

Abstract
Proteolysis-targeting chimeras (PROTACs) are molecules that induce proximity between target proteins and E3 ligases triggering target protein degradation. Pomalidomide, a widely used E3 ligase recruiter in PROTACs, can independently degrade other proteins, including zinc-finger (ZF) proteins, with vital roles in health and disease. This off-target degradation hampers the therapeutic applicability of pomalidomide-based PROTACs, requiring development of PROTAC design rules that minimize off-target degradation. Here we developed a high-throughput platform that interrogates off-target degradation and found that reported pomalidomide-based PROTACs induce degradation of several ZF proteins. We generated a library of pomalidomide analogues to understand how functionalizing different positions of the phthalimide ring, hydrogen bonding, and steric and hydrophobic effects impact ZF protein degradation. Modifications of appropriate size on the C5 position reduced off-target ZF degradation, which we validated through target engagement and proteomics studies. By applying these design principles, we developed anaplastic lymphoma kinase oncoprotein-targeting PROTACs with enhanced potency and minimal off-target degradation.
Imide-based molecular glues (for example, pomalidomide) are proximity-inducing molecules that can simultaneously bind to cereblon (CRBN) and Zn-finger (ZF) proteins. This proximity induction between CRBN, the substrate receptor for an E3 ubiquitin ligase, and ZF proteins triggers the latter’s ubiquitination and degradation1–6. In proteolysis-targeting chimera (PROTAC), pomalidomide is often appended to target protein binder to generate a molecular chimera that induces proximity between the target protein and CRBN ligase complex. Like that of molecule glues, this proximity induction results in target protein ubiquitination and degradation7–9. However, the pomalidomide moiety in these PROTACs can also potentially ubiquitinate and degrade ZF proteins that serve key biological functions in normal development and disease progression10–14. For example, tissue-specific deletion of pomalidomide-degradable ZF protein, ZFP91, in regulatory T cells (Tregs) leads to Treg dysfunction and increases the severity of inflammation-driven colorectal cancer15. Furthermore, numerous other proteins with important roles in cellular function, such as transcription factors, harbour ZF domains16,17. The off-target degradation of these key ZF-containing proteins may have long-term implications, such as the development of new cancers, dysregulation of lymphocyte development, and teratogenic effects18–22. The ability of pomalidomide to degrade other proteins in a PROTAC-independent manner raises concerns about the precariousness of off-target ubiquitination and degradation of these compounds, several of which are already in clinical trials23,24. Thus, there is an urgent need for robust, sensitive and high-throughput methods that can determine off-target degradation of such PROTACs25.
Currently, off-target degradation can be assessed by mass spectrometry (MS)-based methods26–29 that detect protein levels, but these techniques lack sensitivity for low-abundant proteins30. In addition to the expense, the implementation of MS is technically challenging and requires examination of PROTACs across multiple tissue types owing to lineage-specific protein expression31. These analyses are further complicated by the need to perform the off-target assessments under various dose and temporal regimen of PROTAC administration.
In this Article, we report the development of an inexpensive, sensitive, robust and high-throughput imaging platform that profiles the off-target degradation propensities of ZF domains by measuring the decrease in fluorescence of a panel of GFP-tagged various ZF domains upon compound treatment32. We validated this platform using immunoblotting, target engagement and global proteomics experiments. Using this platform, we profiled off-target activities of literature-reported PROTACs with different linker chemistries. Surprisingly, nearly all the profiled PROTACs exhibited off-target degradation of ZF domains. To reduce this, we rationally designed and generated approximately 80 pomalidomide analogues and profiled them for off-target activity. We discovered and validated that substitution at specific positions (C5) on pomalidomide reduced ZF degradation propensities. With these findings, we generated PROTACs that target anaplastic lymphoma kinase (ALK) with reduced off-target ZF degradation and enhanced on-target potencies. Overall, we developed an off-target profiling platform and used that platform to identify pomalidomide analogues with lower off-target propensity and then deployed such analogue to generate a specific PROTAC for ALK.
Results and discussion
Development of an off-target profiling platform for PROTACs
To profile the ZF degradation propensity of pomalidomide and its analogues, we first developed an automated imaging assay (Fig. 1a). For this platform, we selected 23-amino-acid ZF degrons of 11 ZF proteins that are reportedly degraded by pomalidomide and 3 ZFs that are not (Extended Data Table 1)32. We inserted these ZF degrons into a lentiviral degradation reporter vector (cilantro 2)32 to compare the fluorescence of ZF-tagged enhanced green fluorescent protein (eGFP) with untagged mCherry (Fig. 1a). With this assay, wherein compounds are tested against the 14 stable U2OS cell lines containing our tagged ZF proteins, we reliably detected dose-dependent degradation of ZF degron-tagged eGFP by pomalidomide with a robust readout (Z′ = 0.8) (Extended Data Fig. 1a). As expected, pomalidomide degraded all 12 ZF degrons that are sensitive to it in a dose-dependent manner that ranged from 4.3 nM to 6 μM (Fig. 1b). Unlike MS, this reporter-based method is not limited by cell-type-specific expression levels of analyte proteins nor by the accessibility to ZFs in the context of full-length proteins that are engaged in protein complexes. Thus, this method may have enhanced sensitivity over MS-based methods for the detection of pomalidomide-sensitive ZF protein degradation.
Fig. 1 |. Development of a high-throughput assay for evaluating off-target ZF degradation of pomalidomide-based PROTACs.
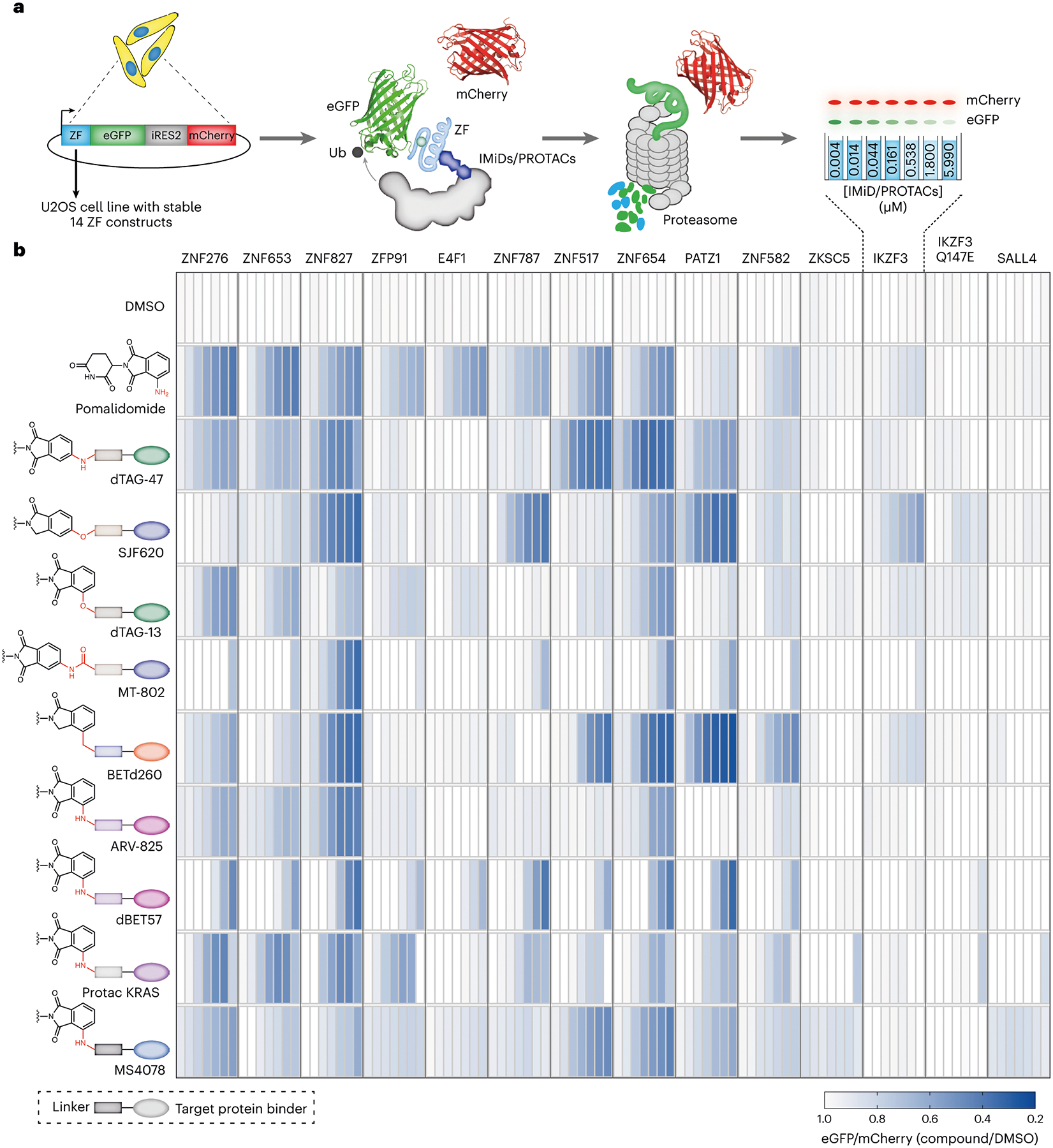
a, An automated imaging screen for the degradation of ZF degrons by pomalidomide analogues and pomalidomide-based PROTACs. Briefly, U2OS cells stably expressing 14 ZF degrons fused to eGFP were treated with PROTACs followed by imaging to assess ZF degradation. Decrease in the eGFP signal is the measure of ZF protein degradation. b, Degradation of validated and pomalidomide-sensitive ZF degrons inside cells by reported PROTACs in a dosing range of 4.3 nM to 6 μM. As shown in the schematic, each rectangle in the heatmap represents a single dose. Chemical structures with rectangle boxes and oval shapes indicate linker chemotype and target protein binding ligands respectively. Data are the mean of at least three independent replicates.
With this assay in hand, we profiled the off-target activity of nine reported PROTACs with varying exit vectors from pomalidomide end and linker lengths (Fig. 1b and Supplementary Fig. 1). We observed degradation of many ZF domains with almost all the nine PROTACs (Fig. 1b). Notably, PROTACs with common exit vectors, such as arylamine, arylether, arylcarbon and arylamide, generally had greater ZF degradation capabilities in similar fashion to pomalidomide (Fig. 1b).
To validate this imaging platform, we determined the relative stability of the ternary complex formed by the PROTACs with CRBN and the off-target ZFP91 using a reported NanoBRET assay (Fig. 2a)33. Here the PROTACs induce a bioluminescence resonance energy transfer (BRET) between luciferase (appended to ZFP91) and a fluorophore (appended to CRBN via a HaloTag domain). We generated a cell line that stably expresses ZFP91–nanoluciferase (NLuc) fusion and transfected these cells with the HaloTag-fused CRBN (HT–CRBN) followed by treatment with a fluorophore (that is, NanoBRET-618 ligand) and PROTACs (Fig. 2a). We observed a BRET signal for the PROTACs, suggesting ternary complex formation between CRBN, off-target ZFP91 and PROTACs (Fig. 2b). Since ternary complex formation does not guarantee target degradation, we also confirmed the off-target degradation of endogenous ZF protein such as ZFP91 by reported PROTACs MS4078 (ALK PROTAC; Fig. 2c and Extended Data Fig. 1b,c)34 and dTAG-13 (FKBP12F36V PROTAC, Fig. 2d)35,36, using immunoblotting and by global proteomic analysis (Fig. 2c,d and Extended Data Fig. 1b–d).
Fig. 2 |. Validation of image-based results using NanoBRET and immunoblotting.

Structural insights and degradation potential of the thalidomide analogues with C4 and C5 amino groups on the phthalimide ring. a, A NanoBRET assay to quantify ternary complex formation between ZFP91, CRBN and imide analogues or PROTACs. b, BRET ratio of reported PROTACs treated at 1 μM dose in a NanoBRET assay. Data are the mean of two independent replicates. c,d, Representative immunoblots (from at least two independent replicates) demonstrating off-target degradation of endogenous ZF protein ZFP91 by MS4078 (ALK PROTAC) (c) and dTAG-13 (FKBP12F36V PROTAC) (d) in a dose-dependent manner in Jurkat cells. e, Crystal structure showing the glutarimide ring deeply buried in the CRBN exposing the 4-amino group of pomalidomide, which makes a crucial water-mediated hydrogen bond between CRBN (E377) and IKZF1 (Q147). Modification on C5 position would potentially bump off the ZF degrons (PDB: 6H0F). f, Structures of pomalidomide (1) and 5-aminothalidomide (8). g, Representative immunoblots (from at least two independent replicates) of endogenous ZF proteins ZFP91 and IKZF3 in MM1.S cells treated with pairs of imide-based analogues with C4 (pomalidomide) and C5 amino modifications on the phthalimide ring.
Next, we queried the influence of exit vectors on pomalidomide-based PROTACs and the degradation of ZFs. Towards this goal, we analysed changes in endogenous ZF proteins from 124 proteomics datasets that were generated for cells treated with pomalidomide-based PROTACs31. In Extended Data Fig. 2, we show the relative abundance of proteins that contained the ZF motif as previously described32 and were detectable in at least one proteomics dataset (that is, 284 ZF proteins). We computed a ZF degradation score for every PROTAC dataset by taking the sum of ZF protein abundance. Analysing the degradation score distribution confirmed that PROTACs had ZF protein degradation activity for amino acetamide and arylamine, arylether and arylcarbon exit vectors (Extended Data Fig. 2). The agreement between our automated imaging assay and proteomics suggests that we can apply our methodology in a high-throughput manner to identify pomalidomide analogues that confer minimal ZF degradation. Furthermore, we can develop a set of rules for PROTAC development to minimize off-target degradation of endogenous ZF proteins.
Library of rationally designed pomalidomide analogues
We next created a library of rationally designed pomalidomide analogues that can provide insights into design of pomalidomide-based PROTACs with minimal off-target ZF degradation. We gained structural insight from the crystal structure of the DDB1–CRBN–pomalidomide complex bound to transcription factor IKZF1 (Protein Data Bank (PDB): 6H0F)32. In the crystal structure, the glutarimide ring of pomalidomide is deeply buried inside CRBN, while the phthalimide ring is accessible for modification. Q147 of IKZF1 forms a water-mediated hydrogen-bonding interaction with the C4 amino group of the compound, while the C5 position is proximal to the ZF domain (Fig. 2e)32. Mutation of Q147, or equivalent residues, has been reported to abrogate IKZF degradation32. We hypothesized that appropriate substitutions at the C4 and/or C5 positions could disrupt the ternary complex of the ZF domains with CRBN while maintaining its interaction with CRBN through glutarimide ring. To investigate this, we first synthesized a thalidomide analogue, 5-aminothalidomide (8, Fig. 2f). The treatment of MM1.S cells with this C5-amino analogue revealed a decrease in overall degradation potency as compared to pomalidomide (1, Fig. 2f,g), suggesting that modifications on the C5 position will ‘bump off’, or eliminate, the endogenous ZFs. Therefore, we generated approximately 80 pomalidomide analogues (Figs. 3 and 4) with increasing size and diverse modifications on the C4 and C5 positions of the phthalimide ring.
Fig. 3 |.

Synthesis of imide analogues via nucleophic aromatic substitution (SNAr) reactions.
Fig. 4 |. Synthesis of imide analogs using bromo- and amino- imide building blocks.

a,b, Synthesis of imide analogues via cross-coupling (a) and amidation (b) chemistries.
To rapidly and systematically construct the library of pomalidomide analogues, we leveraged robust reactions such as nucleophilic aromatic substitution (SNAr), Suzuki, Sonogashira cross-couplings and amidation with commercially available imide compounds for the facile and scalable incorporation of C4 and C5 substitutions on the phthalimide ring (Figs. 3 and 4, and Supplementary Fig. 2). We synthesized the pomalidomide analogues in pairs at C4 and C5 positions and generated a library that can be categorized into three main synthetic groups: C–N (SNAr), C–C (Suzuki/Sonogashira cross-coupling) and N–C (acylation). We employed several aliphatic amines with variable sizes for high-yielding SNAr reactions with 4- and 5-fluorothalidomides. For the SNAr library, we incorporated N-Boc-piperazine and N-Boc diazaspiro[3.3]heptane, which can subsequently be used as handle to append target binder. In addition, we synthesized several pomalidomide analogues with a fluoro substitution at the 6-position of thalidomide (Fig. 3). We utilized Suzuki/Sonogashira cross-coupling reactions on 4- and 5-bromo thalidomides with heterocyclic boronic acids and phenylacetylene (Fig. 4, scheme 2) to prepare the imide analogues (50–61). We carried out amidation with a diverse class of acid chlorides varying from aliphatic to heterocyclic cores. We systematically varied the sizes from a small acetyl group (62 and 72) to the largest cubanoyl group (71 and 81) (Fig. 4). The physicochemical properties of the overall library encompass a reasonable distribution of drug-like properties, such as partition coefficient, or clogP (−2 to 4), molecular weights (250–550 g mol−1), and topological polar surface area (80–140 Å2) (Extended Data Fig. 3d).
ZF protein degradation propensity of pomalidomide analogues
Using our developed off-target profiling platform, we tested the library of pomalidomide analogues to derive rules for the impact of exit vector modifications on pomalidomide and ZF protein degradation. First, we observed that analogues with C5 modifications on the phthalimide ring had reduced ZF degradation relative to identical modifications on the C4 position, particularly for SNAr (C–N) analogues (Fig. 5a and Extended Data Fig. 4a). The trend was less for modifications made with amidation (N–C) and Suzuki/Sonogashira cross-couplings (C–C), partly due to having smaller sets of analogues compared with the SNAr group (Extended Data Fig. 4b,c). Our structure modelling and docking data also suggest that C5 modifications are more likely to create a steric clash between the SNAr exit vectors and the ZF domain than C4 position modifications (Extended Data Fig. 3a–c). Second, analogues lacking H-bond donors immediately attached to the phthalimide ring had reduced ZF degradation compared with those with H-bond donors, regardless of the position of the modification relative to the ring (Fig. 5b), in agreement with ZF degradation pattern observed with the reported PROTACs (Fig. 1b). Furthermore, the immunoblotting studies using 21 and 22 (Fig. 5c) are in agreement with the high-throughput imaging data regarding degradation of endogenous ZF proteins, ZFP91 and IKZF3. Taken together, these data reveal that pomalidomide-based PROTACs with an arylamine exit vector, where NH- is a H-bond donor, induced greater ZF degradation and suggest that PROTACs without H-bond donors will have lower off-target activity. This finding is consistent with reports that pomalidomide can stabilize the ternary complex between ZF and CRBN using H-bonds32. We aimed to further minimize off-target ZF degradation in SNAr analogues with C5 position modifications through the addition of a fluoro group at the C6 position (Figs. 3 and 4, and Supplementary Fig. 2). The fluoro group reduced ZF degradation for most SNAr exit vectors, such as N-acetylpiperazine and morpholine, but not for diazaspiro[3.3]heptane (Extended Data Fig. 4a,d).
Fig. 5 |. Degradation score as metric to nominate the imide analogues with reduced off-targets and cellular target engagement studies.

a, Plot showing pair-wise comparison of GFP degradation levels induced by pairs of SNAr pomalidomide analogues with C4 and C5 modifications (Wilcoxon matchedpairs signed-rank test; P = 0.0029) at the same dose. Data are the mean of at least three independent replicates. Centre of the box plot is median. Minima and maxima are 1.5 times the interquartile range over the 75th and under the 25th percentile, respectively. b, Degradation of pomalidomide-sensitive ZF degrons inside cells by pomalidomide analogues with and without H-bond donor(s) immediately adjacent to the phthalimide ring. Data are the mean of at least three independent replicates. (two tailed Welch’s t-test; P < 0.0001). Centre of the box plot is median. Minima and maxima are 1.5 times the interquartile range over the 75th and under the 25th percentile, respectively. c, Representative immunoblots (from at least two independent replicates) for endogenous ZF proteins ZFP91 and IKZF3 in Jurkat cells treated with pomalidomide analogues (21 and 22) with and without a H-bond donor (NH) immediately attached to the phthalimide ring. d, Scatter dot plot showing ZF degradation scores (that is, −log(degradation sum + 1)) for individual pomalidomide analogues and pomalidomide-based PROTACs investigated in this study. Data are the mean of at least three independent replicates. (C5 -F indicate fluoro substitution on C5) e, A competition-based NanoBRET assay for CRBN engagement in cells by the pomalidomide analogues. f, Dose-dependent CRBN binding curves of selected analogues determined in U2OS cells using NanoBRET-based intracellular CRBN binding assay. Data are the mean of at least four independent replicates. g, BRET ratio for selected analogues treated at 1 μM dose in a NanoBRET based ternary complex formation assay (for the ternary complex analysis of all the imide analogues generated in this study, see Supplementary Fig. 9). Data are the mean two independent replicates.
Next, we aimed to identify exit vector modifications that confer the least off-target ZF degradation. Towards this goal, we derived a degradation score for each pomalidomide analogue, including pomalidomide-based PROTACs, by taking the sum of eGFP degradation values for the ZF degrons at multiple doses for each analogue (Fig. 5d). Compounds with degradation scores close to zero induced the least ZF degradation, whereas compounds with the most negative scores induced the most ZF degradation (Fig. 5d). Analogues with piperazine (39 and 40), and alkyne (55 and 61) exit vectors predominated the group of compounds with degradation scores of 0 (Fig. 5d and Extended Data Fig. 5). Other exit vectors with minimal degradation scores include phenyl (56), azetidine (30), pyrrolidine (31), 3,3-difluoropyrrolidine (32), N-methylcyclohexylamine (36) and N-Boc-diazaspiro[3.3]heptane (37) as well as 6-fluoro-substituted analogues of morpholine (43), N-protected piperazines (44 and 47), 2-oxa-6-azaspiro[3.3]heptane (45) and diazaspiro[5.5]undecane (49) (Fig. 5d and Extended Data Fig. 5). From this study, we established two main rules for designing pomalidomide-based PROTACs to minimize off-target effects. First, exit vectors should predominantly have modifications on the C5 position. Second, the H-bond donors immediately adjacent to the phthalimide ring preferably should be masked.
Validation of results from imaging platform
We first demonstrated CRBN engagement by the imide analogues with close to zero degradation scores in cells using a NanoBRET-based CRBN binding assay. Here the imide analogues compete with the fluorophore-labelled CRBN binder and the decrease the BRET signal (Fig. 5e) correlates with the CRBN engagement. All the analogues showed a dose-dependent decrease in the BRET signal indicative of CRBN engagement (Fig. 5f). We next evaluated the relative stability of the ternary complex formed by these imide analogues with CRBN and the off-target ZFP91 using a NanoBRET assay33. In agreement with the results of our imaging platform, we observed reduced BRET signal for the analogues compared with that of pomalidomide, suggesting reduced ternary complex formation between CRBN, ZFP91 and analogues (Fig. 5g and Extended Data Fig. 6). Furthermore, imide analogues with H-bond donor group (for example, analogues obtained using amidation chemistry) formed stronger ternary complex compared with analogues with C–N bond (SNAr chemistry) or C–C (cross-coupling chemistries) bonds (Extended Data Fig. 6).
We next performed MS-based global proteomics of MOLT4 (Fig. 6) and KELLY cells (Extended Data Fig. 7) treated with imide analogues. These cells were treated with pomalidomide or structurally diverse imides with degradation scores close to zero and the lysates were analysed by MS. Pomalidomide (1) triggered the degradation of several ZF proteins (for example, IKZF1, ZFP91 and SALL4 in KELLY cells) and other off-targets (for example, RAB28, DTWD1, CUTA and POLR2J; Fig. 6a and Extended Data Fig. 7a). Analogues with C5 substitutions with piperazine (39; Fig. 6b and Extended Data Fig. 7b), diazaspiro[3.3] heptane (37; Fig. 6c and Extended Data Fig. 7c) and phenyl (56; Fig. 6d and Extended Data Fig. 8d), 2-oxa-6-azaspiro[3.3]heptane (38; Fig. 6e and Extended Data Fig. 8e), 3,3-difluoropyrrolidine (32; Fig. 6f and Extended Data Fig. 7f), N-methylcyclohexanamine (36; Fig. 6g and Extended Data Fig. 7g), phenylacetylene (61; Fig. 6h and Extended Data Fig. 7h) did not trigger degradation of any common ZF proteins or other off-targets. Taken together, these studies validate the platform and has led to identification of a collection of exit vectors to guide the development of pomalidomide-based PROTACS with reduced off-target ZF degradation.
Fig. 6 |. Validation of image-based platform by global proteomics.

a–h, Global proteomic analysis of MOLT4 cells treated with analogues 1 (a), 39 (b), 37 (c), 56 (d), 38 (e), 32 (f), 36 (g) and 61 (h). FC, fold change.
PROTACs with reduced off-target degradation propensities
As an application of imaging platform and identified analogues with reduced off-targets, we re-engineered the reported ALK PROTAC (MS4078)34, which had a high level of off-target ZF degradation (Figs. 1b and 2c), by altering the exit vectors on pomalidomide to reduce off-target ZF degradation while maintaining on-target degradation. We selected pomalidomide analogues, including alkyne, piperazine on the C5 positions, and 2,6-diazaspiro[3.3]heptane with a C6 fluoro modification, for PROTAC development owing to their near-zero ZF degradation score (Fig. 5d and Extended Data Fig. 5). We rationally designed and synthesized 12 ALK PROTACs with different exit vectors, such as an alkyne (C4: dALK-1 and C5: dALK-2), piperazine with varying alkyl (dALK-3 to dALK-6) and acyl linkers (dALK-7 to 10), and diazaspiro[3.3]heptane (dALK-11 and dALK-12) by Sonogashira cross-coupling, reductive amination and amidation chemistry, respectively (Fig. 7a and Supplementary Figs. 5–7). To investigate the effect of the fluoro group on the on- and off-target propensities, we also synthesized the corresponding fluoro pairs for non-alkyne dALK PROTACs (dALK-4, dALK-6, dALK-8, dALK-10 and dALK-12) (Fig. 7a and Supplementary Figs. 5–7). We also synthesized (5)-MS4078 (Supplementary Fig. 4), which has a C5 substitution (versus C4 substitution) for a direct comparison and performed the off-target analysis of these ALK PROTACs. C5-substituted derivative of MS4078 (that is, (5)-MS4078) reduced the off-targets but retained its ability to degrade ZNF276, ZNF827 and PATZ1 (Fig. 7b). The re-engineered ALK PROTAC with C5 alkyne exit vector (dALK-2) dramatically reduced the off-target effects of the original PROTAC MS4078, reducing the affinity for proteins such as ZNF517, ZNF654, ZNF276, ZNF653 and PATZ1 (Fig. 7b). ALK PROTACs with piperazine exit vectors with and without the fluoro group exhibited minimized off-target effects (Fig. 7b). In agreement with image-based off-target analysis, these re-engineered dALK PROTACs have a reduced ternary complex formation with off-target ZFP91 (Extended Data Fig. 6) and so does ARV110, a PROTAC under clinical trials (Extended Data Fig. 6). In agreement with these findings, the global proteomics studies revealed that, while both MS4078 and dALK-10 induced degradation of ALK (Extended Data Fig. 8a,b), MS4078 also induced degradation of SALL4 (Fig. 7c), an off-target associated with teratogenicity, while dALK-10 did not (Fig. 7d). Finally, we confirmed that the redesigned PROTACs reduced the viability of the ALK-dependent SU-DHL-1 cells (Extended Data Fig. 8c) with seven of the redesigned PROTACs exhibiting lower half-maximal effective concentration (EC50) values than that of MS4078 (Fig. 7e).
Fig. 7 |. Re-engineering of ALK PROTACs based on the design principles.
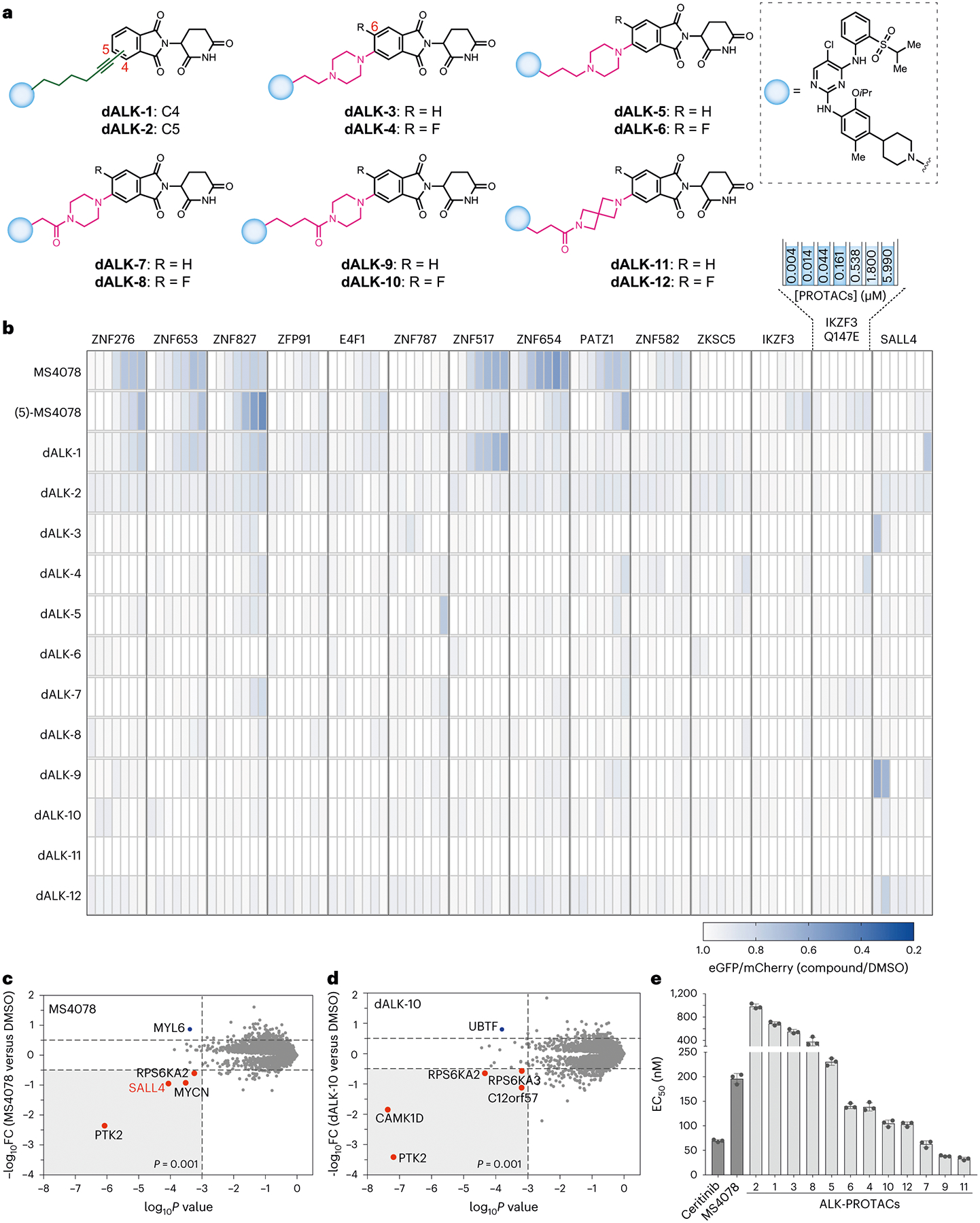
Validation of image-based degradation studies. a, Structures of rationally redesigned ALK PROTACs to minimize off-target ZF degradation. b, Degradation of validated and pomalidomide-sensitive ZF degrons inside cells by redesigned ALK PROTACs in a dosing range of 4.3 nM to 6 μM. Data are the mean of three independent replicates. c,d, Global proteomic analysis of KELLY cells treated with 100 nM of MS4078 (c) or dALK-10 (d), respectively. e, Determination of EC50 values (nM) of SU-DH-L1 cells upon treatment with redesigned ALK PROTACs. Error bars represent mean ± standard deviation from three biological replicates. FC, fold change.
Conclusions
We have developed and validated an image-based high-throughput off-target profiling platform for the systematic evaluation of PROTAC-mediated off-target degradation of ZF proteins, which play crucial roles in biology and disease progression. We leveraged this platform to identify the rules for designing pomalidomide-based PROTACs that minimize off-target degradation of ZF proteins by designing and testing a library of pomalidomide analogues—this library allowed systematic exploration of impact of positional isomerism, non-covalent interactions, and steric and hydrophobic effects on off-target degradation. Here the greatest reduction in off-target ZF degradation was achieved through modification of the exit vectors on the C5 position of the phthalimide ring via nucleophilic aromatic substitution (SNAr). Guided by these designed principles, we re-engineered a reported ALK PROTAC, MS4078 for enhanced potency and reduced off-target degradation. Our collection of pomalidomide analogues with minimal off-target ZF degradation can be widely adopted to generate safer and clinically relevant PROTACs. Notably, recently reported PROTACs in advanced clinical trials by Arvinas (ARV-110 and ARV-471), Nurix Therapeutics (NX-2127) and Foghorn Therapeutics (FHD-609) employ piperazine, pyrrolidine and spirocyclic linkers (Supplementary Fig. 8) that have exhibit reduced off-targets. The rules for pomalidomide-based PROTACs generated in this study can be readily applied to address the crucial need for PROTACs that do not indiscriminately degrade key ZF proteins. While our platform focusses on key off-targets (that is, ZF proteins), future studies will expand our platform to other classes. Regardless, the findings from this study offer opportunities to develop new and safer PROTACs and improve on existent PROTACs with enhanced on-target potency.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-023-01379-8.
Methods
Cell lines
U2OS (ATCC, HTB-96) and 293T (ATCC, CRL-3216) cells were cultured in Dulbecco’s modified Eagle medium (Thermo Fisher Scientific, 12430062), 10% (v/v) foetal bovine serum (FBS; Thermo Fisher Scientific, 16140071) and 100 U ml−1 Antibiotic-Antimycotic (Thermo Fisher Scientific, 15240062). U2OS cells stably expressing the ZF constructures were maintained in full-Dulbecco’s modified Eagle medium medium with 1 μg ml−1 puromycin (Thermo Fisher Scientific, A1113803). MM1.S (ATCC, CRL-2974), Jurkat Clone E6–1 (ATCC, TIB-152), SU-DHL-1 (ATCC, CRL-2955) and H2228 cells (ATCC, CRL-5935) were cultured in RPMI 1640 medium (Thermo Fisher Scientific, 11875119) with 10% (v/v) FBS, and 100 U ml−1 Antibiotic-Antimycotic.
Plasmids
The 14 lentiviral ZF plasmids were generated using the Cilantro 2 degradation reporter vector (Addgene, 74450) as previously described32. The luciferase version of Cilantro 2 degradation reported vector generated by swapping the eGFP with NLuc and mCherry with firefly luciferase followed by cloning the ZFP91 sequence in luciferase vector. The amino sequences of the 14 ZFs are mentioned in Supplementary Table 1.
Lentivirus production and transduction
Viral packaging plasmids psPAX2 (Addgene, 12260) and pMD2.G (Addgene, 12259) together with lentiviral ZF degron plasmids were transfected to 293T cells in a 2:1:3 ratio using TransIT-LT1 transfection reagent (MirusBio, MIR2300) following the manufacturer’s guidelines. Lentiviruses were collected 48 and 72 h after transfection and filtered with 0.45-μm Millex-HP PES filters (Millipore, SLHPM33RS). For transduction in U2OS cells, the viral supernatant was mixed with U2OS culture medium in a 1:1 ratio with 10 μg ml−1 polybrene transfection reagent (Millipore Sigma, TR-1003-G), then selected with 2 μg ml−1 puromycin after at least 24 h of transduction. Finally, antibiotic-selected cells were maintained in 1 μg ml−1 puromycin for routine culture. We chose U2OS for the primary screening as they are large, flat and adherent, making them easy to image. Furthermore, several ZF proteins have low expression in U2OS cells but are abundantly expressed in the myeloid cells, blood cancer cell lines, neuronal cell lines and embryonic stem cells, which were chosen for subsequent validation.
Automated imaging screening and analysis
CellCarrier-384 Ultra Microplates (PerkinElmer, 6057302) were printed with 10 mM of stock compounds in varying volumes using Tecan D300e Digital Dispenser (Tecan) and HP T8+ dispensehead cassettes (F0L59A). U2OS cells stably expressing the ZF degron reporters were seeded in 30 μl of 3,000 cells per well in compound preprinted 384 plates using ThermoFisher combi instrument. After 24 h of incubation, the 384-well plates were washed once with phosphate-buffered saline (PBS) then fixed in 4% paraformaldehyde and stained by HCS NuclearMask Blue stain (Thermo Fisher Scientific, H10325) using ThermoFisher combi instrument. Imaging of eGFP and mCherry was performed for each plate using an Opera Phenix imaging system followed by analysis with Harmony Software v4.9 (PerkinElmer). In-built module was used to calculate the Z′ value using the following formula.
σpc = standard deviation of positive controls
σnc = standard deviation of negative controls
μpc = mean of positive controls
μnc = mean of negative controls
The fluorescence intensity of eGFP was normalized to that of mCherry for every cell. The mean normalized eGFP intensity of all cells in each well was then normalized by that in dimethyl sulfoxide (DMSO)-treated wells to determine the GFP level relative to DMSO for each compound across doses. The GFP level relative to DMSO was used to generate a heatmap using R v4.0.2 or Graphpad Prism 9. Compounds that cause minimal ZF degradation have values close to 1 (that is, DMSO value), whereas compounds that cause extensive ZF degradation have values close to 0. To compute the degradation score for each compound, all DMSO-normalized GFP values greater than 0.9 were excluded to ensure that only ZF degradation values contribute to the score but not those that were associated with increases in GFP or caused minimal to no change in GFP degradation. The remaining DMSO-normalized GFP values were subtracted from 1 to derive the fraction of GFP degradation caused by the compounds. The degradation score was then calculated by taking the sum of the GFP degradation fractions across all doses for each compound.
Proteomics analysis of PROTAC degradation of endogenous ZF proteins
The relative abundance of 811 unique pomalidomide-sensitive ZF proteins32 were extracted from 124 publicly available pomalidomide-based PROTAC proteomics datasets reported previously31. Here, 644 out of 811 ZF proteins were detectable in at least one PROTAC proteomics dataset and are shown in Supplementary Fig. 4. To compute the ZF degradation score for each pomalidomide-based PROTAC dataset, all relative abundance values that are greater than 0 were excluded to ensure that only degradation values contribute to the score but not those that are associated with increases in protein abundance. The degradation score was then calculated by taking the sum of negative abundance values, representing ZF proteins that reduced in abundance when the cells were treated with PROTACs.
Immunoblot analysis
Lysates of cells treated with different compounds were collected with ice-cold M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, 78501) with freshly added phosphatase (Sigma-Aldrich, 04906837001) and protease (Sigma-Aldrich, 04693124001) inhibitors following the manufacturer’s instructions. Next, 20–50 μg of proteins were fractionated with NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific, NP0335BOX) then transferred onto nitrocellulose membranes using iBlot Transfer Stacks (Thermo Fisher Scientific, IB23002) following the manufacturer’s instructions. Membranes were then stained with primary antibodies in 1:1,000 dilutions and secondary fluorescent antibodies in 1:3,000 dilutions using iBind Flex Fluorescent Detection Solution Kit (Thermo Fisher Scientific, SLF2019) following the manufacturer’s instructions.
IKZF3 and ZFP91 are one of the common and important off-targets of pomalidomide-based PROTACs. Furthermore, for these targets, we could easily access detection reagents and cell lines expressing them at high level—both choices allowed us to use western blotting for cross-validation. Primary antibodies used in this study include ZFP91 (Bethyl Laboratories, A303-245A), IKZF3/Aiolos (D1C1E) (Cell Signaling, 15103S), CRBN (D8H3S) (Cell Signaling, 71810S), ALK (D5F3) XP (Cell Signaling, 3633S), phospho-ALK (Tyr1507) (D6F1V) (Cell Signaling, 14678S) and β-actin (8H10D10) mouse mAb (Cell Signaling, 3700S). Fluorescent secondary antibodies used in this study include IRDye 680RD goat anti-mouse IgG (LI-COR Biosciences, 926–68070) and IRDye 800CW goat anti-rabbit IgG (LI-COR Biosciences, 926–32211). Immunoblot blot detection was performed using an Odyssey CLx Imaging System (LI-COR Biosciences). Quantification of the relative area and density values of western blot bands was carried out using ImageJ v2.1.0 following the ImageJ User Guide for gel analysis37. Quantified values were normalized by values for loading controls such as β-actin. For phospho NPM-ALK, quantified values were normalized by the values for total ALK. All the blots were repeated at least twice.
Global proteomic analysis of IMiDs and PROTACs
Sample preparation label-free quantitative MS.
MOLT4 or Kelly or SU-DHL-1 cells were treated with DMSO or IMiD analogues (1 μM) or PROTACs (0.1 μM) for 5 h. Cells were collected by centrifugation and washed with PBS before snap freezing in liquid nitrogen. Cells were lysed by the addition of lysis buffer (8 M urea, 50 mM NaCl, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (EPPS) pH 8.5, protease and phosphatase inhibitors) and homogenization by bead beating (BioSpec) for three repeats of 30 s at 2400. Bradford assay was used to determine the final protein concentration in the clarified cell lysate. Fifty micrograms of protein for each sample was reduced, alkylated and precipitated using methanol/chloroform as previously described21, and the resulting washed precipitated protein was allowed to air dry. Precipitated protein was resuspended in 4 M urea, 50 mM HEPES pH 7.4, followed by dilution to 1 M urea with the addition of 200 mM EPPS, pH 8. Proteins were first digested with LysC (1:50; enzyme:protein) for 12 h at room temperature. The LysC digestion was diluted to 0.5 M urea with 200 mM EPPS pH 8 followed by digestion with trypsin (1:50; enzyme:protein) for 6 h at 37 °C. Sample digests were acidified with formic acid to a pH of 2–3 before desalting using C18 solid phase extraction plates (SOLA, Thermo Fisher Scientific). Desalted peptides were dried in a vacuum, centrifuged and reconstituted in 0.1% formic acid for liquid chromatography–MS analysis. Data were collected using a TimsTOF Pro2 (Bruker Daltonics) coupled to a nanoElute LC pump (Bruker Daltonics) via a CaptiveSpray nano-electrospray source. Peptides were separated on a reversed-phase C18 column (25 cm × 75 μm inner diameter, 1.6 μM, IonOpticks) containing an integrated captive spray emitter. Peptides were separated using a 50 min gradient of 2–30% buffer B (acetonitrile in 0.1% formic acid) with a flow rate of 250 nl min−1 and column temperature maintained at 50 °C.
Data-dependent acquisition (DDA) was performed in Parallel Accumulation-Serial Fragmentation (PASEF) mode to determine effective ion mobility windows for downstream diaPASEF data collection38. The ddaPASEF parameters included the following: 100% duty cycle using accumulation and ramp times of 50 ms each, one thermal ionization mass spectrometry (TIMS)–MS scan and ten PASEF ramps per acquisition cycle. The TIMS–MS survey scan was acquired between 100 and 1,700 m/z and 1/K0 of 0.7–1.3 V s−1 cm−2. Precursors with 1–5 charges were selected, and those that reached an intensity threshold of 20,000 arbitrary units were actively excluded for 0.4 min. The quadrupole isolation width was set to 2 m/z for m/z < 700 and 3 m/z for m/z > 800, with the m/z between 700 and 800 m/z being interpolated linearly. The TIMS elution voltages were calibrated linearly with three points (Agilent ESI-L Tuning Mix Ions; 622, 922 and 1,222 m/z) to determine the reduced ion mobility coefficients (1/K0). To perform data independent acquisition diaPASEF, the precursor distribution in the DDA m/z-ion mobility plane was used to design an acquisition scheme for DIA data collection that included two windows in each 50 ms diaPASEF scan. Data was acquired using 16 of these 25 Da precursor double window scans (creating 32 windows) that covered the diagonal scan line for doubly and triply charged precursors, with singly charged precursors able to be excluded by their position in the m/z-ion mobility plane. These precursor isolation windows were defined between 400 and 1,200 m/z and 1/K0 of 0.7–1.3 V s−1 cm−2.
Liquid chromatography–MS data analysis.
The diaPASEF raw file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides was performed by analysis in DIA-NN 1.8 using library or library free methods. For library analysis: targeted cell-line-specific spectral libraries were generated by searching offline fractionated DDApasef data against a Swissprot human database (January 2021) For library free methods: an in silico digestion of a given protein sequence database is performed alongside deep learning-based predictions to extract the DIA precursor data into a collection of MS2 spectra. The search results are then used to generate a spectral library that is then employed for the targeted analysis of the DIA data searched against a Swissprot human database (January 2021)39. Database search criteria largely followed the default settings for DIA including: tryptic with two missed cleavages, carbomidomethylation of cysteine, and oxidation of methionine and precursor Q-value (false discovery rate) cut-off of 0.01. Precursor quantification strategy was set to Robust LC (high accuracy) with retention time (RT)-dependent cross-run normalization. Proteins with missing values in any of the treatments and with poor-quality data were excluded from further analysis (summed abundance across channels of <100 and mean number of precursors used for quantification <2). Protein abundances were scaled using in-house scripts in the R framework40, and statistical analysis was carried out using the limma package within the R framework41.
NanoBRET assays
CRBN binding assay.
U2OS cells were transfected with a combination of NanoLuc-CRBN fusion vector (1 μg per 100,000 cells) and DDB1 expression vector (4 μg per 100,000 cells) encoded plasmid (Promega, N2910) using Lipofectamine 3000 reagent (Thermo Fisher Scientific, L3000015). After 24 h of transfection, a 384-well white microplate (Corning, 3765) is prepared with doses at 10× concentration printed using 10 mM of stock compounds in varying volumes and Tecan D300e Digital Dispenser (Tecan). Then the transfected cells were washed with PBS, trypsinized, and plated in a 384-well white microplate (Corning, 3765) at 3,000 cells in 34 μl Opti-MEM I reduced serum medium per well density. Then complete 20× NanoBRET Tracer Reagent (2 μl) and dosed of 10× IMiD compounds (4 μl) were dispensed in each well. Then the plates were incubated for 1 h at 37 °C, 5% CO2. After incubation, plates were brought to room temperature and incubated with 3× complete substrate plus inhibitor solution (20 μl) for 3 min at room temperature before taking the BRET signal by an EnVision multilabel plate reader with EnVision manager 1.13 (PerkinElmer).
Ternary complex assay.
U2OS cells stably expressing ZFP91-NLuc were transfected (2 μg per 100,000 cells) with HaloTag–CRBN encoded plasmid (Promega, N2691) using Lipofectamine 3000 reagent (Thermo Fisher Scientific, L3000015). Fourteen hours following transfection, cells were trypsinized and plated at 3,000 cells in 30 μl Opti-MEM I reduced serum medium (no phenol red + 4% FBS) per well in a 384-well white microplate (Corning, 3765). Then the cells were treated with 10 μl HaloTag NanoBRET 618 ligand (Promega, G9801) in Opti-MEM I reduced serum medium (no phenol red + 4% FBS) to a final concentration of 100 nM for 18 h. No HaloTag ligand control wells were added 10 μl above mentioned Opti-MEM I reduced serum media. Post HaloTag reaction, cells were incubated for 15 min with 10 μl of IMiD compound or PROTAC solution in above-mentioned Opti-MEM I reduced serum medium to the final concentration of 1 μM. Followed by compound incubation, 10 μl of NLuc substrate, furimazine (Aobious, AOB36539) in Opti-MEM I reduced serum medium was added at a final concentration of 10 μM and incubated for 5 min before taking the BRET signal by an EnVision multilabel plate reader with EnVision manager 1.13 (PerkinElmer).
NanoBRET values for the CRBN binding or ternary complex formation assay were calculated as follows:
Raw NanoBRET ratio (mBU) = (618 nmEm/460 nmEm) × 1,000
Mean corrected NanoBRET ratio mBU = mean mBU experimental − mean mBU no-ligand control
Note that at each liquid handling step the plates were shaken on a ThermoFisher combi instrument followed by centrifugation at 100g for 1 min.
Cell viability assay
For cell viability assay, 384-well white microplates (Corning, 8867BC) were printed with 10 mM of stock compounds in varying volumes using Tecan D300e Digital Dispenser (Tecan) and HP T8+ dispense head cassettes (F0L59A). SU-DHL-1 were seeded in 30 μl of 3,000 cells per well in compound pre-printed 384-well plates using the ThermoFisher combi instrument. After 24 h, cell viability was determined using a CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7571) following the manufacturer’s instructions. Dose–response curve fitting and EC50 quantification were determined with four-parameter non-linear regression analysis using GraphPad Prism v8.4.2.
Statistical analysis
Statistical tests were conducted using suitable underlying assumptions on variance characteristics and data distribution. Unless otherwise noted, two-tailed Student’s t-tests were used for comparisons between groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Establishment and validation of automated imaging assay for the degradation of ZFs.
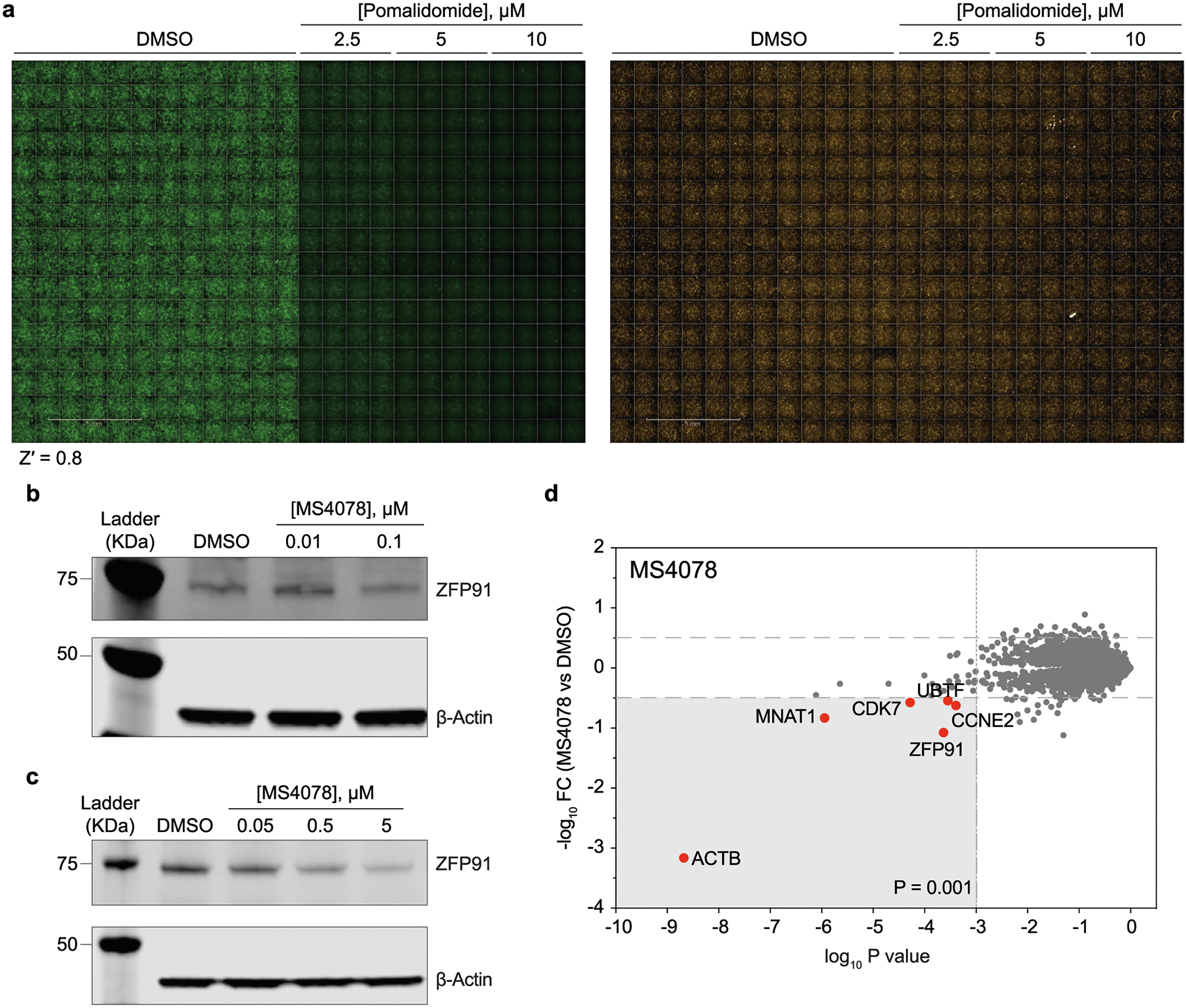
a) Representative readouts of the imaging assay in 384-well plates, demonstrating robust detection of ZF-tagged eGFP degradation as induced by pomalidomide. Shown are images of U2OS cells with stable expression of pomalidomide-sensitive ZFP91 degron reporter. Z′ value was calculated using an in-built module in the Harmony software (See materials and methods). b, c) Immunoblots (from at least two independent replicates) quantifying off-target degradation of endogenous ZF proteins ZFP91 by MS4078 (ALK PROTAC) in a dose-dependent manner across cell lines SU-DHL-1 (b) and H2228 (c). d) Label-free proteomic analysis of MS4078 in MOLT4 cells.
Extended Data Fig. 2 |. Off-target ZF degradation assessed by mass spectrometry-based proteomics.
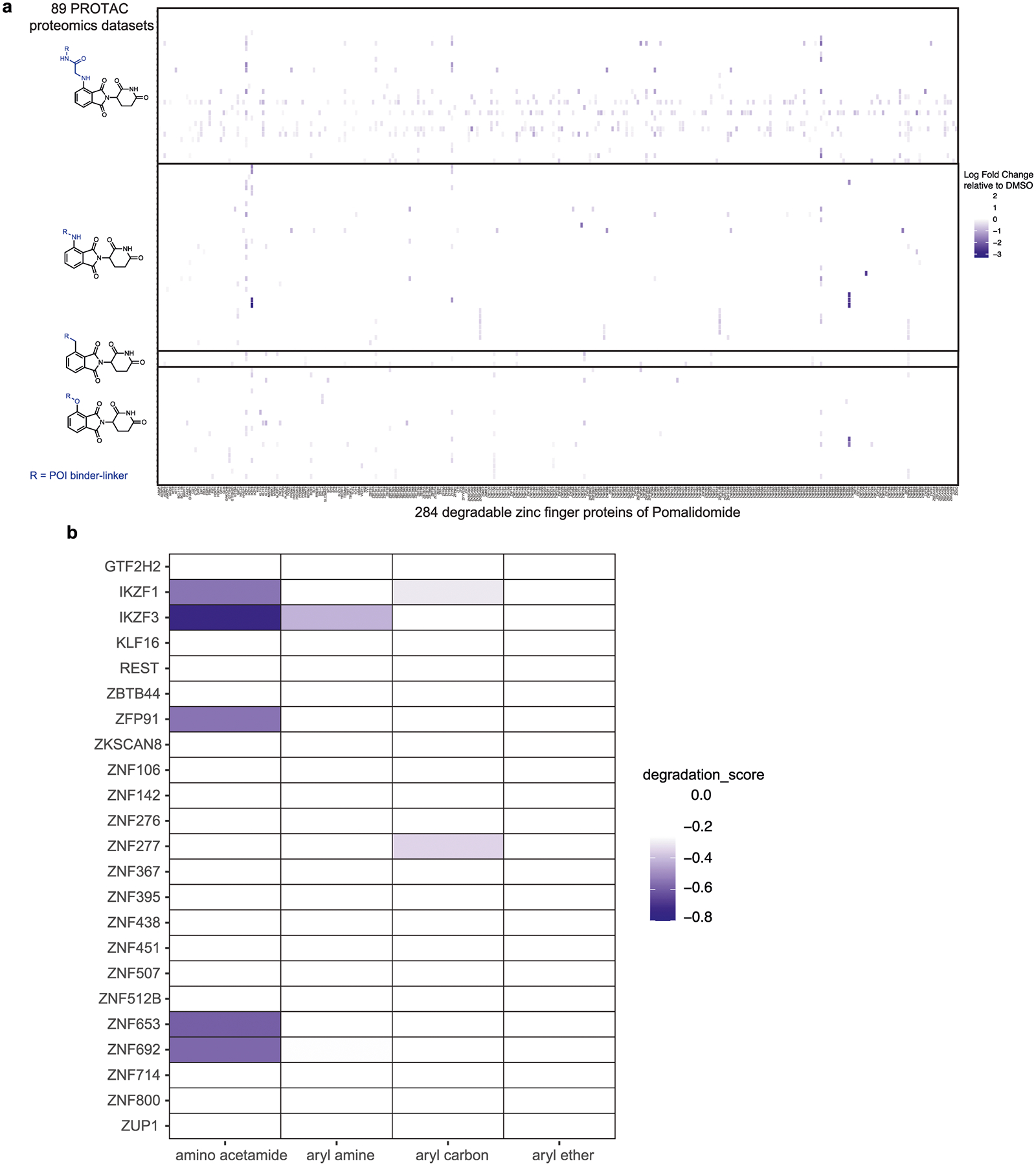
Identification of the same group of exit vectors on pomalidomide with minimal off-target ZF degradation assessed by mass spectrometry-based proteomics for entire data set (a) and top 10 most degraded proteins (b). Relative abundance of endogenous ZF proteins in cells treated with pomalidomide-based PROTACs as arranged based on pomalidomide’s exit vector groups. Data were extracted from proteomics datasets published in Donovan et al.31.
Extended Data Fig. 3 |. Docking scores and topological polar surface area of C4 and C5 pomalidomide analogs.
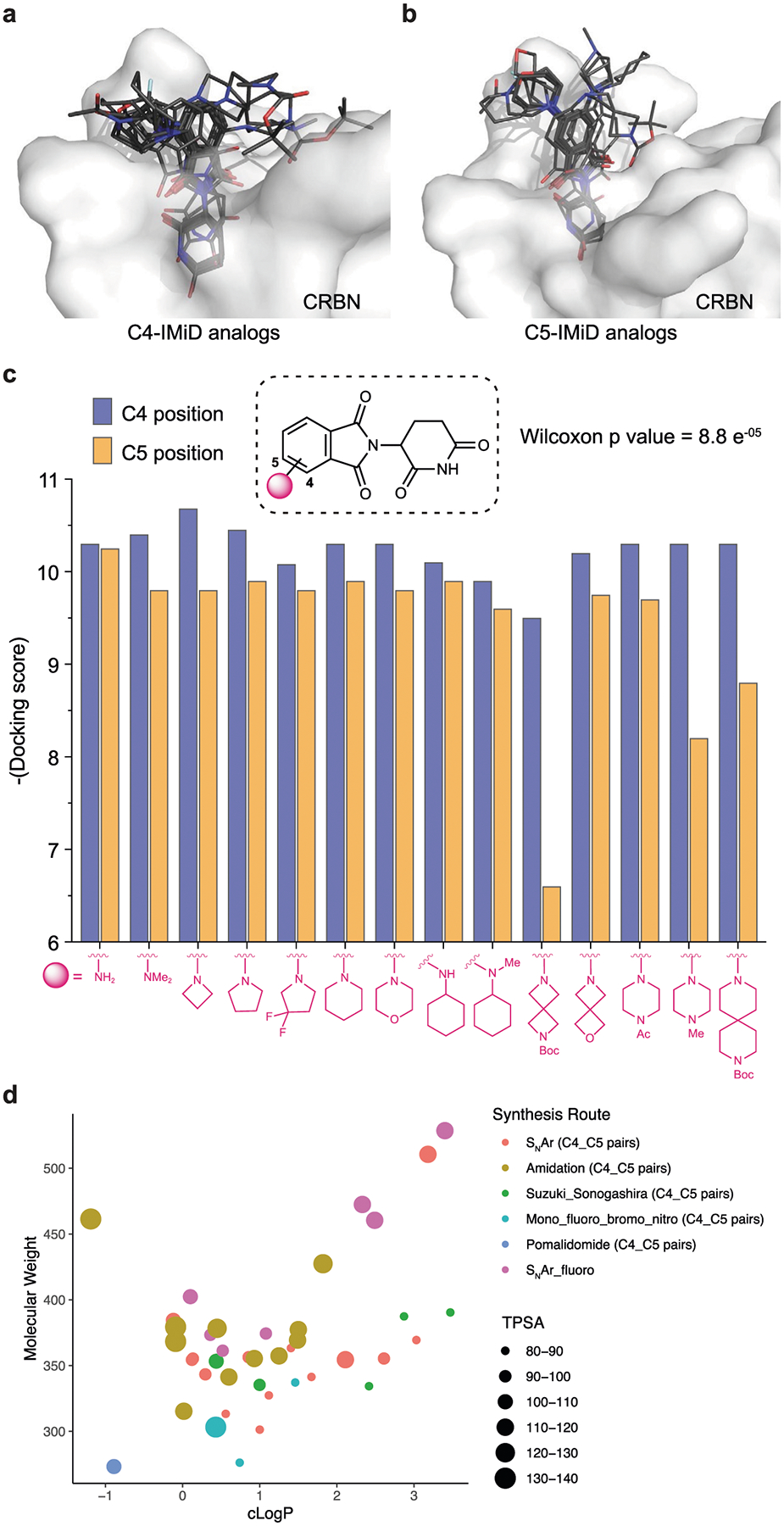
a–c) Structural docking of pomalidomide analogs with C4 (a) and C5 (b) modifications on the phthalimide ring. Docking score (c) of each pair of modifications on C4 and C5 (paired Wilcoxon test, p = 8.8 × e-05). (d) Distribution of the physicochemical properties of the pomalidomide analog library. The topological polar surface area (TPSA) of each molecule is indicated by color and size (see legend). Note that each dot in the scatter plot represents a pair of compounds with the same modification on C4 and C5 positions, except the SNAr fluoro group. Synthetic routes are represented by different shapes shown in the legend).
Extended Data Fig. 4 |. Degradation of validated pomalidomide-sensitive ZF degrons induced by the pomalidomide analogs.

(a) Normalized eGFP intensity in 14 ZF reporter cell lines treated with different doses of 81 pomalidomide analogs ranging from 4.3 nM to 20 μM. Each block of 4.3 nM to 20 μM doses on the x axis represents one ZF reporter cell line. Data are mean of at least 3 independent replicates. (See source data for replicates, mean, SD) (b–d) Box-and-Whisker plots with statistical analysis for pomalidomide analogs arranged in pairs of C4 and C5 modifications such as acylation (b), Suzuki/Sonogashira coupling (c) and effect of -F group (d) on the phthalimide ring. Data are shown for cells treated with 5 μM of each compound (for acylated and Suzuki/Sonogashira couplings) and 20 μM data of each compound for deciphering the -F group effect. For b-d figure panels data points are the mean of at least 3 independent replicates. Centre of the box plot is median. Minima and maxima are 1.5 times the interquartile range over the 75th and under the 25th percentile, respectively.
Extended Data Fig. 5 |.

Structures of IMiD analogs with the least degradation score (close to 0).
Extended Data Fig. 6 |.

NanoBRET based ternary complex analysis of all the IMiD analogs and PROTACs reported in this study.
Extended Data Fig. 7 |.
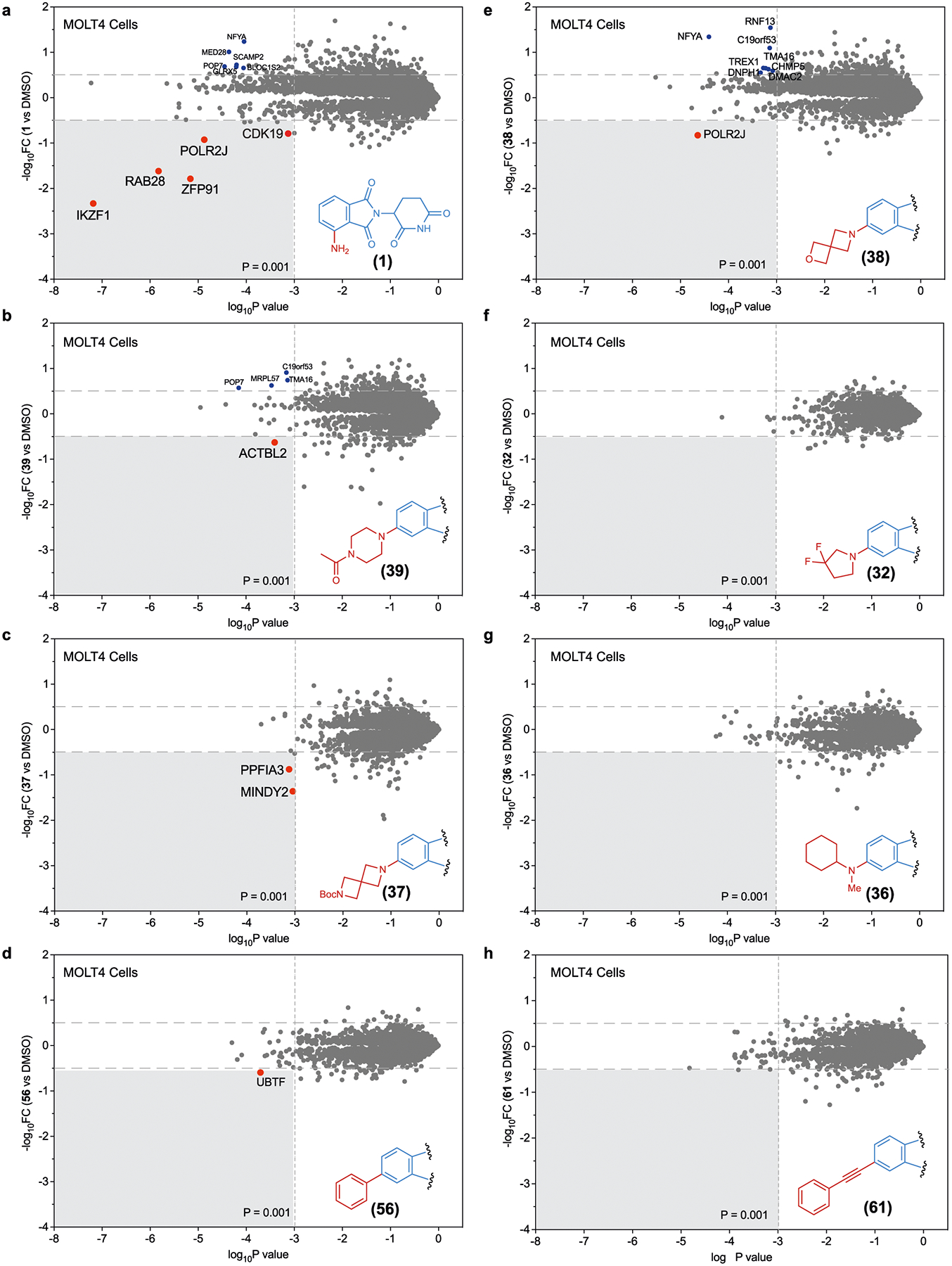
Global Proteomic analysis in KELLY cells. a–h, Global proteomic analysis of Kelly cells treated with analogues 1 (a), 39 (b), 37 (c), 56 (d), 38 (e), 32 (f), 36 (g) and 61 (h). FC, fold change.
Extended Data Fig. 8 |. Cellular characterization of dALK PROTACs.

a, b) Global Proteomic analysis of selected PROTACs, MS4078 (a) and dALK-10 (b) in SU-DHL-1 cells. c) Viability dose curves for the redesigned ALK PROTACs in SU-DHL-1 cells. Data are the mean ± SD of 3 independent replicates.
Extended Data Table 1 |.
Amino acid sequences of the zinc finger motifs cloned in cilantro 2 vector
| Protein (residue numbers) |
Function of the protein | Amino acid sequences |
|---|---|---|
|
ZNF276 (524–546 aa) |
Transcriptional regulation | LQCEVCGFQCRQRASLKYHMTKH |
|
ZNF653 (556–578 aa) |
Transcriptional repressor | LQCEICGYQCRQRASLNWHMKKH |
|
ZNF827 (374–396 aa) |
Transcriptional regulation | FQCPICGLVIKRKSYWKRHMVIH |
|
ZFP91 (400–422 aa) |
Atypical E3 ubiquitin-protein ligase, has key role in cell proliferation, anti-apoptosis | LQCEICGFTCRQKASLNWHMKKH |
|
E4F1 (220–242 aa) |
Transcriptional regulation | HECKLCGASFRTKGSLIRHHRRH |
|
ZNF787 (178–200 aa) |
Transcriptional regulation | FVCPRCGRGFSQPKSLARHLRLH |
|
ZNF517 (452–474 aa) |
Transcriptional regulation | YRCRACGRACSRLSTLIQHQKVH |
|
ZNF654 (25–47 aa) |
Transcriptional regulation | FACVICGRKFRNRGLMQKHLKNH |
|
PATZ1 (383–405 aa) |
chromatin modeling and transcription regulation, has key functions in embryogenesis, senescence, T-cell development, neurogenesis | YSCPVCGLRFKRKDRMSYHVRSH |
|
ZNF582 (395–417 aa) |
Transcriptional regulation | YQCKVCGRAFKRVSHLTVHYRIH |
|
ZKSC5 (430–452 aa) |
Transcriptional regulation | YGCNECGKNFGRHSHLIEHLKRH |
|
IKZF3 (146–168 aa) |
Transcription factor, plays key role in regulation of B-cell differentiation, proliferation and maturation | FQCNQCGASFTQKGNLLRHIKLH |
|
IKZF3 Q147E (146–168 aa) |
Inactive mutant of IKZF3, serves as negative control in image-based assay | FECNQCGASFTQKGNLLRHIKLH |
|
SALL4 (410–432) |
Transcription factor, plays key role in the maintenance and self-renewal of embryonic, hematopoietic stem cells | FVCSVCGHRFTTKGNLKVHFHRH |
Supplementary Material
Acknowledgements
We thank P. Byrne (Broad Institute) for assistance with automated imaging screening experiments. This work was supported by DARPA (N66001-17-2-4055) and NIH (R01 GM137606 and R01 GM132825 to A.C.; NIH CA214608 and CA218278 to E.S.F.; R01 EB031172, R01 EB027793, and R35 GM118062 to D.R.L.). J.A.M.M. is a Ruth L. Kirchstein National Research Service Award Postdoctoral Fellow (F32 GM133088). D.R.L. and B.L.E. are investigators of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests
Broad Institute has filed a patent application including the work described herein. A.C. is a founder and Scientific Advisory Board (SAB) member in Photys therapeutics. E.S.F. is a founder, SAB member, and equity holder in Civetta, Lighthorse, Proximity and Neomorph (board of directors), SAB member and equity holder in Photys and Avilar, and a consultant to Astellas, Novartis, Sanofi, Deerfield and EcoR1. The Fischer lab receives or has received research funding from Novartis, Astellas, Interline and Deerfield. D.R.L. is a consultant and co-founder of Exo Therapeutics, a company that develops small-molecule therapeutics. K.A.D. is a consultant to Kronos Bio and Neomorph Inc. The remaining authors declare no competing financial interests.
Footnotes
Extended data is available for this paper at https://doi.org/10.1038/s41557-023-01379-8.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41557-023-01379-8.
Data availability
Data generated in this study are provided in the manuscript, supplementary information and source data. The raw data corresponding to all the proteomics studies presented in this manuscript are available under the accession code PXD046264. Plasmid from Addgene (plasmid 74450; www.addgene.org/74450/) was used in this study. Structural information from PDB (ID: 6H0F) was used in this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
References
- 1.Chamberlain PP & Cathers BE Cereblon modulators: low molecular weight inducers of protein degradation. Drug Discov. Today Technol 31, 29–34 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Kozicka Z & Thomä NH Haven’t got a glue: protein surface variation for the design of molecular glue degraders. Cell Chem. Biol 28, 1032–1047 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Finley D Recognition and processing of ubiquitin–protein conjugates by the proteasome. Annu. Rev. Biochem 78, 477–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito T et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Krönke J et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain PP & Hamann LG Development of targeted protein degradation therapeutics. Nat. Chem. Biol 15, 937–944 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Sakamoto KM et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winter GE et al. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang B et al. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew. Chem 58, 6321–6326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teng M et al. Development of CDK2 and CDK5 Dual Degrader TMX-2172. Angew. Chem 59, 13865–13870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matyskiela ME et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 535, 252–257 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Petzold G, Fischer ES & Thomä NH Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532, 127–130 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Krönke J et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang A et al. ZFP91 is required for the maintenance of regulatory T cell homeostasis and function. J. Exp. Med 218, e20201217 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu M & Blackshear PJ RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat. Rev. Immunol 17, 130–143 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassandri M et al. Zinc-finger proteins in health and disease. Cell Death Discov 3, 17071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang ES et al. Acute pharmacological degradation of Helios destabilizes regulatory T cells. Nat. Chem. Biol 17, 711–717 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito T, Ando H & Handa H Teratogenic effects of thalidomide: molecular mechanisms. Cell Mol. Life Sci 68, 1569–1579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Therapontos C, Erskine L, Gardner ER, Figg WD & Vargesson N Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation. Proc. Natl Acad. Sci. USA 106, 8573–8578 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donovan KA et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 7, e38430 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matyskiela ME et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol 14, 981–987 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Mullard A Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov 20, 247–250 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Nguyen PA, Born DA, Deaton AM, Nioi P & Ward LD Phenotypes associated with genes encoding drug targets are predictive of clinical trial side effects. Nat. Commun 10, 1579 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deaton AM et al. Rationalizing secondary pharmacology screening using human genetic and pharmacological evidence. Toxicol. Sci 167, 593–603 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang AX et al. The vital role of proteomics in characterizing novel protein degraders. SLAS Discov 26, 518–523 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Beveridge R et al. Native mass spectrometry can effectively predict PROTAC efficacy. ACS Cent. Sci 6, 1223–1230 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grandi P & Bantscheff M Advanced proteomics approaches to unravel protein homeostasis. Drug Discov. Today Technol 31, 99–108 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Liu X et al. A proteomic platform to identify off-target proteins associated with therapeutic modalities that induce protein degradation or gene silencing. Sci. Rep 11, 15856 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reinders J, Lewandrowski U, Moebius J, Wagner Y & Sickmann A Challenges in mass spectrometry-based proteomics. Proteomics 4, 3686–3703 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Donovan KA et al. Mapping the degradable kinome provides a resource for expedited degrader development. Cell 183, 1714–1731.e10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sievers QL et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, eaat0572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riching KM et al. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chem. Biol 13, 2758–2770 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Zhang C et al. Proteolysis targeting chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem 151, 304–314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nabet B et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sreekanth V et al. Chemogenetic system demonstrates that Cas9 longevity impacts genome editing outcomes. ACS Cent. Sci 6, 2228–2237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.ImageJ user guide. (2012) NIH https://imagej.nih.gov/ij/docs/guide/
- 38.Meier F et al. diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 17, 1229–1236 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Demichev V, Messner CB, Vernardis SI, Lilley KS & Ralser M DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.R Development Core Team (R Foundation for Statistical Computing, 2014).
- 41.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are provided in the manuscript, supplementary information and source data. The raw data corresponding to all the proteomics studies presented in this manuscript are available under the accession code PXD046264. Plasmid from Addgene (plasmid 74450; www.addgene.org/74450/) was used in this study. Structural information from PDB (ID: 6H0F) was used in this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.