

Abstract
The chimeric antigen receptor (CAR) T-cell therapy in solid epithelial tumors has been explored, however, with limited success. As much of the preclinical work has relied on xenograft models in immunocompromised animals, the immune-related efficacies and toxicities may have been missed. In this study, we engineered syngeneic murine CAR T cells targeting the tumor form of human mucin-1 (tMUC1) and tested the MUC1 CAR T cells' efficacy and toxicity in the immunocompetent human MUC1-expressing mouse models. The MUC1 CAR T cells significantly eliminated murine pancreatic and breast cancer cell lines in vitro. In vivo, MUC1 CAR T cells significantly slowed the mammary gland tumor progression in the spontaneous PyVMT×MUC1.Tg (MMT) mice, prevented lung metastasis, and prolonged survival. Most importantly, there was minimal short or long-term toxicity with acceptable levels of transient liver toxicity but no kidney toxicity. In addition, the mice did not show any signs of weight loss or other behavioral changes with the treatment. We also report that a single dose of MUC1 CAR T-cell treatment modestly reduced the pancreatic tumor burden in a syngeneic orthotopic model of pancreatic ductal adenocarcinoma given at late stage of an established tumor. Taken together, these findings suggested the further development of tMUC1-targeted CAR T cells as an effective and relatively safe treatment modality for various tMUC1-expressing solid tumors.
Key Words: MUC1, murine CAR T cells, immunotherapy, immunocompetent, toxicity
The success of chimeric antigen receptor (CAR) T-cell therapy achieved in hematological malignancies still has not been replicated in solid tumors.1–4 Whereas, epithelial malignancies account for 80% of all death from cancer. Breast cancer alone is expected to account for 31% of female cancers in the United States in 2023.5 Even though the 5-year relative survival rate for all stages combined with female breast cancer is as high as 90%, finding the cure for metastatic triple-negative breast cancer (TNBC) still remains challenging.6 Pancreatic cance is currently the third leading cause of cancer death in the United States and is projected to rise to second by 2040.7 The 5-year relative survival rate for patients with pancreatic ductal adenocarcinoma (PDA), which comprises over 95% of all pancreatic cancers, is only 11%.8 Thus, CAR T-cell therapy as a new treatment paradigm for solid tumors remains exciting but also highly challenging.
To improve the efficacy and safety of CAR T-cell therapy, it is essential to identify the appropriate and specific tumor antigens. Mucin 1 (MUC1) is a heavily glycosylated transmembrane mucin protein that is normally expressed on the luminal surface of all glandular epithelial cells that form our major organs. Under oncogenesis, MUC1 is overexpressed and aberrantly glycosylated over the entire cell surface of most adenocarcinomas.9 This tumor-associated MUC1 [tumor form of human mucin 1 (tMUC1)] has been predicted to be the second most targetable antigen for the development of targeted immunotherapy.10 Our group has reported on a novel monoclonal antibody TAB004 which only detects tMUC1 with high specificity and spares recognition of normal tissue MUC1.11,12 We used single-chain variable fragment (scFv) from TAB004 and engineered human MUC1 CAR T cells that were highly effective against human TNBC and PDA in an immune-compromised NOD scid gamma (NSG) mouse model.6,13
In the current study, an anti-human MUC1 murine CAR fusion molecule was engineered, structurally similar to the human MUC1 CAR. We generated murine MUC1 CAR T cells and evaluated their tumor lysis efficacy, as well as their potential on-target, off-tumor toxicities in immunocompetent human MUC1.Tg mouse models. The xenograft NSG mouse models have been the most widely used preclinical platform to develop and evaluate human CAR T cells against human cancers.14 However, it is critical to comprehensively evaluate the antitumor activities and adverse effects of CAR T cells, their interaction with host immunity, and their activity within the tumor microenvironment when recipient mice have replete immune systems. There were very few reports of murine CAR T cells using immunocompetent animals, even then, most of those murine CAR T cells targeted mouse tumor antigens.15–19 Here, we utilized 2 immunocompetent human MUC1-expressing tumor mouse models: a syngeneic orthotopic PDA model (KCM cells derived from spontaneous KC mice crossed with human MUC1.Tg mice) and a spontaneous transgenic MMT breast cancer model Polyoma Virus middle T antigen (PyVMT) mice crossed with human MUC1.Tg mice). The PyVMT single transgenic mice had been used as a model to study TNBC.20,21 The molecular and histologic progression in MMT mice was similar to PyVMT mice and highly resembled those in human breast tumors.22,23 In addition, preclinical relevant animal models that express human targets in normal tissues are required by FDA for cellular therapies to reasonably predict human response and off-tumor toxicity.24,25 Both of our mouse models express the full-length human MUC1 in a pattern and level consistent with that observed in humans.26 Thus, our tMUC1-targeting CAR T cells would be investigated in the presence of endogenous human MUC1 both on tumors and on healthy tissues.
Taken together, as an advancement to human MUC1 CAR T-cell study in NSG models as we previously reported,6,13 the murine MUC1 CAR T cells demonstrated significant tumor-killing efficacy in TNBC in vitro and in vivo, with great toxicity tolerance in the presence of intact host immunity. We observed high cytotoxicity against PDA cells in vitro and modest response against late-stage orthotopic PDA in vivo.
MATERIALS AND METHODS
Ethical Approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution and approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Charlotte.
Reagents
The Dulbecco’s Modified Eagle Medium (DMEM) was supplemented with 10% fetal bovine serum (R&D Systems), 1% glutamax, 1% nonessential amino acids, and 1% penicillin/streptomycin as a complete medium. G418 was used for maintaining Neo and MUC1 transfected cell lines. The RPMI1640 was supplemented with 10% heat-inactivated fetal bovine serum, 1% glutamax, 100 mM sodium pyruvate, 50 mM 2-mercaptoethanol, and 1% penicillin/streptomycin. All the medium and supplements were obtained from Thermo Fisher Scientific if not separately mentioned. The human MUC1 core peptide (24mer, TAPPAHGVTSAPDTRPAPGSTAPP)23 was synthesized and biotin was added to the N-term, whereas C-term was left with free acid (AnaSpec, Inc.).
Cell Lines and Mice
The mouse pancreas cell lines were: Panc02.Neo and Panc02.MUC1 (a generous gift from Dr Sandra J Gendler, Mayo Clinic in Arizona), KCKO (MUC1-null), and KCM (expressing full-length human MUC1).27,28 The mouse breast and mammary gland cell lines were: Mtag and Mtag.MUC1, C57mg and C57mg.MUC1.28 Mtag cell line was originally derived from primary tumors spontaneously arising in PyVMT female mice. All MUC1-expressing cell lines had full-length human MUC1. All tumor cell lines were maintained in complete DMEM. Transfected cell lines were maintained in complete DMEM and supplemented with G418 at 150 µg/mL.
The retroviral vector packaging cell Phoenix-Eco was purchased from the American Type Culture Collection (ATCC) and cultured in complete DMEM media.
C57BL/6 mice (8–12 wk old) were purchased from Jackson Laboratory. MUC1.Tg mice26 were in-house bred. Female MUC1.Tg mice were bred with male PyVMT (mouse mammary tumor virus-Polyoma Middle T, Jackson Laboratory)22 to generate human MUC1×PyVMT double transgenic mice, designated as MMT.23 The female MMT mice spontaneously developed multifocal mammary gland tumors with lung and bone metastasis in the presence of human MUC1 which resembles the human disease.23 Most female MMT mice developed palpable tumors by 10–12 weeks of age. All mice were on C57BL/6 background and fully immunocompetent.
Construction of Murine Chimeric Antigen Receptor and Retroviral Vector Production
The second-generation of human tMUC1-specific murine CAR construct was synthesized by subcloning the scFv from TAB004 into the SFG-based retroviral backbone plasmid encoding the transmembrane and intracellular domains of CD28 and the intracellular domain of CD3ζ (kindly provided by Dr John Maher group6). The monoclonal antibody TAB004 was developed against the tumor form of human MUC1.11 Therefore, this TAB004-derived murine CAR was specific for mouse tumor cells that expressed human MUC1.
To produce murine CAR-encoding retroviral supernatants, Phoenix-Eco cells were transfected with the SFG-retroviral plasmid using the calcium phosphate transfection kit following the manufacturer’s instruction (Clontech). Virus supernatants were collected 72 hours after initial transfection, filtered, and stored at −80°C.
Primary Mouse T-Cell Enrichment, CD8+ T-Cell Isolation, and Chimeric Antigen Receptor T-Cell Transduction
Mouse T cells from naïve C57BL/6 mice or MUC1.Tg mice were enriched from pooled spleens using a T-cell enrichment column (R&D systems, Minneapolis, MN). CD8+ T cells were further isolated using beads-based negative selection (Miltenyi Biotec). Enriched T cells or isolated CD8+ T cells (1×106 cells/mL) were activated with anti-mouse CD3/CD28 beads (Thermo Fisher Scientific). On day 2, non–tissue culture–treated 6-well plates were coated with Retronectin (Takara) at 4°C overnight. On day 3, retronectin-coated plates were blocked with 2% bovine serum albumin. Then retroviral supernatants or medium control were added into blocked plates. The T-cell transduction process was referred to by Ma et al.29 The transduction efficiency was determined by its binding to biotinylated human MUC1 peptide. Cells were passaged and expanded as needed and used 3–5 days after initial viral transduction (with ~35%–40% CAR positivity). Activated but nonretroviral transduced cells were included as mock T-cell controls. For in vitro functional assays, MUC1 CAR T cells were generated from enriched T cells. For in vivo mouse tumor treatment, MUC1 CAR T cells were generated from isolated CD8+ T cells in the main text.
Chimeric Antigen Receptor T-Cell Functional Assays
The antigen-specific tumor cell lysis by MUC1 CAR T cells was determined by MTT assay. Briefly, mouse PDA and breast tumor cell lines were plated in 96-well plates overnight. The next day, culture media from the plate was aspirated and MUC1 CAR T cells were added at the indicated effector to target (E:T) ratio for another 24 hours and 48 hours. In the end, T cells were removed and replaced with fresh media containing MTT (500 μg/mL, Sigma) for 3 hours. After the uptake of MTT, supernatants from wells were aspirated, and 150 μL of dimethyl sulfoxide was added to wells to dissolve the formazan crystals. The optical density (OD) value was read at 540 nm. We used the mock T-cell lysis data for calculating percentage lysis. The calculation formula was: (OD of coculture with mock-T−OD of coculture with CAR-T)/OD of coculture with mock-T×100. The antigen-specific cytokine release was performed by coculturing MUC1 CAR T cells with mouse PDA and breast tumor cells at indicated E:T ratios for 24 hours. Supernatants were then collected. The mouse IFN-γ, IL-2, and granzyme B concentrations were determined using Duoset Mouse Cytokine Detection Kits (R&D Systems).
Flow Cytometry
The transduction efficiency of MUC1 CAR in mouse T cells was determined by biotinylated MUC1 core peptide binding, followed by streptavidin-PE staining (BD Biosciences). T-cell phenotypes were determined by staining for CD4-PE/Cy7, CD8-Pacific Blue, CD45-FITC, CD107a-Alexa Fluor 647, and programmed cell death protein 1 (PD-1)-APC (BioLegend). The tMUC1 expression on mouse tumor cells was assessed by TAB004 staining (provided by OncoTab Inc.) conjugated with HiLyte Fluor 647 (Dojindo Molecular Technologies, Inc.), named as TAB004-Fluor 647. To detect tMUC1 on primary tumor cells, spontaneous MMT tumors were resected and digested with Collagenase IV (Life Technologies) plus DNase I (STEMCELL Technologies) to create single-cell suspensions. Cell mixtures were cultured for 2 days and the floating cells were removed. Adherent cells were harvested for staining with TAB004-Fluor 647 (BioLegend). Dead cells were excluded by 7-AAD staining (BD Biosciences). Data were acquired on BD LSRFortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software (v10).
Syngeneic Orthotopic Pancreatic Cancer Mouse Model and Analysis
The pancreas surgical injection with 2×104 KCM-Luciferase (KCM-Luc) tumor cells was performed using immunocompetent male or female MUC1.Tg mice as detailed in.13 Seven days after the surgery, tumor establishment was confirmed using In Vivo Imaging System (PerkinElmer). On day 8, mice were randomly assigned to experimental groups based on their baseline luminescence intensity. On the same day, mice were intravenously injected with 1×107 transduced MUC1 CAR CD8+ T cells (in 200 μL of phosphate-buffered saline). Control mice received mock CD8+ T cells. Tumor growth was monitored by weekly IVIS imaging. Tumor and spleen wet weights were measured at the endpoint.
Spontaneous Mammary Tumor Mouse Model and Analysis
In this efficacy/survival study, immunocompetent spontaneous female MMT mice at the age of ~10–14 weeks old were randomly assigned into 2 groups based on their age and tumor appearance. One day before T-cell adoptive transfer, all MMT mice received cyclophosphamide (CTX, Selleck Chemicals) at 1.5 mg per mouse through intraperitoneal injection. The next day, one group of mice intravenously received 1×107 MUC1 CAR CD8+ T cells, and the other group received mock CD8+ T cells. Three additional intravenous (i.v.) injections of CAR T cells or mock T cells were administered at 2-week intervals. Tumor burden was determined by Caliper measurement and calculated by the formula: tumor volume (mm3)=[length in mm×(width in mm)2]/2. As MMT mammary tumors arose at multiple sites, the final presented tumor volume was the sum of multiple tumors of each individual. Mice were sacrificed at a humane endpoint when at least one of its tumors reached 20 mm in diameter for individual MMT mice. No mouse death was noticed due to T-cell transfer or heavy tumor burdens. The survival curve was plotted according to the humane endpoint.
Immunohistochemistry (IHC) Staining
Tissues were fixed in 10% neutral-buffered formalin. Paraffin-embedded blocks were prepared and 5 μm-thick sections were cut by Pathology Services Core at the University of North Carolina-Chapel Hill (Chapel Hill, NC). Tumor and lung tissues were stained with hematoxylin and eosin (H&E). IHC was performed as described previously.12,30 The antibodies used were as follows: TAB004 (OncoTAB), rabbit anti-mouse CD8α antibody (#98941, Cell Signaling Technology), and HRP-conjugated secondary antibodies (Abcam).
Liver and Renal Toxicity Evaluation
The female MMT mice were grouped and treated as described in the previous spontaneous MMT model. Sera were collected 6 days after the first dose of T-cell i.v. injection (early/acute toxicity) or at the humane endpoint after 4 doses of T-cell injection (late toxicity from above), and stored at −80°C. Sera were also collected from healthy MUC1.Tg mice (~20 wk of age) as normal controls. The serum liver panel (test code: 60405) and renal panel (test code: 60406) were analyzed by IDEXX BioAnalytics.
Clariom S Array
The female MMT mice were treated as described for an early toxicity test. Briefly, 6 days after the first dose of T-cell i.v. injection, tumors were resected from individual mice, and a small fraction was immersed in RNAlater (Thermo Fisher Scientific) and stored at −80°C. To perform gene-level expression profiling, total RNA was extracted and quality-checked. Clariom S Assay Kit (Mouse) was purchased from Thermo Fisher Scientific and the array was performed by Functional Genomics Core at University of North Carolina-Chapel Hill.
Statistical Analysis
Data were analyzed using Prism (version 10; GraphPad Software) and results were presented as mean ± SD or mean ± SEM where indicated. Data were representative of 2 or more independent experiments.
RESULTS
Generation of Murine Mucin 1 Chimeric Antigen Receptor T Cells
We constructed the murine CAR that specifically recognized human tMUC1. A schematic representation of CAR is shown in Figure 1A. MUC1 CAR incorporated the scFv motif derived from TAB004 with the murine CD28/CD3ζ signaling domains, which was closely identical to the human MUC1 CAR except lacking Myc-tag.6 The CAR expression on mouse T cells was determined by its binding to the synthesized peptide from the human MUC1 core. The peptide was tagged with biotin to its N-term (MUC1-biotin); therefore, its capture by CAR was evidenced through secondary binding with PE-conjugated streptavidin. We labeled CAR expression as CAR-PE. The enriched T cells or isolated CD8+ T cells from naïve mice were activated for 3 days and then transduced with MUC1 CAR retrovirus supernatant. Four days after virus transduction, there was 47.8% CAR expression on gated CD8+ T cells (Fig. 1B) and 28.2% CAR expression on gated CD4+ T cells (Supplemental Fig.1, Supplemental Digital Content 1, http://links.lww.com/JIT/A813) when generated from enriched T cells. Within the transduced T-cell population, CD4+ T cells accounted for ~8%–20% (data not shown). There was 38.1% CAR expression on gated CD8+ T cells when generated from isolated CD8+ T cells (Fig. 1C).
FIGURE 1.
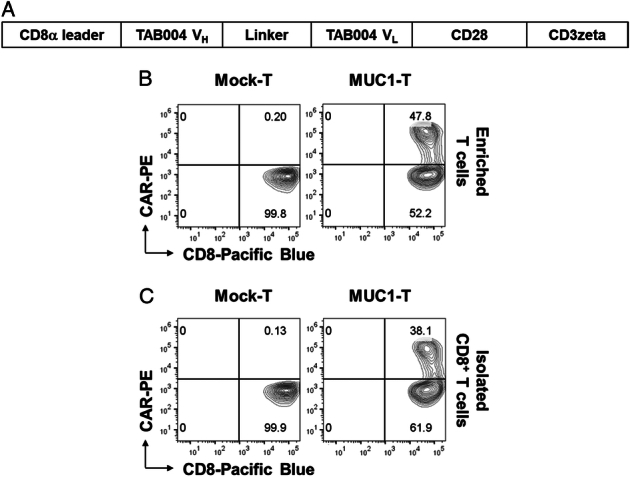
MUC1 CAR expression in transduced murine T cells. A, Schematic of murine MUC1 CAR construct. TAB004 antibody derived ScFv was incorporated in CAR construct for targeting tumor form of human MUC1 (human tMUC1). B and C, MUC1 CAR expression on T cells. Mouse T cells were activated with anti-mouse CD3/CD28 beads for 3 days, followed by overnight CAR transduction with retrovirus supernatant. CAR expression was detected 4 days after transduction. B, Mouse T cells were enriched from naïve mouse spleens. C, Mouse CD8+ T cells were isolated from naïve mouse spleens. The CAR expression was determined by flow cytometry analysis of biotinylated-MUC1 core peptide binding with transduced T cells. Cells were gated on the CD8+ population. Dead cells were excluded by 7-AAD staining. CAR indicates chimeric antigen receptor; MUC1, mucin 1; tMUCI, tumor form of mucin 1.
Mucin 1 Chimeric Antigen Receptor T Cells Recognize Tumor Form of Human Mucin-1 and Lyse Tumor Cells In Vitro
We first determined the percentages of human tMUC1-positive cells in murine pancreatic and breast cancer cells. Flow cytometry data depicted in Figure 2A, shows high specific expression (>90% of the cells) of tMUC1 in KCM, Panc02.MUC1, Mtag.MUC1, and C57mg.MUC1 cell lines while their counterpart control cells were negative for tMUC1. MUC1 CAR T cells generated from enriched T cells were used for the following in vitro studies. The 8 cell lines were cocultured with CAR T cells or mock T cells at the indicated E:T ratios for 24 hours and 48 hours. A CAR T-cell dose-dependent cytotoxicity was observed in all tMUC1-expressing tumor cell lines (Fig. 2B). Significant lysis was observed at E:T ratio of 2:1 in 24-hour coculture and even at E:T ratio as low as 1:1 in the 48-hour coculture (Fig. 2B). Importantly, minimal tumor cell death was observed in tMUC1-negative cell lines when coculture with the same CAR T cells, suggesting the high tumor antigen specificity of MUC1 CAR T cells. All lysis data presented here was normalized to its own mock T-cell lysis. A nearly identical tumor cytotoxicity was found in MUC1 CAR T cells generated using isolated CD8+ T cells (Supplemental Fig. 2, Supplemental Digital Content 1, http://links.lww.com/JIT/A813). Besides tumor cell lysis, the engagement of CAR T cells with tMUC1-expressing tumor cells led to the IFN-γ (Fig. 3A) and IL-2 (Fig. 3B) production in an E:T ratio-dependent manner.
FIGURE 2.
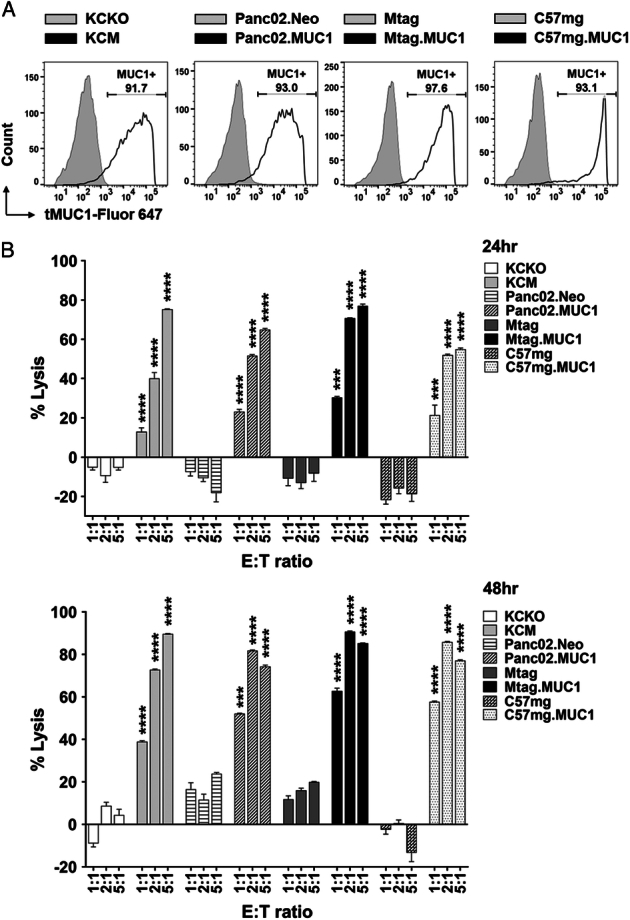
Murine MUC1 CAR T cells target tMUC1-expressing tumor cells for lysis in vitro. A, Percentage of cells expressing human tMUC1, determined by TAB004-Fluor 647 staining and flow cytometry analysis. KCKO and KCM, Panc02.Neo and Panc02.MUC1: pancreatic cancer; Mtag and Mtag.MUC1, C57mg and C57mg.MUC1: breast cancer. The tMUC1 null control cell lines were shown in light gray histograms. B, Percentage of mouse tumor cell lysis by MUC1 CAR T cells. CAR T cells and mouse tumor cell lines were cocultured at the indicated E:T ratio for 24 hours and 48 hours. The lysis of tumor cells was determined by MTT assay. The calculation formula for the percentage of lysis was: (OD of coculture with mock T−OD of coculture with CAR T)/OD of coculture with mock T×100. Data are presented as the mean ± SD from quadruplicate. The statistical comparison was conducted between the MUC1-expressing tumor cell line versus its MUC1-null/low counterpart at the same E:T ratio. ***P <0.001, ****P <0.0001 (multiple unpaired t tests). CAR indicates chimeric antigen receptor; MUC1, mucin 1; tMUCI, tumor form of mucin 1.
FIGURE 3.
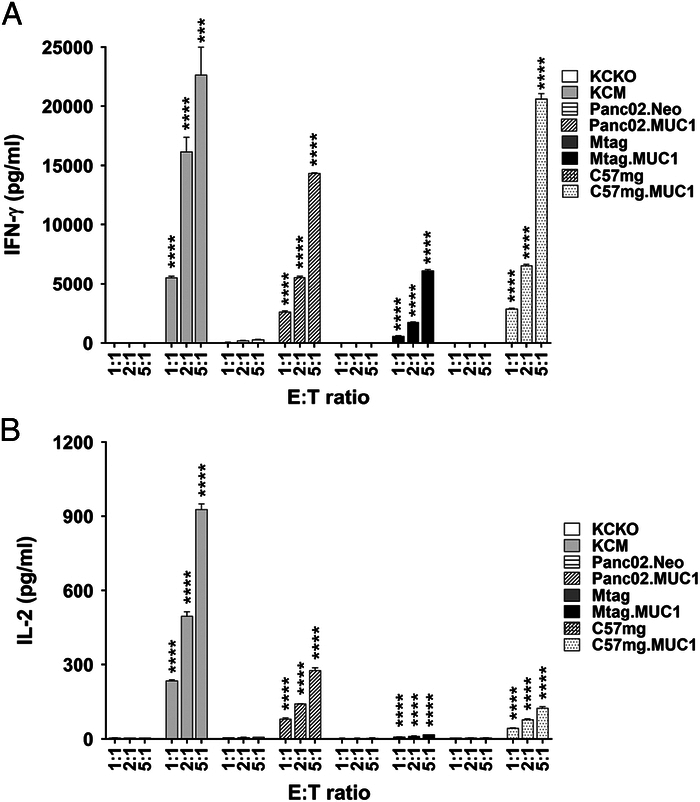
CAR T cells release cytokines while killing tMUC1-positive tumor cells in vitro. A, IFN-γ and (B) IL-2 release in supernatants after coculture for 24 hours as described in Figure 2B. Cytokine levels were determined by ELISA. Data are presented as the mean ± SD. In any cocultures with mock T cells, IFN-γ level was undetectable and IL-2 was in the range of 10 to 20 pg/mL. The statistical comparison was conducted between the MUC1-expressing tumor cell line versus its MUC1-null/low counterpart at the same E:T ratio. ***P <0.001, ****P <0.0001 (multiple unpaired t tests). CAR indicates chimeric antigen receptor; ELISA, enzyme-linked immunosorbent assay; MUC1, mucin 1; tMUCI, tumor form of mucin 1.
The recognition of tMUC1 on tumor cell surface further activated MUC1 CAR T cells, as demonstrated by the significant upregulation of PD-1 on CAR T cells at an E:T ratio of 5:1 after 24-hour coculture with KCM or Mtag.MUC1 cells (Fig. 4A). This engagement of CAR T cells with the tMUC1 activated their cytotoxic activity and caused upregulation of degranulation marker CD107a on CAR T-cell surface (Fig. 4A), as well as increased granzyme B release (Fig. 4B, right). Activated mouse T cells, irrespective of mock T cells or MUC1 CAR T cells, spontaneously produced some granzyme B without further antigen stimulation (Fig. 4B). However, these low granzyme B–producing cells were unable to induce tumor cell death (Fig. 2B). The increases in PD-1 and CD107a expression on CAR T cells were noticed as early as 4-hour coculture (Supplemental Fig. 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A813). Compared with CAR T cells, there was no increase in the expression of activation markers (Supplemental Fig. 4, Supplemental Digital Content 1, http://links.lww.com/JIT/A813) and granzyme B release (Fig. 4B, left) in mock T cells when cocultured with tumor cells with or without tMUC1. In addition, there was a significant loss of CAR+ population (likely CAR+PD-1+) within MUC1 CAR T cells after repetitive tumor antigen challenging through coculture with KCM cells. However, those CAR T cells still maintained strong tumor lysis efficacy (Supplemental Fig. 5, Supplemental Digital Content 1, http://links.lww.com/JIT/A813). Taken together, we generated MUC1 CAR T cells that were highly specific against tMUC1-expressing pancreatic and breast cancer cells.
FIGURE 4.

Engagement of CAR T cells with tMUC1 increases CD107a expression and granzyme B release in vitro. A, Significant increase of PD-1 and CD107a on MUC1 CAR T-cell surface after recognizing tMUC1. CAR T cells were cocultured at an E:T ratio of 5:1 with the indicated tumor cell lines for 24 hours. Cells were then stained for MUC1 CAR expression, as well as for PD-1 and CD107a expressions. Data were analyzed by flow cytometry and gated on live CD8+ T cells. B, Increase of granzyme B release from MUC1 CAR T cells after recognizing tMUC1. Culture supernatants from (A) were assayed for granzyme B level by ELISA. Data are presented as the mean ± SD. Left panel, mock T-cell coculture with tumor cell lines; right panel, MUC1 CAR T-cell coculture with tumor cell lines. Statistical analysis was performed to compare granzyme B level difference between KCM and KCKO, and between Mtag.MUC1 and Mtag. *P <0.05, **P <0.01, ***P <0.001 (multiple unpaired t tests). CAR indicates chimeric antigen receptor; ELISA, enzyme-linked immunosorbent assay; MUC1, mucin 1; tMUCI, tumor form of mucin 1.
Mucin 1 Chimeric Antigen Receptor T Cells Significantly Reduced KCM-Luc Tumor Growth Within the Pancreas in an Immunocompetent Mucin 1.Tg Syngeneic Orthotopic Model
PDA is a highly immunologic cold tumor and is resistant to most treatments.31 To assess the in vivo efficacy of MUC1 CAR T cells on pancreatic tumor growth, we surgically injected KCM cells that had been stably transfected with luciferase gene for live in vivo tracking of tumor progression. The host MUC1.Tg mice were immune-replete and expressed full-length human MUC1 to allow the growth of human MUC1-expressing KCM cells.26 Figure 5A shows the tumor growth for individual mice through IVIS imaging. The bioluminescence intensity was depicted as radiance (Fig. 5A, left panels). The total flux of bioluminescence was quantified to represent the xenograft tumor size. Overall, the tumor progression in CAR T-cell–treated mice appeared slower (Fig. 5A, right panels), which was also confirmed by their tumor wet weight reduction at the endpoint (Fig. 5B). PDA tumors remained high levels of tMUC1 expression post–CAR T-cell treatment (Fig. 5C). The treatment started when the tumor was already at a late stage (day 8 after tumor challenge). By 21 days, these mice had to be euthanized due to large tumors in the pancreas. Given it was promising that a single dose of MUC1 CAR T cells was able to reduce the tumor burden modestly but significantly, new combinations of immunotherapy with chemo/radiation are being explored for future studies. To determine whether MUC1 CAR T cells affected the host secondary lymphoid organs, spleens were resected but their weights had no statistical difference between the two groups (Fig. 5D). In addition, there was a trend of body weight loss in mice receiving mock T cells, but not in CAR T-cell–treated animals at the endpoint (Fig. 5E). This weight loss was likely due to the illness of control mice that developed large pancreas tumor burdens. All the animals survived till the experimental endpoint.
FIGURE 5.

MUC1 CAR T cells reduce KCM-Luc tumor growth in the pancreas. Mouse pancreatic cell line KCM-luciferase (20,000 cells/mouse) was orthotopically injected into the pancreas of MUC1.Tg mice. Seven days postsurgical injection, the presence of a tumor in the pancreas was confirmed by IVIS imaging. On day 8, mice were randomized and received a single i.v. injection of mock T cells as control, or MUC1 CAR T cells. A, Tumor growth was monitored by weekly IVIS imaging. On the right panels, the total flux of bioluminescence was quantified by marking a region of interest covering the tumor xenograft. B, The tumor wet weight for individual mice on day 21 (endpoint; P = 0.0353). C, IHC staining for tumor MUC1 with TAB004 (×100). Brown staining shows tMUC1 positivity (n = 3), three mice from each group were used for sectioning and staining. D, The spleen wet weight of individual mouse at the endpoint. E, The body weight change before and after T-cell treatment. No significance is observed between the mock-T group and the MUC1 T-cell group. n = 5 mice per group. The horizontal bar marks the mean value in each group. Two experiments are conducted with one representative data being shown here. *P <0.05 (unpaired t test). CAR indicates chimeric antigen receptor; i.p., intraperitoneal; i.v., intravenous; MUC1, mucin 1; tMUCI, tumor form of mucin 1.
Given the aggressive nature of the orthotopic PDA model, we focused on the rest of the in vivo studies using the MMT mice.
Mucin 1 Chimeric Antigen Receptor T Cells Delay Spontaneous MMT Breast Tumor Progression In Vivo
To the best of our knowledge, this would be the first MUC1 CAR T-cell efficacy and toxicity evaluation using a spontaneous immune intact mouse model with appropriate genetic and phenotypic heterogeneities and expressing a human antigen, MUC1, in its entire normal epithelia. First, we confirmed the presence of tMUC1 in 2 fresh ex vivo MMT tumors without treatment (Fig. 6A). The immunocompetent MMT mice spontaneously develop multifocal palpable mammary gland tumors at 10–12 weeks of age.23 Therefore, starting at ~10–14 weeks of age, MMT mice received MUC1 CAR T-cell treatment after lymphodepletion with cyclophosphamide. Despite the variation in tumor growth kinetics within each group (Fig. 6B), the tumor progression in CAR T-cell–treated mice was dramatically slower than those of age-matched control mice (Fig. 6B). All MMT mice remained alive till their humane endpoint for survival tracking and tissue collection. Their survival profile is shown in Figure 6C. MUC1 CAR T-cell treatment significantly prolonged the lifespan of tumor-bearing MMT mice. The H&E staining of fixed lungs showed the presence of multiple tumor metastasis in 3 out of 3 mice receiving mock T cells, compared with only 1 out of 3 mice in those receiving MUC1 CAR T cells with fewer lung metastasis (Fig. 6D). The histology of resected tumors was compared in Figure 6E. The H&E staining showed a distinct tumor tissue morphology between mock T-cell–treated tumors and CAR T-cell–treated tumors. While the MUC1 CAR T-cell–treated tumors maintained a clear tissue and adenocarcinoma structure, the mock T-cell–treated tumor had a massive undifferentiated-like structure. By IHC staining, tMUC1 was positive in both treatment groups detected by TAB004 (Fig. 6E, middle panels). To assess the mechanism of action, histologic analysis of T-cell infiltration into the tumor was performed. There were sparse CD8+ cells located at the tumor margin area in the MUC1 CAR T-cell group, but they were totally absent in the mock T-cell group (Fig. 6E, bottom panels). Those areas with CD8+ staining were quantitated (Fig. 6E, right). The presence of CD8+ cells within the MMT tumor led us to further evaluate the persistence of MUC1 CAR T cells using Mtag.MUC1 cell–injected TNBC model (Supplemental Fig. 6, Supplemental Digital Content 1, http://links.lww.com/JIT/A813). From the 6 time points of choice, the donor CAR T cells reached the peak frequency at 24 hours in peripheral blood, at 24–48 hours in the spleen, and at 48 hours in the tumor. The presence of the donor CAR T cells significantly dropped in a week. Instead of completely disappearing, donor CAR T cells resided in these selected tissues at very low frequencies.
FIGURE 6.

MUC1 CAR T cells control spontaneous MMT tumor growth. A, The tMUC1 expression in MMT tumors. Tumors from 2 female MMT mice (~20 wk of age) were resected to make single-cell suspensions. Cell mixtures were cultured and attached for 2 days, and then stained with TAB004-Fluor 647 and analyzed for tMUC1 level. B, Significantly delayed MMT tumor progression in MUC1 CAR T-cell–treated mice. Female MMT mice at ~10–14 weeks of age were pooled and randomized into 2 treatment groups. All MMT mice received one dose of cyclophosphamide (CTX, 1.5 mg per mouse, intraperitoneal injection) the day before T-cell adoptive transfer. The next day, the mock-T group received i.v. injection of mock T cells (10×106 per mouse), and MUC1-T group received i.v. injection of murine MUC1 CAR T cells (10×106 per mouse). Three additional T-cell injections at the same dose were administered at a 2-week interval afterward, with CTX pretreatment the day before. Data show the tumor volume up to 22 weeks of age. The blue curve represents the average tumor volume of live mice within the mock T-cell treatment group; the red curve represents the average tumor volume of live mice within the CAR T-cell treatment group. n = 9 mice for mock T-cell group; n = 10 mice for the MUC1 CAR T-cell group. Two experiments are conducted with one representative data shown here. C, Improved survival in CAR T-cell group. The survival was monitored for mice from (B). All MMT mice survived till their tumors reached the humane endpoint. Lungs (D) and tumors (E) were harvested at the endpoint from (B). n = 3 mice from each group were used for sectioning and staining. D, Dramatic decrease of tumor-distant metastasis into the lung. Left, H&E staining of lung sections. The black arrow points to the metastasis tumor. Magnification = ×400. Right, the number of tumor metastasis in the lung. The horizontal bar marks the mean value in each group. E, Top panels, tumor tissue morphology by H&E staining (×100). The boxed areas are shown in ×400 magnification. Middle panels, IHC staining for tumor MUC1 with TAB004 (×100, same area as for H&E). Brown staining shows tMUC1 positivity. Bottom panels, IHC staining for mouse CD8 T cells (×250). Brown staining shows CD8 positivity, as pointed out by the black arrows. The number of distinct areas with CD8+ staining is shown on the right scatter plot (P = 0.0782, unpaired t test). *P <0.05 (P = 0.0241, unpaired t test); ***P <0.001 (P = 0.0009, log-rank test); ****P <0.0001 (2-way ANOVA). ANOVA indicates analysis of variance; CAR, chimeric antigen receptor; H&E, hematoxylin and eosin; i.p., intraperitoneal; i.v., intravenous; MUC1, mucin 1; tMUCI, tumor form of mucin 1.
Mucin 1 Chimeric Antigen Receptor T Cells Are Well Tolerated in MMT Mice In Vivo
The liver and renal toxicity of MUC1 CAR T-cell treatment was evaluated in spontaneous MMT mice 6 days after a single dose of CAR T-cell injection (early/acute toxicity) or at the survival endpoint after 4 rounds of CAR T-cell injections (late toxicity). First of all, there was no mouse death before endpoint studies. Within the liver panel, the only significance noticed was in creatine kinase (Fig. 7A). The level of creatine kinase was lower in the mock T-cell group from the survival study (mock-T, late) when compared with normal MUC1.Tg group and to its respective CAR T-cell group. Within the renal panel, the changes with significance were marked (Fig. 7B). Additional data were provided in Supplemental Figure 7 (Supplemental Digital Content 1, http://links.lww.com/JIT/A813). As those significant changes happened only between T-cell treatment groups versus normal healthy control, this was very likely due to disease progression in MMT mice, not due to interference by T cells. Most importantly, there was no significant difference between the MUC1 CAR T-cell group and their respective mock T-cell control, despite T-cell treatment being acute or prolonged. The histologic evaluation on liver and kidney tissues further demonstrated that MUC1 CAR T cells appeared safe to use in the immunocompetent MMT mice (Supplemental Fig. 8, Supplemental Digital Content 1, http://links.lww.com/JIT/A813). The body weights were monitored and no statistical significance was noted for each individual MMT mouse over the T-cell treatment period (Fig. 7C). Thus, overall, the MUC1 CAR T cells were well tolerated in the immunocompetent human MUC1-expressing spontaneous breast cancer model.
FIGURE 7.

MUC1 CAR T cells are well tolerated in spontaneous MMT mice. A and B, The mouse sera were collected to evaluate CAR T-cell toxicity for liver panel (A) and renal panel (B) as described in “Materials and Methods.” Normal, sera from normal MUC1.Tg mice; n = 5. Early, sera from spontaneous MMT mice 6 days after receiving one dose of T cells; n = 6 for each group. Late, sera from spontaneous MMT mice at the survival endpoint as in Figure 6B; n = 5 picked from each group. The statistical comparison was performed between normal and each T-cell treatment group, and between mock-T and MUC1-T at an early stage or at a late stage. The horizontal bar marks the median value in each group. The legends for (A) and (B) are shared and shown at the bottom (A). C, No significant body weight changes after T-cell treatment. Mice were the same as for (A and B) and no statistical difference was achieved. Left: mice were injected with mock T cells or MUC1 CAR T cells. Body weights were recorded right before T-cell injection (before injection) and on day 6 endpoint (after injection). Right: mice were from Figure 6B and body weights were recorded during the T-cell treatment course. *P <0.05, **P <0.01 (unpaired t tests). CAR indicates chimeric antigen receptor; MUC1, mucin 1. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen.
Mucin 1 Chimeric Antigen Receptor T Cells Alter the Gene Landscape in Spontaneous MMT Mice In Vivo
To reveal the underlying mechanism for MMT tumor reduction by MUC1 CAR T cells in vivo, gene expression profiling was conducted using a mouse Clariom S array which was based on all known well-annotated genes. The differential expression analysis identified 725 genes with significance in MUC1 CAR T-cell–treated tumors when compared with mock T-cell–treated tumors (Fig. 8). Those differentially expressed genes in the CAR T-cell group were input into ingenuity pathway analysis and then those gene-associated top pathways were profiled as shown in Figure 8A. It is interesting to note that multiple biosynthesis pathways were on the list, as detailed in Figure 8A, which probably changed the metabolism within the tumor microenvironment. Another noticeable change was in the pathways involved in cell/tissue structure, including actin cytoskeleton signaling pathway and remodeling of epithelial adherens junctions, suggesting their role in tumor tissue morphology change or tumor metastasis. Out of the 725 genes, selected genes were plotted as heatmap (Fig. 8B). Those genes were involved in tumor cell signaling (Areg, Cadm1, Bmp8b), tumor metabolisms (Glul), tissue structure (Dsg3), matrix protein regulation (Mmp8, Slpi), tumor metastasis (Il20ra, Cd302), and cell infiltration and function (S100a8, Cxcl15, H2-Bl). Furthermore, 610 out of 725 genes were able to be compared with immunity pathway genes to determine the immune responses altered by CAR T cells. In MUC1 CAR T-cell–treated tumors, 53 out of 610 of the differentially expressed genes were involved in immunity pathways as outlined in Figure 8C.
FIGURE 8.

MUC1 CAR T cells regulate differential gene expression in spontaneous MMT mice. The tumors were collected from spontaneous MMT mice 6 days after receiving only one dose of T cells; n = 5 tumor RNA samples for each group. The RNAs were isolated from whole tumor tissues for the Clariom S Array. A, Top pathways affected by MUC1 CAR T cells. Differential expression analysis was conducted using limma between the CAR T-cell group and the mock T-cell group. Genes with adjusted P value <0.05 were input into IPA to identify top-associated pathways. In each pathway, the green bar shows the downregulated genes, whereas the red bar shows the upregulated genes. The numbers of affected genes are labeled on the bars. B, Heatmap of selected genes of interest. C, Comparison of MUC1 CAR T-cell altered genes with known immunity pathway genes. DEGs showed significant differences between the mock-T group and MUC1-T group; IPMs, immunity pathway molecules. CAR indicates chimeric antigen receptor; DEGs, differential expression genes; IPA, ingenuity pathway analysis; MUC1, mucin 1. BMP, bone morphogenetic protein; FXR, farnesoid X receptor; LPS, lipopolysaccharide; LXR, liver X receptor; NRF-2, nuclear factor erythroid 2-related factor 2; RXR, retinoid X receptor; TGF, transforming growth factor.
DISCUSSION
We had discussed in our recent publications6,13 that MUC1 remains an excellent tumor-associated antigen for developing CAR T-cell immunotherapies against solid tumors, including TNBC and PDA in immunocompromised NSG models. Here, we report the efficacy and toxicity of the novel human MUC1-targeted murine CAR T cells in vitro and in our unique human MUC1-expressing immunocompetent PDA model and spontaneous breast cancer model. This second-generation MUC1 CAR T-cell exhibited potent tumor cytotoxicity in vitro, and their lysis of tumor cells required the presence of tumor antigen tMUC1 (Fig. 2).
In vivo data show that the MUC1 CAR T-cell moderately albeit significantly reduced PDA tumor growth in an immunocompetent orthotopic mouse model (Fig. 5). This moderate response was primarily because the KCM tumors were too large by the time the treatment was started because of its aggressive growth kinetics within the pancreas. Thus, a single dose of CAR T cells given as late intervention was possibly not enough. Nevertheless, the fact that there was any effect warrants further study for a disease that currently has very few treatment options. Another factor for incomplete eradication of the tumor could be the low “persistence” of mouse CAR T cells in vivo. (Supplemental Fig. 5, Supplemental Digital Content 1, http://links.lww.com/JIT/A813). Therefore, repeated infusions of CAR T cells may be needed, or combining with modality may improve efficacy. We have identified a few candidates and, in the future, we will continue to explore these target candidates, as well as chemo/radiation for combinations with CAR T cells.
In the MMT model, we observed a significant and sustained delay in mammary gland tumor progression (Fig. 6), prevented lung metastasis, and improved survival. Importantly, there was negligible or no liver and kidney toxicity in MMT mice receiving up to 4 injections of MUC1 CAR T cells, suggesting their safe use in vivo (Fig. 7). The mice also showed no signs of weight loss or other adverse health effects. We recognize it is possible that the marginal/low toxicity might be partially due to the short persistence of mouse CAR T cells in vivo, nevertheless, given the efficacy of the CAR T cells in the MMT mice, we consider the treatment to be effective and safe in these conditions.
We noticed that out of the 4 pairs of tumor cell lines, Mtag.MUC1 was highly sensitive to MUC1 CAR T-cell–induced lysis. Based on the upregulation of PD-1/CD107a and the release of granzyme B/IFN-γ/IL-2 in CAR T cells when cocultured with Mtag.MUC1 cells, the high sensitivity of Mtag.MUC1 cells to MUC1 CAR T-cell killing might be ascribed to its own intrinsic sensitive characteristics, which would not cause strong immunogenic responses.
It was rather interesting to observe the distinct tumor tissue morphology and CD8 T-cell infiltration between CAR T-cell–treated mice and mock-T–treated MMT mice. This might suggest the delayed tumor progression towards its advanced stages by MUC1 CAR T cells. The mortality of advanced breast cancer was closely associated with lung metastasis, which was previously considered incurable.32,33 It was highly encouraging that lung metastasis was reduced in MUC1 CAR T-cell–treated MMT mice (Fig. 6D), which corresponded with better survival (Fig. 6C). Another promising aspect was the presence of CD8+ T cells in CAR T-cell–treated tumors even 4–5 weeks after the last T-cell transfer. These persistent CD8+ T cells within the tumor microenvironment might correspond with the overall slower growth of the primary MMT tumor and reduced metastasis.
The in vitro efficacy of MUC1 CAR T cells was highly potent. However, although the in vivo tumor growth and progression were decreased by CAR T cells, it was not completely eradicated. The short persistence of mouse MUC1 CAR T cells might be one of the limitations. Thus, in MMT mice, multiple injections of MUC1 CAR T cells were needed to stabilize the disease. This is different from the human MUC1 CAR T cells, in which a single dose was already able to significantly reduce human TNBC tumor progression in NSG mice for 81 days till their control group reached a humane endpoint.6 Future studies will focus on improving the penetration of MUC1 CAR T cells to reverse the scarcity of CAR T cells within the tumor microenvironment. In clinic and preclinical models, lymphodepletion with cyclophosphamide improved CAR T-cell expansion in the peripheral blood.34–37 In this study, we used cyclophosphamide to improve the acceptance of CAR T-cell adoptive transfer. Neoadjuvant therapy or chemotherapies could be introduced to make the tumor inflamed or leaky before MUC1 CAR T-cell injection to improve immune cell tumor infiltration. We are also exploring the combination of MUC1 CAR T cells with immune checkpoint inhibitors to improve their intratumor activities. Besides adding treatment frequency and boosting with combination to improve CAR T-cell efficacy, the optimal dosing schedule will also be taken into consideration. This has been evidenced in a recent syngeneic mouse CAR-T study, in which the efficacy of the same CAR T cells differed significantly depending on their injection schedule post-tumor cell inoculation.19
Based on gene profiling, MUC1 CAR T cells remodeled the tumor microenvironment in the spontaneously arising MMT tumors (Fig. 8). Further analysis could reveal other novel targets for combination treatments. MUC1 CAR T cells regulated a few biosynthesis pathways involved in tumor metabolism, which overall might reduce tumor growth. The 2 pathways for actin signaling and epithelial adherens junction remodeling might explain the different tissue morphology between CAR T-cell–treated tumors and mock T-cell–treated tumors, as well as the lower tumor metastasis (Fig. 6). Gene profiling study was conducted 1 week post-MUC1 CAR T-cell treatment. Further study is under consideration to compare the change of gene landscape with different durations of CAR T-cell treatment in the spontaneous MMT model, which might also help identify novel candidates for combinations with MUC1 CAR T cells to further improve efficacy.
CONCLUSION
Our data complement other promising studies using CAR T cells directed against other MUC1 epitopes, as well as MUC1 Tn.38–41 Our studies go beyond those studies by using the appropriate MMT mouse model and the orthotopic late-stage PDA model. These comprehensive studies in immunocompetent appropriate mouse models are critical for the transition of tMUC1-targeted CAR T-cell therapy into the clinic. The MUC1 CAR T cells effectively and safely target human tMUC1 in breast cancer and PDA. This is highly applicable to the majority of solid tumors, which are epithelial malignancies and express tMUC1. The strategies to improve the efficacy of MUC1 CAR T-cell and to find its optimal combinations are under exploration.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all the staff members in the vivarium for their assistance with training and animal care.
Conflicts of Interest/Financial Disclosures
This study was supported by DOD W81XWH-18-1-0711; Belk Endowment, University of North Carolina at Charlotte. P.M. is a board member of OncoTAB, Inc.
None reported. All authors have declared that there are no financial conflicts of interest with regard to this work.
Footnotes
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.
Contributor Information
Ru Zhou, Email: rzhou@charlotte.edu.
Shu-ta Wu, Email: shutawu@gmail.com.
Mahboubeh Yazdanifar, Email: Myazdanifar@gmail.com.
Chandra Williams, Email: Chandra.williams@pfizer.com.
Alexa Sanders, Email: jsande50@uncc.edu.
Cory Brouwer, Email: Cory.Brouwer@uncc.edu.
John Maher, Email: john.maher@kcl.ac.uk.
Pinku Mukherjee, Email: pmukherj@charlotte.edu.
REFERENCES
- 1.Feigal EG, DeWitt ND, Cantilena C, et al. At the end of the beginning: immunotherapies as living drugs. Nat Immunol. 2019;20:955–962. [DOI] [PubMed] [Google Scholar]
- 2.Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. 2020;38:473–488. [DOI] [PubMed] [Google Scholar]
- 3.O’Leary MC, Lu X, Huang Y, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia FDA approval summary: tisagenlecleucel for R/R BCP ALL. Clin Cancer Res. 2019;25:1142–1146. [DOI] [PubMed] [Google Scholar]
- 4.Bouchkouj N, Kasamon YL, de Claro RA, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25:1702–1708. [DOI] [PubMed] [Google Scholar]
- 5.Siegel RL, Miller KD, Wagle NS, et al. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. [DOI] [PubMed] [Google Scholar]
- 6.Zhou R, Yazdanifar M, Roy LD, et al. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front Immunol. 2019;10:1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahib L, Wehner MR, Matrisian LM, et al. Estimated projection of US cancer incidence and death to 2040. JAMA Netw Open. 2021;4:e214708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 9.Vlad AM, Kettel JC, Alajez NM, et al. MUC1 immunobiology: from discovery to clinical applications. Adv Immunol. 2004;82:249–293. [DOI] [PubMed] [Google Scholar]
- 10.Kimura T, Finn OJ. MUC1 immunotherapy is here to stay. Expert Opin Biol Ther. 2013;13:35–49. [DOI] [PubMed] [Google Scholar]
- 11.Curry JM, Thompson KJ, Rao SG, et al. The use of a novel MUC1 antibody to identify cancer stem cells and circulating MUC1 in mice and patients with pancreatic cancer. J Surg Oncol. 2013;107:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roy LD, Dillon LM, Zhou R, et al. A tumor-specific antibody to aid breast cancer screening in women with dense breast tissue. Genes Cancer. 2017;8:536–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yazdanifar M, Zhou R, Grover P, et al. Overcoming immunological resistance enhances the efficacy of a novel anti-tMUC1-CAR T-cell treatment against pancreatic ductal adenocarcinoma. Cells. 2019;8:1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegler EL, Wang P. Preclinical models in chimeric antigen receptor–engineered T-cell therapy. Hum Gene Ther. 2018;29:534–546. [DOI] [PubMed] [Google Scholar]
- 15.Lanitis E, Rota G, Kosti P, et al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med. 2021;218:e20192203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu B, Ren J, Luo Y, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20:3025–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li P, Yang L, Li T, et al. The third generation anti-HER2 chimeric antigen receptor mouse T cells alone or together with anti-PD1 antibody inhibits the growth of mouse breast tumor cells expressing HER2 in vitro and in immune-competent mice. Front Oncol. 2020;10:1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin D, Li D, Zhang B, et al. Potential lung attack and lethality generated by EpCAM-specific CAR-T cells in immunocompetent mouse models. Oncoimmunology. 2020;9:1806009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato D, Yaguchi T, Iwata T, et al. GPC1-specific CAR-T cells eradicate established solid tumor without adverse effects and synergize with anti-PD-1 Ab. eLife. 2020;9:e49392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamballe F, Ahmad F, Vinik Y, et al. Modeling heterogeneity of triple-negative breast cancer uncovers a novel combinatorial treatment overcoming primary drug resistance. Adv Sci. 2021;8:2003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christenson JL, Butterfield KT, Spoelstra NS, et al. MMTV-PyMT and derived Met-1 mouse mammary tumor cells as models for studying the role of the androgen receptor in triple-negative breast cancer progression. Horm Cancer. 2017;8:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Attalla S, Taifour T, Bui T, et al. Insights from transgenic mouse models of PyMT-induced breast cancer: recapitulating human breast cancer progression in vivo. Oncogene. 2021;40:475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukherjee P, Madsen CS, Ginardi AR, et al. Mucin 1-specific immunotherapy in a mouse model of spontaneous breast cancer. J Immunother. 2003;26:47–62. [DOI] [PubMed] [Google Scholar]
- 24.Castellarin M, Sands C, Da T, et al. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI Insight. 2020;5:e136012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Health USDo, Human Services F, Drug Administration Center for Biologics E, et al . Guidance for industry: guidance for human somatic cell therapy and gene therapy. Hum Gene Ther. 2001;12:303–314. [DOI] [PubMed] [Google Scholar]
- 26.Rowse GJ, Tempero RM, VanLith ML, et al. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998;58:315–321. [PubMed] [Google Scholar]
- 27.Besmer DM, Curry JM, Roy LD, et al. Pancreatic ductal adenocarcinoma mice lacking mucin 1 have a profound defect in tumor growth and metastasis. Cancer Res. 2011;71:4432–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lakshminarayanan V, Supekar NT, Wei J, et al. MUC1 vaccines, comprised of glycosylated or non-glycosylated peptides or tumor-derived MUC1, can circumvent immunoediting to control tumor growth in MUC1 transgenic mice. PLoS One. 2016;11:e0145920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma L, Dichwalkar T, Chang JYH, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou R, Curry JM, Roy LD, et al. A novel association of neuropilin-1 and MUC1 in pancreatic ductal adenocarcinoma: role in induction of VEGF signaling and angiogenesis. Oncogene. 2016;35:5608–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ullman NA, Burchard PR, Dunne RF, et al. Immunologic strategies in pancreatic cancer: making cold tumors hot. J Clin Oncol. 2022;40:2789–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harbeck N, Penault-Llorca F, Cortes J, et al. Breast cancer. Nat Rev Dis Primers. 2019;5:66. [DOI] [PubMed] [Google Scholar]
- 33.Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med. 2013;274:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haas AR, Tanyi JL, O’Hara MH, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther. 2019;27:1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell. 2021;39:193–208. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bot A, Rossi JM, Jiang Y, et al. Cyclophosphamide and fludarabine conditioning chemotherapy induces a key homeostatic cytokine profile in patients prior to CAR T-cell therapy. Blood. 2015;126:4426. [Google Scholar]
- 37.Neelapu SS. CAR-T efficacy: is conditioning the key? Blood. 2019;133:1799–1800. [DOI] [PubMed] [Google Scholar]
- 38.Wilkie S, Picco G, Foster J, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901–4909. [DOI] [PubMed] [Google Scholar]
- 39.Posey AD, Jr, Schwab RD, Boesteanu AC, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44:1444–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkie S, van Schalkwyk MC, Hobbs S, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–1070. [DOI] [PubMed] [Google Scholar]
- 41.Posey AD, Jr, Clausen H, June CH. Distinguishing truncated and normal MUC1 Glycoform targeting from Tn-MUC1-specific CAR T Cells: specificity is the key to safety. Immunity. 2016;45:947–948. [DOI] [PMC free article] [PubMed] [Google Scholar]