

Abstract
α-Synuclein is a pleiotropic protein underlying a group of progressive neurodegenerative diseases, including Parkinson’s disease and dementia with Lewy bodies. Together, these are known as synucleinopathies. Like all neurological diseases, understanding of disease mechanisms is hampered by the lack of access to biopsy tissues, precluding a real-time view of disease progression in the human body. This has driven researchers to devise various experimental models ranging from yeast to flies to human brain organoids, aiming to recapitulate aspects of synucleinopathies. Studies of these models have uncovered numerous genetic modifiers of α-synuclein, most of which are evolutionarily conserved. This review discusses what we have learned about disease mechanisms from these modifiers, and ways in which the study of modifiers have supported ongoing efforts to engineer disease-modifying interventions for synucleinopathies.
Keywords: Drug targets, Lewy bodies, Parkinson’s disease, SNCA, α-synuclein, Genetic modifiers
INTRODUCTION
Synucleinopathies refers to a group of progressive neurodegenerative diseases that are characterized by proteinaceous inclusion bodies enriched in α-synuclein (α-syn) [1]. These include Parkinson's disease (PD), Dementia with Lewy Bodies (DLB), Alzheimer's disease (AD) and multiple system atrophy (MSA) [2–6]. In PD, DLB and subsets of AD, α-syn inclusion bodies are mostly found in neurons, while in MSA, they are found in oligodendrocytes. Clinically, PD is defined by the presence of levodopa-responsive motor symptoms like bradykinesia with either tremor or rigidity or both [7]; PD patients often live many years with the disease and develop dementia at later stages. DLB is characterized by the co-occurrence of dementia and motor symptoms within a year of the appearance of either symptom [8]. On the other hand, MSA is characterized by autonomic failure and poorly levodopa-responsive motor symptoms [9]. Individual patients with each type of synucleinopathy show considerable variations in how their symptoms manifest and progress.
α-syn is a small protein of 140 amino acids, which is encoded by the SNCA gene. Genome-wide association studies (GWAS) of non-familial forms of PD and DLB show strong associations of single nucleotide polymorphisms (SNPs) within or near SNCA with the respective diseases [10,11]. In addition, rare mutations in the SNCA gene have been associated with the familial forms of various synucleinopathies. Hence, missense mutations A53T, A30P, E46K, G51D and H50Q have been found in human pedigrees with PD [12–17]. In addition, rare duplications and triplications of the wildtype SNCA gene are also associated with familial PD [18–20], indicating that increased levels of wild type α-syn protein is sufficient to cause the disease. Interestingly, the E46K missense mutation has also been associated with DLB [21] and the G51D mutation with MSA [22]. While mutations in SNCA can underlie multiple forms of synucleinopathies, the distinct pathology of each disease suggests that additional genetic or environmental modifiers may influence how the pathology unfolds.
The functions of α-syn are still a matter of much debate. In the human brain, SNCA transcripts are found mainly in neurons and oligodendrocytes; in addition, moderate levels of transcripts are found in microglia and minute levels in astrocytes [23]. In neurons, α-syn has been localized mainly to the presynaptic compartment and neurites [24], but is also associated with mitochondria, synaptic vesicles and other subcellular compartments [25]. The normal neuronal functions of α-syn are thought to include synaptic vesicle trafficking, synaptic plasticity and mitochondrial function; the reader is referred to excellent reviews that discuss normal functions of α-syn [24–27].
α-syn is known to form pathological aggregates in neuronal cell bodies and neurites, known as Lewy bodies and Lewy neurites, respectively [28]. This is favored by its high abundance at synapses, where its concentration (~ 22 μM) is similar to those of clathrin heavy chain and SV2A [29]. Interestingly, genetic evidence suggests that further increases in the concentration of α-syn are a driving force for neuronal toxicity [18,30–32]. In vivo factors that are thought to drive the misfolding of α-syn into pathological aggregates are: (1) total α-syn levels, (2) mitochondria impairment and (3) membrane associations, as summarized in a recent review [33]. In vitro studies have demonstrated that high concentrations of α-syn can progressively form aggregates such as the smaller soluble oligomers and the higher-order insoluble fibrils [34]; such fibrils are thought to be the precursors of Lewy bodies [35]. Meanwhile, the mechanisms of α-syn neurotoxicity is currently debated; while the prevailing view is that both oligomers and fibrils contribute to toxicity [36], an alternative view suggests that excessive membrane binding by monomeric α-syn also contributes to pathology [37]. Besides causing pathologies within a given cell, α-syn aggregates are known to spread from one cell to another to seed further aggregations and spread the pathology across brain regions [33,38].
The occurrence of α-syn spreading in experimental models is consistent with Braak's hypothesis which states that for at least a subset of PD cases, Lewy pathology spreads from the olfactory system or the enteric nervous system to the central nervous system [28]; this hypothesis is based on the analysis of Lewy pathology in postmortem brains from patients at various stages of PD [39]. Subsequent work has resulted in the view that Lewy pathology in the periphery and brainstem may be responsible for prodromal symptoms such as rapid eye movement (REM) sleep behavior disorder, constipation and anosmia that may emerge before the diagnosis of motor symptoms in PD, in a decades-long process of disease progression [40–42].
As summarized above, genetic, biochemical and pathological evidence point to key roles for α-syn in the pathogenesis of synucleinopathies. However, currently known GWAS variants account for only up to a third of the genetic risk of PD [10]. One way to expand our understanding is through the characterization of genetic modifiers of SNCA that alter the risk of synucleinopathies [43,44]. While GWASes of genetic modifiers of LRRK2 and GBA with respect to PD have been performed in human populations [45,46], there is no similar human study for SNCA, which shows the strongest association with PD. However, studies done using model organisms and cell lines to discover SNCA modifiers have revealed much about potential mechanisms of synucleinopathies; the current review will discuss these findings and their implications for novel therapeutic interventions.
GENETIC MODIFIERS OF SNCA
Genetic modifiers are genes that modify a clinical phenotype of a disease, i.e. the measure of the clinical variability in the population of affected individuals [47]. In the case of synucleinopathies, this may simply be a measure of SNCA-associated neurodegeneration or α-syn aggregation. The search for these modifiers can help elucidate genetic interactions of SNCA, hence shedding light on pathways important in causing disease in synucleinopathies. Once validated, these target proteins and pathways may then be exploited to develop therapeutics.
A Drosophila screen for SNCA modifiers
In our recent work, we described a screen in flies that identified twelve genetic modifiers in a model of SNCA-associated neurodegeneration (Table 1) [48]. Three-quarters of the modifiers have known roles in metabolism. ABCB7, CG4553/NGRN, GlyRS/GARS1, COX20 and ND-39/NDUFA9 encode mitochondrial proteins [49–55], while knockdown of the Drosophila nuclear protein MTA1-like led to mitochondrial defects [56]. On the other hand, MED13 and CDC27 encode subunits of protein complexes that regulate different aspects of glycolysis [57–59]. Lastly, the human orthologs of Eip75b, NR1D1 and NR1D2 regulate many aspects of energy metabolism [60,61]. The large proportion of modifiers with metabolic functions underscores the importance of energy metabolism in synucleinopathies, as observed previously [62,63].
Table 1. Human orthologs of SNCA modifiers from a fly genetic screen conducted by Ren et al., (2022) [48].
| Gene | Disease Association | Molecular Function | Perturbations in PD Brains |
|---|---|---|---|
| CDC27 | 328 kb from PD GWAS SNP rs11658976 [10] | Adaptor protein for a ubiquitin ligase complex [323] | mRNA decreased in PD brains [65] |
| ABCB7 | X-linked sideroblastic anemia with ataxia [323] | Mitochondrial transporter for Fe-S cluster [49] | Protein upregulated in MPTP mouse model [68] |
| CDAN1 | 1 kb from bipolar type 1 GWAS SNP rs112968809 [324] | Role in nuclear envelope integrity [323] | No data |
| MTA1, 2, 3 |
MTA1: CNV associated with inflammatory bowel disease [325] MTA3: Insomnia GWAS SNP rs6734957 in intron [269] |
Transcriptional co-activator/repressor [323] | MTA1 mRNA and MTA1 protein decreased in PD brains [66] |
| MED13, MED13L | MED13 is 103 kb from PD GWAS SNP rs61169879 [10] | Subunit of Mediator kinase module [326] | Protein upregulated in fly and mouse SNCA models [48] |
| NGRN | Implicated in mitochondrial translation [51] | No data | |
| HECA | Common variants close to genome-wide significance in association with coronary artery disease in diabetes [327]; rare variants associated with congenital heart disease [328] | Regulates insulin signaling in flies [128] | No data |
| GARS | Charcot–Marie–Tooth Disease Type 2D and Distal Hereditary Motor Neuronopathy Type VA [323] | Glycyl-tRNA synthetase 1 [323] | mRNA decreased in PD brains [64,65] |
| COX20 | Mitochondrial complex IV deficiency nuclear type 11 [323] | Cytochrome c oxidase assembly factor [323] | No data |
| RBPMS, RBPMS2 | RBPMS2 is 8 kb from coronary artery disease GWAS SNP rs6494488 [329] | RNA binding protein [323] | No data |
| NDUFA9 | Mitochondrial complex I deficiency, nuclear type 26 [323] | Mitochondrial complex I component [323] | mRNA decreased in PD brains [64,65] |
| NR1D1, NR1D2 | NR1D1 intron contains multiple sclerosis GWAS SNP rs883871 [330] | Homolog of nuclear receptor implicated in circadian rhythm regulation [267] | NR1D1 mRNA and NR1D1 protein decreased in rat brains treated with LPS and rotenone [331] |
Since all 12 modifiers were loss-of-function mutations that enhance SNCA-associated neurodegeneration, we propose that subsets of their mammalian orthologs show expression changes in two plausible scenarios (Table 1): (1) a constitutively expressed neuroprotective gene that is downregulated through a pathological mechanism leading to enhanced neurodegeneration; (2) a neuroprotective gene that is induced upon neuronal stress to compensate for the primary pathological event. Genes that fit Scenario 1 are CDC27, GARS1, NDUFA9 and MTA1. Transcripts of CDC27, GARS1 and NDUFA9 were found to be significantly downregulated in meta-analyses of transcriptomes of PD brains [64,65]. Similarly, MTA1 mRNA and its associated protein are downregulated in PD substantia nigra samples [66]. In addition, MTA1 is a coactivator that drives transcription of the TH gene [67], which is essential for dopaminergic function. Genes that fit Scenario 2 are ABCB7 and MED13. ABCB7 was upregulated in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD [68], suggesting that its anti-apoptotic function may be induced by neuronal stress [69]. In our work, MED13 was induced in flies and mice with elevated levels of SNCA expression and was significantly associated with PD in eQTL analysis and TWAS [10,70,71]. Hence, the mammalian orthologs of six SNCA modifiers show perturbations in expression levels in PD brains and, in some cases, the relevant animal models.
SNCA modifiers from other experimental models
Since the discovery of the central role of SNCA in both familial PD and sporadic PD [2,12,72], there have been many efforts to identify genetic modifiers of this gene. Our work summarized above is just the latest in the field. An overview reveals that a number of pathways are important in causing disease in synucleinopathies. In this review, we have compiled a list of genetic modifiers of SNCA, classified into two types (Table 2). A gene is a Type 1 Modifier when its function is inversely correlated with the level of α-syn pathology; a Type 1 Modifier has a neuroprotective function. On the other hand, a Type 2 Modifier has functions that are directly correlated with the level of α-syn pathology; inhibiting a Type 2 Modifier is likely to protect neurons. For the sake of consistency, we will use the name of the human ortholog to refer to each modifier where feasible.
Table 2. Genetic Modifiers of SNCA.
| Pathway | Modifier Type | Genes |
|---|---|---|
| Mitochondria | 1 | Protein transport: TOMM20 [104]; TIMM9 [332]; CSNK2B [333] Mitochondrial protein synthesis: PPARGC1A (PGC-1α) [101–103]; REST [103]; NGRN, GARS1 [48] Mitochondrial chaperone: TRAP1 [105] Mitochondrial kinase: PINK1 [334–336] Fusion: OPA1 [87]; PINK1, PARK7 (DJ-1), PRKN (Parkin) [337] Fission: DNM1L (Drp1) [83]; Fragmentation: PAPSS1, PAPSS2, PTPN23 [333] Mitophagy: CNTNAP4 [338]; TMEM175 [339]; FBXO5 [340]; CDC37 [333] Electron transport chain: COX20, NDUFA9 [48] Calcium: CAMK2D, ITPKB [341] ER-mitochondria calcium transport: ITPKB [252] ROS: NCEH1, SOAT1 [191]; GSH: MED13 [48] NAD+: FBXO5 [340] Iron–sulfur cluster transport: ABCB7 [48] |
| 2 | Mitochondrial function: CHCHD2/10 [342] Fusion: PDE4B [343] Fission: DNM1L (Drp1) [86] Mitophagy: SIRT1 [250] Calcium: TLK2 [253]; MICU3 [341] |
|
| Glycolysis | 1 | GPI [48,106]; MED13, PFKM, HIF1A [48]; GAPDH [106] |
| 2 | LDHA [48]; AMPK [344]; | |
| Antioxidant/glutathione metabolism | 1 | Oxidative stress: PARK7 (DJ-1) [114,332]; UBE2I (Ubc9) [345]; FOXO3 [346]; NFE2L2 (Nrf2) [112,113,347,348] [112,113], MAFK [113]; PPARGC1A (PGC-1α) [102]; TXN [332]; CTH, GCLM, HPGDS [116] Nitrosative stress: COX4I1/2 [349]; KLF11, KLF15 [350] Lipid peroxidation: RSPA [136] Detoxification: CYP2B6 [249] |
| 2 | Oxidative stress: PREP [351]; KEAP1 [113] Nitrosative stress: COX4I2 [349] |
|
| Insulin-like signalling (IIS) pathway | 1 | AMPK [352]; IGF1, AKT1 [120]; FOXO, TFPI [106]; KDM1A [136]; RPS6 [353]; IDE [354]; HECA [48]; MLST8 [350] |
| 2 | IGF1R, IRS1 [106]; MTOR [355] | |
| Cytoskeleton | 1 | SPTAN1 (Spectrin) [83]; LRRK2 [237]; PFDN5 (Prefoldin), TBCA [356]; TTC7A, TTC7B [357]; CFL1 [135], GSN [83,135]; ACTG1 (Actin) [136]; HDAC6 [358]; ITGA8 [340] |
| 2 | ACTB (Actin) [83,359] [83]; FHOD1, FHOD3 [83]; MAPT (Tau) [83]; TPPP [360]; SIRT2 [245]; LRRK2 [237] | |
| α-syn accumulation (transcription) | 1 | ZSCAN21 [146]; CEBPD [141]; TRIM41 [144] |
| 2 | ZSCAN21 [144–146]; GATA2 [142]; TP53 (p53) [143]; TRIM17 [144]; HMOX1 [361]; CEBPB [362] | |
| α-syn accumulation (Degradation pathway unknown) | 1 | Target for degradation: STUB1 (CHIP) [363,364]; USP8, USP13 [365] ER stress: GBA1 [366]; SIRT1 [249]; COX4I1/2 [349]; MANF [367]; PISD [368] Protein misfolding: VPS35 [369] |
| 2 | Ubiquitination: BAG5, HSPA1A (Hsp70) [364]; USP9X [365] ER stress: CASP12 [370]; COX4I2 [349] |
|
| α-syn accumulation (autophagy) | 1 |
AMPK [352]; ATG5 [87,371]; VPS41 [372,373]; STUB1 (CHIP) [374]; BECN1 [375]; MANF [367]; NFE2L2 (Nrf2) [376]; PFDN2, PFDN5 [377]; PINK1 [378]; SQSTM1 (p62) [371]; SIRT1 [243]; TFEB [177,243,379]; TMEM175 [339]; TERT [380]; TTC7A, TTC7B [357]; HSPA1B (Hsp70) [381]; CREBRF [253]; SNCAIP (Synphilin-1) [152,154]; SCARB2 (LIMP-2) [382]; VPS35 [383]; SMPD1 [197]; ATP13A2 (PARK9) [384–387]; PLD1 [388]; NSF [341,389]; RAB27B [390]; RAB7A [391]; RAC1 [392]; PSAP [393]; CTSD [394,395]; HMGB1 [375,396]; FOXO3 [346]; GBA1 [175,176,199,366,397,398]; CFL1, GSN [135]; LRRK2 [314,399,400]; CNTNAP4 [338]; NSFL1C [401]; GBA1 [402]; HDAC6 [358]; TAX1BP1 [340]; VPS41, GIPC1, ATG7 [403]; BAG3 [404]; RAB1A [405] Ubiquitination: NEDD4 [168,406] Deubiquitination: USP9X [173] SUMOylation: UBE2I (Ubc9) [345] |
| 2 |
LPCAT1 [407]; NLRP3 [408]; UCHL1 [409]; PREP [351]; DCLK1 [410]; PARP1 [243]; SIRT1 [250]; MTOR [355]; BAG5 [411]; LGALS3BP, LOXL1 [412]; ATG5 [404]; TLK2 [253] Deacetylation: SIRT2 [246] Deubiquitinaition: USP8 [413] Calcium: GSK3B [414] |
|
| α-syn accumulation (proteasomal degradation) | 1 |
STUB1 (CHIP) [374]; NFE2L2 (Nrf2) [415]; RER1 [416]; ATP13A2 (PARK9) [386]; PINK1 [336]; PRKN (Parkin) [417,418]; CTSD [395]; PSMA6 [353]; NSFL1C [401]; PPP6C [401]; USP9X [173] Protease: ECE1 [419]; USP10 [308]; ADAMTS19 [340]; HTRA2 (Omi) [178] |
| 2 |
UCHL1 [420]; PREP [351]; PIAS2 [421]; USP13 [422] Protease: CAPN1 (Calpain) [423] |
(Continued)
Table 2. Continued.
| Pathway | Modifier Type | Genes |
|---|---|---|
| Chaperone | 1 |
ARSA [424]; HSPA1A (Hsp70) [147,425–432]; HSPA8 (Hsc70) [147,433]; DNAJB6 [425]; DNAJB1, HSPA4 (Hsp110 family), [433]; DNAJA1 (Hsp40), TOR1A (TorsinA) [426]; RPS3A [434]; CRYAB (HspB5; αB-c), HSPB1 (Hsp27) [435,436]; IDE [354]; HSP90AA1 (Hsp90) [437]; HSPA4L (Hsp110) [438]; CLPB (Hsp104) [439]; DNAJB1 (Hsp40), PARK7 (DJ-1) [157,332]; PSMA6 [353]; YWHAH (14–3-3η) [440]; YWHAQ (14–3-3θ) [441,442]; CACYBP (SIP) [443]; PCSK1N (proSAAS) [444] Extracellular: HSPA1A (Hsp70) [445] Upstream: HSF1 [446]; SIRT1 [447]; HDAC6 [448]; PPP1R3C [350]; KDELR2 [412] |
| Direct interaction | 1 | SNCB (β-syn) [449–453]; SNCAIP (Synphilin-1) [152,153,155]; HDAC6 [454] |
| 2 | TARDBP (TDP-43) [455,456]; DDX10 [457]; DUSP11 [412]; TPPP [360]; MAPT (Tau) [458–460]; TGM2 (tTGase; TG2) [461]; SNCAIP (Synphilin-1) [156,462–464]; PREP [465]; PARP1 [466] | |
| Post-translational modifications | 1 | Ubiquitination: NEDD4 [467]; ATP13A2 (PARK9) [386]; SYVN1 [308] Phosphorylation: CDK4, CDK6, STK3 [187]; PTPA [401]; SEPTIN4 [468]; PRKN (Parkin) [469]; CSNK1G2 (CKI) [470] SUMOylation: CBX4 (Pc2) [471] |
| 2 | Ubiquitination: TRAF6 [472]; SIAH1 [171,172]; SIAH2 [171] Phosphorylation: GRK3, GRK5 [249] Y39: ABL1 [149]; Y125: FYN [473,474]; SRC [474] S129: CSNK1G2 (CKI) [188,475]; CSNK2A2 (CK2) [475–478]; GRK2 [479]; GRK5 [479,480]; LRRK2 [481]; PLK1 [482]; PLK2 [482–484]; PLK3 [484]; SGK2 [188] S87: DYRK1A [485] N-terminal acetylation: NAA20 (NAT5) [188] Not phosphorylate: CDK14 [486] |
|
| Vesicular/membrane trafficking | 1 | ER-Golgi: YKT6 [350,487]; RAB1A [184–186,350,405]; RAB3A, RAB8A [186]; CHMP2B [106]; PPP6C [401]; YIF1A [358]; SEC22B [403]; VPS35 (PARK17) [342] Endocytic trafficking: RAB11A, RAB13 [488]; RAB29 (PARK16/RAB7L1), SYNJ1 (PARK20) [342]; ALS2 [332]; NSF [341]; GIPC1 [403] Endocytosis: AP2A2 (AP-2), EPS8L2, RAB7A [187]; AAK1 [358]; GAK [489]; SORL1 [342] Golgi-endosome: GGA1 [332]; Golgi clearance and turnover: GAK, DNAJC6 [490] |
| 2 | Endocytosis: BIN3, CLTC [188]; CHMP4B [412] Endosome maturation: VPS33A [188] |
|
| Lipid metabolism | 1 | Phospholipid: CHKB [136]; SYNJ1 (PARK20) [342]; INPP5F [341] Sterol metabolism: NCEH1, SOAT1 [191]; AMFR, GPI [106]; LIPA, NPC1 [190]; OSBP, OSBPL7 [192,350]; SIGMAR1 [188]; SYVN1 [350] Very long chain fatty acid synthesis: ELOVL1/4/6/7 [203] Sphingolipid/ceramide: PLA2G6 (PARK14) [196,201]; B4GALNT1 [200]; GBA1 [190,199]; SCARB2, SMPD1 [190]; CERS2 [249] Glycosaminoglycan: GLB1, IDUA, MAN2B1, MANBA [190] |
| 2 | Phospholipid: LPCAT1 [407] Sterol metabolism: OSBP, OSBPL7 [192]; Long-chain fatty acid: ACSBG1 [412] Mevalonate-ergosterol: DOLPP1, RABGGTA [350] |
|
| α-syn spread | 1 |
YWHAQ (14–3-3θ) [442]; ATP13A2 (PARK9), LRRK1, PARK7 (DJ-1), PRKN (Parkin), VPS35 [214]; HSPA4L (Hsp110) [438]; VCP [491]; RAB8B, RAB13, SYTL5 [488]; CTSD [394]; GBA1 [398]; ADAMTS19, FBXO5, ITGA8, SLC30A3, TAX1BP1 [340]; CHRNA7 [492] Uptake: CDNF [493] Secretion: GABBR1 [223]; ARSA [424]; GBA1 [198,494]; ATP13A2 (PARK9) [225,386]; HDAC6 [224]; RAB11A [495,496]; RAB27B [390] |
| 2 |
APOE4 [497,498]; LPCAT1 [407]; LRRK2 [215]; PARP1 [466]; PREP [351]; NFX1, POLR2A [340]; PRNP [499] Uptake: LRP1 [500]; LAG3 [216]; SLC35B2, MYO7B [501]; RAB5A [502] Secretion: ABCC8 (SUR1) [223]; PIAS2 [421]; MAOB [503]; TPPP (p25α), RAB1A, RAB7A, RAB8A, ATG5 [224]; PIKFYVE [213] |
|
| Synaptic impairment | 1 |
RAB11A [504]; TOR1A [505] Dopaminergic neurotransmission: SEPTIN4 [468]; PRKN (Parkin) [506] |
| 2 |
SLC6A2 (DAT-1) [505]. SYN3 [507]; ADORA2A [508]; PRNP [509]; FYN [473,474]; SRC [474] Dopaminergic neurotransmission: PREP [351]; TNK2 (ACK1) [510] |
|
| Neuroinflammatory responses | 1 | NR4A2 (Nurr1) [232]; PRKN (Parkin) [469]; NR1D1 [48] |
| 2 | LRRK2 [215,239,511] [215,511]; NLRP3 [408]; TGM2 (tTGase; TG2) [231] | |
| Calcium homeostasis | 1 | PPP3CA, PPP3CB, PPP3R1 [401]; CRTC2, PPP3CC [254] |
| 2 | NFATC4, PPP3CC [254]; FKBP1A (FKBP12) [512] | |
| Cell cycle | 1 | PLK2 [308]; PPP6C [401]; PPP2R2B (ATXN12) [342]; CDC27, HECA [48]; RNR1 [513] |
| Circadian rhythm | 1 | MTA1/2/3, NR1D1/2 [48] |
(Continued)
Table 2. Continued.
| Pathway | Modifier Type | Genes |
|---|---|---|
| Other pathways | 1 | Translation: ATXN2, EIF4G1 (PARK18) [342]; RPSA [136]; RPS6 [353]; PABPC1 [342]; GARS1 [48]; RSPA [136] Transcription: ATXN7 [342]; MTA1/2/3, NR1D1/2, RBPMS, RBPMS2 [48]; EXOSC9 [333] Apoptotic pathway: HSPB1 (HSP27) [514]; SNCAIP (Synphilin-1) [152]; ATP13A2 (PARK9) [387] α-syn sequestration on intracellular membranes: GDI1, RAB3A [515] Ion homeostasis: DRD4, NPY1R [358]; ATP13A2 (PARK9) [342] Potassium homeostasis: PPP6C, PPP6R1, PPP6R2, PPP6R3 [401] Zinc homeostasis: ADAMTS19, SLC30A3 [340] Iron: ABCB7 [48] Mn2+: ATP13A2 (PARK9) [308] Acetylcholine signaling: ACTG1, DPYSL2 (CRMP2), KDM1A [136] Phosphodiesterase: PDE9A, PDE8B [308] Neurotrophic factor signalling: NR4A2 (Nurr1) [516,517]; RET [517] |
| 2 | Translation: TARDBP (TDP-43) [455]; EIF4G1 (PARK18) [369]; POLR2A [340] Apoptotic pathway: PDE4B [343]; CASP3, CASP9, CASP12 [370]; FOXO3 [346] Parthanatos: PARP1 [466] α-syn sequestration on intracellular membranes: Hsp90AA1 (Hsp90) [515] Polyamine pathway: SLC22A2 [518] |
|
| Not mentioned in text/No clear category | 1 | RTCB [403]; SUGT1 (upregulate PINK1 and PARK9) [519]; ATP13A2 [403]; GBA1 [520]; GAK, DNAJC6, RIT2, VPS13C [399,521]; PRKN (Parkin) [522,523]; TRIM28 [524]; DNAJC5 [190]; RAB39B [525] |
| 2 | PRKN (Parkin) [526]; MAPT (Tau) [527]; PARK7 (DJ-1) [528]; TARDBP (TDP-43) [529]; SIDT1, SIDT2 [510]; DCLK1 [412]; LRRK2, RAB29 [341]; SIN3A [530]; SERF1A, SERF2 [531] |
The human orthologs of genetic modifiers of SNCA identified in various experimental models are listed by the pathways they are involved in. We have classified the modifiers into two types. A gene is a Type 1 Modifier when its function is inversely correlated with the level of α-syn pathology; such a gene can be inferred to possess neuroprotective function. On the other hand, a gene is a Type 2 Modifier when its function is directly correlated with the level of α-syn pathology; such a gene is a candidate target for pharmacological inhibitors for neuroprotection. In brackets are alternative protein and gene names used in literature.
Mitochondria functions
Mitochondria are powerhouses of the cell, while also important in cell development, cell cycle and cell death [73]. The important role of mitochondria in synucleinopathies is highlighted by the reduction of mitochondria copy number detected in postmortem PD brains [74]. This is consistent with key genes underlying familial PD, such as PINK1 and PRKN/parkin, which are important for mitochondria homeostasis [75]. In addition, α-syn has been shown to disrupt mitochondria functions via multiple mechanisms [76–79]. Accordingly, many SNCA modifiers identified in experimental models are important for mitochondrial function.
Mitochondrial fusion and fission events are dysregulated in synucleinopathies, correlating with impaired mitochondrial bioenergetics. Mitochondrial fission can lead to either proliferation or degradation of damaged components via mitophagy, depending on the physiological context and the molecular machinery that is recruited for the fission event [80]. Conversely, fusion can promote content exchange and maintenance of mitochondrial DNA [81]. The dynamic balance between the two opposing processes is probably more important than the level of either process alone. For example, mice deficient in fission through disruption of Mff, or those deficient in fusion through disruption of Mfn1, suffer early lethality, but double mutants are largely normal [82]. Various experimental models expressing α-syn have shown alterations of mitochondrial morphology in the directions of either excessive fission or fusion. For example, a fly model with a high level of wild type human α-syn expression and a mouse model expressing an A53T mutant version of human α-syn both shows increased volumes of mitochondria residing at neuronal cell bodies in the respective brains [83,84]. In contrast, a different fly model expressing wild type human α-syn at a relatively low level, primary cultured neurons from A53T α-syn transgenic mice and several human cell line α-syn transfection models show mitochondrial fragmentation in either axons or cell bodies [76,85–88]. While many factors can potentially account for the contrasting effects of α-syn on mitochondrial morphology, a plausible factor could be the different levels of α-syn expression. Hence, increased mitochondrial volumes are associated with relatively higher levels of α-syn (in both flies and mice), while decreased mitochondrial volumes are associated with relatively low α-syn levels; transfected cell lines and primary neuronal cultures may express α-syn at levels lower than that found in mature neurons [29].
Just as there are contrasting changes to mitochondrial morphology in response to α-syn expression, a key fission protein, dynamin-related protein 1 (Drp1), encoded by DNM1L, has been found to modify α-syn toxicity in opposite directions. In the fly model expressing high levels of α-syn and showing enlarged mitochondria, co-overexpression of DNM1L partially suppressed neuronal loss, mitochondrial reactive oxygen species (ROS) and locomotor decline [83]. In the fly model expressing relatively lower of α-syn and showing mitochondrial fragmentation, reduction of DNM1L function using the drug, Mdivi-1, ameliorated mitochondrial aging, as measured by the MitoTimer transgene [85,89]. Similarly, in SH-SY5Y cell lines transfected with α-syn that show mitochondrial fragmentation, inhibiting fission via DNM1L knockdown or promoting fusion through OPA1 overexpression suppressed cell death and ROS levels [86,87]. Currently, it is unclear whether the contrasting effects of α-syn expression on mitochondrial morphology and the opposing effects of DNM1L on α-syn toxicity result from experimental artifacts or these different results reflect different manifestations of α-syn toxicity in different neuron types or stages of disease. Careful examination of Drp1 levels in various postmortem brain regions of PD patients at different stages of the disease may help resolve this issue.
Mitochondrial fragmentation is known to facilitate mitophagy [90], and evidence shows that α-syn may be associated with reduced mitophagy. Miro1, encoded by RHOT1, is an outer mitochondrial membrane protein important in mitochondrial transport through its interactions with the microtubule motor kinesin [91–93]. For mitophagy to occur, Miro1 must be removed from damaged mitochondrial surfaces to arrest mitochondrial motility [94,95]. This process seems to be disrupted in Parkinson’s disease, as α-syn and Miro1 were both found to be upregulated in PD postmortem brains [96]. Conversely, Miro1 knockdown in iPSC-derived neurons expressing α-syn increased mitophagy as measured by the fusion of mitochondria-containing autophagosomes with acidic lysosomes [96]. Indeed, reducing Miro1 suppresses dopaminergic neurodegeneration in flies expressing α-syn [96,97] and reduces α-syn toxicity in yeast [98]. A small molecule that promotes Miro1 degradation has been identified, which rescues the mitochondrial motility and clearance of defective mitochondria in iPSC-derived neurons [97], indicating the potential of targeting Miro1 as a therapeutic for synucleinopathology.
Furthermore, mitochondrial biogenesis is also important to maintain an adequate and functional mitochondrial population (Figure 1). PGC-1α, encoded by PPARGC1A, and a master regulator of mitochondrial biogenesis and ROS scavenging [99]. Using a systems biology approach, PGC-1α transcriptional targets were found to be under-expressed in PD brains [100]; it was demonstrated to be an SNCA modifier when its overexpression suppressed SNCA-associated death of cultured dopaminergic neurons [100]. Indeed, overexpressing PGC-1α also rescues α-syn toxicity in mice and a zebrafish model exhibiting mitochondrial pathology [101,102]. Interestingly, REST, which induces PGC-1α expression, was downregulated in iPSC-derived dopaminergic neurons carrying the SNCA triplication, while its overexpression reduced cell death in response to MPTP-induced dysfunction of mitochondrial complex I in SH-SY5Y cells [103]. Hence, the evidence suggests that upregulation of mitochondrial biogenesis may be neuroprotective.
Figure 1.

Pathways by which genes important for mitochondrial biogenesis that modify α-syn pathology. PGC-1α, encoded by PPARGC1A, can stimulate transcription of genes for mitochondrial biogenesis. These genes are downregulated in PD brains. REST induces PGC-1α expression and is downregulated in iPSC-derived dopaminergic neurons carrying the SNCA triplication. Created with BioRender.com
In addition, other SNCA modifiers are involved in other aspects of mitochondria function, such as COX20 and NDUFA9 (electron transport chain) [48], TOMM20 (protein transport) [104], GARS and NGRN (mitochondrial protein synthesis) [48] and TRAP1 (mitochondrial chaperone) [105]. Evidently, the association between α-syn and mitochondrial dysfunction is significant and reducing this dysfunction can play a major role in treatment of synucleinopathies.
Glycolysis and glutathione synthesis
A recent review summarizes various studies indicating that elevation of glucose metabolism is neuroprotective in neurodegenerative diseases [63]. In flies overexpressing SNCA, overexpression of either of the two glycolytic enzymes, GPI and PFK, partially rescued neurodegeneration [48]. Conversely, knockdown of GPI worsened neurodegeneration in worms and flies overexpressing SNCA and led to accumulation of α-syn protein in cultured mouse neurons [106]. Interestingly, terazosin, a hypertension medication that is secondarily found to activate another glycolytic enzyme, phosphoglycerate kinase (PGK), is now considered a potential drug for PD [107,108]; here terazosin was found to alleviate α-syn accumulation and neurotoxicity in a few PD models [107].
How is glycolysis involved in synucleinopathies? A recent study revealed that disruption of mitochondrial complex I in mice leads to parkinsonian phenotypes and upregulation of glycolysis [109]. Given that high levels of α-syn impair mitochondria functions, we examined fly and mouse models of synucleinopathy. Indeed, SNCA overexpression in flies and mice leads to upregulation of a subset of glycolytic enzymes [48]. While glycolysis generates metabolites for many cellular functions, one notable process is de novo glutathione synthesis, which involves two ATP-consuming steps [110] (Figure 2). Interestingly, metabolomics and isotope tracing by Patten et al. showed that in cells with impaired mitochondria, de novo glutathione synthesis increases in a manner that is dependent on the ability of the cell to upregulate glycolysis [111]. We thus examined the ratio of oxidized to reduced glutathione (GSSG:GSH) in fly neurons and found that neurons co-overexpressing SNCA and GPI showed lower GSSG:GSH ratio than those overexpressing only SNCA [48]; hence, these neurons likely channeled ATP from glycolysis towards GSH synthesis.
Figure 2.
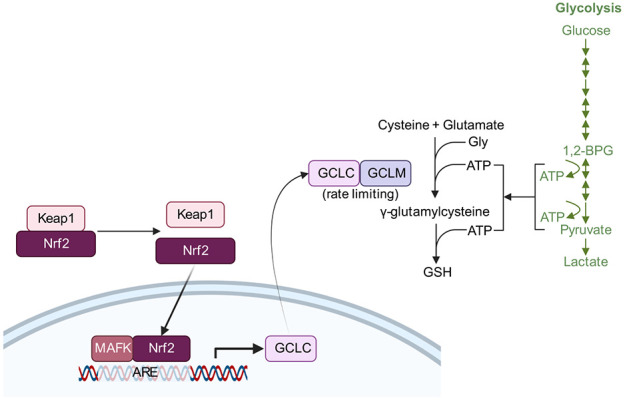
Pathways by which genetic modifiers of α-syn pathology may act. Nrf2, encoded by NFE2L2, can interact with MAFK and bind to the antioxidant responsive element (ARE) to drive transcription of GCLC, encoding a subunit of GCL (glutamate cysteine ligase). GCL catalyzes the rate-limiting step in glutathione (GSH) biosynthesis. Keap1 inhibits Nrf2. ATP required for the de novo synthesis of GSH is provided for by glycolysis (green). Created with BioRender.com
Besides glycolysis, antioxidant pathways have also been shown to be an adaptive response toward synucleinopathies. NFE2L2 encoding Nrf2, a transcription factor regulating cellular antioxidant responses, and genes in its pathway have been demonstrated to modify α-syn-associated neurodegeneration in Drosophila, by increasing transcription of the catalytic subunit of GCL [112,113] (Figure 2). PARK7 apparently also confers its protection against α-syn-mediated toxicity by increasing GCL expression [114]. Phase II detoxification enzymes involved in glutathione metabolism were also identified as enhancers of α-syn-associated toxicity in a yeast screen [115], with further validation in the fly, including the fly homolog of GCLM encoding the modifier subunit of GCL [116]. Hence, glutathione biosynthesis pathways are implicated as modifiers of α-syn, indicating the role of oxidative stress in α-syn pathophysiology.
Insulin, insulin-like growth factors and mTOR signaling
The insulin signaling pathway has been found to confer protection against α-syn toxicity in some experimental models. Using a human SH-SY5Y neuroblastoma cell line overexpressing SNCA, Kao showed that insulin-like growth factor 1 (IGF1) application rescues α-syn toxicity and aggregation [120]. Similarly, a different study using 6-hydroxydopamine (6-OHDA) treatment of SH-SY5Y cells and mouse to model PD showed that insulin-like growth factor 2 (IGF2) rescues α-syn toxicity and aggregation [121]. Both studies demonstrated that the rescue of synucleinopathy by IGFs requires a functional PI3K/AKT pathway. IGF1 is thought to rescue neurotoxicity by activating mTOR and preventing excessive autophagy in an MPTP mouse PD model [122]. Besides blocking neuronal apoptosis at the level of neuronal cell bodies, activation of AKT/mTOR signaling has also been shown to suppress axon degeneration by inhibiting macroautophagy [123]. Together these studies suggest that activating PI3K/AKT/mTOR signaling may be neuroprotective (Figure 3B).
Figure 3.

Genetic modifiers of α-syn pathology from the insulin-like signaling pathway and glycolysis pathways. (A) Disruption (blue stroke) of insulin receptor (InR encoded by daf-2) in worms leads to derepression of FOXO and upregulation of PGI, both of which contribute to neuroprotection; PGI upregulation in daf-2 mutants occurs via an indirect mechanism that does not involve FOXO [106,117]. (B) Application of IGFs in mammalian cells or rodent models is neuroprotective. (C) GLP1 can potentially enhance insulin sensitivity via SIRT1 and IRS [118,119].
Interestingly, glucagon-like peptide-1 (GLP1), a peptide hormone that enhances insulin sensitivity [118,124,125] (Figure 3C), is associated with neuroprotective activities [126]. A brain-penetrant GLP1 analog is shown to protect against dopaminergic neuronal death in an α-syn preformed fibril mouse model of PD [127]; in this study, the GLP1 analog prevents microglia from converting astrocytes to a neurotoxic phenotype, hence preventing the death of dopaminergic neurons.
While the above studies suggest that activation of the insulin signaling pathway may protect neurons, others below suggest the opposite is true, at least in the following contexts. In worms and flies, disruption of insulin receptor and the insulin receptor substrate homologs alleviates dopaminergic neuron death [106] (Figure 3A). Similarly, in the SNCA modifier screen performed in our laboratory [48], heterozygous loss-of-function mutations in headcase (hdc)/HECA, a negative regulator of insulin/mTOR signaling [128,129], was found to enhance α-syn-associated neurodegeneration. In contrast to findings discussed in the previous two paragraphs, this suggests that reducing insulin/mTOR protects neurons against α-syn toxicity. It is interesting that the findings from Knight et al. and Ren et al. are based on chronic disruption of insulin/mTOR signaling, while those in the preceding two paragraphs are based on acute stimulation with IGFs or GLP1 analogs. It is possible that genetic or epigenetic compensation may result from chronic disruption leading to different outcomes from acute manipulation of this pathway. Further investigations will be necessary to reconcile these contrasting findings.
Cytoskeleton
An increasing amount of evidence points towards the cytoskeleton as a mechanism by which α-syn causes neurotoxicity in synucleinopathies [130]. A recent paper found that reducing the levels of actin partially ameliorates α-syn-driven locomotor decline and neuronal loss in flies [83]; conversely, increasing the levels of α-spectrin suppressed α-syn-associated neurodegeneration. A bidirectional interaction between α-syn and actin was suspected, when the authors found rod-shaped actin-rich inclusions (actin rods) in fly and mouse brains overexpressing α-syn, as well as postmortem brain samples of synucleinopathy patients. Interestingly, α-spectrin overexpression in fly neurons abolished the formation of actin rods that are associated with α-syn expression, further suggesting an antagonistic relationship between actin and α-spectrin in synucleinopathy.
How do cytoskeletal aberrations affect neurodegeneration? As actin cytoskeleton plays a role in mitochondrial fission via a Drp1-dependent pathway [131–134], Ordonez et al. investigated the relationship between cytoskeleton and mitochondria in α-syn-expressing flies [83]. Their work suggests that α-syn binds to α-spectrin and disrupts its regular subplasmalemmal network, leading to abnormal stabilization of the actin cytoskeleton, which reduces the mitochondrial localization of Drp1, leading to mitochondrial enlargement and dysfunction, subsequently resulting in neuronal death [83] (Figure 4).
Figure 4.

Pathways by which genetic modifiers of α-syn pathology may lead to cytoskeletal dysregulation, mainly by the abnormal stabilization of the actin cytoskeleton, resulting in altered mitochondrial dynamics and mitochondrial dysfunction. Created with BioRender.com
Besides the mitochondrial fission-fusion cycle, abnormal actin stabilization by α-syn also impairs autophagy. Fly neurons expressing α-syn accumulate aberrantly enlarged autophagosomes [135]. Overexpression of genes encoding actin severing proteins, GSN [83,135] CFL1 [135], reverse abnormal actin stabilization and restore normal autophagy and mitophagy in fly neurons expressing α-syn.
Although reducing Act5C levels in fly neurons ameliorates synucleinopathy, act-5 knockdown in worms is associated with increased α-syn aggregation and decreased mitochondrial content [136]. We also note that α-syn was expressed in muscles in the worm study, while it was expressed in neurons in the fly papers. It is possible the different structural and functional roles of actin in muscles and neurons, rather than species differences, is responsible for the contrasting results with regards to the role of cytoskeleton in synucleinopathy. Additionally, act-5 is the most divergent of the five actin isoforms in the worm, with a specialized function in the microvilli and does not appear to have a clear ortholog in flies or humans [137,138]. Therefore, unlike Act5C, it is not possible to infer a conserved SNCA modifier role for act-5 in PD in humans.
α-Syn accumulation
Given that total α-syn level is thought to contribute to the misfolding of α-syn into pathological aggregates, transcriptional upregulation of α-syn would naturally be thought to lead to disease. In rare cases of familial synucleinopathy involving duplication and triplication of the SNCA locus, SNCA dosage and α-syn levels are correlated with age of onset and disease severity [19,20,30,139,140]. Similarly for sporadic PD, analysis of an intronic SNP in SNCA identified it as a cis-regulatory element that binds brain-specific transcription factors, EMX2 and NKX6–1, to reduce SNCA transcription [32]. Additional work by others have identified CCAAT/enhancer binding protein δ (C/EBPδ) as a transcriptional repressor of SNCA [141], while p53 and GATA-2 are transcriptional activators [142,143]. Furthermore, ZSCAN21 has been identified to activate SNCA expression [144,145], but the lack of effect on SNCA expression after ZSCAN21 knockdown in the rat brain suggests that its function may be dependent on the developmental stage of the neurons [146]. As postmitotic neurons live a long time before dying in cases of synucleinopathy, even minor differences in SNCA expression levels may have cumulative effects of clinical significance. Hence, further investigations into the transcriptional regulation of SNCA is likely to yield more disease-relevant insights.
As neurotoxicity of elevated α-syn largely depends on its misfolding and aggregation, chaperones play important protective roles. It is therefore pertinent that one of the first genetic modifiers of SNCA is HSP70; co-overexpression of HSP70 and SNCA in fly dopaminergic neurons promoted neuronal survival, while HSP70 and its partner HSP40 were found localized to Lewy bodies in PD brains [147]. Recent NMR characterization revealed that diverse classes of chaperones interact predominantly with α-syn at its N-terminus and the region around tyrosine 39 and prevent aggregation [148]. Interestingly, pharmacological or shRNA inhibition of the chaperones caused relocalization of α-syn to mitochondria, where it presumably forms aggregates. Burmann et al. also found that phosphorylation of tyrosine 39 disrupts chaperone-α-syn interactions, supporting a possible mechanism by which phosphorylation at this residue by the Abelson kinase (ABL1) drives synucleinopathies [149]; we note here that nitration at tyrosine 39 that occurs in response to nitrative insult has also been found to induce aggregation [150,151]. Besides classical chaperones of the HSP70 and HSP90 families, other molecules like DJ-1 (encoded by the PD gene PARK7) and synphilin-1 (encoded by SNCAIP) have been shown to display chaperone-like activities that reduce α-syn toxicity [152–157].
Defects in protein degradation may also lead to the pathological accumulation of α-syn. Degradation of α-syn may occur via both major protein degradation pathways, the proteasomal degradation pathway and the autophagy-lysosomal pathway (referred to as autophagy henceforth) [158–161]. Within the umbrella of autophagy, α-syn is known to be degraded through either macroautophagy or chaperone-mediated autophagy [162,163]; the latter type of autophagy involves a translocation of unfolded polypeptides into lysosome for degradation, with the help of HSP70. A large volume of research on α-syn degradation and the associated post-translational modifications has been covered by excellent reviews [164–166]. Below, we will highlight a few key findings.
There has been some debate on the relative importance of proteasomal degradation and autophagy on α-syn degradation [163,167]. By performing live brain imaging on transgenic mice expressing GFP-tagged α-syn through cranial windows, Ebrahimi-Fakhari et al. showed that the administration of inhibitors of either proteasomes or autophagy induced accumulation of the α-syn-GFP fusion protein [167]; the authors further showed that normal, endogenous levels of α-syn in non-transgenic mice are maintained by proteasomes, but not autophagy, while elevated levels of α-syn in transgenic mice is prevented from accumulating by a combination of proteasomal degradation and autophagy.
As ubiquitination is a major mechanism by which α-syn is targeted for degradation, ubiquitin ligases have been identified as genetic modifiers of α-syn. For example, NEDD4, a druggable E3 ubiquitin ligase, and its yeast homolog have been found responsible for ubiquitination of α-syn and targeting it for autophagic degradation [168,169]. Two E3 ligases, SIAH1 and SIAH2, monoubiquitinate α-syn to drive it towards either proteasome degradation or aggregation [170–173].
Besides genes that directly target α-syn for proteasomal degradation and autophagy, additional genetic modifiers have regulatory roles in influencing α-syn degradation. One of the most prominent modifiers of α-syn accumulation is glucocerebrosidase, GBA, one of the most common genetic risk factors of PD [174]. Mutant GBA impairs lysosomal degradation of α-syn by causing aberrant accumulation of sphingolipids in the lysosome (Figure 5) and blocking chaperone-mediated autophagy [175,176]. Consistent with the importance of autophagy in buffering against α-syn toxicity, transcription factor EB (TFEB) regulates the autophagy pathway through the mTOR pathway and promotes the clearance of α-syn toxic oligomers [177].
Figure 5.

Genetic modifiers of α-syn pathology involved in membrane trafficking (green) ceramide/sphingolipid metabolism (blue). Created with BioRender.com
Other than the generic protein clearance pathways, there is a protease that targets toxic α-syn species. HTRA2, encoding a serine protease called Omi, can specifically recognize and precisely degrade toxic α-syn oligomers, but not its monomers thought to be important for normal function at the synapse [24,178–180]. In the fly, Omi expression in all neurons rescued locomotor defects and increased survivability due to α-syn expression, showing its potential as a candidate for therapeutics [178]. Interestingly, Omi was previously identified as a target of phosphorylation by PINK1, which may increase its protease activity [181]. Hence, targeting such proteases with specificity for toxic α-syn species is a viable option for therapeutics.
The importance of manipulating α-syn abundance as a candidate therapeutic strategy cannot be overstated. The above section only provides a glimpse of modifiers in this category and the reader may refer to excellent reviews here [182,183].
Membrane trafficking and lipid metabolism
Numerous genes involved in membrane trafficking are genetic modifiers of α-syn (Table 2; Figure 5). Proteins with neuroprotective function include Rab1a [184–186], Rab3a and Rab8a [186], which regulate ER-to-Golgi trafficking, post-Golgi trafficking and neurotransmitter tethering and docking, respectively. In addition, AP-2, which is encoded by AP2A2, is essential for the formation of endocytic pits and vesicles [187]. On the other hand, the yeast homolog of VPS33A is important in vacuolar biogenesis; its depletion is associated with increased α-syn inclusion formation [188].
Endolysosomal membrane trafficking is known to depend on optimal metabolism of sphingolipids and cholesterols (Figure 5) [189]. In a recent genetic modifier screen that is not yet peer-reviewed, fly homologs of human lysosomal storage disorder (LSD) genes were considered, yielding homologs of 14 human LSD genes that enhanced defects in locomotion of flies expressing α-syn upon knockdown [190]. Among the genes recovered, two cause human cholesterol storage disorders, LIPA and NPC1, implicating cholesterol accumulation in pathways leading to α-syn toxicity [190]. In a screen performed in worms, the worm homolog of neutral cholesterol ester hydrolase 1 (NCEH1) was found to protect dopaminergic neurons against α-syn toxicity; NCEH1 appears to act via LDLR-mediated cholesterol endocytosis, cholesterol efflux and the synthesis of neurosteroid that lowers mitochondrial ROS [191]. This finding supports a connection between cholesterol metabolism and the mitochondrial dysfunction observed in synucleinopathies. While lower concentrations of cholesterol fed to worms are correlated with lower levels of α-syn-associated dopaminergic neurodegeneration, toxicity due to higher levels of cholesterol supplementation is likely a result of accumulation of cholesterol ester [191]. On the other hand, treatment with statins to inhibit sterol production showed that higher membrane sterol concentrations may be protective in yeast by promoting the binding of α-syn to the plasma membrane [192]. While the yeast study suggests the interesting possibility of using statins as a prophylactic against PD, meta-analyses suggest that the relationship between statin use and PD risk is still controversial [193,194].
Metabolism of ceramides, which include sphingolipids and gangliosides, are implicated in α-syn pathology (Figure 5), perhaps due to their roles in cell signaling and membrane trafficking [189,195]. Ceramide accumulation has been implicated to play a role in α-syn–mediated neurodegeneration [196], while loss of enzymes found upstream in ceramide catabolism pathways, including GBA1 and SMPD1, is described to enhance α-syn pathology [197–199], potentially due to downregulation of the salvage pathway where sphingolipids are recycled to form ceramides that can then be used to produce the various gangliosides needed for proper cell function. Indeed, treatment with exogenous ceramide or inhibition of acid ceramidase to restore ceramide levels reverses defects associated with loss of GBA1 [198]. Interestingly, selective depletion of subsets of gangliosides by disrupting the gene encoding a ganglioside biosynthetic enzyme, B4galnt1, leads to aberrant accumulation of α-syn oligomers, dopaminergic neuron death and motor deficits in mice [200]; furthermore, these phenotypes could be rescued by a brain-permeant analog of GM1 ganglioside. Hence, supplementation of a ceramide species can be neuroprotective in cells with specific deficiencies in related metabolic pathways.
In contrast, reducing the overall level of ceramides may be protective in a PD model. In two related studies, fly models of the PARK14/PLA2G6 monogenic form of PD show neurodegeneration associated with α-syn aggregation and ceramide accumulation [196,201]. Here, inhibiting de novo synthesis of ceramides using myriocin was neuroprotective in flies overexpressing α-syn [196]. Consistent with these data, a subsequent work using human SH-SY5Y cells showed that myriocin alleviated oxidative stress and α-syn inclusions induced by application of α-syn preformed fibrils (PFFs) in the culture medium [202]. However, while myriocin suppressed α-syn toxicity in flies and a human neural cell line, it was instead toxic to yeast cells expressing α-syn [203]. Intriguingly, the yeast synthetic lethality associated with α-syn and myriocin requires only 1 μM myriocin, while SH-SY5Y cells subjected to α-syn PFFs were alleviated α-syn toxicity at 50 μM myriocin. As myriocin possesses powerful antifungal properties [204,205], the synthetic lethality in yeast caused by α-syn and myriocin may be due to the exquisite susceptibility of fungal metabolism to myriocin. Nevertheless, myriocin has shown beneficial effects in non-PD mouse models of neurodegeneration [206,207], suggesting that reduction of ceramide levels might have general neuroprotective effects.
Genetic modifiers may also exert their protective effects by remodeling the lipid membrane, hence influencing the membrane binding propensity of α-syn [208]. As a membrane-driven process, autophagy may also be affected by abnormal cellular lipid profiles, resulting in accumulation of α-syn aggregates due to reduced clearance [209]. In summary, several genes encoding proteins involved in lipid metabolism are implicated in α-syn accumulation due to impaired clearance, leading to greater toxicity.
α-Syn spread
Prion-like spreading of α-syn has been hypothesized and observed in vitro in cell culture, and in animal models, as previously reviewed [210–212]. Screens have been conducted to elucidate the pathways involved in α-syn spreading. A useful screening tool, bimolecular fluorescence complementation (BiFC) has been used to track the spread of α-syn aggregation, whereby α-syn is fused to two different fluorescent proteins and their fusion is triggered by addition of α-syn fibrils [213,214]. In an article that has not been peer-reviewed, a genome-wide screen in HEK293T cells was described which identified PIKFYVE and the phosphatidylinositol pathway as modifiers of the spread of α-syn aggregation, showing that PIKfyve inhibition prevents α-syn fibrils from reaching the lysosome [213] (Figure 6). Given the involvement of cellular trafficking and autophagy pathways in α-syn spread, genetic and pharmacological modulation of autophagy was predictably found to alter α-syn transmission in worms [214].
Figure 6.

Pathways by which genetic modifiers of α-syn pathology may modulate α-syn spread. Created with BioRender.com
Two key steps of α-syn spread are the uptake of synuclein fibrils by the recipient neuron and the subsequent aggregate formation. In a small genetic screen in which neurons exposed to synuclein PFFs were screened with lentiviral knockdown vectors expressing shRNA targeting selected genes, LRRK2 was found to promote the formation of α-syn aggregates after PFF uptake [215]. Using a different strategy, Mao et al. sought to identify the cell surface receptor responsible for the uptake of PFFs. In this screen from a library of transmembrane proteins in SH-SY5Y cells, LAG3 was identified to preferentially bind PFFs over α-syn monomers (Figure 6) [216]. Upon further characterization, α-syn binding to LAG3 was shown to be required for clathrin-mediated endocytosis of α-syn; the loss of LAG3 rescued neurodegeneration and motor defects in mice injected with α-syn PFF [216]. Hence, this study identifies LAG3 as a candidate therapeutic target for synucleinopathies.
Besides uptake, α-syn spread is also mediated by its secretion. Interestingly, two genetic modifiers of α-syn secretion are the H50Q and G51D mutations in the protein itself [217,218]. Multiple reports indicate that inhibition of autophagy leads to increased α-syn secretion [219–222], hence it is possible that the above two SNCA mutations enhance secretion at least in part through the disruption of autophagy. Another report implicates ABCC8, which encodes SUR1, as a genetic modifier of α-syn transfer [223]. Specifically, it was shown that activation of SUR1-KATP channels leads to hyperpolarized membranes in GABAergic neurons, decreasing GABA release and activation of presynaptic GABAB receptors, reducing the inhibition of Ca2+ channels, such that the increase in intracellular Ca2+ triggers α-syn release from the neuron [223]. Additional studies have implicated tubulin polymerization-promoting protein (TPPP/p25α) and ATP13A3/PARK9 as modifiers of α-syn secretion [224,225].
As α-syn spreading is a topic of intense research, we expect a better understanding of this process in the near future. Hopefully, this would translate into further therapeutic strategies for synucleinopathies.
Neuroinflammatory responses
Besides neurons, the spread of extracellular α-syn affects microglia, the resident innate immune cells of the brain and astrocytes, hence triggering neuroinflammatory responses [226–228]. A number of genes involved in neuroinflammatory responses have been identified as genetic modifiers of α-syn, mainly from the TLR4/NF-κB signaling pathway. Deletion of TGM2, encoding the transglutaminase 2, TG2 which is involved in NF-κB-mediated inflammation [229,230], attenuates neuroinflammatory responses to α-syn in transgenic mice [231]. A different work showed that Nurr1 overexpression inhibited nuclear translocation of NF-κB, and partially attenuated the increased production of cytokine TNF-α induced by α-syn [232] (Figure 7). Besides this, Rev-erbα, encoded by NR1D1 that was recovered from our screen [48], was also recently shown to attenuate neuroinflammation in an MPTP-induced mice PD model, likely by reducing NLRP3 inflammasome activation by NF-κB [233].
Figure 7.

Pathways by which genetic modifiers of α-syn pathology may lead to neuroinflammatory responses such as the production of cytokine TNF-α. created with BioRender.Com
LRRK2 and the sirtuins—pleiotropic modifiers of SNCA
LRRK2 mutations are the most common genetic causes of PD [174]. It encodes a large ~280 kD protein with two enzymatic domains—a Ras of complex (ROC) GTPase domain and a serine/threonine kinase domain [234]. It has been implicated in a wide range of cellular functions, based on its kinase substrates that include proteins involved in synaptic vesicle endocytosis, microtubule network, protein translation and several members of the Rab GTPases that function in various cellular processes [234]. Brain RNA Seq data indicate that LRRK2 is expressed mainly in microglia and oligodendrocytes, with lower levels of expression in neurons and astrocytes [23,235]. Knockin mice expressing the pathogenic R1441G mutant protein from the endogenous promoter accumulates increased levels of intracellular and extracellular α-syn, indicating that LRRK2 is a SNCA modifier. Although much of LRRK2 is expressed outside neurons, many studies examined neuron overexpression of gain-of-function mutations of LRRK2. For example, mouse brains co-expressing the G2019S LRRK2 mutant with the A53T SNCA mutant showed accumulation of high molecular weight species of α-syn, enhanced striatal neuron loss, synergistic disruption of Golgi network, microtubule network and mitochondrial functions [236]. Similarly, co-expression of LRRK2 mutants and SNCA in fly neurons enhanced neurodegeneration [237]. A difference between the fly and mouse studies was that human A53T SNCA expression combined with LRRK2 loss-of-function mutation in mouse suppressed high molecular weight α-syn accumulation and neuron loss, while LRRK2 loss-of-function in SNCA-expressing flies enhanced neurodegeneration phenotypes [236,237]; it is possible that some of the pleiotropic functions of LRRK2 may have diverged between flies and mice. Finally, the PD risk gene LRRK2 is found to be highly associated with microglia in the single-nuclei transcriptome of sporadic PD midbrain, suggesting a role in neuroinflammation [238]. Indeed, upon exposure to α-syn PFFs, primary microglia cells with a LRRK2 loss-of-function mutation shows attenuated activation compared to controls [239], while gain-of-function mutations in mice injected with PFFs display increased microglia activation. Hence, this finding suggests that LRRK2 may be a drug target for mitigating neuroinflammation in synucleinopathies.
The sirtuins are a family of genes encoding NAD+-dependent histone deacetylases consisting of SIRT1–7 in humans, each implicated with diverse functions. For this review, we will discuss only SIRT1, 2 and 3, focusing on the characterization of these 3 genes in PD models. While SIRT1 is localized to the cytoplasm in the adult mouse brain, SIRT2 is known to undergo nuclear-cytoplasmic shuttling and SIRT3 is localized to mitochondria [240,241]. Studies of sirtuins in a variety of experimental models have suggested pro-aging or anti-aging, as well as, pro-neurodegeneration or neuroprotective functions in various contexts [240,242].
SIRT1 has been shown to be neuroprotective in mammalian models of synucleinopathy. Using viral overexpression of SNCA in mouse primary neurons and a mouse dopaminergic cell line, SIRT1 activation by a selective agonist, SRT2104, was shown to reduce cell death; this neuroprotective effect of SRT2104 was attributable to SIRT1 as it could be reversed by a SIRT1 inhibitor, EX527 [243]. Additional investigation suggests that SIRT1 activation was associated with restoration of mitochondrial function and autophagy that were impaired by SNCA overexpression. Specifically, pharmacological activation of SIRT1 was associated with increased nuclear localization of TFEB, a transcription factor that upregulates autophagy, and various markers of autophagy flux. Another group reported that activation of SIRT1 by resveratrol reduced dopaminergic neuron loss and restored locomotor functions in mice injected with MPTP in the substantia nigra; the authors attributed the neuroprotective action of resveratrol to SIRT1 by reversing them with the SIRT1 inhibitor, EX527 [244]. Interestingly, they found that the above SIRT1 activation correlated with deacetylation of the autophagy protein LC3 in the striatum, where axons from substantia nigra terminate; this suggests that LC3 may a direct target of the deacetylase activity of SIRT1.
Unlike SIRT1, SIRT2 has been shown to be pro-neurodegeneration in PD models. A chemogenetic study identified a series of small molecule inhibitors of SIRT2 that specifically inhibit SIRT2, but do not affect SIRT3 activities [245]. These inhibitors were shown to suppress α-syn toxicity in human H4 neuroglioma cell line and cultured rat midbrain primary neurons that were transfected with SNCA; they also suppressed dopaminergic neuron loss in transgenic Drosophila overexpressing SNCA. A follow-up study by the Outeiro group suggests that SIRT2 may promote α-syn toxicity, at least in part, through the removal of acetyl groups from lysine residues on α-syn [246]. They showed that endogenous α-syn is acetylated and can be deacetylated by SIRT2. They further compared two artificial α-syn mutations at K6 and K10 that either mimic acetylation or the non-acetylated states; the acetylation mimic version was less toxic than the acetylation-resistant one when overexpressed in cultured rat primary neurons and in rat substantia nigra using viral vectors. Taken together, these findings suggest that deacetylation of α-syn at lysine residues by SIRT2 may promote neurodegeneration.
The mitochondrial localized SIRT3 has been shown to be neuroprotective. In rats that were unilaterally injected with viral vectors in the substantia nigra, co-expression of SIRT3 with SNCA partially suppressed dopaminergic neuron loss and locomotor defects [247]; this neuroprotective function depends on the deacetylase activity as the catalytic mutant of SIRT3 was unable to rescue the α-syn toxicity. Consistent with this finding, SIRT3 protein levels are downregulated in post mortem brains with Lewy body disease and rat brains overexpressing α-syn [248].
Besides studies performed in mammalian systems, sirtuins have been found to be SNCA modifiers in worms and yeast. A large unbiased screen in worms using overexpression of GFP-tagged α-syn in muscles identified sir-2.1, whereby disruption of this gene increased the number of α-syn inclusions [249]. In contrast, a yeast study found that a candidate SIRT1 ortholog, sir2, is essential for SNCA toxicity and deletion of this gene suppressed autophagy and rescued the yeast cells [250]. While both the worm sir-2.1 gene and the yeast sir2 gene are candidate orthologs of SIRT1, it seems that one has neuroprotective function while the other is pro-neurodegeneration. Given that there are 4 worm sirtuins and 5 yeast sirtuins compared to 7 human counterparts, one must use caution when inferring orthology between human sirtuins and their yeast or worm counterparts, due to the relatively low levels of sequence conservation. In addition, it is intriguing that in yeast study on sir2, suppression of excessive autophagy was protective against cell death, in contrast to the findings on SIRT1 and others in the preceding section on α-syn accumulation. It is possible that yeast cells are less equipped than neuronal cells to high levels of α-syn, given that the latter are adapted to high endogenous levels of this protein [29] and are able to upregulate autophagy for degrading excess α-syn [167].
Other pathways
Pathological α-syn aggregates have shown to disrupt calcium (Ca2+) homeostasis [251]. This may be through its modulation of voltage-gated Ca2+ channels, intracellular Ca2+ channels, formation of Ca2+ permeable pores, disruption in lipid membrane composition and packing, even ER or mitochondrial dysfunction [252]. As mentioned above, calcium overload due to α-syn overexpression leads to dysfunctions in mitochondria and autophagy, resulting in cell death, likely by acting through TLK2 [253]. Besides this, calcium was reported to bind and activate calcineurin and calmodulin, which dephosphorylates NFATC4, which then localizes to the nucleus to activate a toxic program [254]. However, both deletion and overexpression of calcineurin were found to exacerbate α-syn toxicity in yeast [254]. Calcineurin likely confers its protective effects through cAMP-responsive element-binding protein-regulated transcription coactivator 2 (CRTC2) [255], which can be achieved by limiting but not eliminating the availability of calmodulin, calcineurin and their substrates [254]. Hence, Ca2+ homeostasis and downstream pathways including that involving calcineurin are also important in synucleinopathies.
Dysregulation of the cell cycle has been demonstrated in synucleinopathies and in PD models [256–259]. The human orthologs of two genes from our SNCA modifier screen have been implicated in this pathway elsewhere (CDC27, HECA) [48,129,260,261], perhaps implicating its relevance to cause disease in synucleinopathies. While few other genetic modifiers of α-syn seem to be involved in this pathway (Table 2), PARK7 was also recently found to cause aberrant cell cycle re-entry, thus resulting in neuronal death [262].
Another pathway poorly represented in our list is circadian rhythm (Table 2), despite being one of the most common non-motor symptoms of PD [263] and associated with PD risk [264], even suggested to cause neurodegenerative diseases [265,266]. One of the fly SNCA modifiers from our screen, Eip75b, is homologous to NR1D1 and NR1D2, which are core components of the circadian machinery and are involved in dopaminergic regulation [61,267]. In addition, Drosophila MTA1-like which is found in our SNCA modifier screen [48], has two human orthologs which are implicated with circadian rhythm-related functions: MTA1, which is an integral component of the circadian transcriptional circuit [268] and MTA3, which is associated with insomnia in a large GWAS meta-analysis [269].
SNCA MODIFIERS AND PATHWAYS TARGETED IN PD CLINICAL TRIALS
How might the discovery of SNCA modifiers help in the development of therapeutic interventions? An analysis of previous drug development pipelines estimates that drugs with targets that are supported by human genetics evidence are twice as likely as those with none to progress from Phase I clinical trials to approval [270]. For example, the discovery of PCSK9 has led to the development and approval of Inclisiran for hypercholesterolemia [271]. Inspired by such findings, a few biotech startups now aim to screen genetic modifiers for druggable pathways [272–274]. While no disease-modifying interventions have been approved for PD, several candidates are currently in clinical trials. In this section, we shall discuss selected clinical trials in light of the above findings on SNCA modifiers (Table 3), with a focus on PD where an increasing number of interventions are emerging in the pipeline.
Table 3. PD clinical trials of disease-modifying interventions.
| Drug | Drug action | Main outcome | Phase of clinical trial | References | Year(s) of publication |
|---|---|---|---|---|---|
| Exenatide | Glp-1 receptor agonist | Positive effects on motor scores. | 2 | [278–280] | 2014, 2017, 2019 |
| Pioglitazone | PPARα & PPARγ agonist | Did not modify progression in early PD. | 2 | [282] | 2015 |
| GM1 ganglioside | Modulator of sphingolipid metabolism | Reduced worsening of motor symptoms. | Single center RCT | [285–287] | 2010, 2013, 2015 |
| Recombinant GBA1 | Catabolism of glucosylceramide | GBA was permeable through BBB. Demonstrated safety. | 1 | [288] | 2022 |
| Venglustat | Glucosylceramide synthase inhibitor | Failed to provide benefit for motor symptoms (Sanofi press release). | 2 | [289–291] | 2021 |
| Prasinezumab | Anti-aggregated α-syn antibody | No difference from placebo in motor scores and in dopamine transporter levels in putamen. | 2 | [292] | 2022 |
| UCB0599 | Inhibitor of α-syn misfolding | Demonstrated safety. | 1 | [532] | 2022 |
| Anle138b | Inhibitor of α-syn misfolding | Demonstrated safety. | 1 | [533] | 2022 |
| Deferiprone | Iron chelator | Motor symptoms worsened despite reduction of iron in SNc. | 2 | [295] | 2022 |
| Nilotinib | ABL kinase inhibitor | Biomarker engagement data conflicting between 2 studies. Motor symptoms showed negative trend in Simuni et al. (2021) | 2 | [534,535] | 2020, 2021 |
| Intranasal glutathione | Antioxidant | No symptomatic benefit compared to placebo. | 2b | [296,297] | 2015, 2017 |
| Isradipine | L-type calcium channel blocker | Did not slow progression of early PD. | 2 | [298,536] | 2013, 2020 |
| Nicotinamide Riboside (NR) | Precursor to NAD, a cofactor of sirtuins | Oral NR therapy increases brain NAD levels and impacts cerebral metabolism in PD. | 1 | [537] | 2022 |
| Infusion of GDNF into putamen | Neuroprotective trophic factor | No motor improvement over placebo, but 18F-DOPA uptake increased in putamen. | Single center RCT | [299–301] | 2006, 2007, 2019 |
RCT, randomized control trial
Insulin signaling
One candidate that has shown promising clinical data is exenatide, a glucagon-like peptide 1 (GLP1) analog, which is currently used for treating type 2 diabetes. Endogenous GLP1 is known to augment insulin signaling and have neuroprotective functions [126,275]; the likely mode of action of GLP1 and its analogs is by enhancing insulin sensitivity through the action of SirT1 on IRS [118,124,125]. As PD has been associated with brain biomarkers of insulin resistance in humans [276,277], it is thought that relieving brain insulin resistance may ameliorate the progression of PD pathology. A single-center Phase 2 trial of exenatide on PD showed improvement of motor scores that was sustained beyond the period of exposure [278,279]. Furthermore, analysis of neuron-derived exosomes from subjects in this trial revealed exenatide treatment had augmented tyrosine phosphorylation of IRS1, suggesting that activation of neuronal insulin signaling by exenatide is correlated with locomotor benefits [280]. Consistent with the above PD trial, GLP1 analogs have been found to benefit dementia patients in another clinical study [281]. Hence, GLP1 analogs are likely to provide a protective function against synucleinopathy and perhaps neurodegeneration, in general, by alleviating brain insulin resistance.
While exenatide has shown promising data in the above study, another diabetes drug, pioglitazone, did not modify progression in early PD in a different Phase 2 trial published in 2015 [282]. The mechanism of action of pioglitazone differs from that of exenatide in that the former is an agonist of peroxisome proliferator-activated receptors (PPARs) PPARα and PPARγ [283]. Currently, it is not clear whether the difference in efficacies against PD between pioglitazone and exenatide is due to the two drugs targeting different aspects of insulin signaling or other less obvious reasons.
Notwithstanding negative results from the pioglitazone trial, promising data from the exenatide studies suggests that enhancing the insulin/IGF signaling pathway in the brain is a viable target. This is consistent with the identification of IGF1 and IGF2 as SNCA modifiers that ameliorate α-syn accumulation and cytotoxicity in cell lines and a 6-OHDA mouse model of PD [120,121]. While there are contrasting data from worm and fly models of synucleinopathy showing that reduced insulin signaling is neuroprotective [106], it is possible that different PD models may reflect different stages of synucleinopathy that may or may not match those of the PD patients who participated in the clinical trials.
Ceramide metabolism
Ceramides, sphingolipids and gangliosides play important roles in cellular signaling and membrane trafficking [189,195]. Based on the observation that GM1 ganglioside level is reduced in PD patients [200,284], Jay Schneider and colleagues conducted a randomized control trial and showed that supplementation of GM1 ganglioside can delay the progression of motor symptoms in PD [285,286]. Positron emission tomography imaging of dopamine transporter binding by a tracer suggests that GM1-treated subjects showed slower loss of striatal dopaminergic synapses, suggesting a disease-modifying effect of GM1 on PD [287].
Two other drug trials that target ceramide metabolism involve putaminal recombinant GBA1 delivery and Venglustat. The GBA1 Phase 1 trial demonstrated that a sophisticated ultrasound technique is able to deliver GBA1 across the blood–brain barrier safely with signs of target engagement [288]. Venglustat is a glucosylceramide synthase inhibitor developed by Sanofi to alleviate the accumulation of glycosphingolipids, including glucosylceramide, which is thought to impair lysosome function. The Venglustat trial for PD has failed to demonstrate benefits to motor function even though glucosylceramide levels decreased in cerebrospinal fluids, indicating successful target engagement [289–291].
The preliminary success of the PD trials with GM1 ganglioside and the failure of Venglustat suggest that further understanding of ceramide metabolism and its role in brain physiology is needed. Nevertheless, this pathway appears to provide a fruitful avenue for more investigations on potential PD interventions.
Other pathways
Table 3 lists additional trials of candidate disease-modifying treatments, including those that investigate drug safety profiles and those that tested efficacies. The following are some trials that have failed to demonstrate efficacy.
Anti-aggregated α-syn antibody, Prasinezumab: This trial aimed to prevent the spread of aggregated α-syn. However, the motor scores and dopamine transporter levels in the putamen were not different from placebo [292].
Iron chelator deferiprone: Potential mechanisms of action—ameliorate iron over-accumulation [293] or activate HIF1A [294]. Motor symptoms worsened despite reduction of iron in SNc [295]; possibly due to depletion of the iron co-factor from tyrosine hydroxylase.
In addition, the following PD trials also failed to demonstrate efficacy: intranasal glutathione delivery, intraputaminal delivery of GDNF and Isradipine, an L-type calcium channel blocker [296–301].
While disappointing, it is worth investigating how each trial might have failed (or succeeded) to demonstrate efficacy. One could ask whether target engagement was successful. Was the hypothesis that guided the intervention correct? Was the stage of PD at which the intervention was administered allow for the hypothesized mechanism to occur? Could the pleiotropic nature of a drug, such as deferiprone, have resulted in negative actions outweighing positive ones?
While experimental models of PD have allowed researchers to gain many mechanistic insights, there are many aspects of synucleinopathies that are not captured by models. Hence, it is important to go beyond clinical outcomes in these trials to investigate underlying mechanisms of the interventions, using biomarkers and brain imaging methods.
FUTURE PERSPECTIVES
Challenges in modeling synucleinopathies
In trying to model progressive neurodegeneration with complex etiologies, experimental models of synucleinopathies face several challenges. First, it is a challenge to model the effect of aging on synucleinopathy; disease progression occurs in time scales of decades in humans, while experimental models like yeast, worms, flies and mice have lifespans ranging from days to three years. When modeling synucleinopathy in cultured cells or organoids, it is unclear what in vitro correlate of biological aging should be [302]; in cases where proliferating cells are used to model post-mitotic neurons, it is possible that differences in cellular physiology may be a confounding factor [303]. Second, in attempting to accelerate neurodegeneration in models, experimenters may be applying genetic or chemical insults that are more intense than what occurs in humans. It is not clear whether biological responses to long-term mild insults are different from those to acute strong insults. Third, species differences between human and model organisms may be yet another confounding factor [304]. Although organisms with nervous systems possess sophisticated cell–cell interactions in their brains, that could still be challenging to model using human-derived neurons [305].
Notwithstanding the above limitations, the identification of SNCA modifiers in model organisms that are homologous to genes underlying monogenic forms of PD and sporadic PD suggests that substantial genetic contributions in synucleinopathies are evolutionarily conserved [48,306–308]. The diverse characteristics of experimental models may allow different but complementary sets of genes to be discovered, thus expanding our knowledge of synucleinopathies.
A multi-step hypothesis of PD progression
The increase of PD incidence with age has prompted researchers to propose a multi-hit hypothesis that is analogous to that for cancer progression [309–311]. Using robust, nationwide PD incidence data from New Zealand, Le Heron et al. found that the way PD incidence varies with age is consistent with a multi-step disease progression hypothesis; six steps and eight steps for early-onset and late-onset PD, respectively [309]. In experiments done on rodents, sequential application of lipopolysaccharides or a combination of paraquat and maneb have led to progressive nigral dopaminergic neuron loss in rodents after cessation of toxin exposure [312,313]; this is despite the fact that acute toxin applications typically do not yield progressive degeneration [310].
How do SNCA modifiers fit into a multi-step PD progression model?
If the progression of synucleinopathies involves multiple steps, how do genetic modifiers contribute to this process? For sporadic synucleinopathies, it is possible that individual patients may carry multiple variants, each with small effect sizes, which contribute to disease risk. Genes near some PD GWAS have been identified independently or verified in experimental models as SNCA modifiers, such as GBA, LRRK2, MED13, CDC27, [48,175,314]. However, many other SNCA modifiers discussed in this review do not have human orthologs lying close to genetic variants associated with PD or DLB. Perhaps, the identification of more GWAS loci would show if more SNCA modifiers are indeed genetic predisposition factors. Alternatively, epigenetic modifications instead of genetic mutations at SNCA modifiers could contribute to disease progression. In this scenario, it is possible that an environmental insult could contribute to disease progression through the epigenetic modification of an SNCA modifier. Here, we take epigenetic changes to broadly mean any mechanism from chromatin modifications to transcriptomic changes to metabolic reprogramming. Hence, it is conceivable that a genetic mutation or an epigenetic change to an SNCA modifier could contribute to disease progression.
How could one test whether genetic modifiers of SNCA act as sequential hits in a multi-hit experimental model? For example, one could have an experimental model in which one SNCA modifier is constitutively mutated while a second modifier is conditionally knocked down in a temporally controlled manner. Such experiments may help establish a sequence of events that may be theoretically possible during the progression of synucleinopathy.
How would one investigate whether there is a sequence of gene actions that contribute to progression in humans? If resources are not limited, one way to address this is to investigate epigenetic changes associated with different brain cell types and brain regions from postmortem samples at different stages of disease progression, as defined by abundance and distribution of Lewy pathology [28,315]. One could then ask whether a group of SNCA modifiers consistently undergoes epigenetic changes in the same sequence across samples. Given the heterogeneity of synucleinopathies [316,317], such an approach may even be used to determine whether distinct sequences of epigenetic changes occur in different forms or subtypes of synucleinopathies. Currently, there are many efforts to characterize chromatin modifications or transcriptomic changes in synucleinopathies using bulk tissue or single-nuclei approaches [238,318–321]. Interestingly, single-nuclei transcriptomics create opportunities to computationally reconstruct gene expression changes in specific cell populations, as performed recently on PD brains [238]. Similar strategies may guide efforts to place particular SNCA modifiers to specific stages (and cell types) during disease progression.
How would a multi-step model guide design of therapeutic interventions?
As synucleinopathies like PD progress in humans, different symptoms emerge that likely reflect changes in the underlying cellular processes. For example, postmortem data show that dopaminergic synapses in striatum suffer larger losses compared to the corresponding cell bodies in substantia nigra, suggesting that neuronal pathology occurs in a synapse-to-soma direction [322]. This suggests that efforts to save the synaptic terminals need to be applied at a relatively early stage of the disease. Similarly, a better knowledge of how synucleinopathy spreads across brain regions correlates with clinical symptoms will allow investigators to infer stage of disease progression of each patient. Being able to place SNCA modifiers at different stages of disease progression may allow the identification of druggable targets that are appropriate to specific clinical stages of the diseases. Hopefully, such knowledge would guide investigators towards successful clinical trials for disease-modifying interventions.
Supplementary Material
Acknowledgments
The authors thank Wilson Goh and Teddy Tng for helpful discussions.
Contributor Information
Rachel Min Qi Lee, Temasek Life Sciences Laboratory, 1 Research Link, Singapore, 117604, Singapore.
Tong-Wey Koh, Temasek Life Sciences Laboratory, 1 Research Link, Singapore, 117604, Singapore; Department of Biological Sciences, National University of Singapore, Block S3 #05-01, 16 Science Drive 4, Singapore, 117558, Singapore.
Funding
This work was supported by Temasek Life Sciences Laboratory Limited (CC2200).
Conflict of Interest
Both authors have no conflict of interest to declare.
Author Contributions
Rachel Min Qi Lee: Data curation, Wr iting (original draft), Writing (review & editing). Tong-Wey Koh: Data curation, Writing (original draft), Writing (review & editing), Supervision
Data Availability Statement
All data mentioned in this article has been published elsewhere as referenced.
References
- 1.Goedert M, Spillantini MG, Del Tredici Ket al. 100 years of Lewy pathology. Nat Rev Neurol 2013;9:13–24 [DOI] [PubMed] [Google Scholar]
- 2.Spillantini , Schmidt ML, Lee VM-Yet al. α-Synuclein in Lewy bodies. Nature 1997;388:839–40 [DOI] [PubMed] [Google Scholar]
- 3.Ueda K, Fukushima H, Masliah Eet al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci 1993;90:11282–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spillantini , Crowther RA, Jakes Ret al. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 1998;251:205–8 [DOI] [PubMed] [Google Scholar]
- 5.Tu PH, Galvin JE, Baba Met al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol 1998;44:415–22 [DOI] [PubMed] [Google Scholar]
- 6.Wakabayashi K, Yoshimoto M, Tsuji Set al. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 1998;249:180–2 [DOI] [PubMed] [Google Scholar]
- 7.Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet 2021;397:2284–303 [DOI] [PubMed] [Google Scholar]
- 8.Armstrong MJ. Advances in dementia with Lewy bodies. Ther Adv Neurol Disord 2021;14:17562864211057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stankovic I, Fanciulli A, Sidoroff Vet al. A review on the clinical diagnosis of multiple system atrophy. Cerebellum 2022. 10.1007/s12311-022-01453-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nalls MA, Blauwendraat C, Vallerga CLet al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019;18:1091–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chia R, Sabir MS, Bandres-Ciga Set al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 2021;53:294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polymeropoulos MH, Lavedan C, Leroy Eet al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045–7 [DOI] [PubMed] [Google Scholar]
- 13.Krüger R, Kuhn W, Müller Tet al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998;18:106–8 [DOI] [PubMed] [Google Scholar]
- 14.Lesage S, Anheim M, Letournel Fet al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 2013;73:459–71 [DOI] [PubMed] [Google Scholar]
- 15.Appel-Cresswell S, Vilarino-Guell C, Encarnacion Met al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 2013;28:811–3 [DOI] [PubMed] [Google Scholar]
- 16.Proukakis C, Dudzik CG, Brier Tet al. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013;80:1062–4. 10.1212/WNL.0b013e31828727ba [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pimentel MMG, Rodrigues FC, Leite MAAet al. Parkinson disease: α-synuclein mutational screening and new clinical insight into the p.E46K mutation. Parkinsonism Relat Disord 2015;21:586–9 [DOI] [PubMed] [Google Scholar]
- 18.Singleton AB, Farrer M, Johnson Jet al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841–1 [DOI] [PubMed] [Google Scholar]
- 19.Farrer M, Kachergus J, Forno Let al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 2004;55:174–9 [DOI] [PubMed] [Google Scholar]
- 20.Fuchs J, Nilsson C, Kachergus Jet al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 2007;68:916–22 [DOI] [PubMed] [Google Scholar]
- 21.Zarranz JJ, Alegre J, Gómez-Esteban JCet al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164–73 [DOI] [PubMed] [Google Scholar]
- 22.Kiely AP, Asi YT, Kara Eet al. α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 2013;125:753–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Sloan SA, Clarke LEet al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016;89:37–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burré J. The synaptic function of α-synuclein. J Parkinsons Dis 2015;5:699–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MAet al. Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci 2019;13:1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodin L, Milovanovic D, Rizzoli SOet al. α-synuclein in the synaptic vesicle liquid phase: active player or passive bystander? Front Mol Biosci 2022;9:891508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron 2013;79:1044–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis 2017;7:S71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilhelm BG, Mandad S, Truckenbrodt Set al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014;344:1023–8 [DOI] [PubMed] [Google Scholar]
- 30.Chartier-Harlin , Kachergus J, Roumier Cet al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004;364:1167–9 [DOI] [PubMed] [Google Scholar]
- 31.Ibáñez P, Bonnet A-M, Débarges Bet al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004;364:1169–71 [DOI] [PubMed] [Google Scholar]
- 32.Soldner F, Stelzer Y, Shivalila CSet al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016;533:95–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener 2020;15:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cremades N, Cohen SIA, Deas Eet al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012;149:1048–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fares MB, Jagannath S, Lashuel HA. Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci 2021;22:111–31 [DOI] [PubMed] [Google Scholar]
- 36.Cascella R, Bigi A, Cremades Net al. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell Mol Life Sci 2022;79:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fanning S, Selkoe D, Dettmer U. Parkinson’s disease: proteinopathy or lipidopathy? NPJ Parkinsons Dis 2020;6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmqvist S, Chutna O, Bousset Let al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 2014;128:805–20 [DOI] [PubMed] [Google Scholar]
- 39.Braak H, Rüb U, Gai WPet al. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 2003;110:517–36 [DOI] [PubMed] [Google Scholar]
- 40.Jellinger KA. Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord 2012;27:8–30 [DOI] [PubMed] [Google Scholar]
- 41.Borghammer P, Van Den Berge N. Brain-first versus gut-first Parkinson’s disease: a hypothesis. J Parkinsons Dis 2019;9:S281–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalia LV, Lang AE. Parkinson’s disease. Lancet 2015;386:896–912 [DOI] [PubMed] [Google Scholar]
- 43.Fang G, Wang W, Paunic Vet al. Discovering genetic interactions bridging pathways in genome-wide association studies. Nat Commun 2019;10:4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuk O, Hechter E, Sunyaev SRet al. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 2012;109:1193–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blauwendraat C, Reed X, Krohn Let al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020;143:234–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai D, Alipanahi B, Fontanillas Pet al. Genomewide association studies of LRRK2 modifiers of Parkinson’s disease. Ann Neurol 2021;90:76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Génin E, Feingold J, Clerget-Darpoux F. Identifying modifier genes of monogenic disease: strategies and difficulties. Hum Genet 2008;124:357–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ren , Yang Y, Heng KHYet al. MED13 and glycolysis are conserved modifiers of α-synuclein-associated neurodegeneration. Cell Rep 2022;41:111852 [DOI] [PubMed] [Google Scholar]
- 49.Pondarré C, Antiochos BB, Campagna DRet al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum Mol Genet 2006;15:953–64 [DOI] [PubMed] [Google Scholar]
- 50.Arroyo JD, Jourdain AA, Calvo SEet al. A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab 2016;24:875–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antonicka H, Choquet K, Lin Z-Yet al. A pseudouridine synthase module is essential for mitochondrial protein synthesis and cell viability. EMBO Rep 2017;18:28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Merz S, Westermann B. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol 2009;10:R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griffin LB, Sakaguchi R, McGuigan Det al. Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutations. Hum Mutat 2014;35:1363–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szklarczyk R, Wanschers BFJ, Nijtmans LGet al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet 2013;22:656–67 [DOI] [PubMed] [Google Scholar]
- 55.van den Bosch BJC, Gerards M, Sluiter Wet al. Defective NDUFA9 as a novel cause of neonatally fatal complex I disease. J Med Genet 2012;49:10–5 [DOI] [PubMed] [Google Scholar]
- 56.Zhou J, Xu L, Duan Xet al. Large-scale RNAi screen identified Dhpr as a regulator of mitochondrial morphology and tissue homeostasis. Sci Adv 2019;5:eaax0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galbraith MD, Allen MA, Bensard CLet al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013;153:1327–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.King RW, Peters JM, Tugendreich Set al. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 1995;81:279–88 [DOI] [PubMed] [Google Scholar]
- 59.Herrero-Mendez A, Almeida A, Fernández Eet al. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 2009;11:747–52 [DOI] [PubMed] [Google Scholar]
- 60.Solt LA, Wang Y, Banerjee Set al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012;485:62–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cho H, Zhao X, Hatori Met al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012;485:123–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–95 [DOI] [PubMed] [Google Scholar]
- 63.Tang BL. Glucose, glycolysis, and neurodegenerative diseases. J Cell Physiol 2020;235:7653–62 [DOI] [PubMed] [Google Scholar]
- 64.Wang Q, Zhang Y, Wang Met al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat Commun 2019;10:5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kelly J, Moyeed R, Carroll Cet al. Gene expression meta-analysis of Parkinson’s disease and its relationship with Alzheimer's disease. Mol Brain 2019;12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar AS, Jagadeeshan S, Subramanian Aet al. Molecular mechanism of regulation of MTA1 expression by granulocyte colony-stimulating factor. J Biol Chem 2016;291:12310–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reddy SDN, Rayala SK, Ohshiro Ket al. Multiple coregulatory control of tyrosine hydroxylase gene transcription. Proc Natl Acad Sci U S A 2011;108:4200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chi H, Tang W, Bai Y. Molecular evidence of impaired iron metabolism and its association with Parkinson’s disease progression. 3 Biotech 2020;10:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim JY, Kim J-K, Kim H. ABCB7 simultaneously regulates apoptotic and non-apoptotic cell death by modulating mitochondrial ROS and HIF1α-driven NFκB signaling. Oncogene 2020;39:1969–82 [DOI] [PubMed] [Google Scholar]
- 70.Li YI, Wong G, Humphrey Jet al. Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat Commun 2019;10:994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grenn FP, Kim JJ, Makarious MBet al. The Parkinson’s disease genome-wide association study locus browser. Mov Disord 2020;35:2056–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nalls MA, Pankratz N, Lill CMet al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014;46:989–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Altieri DC. Mitochondrial dynamics and metastasis. Cell Mol Life Sci 2019;76:827–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pyle A, Anugrha H, Kurzawa-Akanbi Met al. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol Aging 2016;38:216.e7–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ryan BJ, Hoek S, Fon EAet al. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 2015;40:200–10 [DOI] [PubMed] [Google Scholar]
- 76.Nakamura K, Nemani VM, Azarbal Fet al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem 2011;286:20710–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ludtmann MHR, Angelova PR, Horrocks MHet al. α-Synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun 2018;9:2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feng S-T, Wang Z-Z, Yuan Y-Het al. Update on the association between alpha-synuclein and tau with mitochondrial dysfunction: implications for Parkinson’s disease. Eur J Neurosci 2021;53:2946–59 [DOI] [PubMed] [Google Scholar]
- 79.Deas E, Cremades N, Angelova PRet al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid Redox Signal 2016;24:376–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kleele T, Rey T, Winter Jet al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021;593:435–9 [DOI] [PubMed] [Google Scholar]
- 81.Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol 2020;15:235–59 [DOI] [PubMed] [Google Scholar]
- 82.Chen H, Ren S, Clish Cet al. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol 2015;211:795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ordonez DG, Lee MK, Feany MB. α-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 2018;97:108–124.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Portz P, Lee MK. Changes in Drp1 function and mitochondrial morphology are associated with the α-Synuclein pathology in a transgenic mouse model of Parkinson’s disease. Cell 2021;10:885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krzystek TJ, Banerjee R, Thurston Let al. Differential mitochondrial roles for α-synuclein in DRP1-dependent fission and PINK1/Parkin-mediated oxidation. Cell Death Dis 2021;12:796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gui Y-X, Wang X-Y, Kang W-Yet al. Extracellular signal-regulated kinase is involved in alpha-synuclein-induced mitochondrial dynamic disorders by regulating dynamin-like protein 1. Neurobiol Aging 2012;33:2841–54 [DOI] [PubMed] [Google Scholar]
- 87.Martinez JH, Alaimo A, Gorojod RMet al. Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol Cell Neurosci 2018;88:107–17 [DOI] [PubMed] [Google Scholar]
- 88.Choubey V, Safiulina D, Vaarmann Aet al. Mutant A53T α-Synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011;286:10814–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Laker , Xu P, Ryall KAet al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem 2014;289:12005–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Twig G, Elorza A, Molina AJAet al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008;27:433–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang X, Schwarz TL. The mechanism of Ca2+ −dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009;136:163–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo X, Macleod GT, Wellington Aet al. The GTPase dMiro is required for axonal transport of mitochondria to drosophila synapses. Neuron 2005;47:379–93 [DOI] [PubMed] [Google Scholar]
- 93.Glater EE, Megeath LJ, Stowers RSet al. Axonal transport of mitochondria requires Milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 2006;173:545–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ashrafi G, Schlehe JS, LaVoie MJet al. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 2014;206:655–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang X, Winter D, Ashrafi Get al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011;147:893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shaltouki A, Hsieh C-H, Kim MJet al. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol 2018;136:607–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hsieh C-H, Li L, Vanhauwaert Ret al. Miro1 Marks Parkinson’s disease subset and Miro1 reducer rescues neuron loss in Parkinson’s models. Cell Metab 2019;30:1131–1140.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Melo TQ, Palma FR, Gomes Fet al. Absence of Gem1 (mammalian Miro/Rhot) mitigates alpha-synuclein toxicity in a yeast model of Parkinson’s disease. Mol Cell Neurosci 2022;122:103757 [DOI] [PubMed] [Google Scholar]
- 99.Jamwal S, Blackburn JK, Elsworth JD. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol Ther 2021;219:107705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zheng B, Liao Z, Locascio JJet al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2010;2:52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.O’Donnell KC, Lulla A, Stahl MCet al. Axon degeneration and PGC-1α-mediated protection in a zebrafish model of α-synuclein toxicity. Dis Model Mech 2014;7:571–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ciron C, Zheng L, Bobela Wet al. PGC-1α activity in nigral dopamine neurons determines vulnerability to α-synuclein. Acta Neuropathol Commun 2015;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ryan BJ, Bengoa-Vergniory N, Williamson Met al. REST protects dopaminergic neurons from mitochondrial and α-synuclein oligomer pathology in an alpha synuclein overexpressing BAC-transgenic mouse model. J Neurosci 2021;41:3731–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.De Miranda BR, Rocha EM, Castro SLet al. Protection from α-Synuclein induced dopaminergic neurodegeneration by overexpression of the mitochondrial import receptor TOM20. NPJ Parkinsons Dis 2020;6:38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Butler EK, Voigt A, Lutz AKet al. The mitochondrial chaperone protein TRAP1 mitigates α-Synuclein toxicity. PLoS Genet 2012;8:e1002488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Knight AL, Yan X, Hamamichi Set al. The glycolytic enzyme, GPI, is a functionally conserved modifier of dopaminergic neurodegeneration in Parkinson’s models. Cell Metab 2014;20:145–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cai R, Zhang Y, Simmering JEet al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J Clin Invest 2019;129:4539–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen X, Zhao C, Li Xet al. Terazosin activates Pgk1 and Hsp90 to promote stress resistance. Nat Chem Biol 2015;11:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.González-Rodríguez P, Zampese E, Stout KAet al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021;599:650–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schmidt MM, Dringen R. Glutathione (GSH) Synthesis and Metabolism. In: Choi I-Y, Gruetter R, (eds.), Neural Metabolism In Vivo. Boston, MA: Springer US, 2012, 1029–50 [Google Scholar]
- 111.Patten DA, McGuirk S, Anilkumar Uet al. Altered mitochondrial fusion drives defensive glutathione synthesis in cells able to switch to glycolytic ATP production. Biochim Biophys Acta Mol Cell Res 2021;1868:118854 [DOI] [PubMed] [Google Scholar]
- 112.Wang B, Liu Q, Shan Het al. Nrf2 inducer and cncC overexpression attenuates neurodegeneration due to α-synuclein in drosophila. Biochem Cell Biol 2015;93:351–8 [DOI] [PubMed] [Google Scholar]
- 113.Barone MC, Sykiotis GP, Bohmann D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a drosophila model of Parkinson’s disease. Dis Model Mech 2011;4:701–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou W, Freed CR. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T α-Synuclein toxicity. J Biol Chem 2005;280:43150–8 [DOI] [PubMed] [Google Scholar]
- 115.Willingham S, Outeiro TF, DeVit MJet al. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 2003;302:1769–72 [DOI] [PubMed] [Google Scholar]
- 116.Trinh K, Moore K, Wes PDet al. Induction of the phase II detoxification pathway suppresses neuron loss in drosophila models of Parkinson’s disease. J Neurosci 2008;28:465–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dong M-Q, Venable JD, Au Net al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 2007;317:660–3 [DOI] [PubMed] [Google Scholar]
- 118.Jeon JY, Choi S-E, Ha ESet al. GLP-1 improves palmitate-induced insulin resistance in human skeletal muscle via SIRT1 activity. Int J Mol Med 2019;44:1161–71 [DOI] [PubMed] [Google Scholar]
- 119.Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem 2007;282:34356–64 [DOI] [PubMed] [Google Scholar]
- 120.Kao S-Y. Rescue of α-synuclein cytotoxicity by insulin-like growth factors. Biochem Biophys Res Commun 2009;385:434–8 [DOI] [PubMed] [Google Scholar]
- 121.Zhang H-Y, Jiang Y-C, Li J-Ret al. Neuroprotective effects of insulin-like growth factor-2 in 6-hydroxydopamine-induced cellular and mouse models of Parkinson’s disease. Neural Regeneration Res 2022;18:1099–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang X-W, Yuan L-J, Yang Yet al. IGF-1 inhibits MPTP/MPP+-induced autophagy on dopaminergic neurons through the IGF-1R/PI3K-Akt-mTOR pathway and GPER. Am J Physiol Endocrinol Metab 2020;319:E734–43 [DOI] [PubMed] [Google Scholar]
- 123.Cheng H-C, Kim SR, Oo TFet al. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci 2011;31:2125–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu F, Li Z, Zheng Xet al. SIRT1 mediates the effect of GLP-1 receptor agonist exenatide on ameliorating hepatic steatosis. Diabetes 2014;63:3637–46 [DOI] [PubMed] [Google Scholar]
- 125.Yoshizaki T, Milne JC, Imamura Tet al. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol 2009;29:1363–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.During MJ, Cao L, Zuzga DSet al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 2003;9:1173–9 [DOI] [PubMed] [Google Scholar]
- 127.Yun SP, Kam T-I, Panicker Net al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 2018;24:931–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Avet-Rochex A, Carvajal N, Christoforou CPet al. Unkempt is negatively regulated by mTOR and uncouples neuronal differentiation from growth control. PLoS Genet 2014;10:e1004624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li N, Liu Q, Xiong Yet al. Headcase and unkempt regulate tissue growth and cell cycle progression in response to nutrient restriction. Cell Rep 2019;26:733–747.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oliveira da Silva MI, Liz MA. Linking alpha-Synuclein to the actin cytoskeleton: consequences to neuronal function. Front cell. Dev Biol 2020;8:787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.DuBoff B, Götz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012;75:618–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013;339:464–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hatch AL, Gurel PS, Higgs HN. Novel roles for actin in mitochondrial fission. J Cell Sci 2014;127:4549–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moore AS, Wong YC, Simpson CLet al. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat Commun 2016;7:12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sarkar S, Olsen AL, Sygnecka Ket al. α-Synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet 2021;17:e1009359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jadiya P, Fatima S, Baghel Tet al. A systematic RNAi screen of neuroprotective genes identifies novel modulators of alpha-synuclein-associated effects in transgenic Caenorhabditis elegans. Mol Neurobiol 2016;53:6288–300 [DOI] [PubMed] [Google Scholar]
- 137.MacQueen AJ, Baggett JJ, Perumov Net al. ACT-5 is an essential Caenorhabditis elegans actin required for intestinal microvilli formation. Mol Biol Cell 2005;16:3247–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hu Y, Flockhart I, Vinayagam Aet al. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 2011;12:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miller DW, Hague SM, Clarimon Jet al. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 2004;62:1835–8 [DOI] [PubMed] [Google Scholar]
- 140.Konno T, Ross OA, Puschmann Aet al. Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat Disord 2016;22:S1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Valente T, Dentesano G, Ezquerra Met al. CCAAT/enhancer binding protein δ is a transcriptional repressor of α-synuclein. Cell Death Differ 2020;27:509–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Scherzer CR, Grass JA, Liao Zet al. GATA transcription factors directly regulate the Parkinson’s disease-linked gene α-synuclein. Proc Natl Acad Sci 2008;105:10907–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Duplan E, Giordano C, Checler Fet al. Direct α-synuclein promoter transactivation by the tumor suppressor p53. Mol Neurodegener 2016;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lassot I, Mora S, Lesage Set al. The E3 ubiquitin ligases TRIM17 and TRIM41 modulate α-Synuclein expression by regulating ZSCAN21. Cell Rep 2018;25:2484–2496.e9 [DOI] [PubMed] [Google Scholar]
- 145.Clough RL, Dermentzaki G, Stefanis L. Functional dissection of the alpha-synuclein promoter: transcriptional regulation by ZSCAN21 and ZNF219. J Neurochem 2009;110:1479–90 [DOI] [PubMed] [Google Scholar]
- 146.Dermentzaki G, Paschalidis N, Politis PKet al. Complex effects of the ZSCAN21 transcription factor on transcriptional regulation of α-Synuclein in primary neuronal cultures and in vivo. J Biol Chem 2016;291:8756–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Auluck PK, Chan HYE, Trojanowski JQet al. Chaperone suppression of alpha-synuclein toxicity in a drosophila model for Parkinson’s disease. Science 2002;295:865–8 [DOI] [PubMed] [Google Scholar]
- 148.Burmann BM, Gerez JA, Matečko-Burmann Iet al. Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020;577:127–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Brahmachari S, Ge P, Lee SHet al. Activation of tyrosine kinase c-Abl contributes to α-synuclein-induced neurodegeneration. J Clin Invest 2016;126:2970–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Paxinou E, Chen Q, Weisse Met al. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 2001;21:8053–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Giasson BI, Duda JE, Murray IVet al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000;290:985–9 [DOI] [PubMed] [Google Scholar]
- 152.Smith WW, Liu Z, Liang Yet al. Synphilin-1 attenuates neuronal degeneration in the A53T α-synuclein transgenic mouse model. Hum Mol Genet 2010;19:2087–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hernández-Vargas R, Fonseca-Ornelas L, López-González Iet al. Synphilin suppresses α-synuclein neurotoxicity in a Parkinson’s disease drosophila model. Genesis 2011;49:392–402 [DOI] [PubMed] [Google Scholar]
- 154.Casadei N, Pöhler A-M, Tomás-Zapico Cet al. Overexpression of synphilin-1 promotes clearance of soluble and misfolded alpha-synuclein without restoring the motor phenotype in aged A30P transgenic mice. Hum Mol Genet 2014;23:767–81 [DOI] [PubMed] [Google Scholar]
- 155.Tanaka M, Kim YM, Lee Get al. Aggresomes formed by α-synuclein and synphilin-1 are cytoprotective. J Biol Chem 2004;279:4625–31 [DOI] [PubMed] [Google Scholar]
- 156.Engelender K, Guo S, Amaravi Ket al. Synphilin-1 associates with α-synuclein and promotes the formation of cytosolic inclusions. Nat Genet 1999;22:110–4 [DOI] [PubMed] [Google Scholar]
- 157.Zondler L, Miller-Fleming L, Repici Met al. DJ-1 interactions with α-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis 2014;5:e1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 2000;275:35661–4 [DOI] [PubMed] [Google Scholar]
- 159.Vogiatzi T, Xilouri M, Vekrellis Ket al. Wild type α-Synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells*. J Biol Chem 2008;283:23542–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bennett MC, Bishop JF, Leng Yet al. Degradation of α-synuclein by proteasome. J Biol Chem 1999;274:33855–8 [DOI] [PubMed] [Google Scholar]
- 161.Webb JL, Ravikumar B, Atkins Jet al. α-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003;278:25009–13 [DOI] [PubMed] [Google Scholar]
- 162.Lee H-J, Khoshaghideh F, Patel Set al. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 2004;24:1888–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cuervo AM, Stefanis L, Fredenburg Ret al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science Jiang Y-C;305:1292–5 [DOI] [PubMed] [Google Scholar]
- 164.Sahoo S, Padhy AA, Kumari Vet al. Role of ubiquitin–proteasome and autophagy-lysosome pathways in α-Synuclein aggregate clearance. Mol Neurobiol 2022;59:5379–407 [DOI] [PubMed] [Google Scholar]
- 165.Xilouri M, Brekk OR, Stefanis L. α-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol 2013;47:537–51 [DOI] [PubMed] [Google Scholar]
- 166.Zhang J, Li X, Li J-D. The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front Neurosci 2019;13:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Zet al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J Neurosci 2011;31:14508–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tofaris GK, Kim HT, Hourez Ret al. Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal–lysosomal pathway. Proc Natl Acad Sci 2011;108:17004–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tardiff DF, Jui NT, Khurana Vet al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science 2013;342:979–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liani E, Eyal A, Avraham Eet al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc Natl Acad Sci U S A 2004;101:5500–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rott R, Szargel R, Haskin Jet al. Monoubiquitylation of α-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem 2008;283:3316–28 [DOI] [PubMed] [Google Scholar]
- 172.Lee JT, Wheeler TC, Li Let al. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum Mol Genet 2008;17:906–17 [DOI] [PubMed] [Google Scholar]
- 173.Rott R, Szargel R, Haskin Jet al. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A 2011;108:18666–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol 2020;19:170–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fishbein I, Kuo Y-M, Giasson BIet al. Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain 2014;137:3235–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kuo S-H, Tasset I, Cheng MMet al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv 2022;8:eabm6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Decressac M, Mattsson B, Weikop Pet al. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A 2013;110:E1817–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chung H-J, Islam MS, Rahman MMet al. Neuroprotective function of Omi to α-synuclein-induced neurotoxicity. Neurobiol Dis 2020;136:104706 [DOI] [PubMed] [Google Scholar]
- 179.Bellani S, Sousa VL, Ronzitti Get al. The regulation of synaptic function by α-synuclein. Integr Biol 2010;3:106–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Scott D, Roy S. α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci 2012;32:10129–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Plun-Favreau H, Klupsch K, Moisoi Net al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol 2007;9:1243–52 [DOI] [PubMed] [Google Scholar]
- 182.Fleming A, Bourdenx M, Fujimaki Met al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022;110:935–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fields CR, Bengoa-Vergniory N, Wade-Martins R. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front Mol Neurosci 2019;12:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cooper AA, Gitler AD, Cashikar Aet al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006;313:324–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Coune PG, Bensadoun JC, Aebischer Pet al. Rab1A over-expression prevents Golgi apparatus fragmentation and partially corrects motor deficits in an alpha-synuclein based rat model of Parkinson’s disease. J Parkinsons Dis 2011;1:373–87 [DOI] [PubMed] [Google Scholar]
- 186.Gitler AD, Bevis BJ, Shorter Jet al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A 2008;105:145–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kuwahara T, Koyama A, Koyama Set al. A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum Mol Genet 2008;17:2997–3009 [DOI] [PubMed] [Google Scholar]
- 188.Zabrocki P, Bastiaens I, Delay Cet al. Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson. Biochim Biophys Acta 2008;1783:1767–80 [DOI] [PubMed] [Google Scholar]
- 189.Breiden B, Sandhoff K. Ganglioside metabolism and its inherited diseases. Methods Mol Biol 2018;1804:97–141 [DOI] [PubMed] [Google Scholar]
- 190.Yu M, Ye H, De-Paula RBet al. Functional screening of lysosomal storage disorder genes identifies modifiers of alpha-synuclein mediated neurodegeneration. bioRxiv 2022. 10.1101/2022.07.23.501240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang S, Glukhova SA, Caldwell KAet al. NCEH-1 modulates cholesterol metabolism and protects against α-synuclein toxicity in a C. elegans model of Parkinson’s disease. Hum Mol Genet 2017;26:3823–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Valastyan JS, Termine DJ, Lindquist S. Splice isoform and pharmacological studies reveal that sterol depletion relocalizes α-synuclein and enhances its toxicity. Proc Natl Acad Sci U S A 2014;111:3014–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bykov K, Yoshida K, Weisskopf MGet al. Confounding of the association between statins and Parkinson disease: systematic review and meta-analysis. Pharmacoepidemiol Drug Saf 2017;26:294–300 [DOI] [PubMed] [Google Scholar]
- 194.Sheng Z, Jia X, Kang M. Statin use and risk of Parkinson’s disease: a meta-analysis. Behav Brain Res 2016;309:29–34 [DOI] [PubMed] [Google Scholar]
- 195.Ledeen RW, Wu G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem Sci 2015;40:407–18 [DOI] [PubMed] [Google Scholar]
- 196.Lin G, Lee P-T, Chen Ket al. Phospholipase PLA2G6, a parkinsonism-associated gene, affects Vps26 and Vps35, Retromer function, and ceramide levels, similar to α-Synuclein gain. Cell Metab 2018;28:605–618.e6 [DOI] [PubMed] [Google Scholar]
- 197.Alcalay RN, Mallett V, Vanderperre Bet al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov Disord 2019;34:526–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kim MJ, Jeon S, Burbulla LFet al. Acid ceramidase inhibition ameliorates α-synuclein accumulation upon loss of GBA1 function. Hum Mol Genet 2018;27:1972–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Glajch KE, Moors TE, Chen Yet al. Wild-type GBA1 increases the α-synuclein tetramer–monomer ratio, reduces lipid-rich aggregates, and attenuates motor and cognitive deficits in mice. Proc Natl Acad Sci 2021;118:e2103425118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wu G, Lu Z-H, Kulkarni Net al. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J Neurosci Res 2012;90:1997–2008 [DOI] [PubMed] [Google Scholar]
- 201.Mori A, Hatano T, Inoshita Tet al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc Natl Acad Sci 2019;116:20689–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mingione A, Pivari F, Plotegher Net al. Inhibition of ceramide synthesis reduces α-Synuclein Proteinopathy in a cellular model of Parkinson’s disease. Int J Mol Sci 2021;22:6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lee YJ, Wang S, Slone SRet al. Defects in very long chain fatty acid synthesis enhance alpha-synuclein toxicity in a yeast model of Parkinson’s disease. PLoS One 2011;6:e15946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kluepfel D, Bagli J, Baker Het al. Myriocin, a new antifungal antibiotic from Myriococcum albomyces. J Antibiot 1972;25:109–15 [DOI] [PubMed] [Google Scholar]
- 205.Shao J, Pei Z, Jing Het al. Antifungal activity of myriocin against fusarium graminearum and its inhibitory effect on deoxynivalenol production in wheat grains. Physiol Mol Plant Pathol 2021;114:101635 [Google Scholar]
- 206.Strettoi E, Gargini C, Novelli Eet al. Inhibition of ceramide biosynthesis preserves photoreceptor structure and function in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci U S A 2010;107:18706–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Petit CS, Lee JJ, Boland Set al. Inhibition of sphingolipid synthesis improves outcomes and survival in GARP mutant wobbler mice, a model of motor neuron degeneration. Proc Natl Acad Sci U S A 2020;117:10565–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kiechle M, Grozdanov V, Danzer KM. The role of lipids in the initiation of α-Synuclein Misfolding. Front Cell Dev Biol 2020;8:562241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J 2010;24:3052–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ma J, Gao J, Wang Jet al. Prion-like mechanisms in Parkinson’s disease. Front Neurosci 2019;13:552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jan A, Gonçalves NP, Vaegter CBet al. The prion-like spreading of alpha-Synuclein in Parkinson’s disease: update on models and hypotheses. Int J Mol Sci 2021;22:8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Brundin P, Melki R. Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 2017;37:9808–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.See SK, Chen M, Bax Set al. PIKfyve inhibition blocks endolysosomal escape of α-synuclein fibrils and spread of α-synuclein aggregation. bioRxiv 2021. 10.1101/2021.01.21.427704 [DOI] [Google Scholar]
- 214.Tyson T, Senchuk M, Cooper JFet al. Novel animal model defines genetic contributions for neuron-to-neuron transfer of α-synuclein. Sci Rep 2017;7:7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bieri G, Brahic M, Bousset Let al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol 2019;137:961–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mao X, Ou MT, Karuppagounder SSet al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016;353:aah3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Fares MB, Ait-Bouziad N, Dikiy Iet al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet 2014;23:4491–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Khalaf O, Fauvet B, Oueslati Aet al. The H50Q mutation enhances α-Synuclein aggregation, secretion, and toxicity*. J Biol Chem 2014;289:21856–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Poehler A-M, Xiang W, Spitzer Pet al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014;10:2171–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Minakaki G, Menges S, Kittel Aet al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018;14:98–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fussi N, Höllerhage M, Chakroun Tet al. Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis 2018;9:757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lee H-J, Cho E-D, Lee KWet al. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 2013;45:e22–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Emmanouilidou E, Minakaki G, Keramioti MVet al. GABA transmission via ATP-dependent K+ channels regulates α-synuclein secretion in mouse striatum. Brain 2016;139:871–90 [DOI] [PubMed] [Google Scholar]
- 224.Ejlerskov P, Rasmussen I, Nielsen TTet al. Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-Synuclein through Exophagy by impairing autophagosome-lysosome fusion*. J Biol Chem 2013;288:17313–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tsunemi T, Hamada K, Krainc D. ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J Neurosci 2014;34:15281–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Fellner L, Irschick R, Schanda Ket al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013;61:349–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Kim C, Ho D-H, Suk J-Eet al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 2013;4:1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Guo M, Wang J, Zhao Yet al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 2020;143:1476–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ientile R, Currò M, Caccamo D. Transglutaminase 2 and neuroinflammation. Amino Acids 2015;47:19–26 [DOI] [PubMed] [Google Scholar]
- 230.Yakubov B, Chelladurai B, Schmitt Jet al. Extracellular tissue transglutaminase activates noncanonical NF-κB signaling and promotes metastasis in ovarian cancer. Neoplasia 2013;15:609–IN8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zhang J, Grosso Jasutkar H, Yan Ret al. Transglutaminase 2 depletion attenuates α-Synuclein mediated toxicity in mice. Neuroscience 2020;441:58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shao Q-H, Yan W-F, Zhang Zet al. Nurr1: a vital participant in the TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. Neuropharmacology 2019;144:388–99 [DOI] [PubMed] [Google Scholar]
- 233.Kou L, Chi X, Sun Yet al. The circadian clock protein rev-erbα provides neuroprotection and attenuates neuroinflammation against Parkinson’s disease via the microglial NLRP3 inflammasome. J Neuroinflammation 2022;19:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jeong GR, Lee BD. Pathological functions of LRRK2 in Parkinson’s disease. Cell 2020;9:2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhang Y, Chen K, Sloan SAet al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014;34:11929–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lin X, Parisiadou L, Gu X-Let al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 2009;64:807–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sarkar S, Bardai F, Olsen ALet al. Oligomerization of Lrrk controls actin severing and α-synuclein neurotoxicity in vivo. Mol Neurodegener 2021;16:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Smajić S, Prada-Medina CA, Landoulsi Zet al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2022;145:964–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Russo I, Berti G, Plotegher Net al. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J Neuroinflammation 2015;12:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Manjula R, Anuja K, Alcain FJ. SIRT1 and SIRT2 activity control in neurodegenerative diseases. Front Pharmacol 2020;11:585821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li Y, Xu W, McBurney MWet al. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab 2008;8:38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kaeberlein M. The ongoing saga of sirtuins and aging. Cell Metab 2008;8:4–5 [DOI] [PubMed] [Google Scholar]
- 243.Mao K, Chen J, Yu Het al. Poly (ADP-ribose) polymerase 1 inhibition prevents neurodegeneration and promotes α-synuclein degradation via transcription factor EB-dependent autophagy in mutant α-synucleinA53T model of Parkinson’s disease. Aging Cell 2020;19:e13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Guo Y-J, Dong S-Y, Cui X-Xet al. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of α-synuclein via SIRT1-deacetylated LC3. Mol Nutr Food Res 2016;60:2161–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Outeiro TF, Kontopoulos E, Altmann SMet al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007;317:516–9 [DOI] [PubMed] [Google Scholar]
- 246.de Oliveira RM, Vicente Miranda H, Francelle Let al. The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol 2017;15:e2000374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gleave JA, Arathoon LR, Trinh Det al. Sirtuin 3 rescues neurons through the stabilisation of mitochondrial biogenetics in the virally-expressing mutant α-synuclein rat model of parkinsonism. Neurobiol Dis 2017;106:133–46 [DOI] [PubMed] [Google Scholar]
- 248.Park J-H, Burgess JD, Faroqi AHet al. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol Neurodegener 2020;15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.van Ham TJ, Thijssen KL, Breitling Ret al. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet 2008;4:e1000027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Sampaio-Marques B, Felgueiras C, Silva Aet al. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 2012;8:1494–509 [DOI] [PubMed] [Google Scholar]
- 251.Zaichick SV, McGrath KM, Caraveo G. The role of Ca2+ signaling in Parkinson’s disease. Dis Model Mech 2017;10:519–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Apicco DJ, Shlevkov E, Nezich CLet al. The Parkinson’s disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release. Proc Natl Acad Sci U S A 2021;118: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gong F, Chen H, Xiong Yet al. α-Synucleinopathy associated calcium overload and autophagy failure is regulated by gain-of-function of tousled-like kinase. bioRxiv 2022. 10.1101/2022.07.17.500360 [DOI] [Google Scholar]
- 254.Caraveo G, Auluck PK, Whitesell Let al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc Natl Acad Sci U S A 2014;111:E3544–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Screaton RA, Conkright MD, Katoh Yet al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 2004;119:61–74 [DOI] [PubMed] [Google Scholar]
- 256.Höglinger GU, Breunig JJ, Depboylu Cet al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proc Natl Acad Sci U S A 2007;104:3585–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Lee SS, Kim YM, Junn Eet al. Cell cycle aberrations by α-synuclein over-expression and cyclin B immunoreactivity in Lewy bodies. Neurobiol Aging 2003;24:687–96 [DOI] [PubMed] [Google Scholar]
- 258.Paiva I, Pinho R, Pavlou MAet al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum Mol Genet 2017;26:2231–46 [DOI] [PubMed] [Google Scholar]
- 259.Findeiss E, Schwarz SC, Evsyukov Vet al. Comprehensive miRNome-wide profiling in a neuronal cell model of Synucleinopathy implies involvement of cell cycle genes. Front Cell Dev Biol 2021;9:561086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Veas-Pérez de Tudela M, Maestre C, Delgado-Esteban Met al. Cdk5-mediated inhibition of APC/C-Cdh1 switches on the cyclin D1-Cdk4-pRb pathway causing aberrant S-phase entry of postmitotic neurons. Sci Rep 2015;5:18180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Dowejko A, Bauer RJ, Müller-Richter UDAet al. The human homolog of the drosophila headcase protein slows down cell division of head and neck cancer cells. Carcinogenesis 2009;30:1678–85 [DOI] [PubMed] [Google Scholar]
- 262.López-Grueso MJ, Padilla CA, Bárcena JAet al. Deficiency of Parkinson’s related protein DJ-1 alters Cdk5 Signalling and induces neuronal death by aberrant cell cycle re-entry. Cell Mol Neurobiol 2022;43:757–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Barone P, Antonini A, Colosimo Cet al. The PRIAMO study: a multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord 2009;24:1641–9 [DOI] [PubMed] [Google Scholar]
- 264.Leng Y, Musiek ES, Hu Ket al. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol 2019;18:307–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016;354:1004–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Fifel K. Alterations of the circadian system in Parkinson’s disease patients. Mov Disord 2017;32:682–92 [DOI] [PubMed] [Google Scholar]
- 267.Jager J, O’Brien WT, Manlove Jet al. Behavioral changes and dopaminergic dysregulation in mice lacking the nuclear receptor rev-erb alpha. Mol Endocrinol 2014;28:490–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Li D-Q, Pakala SB, Reddy SDNet al. Metastasis-associated protein 1 is an integral component of the circadian molecular machinery. Nat Commun 2013;4:2545. [DOI] [PubMed] [Google Scholar]
- 269.Watanabe K, Jansen PR, Savage JEet al. Genome-wide meta-analysis of insomnia prioritizes genes associated with metabolic and psychiatric pathways. Nat Genet 2022;54:1125–32 [DOI] [PubMed] [Google Scholar]
- 270.Nelson MR, Tipney H, Painter JLet al. The support of human genetic evidence for approved drug indications. Nat Genet 2015;47:856–60 [DOI] [PubMed] [Google Scholar]
- 271.Libby P, Tokgözoğlu L. Chasing LDL cholesterol to the bottom—PCSK9 in perspective. Nat Cardiovasc Res 2022;1:554–61 [DOI] [PubMed] [Google Scholar]
- 272.Sheridan C. Unicorn startup trawls databases for protective genetic modifiers. Nat Biotechnol 2019;37:487–9 [DOI] [PubMed] [Google Scholar]
- 273.Nijman S. Genetic modifiers: Why drug developers should care. In: Drug Discovery World. 18 Jan 2021. [cited 11 Oct 2022]. https://www.ddw-online.com/genetic-modifiers-why-drug-developers-should-care-9145-202101/
- 274.Eisenstein M. Adrestia therapeutics - gene networks to the rescue. Nat Biotechnol 2022. 10.1038/d41587-022-00011-3 [DOI] [PubMed] [Google Scholar]
- 275.McLean BA, Wong CK, Campbell JEet al. Revisiting the complexity of GLP-1 action from sites of synthesis to receptor activation. Endocr Rev 2021;42:101–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Chou S-Y, Chan L, Chung C-Cet al. Altered insulin receptor substrate 1 phosphorylation in blood neuron-derived extracellular vesicles from patients with Parkinson’s disease. Front Cell Dev Biol 2020;8:564641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Bassil F, Delamarre A, Canron M-Het al. Impaired brain insulin signalling in Parkinson’s disease. Neuropathol Appl Neurobiol 2022;48:e12760 [DOI] [PubMed] [Google Scholar]
- 278.Aviles-Olmos I, Dickson J, Kefalopoulou Zet al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis 2014;4:337–44 [DOI] [PubMed] [Google Scholar]
- 279.Athauda D, Maclagan K, Skene SSet al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 2017;390:1664–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Athauda D, Gulyani S, Karnati HKet al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: a secondary analysis of the Exenatide-PD trial. JAMA Neurol 2019;76:420–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Nørgaard CH, Friedrich S, Hansen CTet al. Treatment with glucagon-like peptide-1 receptor agonists and incidence of dementia: data from pooled double-blind randomized controlled trials and nationwide disease and prescription registers. Alzheimers Dement 2022;8:e12268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators . Pioglitazone in early Parkinson’s disease: a phase 2, multicentre, double-blind, randomised trial. Lancet Neurol 2015;14:795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Bordet R, Ouk T, Petrault Oet al. PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochem Soc Trans 2006;34:1341–6 [DOI] [PubMed] [Google Scholar]
- 284.Huebecker M, Moloney EB, van der Spoel ACet al. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol Neurodegener 2019;14:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Schneider JS, Sendek S, Daskalakis Cet al. GM1 ganglioside in Parkinson’s disease: results of a five year open study. J Neurol Sci 2010;292:45–51 [DOI] [PubMed] [Google Scholar]
- 286.Schneider JS, Gollomp SM, Sendek Set al. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J Neurol Sci 2013;324:140–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Schneider JS, Cambi F, Gollomp SMet al. GM1 ganglioside in Parkinson’s disease: pilot study of effects on dopamine transporter binding. J Neurol Sci 2015;356:118–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Meng Y, Pople CB, Huang Yet al. Putaminal recombinant Glucocerebrosidase delivery with magnetic resonance-guided focused ultrasound in Parkinson’s disease: a phase I study. Mov Disord 2022;37:2134–9 [DOI] [PubMed] [Google Scholar]
- 289.Peterschmitt MJ, Saiki H, Hatano Tet al. Safety, pharmacokinetics, and pharmacodynamics of Oral Venglustat in patients with Parkinson’s disease and a GBA mutation: results from part 1 of the randomized, double-blinded, placebo-controlled MOVES-PD trial. J Parkinsons Dis 2022;12:557–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Venglustat. [cited 22 Dec 2022]. https://www.alzforum.org/therapeutics/venglustat
- 291.Sanofi . Sanofi delivered close to double-digit Q4 2020 business EPS(1) growth at CER. Sanofi; 2021 Feb. [Google Scholar]
- 292.Pagano G, Taylor KI, Anzures-Cabrera Jet al. Trial of Prasinezumab in early-stage Parkinson’s disease. N Engl J Med 2022;387:421–32. 10.1056/NEJMoa2202867 [DOI] [PubMed] [Google Scholar]
- 293.Ndayisaba A, Kaindlstorfer C, Wenning GK. Iron in neurodegeneration - cause or consequence? Front Neurosci 2019;13:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Bukowiecki J, Pförringer D, Thor Det al. HIF-1α stimulators function equally to leading hair loss agents in enhancing dermal papilla growth. Skin Pharmacol Physiol 2021;33:309–16 [DOI] [PubMed] [Google Scholar]
- 295.Devos D, Labreuche J, Rascol Oet al. Trial of Deferiprone in Parkinson’s disease. N Engl J Med 2022;387:2045–55 [DOI] [PubMed] [Google Scholar]
- 296.Mischley LK, Leverenz JB, Lau RCet al. A randomized, double-blind phase I/IIa study of intranasal glutathione in Parkinson’s disease. Mov Disord 2015;30:1696–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Mischley LK, Lau RC, Shankland EGet al. Phase IIb study of intranasal glutathione in Parkinson’s disease. J Parkinsons Dis 2017;7:289–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Parkinson Study Group STEADY-PD III Investigators . Isradipine versus placebo in early Parkinson disease: a randomized trial. Ann Intern Med 2020;172:591–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Lang AE, Gill S, Patel NKet al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 2006;59:459–66 [DOI] [PubMed] [Google Scholar]
- 300.Slevin JT, Gash DM, Smith CDet al. Unilateral intraputamenal glial cell line-derived neurotrophic factor in patients with Parkinson disease: response to 1 year of treatment and 1 year of withdrawal. J Neurosurg 2007;106:614–20 [DOI] [PubMed] [Google Scholar]
- 301.Whone A, Luz M, Boca Met al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019;142:512–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Ogrodnik M. Cellular aging beyond cellular senescence: markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell 2021;20:e13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Zheng X, Boyer L, Jin Met al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. elife 2016;5:1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci 2018;21:1370–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kim J, Sullivan GJ, Park I-H. How well do brain organoids capture your brain? iScience 2021;24:102063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Tardiff DF, Khurana V, Chung CYet al. From yeast to patient neurons and back again: powerful new discovery platform. Mov Disord 2014;29:1231–40 [DOI] [PubMed] [Google Scholar]
- 307.Ma M, Moulton MJ, Lu Set al. “Fly-ing” from rare to common neurodegenerative disease mechanisms. Trends Genet 2022;38:972–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Gitler AD, Chesi A, Geddie MLet al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 2009;41:308–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Le Heron C, MacAskill M, Mason Det al. A multi-step model of Parkinson’s disease pathogenesis. Mov Disord 2021;36:2530–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Carvey PM, Punati A, Newman MB. Progressive dopamine neuron loss in Parkinson’s disease: the multiple hit hypothesis. Cell Transplant 2006;15:239–50 [DOI] [PubMed] [Google Scholar]
- 311.Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci 2007;30:244–50 [DOI] [PubMed] [Google Scholar]
- 312.Ling Z, Zhu Y, Tong CWet al. Progressive dopamine neuron loss following supra-nigral lipopolysaccharide (LPS) infusion into rats exposed to LPS prenatally. Exp Neurol 2006;199:499–512 [DOI] [PubMed] [Google Scholar]
- 313.Thiruchelvam M, Richfield EK, Goodman BMet al. Developmental exposure to the pesticides paraquat and maneb and the Parkinson’s disease phenotype. Neurotoxicology 2002;23:621–33 [DOI] [PubMed] [Google Scholar]
- 314.Volpicelli-Daley LA, Abdelmotilib H, Liu Zet al. G2019S-LRRK2 expression augments α-Synuclein sequestration into inclusions in neurons. J Neurosci 2016;36:7415–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Borghammer P, Horsager J, Andersen Ket al. Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol Dis 2021;161:105557 [DOI] [PubMed] [Google Scholar]
- 316.Peng C, Gathagan RJ, Covell DJet al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018;557:558–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Pedersen CC, Lange J, Førland MGGet al. A systematic review of associations between common SNCA variants and clinical heterogeneity in Parkinson’s disease. NPJ Parkinson’s Disease 2021;7:54–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Nido GS, Dick F, Toker Let al. Common gene expression signatures in Parkinson’s disease are driven by changes in cell composition. Acta Neuropathol Commun 2020;8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Kamath T, Abdulraouf A, Burris SJet al. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat Neurosci 2022;25:588–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Toker L, Tran GT, Sundaresan Jet al. Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson’s disease brain. Mol Neurodegener 2021;16:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Corces MR, Shcherbina A, Kundu Set al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson's diseases. Nat Genet 2020;52:1158–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Cheng H-C, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.OMIM . OMIM®. Online Mendelian Inheritance in Man. Baltimore, MD: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; https://omim.org/. [Google Scholar]
- 324.Stahl EA, Breen G, Forstner AJet al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 2019;51:793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Frenkel S, Bernstein CN, Sargent Met al. Genome-wide analysis identifies rare copy number variations associated with inflammatory bowel disease. PLoS One 2019;14:e0217846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Dannappel MV, Sooraj D, Loh JJet al. Molecular and in vivo functions of the CDK8 and CDK19 kinase modules. Front Cell Dev Biol 2018;6:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Song Y, Choi J-E, Kwon Y-Jet al. Identification of susceptibility loci for cardiovascular disease in adults with hypertension, diabetes, and dyslipidemia. J Transl Med 2021;19:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Li T, Wu Y, Chen W-Cet al. Functional analysis of HECA variants identified in congenital heart disease in the Chinese population. J Clin Lab Anal 2022;36:e24649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res 2018;122:433–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.IMSGC . Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019;365:eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Li H, Song S, Wang Yet al. Low-grade inflammation aggravates rotenone neurotoxicity and disrupts circadian clock gene expression in rats. Neurotox Res 2019;35:421–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Chen Y-C, Farzadfard F, Gharaei Net al. Randomized CRISPR-Cas transcriptional perturbation screening reveals protective genes against alpha-Synuclein toxicity. Mol Cell 2017;68:247–257.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Moreno A, Taffet A, Tjahjono Eet al. A novel Caenorhabditis elegans gene network uncovers mechanisms of mitochondrial maintenance. bioRxiv 2022. 10.1101/2022.04.06.487352 [DOI] [Google Scholar]
- 334.Todd AM, Staveley BE. Pink1 suppresses alpha-synuclein-induced phenotypes in a drosophila model of Parkinson’s disease. Genome 2008;51:1040–6 [DOI] [PubMed] [Google Scholar]
- 335.Todd AM, Staveley BE. Expression of Pink1 with α-synuclein in the dopaminergic neurons of drosophila leads to increases in both lifespan and healthspan. Genet Mol Res 2012;11:1497–502 [DOI] [PubMed] [Google Scholar]
- 336.Liu W, Vives-Bauza C, Acín-Peréz- Ret al. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS One 2009;4:e4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Kamp F, Exner N, Lutz AKet al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 2010;29:3571–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Zhang W, Zhou M, Lu Wet al. CNTNAP4 deficiency in dopaminergic neurons initiates parkinsonian phenotypes. Theranostics 2020;10:3000–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Jinn S, Drolet RE, Cramer PEet al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci U S A 2017;114:2389–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Kara E, Crimi A, Wiedmer Aet al. An integrated genomic approach to dissect the genetic landscape regulating the cell-to-cell transfer of α-synuclein. Cell Rep 2021;35:109189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Vozdek R, Pramstaller PP, Hicks AA. Functional screening of Parkinson’s disease susceptibility genes to identify novel modulators of α-Synuclein neurotoxicity in Caenorhabditis elegans. Front Aging Neurosci 2022;14:806000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Khurana , Peng J, Chung CYet al. Genome-scale networks link neurodegenerative disease genes to α-synuclein through specific molecular pathways. Cell Syst 2017;4:157–170.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Zhong J, Li M, Xu Jet al. Roflupram attenuates α-synuclein-induced cytotoxicity and promotes the mitochondrial translocation of Parkin in SH-SY5Y cells overexpressing A53T mutant α-synuclein. Toxicol Appl Pharmacol 2022;436:115859 [DOI] [PubMed] [Google Scholar]
- 344.Jiang P, Gan M, Ebrahim ASet al. Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of α-synuclein oligomers and decrease of neurites. Neurobiol Aging 2013;34:1504–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Verma DK, Ghosh A, Ruggiero Let al. The SUMO Conjugase Ubc9 protects dopaminergic cells from cytotoxicity and enhances the stability of α-Synuclein in Parkinson’s disease models. eNeuro 2020;7:ENEURO.0134–20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Pino E, Amamoto R, Zheng Let al. FOXO3 determines the accumulation of α-synuclein and controls the fate of dopaminergic neurons in the substantia nigra. Hum Mol Genet 2014;23:1435–52 [DOI] [PubMed] [Google Scholar]
- 347.Delaidelli A, Richner M, Jiang Let al. α-Synuclein pathology in Parkinson disease activates homeostatic NRF2 anti-oxidant response. Acta Neuropathol Commun 2021;9:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Anandhan A, Nguyen N, Syal Aet al. NRF2 loss accentuates parkinsonian pathology and behavioral dysfunction in human α-Synuclein overexpressing mice. Aging Dis 2021;12:964–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Chung CY, Khurana V, Auluck PKet al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 2013;342:983–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Yeger-Lotem E, Riva L, Su LJet al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat Genet 2009;41:316–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Svarcbahs R, Julku UH, Norrbacka Set al. Removal of prolyl oligopeptidase reduces alpha-synuclein toxicity in cells and in vivo. Sci Rep 2018;8:1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Bobela W, Nazeeruddin S, Knott Get al. Modulating the catalytic activity of AMPK has neuroprotective effects against α-synuclein toxicity. Mol Neurodegener 2017;12:80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Narayanan A, Kabir MA. Human ribosomal protein and proteasomal subunit suppress cct mutations and reduce alpha-synuclein toxicity in Saccharomyces cerevisiae. Gene Rep 2021;24:101280 [Google Scholar]
- 354.Sharma SK, Chorell E, Steneberg Pet al. Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci Rep 2015;5:12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Guedes A, Ludovico P, Sampaio-Marques B. Caloric restriction alleviates alpha-synuclein toxicity in aged yeast cells by controlling the opposite roles of Tor1 and Sir2 on autophagy. Mech Ageing Dev 2017;161:270–6 [DOI] [PubMed] [Google Scholar]
- 356.Kim M, Jung W, Lee I-Het al. Impairment of microtubule system increases α-synuclein aggregation and toxicity. Biochem Biophys Res Commun 2008;365:628–35 [DOI] [PubMed] [Google Scholar]
- 357.Flower TR, Clark-Dixon C, Metoyer Cet al. YGR198w (YPP1) targets A30P alpha-synuclein to the vacuole for degradation. J Cell Biol 2007;177:1091–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Usenovic M, Knight AL, Ray Aet al. Identification of novel ATP13A2 interactors and their role in α-synuclein misfolding and toxicity. Hum Mol Genet 2012;21:3785–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Bellani S, Mescola A, Ronzitti Get al. GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ 2014;21:1971–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Hasegawa T, Baba T, Kobayashi Met al. Role of TPPP/p25 on α-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy. Neurochem Int 2010;57:857–66 [DOI] [PubMed] [Google Scholar]
- 361.Cressatti M, Schipper HM. Dysregulation of a heme oxygenase–Synuclein Axis in Parkinson disease. NeuroSci 2022;3:284–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Gómez-Santos C, Barrachina M, Giménez-Xavier Pet al. Induction of C/EBP beta and GADD153 expression by dopamine in human neuroblastoma cells. Relationship with alpha-synuclein increase and cell damage. Brain Res Bull 2005;65:87–95 [DOI] [PubMed] [Google Scholar]
- 363.Tetzlaff JE, Putcha P, Outeiro TFet al. CHIP targets toxic α-Synuclein oligomers for degradation. J Biol Chem 2008;283:17962–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Kalia LV, Kalia SK, Chau Het al. Ubiquitinylation of α-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS One 2011;6:e14695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Vajhøj C, Schmid B, Alik Aet al. Establishment of a human induced pluripotent stem cell neuronal model for identification of modulators of A53T α-synuclein levels and aggregation. PLoS One 2021;16:e0261536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Fernandes HJR, Hartfield EM, Christian HCet al. ER stress and Autophagic perturbations Lead to elevated extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports 2016;6:342–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Zhang Z, Shen Y, Luo Het al. MANF protects dopamine neurons and locomotion defects from a human α-synuclein induced Parkinson’s disease model in C. elegans by regulating ER stress and autophagy pathways. Exp Neurol 2018;308:59–71 [DOI] [PubMed] [Google Scholar]
- 368.Wang S, Zhang S, Liou L-Cet al. Phosphatidylethanolamine deficiency disrupts α-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc Natl Acad Sci U S A 2014;111:E3976–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Dhungel N, Eleuteri S, Li L-Bet al. Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron 2015;85:76–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Smith WW, Jiang H, Pei Zet al. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet 2005;14:3801–11 [DOI] [PubMed] [Google Scholar]
- 371.Watanabe Y, Tatebe H, Taguchi Ket al. p62/SQSTM1-dependent autophagy of Lewy body-like α-synuclein inclusions. PLoS One 2012;7:e52868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Harrington AJ, Yacoubian TA, Slone SRet al. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J Neurosci 2012;32:2142–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Ruan Q, Harrington AJ, Caldwell KAet al. VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson’s disease. Neurobiol Dis 2010;37:330–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Shin Y, Klucken J, Patterson Cet al. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates α-Synuclein degradation decisions between proteasomal and lysosomal pathways*. J Biol Chem 2005;280:23727–34 [DOI] [PubMed] [Google Scholar]
- 375.Wang K, Huang J, Xie Wet al. Beclin1 and HMGB1 ameliorate the α-synuclein-mediated autophagy inhibition in PC12 cells. Diagn Pathol 2016;11:15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Gan L, Vargas MR, Johnson DAet al. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci 2012;32:17775–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Takano M, Tashiro E, Kitamura Aet al. Prefoldin prevents aggregation of α-synuclein. Brain Res 2014;1542:186–94 [PubMed] [Google Scholar]
- 378.Liu J, Wang X, Lu Yet al. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis 2017;8:e3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Arotcarena M-L, Bourdenx M, Dutheil Net al. Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight 2019;4:e129719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Wan T, Weir EJ, Johnson Met al. Increased telomerase improves motor function and alpha-synuclein pathology in a transgenic mouse model of Parkinson’s disease associated with enhanced autophagy. Prog Neurobiol 2021;199:101953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Gupta A, Puri A, Singh Pet al. The yeast stress inducible Ssa Hsp70 reduces α-synuclein toxicity by promoting its degradation through autophagy. PLoS Genet 2018;14:e1007751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Rothaug M, Zunke F, Mazzulli JRet al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci U S A 2014;111:15573–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Tang F-L, Erion JR, Tian Yet al. VPS35 in dopamine neurons is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-Synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J Neurosci 2015;35:10613–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Schultheis PJ, Fleming SM, Clippinger AKet al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet 2013;22:2067–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Usenovic M, Tresse E, Mazzulli JRet al. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci 2012;32:4240–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Si J, Van den Haute C, Lobbestael Eet al. ATP13A2 regulates cellular α-Synuclein Multimerization, membrane association, and externalization. Int J Mol Sci 2021;22:2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Gao H, Sun H, Yan Net al. ATP13A2 declines zinc-induced accumulation of α-Synuclein in a Parkinson’s disease model. Int J Mol Sci 2022;23:8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Bae E-J, Lee H-J, Jang Y-Het al. Phospholipase D1 regulates autophagic flux and clearance of α-synuclein aggregates. Cell Death Differ 2014;21:1132–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Babcock DT, Shen W, Ganetzky B. A neuroprotective function of NSF1 sustains autophagy and lysosomal trafficking in drosophila. Genetics 2015;199:511–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Underwood R, Wang B, Carico Cet al. The GTPase Rab27b regulates the release, autophagic clearance, and toxicity of α-synuclein. J Biol Chem 2020;295:8005–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Dinter E, Saridaki T, Nippold Met al. Rab7 induces clearance of α-synuclein aggregates. J Neurochem 2016;138:758–74 [DOI] [PubMed] [Google Scholar]
- 392.Kim H, Calatayud C, Guha Set al. The small GTPase RAC1/CED-10 is essential in maintaining dopaminergic neuron function and survival against α-Synuclein-induced toxicity. Mol Neurobiol 2018;55:7533–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Kojima R, Zurbruegg M, Li Tet al. Prosaposin reduces α-Synuclein in cells and Saposin C dislodges it from glucosylceramide-enriched lipid membranes. J Mol Neurosci 2022;72:2313–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Bae E-J, Yang NY, Lee Cet al. Haploinsufficiency of cathepsin D leads to lysosomal dysfunction and promotes cell-to-cell transmission of α-synuclein aggregates. Cell Death Dis 2015;6:e1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Qiao L, Hamamichi S, Caldwell KAet al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 2008;1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Guan Y, Li Y, Zhao Get al. HMGB1 promotes the starvation-induced autophagic degradation of α-synuclein in SH-SY5Y cells Atg 5-dependently. Life Sci 2018;202:1–10 [DOI] [PubMed] [Google Scholar]
- 397.Du T-T, Wang L, Duan C-Let al. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015;11:1803–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Bae E-J, Yang N-Y, Song Met al. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun 2014;5:4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Olsen AL, Feany MB. Parkinson’s disease risk genes act in glia to control neuronal α-synuclein toxicity. Neurobiol Dis 2021;159:105482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Bae E-J, Kim D-K, Kim Cet al. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat Commun 2018;9:3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Brás IC, Tenreiro S, Silva AMet al. Identification of novel protein phosphatases as modifiers of alpha-synuclein aggregation in yeast. FEMS Yeast Res 2018;18:foy108 [DOI] [PubMed] [Google Scholar]
- 402.Yang S-Y, Gegg M, Chau Det al. Glucocerebrosidase activity, cathepsin D and monomeric α-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol Dis 2020;134:104620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Hamamichi S, Rivas RN, Knight ALet al. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc Natl Acad Sci U S A 2008;105:728–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Cao Y-L, Yang Y-P, Mao C-Jet al. A role of BAG3 in regulating SNCA/α-synuclein clearance via selective macroautophagy. Neurobiol Aging 2017;60:104–15 [DOI] [PubMed] [Google Scholar]
- 405.Mazzulli JR, Zunke F, Isacson Oet al. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 2016;113:1931–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Davies SE, Hallett PJ, Moens Tet al. Enhanced ubiquitin-dependent degradation by Nedd4 protects against α-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol Dis 2014;64:79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Nicholatos JW, Tran D, Liu Yet al. Lysophosphatidylcholine acyltransferase 1 promotes pathology and toxicity in two distinct cell-based alpha-synuclein models. Neurosci Lett 2022;772:136491 [DOI] [PubMed] [Google Scholar]
- 408.Ou Z, Zhou Y, Wang Let al. NLRP3 Inflammasome inhibition prevents α-Synuclein pathology by relieving autophagy dysfunction in chronic MPTP-treated NLRP3 knockout mice. Mol Neurobiol 2021;58:1303–11 [DOI] [PubMed] [Google Scholar]
- 409.Liu Z, Meray RK, Grammatopoulos TNet al. Membrane-associated farnesylated UCH-L1 promotes α-synuclein neurotoxicity and is a therapeutic target for Parkinson’s disease. Proc Natl Acad Sci 2009;106:4635–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Vazquez-Velez GE, Gonzales KA, Revelli J-Pet al. Doublecortin-like kinase 1 regulates -Synuclein levels and toxicity. Neurobiol Dis 2020;40:459–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Friesen EL, Zhang YT, Earnshaw Ret al. BAG5 promotes alpha-Synuclein oligomer formation and functionally interacts with the autophagy adaptor protein p62. Front Cell Dev Biol 2020;8:716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Rousseaux MWC, Vázquez-Vélez GE, Al-Ramahi Iet al. A Druggable genome screen identifies modifiers of α-Synuclein levels via a tiered cross-species validation approach. J Neurosci 2018;38:9286–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Alexopoulou Z, Lang J, Perrett RMet al. Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc Natl Acad Sci U S A 2016;113:E4688–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Yuan Y-H, Yan W-F, Sun J-Det al. The molecular mechanism of rotenone-induced α-synuclein aggregation: emphasizing the role of the calcium/GSK3β pathway. Toxicol Lett 2015;233:163–71 [DOI] [PubMed] [Google Scholar]
- 415.Skibinski G, Hwang V, Ando DMet al. Nrf2 mitigates LRRK2- and α-synuclein-induced neurodegeneration by modulating proteostasis. Proc Natl Acad Sci U S A 2017;114:1165–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Park H-J, Ryu D, Parmar Met al. The ER retention protein RER1 promotes alpha-synuclein degradation via the proteasome. PLoS One 2017;12:e0184262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Petrucelli L, O’Farrell C, Lockhart PJet al. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 2002;36:1007–19 [DOI] [PubMed] [Google Scholar]
- 418.Haywood AFM, Staveley BE. Parkin counteracts symptoms in a drosophila model of Parkinson’s disease. BMC Neurosci 2004;5:14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Chen AY-Y, Tully T. A stress-enhanced model for discovery of disease-modifying gene: Ece1-suppresses the toxicity of α-synuclein A30P. Neurobiol Dis 2018;114:153–63 [DOI] [PubMed] [Google Scholar]
- 420.Liu Y, Fallon L, Lashuel HAet al. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 2002;111:209–18 [DOI] [PubMed] [Google Scholar]
- 421.Rott R, Szargel R, Shani Vet al. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc Natl Acad Sci U S A 2017;114:13176–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Liu X, Hebron M, Shi Wet al. Ubiquitin specific protease-13 independently regulates parkin ubiquitination and alpha-synuclein clearance in alpha-synucleinopathies. Hum Mol Genet 2019;28:548–60 [DOI] [PubMed] [Google Scholar]
- 423.Dufty BM, Warner LR, Hou STet al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol 2007;170:1725–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Lee JS, Kanai K, Suzuki Met al. Arylsulfatase a, a genetic modifier of Parkinson’s disease, is an α-synuclein chaperone. Brain 2019;142:2845–59 [DOI] [PubMed] [Google Scholar]
- 425.Aprile FA, Källstig E, Limorenko Get al. The molecular chaperones DNAJB6 and Hsp70 cooperate to suppress α-synuclein aggregation. Sci Rep 2017;7:9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Mc Lean PJ, Kawamata H, Shariff Set al. TorsinA and heat shock proteins act as molecular chaperones: suppression of a-synuclein aggregation. J Neurochem 2002;83:846–54 [DOI] [PubMed] [Google Scholar]
- 427.Tao J, Berthet A, Citron YRet al. Hsp70 chaperone blocks α-synuclein oligomer formation via a novel engagement mechanism. J Biol Chem 2021;296:100613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Flower TR, Chesnokova LS, Froelich CAet al. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J Mol Biol 2005;351:1081–100 [DOI] [PubMed] [Google Scholar]
- 429.Klucken J, Shin Y, Masliah Eet al. Hsp70 reduces α-synuclein aggregation and toxicity. J Biol Chem 2004;279:25497–502 [DOI] [PubMed] [Google Scholar]
- 430.Dedmon MM, Christodoulou J, Wilson MRet al. Heat shock protein 70 inhibits α-Synuclein fibril formation via preferential binding to prefibrillar species*. J Biol Chem 2005;280:14733–40 [DOI] [PubMed] [Google Scholar]
- 431.Moloney TC, Hyland R, O’Toole Det al. Heat shock protein 70 reduces α-synuclein-induced predegenerative neuronal dystrophy in the α-synuclein viral gene transfer rat model of Parkinson’s disease. CNS Neurosci Ther 2014;20:50–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Outeiro TF, Putcha P, Tetzlaff JEet al. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One 2008;3:e1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Gao X, Carroni M, Nussbaum-Krammer Cet al. Human Hsp70 Disaggregase reverses Parkinson’s-linked α-Synuclein amyloid fibrils. Mol Cell 2015;59:781–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.De Graeve S, Marinelli S, Stolz Fet al. Mammalian ribosomal and chaperone protein RPS3A counteracts α-synuclein aggregation and toxicity in a yeast model system. Biochem J 2013;455:295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Outeiro TF, Klucken J, Strathearn KEet al. Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem Biophys Res Commun 2006;351:631–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Cox D, Selig E, Griffin MDWet al. Small heat-shock proteins prevent α-Synuclein aggregation via transient interactions and their efficacy is affected by the rate of aggregation *. J Biol Chem 2016;291:22618–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Daturpalli S, Waudby CA, Meehan Set al. Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers. J Mol Biol 2013;425:4614–28 [DOI] [PubMed] [Google Scholar]
- 438.Taguchi YV, Gorenberg EL, Nagy Met al. Hsp110 mitigates α-synuclein pathology in vivo. Proc Natl Acad Sci U S A 2019;116:24310–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Lo Bianco C, Shorter J, Régulier Eet al. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest 2008;118:3087–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Plotegher N, Kumar D, Tessari Iet al. The chaperone-like protein 14-3-3η interacts with human α-synuclein aggregation intermediates rerouting the amyloidogenic pathway and reducing α-synuclein cellular toxicity. Hum Mol Genet 2014;23:5615–29 [DOI] [PubMed] [Google Scholar]
- 441.Underwood R, Gannon M, Pathak Aet al. 14-3-3 mitigates alpha-synuclein aggregation and toxicity in the in vivo preformed fibril model. Acta Neuropathol Commun 2021;9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Wang B, Underwood R, Kamath Aet al. 14-3-3 proteins reduce cell-to-cell transfer and propagation of pathogenic α-synuclein. J Neurosci 2018;38:8211–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Bohush A, Filipek A. HSP90 co-chaperone, CacyBP/SIP, protects α-Synuclein from aggregation. Cell 2020;9:2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Jarvela TS, Lam HA, Helwig Met al. The neural chaperone proSAAS blocks α-synuclein fibrillation and neurotoxicity. Proc Natl Acad Sci U S A 2016;113:E4708–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Danzer KM, Ruf WP, Putcha Pet al. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J 2011;25:326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Liangliang X, Yonghui H, Shunmei Eet al. Dominant-positive HSF1 decreases alpha-synuclein level and alpha-synuclein-induced toxicity. Mol Biol Rep 2010;37:1875–81 [DOI] [PubMed] [Google Scholar]
- 447.Donmez G, Arun A, Chung C-Yet al. SIRT1 protects against α-synuclein aggregation by activating molecular chaperones. J Neurosci 2012;32:124–32 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 448.Du Y, Wang F, Zou Jet al. Histone deacetylase 6 regulates cytotoxic α-synuclein accumulation through induction of the heat shock response. Neurobiol Aging 2014;35:2316–28 [DOI] [PubMed] [Google Scholar]
- 449.Janowska MK, Wu K-P, Baum J. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci Rep 2015;5:15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Hashimoto M, Rockenstein E, Mante Met al. Beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 2001;32:213–23 [DOI] [PubMed] [Google Scholar]
- 451.Park J-Y, Lansbury PT Jr. Beta-synuclein inhibits formation of alpha-synuclein protofibrils: a possible therapeutic strategy against Parkinson’s disease. Biochemistry 2003;42:3696–700 [DOI] [PubMed] [Google Scholar]
- 452.Uversky VN, Li J, Souillac Pet al. Biophysical properties of the Synucleins and their propensities to fibrillate: Inhibition of α-synuclein assembly by β- and γ-synucleins. J Biol Chem 2002;277:11970–8 [DOI] [PubMed] [Google Scholar]
- 453.Williams JK, Yang X, Atieh TBet al. Multi-pronged interactions underlie inhibition of α-Synuclein aggregation by β-Synuclein. J Mol Biol 2018;430:2360–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Du G, Liu X, Chen Xet al. Drosophila histone deacetylase 6 protects dopaminergic neurons against α-synuclein toxicity by promoting inclusion formation. Mol Biol Cell 2010;21:2128–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Shen L, Wang C, Chen Let al. TDP-1/TDP-43 potentiates human α-Synuclein (HASN) neurodegeneration in Caenorhabditis elegans. Biochim Biophys Acta Mol basis Dis 2020;1866:165876 [DOI] [PubMed] [Google Scholar]
- 456.Dhakal S, Wyant CE, George HEet al. Prion-like C-terminal domain of TDP-43 and α-Synuclein interact synergistically to generate neurotoxic hybrid fibrils. J Mol Biol 2021;433:166953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Popova B, Wang D, Pätz Cet al. DEAD-box RNA helicase Dbp4/DDX10 is an enhancer of α-synuclein toxicity and oligomerization. PLoS Genet 2021;17:e1009407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Giasson BI, Forman MS, Higuchi Met al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003;300:636–40 [DOI] [PubMed] [Google Scholar]
- 459.Badiola N, de Oliveira RM, Herrera Fet al. Tau enhances α-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One 2011;6:e26609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Pan L, Li C, Meng Let al. Tau accelerates α-synuclein aggregation and spreading in Parkinson’s disease. Brain 2022;145:3454–71 [DOI] [PubMed] [Google Scholar]
- 461.Junn E, Ronchetti RD, Quezado MMet al. Tissue transglutaminase-induced aggregation of alpha-synuclein: implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 2003;100:2047–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Büttner S, Delay C, Franssens Vet al. Synphilin-1 enhances α-synuclein aggregation in yeast and contributes to cellular stress and cell death in a Sir2-dependent manner. PLoS One 2010;5:e13700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Smith WW, Margolis RL, Li Xet al. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci 2005;25:5544–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Xie Y-Y, Zhou C-J, Zhou Z-Ret al. Interaction with synphilin-1 promotes inclusion formation of alpha-synuclein: mechanistic insights and pathological implication. FASEB J 2010;24:196–205 [DOI] [PubMed] [Google Scholar]
- 465.Savolainen MH, Yan X, Myöhänen TTet al. Prolyl Oligopeptidase enhances α-Synuclein dimerization via direct protein-protein interaction*. J Biol Chem 2015;290:5117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Kam T-I, Mao X, Park Het al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018;362:eaat8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Mund T, Masuda-Suzukake M, Goedert Met al. Ubiquitination of alpha-synuclein filaments by Nedd4 ligases. PLoS One 2018;13:e0200763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Ihara M, Yamasaki N, Hagiwara Aet al. Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of alpha-synuclein neurotoxicity. Neuron 2007;53:519–33 [DOI] [PubMed] [Google Scholar]
- 469.Khandelwal PJ, Dumanis SB, Feng LRet al. Parkinson-related parkin reduces α-Synuclein phosphorylation in a gene transfer model. Mol Neurodegener 2010;5:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Sancenon V, Lee S-A, Patrick Cet al. Suppression of α-synuclein toxicity and vesicle trafficking defects by phosphorylation at S129 in yeast depends on genetic context. Hum Mol Genet 2012;21:2432–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Oh Y, Kim YM, Mouradian MMet al. Human Polycomb protein 2 promotes α-synuclein aggregate formation through covalent SUMOylation. Brain Res 2011;1381:78–89 [DOI] [PubMed] [Google Scholar]
- 472.Zucchelli S, Codrich M, Marcuzzi Fet al. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum Mol Genet 2010;19:3759–70 [DOI] [PubMed] [Google Scholar]
- 473.Nakamura T, Yamashita H, Takahashi Tet al. Activated Fyn phosphorylates alpha-synuclein at tyrosine residue 125. Biochem Biophys Res Commun 2001;280:1085–92 [DOI] [PubMed] [Google Scholar]
- 474.Ellis CE, Schwartzberg PL, Grider TLet al. Alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J Biol Chem 2001;276:3879–84 [DOI] [PubMed] [Google Scholar]
- 475.Okochi M, Walter J, Koyama Aet al. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J Biol Chem 2000;275:390–7 [DOI] [PubMed] [Google Scholar]
- 476.Ishii A, Nonaka T, Taniguchi Set al. Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human alpha-synuclein: implication for alpha-synucleinopathies. FEBS Lett 2007;581:4711–7 [DOI] [PubMed] [Google Scholar]
- 477.Waxman EA, Giasson BI. Specificity and regulation of casein kinase-mediated phosphorylation of α-Synuclein. J Neuropathol Exp Neurol 2008;67:402–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Wakamatsu M, Ishii A, Ukai Yet al. Accumulation of phosphorylated alpha-synuclein in dopaminergic neurons of transgenic mice that express human alpha-synuclein. J Neurosci Res 2007;85:1819–25 [DOI] [PubMed] [Google Scholar]
- 479.Pronin AN, Morris AJ, Surguchov Aet al. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem 2000;275:26515–22 [DOI] [PubMed] [Google Scholar]
- 480.Chen L, Feany MB. α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a drosophila model of Parkinson disease. Nat Neurosci 2005;8:657–63 [DOI] [PubMed] [Google Scholar]
- 481.Qing H, Wong W, McGeer EGet al. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem Biophys Res Commun 2009;387:149–52 [DOI] [PubMed] [Google Scholar]
- 482.Waxman EA, Giasson BI. Characterization of kinases involved in the phosphorylation of aggregated α-synuclein. J Neurosci Res 2011;89:231–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Basso E, Antas P, Marijanovic Zet al. PLK2 modulates α-synuclein aggregation in yeast and mammalian cells. Mol Neurobiol 2013;48:854–62 [DOI] [PubMed] [Google Scholar]
- 484.Mbefo MK, Paleologou KE, Boucharaba Aet al. Phosphorylation of synucleins by members of the polo-like kinase family. J Biol Chem 2010;285:2807–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Kim EJ, Sung JY, Lee HJet al. Dyrk1A phosphorylates α-Synuclein and enhances intracellular inclusion formation. J Biol Chem 2006;281:33250–7 [DOI] [PubMed] [Google Scholar]
- 486.Parmasad J-LA, Ricke KM, Stykel MGet al. Genetic and pharmacological reduction of CDK14 mitigates synucleinopathy. bioRxiv 2022. 10.1101/2022.05.02.490309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Thayanidhi N, Helm JR, Nycz DCet al. α-Synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell 2010;21:1850–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Gonçalves SA, Macedo D, Raquel Het al. shRNA-based screen identifies endocytic recycling pathway components that act as genetic modifiers of alpha-Synuclein aggregation, secretion and toxicity. PLoS Genet 2016;12:e1005995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Dumitriu A, Pacheco CD, Wilk JBet al. Cyclin-G-associated kinase modifies α-synuclein expression levels and toxicity in Parkinson’s disease: results from the GenePD study. Hum Mol Genet 2011;20:1478–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Song L, He Y, Ou Jet al. Auxilin underlies progressive locomotor deficits and dopaminergic neuron loss in a drosophila model of Parkinson’s disease. Cell Rep 2017;18:1132–43 [DOI] [PubMed] [Google Scholar]
- 491.Zhu J, Pittman S, Dhavale Det al. VCP suppresses proteopathic seeding in neurons. Mol Neurodegener 2022;17:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Zhao J, Li Y, Li Yet al. Activation of α7-nAChRs promotes the clearance of α-Synuclein and protects against apoptotic cell death induced by exogenous α-Synuclein fibrils. Front Cell Dev Biol 2021;9:637319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Albert K, Raymundo DP, Panhelainen Aet al. Cerebral dopamine neurotrophic factor reduces α-synuclein aggregation and propagation and alleviates behavioral alterations in vivo. Mol Ther 2021;29:2821–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Papadopoulos VE, Nikolopoulou G, Antoniadou Iet al. Modulation of β-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum Mol Genet 2018;27:1696–710 [DOI] [PubMed] [Google Scholar]
- 495.Chutna O, Gonçalves S, Villar-Piqué Aet al. The small GTPase Rab11 co-localizes with α-synuclein in intracellular inclusions and modulates its aggregation, secretion and toxicity. Hum Mol Genet 2014;23:6732–45 [DOI] [PubMed] [Google Scholar]
- 496.Liu J, Zhang J-P, Shi Met al. Rab11a and HSP90 regulate recycling of extracellular alpha-synuclein. J Neurosci 2009;29:1480–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Davis AA, Inman CE, Wargel ZMet al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med 2020;12:eaay3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Jin Y, Li F, Sonoustoun Bet al. APOE4 exacerbates α-synuclein seeding activity and contributes to neurotoxicity in Alzheimer’s disease with Lewy body pathology. Acta Neuropathol 2022;143:641–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Urrea L, Segura-Feliu M, Masuda-Suzukake Met al. Involvement of cellular prion protein in α-Synuclein transport in neurons. Mol Neurobiol 2018;55:1847–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Chen K, Martens YA, Meneses Aet al. LRP1 is a neuronal receptor for α-synuclein uptake and spread. Mol Neurodegener 2022;17:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Zhang Q, Xu Y, Lee Jet al. A myosin-7B-dependent endocytosis pathway mediates cellular entry of α-synuclein fibrils and polycation-bearing cargos. Proc Natl Acad Sci U S A 2020;117:10865–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Sung JY, Kim J, Paik SRet al. Induction of neuronal cell death by Rab5A-dependent endocytosis of α-Synuclein*. J Biol Chem 2001;276:27441–8 [DOI] [PubMed] [Google Scholar]
- 503.Nakamura Y, Arawaka S, Sato Het al. Monoamine oxidase-B inhibition facilitates α-Synuclein secretion in vitro and delays its aggregation in rAAV-based rat models of Parkinson’s disease. J Neurosci 2021;41:7479–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Breda C, Nugent ML, Estranero JGet al. Rab11 modulates α-synuclein-mediated defects in synaptic transmission and behaviour. Hum Mol Genet 2015;24:1077–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Cao S, Gelwix CC, Caldwell KAet al. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci 2005;25:3801–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Moszczynska A, Saleh J, Zhang Het al. Parkin disrupts the alpha-synuclein/dopamine transporter interaction: consequences toward dopamine-induced toxicity. J Mol Neurosci 2007;32:217–27 [DOI] [PubMed] [Google Scholar]
- 507.Faustini G, Longhena F, Varanita Tet al. Synapsin III deficiency hampers α-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson’s disease. Acta Neuropathol 2018;136:621–39 [DOI] [PubMed] [Google Scholar]
- 508.Ferreira DG, Batalha VL, Vicente Miranda Het al. Adenosine A2A receptors modulate α-Synuclein aggregation and toxicity. Cereb Cortex 2017;27:718–30 [DOI] [PubMed] [Google Scholar]
- 509.Ferreira DG, Temido-Ferreira M, Vicente Miranda Het al. α-Synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci 2017;20:1569–79 [DOI] [PubMed] [Google Scholar]
- 510.Gaeta AL, Nourse JB Jr, Willicott Ket al. Systemic RNA interference defective (SID) genes modulate dopaminergic neurodegeneration in C. elegans. PLoS Genet 2022;18:e1010115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Daher JPL, Volpicelli-Daley LA, Blackburn JPet al. Abrogation of α-synuclein–mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci 2014;111:9289–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Caraveo G, Soste M, Cappelleti Vet al. FKBP12 contributes to α-synuclein toxicity by regulating the calcineurin-dependent phosphoproteome. Proc Natl Acad Sci U S A 2017;114:E11313–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Sampaio-Marques B, Guedes A, Vasilevskiy Iet al. α-Synuclein toxicity in yeast and human cells is caused by cell cycle re-entry and autophagy degradation of ribonucleotide reductase 1. Aging Cell 2019;18:e12922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Zourlidou A, Payne Smith MD, Latchman DS. HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem 2004;88:1439–48 [DOI] [PubMed] [Google Scholar]
- 515.Chen RHC, Wislet-Gendebien S, Samuel Fet al. α-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J Biol Chem 2013;288:7438–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Decressac M, Kadkhodaei B, Mattsson Bet al. α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med 2012;4:163ra156 [DOI] [PubMed] [Google Scholar]
- 517.Volakakis N, Tiklova K, Decressac Met al. Nurr1 and retinoid X receptor ligands stimulate ret signaling in dopamine neurons and can alleviate α-Synuclein disrupted gene expression. J Neurosci 2015;35:14370–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Lewandowski NM, Ju S, Verbitsky Met al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc Natl Acad Sci U S A 2010;107:16970–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Bohush A, Góral A, Sierant Met al. Sgt1 regulates α-Synuclein subcellular localization and expression of Parkinson’s disease related genes, PINK1 and PARK9. Biomol Ther 2021;11:1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Tayebi N, Parisiadou L, Berhe Bet al. Glucocerebrosidase haploinsufficiency in A53T α-synuclein mice impacts disease onset and course. Mol Genet Metab 2017;122:198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Jansen IE, Ye H, Heetveld Set al. Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol 2017;18:22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Oluwatosin-Chigbu Y, Robbins A, Scott CWet al. Parkin suppresses wild-type alpha-synuclein-induced toxicity in SHSY-5Y cells. Biochem Biophys Res Commun 2003;309:679–84 [DOI] [PubMed] [Google Scholar]
- 523.Yang Y, Nishimura I, Imai Yet al. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in drosophila. Neuron 2003;37:911–24 [DOI] [PubMed] [Google Scholar]
- 524.Rousseaux MW, de Haro M, Lasagna-Reeves CAet al. TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. elife 2016;5:e19809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Wilson GR, Sim JCH, McLean Cet al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am J Hum Genet 2014;95:729–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Fournier M, Vitte J, Garrigue Jet al. Parkin deficiency delays motor decline and disease manifestation in a mouse model of synucleinopathy. PLoS One 2009;4:e6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Zabrocki P, Pellens K, Vanhelmont Tet al. Characterization of a-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J 2005;272:1386–400 [DOI] [PubMed] [Google Scholar]
- 528.Batelli S, Invernizzi RW, Negro Aet al. The Parkinson’s disease-related protein DJ-1 protects dopaminergic neurons in vivo and cultured cells from alpha-synuclein and 6-hydroxydopamine toxicity. Neurodegener Dis 2015;15:13–23 [DOI] [PubMed] [Google Scholar]
- 529.Tian T, Huang C, Tong Jet al. TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int J Biol Sci 2011;7:234–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 2006;15:3012–23 [DOI] [PubMed] [Google Scholar]
- 531.van Ham TJ, Holmberg MA, van der Goot ATet al. Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 2010;142:601–12 [DOI] [PubMed] [Google Scholar]
- 532.Smit JW, Basile P, Prato MKet al. Phase 1/1b studies of UCB0599, an Oral inhibitor of α-Synuclein Misfolding, including a randomized study in Parkinson’s disease. Mov Disord 2022;37:2045–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Levin J, Sing N, Melbourne Set al. Safety, tolerability and pharmacokinetics of the oligomer modulator anle138b with exposure levels sufficient for therapeutic efficacy in a murine Parkinson model: a randomised, double-blind, placebo-controlled phase 1a trial. EBioMedicine 2022;80:104021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Pagan FL, Hebron ML, Wilmarth Bet al. Nilotinib effects on safety, tolerability, and potential biomarkers in Parkinson disease: a phase 2 randomized clinical trial. JAMA Neurol 2020;77:309–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Simuni T, Fiske B, Merchant Ket al. Efficacy of Nilotinib in patients with moderately advanced Parkinson disease: a randomized clinical trial. JAMA Neurol 2021;78:312–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Parkinson Study Group . Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson’s disease (STEADY-PD). Mov Disord 2013;28:1823–31 [DOI] [PubMed] [Google Scholar]
- 537.Brakedal B, Dölle C, Riemer Fet al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab 2022;34:396–407.e6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data mentioned in this article has been published elsewhere as referenced.