteasome activity assay using purified human 20S proteasome (Boston Biochem: constitutive proteasome, Cat No. E-360-050; immunoproteasome, Cat No E-370-025), 50 nanograms of diluted purified human 20S proteasomes in 20S proteasome assay buffer (20 mM Tris-HCl, 0.5 mM EDTA, 0.035% SDS, pH 8.0) were transferred to a 96-well microtiter plate and incubated with 8 concentrations of inhibitors (3,10, 30, 100, 300, 1,000, 3,000, 10,000 nM) and control (DMSO only control) for 1 h. For in-cellulo proteasome activity assay using cells, cells were treated with proteasome inhibitors for 4 hours and subsequently lysed using the passive lysis buffer (Promega). Cell lysates were centrifuged at 14,000 g for 20 min at 4 °C. Cell lysates containing five micrograms of total protein (in 20S proteasome assay buffer; 20 mM Tris-HCl, 0.5 mM EDTA, 0.035% SDS, pH 8.0) were added to a 96-well microplate. To measure the proteasome subunit activity for purified proteasome or cell lysates, the fluorescence substrates were incubated, and fluorescence (360 nm excitation, 460 nm emission) was monitored on a SpectraMax M5 microplate reader every minute for 1 h. For individual proteasome subunit activity, the fluorescence substrates were used as follows: Suc-LLVY-AMC (CT-L activity, 100 μM), Ac-PAL-AMC (LMP2 activity, 100 μM), Ac-nLPnLD-AMC (β1 activity,100 μM), Ac-ANW-AMC (LMP7 activity, 100 μM), and Ac-WLA-AMC (β5 activity, 20 μM).
In vitro metabolic stability assays.
The whole blood and the liver were collected from ICR mice (male, 7-week-old; DBL (Eumseong, Korea)). The whole blood was treated with 25 IU/mL heparin, and the liver was rinsed and homogenized in ice-cold phosphate-buffered saline (PBS, 2.5 mL/g tissue). Following pre-incubation at 37°C, aliquots of whole blood or liver homogenates (396 μL) were mixed with the stock solutions of the test compounds (4 μL) to make the final concentration of 1 μM. From the reaction mixture, an aliquot (40 μL) was taken at the pre-determined time points: 0, 10, and 30 min (for whole blood) and 0, 10, and 20 min (for liver homogenates). Samples were immediately mixed with acetonitrile (300 μL) containing 100 nM carfilzomib as an internal standard. After vortexing and centrifugation (13,000 rpm, 10 min, 4°C), the supernatant's drug levels were quantified via LC-MS/MS. Differences between the groups were assessed using Student’s t-test (GraphPad Prism, v.8.4.1), and P values less than 0.05 were considered statistically significant.
Quantitation of drug levels via LC-MS/MS.
YU102 and DB-60 (20) were quantified by HPLC 1290 infinity II (Agilent, Palo Alto, CA, USA) coupled to Agilent 6460 triple quadrupole (Agilent, Palo Alto, CA, USA) equipped with jet stream technology ion source interface operated in the positive ion mode. The chromatographic separation was performed on a Phenomenex Luna C18 column (50 x 2.0 mm id, 3 μm) with a mobile phase that consisted of 0.1% formic acid in water(A)-0.1% formic acid in acetonitrile(B) at a flow rate of 0.3 mL/min. The gradient was set up as follows: 0.5 min 85% A, 15% B; 3.5 min 100% B; 5.5 min 100% B; 5.6 min 85% A, 15% B; 7.5 min 85% A, 15% B. The optimized parameters for the source-dependent mass spectrometry were as follows: gas temperature of 325°C; a gas flow of 11 L/min; nebulizing gas 35 psi; sheath gas flow 12 L/min; capillary voltage of 4500 V. The compound-dependent parameters, fragment voltage, collision energy (qualifier/quantifier), and cell accelerator voltage were set as follows: for YU102, 150 V, 25 V/20 V, and 4 V; for DB-60 (20), 176 V, 40 V/48 V, and 4 V; for carfilzomib 192 V, 29 V/29 V, and 4 V. Quantification was carried out in the selected reaction monitoring (SRM) mode using the following transition: for YU102, m/z 515.3>217.1 (qualifier), m/z 515.3>197.1 (quantifier); for DB-60 (20) m/z 590.3>260.2 (qualifier), m/z 590.3>70.2 (quantifier); for carfilzomib, m/z 720.4>402.3 (qualifier), m/z 720.4> 289.2 (qualifier). Linearity was verified in the following concentration ranges: for whole blood, 10~2,500 nM; for liver homogenates, 1~2,000 nM.
ABCG2 interaction assays.
The inhibitory interactions with ABCG2 were assessed by measuring the changes in the cellular accumulation of PhA (a fluorescent ABCG2 substrate) using MDCKII cells stably expressing ABCG2 (established in our previous study41) and parental cells. The known ABCG2 inhibitor Ko143 was used as a positive control. Cells were preincubated in the complete medium containing pheophorbide A (1 μM) with Ko143 (0.2 μM) or DB-60 (20) (2 or 5 μM) at 37°C for 30 min on a rotary shaker. Cells were then washed with an ice-cold medium and resuspended in the medium containing only Ko143 or DB-60 (20). After incubation for 45 min at 37°C on a rotary shaker, cells were washed with 1 mL of ice-cold PBS containing 2% fetal bovine serum. After centrifugation, cell pellets were kept on ice until the fluorescent signal measurement via flow cytometry (FACSCalibur Flow Cytometry System, BD Biosciences, San Jose, CA, USA) using the excitation and emission wavelengths at 635 and 670 nm, respectively.
Cytokine enzyme-linked immunosorbent assay (ELISA).
This experiment was performed following a procedure reported previously by us.23 Briefly, standards were diluted by seven serial twofold dilutions for the generation of a standard curve. Prepared standards and supernatants from BV-2 cells were incubated on a capture antibody coated plate overnight at 4 °C. For IL-1α levels measurement in Tg2576 mice serum, mouse serum samples were diluted 1:5 in the diluent before the ELISA, and IL-1α standard and diluted serum samples were incubated at room temperature for 2 h. Then the plates were washed five times with the washing solution. Next, a detection antibody was added, and the plate was then incubated for 1 h, followed by the addition of avidin conjugated with horseradish peroxidase for 30 minutes. The plates were then washed five times and developed by adding substrate solution to each well, and then the reaction was stopped by the addition of stop solution (2N H2SO4). Absorbance at 450 nm wavelength was measured using a microplate reader.
Morris water maze test.
Morris water maze test was performed following a procedure described previously.22 Briefly, the water inside a circular plastic pool was warmed at 25 ± 1 C°, and the escape platform was hidden 1-1.5 cm under the water level. The mice were released into the water with their head pointing to the pool wall and trained three times a day with randomized starting points over 5 days to promote the acquisition of the location of an escape platform in the pool (trial duration: 60 sec). A camera was positioned directly above the center of the pool, which recorded the behaviors of mice. The video was analyzed, and swimming speed, escape latency and escape distance on every trial was calculated by the SMART-LD program (Panlab, Spain). A probe test was performed 24 h after the water maze test (i.e., Day 6) to assess spatial memory consolidation based on the preference for the quadrant where the platform was placed. The swimming patterns of each mouse for 60 s were tracked and analyzed by the SMART-LD program.
Passive avoidance.
As previously described by our group,22 the effects of proteasome inhibitors on learning and memory function in mice were evaluated using a step-through passive avoidance test. The passive avoidance test was performed 48 h after the probe test. A “step-through” apparatus (Med Associates Inc., Vermont, USA) consists of two connected chambers (a lighted chamber and a dark chamber) by a small gate. On the first day (i.e., Day 7 of the behavior study), the mice were placed in the illuminated compartment facing away from the dark chamber for the learning trial. As soon as they entered the darkroom, Mice received a punishing electric shock (0.45 mA, 3 s duration). The testing trial was given 24 h after termination of the learning trial (i.e., Day 8) using the same paradigm but without the electric shock. Each mouse was released in the lighted compartment, and the entrance latency until the mice re-entered the dark chamber was determined and defined as the step-through latency. The maximum latency time for entering the dark compartment was 120 seconds.
RPE flat mounts.
RPE flat mounts were performed following the previously published protocols by our group.22 Briefly, surgically isolated eyes of Tg2576 transgenic mice were first fixed in 4% paraformaldehyde for 1 h at room temperature. Next, anterior eyecups were dissected on ice-cold PBS, followed by the removal of the retina. The dissected RPE sheets were permeabilized with 0.2% Triton X-100 for 15 min and then blocked with 3% BSA for 1 h at room temperature. The sections were then incubated with β-catenin (1:100; Abcam, ab19381) overnight at 4 °C. After being rinsed in PBS containing 0.5% BSA, the tissues were incubated further with secondary antibodies coupled to Alexa 555 (1:1000; Invitrogen, A21422) for 2 h at room temperature and mounted. The RPE sheets were imaged using a confocal microscope (Carl Zeiss, LSM 800).
Computational docking.
AutoDock Vina was used to docking DB-310 into the atomic structure of mouse LMP2 (PDB ID: 3UNF).23 A 3D docking box of 30-Å width covering the ligand-binding site was used (For PDB validation report, see Supporting Information). The highest-scoring, predicted ligand model, with the epoxy ketone positioned near the N-terminal threonine for possible covalent interaction, was chosen for our investigations. The chosen model shows a 95% calculated affinity compared to the highest scoring model, with no covalent bond formation.
Supplementary Material
ACKNOWLEDGMENT
We want to thank the National Institutes of Health (R01 CA188354 to K.B.K. and R01 GM111084 to S.L.), National Research Foundation of Korea (2017R1E1A1A01074656 to D-E.K. and MRC2017R A5A2015541 to J.T.H), and the Creative-Pioneering Researchers Program through Seoul National University (to W.L.) for financially supporting this work.
ABBREVIATIONS
- AD
Alzheimer’s disease
- Aβ
amyloid-beta
- COX
cyclooxygenase
- TNF-α
tumor necrosis factor-α
- iP
immunoproteasome
- LMP2
low molecular mass polypeptide-2
- MECL-1
multicatalytic endopeptidase complex subunit-1
- LMP7
low molecular mass polypeptide-7
- RPE
retinal pigment epithelium
- CNS
central nervous system
- BBB
blood-brain barrier
- CT-L
chymotrypsin-like
- LPS
lipopolysaccharide
- cP
constitutive proteasome
- AMD
age-related macular degeneration
- APP
amyloid precursor protein
- PhA
pheophorbide A
Footnotes
ASSOCIATED CONTENT
The following supporting information is available free of charge via the Internet at http://pubs.acs.org.
Proteasome catalytic subunit inhibition of DB-60 (20) in different cell lines
Molecular formula strings (CSV)
Molecular docking models (PDB) file
1H/13C NMR spectra of all macrocyclic compounds (PDF) and HPLC elution profile of the lead compound DB-60 (20)
The authors declare no competing financial interest.
REFERENCES
- 1.Yiannopoulou KG; Anastasiou AI; Zachariou V; Pelidou SH, Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long JM; Holtzman DM, Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179 (2), 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummings J; Blennow K; Johnson K; Keeley M; Bateman RJ; Molinuevo JL; Touchon J; Aisen P; Vellas B, Anti-Tau Trials for Alzheimer's Disease: A Report from the EU/US/CTAD Task Force. J Prev Alzheimers Dis 2019, 6 (3), 157–163. [DOI] [PubMed] [Google Scholar]
- 4.Congdon EE; Sigurdsson EM, Tau-targeting therapies for Alzheimer's disease. Nat Rev Neurol 2018, 14 (7), 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGeer PL; Rogers J; McGeer EG, Inflammation, anti-inflammatory agents, and Alzheimer's disease: the last 22 years. J Alzheimers Dis 2016, 54 (3), 853–857. [DOI] [PubMed] [Google Scholar]
- 6.Regen F; Hellmann-Regen J; Costantini E; Reale M, Neuroinflammation and Alzheimer's disease: implications for microglial activation. Curr Alzheimer Res 2017, 14 (11), 1140–1148. [DOI] [PubMed] [Google Scholar]
- 7.Liddelow SA; Guttenplan KA; Clarke LE; Bennett FC; Bohlen CJ; Schirmer L; Bennett ML; Munch AE; Chung WS; Peterson TC; Wilton DK; Frouin A; Napier BA; Panicker N; Kumar M; Buckwalter MS; Rowitch DH; Dawson VL; Dawson TM; Stevens B; Barres BA, Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541 (7638), 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ising C; Venegas C; Zhang S; Scheiblich H; Schmidt SV; Vieira-Saecker A; Schwartz S; Albasset S; McManus RM; Tejera D; Griep A; Santarelli F; Brosseron F; Opitz S; Stunden J; Merten M; Kayed R; Golenbock DT; Blum D; Latz E; Buee L; Heneka MT, NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575 (7784), 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta PP; Pandey RD; Jha D; Shrivastav V; Kumar S, Role of traditional nonsteroidal anti-inflammatory drugs in Alzheimer's disease: a meta-analysis of randomized clinical trials. Am J Alzheimers Dis Other Demen 2015, 30 (2), 178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou RC; Kane M; Ghimire S; Gautam S; Gui J, Treatment for rheumatoid arthritis and risk of Alzheimer's disease: a nested case-control analysis. CNS Drugs 2016, 30 (11), 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Group, A. D. C.; Bentham P; Gray R; Sellwood E; Hills R; Crome P; Raftery J, Aspirin in Alzheimer's disease (AD2000): a randomised open-label trial. Lancet Neurol 2008, 7 (1), 41–49. [DOI] [PubMed] [Google Scholar]
- 12.Group, A. s. D. A.-i. P. T. R., Results of a follow-up study to the randomized Alzheimer's disease anti-inflammatory prevention trial (ADAPT). Alzheimers Dement 2013, 9 (6), 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller Z; Ao L; Kim KB; Lee W, Inhibitors of the immunoproteasome: current status and future directions. Curr Pharm Des 2013, 19 (22), 4140–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basler M; Mundt S; Bitzer A; Schmidt C; Groettrup M, The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol 2015, 33 (4 Suppl 92), S74–S79. [PubMed] [Google Scholar]
- 15.Ogorevc E; Schiffrer ES; Sosic I; Gobec S, A patent review of immunoproteasome inhibitors. Expert Opin Ther Pat 2018, 28 (7), 517–540. [DOI] [PubMed] [Google Scholar]
- 16.Johnson HWB; Lowe E; Anderl JL; Fan A; Muchamuel T; Bowers S; Moebius D; Kirk C; McMinn DL, A required immunoproteasome subunit inhibition profile for anti-inflammatory efficacy and clinical candidate KZR-616 ((2S,3R)-N-((S)-3-(cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2 -yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)pr openamide). J Med Chem 2018, 61 (24), 11127–11143. [DOI] [PubMed] [Google Scholar]
- 17.Orre M; Kamphuis W; Dooves S; Kooijman L; Chan ET; Kirk CJ; Dimayuga Smith V; Koot S; Mamber C; Jansen AH; Ovaa H; Hol EM, Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain 2013, 136 (5), 1415–1431. [DOI] [PubMed] [Google Scholar]
- 18.Mishto M; Bellavista E; Santoro A; Stolzing A; Ligorio C; Nacmias B; Spazzafumo L; Chiappelli M; Licastro F; Sorbi S; Pession A; Ohm T; Grune T; Franceschi C, Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains. Neurobiol Aging 2006, 27 (1), 54–66. [DOI] [PubMed] [Google Scholar]
- 19.Mishto M; Bonafe M; Salvioli S; Olivieri F; Franceschi C, Age dependent impact of LMP polymorphisms on TNF-alpha-induced apoptosis in human peripheral blood mononuclear cells. Exp Gerontol 2002, 37 (2-3), 301–308. [DOI] [PubMed] [Google Scholar]
- 20.Wagner LK; Gilling KE; Schormann E; Kloetzel PM; Heppner FL; Kruger E; Prokop S, Immunoproteasome deficiency alters microglial cytokine response and improves cognitive deficits in Alzheimer's disease-like APPPS1 mice. Acta Neuropathol Commun 2017, 5 (1), 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moritz KE; McCormack NM; Abera MB; Viollet C; Yauger YJ; Sukumar G; Dalgard CL; Burnett BG, The role of the immunoproteasome in interferon-gamma-mediated microglial activation. Sci Rep 2017, 7 (1), 9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeo IJ; Lee MJ; Baek A; Miller Z; Bhattarai D; Baek YM; Jeong HJ; Kim YK; Kim DE; Hong JT; Kim KB, A dual inhibitor of the proteasome catalytic subunits LMP2 and Y attenuates disease progression in mouse models of Alzheimer's disease. Sci Rep 2019, 9 (1), 18393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattarai D; Lee MJ; Baek A; Yeo IJ; Miller Z; Baek YM; Lee S; Kim DE; Hong JT; Kim KB, LMP2 Inhibitors as a Potential Treatment for Alzheimer's Disease. J Med Chem 2020, 63 (7), 3763–3783. [DOI] [PubMed] [Google Scholar]
- 24.Dong ZZ; Li J; Gan YF; Sun XR; Leng YX; Ge J, Amyloid beta deposition related retinal pigment epithelium cell impairment and subretinal microglia activation in aged APPswePS1 transgenic mice. Int J Ophthalmol 2018, 11 (5), 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez SE; Lumayag S; Kovacs B; Mufson EJ; Xu S, Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Invest Ophthalmol Vis Sci 2009, 50 (2), 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanke NT; Imler E; Marron MT; Seligmann BE; Garland LL; Baker AF, Characterization of carfilzomib-resistant non-small cell lung cancer cell lines. J Cancer Res Clin Oncol 2018, 144 (7), 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson HWB; Anderl JL; Bradley EK; Bui J; Jones J; Arastu-Kapur S; Kelly LM; Lowe E; Moebius DC; Muchamuel T; Kirk C; Wang Z; McMinn D, Discovery of highly selective inhibitors of the immunoproteasome low molecular mass polypeptide 2 (LMP2) subunit. ACS Med Chem Lett 2017, 8 (4), 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deshmukh RR; Kim S; Elghoul Y; Dou QP, P-Glycoprotein Inhibition Sensitizes Human Breast Cancer Cells to Proteasome Inhibitors. J Cell Biochem 2017, 118 (5), 1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verbrugge SE; Assaraf YG; Dijkmans BA; Scheffer GL; Al M; den Uyl D; Oerlemans R; Chan ET; Kirk CJ; Peters GJ; van der Heijden JW; de Gruijl TD; Scheper RJ; Jansen G, Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 2012, 341 (1), 174–182. [DOI] [PubMed] [Google Scholar]
- 30.Lee MJ; Bhattarai D; Yoo J; Miller Z; Park JE; Lee S; Lee W; Driscoll JJ; Kim KB, Development of novel epoxyketone-based proteasome inhibitors as a strategy to overcome cancer resistance to carfilzomib and bortezomib. J Med Chem 2019, 62 (9), 4444–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park JE; Miller Z; Jun Y; Lee W; Kim KB, Next-generation proteasome inhibitors for cancer therapy. Transl Res 2018, 198, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marsault E; Peterson ML, Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J Med Chem 2011, 54 (7), 1961–2004. [DOI] [PubMed] [Google Scholar]
- 33.Driggers EM; Hale SP; Lee J; Terrett NK, The exploration of macrocycles for drug discovery--an underexploited structural class. Nat Rev Drug Discov 2008, 7 (7), 608–624. [DOI] [PubMed] [Google Scholar]
- 34.Bhat A; Roberts LR; Dwyer JJ, Lead discovery and optimization strategies for peptide macrocycles. Eur J Med Chem 2015, 94, 471–479. [DOI] [PubMed] [Google Scholar]
- 35.White AM; Craik DJ, Discovery and optimization of peptide macrocycles. Expert Opin Drug Discov 2016, 11 (12), 1151–1163. [DOI] [PubMed] [Google Scholar]
- 36.Cacciatore I; Fornasari E; Di Stefano A; Marinelli L; Cerasa LS; Turkez H; Aydin E; Moretto A; Ferrone A; Pesce M; di Giacomo V; Reale M; Costantini E; Di Giovanni P; Speranza L; Felaco M; Patruno A, Development of glycine-alpha-methyl-proline-containing tripeptides with neuroprotective properties. Eur J Med Chem 2016, 108, 553–563. [DOI] [PubMed] [Google Scholar]
- 37.Bach A; Eildal JN; Stuhr-Hansen N; Deeskamp R; Gottschalk M; Pedersen SW; Kristensen AS; Stromgaard K, Cell-permeable and plasma-stable peptidomimetic inhibitors of the postsynaptic density-95/N-methyl-D-aspartate receptor interaction. J Med Chem 2011, 54 (5), 1333–1346. [DOI] [PubMed] [Google Scholar]
- 38.Chua KC; Pietsch M; Zhang X; Hautmann S; Chan HY; Bruning JB; Gutschow M; Abell AD, Macrocyclic protease inhibitors with reduced peptide character. Angew Chem Int Ed Engl 2014, 53 (30), 7828–7831. [DOI] [PubMed] [Google Scholar]
- 39.Besse A; Stolze SC; Rasche L; Weinhold N; Morgan GJ; Kraus M; Bader J; Overkleeft HS; Besse L; Driessen C, Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32 (2), 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ao L; Wu Y; Kim D; Jang ER; Kim K; Lee DM; Kim KB; Lee W, Development of peptide-based reversing agents for p-glycoprotein-mediated resistance to carfilzomib. Mol Pharm 2012, 9 (8), 2197–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song YK; Park JE; Oh Y; Hyung S; Jeong YS; Kim MS; Lee W; Chung SJ, Suppression of Canine ATP Binding Cassette ABCB1 in Madin-Darby Canine Kidney Type II Cells Unmasks Human ABCG2-Mediated Efflux of Olaparib. J Pharmacol Exp Ther 2019, 368 (1), 79–87. [DOI] [PubMed] [Google Scholar]
- 42.Abuznait AH; Kaddoumi A, Role of ABC transporters in the pathogenesis of Alzheimer's disease. ACS Chem Neurosci 2012, 3 (11), 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding JD; Johnson LV; Herrmann R; Farsiu S; Smith SG; Groelle M; Mace BE; Sullivan P; Jamison JA; Kelly U; Harrabi O; Bollini SS; Dilley J; Kobayashi D; Kuang B; Li W; Pons J; Lin JC; Bowes Rickman C, Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci U S A 2011, 108 (28), E279–E287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li D; Zhang X; Ma X; Xu L; Yu J; Gao L; Hu X; Zhang J; Dong X; Li J; Liu T; Zhou Y; Hu Y, Development of Macrocyclic Peptides Containing Epoxyketone with Oral Availability as Proteasome Inhibitors. J Med Chem 2018, 61 (20), 9177–9204. [DOI] [PubMed] [Google Scholar]
- 45.Pogue AI; Dua P; Hill JM; Lukiw WJ, Progressive inflammatory pathology in the retina of aluminum-fed 5xFAD transgenic mice. J Inorg Biochem 2015, 152, 206–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai Y; Lu B; Ljubimov AV; Girman S; Ross-Cisneros FN; Sadun AA; Svendsen CN; Cohen RM; Wang S, Ocular changes in TgF344-AD rat model of Alzheimer's disease. Invest Ophthalmol Vis Sci 2014, 55 (1), 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu B; Rasool S; Yang Z; Glabe CG; Schreiber SS; Ge J; Tan Z, Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer's transgenic mice. Am J Pathol 2009, 175 (5), 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masuzzo A; Dinet V; Cavanagh C; Mascarelli F; Krantic S, Amyloidosis in Retinal Neurodegenerative Diseases. Front Neurol 2016, 7, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sivak JM, The aging eye: common degenerative mechanisms between the Alzheimer's brain and retinal disease. Invest Ophthalmol Vis Sci 2013, 54 (1), 871–880. [DOI] [PubMed] [Google Scholar]
- 50.Kaarniranta K; Salminen A; Haapasalo A; Soininen H; Hiltunen M, Age-related macular degeneration (AMD): Alzheimer's disease in the eye? J Alzheimers Dis 2011, 24 (4), 615–631. [DOI] [PubMed] [Google Scholar]
- 51.Ohno-Matsui K, Parallel findings in age-related macular degeneration and Alzheimer's disease. Prog Retin Eye Res 2011, 30 (4), 217–238. [DOI] [PubMed] [Google Scholar]
- 52.Chiquita S; Rodrigues-Neves AC; Baptista FI; Carecho R; Moreira PI; Castelo-Branco M; Ambrosio AF, The Retina as a Window or Mirror of the Brain Changes Detected in Alzheimer's Disease: Critical Aspects to Unravel. Mol Neurobiol 2019, 56 (8), 5416–5435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.