

Abstract
The reconstruction of ancestral sequences can offer a glimpse into the fascinating process of molecular evolution by exposing the adaptive pathways that shape the proteins found in nature today. Here, we track the evolution of the carbohydrate-active enzymes responsible for the synthesis and turnover of mannogen, a critical carbohydrate reserve in Leishmania parasites. Biochemical characterization of resurrected enzymes demonstrated that mannoside phosphorylase activity emerged in an ancestral bacterial mannosyltransferase, and later disappeared in the process of horizontal gene transfer and gene duplication in Leishmania. By shuffling through plausible historical sequence space in an ancestral mannosyltransferase, we found that mannoside phosphorylase activity could be toggled on through various combinations of mutations at positions outside of the active site. Molecular dynamics simulations showed that such mutations can affect loop rigidity and shield the active site from water molecules that disrupt key interactions, allowing α-mannose 1-phosphate to adopt a catalytically productive conformation. These findings highlight the importance of subtle distal mutations in protein evolution and suggest that the vast collection of natural glycosyltransferases may be a promising source of engineering templates for the design of tailored phosphorylases.
Keywords: enzyme engineering, enzyme evolution, ancestral sequence reconstruction, glycoside phosphorylase, GT108, Leishmania, mannogen
Introduction
To orchestrate the beautiful biochemistry of carbohydrates and their derivatives, nature has evolved a large and diverse collection of biocatalysts known as carbohydrate-active enzymes (CAZymes). The most common and most extensively studied CAZymes are glycoside hydrolases (GHs) and glycosyltransferases (GTs), which catalyze the breakdown and synthesis of glycosidic bonds, respectively. Among the more peculiar products of CAZyme evolution are the glycoside phosphorylases (GPs), which appear to combine the characteristics of GHs and GTs.1 GPs have evolved to degrade glycosidic bonds using inorganic phosphate instead of water as their nucleophile of choice, resulting in the release of a glycosyl phosphate. However, since the phosphorolysis reaction is easily reversible, GPs can be applied for the synthesis of glycosidic bonds as well.2 From that perspective, these enzymes can also be regarded as transferases that have evolved to not use nucleoside diphosphate sugars (e.g. GDP-mannose) but the shorter corresponding glycosyl phosphates (e.g. α-mannose 1-phosphate) as their glycosyl donor of choice. The great functional flexibility of GPs makes them particularly attractive catalysts for various biotechnological applications, from the production of specialty sugars and glycosides3,4 to the development of more energy-efficient microbial cell factories.5
The obvious similarities between GPs and other CAZymes are not just limited to their function but also extend to their structure and mechanism. GPs are scattered across various GH and GT families in the CAZy database classification,6 which suggests that several ancestral hydrolases and transferases have acquired phosphorylase activity through independent evolutionary itineraries. We previously demonstrated that it is indeed possible to introduce significant phosphorolytic activity in a hydrolase.7 By thorough analysis of the conserved sequence patterns in β-N-acetylglucosaminide hydrolases and β-glucoside phosphorylases from family GH3, three active site positions were found to be involved in phosphate recognition. When those positions were targeted for mutagenesis, β-N-acetylglucosaminidases could be converted into β-N-acetylglucosaminide phosphorylases. A switch from hydrolase to phosphorylase activity was also accomplished in N-acetylglucosaminide hydrolases from family GH84 by Teze et al. after mutation of a catalytic residue.8 In contrast, a switch between transferase and phosphorylase activity has not yet been reported. Intriguingly, GTs show no activity whatsoever on glycosyl phosphates as (shorter) donor substrate, and attempts to unravel how GPs may have diverged from them have remained unsuccessful.1,9−11
The functional and evolutionary parallels between GTs and GPs are particularly obvious in a group of highly homologous mannosyltransferase/phosphorylases (MTPs) that was recently discovered in the parasite Leishmania mexicana.12 These enzymes from family GT108 play a central role in the biosynthesis and cycling of linear β-1,2-mannans known as mannogen, constituting a noncanonical carbohydrate reserve pathway that is thought to have been essential in enabling Leishmania to colonize new insect and mammalian host niches (Figure 1A).12−15 Two of the enzymes, i.e. MTP1 and MTP2, are strict GTs that prime mannogen synthesis using GDP-mannose. Four other enzymes, i.e. MTP3, MTP4, MTP6, and MTP7, can catalyze two different reactions. They exclusively act as mannogen phosphorylases in vivo, catalyzing the reversible phosphorolysis of mannogen in the presence of phosphate with production of α-d-mannose 1-phosphate. However, these enzymes remarkably also show transferase activity in vitro.12 Furthermore, the donor substrates for both reaction types are accommodated in the same active site (α-mannose 1-phosphate and GDP-mannose for GP and GT, respectively). The unique dual activity of these leishmanial enzymes may be a curious remnant of the common evolutionary history of mannogen GTs and GPs.
Figure 1.

Characterization of ancestral enzymes. (a) Transferase and (reverse) phosphorylase reactions catalyzed by enzymes from GT108 for the synthesis and degradation of mannogen (Pi, inorganic phosphate). (b) Cartoon representation of the phylogeny of the CAZy family GT108. The full phylogenetic tree is shown in Figure S1. The ancestral nodes that were studied in this work (AncX to AncZ and AncA to AncC) are labeled (spheres) and colored by their function (blue, strict GT; yellow, bifunctional GP/GT). MTP2 and MTP4 are representative extant proteins from the collapsed clades of enzymes from Leishmania species. AsGT is the closest bacterial homologue of the leishmanial enzymes. (c) Apparent folding transition temperatures for the ancestral and extant enzymes characterized in this work, as determined by differential scanning fluorimetry. (d) Catalytic efficiencies of ancestral and extant enzymes from GT108 toward GDP-mannose (blue bars) and α-mannose 1-phosphate (yellow bars) at 27 °C and pH 6.5. An overview of the apparent kinetic parameters is provided in Table S2.
In this work, we set out to better understand how phosphorylase activity may emerge in GTs by taking a closer look at the evolutionary trajectory of CAZymes in GT108. We combine ancestral sequence reconstruction with detailed functional characterization to identify ancestral enzymes that are situated at an interesting crossroad of evolution. By shuffling through the sequence space of ancestral mutations, we pinpoint several positions outside the active site that can contribute to the adaptive process of switching phosphorylase activity off and on. Finally, we perform structural analyses to uncover how those distal positions affect the interactions and conformational dynamics in the catalytic pocket.
Results
Ancestral Protein Reconstruction and Resurrection
The evolutionary history of the strict GTs and bifunctional GPs/GTs from L. mexicana was reconstructed by inferring the maximum-likelihood phylogeny of family GT108, and subsequently using the ancestral sequence reconstruction tool GRASP to infer the most likely ancestral amino acid sequence at key nodes in the phylogenetic tree16 (Figures 1B, S1). Node AncA represents the common ancestor of all leishmanial enzymes of interest, which was acquired from bacteria by horizontal gene transfer. Its daughter nodes AncB and AncC represent the ancestors of the clades containing the confirmed bifunctional GPs/GTs or the confirmed strict GTs, respectively. A few intermediates in the evolution from the root of family GT108 to AncA were sampled as well and designated as AncX, AncY, and AncZ. The latter is the common ancestor of AncA and the closest bacterial homologue of the leishmanial enzymes, originating from Anaerocolumna sedimenticola (AsGT). All ancestral sequences at the targeted nodes could be derived with reasonably high confidence, containing between 12% and 20% ambiguous positions (i.e. positions where two or more amino acid states are plausible with posterior probabilities >0.2) (Table S1). The sequence identity to their extant homologues from Leishmania ranged from ∼40% for AncX to ∼70% for AncB and AncC.
All selected ancestors could be expressed in the soluble form in Escherichia coli. To evaluate their thermostability, the apparent folding transition temperatures (Tm,app) were measured via differential scanning fluorimetry (Figure 1C). The ancestral enzymes were considerably more thermostable than three reference wild-type enzymes (MTP2, MTP4, and AsGT), with an average Tm,app difference of 24 °C (average Tm,app of ancestors = 63 ± 10 °C; average Tm,app of wild-types = 39 ± 5 °C). These findings were further confirmed by comparing the kinetic stability of AncB and AncC to that of their respective extant descendants, MTP4 and MTP2 (Figure S2). After 15 min of incubation at 50 °C, MTP4 lost half of its initial activity and MTP2 was fully inactivated. In contrast, their ancestors retained all activity after incubation at temperatures up to 65 °C. These results are consistent with numerous earlier studies, which have shown that ancestral proteins tend to be more stable than their extant counterparts.17,18
We tracked the functional evolution of the resurrected enzymes by determining the kinetic parameters for their synthesis reactions with each of the relevant glycosyl donors, GDP-mannose and α-mannose 1-phosphate (Figure 1D, Table S2). These parameters were also obtained for the reference wild-type enzymes MTP2 and MTP4, corroborating the activity profiles described by Sernee et al.12 The oldest ancestor AncX was found to be a strict GT that can prime oligosaccharide synthesis from GDP-mannose, but not from α-mannose 1-phosphate. GP-type activity was first established in AncY, which showed largely identical catalytical efficiencies for both donors in vitro, and this dual activity was retained in AncZ. Since none of the bacterial descendants of AncY and AncZ had been characterized before, we also determined the kinetic parameters of AsGT to find that it too is a bifunctional GP/GT. AncB and AncC exhibited dual GP/GT and strict GT activity, respectively, which is in agreement with the function of their characterized descendants from Leishmania mexicana. However, their most recent common ancestor AncA showed only GT activity.
A common issue in ancestral protein reconstruction is the unavoidable statistical uncertainty that causes some sites in the sequence to be ambiguously inferred. As mentioned above, the ancestral sequences in this study contain various positions in which two or more amino acids were found to be statistically plausible. To evaluate the robustness of their function to this uncertainty, we also resurrected a so-called “worst plausible case” ancestor for every node of interest.19 These proteins contain the second most likely amino acid at every ambiguously reconstructed site. All alternative ancestors were found to display the same function as their maximum-likelihood counterparts, except for the alternative version of AncA (AncA-Alt). While AncA was a strict GT, AncA-Alt surprisingly also showed significant GP activity (Figure 1D, Table S2 and Figure S3). As a result, the true function of the common ancestor of all leishmanial enzymes from GT108 cannot be determined conclusively.
Our paleoenzymology study thus suggests that the high GP activity of the bifunctional enzymes from the family first emerged in an ancestral bacterial mannosyltransferase. The ability to catalyze the reversible phosphorolysis of mannans appears to have been lost at some point in time in an early ancestor of leishmanial GT108 family members. In the maximum-likelihood scenario, it was lost in their last common ancestor AncA and, subsequently, reinstalled in a daughter paralogue after one of the duplication events that led to the tandem gene array for mannogen metabolism currently found in Leishmania.12 In an alternative scenario that is statistically plausible and more parsimonious, GP activity was still present in the preduplication ancestor that was acquired from bacteria by horizontal gene transfer, after which a paralogous protein copy diverged to become dedicated to the synthesis of mannogen instead, as MTP1 and MTP2 are today.12
Catalytic Site Residues Alone are Not Responsible for Establishing Phosphorylase Activity
Based on structural analysis, one key mutation in the catalytic site was initially hypothesized to be responsible for the difference in donor specificity among GT108 enzymes.12 Extant enzymes that preferentially exhibit phosphorolytic activity contain an Arg residue in the phosphate-binding pocket (Arg150 in MTP4), whereas those that primarily act as mannosyltransferases contain a His at the corresponding position (His161 in MTP2) (Figure 2A). However, the sequence–function relationships in the ancestral enzymes shed new light on this theory. An Arg residue was unambiguously inferred to be present at the putative phosphate-binding position in the ancestral strict GTs AncA and AncX (posterior probability = 96% and 99%, respectively), demonstrating that it is, in fact, not indicative of significant GP activity (Figure 2B). Further, we designed the H161R variant of MTP2, confirming that this active site mutation indeed does not introduce measurable GP activity. Moreover, no other candidate function-switching mutations can be distinguished at any of the other positions in the donor subsite of ancestral or extant enzymes (Figure S4). The observation that AncA and AncA-Alt show a different donor specificity despite sharing the same active site architecture is especially intriguing. Therefore, it appears that more remote positions must critically contribute to the presence or absence of activity on α-mannose 1-phosphate.
Figure 2.
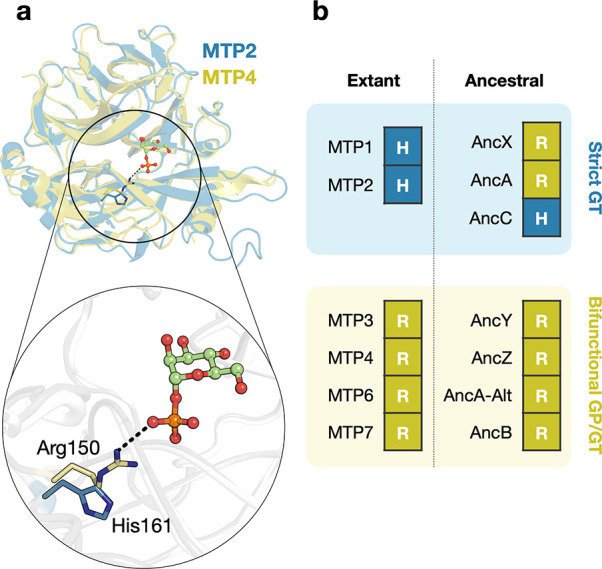
Analysis of the putative function-determining active site position in various extant and ancestral enzymes from GT108. (a) Superposition of the structures of MTP2 (PDB id: 6Q4X) and MTP4 (PDB id: 6Q50) with α-mannose 1-phosphate modeled in the active site. The hypothesized functional determinants His161 (in MTP2, blue) and Arg150 (in MTP4, yellow) are shown as sticks. (b) Overview of the identity of the residue that is found at the corresponding positions in the characterized extant and ancestral enzymes.
Shuffling the Ancestral Sequence Ambiguity of AncA
The discrepancy between the inferred functions of AncA and AncA-Alt slightly obscures the true path of functional evolution in GT108. However, we hypothesized that the statistical ambiguity in the reconstructed sequence of AncA may be used to our advantage by narrowing the search for the mutations that are responsible for toggling GP activity in between the most likely and worst plausible states of this ancestor. Indeed, AncA and AncA-Alt show a higher degree of identity at the sequence level (85%) than other extant or functionally robust ancestral pairs of strict GTs and bifunctional GP/GTs (e.g. 48% for MTP2 and MTP4, 50% for AncX and AncY). We sampled through the ensemble of plausible alternate states of AncA by designing a shuffling library with AncA and AncA-Alt as the parental genes. All variants in this library thus carried either the most probable or the second-most probable ancestral residue at each of the 46 ambiguously reconstructed sites (Figures 3A, S5). Clarified cell lysates of ∼400 clones were screened for activity in reactions with α-mannose 1-phosphate as glycosyl donor and mannose as acceptor, allowing us to identify several variants with GP activity significantly above the background (Figure 3B, Table S3).
Figure 3.

Shuffling between AncA and AncA-Alt. (a) Model of AncA with 46 positions (yellow spheres) that were ambiguously reconstructed. The catalytic residue (Asp86), the residue previously hypothesized to be responsible for GP activity (Arg153), and a phosphate molecule bound to the active site are shown for reference. (b) Phosphate released from α-mannose 1-phosphate by lysates containing shuffled variants of which AncA and AncA-Alt were the parental genes. The variants are ranked in order of ascending GP activity, and the top 15 hits are indicated in red. (c) Observed amino acid distribution at 3 of the 46 variable positions in the top 15 hits from the shuffling library. These positions were strongly enriched for the alternative residue from AncA-Alt. The most probable residue (from AncA) is shown in black, and the second-most probable residue (from AncA-Alt) is shown in yellow. (d) Model of AncA showing the three positions (106, 187, 245; black sticks) that were most strongly enriched for the alternative residue from AncA-Alt in the hits from the shuffling library.
Among the 15 AncA variants exhibiting the highest GP activity, the alternative residue originating from AncA-Alt was strongly enriched at several sites (Figures 3C,D, S6). Most notable was position 187, located in a peripheral pocket, which always carried Val instead of Thr. However, when a variant of AncA containing only this T187V mutation was constructed and purified, it did not exhibit measurable GP activity. Other enriched mutations toward the lower probability state were N106T and F245L, each present in 14 out of 15 hits. Position 106 is located at the surface, whereas position 245 is in close contact with position 187. These substitutions also could not establish significant GP activity alone nor when paired with T187V. However, the triple mutant N106T+T187V+F245L did exhibit measurable Michaelis–Menten kinetics on α-mannose 1-phosphate (kcat/KM = 0.9 mM–1min–1). The observation that not all hits from the library screening effort contain this minimally viable set of three mutations indicates that multiple combinations of mutations are capable of switching GP activity on or off in AncA, although position 187 does appear to play a central role. A closer inspection of the corresponding sites in ancestors at other nodes and in characterized modern-day enzymes revealed that the sequence–function relationships at position 187 indeed follow a clear trend that matches the results from the screening effort. A hydrophobic residue (Ile/Leu/Val/Phe) is present in all bifunctional enzymes, whereas strict GTs carry a hydrophilic residue (Arg/Gln/Thr) (Figure S7). Curiously, no such evident patterns can be identified at positions 106 and 245, supporting the finding that there are multiple possible sets of mutations at distal positions that can cooperatively enable or disable activity on α-mannose 1-phosphate.
Shuffling Between Ancestral Nodes
The search through the plausible ancestral sequence space of AncA suggests that the evolution and loss of phosphorylase activity in transferases from GT108 cannot be attributed to a single defined set of specificity-determining sequence fingerprints. We sought to further strengthen this hypothesis by attempting to introduce GP activity in AncA using a different collection of possible mutations, this time originating from the ancestor of its bifunctional daughter lineage AncB. We focused on a cluster of seven variable residues that shape the peripheral pocket that the critical residue at position 187 is located in (Ile184, His186, Thr187, His213, Thr231, Phe235, Phe245) (Figure 4A). A combinatorial binary library was constructed in which each of these seven positions matches the state of either AncA or AncB, resulting in 27 possible combinations. Approximately 300 clarified lysates were screened for GP activity, and eight unique hits were identified with product release rates over the background (Figure 4B). All hits contained the residue of AncB at positions 187, 231, and 235, and the same was true for all but one hit at position 245 (Figure 4C). Measurements using purified variants confirmed these findings (Table S4). Variants containing just one of the three mutations that were fully enriched among the eight hits (T187V, T231Y, or F235L) did not show measurable activity on α-mannose 1-phosphate, while the triple mutant did (kcat/KM = 0.1 mM–1min–1). This activity could be increased considerably by adding the fourth strongly enriched mutation F245A (kcat/KM = 0.4 mM–1min–1). The deletion of His186 that occurred between AncA and AncB is not necessary for establishing detectable GP activity, but the mutation was present in the best-performing variants. Indeed, the deletion causes an additional 10-fold improvement in catalytic efficiency for α-mannose 1-phosphate when compared to the quadruple mutant (kcat/KM = 5.2 mM–1min–1). Further, the screening results indicated that the mutations at positions 184 and 213 do not contribute to establishing GP activity. The variant where all seven positions were in the state of AncB even showed a significantly lower catalytic efficiency (kcat/KM = 1.0 mM–1min–1) than the variant without those two unnecessary mutations.
Figure 4.

Shuffling between AncA and AncB. (a) Model of AncA with the cluster of seven positions that was targeted in a combinatorial binary library (yellow spheres and black sticks). Each variant in the library contained the residue from AncA or AncB at those positions. The catalytic residue (Asp86), a putative phosphate-binding residue (Arg153), and a phosphate molecule bound to the active site are shown for reference. (b) Phosphate released from α-mannose 1-phosphate by lysates containing variants from the combinatorial binary library. The variants are ranked in order of ascending GP activity, and the hits are indicated in red. (c) Observed amino acid distribution at the seven variable positions in the hits. The residue from AncA is shown in black, and the residue from AncB is shown in yellow.
A second minimal set of mutations that is sufficient to establish GP activity in AncA could thus be found among the alternate collection of historical mutations that may have occurred between AncA and AncB. Both sets share only the T187V substitution. The mutation of Thr231 was enriched only in this library, where it was exchanged for the residue from AncB (Tyr), but not in the previous shuffling library where it was exchanged for the residue from AncA-Alt (Met). Notably, the third important mutation at Phe235 certainly could not have contributed to the functional difference between AncA and AncA-Alt, as this position is occupied by the same residue in those two ancestors. In summary, it appears that the adaptive process of acquiring GP activity can be accomplished in various ways involving different positions. None of the identified adaptive paths require any modification of active site residues.
Finally, we wondered whether the His/Arg residue in the phosphate-binding pocket, which was initially suggested to be diagnostic for the preference for phosphorolytic versus transferase activity, has any impact on the effect of the discovered adaptive mutations. The newly installed GP activity of the most active AncA variant (H186del+T187V+ T231Y+F235L+F245A) could be abolished completely by replacing Arg153 for His, i.e. the residue that is present in extant GTs. In other words, even though the presence of Arg at this active site position is not necessarily indicative of activity on α-mannose 1-phosphate, the presence of His can epistatically hinder the adaptive effect of distal mutations toward acquiring such activity.
Structural Basis for the Emergence of GP Activity
The structures of the ancestral proteins were generated using AlphaFold2,20 and were found to show high similarity to the crystal structures of their extant homologues MTP2 and MTP4 (root-mean-square deviation <0.75 Å) (Figure S8). However, a notable exception was observed in the loop containing residue 187 (AncA numbering), hereafter referred to as L1. This loop adopts a solvent-exposed conformation in strict GTs, while it faces inward in ancestors with GP activity (Figure 5A). The crystal structures of the extant homologues match this observation, with L1 exposed to the solvent in MTP2 and enclosed in a hydrophobic cavity in MTP4. Therefore, we hypothesized that the conformation of L1 may be a determinant of the GP activity. In addition to the differences in L1, the oldest bifunctional ancestors AncX and AncZ display a structured α-helix in a loop that is situated in proximity to the previously suggested function determinant Arg/His, hereafter referred to as L2 (Figures 5A, S8).
Figure 5.

Structural comparison of ancestral enzymes from the family GT108. (a) Structural superposition of ancestral enzyme models (blue, strict GTs; yellow, bifunctional GP/GTs). In loops L1 and L2, structural variations are observed between enzymes with different activities. (b) Structural superposition of AncA (blue) and its GP-active variant with mutations H186del+T187V+T231Y+F235L+F245A (yellow). The five mutated positions are indicated by spheres. Residue T187 of AncA and the corresponding residue V186 of the variant, which have different position numbers due to the deletion of H186, are shown as sticks. (c) Solvent-accessible surface area analysis for loop L1 in AncA (blue) and the GP-active variant (yellow).
To further assess whether there is a correlation between the conformation of these loops and the presence of GP activity, we generated the structure of the AncA variant that showed the highest activity on α-mannose 1-phosphate (AncA H186del+T187V+T231Y+F235L+F245A; kcat/KM = 5.2 mM–1min–1). Despite the great structural similarity between AncA and the variant (root-mean-square deviation = 0.102 Å), the variant presents a distinct L1 conformation where the loop is directed toward a hydrophobic pocket, with Val186 assuming an inward-facing orientation. In contrast, L1 is water-exposed in AncA, and the corresponding residue Thr187 is oriented toward the solvent (Figure 5B,C).
Molecular dynamics (MD) simulations (600 ns, in 3 replicas) were performed for the unliganded structures of both AncA and its GP-active variant to study the effect of the above mutations on loop dynamics (Figure S9). Analysis of the root-mean-square fluctuations (RMSF) indicated that loops L1 and L2 are considerably more rigid in the GP-active variant than in AncA (Figure S10). Mutations H186del, T187V and F245A contribute to this behavior by promoting the insertion of L1, as shown by solvent-accessible surface area analysis (619.90 ± 57.17 Å2 for AncA; 533.31 ± 31.33 Å2 for the AncA variant) (Figure 5C). In turn, L2 becomes more exposed to the solvent, and the hydrophobic residues Ala160 and Leu161 in L2 adopt new orientations to avoid solvent contact, resulting in increased rigidity.
Next, we constructed Michaelis complexes of both AncA and the GP-active variant with α-mannose 1-phosphate, unveiling an extensive network of predicted hydrogen bonds and salt bridges. Notably, residues Lys136, Arg153, His212, and Tyr229 appear to establish interactions with the phosphate moiety of the glycosyl donor, while residues Asp86 and Asp289 are predicted to interact with the mannosyl moiety. MD simulations (3.0 μs, in 3 replicas) confirmed the stability of the complex (Figure S11). Consistent with our observations with the apo structure, RMSF analysis indicated that loops L1 and L2 are less flexible in the GP-active variant (Figure 6A). These changes influence the accessible volume of the active site pocket and limit the number of water molecules capable of disrupting donor–protein interactions (Figure 6). Interestingly, there is a persistent interaction between α-mannose 1-phosphate and the catalytic base Asp86 in the simulations for the GP-active variant, whereas this interaction is easily lost in the simulations for AncA (Figures 6B,S11).
Figure 6.

Molecular dynamics simulation of AncA (blue) and its GP-active variant with mutations H186del+T187V+T231Y+F235L+F245A (yellow) in complex with α-mannose 1-phosphate (Man1P). (a) Root-mean-square fluctuation per residue. (b) Distribution of the distance between HO3Man1P and an oxygen atom of catalytic base Asp86. (c) Solvent-accessible surface area (SASA) within the pocket (residues within 6.5 Å from the donor) and its correlation with the number of water molecules in the cavity close to residues Asp86 and Asp289. (d) Representative snapshots of the structures of AncA and the GP-active variant, showing the difference in exposure of Man1P to the solvent.
The different hydrogen bond profiles observed for AncA and its GP-active variant result in changes in the conformations adopted by the sugar moiety of the donor (Figure S12). In AncA, the mannosyl group of α-mannose 1-phosphate adopts 4C1, B3,O, 1S3 and 1C4 conformations. In contrast, the GP-active variant predominantly visits conformations between B2,5 and OS2. Only the latter conformations result in productive binding that could lead to catalysis, as shown by the analysis of catalytic distances in a model ternary complex of the enzyme with α-mannose 1-phosphate and mannose (Figure S13). Our present findings are consistent with recent studies on a bacterial mannoside phosphorylase from CAZy family GH130, where α-mannose 1-phosphate also adopts the OS2 conformation.21 Structurally, the catalytic pockets of GT108 and GH130 enzymes show striking similarity, and both families are believed to employ the same reaction mechanism.12
In summary, our results suggest that the emergence of phosphorylase activity in the family GT108 is driven by the effect of distal mutations on conformational dynamics. An increased structural rigidity and a reduced accessibility of the active site for water molecules ensure that important interactions between the sugar moiety of the donor and the active site residues, including the catalytic base, are maintained. In turn, these interactions effectively keep α-mannose 1-phosphate in a catalytically productive conformation.
Discussion
Recent years have seen great successes in applying ancestral sequence reconstruction to help solve evolutionary puzzles, from unveiling how hydrolases and monooxygenases adapted to xenobiotics to elucidating the origins of fold switching and catalysis.22−28 Here, the evolution of function in a recently discovered group of CAZymes was reconstructed to track the origins of phosphorylase activity. We found that GP activity in family GT108 was established and lost at different points in time and that multiple combinations of mutations at positions located outside of the active site are capable of introducing GP activity in an ancestral GT template. The observed functional plasticity of these enzymes is likely to have facilitated the emergence of the unusual mannogen biosynthesis and cycling pathway that is tightly linked to the unique survival and virulence strategy of Leishmania parasites. These protists accumulate mannogen as a reserve carbohydrate that enables them to navigate the fluctuating conditions they experience during their complex life cycle, from the nutrient-rich surroundings in their sandfly vector after a bloodmeal to the hostile environment in the phagolysosome compartments of mammalian macrophages where they switch to a metabolically quiescent state.13,15,29−31 The MTPs that are collectively responsible for tuning this metabolic rheostat by regulating the dynamic equilibrium between mannogen and monosaccharides have all evolved from a single ancestor that was acquired from bacteria by horizontal gene transfer, represented by AncA. Our mutational study demonstrates that the specialization of AncA paralogues into separate dedicated catalysts for either the synthesis or phosphorolysis of mannogen did not require drastic adaptation and optimization at the sequence level. The overall evolutionary trajectory of some of these paralogues also presents an uncommon example of reversal of function in enzyme evolution, as it encompasses the transition from an ancestral mannosyltransferase to a bifunctional MTP and then back to a strict mannosyltransferase.
Further, our results suggest that transferases may be viable starting points for the development of novel phosphorylases that fulfill specific biotechnological needs. Over the years, various GPs with tailored substrate specificities or selectivities have already been obtained by directed evolution and (semi)rational engineering efforts, each of them starting from one of the few dozen natural phosphorylases that have been discovered to date.4,32−35 Unfortunately, given the small size of the pool of known GPs, it is not always possible to find an appropriate engineering template that can be adapted to recognize a substrate of interest without requiring extensive and challenging modifications to the active site. In those scenarios, it may be worth exploring the alternative strategy of scanning the enormous collection of characterized GTs for a template enzyme that already shows the desired specificity and subsequently evolving its glycosyl donor leaving group preference from nucleoside diphosphate to phosphate instead. A particularly appealing and rich source of templates for making such a switch would be CAZy family GT4, where at least one GP specificity is known to have naturally emerged among the large and diverse set of GTs.36
The introduction of phosphorylase activity in a hydrolase has previously been achieved by simply targeting positions near the putative phosphate-binding pocket.7,8 In contrast, our findings show that attempts to introduce phosphorylase activity in a transferase should consider the possibility that the required function-switching mutations may be situated outside the active site. Structural analyses demonstrated that distal mutations in AncA can indirectly alter the conformational dynamics of the sugar moiety of the glycosyl phosphate by affecting loop rigidity and shielding the active site from excess water molecules. Rationally predicting the effect of such distal mutations in different templates would be far from trivial, but promising computational strategies for identifying mutation hotspots across the entire protein scaffold have recently been developed.37 However, another option is to resort to the more traditional approach of directed evolution, combining random mutagenesis with one of the convenient colorimetric assays that are available for detecting the reaction products of phosphorylase activity.38−40
Finally, our study further highlights the importance of paying special attention to ambiguously reconstructed sites in ancestral sequences. It is common practice to sample alternative amino acid states from the posterior distribution at such sites to examine the robustness of the inferred ancestral function to statistical uncertainty. However, this strategy also seems to be highly useful for identifying nonobvious positions that have played a significant role in protein evolution. Even though the most likely and worst plausible sequence states of AncA exhibited inconsistent phenotypes, shuffling between those states conveniently allowed us to search the cloud of possible historical mutations for those that can be involved in functional adaptation. It thus appears that even the uncertainties that may arise during ancestral sequence reconstruction can be an unexpected and helpful source of inspiration for protein engineers.
Methods
Chemicals
All chemicals were purchased from Merck, unless otherwise noted. Ultrapure GDP-mannose was purchased from Promega.
Phylogenetic Analysis and Ancestral Sequence Reconstruction
All proteins classified in family GT108 were extracted from the CAZy database (http://www.cazy.org; accessed in May 2020) and their amino acid sequences were retrieved from GenBank. Redundant sequences were removed using CD-HIT with a sequence identity threshold of 98% and default parameters.41 The remaining 127 sequences were aligned using Clustal Omega with default parameters.42 The most appropriate substitution model for this sequence dataset was found to be LG+I+G via the Smart Model Selection tool.43 Maximum-likelihood phylogenetic analysis was performed using RAxML v8.2.12 with 1,000 rapid bootstrap inferences followed by a search for the best-scoring maximum-likelihood tree.44 The obtained tree was visualized and annotated using iTOL v5.45
Graphical Representation of Ancestral Sequence Predictions (GRASP) was used to infer the amino acid sequences at selected ancestral nodes in the phylogeny via marginal reconstruction.16 The posterior probability of each character at each position was obtained as output. The full-length ancestral sequences contained the most probable characters at each position. Sites were labeled as ambiguous when more than one character had a posterior probability >0.2. At each node of interest, a worst-plausible case or AltAll ancestor was designed which contained the second-most likely character at every ambiguously reconstructed site.19
Cloning, Expression and Purification
The sequences of MTP2 (UniProt: E9AND8), MTP4 (UniProt: E9ANE0), AsGT (UniProt: A0A6P1TG37) and the ancestral proteins (Table S5) were codon-optimized for E. coli, synthesized and subcloned into the NheI and XhoI recognition sites of a pET21a vector by Life Technologies (Belgium). As a result, all genes contained a C-terminal His6-tag. The plasmids were used for transformation of electrocompetent E. coli BL21(DE3) agp- cells.
For recombinant protein expression, an overnight culture (2%) of cells carrying the desired pET21a expression plasmid was used to inoculate 500 mL of LB medium (10 g·L–1 tryptone, 5 g·L–1 NaCl, and 5 g·L–1 yeast extract) containing 100 μg·mL–1 ampicillin. The culture was incubated at 37 °C and 200 rpm until OD600 reached 0.8, after which the temperature was lowered to 18 °C for 2 h. Then, expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM and the culture was incubated at 18 °C for 16 h, after which cells were harvested by centrifugation. The obtained cell pellets were frozen at −20 °C for at least 4 h.
Cell pellets of a 250 mL culture were resuspended in 8 mL of lysis buffer (10 mM imidazole, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mg·mL–1 lysozyme from chicken egg white, 50 mM phosphate-buffered saline; pH 7.4) and incubated on ice for 30 min. The lysate was sonicated 3 times 3 min (Branson sonifier 450, level 3, 50% duty cycle) and the cell extract was separated from the cell debris by centrifugation (20,000g for 1 h at 4 °C). The extract was purified by nickel-nitrilotriacetic acid chromatography following the instructions of the supplier (HisPur Ni-NTA; Thermo Fisher Scientific). After elution of purified protein fractions, the buffer was exchanged to 20 mM 2-morpholinoethanesulfonic acid (MES) at pH 6.5 using an Amicon centrifugal filter unit with a 30 kDa cutoff (Merck Millipore). The extinction coefficient and molecular weight of all enzymes were calculated using the ProtParam tool on the ExPASy server (http://web.expasy.org/protparam) and protein concentrations were measured using a NanoDrop ND-1000 at 280 nm (Thermo Fisher Scientific). Purified enzyme preparations were stored at −20 °C.
Enzyme Kinetics
The kinetic parameters toward α-mannose 1-phosphate and GDP-mannose were determined by incubating purified enzymes with 25 mM mannose and varying donor substrate concentrations in 20 mM MES at pH 6.5 and 27 °C. One unit of activity was defined as the amount of enzyme that produces one μmol of the relevant product (phosphate or GDP, respectively) per minute under the specified conditions. All reactions were performed in triplicate. Kinetic parameters were calculated by fitting initial reaction rates to the Michaelis–Menten equation in SigmaPlot 11.
Reverse phosphorylase activity was measured by following the release of inorganic phosphate from α-mannose 1-phosphate with the phosphomolybdate assay.40 Reaction samples (50 μL) were added to 150 μL of assay solution consisting of reagents A (12% (w/v) l-ascorbic acid in 1 N HCl) and B (2% (w/v) ammonium molybdate tetrahydrate in ddH2O) in a 2:1 ratio. After incubation for 5 min at room temperature, color development was terminated by adding 150 μL of stop solution (2% (w/v) sodium citrate tribasic dihydrate and 2% (w/v) acetic acid in ddH2O). After 15 min, absorbance was measured at 655 nm in an Anthos Zenyth 200rt microplate reader (Biochrom).
Glycosyltransferase activity was measured by following the release of GDP from GDP-mannose with the GDP-Glo assay (Promega) according to the manufacturer’s instructions. Luminescence was measured in an Infinite M200 plate reader (Tecan).
Enzyme Stability
Apparent folding transition temperatures were measured by differential scanning fluorimetry in a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) by using the FRET channel. SYPRO Orange Protein Stain (5000X in DMSO) was diluted to 50× in water and 2.5 μL of this solution was added to 22.5 μL purified protein (0.5 mg·mL–1 in 20 mM MES, pH 6.5). The temperature was gradually increased from 20 to 99 °C with increments of 0.5 °C/min. Fluorescence emissions were plotted as a function of temperature, and the Tm,app values were determined at the inflection point as the minimum of the first derivative. All measurements were performed in triplicate.
To determine kinetic stability, purified enzyme (0.1 mg·mL–1 in 20 mM MES, pH 6.5) was incubated at various temperatures between 25 and 80 °C (5 °C intervals) for 15 min and then cooled on ice. The residual specific activity in each sample was determined in reactions with 1 mM GDP-mannose and 10 mM mannose using the GDP-Glo assay and compared to the activity of untreated enzyme preparations. Three independent replicates were performed for each enzyme.
High-Performance Anion Exchange Chromatography
Reactions could be monitored by high-performance anion exchange chromatography (Dionex ICS-6000; Thermo Scientific) with CarboPac PA20 pH-stable columns and pulsed amperometric detection. A constant flow rate of 0.5 mL·min–1 was applied at 30 °C, using a three-eluent system with ultrapure water (eluent A), 100 mM NaOH (eluent B) and 1 M sodium acetate and 100 mM NaOH (eluent C). The elution profile was as follows: 0–2 min, 80% A and 20% B; 2–4 min, linear gradient to 100% B; 4–20 min, linear gradient to 85% B and 15% C; 20–20.5 min, linear gradient to 60% B and 40% C; 20.5–24 min, 60% B and 40% C; 24–24.5 min, linear gradient to 80% A and 20% B; 24.5–30 min, 80% A and 20% B.
Mutagenesis and Library Construction
The H161R mutation was introduced in MTP2 as described in the manual of the QuickChange II Site-Directed Mutagenesis Kit (Agilent) using the primers in Table S6. Mutations N106T, T187V, and F245L were introduced in AncA as described in the manual of the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) using the primers in Table S6. The library for shuffling between AncA and AncA-Alt was constructed by Life Technologies. The library for shuffling between AncA and AncB at seven positions was constructed by assembly of two DNA fragments by circular polymerase extension cloning (CPEC).46 The mutagenic library fragment (Table S7) was ordered from Telesis Bio. The backbone fragment containing the vector was amplified from a pET21a plasmid containing the AncA gene using PrimeStar GXL DNA Polymerase (Takara Bio) and primers Fw_AncA_300 and Rv_AncA_147 in Table S6. The assembly reaction (25 μL) contained 100 ng of the backbone fragment, 20 ng of the library fragment, 0.4 mM dNTPs, 0.75 μL dimethyl sulfoxide, 1X Q5 Reaction buffer, and 0.04 U/μL Q5 High-Fidelity DNA Polymerase (New England Biolabs). The cycling conditions were as follows: initial denaturation at 98 °C for 30 s followed by 15 cycles of denaturation (98 °C, 10 s), annealing (64 °C, 30 s) and extension (72 °C, 100 s), and a final extension step at 72 °C for 10 min. Characterized variants that could not be isolated after sequencing the hits from this library were generated by using the same CPEC method with mutagenic fragments ordered from Integrated DNA Technologies (Table S7).
Library Screening
After transformation of E. coli BL21(DE3) agp-, individual colonies were picked from solid LB medium supplemented with 100 μg·mL–1 ampicillin with an automated colony picking system (K63 picker; Kbiosystems) and transferred to 175 μL liquid LB medium with ampicillin in sterile flat-bottomed 96-well microtiter plates (Greiner). After overnight incubation at 37 °C and 250 rpm, the plates were replicated by transferring 1 μL of each culture to fresh medium in new plates. The initial plates were stored at −20 °C after addition of 125 μL of 70% glycerol, whereas the fresh plates were incubated at 37 °C and 250 rpm for 2.5 h. The temperature was lowered to 20 °C for 0.5 h, and expression was subsequently induced by addition of IPTG to a final concentration of 0.1 mM. After 16 h of further incubation, cells were harvested by centrifugation (10,000g, 30 min), the supernatant was discarded, and the obtained pellets were frozen at −20 °C for 2 h. Pellets were resuspended in 150 μL lysis buffer containing 50 mM MES, 0.1 mM PMSF, 1 mg·mL–1 lysozyme, 1 mM ethylenediaminetetraacetic acid, 4 mM MgSO4, and 50 mM Na2SO4, pH 6.5. After incubation at 30 °C for 1 h, cell debris was removed by centrifugation (10,000g, 30 min).
Reactions were initiated by transferring 50 μL of the lysates to new microtiter plates containing 50 μL of substrate solution to obtain a final concentration of 5 mM α-mannose 1-phosphate and 10 mM mannose. The reactions were incubated at 30 °C for 1 h, after which the phosphate concentration was measured by analyzing a 50 μL sample using the phosphomolybdate assay as described above. To visualize the amino acid distribution at the variable position in the hits, a sequence logo was constructed using WebLogo 3.47
Modeling of the Ancestral Enzymes
The generation of all ancestral protein models was conducted utilizing ColabFold v1.5.3, specifically AlphaFold 2 (AF2) employing MMseqs2 through a collaborative notebook.20 Throughout the modeling process, default settings were maintained for both the multiple sequence alignment options and the advanced settings. The analysis exclusively considered models ranking first for each respective ancestor.
Molecular Dynamics Simulations
Molecular dynamics simulations were executed utilizing the AF2 models of AncA and its GP-active variant (H186del+T187V+T231Y+F235L+F245A), both in unbound form and complexed with α-mannose 1-phosphate. For the construction of the complex, structures of MTP2 in complex with mannose (PDB ID: 6Q4Y) and MTP4 with phosphate (PDB ID: 6Q50) were superimposed onto the AF2 models using PyMOL (Shrödinger), preserving the mannose and inorganic phosphate moieties to form α-mannose 1-phosphate. Simulations were carried out at pH 6.5, with all arginine and lysine residues positively charged, while aspartate and glutamate residues, along with the phosphate group of the donor, were negatively charged. Protonation states of histidine residues were determined using the H+2 server48 and guided by chemical intuition.
Each system was positioned at the center of a periodically repeated rectangular box, maintaining a minimum distance of 10 between the solute surface and the box edge. The dimensions varied for different configurations: 79.094 × 79.666 × 78.018 Å3 for AncA, 79.094 × 77.891 × 78.411 Å3 for the GP-active variant, 78.672 × 79.666 × 80.340 Å3 for AncA in complex with the donor, and 77.061 × 79.666 × 80.340 Å3 for the GP-active variant in complex with the donor. Solvation of AncA and variant boxes included 11,706 and 11,412 TIP3P water molecules in plain systems (i.e., unliganded enzyme), and 11,973 and 11,610 TIP3P water molecules in complex systems.49 Sodium ions were introduced to maintain box neutrality, comprising 13 ions in plain systems and 15 ions in complex systems. The protein was characterized using the Amber ff14SB force field,50 while the GLYCAM06 force field51 described the mannose unit in both complex systems. Atomic partial charges (RESP) for the phosphate portion in both complex systems were computed at the HF/6-31G* level of theory using Gaussian09.52 Topology and coordinate files for classical MD simulations were generated with the LEaP module of AmberTools21.53 Simulations were carried out using Amber2053 with the CUDA version of the PMEMD module.54 The solvated system underwent energy minimization with a two-step protocol. First, the solvent was minimized with 5000 steps of steepest descent and 5000 steps of conjugate gradient minimization with positional restraints on the rest of the system. Subsequently, the entire system was minimized with 15,000 steps of steepest descent and conjugate gradient (7500 each). For complex systems, an additional intermediate step of minimization was added to relax both the solvent and the ligand, while the protein was fixed. The system was gradually heated to 300 K in the NVT ensemble using the Berendsen thermostat, with positional restraints applied to the heavy atoms of the protein in plain simulations and to both the protein and the donor in complex simulations. The restraints were gradually decreased over three 500 ps steps, followed by 1000 ps of NPT equilibration by using the Berendsen thermostat and barostat to maintain the system at 300 K and 1 atm for density equilibration. Triplicate MD runs were performed for all systems, with plain systems simulated for 200 ns (600 ns for both AncA and the variant) and complex systems simulated for 1 μs (3 μs for both AncA and the variant). Root-mean-squared deviation analysis was employed to evaluate the system stability. The SHAKE algorithm55 constrained all bonds involving hydrogen atoms, and the time step was set to 2 fs. Production runs in the NPT ensemble were controlled by a Langevin thermostat and a Berendsen barostat. All MD analyses were conducted employing the CPPTRAJ module of AmberTools21, complemented by VMD tools.56 Postprocessing was carried out in a Python Jupyter Notebook. Puckering analysis was performed by utilizing a custom Python-based script developed in-house. Visualization and rendering of molecular structures were done by using PyMOL and VMD.
Availability of Expression Plasmids
The codon-optimized nucleotide sequences encoding the characterized ancestral and extant sequences have been deposited in the Genbank database under accession codes: OR742801 (AncA), OR742802 (AncA-Alt), OR742803 (AncB), OR742804 (AncC), OR742805 (AncX), OR742806 (AncY), OR742807 (AncZ), OR742808 (MTP2), OR742809 (MTP4), and OR742810 (AsGT). The plasmids for expression of these enzymes have been made publicly available at the BCCM/GeneCorner plasmid collection.
Acknowledgments
This work was supported by Research Foundation – Flanders (FWO-Vlaanderen; postdoctoral fellowship J.F. (grant nos. 12ZD821N and 1226424N); doctoral fellowship J.L. (grant no. 1155922N), the Spanish Ministry of Science and Innovation (MICINN/AEI/FEDER, UE, PID2020-118893GB-100, to C.R.), the Spanish Structures of Excellence María de Maeztu (CEX2021-001202-M, to C.R.), the Agency for Management of University and Research Grants of Catalonia (AGAUR, 2021-SGR-00680, to C.R.), and the European Research Council (ERC-2020-SyG-951231 “Carbocentre”, to C.R.). J.P.R.F. was supported by MICINN (FPI fellowship PRE2021-097898). We would like to thank the Ghent University Core Facility “HTS for Synthetic Biology” for training, support and access to the instrument park.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c05819.
Phylogenetic tree of GT108; evaluation of kinetic stability and product profiles; multiple sequence alignment and comparison of active site residues; overview of the obtained hits; analyses of molecular dynamics simulations; apparent kinetic parameters; sequences of primers and DNA fragments (PDF)
Author Contributions
J.F. and T.D. conceived the study. J.F. performed the ancestral sequence reconstruction. J.F. and J.L. performed the experimental work including the expression, purification and characterization of extant and ancestral enzymes, as well as the design, construction and screening of libraries. J.P.R.F. performed the structural analyses and molecular dynamics simulations. T.D. and C.R. supervised the work and provided critical feedback. J.F. wrote the initial draft of the manuscript, except for the section on structural analyses which was written by J.P.R.F. All authors contributed to the further editing of the manuscript. All authors approved of the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Franceus J.; Lormans J.; Desmet T. Building Mutational Bridges between Carbohydrate-Active Enzymes. Curr. Opin. Biotechnol. 2022, 78, 102804. 10.1016/j.copbio.2022.102804. [DOI] [PubMed] [Google Scholar]
- Awad F. N. Glycoside Phosphorylases for Carbohydrate Synthesis: An Insight into the Diversity and Potentiality. Biocatal. Agric. Biotechnol. 2021, 31, 101886. 10.1016/j.bcab.2020.101886. [DOI] [Google Scholar]
- Nishimoto M. Large Scale Production of Lacto-N-Biose I, a Building Block of Type I Human Milk Oligosaccharides, Using Sugar Phosphorylases. Biosci. Biotechnol. Biochem. 2020, 84 (1), 17–24. 10.1080/09168451.2019.1670047. [DOI] [PubMed] [Google Scholar]
- Ubiparip Z.; Moreno D. S.; Beerens K.; Desmet T. Engineering of Cellobiose Phosphorylase for the Defined Synthesis of Cellotriose. Appl. Microbiol. Biotechnol. 2020, 104, 8327. 10.1007/s00253-020-10820-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Gu Y.; Quan Y.; Gao W.; Dang Y.; Cao M.; Lu X.; Wang Y.; Song C.; Wang S. Construction of Energy-Conserving Sucrose Utilization Pathways for Improving Poly-γ-Glutamic Acid Production in Bacillus Amyloliquefaciens. Microb. Cell Factories 2017, 16 (1), 98. 10.1186/s12934-017-0712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drula E.; Garron M.; Dogan S.; Lombard V.; Henrissat B.; Terrapon N. The Carbohydrate-Active Enzyme Database: Functions and Literature. Nucleic Acids Res. 2022, 50 (D1), D571–D577. 10.1093/nar/gkab1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceus J.; Lormans J.; Cools L.; Desmet T. Evolution of Phosphorylases from N-Acetylglucosaminide Hydrolases in Family GH3. ACS Catal. 2021, 11 (10), 6225–6233. 10.1021/acscatal.1c00761. [DOI] [Google Scholar]
- Teze D.; Coines J.; Raich L.; Kalichuk V.; Solleux C.; Tellier C.; André-Miral C.; Svensson B.; Rovira C. A Single Point Mutation Converts GH84 O-GlcNAc Hydrolases into Phosphorylases: Experimental and Theoretical Evidence. J. Am. Chem. Soc. 2020, 142 (5), 2120–2124. 10.1021/jacs.9b09655. [DOI] [PubMed] [Google Scholar]
- Gibson R. P.; Turkenburg J. P.; Charnock S. J.; Lloyd R.; Davies G. J. Insights into Trehalose Synthesis Provided by the Structure of the Retaining Glucosyltransferase OtsA. Chem. Biol. 2002, 9 (12), 1337–1346. 10.1016/S1074-5521(02)00292-2. [DOI] [PubMed] [Google Scholar]
- Goedl C.; Griessler R.; Schwarz A.; Nidetzky B. Structure–Function Relationships for Schizophyllum Commune Trehalose Phosphorylase and Their Implications for the Catalytic Mechanism of Family GT-4 Glycosyltransferases. Biochem. J. 2006, 397 (3), 491–500. 10.1042/BJ20060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill E. C.; Field R. A. Enzymatic Synthesis Using Glycoside Phosphorylases. Carbohydr. Res. 2015, 403, 23–37. 10.1016/j.carres.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sernee M. F.; Ralton J. E.; Nero T. L.; Sobala L. F.; Kloehn J.; Vieira-Lara M. A.; Cobbold S. A.; Stanton L.; Pires D. E. V.; Hanssen E.; Males A.; Ward T.; Bastidas L. M.; van der Peet P. L.; Parker M. W.; Ascher D. B.; Williams S. J.; Davies G. J.; McConville M. J. A Family of Dual-Activity Glycosyltransferase-Phosphorylases Mediates Mannogen Turnover and Virulence in Leishmania Parasites. Cell Host Microbe 2019, 26 (3), 385–399. 10.1016/j.chom.2019.08.009. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Beverley S. M. Mannogen-Ing Central Carbon Metabolism by Leishmania. Trends Parasitol 2019, 35 (12), 947–949. 10.1016/j.pt.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralton J. E.; Naderer T.; Piraino H. L.; Bashtannyk T. A.; Callaghan J. M.; McConville M. J. Evidence That Intracellular Β1–2 Mannan Is a Virulence Factor in Leishmania Parasites. J. Biol. Chem. 2003, 278 (42), 40757–40763. 10.1074/jbc.M307660200. [DOI] [PubMed] [Google Scholar]
- Ralton J. E.; Sernee M. F.; McConville M. J. Evolution and Function of Carbohydrate Reserve Biosynthesis in Parasitic Protists. Trends Parasitol 2021, 37 (11), 988–1001. 10.1016/j.pt.2021.06.005. [DOI] [PubMed] [Google Scholar]
- Foley G.; Mora A.; Ross C. M.; Bottoms S.; Sützl L.; Lamprecht M. L.; Zaugg J.; Essebier A.; Balderson B.; Newell R.; Thomson R. E. S.; Kobe B.; Barnard R. T.; Guddat L.; Schenk G.; Carsten J.; Gumulya Y.; Rost B.; Haltrich D.; Sieber V.; Gillam E. M. J.; Bodén M. Engineering Indel and Substitution Variants of Diverse and Ancient Enzymes Using Graphical Representation of Ancestral Sequence Predictions (GRASP). PLOS Comput. Biol. 2022, 18 (10), e1010633 10.1371/journal.pcbi.1010633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R. E. S.; Carrera-Pacheco S. E.; Gillam E. M. J. Engineering Functional Thermostable Proteins Using Ancestral Sequence Reconstruction. J. Biol. Chem. 2022, 298 (10), 102435. 10.1016/j.jbc.2022.102435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau D. L.; Kaltenbach M.; Tawfik D. S. On the Potential Origins of the High Stability of Reconstructed Ancestral Proteins. Mol. Biol. Evol. 2016, 33 (10), 2633–2641. 10.1093/molbev/msw138. [DOI] [PubMed] [Google Scholar]
- Eick G. N.; Bridgham J. T.; Anderson D. P.; Harms M. J.; Thornton J. W. Robustness of Reconstructed Ancestral Protein Functions to Statistical Uncertainty. Mol. Biol. Evol. 2017, 34 (2), 247–261. 10.1093/molbev/msw223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M.; Schütze K.; Moriwaki Y.; Heo L.; Ovchinnikov S.; Steinegger M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19 (6), 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Prieto M.; Cuxart I.; Potocki-Véronèse G.; André I.; Rovira C. Substrate-Assisted Mechanism for the Degradation of N -Glycans by a Gut Bacterial Mannoside Phosphorylase. ACS Catal. 2023, 13 (7), 4283–4289. 10.1021/acscatal.3c00451. [DOI] [Google Scholar]
- Gumulya Y.; Gillam E. M. J. Exploring the Past and the Future of Protein Evolution with Ancestral Sequence Reconstruction: The’retro Approach to Protein Engineering. Biochem. J. 2017, 474 (1), 1–19. 10.1042/BCJ20160507. [DOI] [PubMed] [Google Scholar]
- Kaltenbach M.; Burke J. R.; Dindo M.; Pabis A.; Munsberg F. S.; Rabin A.; Kamerlin S. C. L.; Noel J. P.; Tawfik D. S. Evolution of Chalcone Isomerase from a Noncatalytic Ancestor. Nat. Chem. Biol. 2018, 14 (6), 548–555. 10.1038/s41589-018-0042-3. [DOI] [PubMed] [Google Scholar]
- Clifton B. E.; Kaczmarski J. A.; Carr P. D.; Gerth M. L.; Tokuriki N.; Jackson C. J. Evolution of Cyclohexadienyl Dehydratase from an Ancestral Solute-Binding Protein. Nat. Chem. Biol. 2018, 14 (6), 542–547. 10.1038/s41589-018-0043-2. [DOI] [PubMed] [Google Scholar]
- Yang G.; Anderson D. W.; Baier F.; Dohmen E.; Hong N.; Carr P. D.; Kamerlin S. C. L.; Jackson C. J.; Bornberg-Bauer E.; Tokuriki N. Higher-Order Epistasis Shapes the Fitness Landscape of a Xenobiotic-Degrading Enzyme. Nat. Chem. Biol. 2019, 15 (11), 1120–1128. 10.1038/s41589-019-0386-3. [DOI] [PubMed] [Google Scholar]
- Bailleul G.; Yang G.; Nicoll C. R.; Mattevi A.; Fraaije M. W.; Mascotti M. L. Evolution of Enzyme Functionality in the Flavin-Containing Monooxygenases. Nat. Commun. 2023, 14 (1), 1042. 10.1038/s41467-023-36756-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joho Y.; Vongsouthi V.; Spence M. A.; Ton J.; Gomez C.; Tan L. L.; Kaczmarski J. A.; Caputo A. T.; Royan S.; Jackson C. J.; Ardevol A. Ancestral Sequence Reconstruction Identifies Structural Changes Underlying the Evolution of Ideonella Sakaiensis PETase and Variants with Improved Stability and Activity. Biochemistry 2023, 62 (2), 437–450. 10.1021/acs.biochem.2c00323. [DOI] [PubMed] [Google Scholar]
- Dishman A. F.; Tyler R. C.; Fox J. C.; Kleist A. B.; Prehoda K. E.; Babu M. M.; Peterson F. C.; Volkman B. F. Evolution of Fold Switching in a Metamorphic Protein. Science 2021, 371 (6524), 86–90. 10.1126/science.abd8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders E. C.; De Souza D. P.; Naderer T.; Sernee M. F.; Ralton J. E.; Doyle M. A.; Macrae J. I.; Chambers J. L.; Heng J.; Nahid A.; Likic V. A.; Mcconville M. J. Central Carbon Metabolism of Leishmania Parasites. Parasitology 2010, 137 (9), 1303–1313. 10.1017/S0031182010000077. [DOI] [PubMed] [Google Scholar]
- McConville M. J.; Saunders E. C.; Kloehn J.; Dagley M. J. Leishmania Carbon Metabolism in the Macrophage Phagolysosome- Feast or Famine?. F1000Research 2015, 4, 938. 10.12688/f1000research.6724.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders E. C.; Sernee M. F.; Ralton J. E.; McConville M. J. Metabolic Stringent Response in Intracellular Stages of Leishmania. Curr. Opin. Microbiol. 2021, 63, 126–132. 10.1016/j.mib.2021.07.007. [DOI] [PubMed] [Google Scholar]
- Sun S.; You C. Disaccharide Phosphorylases: Structure, Catalytic Mechanisms and Directed Evolution. Synth. Syst. Biotechnol. 2021, 6 (1), 23–31. 10.1016/j.synbio.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T.; Nishimoto T.; Chaen H.; Fukuda S. Improvement of the Enzymatic Properties of Kojibiose Phosphorylase from Thermoanaerobacter Brockii by Random Mutagenesis and Chimerization. J. Appl. Glycosci. 2006, 53 (2), 123–129. 10.5458/jag.53.123. [DOI] [Google Scholar]
- De Groeve M. R. M.; De Baere M.; Hoflack L.; Desmet T.; Vandamme E. J.; Soetaert W. Creating Lactose Phosphorylase Enzymes by Directed Evolution of Cellobiose Phosphorylase. Protein Eng. Des. Sel. PEDS 2009, 22 (7), 393–399. 10.1093/protein/gzp017. [DOI] [PubMed] [Google Scholar]
- Franceus J.; Desmet T. Sucrose Phosphorylase and Related Enzymes in Glycoside Hydrolase Family 13: Discovery, Application and Engineering. Int. J. Mol. Sci. 2020, 21 (7), 2526. 10.3390/ijms21072526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchart V. Glycoside Phosphorylases: Structure, Catalytic Properties and Biotechnological Potential. Biotechnol. Adv. 2015, 33 (2), 261–276. 10.1016/j.biotechadv.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Osuna S. The Challenge of Predicting Distal Active Site Mutations in Computational Enzyme Design. WIREs Comput. Mol. Sci. 2021, 11, 3. 10.1002/wcms.1502. [DOI] [Google Scholar]
- Arnold F. H. Innovation by Evolution: Bringing New Chemistry to Life (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2019, 58 (41), 14420–14426. 10.1002/anie.201708408. [DOI] [PubMed] [Google Scholar]
- Silverstein R.; Voet J.; Reed D.; Abeles R. Purification and Mechanism of Action of Sucrose Phosphorylase. J. Biol. Chem. 1967, 242 (6), 1338–1346. 10.1016/S0021-9258(18)96185-5. [DOI] [PubMed] [Google Scholar]
- Gawronski J. D.; Benson D. R. Microtiter Assay for Glutamine Synthetase Biosynthetic Activity Using Inorganic Phosphate Detection. Anal. Biochem. 2004, 327 (1), 114–118. 10.1016/j.ab.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Niu B.; Gao Y.; Fu L.; Li W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 2010, 26 (5), 680–682. 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Söding J.; Thompson J. D.; Higgins D. G. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort V.; Longueville J.-E.; Gascuel O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34 (9), 2422–2424. 10.1093/molbev/msx149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30 (9), 1312–1313. 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I.; Bork P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan J.; Tian J. Circular Polymerase Extension Cloning for High- Throughput Cloning of Complex and Combinatorial DNA Libraries. Nat. Protoc. 2011, 6 (2), 242–251. 10.1007/978-1-62703-764-8_8. [DOI] [PubMed] [Google Scholar]
- Crooks G.; Hon G.; Chandonia J.; Brenner S. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. 10.1101/gr.849004.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandakrishnan R.; Aguilar B.; Onufriev A. V. H++ 3.0: Automating pK Prediction and the Preparation of Biomolecular Structures for Atomistic Molecular Modeling and Simulations. Nucleic Acids Res. 2012, 40 (W1), W537–W541. 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner K. N.; Yongye A. B.; Tschampel S. M.; González-Outeiriño J.; Daniels C. R.; Foley B. L.; Woods R. J. GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates. J. Comput. Chem. 2008, 29 (4), 622–655. 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.. Gaussian 09, Revision B.01; Gaussian, Inc; Wallingford CT, 2009. [Google Scholar]
- Case D. A.; Aktulga H. M.; Belfon K.; Ben-Shalom I.; Brozell S. R.; Cerutti D. S.; Cheatham III T. E.; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.. Amber 2021; University of California; San Francisco, 2021. [Google Scholar]
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8 (5), 1542–1555. 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. Visual Molecular Dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.