

Abstract
The efficient management of skin wounds for rapid and scarless healing represents a major clinical unmet need. Nonhealing skin wounds and undesired scar formation impair quality of life and result in high healthcare expenditure worldwide. The skin-colonizing microbiota contributes to maintaining an intact skin barrier in homeostasis, but it also participates in the pathogenesis of many skin disorders, including aberrant wound healing, in many respects. This review focuses on the composition of the skin microbiome in cutaneous wounds of different types (i.e. acute and chronic) and with different outcomes (i.e. nonhealing and hypertrophic scarring), mainly based on next-generation sequencing analyses; furthermore, we discuss the mechanistic insights into host–microbe and microbe–microbe interactions during wound healing. Finally, we highlight potential therapeutic strategies that target the skin microbiome to improve healing outcomes.
Keywords: Skin microbiome, 16S Sequencing, Wound healing, Scarring
Highlights.
This paper summarizes recent research regarding the association of the skin microbiome with wound healing over different time courses and leading to different healing results.
This paper also addresses the pathogenesis of microbiota dysbiosis in nonhealing or aberrantly healed wounds that could guide novel wound treatment modalities.
Background
The skin microbiota refers to the community of microorganisms residing on the skin, that together with their collective genomes, which have become detectable with the advent of next-generation sequencing technology targeting microbial-specific genes, are known as the microbiome. As skin is the largest and most exposed organ in the human body, the skin microbiota contains trillions of microorganisms, including bacteria, fungi and viruses, which intimately interact with the host to regulate host health and diseases.
Skin wounds are a broad category of injuries ranging from minor cuts and scratches to surgical incisions, burns and serious trauma. Minor and superficial injuries usually heal well in healthy individuals; however, larger and deeper injuries may result in a variety of pathological healing modalities, including chronic nonhealing ulcers and overly healed hypertrophic scars and keloids. Breakage of the skin barrier during injury allows for colonization of microbes that are not usually found on the skin and/or translocation of constituents of microbiota to places where they do not normally appear. Studies show that the microbiota might be beneficial for wound healing by modulating the innate immune response and facilitating regeneration [1–4]. However, the entrance of defined microorganisms with high proliferative and inflammatory potentials into the underlying skin tissue may induce a broad recall response by activating skin-resident microbiota-specific T cells and cause sustained inflammation and hence delayed healing [5].
In this review, we describe our current understanding of the skin microbiota and the physiology of wound healing. We highlight the association of the wound microbiome with clinical outcomes and gather mechanistic insights into the role of the microbiome in aiding or hampering wound healing. We also discuss how, based on an improved understanding of the microbe–host interactions in wounds, microbiota-based mechanisms may inform future therapeutic interventions.
Review
Skin microbiota and its role in homeostasis and diseases
The skin microbiota
Using 16S rRNA gene sequencing, at least 19 bacterial phyla and over 1000 bacterial species have been identified on the human skin surface [6]. In general, skin bacteria predominantly belong to three phyla, Actinobacteria (51.8%), Firmicutes (24.4%) and Proteobacteria (16.5%), with the most represented genera being Corynebacteria, Cutibacteria (formerly Propionibacteria) and Staphylococci [6]. Both culture-based methods and genomic methodologies have confirmed that Malassezia spp. predominate among fungal communities on human skin, especially on the torso and the arm, whereas the foot is colonized by more diverse types of fungi [7]. For the skin viral community (virome), metagenomics has identified a large number of Caudovirales bacteriophage and single-stranded DNA viruses, including polyomaviruses, papillomaviruses and circoviruses [8–11]. In contrast to bacteria and fungi, viruses show little site specificity in skin colonization [12].
The initial colonization of skin microbes depends on the mode of delivery, with vaginally delivered babies acquiring microorganisms similar to their mother’s vagina and babies delivered by Cesarean sections colonized with microbiota similar to that associated with the skin [13]. As the babies grow up, their skin microbiome gradually achieves a typical profile with site-specific colonization (Table 1), and most sites remain surprisingly stable over time [6, 12]. The major shift occurs at puberty when sex hormones drive the maturation of the sebaceous glands and initiate sebum production, favoring the expansion of lipophilic Cutibacterium spp., Staphylococcus spp. and Malassezia spp. on oily sites. The pilosebaceous unit is especially suitable for the existence of the anaerobe Cutibacterium acnes (C. acnes), which hydrolyses triglycerides in the sebum and releases free fatty acids to promote adherence [14, 15]. These bacteria also produce propionic acid to maintain an acidic pH in the pilosebaceous follicles and bacteriocins that prevent other microbes from colonizing the sebaceous ducts [16]. Skin sites with higher moisture and occlusion (e.g. the groin, axilla and umbilicus) are preferentially enriched with Corynebacterium spp. and Staphylococcus spp. [6]. Staphylococcus epidermidis (S. epidermidis) tolerates high salt concentrations and antibacterial molecules in sweat and may even use urea within sweat as a source of nutrients. The microbial profile of dry sites such as the forearm, hypothenar palm and buttock is more diverse and unstable, with a greater prevalence of β-Proteobacteria and Flavobacteriales [6]. In addition to intrinsic factors such as age, gender and body site, constituents of the skin microbiome vary according to the external environment, such as place of residence [17].
While the epidermal compartment harbors the vast majority of skin microorganisms, recent studies have shown that bacteria are also in the deeper dermis and dermal adipose tissue in healthy people, allowing for physical contact between microbes and immune cells, including innate immune cells, innate lymphoid cells and adaptive immune cells [18, 19]. The dermal microbiome exhibits a distinct profile compared to the epidermis, albeit with evident interpersonal variations. A higher proportion of Proteobacteria and Actinobacteria and a lower abundance of Firmicutes were observed in facial and palm skin samples from 6 healthy individuals [18], and Clostridiales and Bacteroidetes were found to be enriched in deep skin biopsies from 16 patients with noninflammatory skin diseases [20]. How these microbes that usually reside on the superficial skin translocate across the epidermal layer remains to be determined.
The microbiota helps maintain skin barrier function in homeostasis
The skin establishes a symbiotic relationship with its resident microbiota that forms a first line of host defense against pathogens through several strategies (Figure 1). These include the cornified, stratified layers of keratinocytes as a physical barrier, an acidic or salty niche as a chemical barrier, surveillance by tissue-resident immune cells, and the microbial barrier itself [21]. The most abundant and stable skin microbes are beneficial for skin homeostasis, as they compete with other microbes for space and nutrients, secrete antimicrobial proteins and proteases, and have evolved mechanisms to interfere with pathways vital for their rivals’ colonization [21] (colonization resistance).
Figure 1.

The microbiota in skin homeostasis and diseases. Skin commensals constitute an important microbial barrier of the skin in steady state. Microbiota-derived metabolites together with sweat create an uninhabitable chemical environment for other microbes on the skin surface. They directly produce or stimulate keratinocyte production of AMPs. Commensal-derived LTA attenuates keratinocyte-mediated cutaneous inflammation. Skin microbiobes also stimulate skin dendritic cell production of anti-inflammatory cytokines and IL-1 to prime CD17+ T cells for defense against pathogens such as Leishmania major and C. albicans. Bacteria are also found in the deep dermis in healthy skin, but their roles remain unknown. When the skin barrier is compromised due to the loss of an intact physical barrier (stratified keratinocytes), a change in the chemical niche or an overgrowth of pathogens, neutrophils and macrophages at early phases and effector T cells at later phases are recruited and activated to elicit skin inflammation. S. aureus amplifies skin sensitization to allergens, upregulates IL-36 and IgE expression and eventually contributes to a heightened risk of developing allergic lung inflammation. C. acnes Cutibacterium acnes, S. epidermidis Staphylococcus epidermidis, S. aureus Staphylococcus aureus, P. aeruginosa Pseudomonas aeruginosa, C. albicans Candida albicans, LTA lipoteichoic acid, AMP antimicrobial peptides, SCFA short-chain fatty acid, DC dendritic cell, IL-1 interleukin-1, IgE immunoglobulin E
Additionally, although the skin microbiota is not required for the seeding of immune cells residing in the skin due to the role of the gut microbiota in promoting gut-associated lymphoid structure development, it provides protective immunity for cutaneous infections as an adjuvant to the host’s immune system [22]. For instance, skin microbes directly produce or regulate keratinocyte and immune cell expression of innate factor antimicrobial peptides (AMPs), such as cathelicidins and β-defensins [23, 24]. They educate and prime skin-resident T cells for a better response to limit pathogen invasion. S. epidermidis-loaded dendritic cell-derived interleukin-1 (IL-1) could polarize skin-homing T cells into IL-17A-producing CD8+ T (Tc17) cells, which are important in promoting the keratinocyte production of AMPs [S100 calcium binding protein A8–A9 complex (S100A8/9)] to fight against pathogens such as Leishmania major and Candida albicans (C. albicans) [5, 22]. S. epidermidis can also prevent exaggerated inflammation by promoting monocyte-derived dendritic cell (DC) secretion of the anti-inflammatory cytokine IL-10 [25] and by producing lipoteichoic acid (LTA), which decreases keratinocyte cytokine production triggered by RNA released from damaged cells [1]. Intriguingly, in sharp contrast to immunity against pathogen invasion, such ‘homeostatic immunity’ happens silently without actively recruiting immune cells to the skin; thus, no inflammation occurs [2, 5, 26].
Microbiota dysbiosis is associated with diseases
A delicate balance is achieved between the host and skin microbiota in the steady state, and various skin diseases can occur upon perturbation of the balance by either side (Figure 1). Normal constituents of the commensal microbiota can cause infections in certain cases (defined as opportunistic infection) and contribute to the initiation and/or amplification of skin disorders. Whether these microbes are friends or foes to humans is highly context dependent, profoundly influenced by the host’s immune system, metabolism and genetic predisposition, bioburden, virulence and location of the microorganism, as well as microbe–microbe interactions. For instance, after the acute infection is resolved, herpes simplex virus 1 usually lurks in the host and establishes lifelong latency but can be reactivated when the host is immunocompromised [27]. As frequent opportunists of the skin, certain C. acnes strains with high inflammatory potential cause acne lesions, and S. aureus, when overwhelming the commensal microbiota, is responsible for most bacterial skin and soft tissue infections (e.g. furuncles and cellulitis) [28] and is also associated with immune diseases, including atopic dermatitis (AD) [24, 29] and psoriasis [30].
In addition to skin-specific disorders, the skin microbiota has been associated with systemic inflammatory diseases. Repetitive exposure to skin allergens due to skin barrier defects, such as those seen in AD patients with dysfunctional filaggrin, at early life stages leads to allergen sensitization and subsequent development of systemic allergies, including rhinitis and asthma [31]. This process is dependent on the recruitment of skin microbiota-induced antigen-presenting cells, as allergic inflammation was largely reduced in the absence of skin antigen-presenting cells in neonatal mice and adult germ-free mice [32]. The most common colonizer on AD skin, S. aureus, also plays important roles. Superantigen secreted by S. aureus augments ovalbumin-induced allergic lung inflammation by upregulating the IL-17A-associated immune response [33]. Epicutaneous exposure to S. aureus induces keratinocyte release of IL-36, increasing serum immunoglobulin E (IgE) levels and eventually leading to allergen-specific lung inflammation [34]. Through an undefined mechanism, chronic skin inflammation in psoriasis has been linked to an increased risk of atherosclerotic cardiovascular diseases [35–37]. Therefore, it is plausible that the unique skin microbiome in psoriasis patients might play a role in this [30, 38], which remains an interesting question and further studies are needed.
Wound healing processes
The cutaneous wound healing process is traditionally classified into the following three overlapping phases: inflammation, tissue regeneration/proliferation and remodeling [39]. Upon full-thickness skin injury, a blood clot forms to plug the defect as a temporary repair, which also initiates inflammation. The coagulation factors trigger classical and alternative complement cascades, and activated platelets release cytokines and growth factors that attract circulating neutrophils and monocytes to the injured site and activate them [40]. Neutrophils are phagocytosed by macrophages (i.e. efferocytosis) quickly once they eliminate infectious agents and devitalized host tissue, unless pathogens remain [40]. Monocyte-derived and tissue-resident macrophages, with the help of transforming growth factor (TGF)-β, degrade and remove injured tissue debris and apoptotic neutrophils [41]. These macrophages demonstrate a pro-inflammatory M1 phenotype [producing IL-1, tumor necrosis factor (TNF), inducible nitric oxide synthase (iNOS) and reactive oxygen species (ROS)] at the acute phase and convert to an anti-inflammatory M2 profile [producing IL-10, matrix metalloproteinase (MMP), TGF-β, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and epidermal growth factor (EGF)] at later stages, which is favorable for constructive tissue remodeling and guides tissue repair in a regenerative manner [42].
A few hours after injury, keratinocytes change their gene expression profile to initiate re-epithelialization, which is stimulated by keratinocyte growth factor 1 that is mainly produced by fibroblasts [43]. They migrate to damaged sites and proliferate in response to chemotactic cytokines and growth factors, including epidermal growth factor receptor (EGFR) ligands, the FGF family and TGF-α/β [44]. Meanwhile, granulation tissue consisting of macrophages, fibroblasts, loose connective tissue and new blood vessels is formed to replace the initial clot and start fibroplasia and neovascularization [40]. At this stage, growth factors, especially TGF-β, stimulate fibroblasts to proliferate and secrete collagen, fibronectin, glycosaminoglycans, proteoglycans and hyaluronic acid [45]. After granulation tissue formation, fibroblasts differentiate into α-smooth muscle actin-expressing myofibroblasts, which is a result of the increased stiffness of the wound compared to normal skin [46]. Then, large numbers of T cells infiltrate at the late proliferative phase and regulate the transition from inflammation to tissue remodeling [47, 48].
In the third phase, the cellularity of the wound gradually decreases. Macrophages, endothelial cells and myofibroblasts undergo apoptosis, representing the resolution of inflammation, involvement of vascularization and cessation of contraction, respectively [49, 50]. Under the regulation of MMPs and their inhibitors, collagen type III in the extracellular matrix (ECM) is replaced by collagen type I, the hyaluronan and glycosaminoglycan content decreases, and the more resilient and stiff molecules of the proteoglycan family increase [51]. Finally, a scar is formed that resembles normal skin tissue but is less elastic and resilient due to the altered organization and composition of the ECM. Not all wounds heal in a timely manner. Continuous infection exaggerates local inflammation and prevents the stage transition from inflammation to proliferation. In the case of redundant collagen deposition, dysfunctional fibrotic tissues are formed, namely, hypertrophic scars or keloids, according to the extent of fibroplasia.
The microbiome in acute wounds
Bacterial and fungal communities in acute wounds
Because of the good prognosis, acute wounds are often overlooked in the field of wound microbiome study, and only limited information is available. Burn injury provides a typical acute wound model. It increases epithelial permeability, and thus, skin microbes are more likely to translocate to deeper tissue and cause infection. Burns can significantly alter the skin microbial profile, even in unaffected areas, increasing the abundance of thermophile microbes such as Aeribacillus, Caldalkalibacilus and Nesterenkonia while decreasing skin commensals such as Cutibacteria, Staphylococci and Corynebacteria [52, 53]. These changes are associated with certain outcomes of wound healing. The abundance of Corynebacterium correlates positively with wound infections, whereas Staphylococci and Cutibacteria have a negative correlation with postburn infections [52]. In terms of changes in bacterial diversity, contradictory results have been obtained by different studies, possibly due to the small sample size and use of antimicrobial agents [52, 53]. Investigation of recently healed burn wounds also revealed an altered composition, with Staphylococcus overgrowth, reduced community richness and increased bacterial diversity compared to healthy individuals [54]. Other forms of acute wounds also change skin microbial diversity and composition depending on the type of injury, and longitudinal observations suggest that those changes are dynamic in nature. Nevertheless, the microbiome in acute wounds gradually becomes similar to the adjacent skin microbiome with healing [55, 56]. Such a temporal and dynamic microbiome and its metabolome may be exploited to identify novel biomarkers that correspond to specific wound-healing processes [57].
As a known serious complication of traumatic wounds, the emergence of invasive fungal infection was identified in combat-related wounds of military personnel during the Afghanistan War [58], and it is associated with high mortality and morbidity, including residual limb shortening and high-level amputations (defined by either a hemipelvectomy or hip disarticulation) [58–60]. The presence of invasive molds (such as those of the order Mucorales, Aspergillus and Fusarium spp.) significantly delayed wound closure compared to wounds without fungal infection with similar injury pattern [59, 60], as measured by wound cultures. The coexistence of bacteria such as Enterococci is usually observed in invasive fungal infection, increasing the complexity of traumatic wounds [59]. However, no sequencing data regarding the fungal community in traumatic wounds and thus no comprehensive fungal profile are available to date.
Skin commensals may promote wound healing
Sometimes, bacteria inhabit the wound bed as passive residents without arousing active infection and leave the healing process uninterrupted. Moreover, there is a growing body of evidence showing that a mixed population of skin commensals can actively interact with the immune system to promote wound healing. Constant encounter with symbionts, even a low level of bacteria, in the steady state leads to induction of microbiota-specific T-cell responses in the absence of inflammation, which are important in antimicrobial defense and tissue repair upon skin injury [2, 5, 26]. During skin injury, commensal bacteria stimulate neutrophils to express CXC ligand 10 (CXCL10), which eradicates wound-colonizing microbes and recruits activated plasmacytoid DCs (pDCs). pDC-derived type I interferons then accelerate wound repair by inducing growth factor production in fibroblasts and macrophages [3]. This result may alter our current understanding that inflammation in noninfected wounds is triggered by dying host cells; instead, this data suggests that commensals are required to elicit innate immune responses. In a separate study, resident commensals and even pathogenic S. aureus enhanced skin regeneration and wound-induced hair follicle neogenesis by inducing IL-1β and keratinocyte-dependent IL-1R-myeloid differentiation primary response 88 (MyD88) signaling [4]. In addition, S. aureus potently induces keratinocyte production of AMPs in a Toll-like receptor (TLR) 2-independent manner and, together with commensal-induced AMPs, may synergistically promote innate immune responses in wound healing [61]. When skin commensals were disturbed by oral vancomycin treatment, mice demonstrated decreased skin bacterial diversity and altered composition. Compared to the control, these mice showed delayed wound healing, possibly associated with downregulation of IL-17 and the regeneration of islet-derived protein-III γ, which are important for keratinocyte differentiation and proliferation [62].
However, when Canesso et al. compared the healing process in germ-free Swiss mice and conventional Swiss mice, they found that wound healing was accelerated and scarless in the absence of a commensal microbiota because of controlled inflammation, characterized by low accumulation of neutrophils and high levels of alternatively activated macrophages as well as increased angiogenesis at wound sites. Restoration of the skin microbiota deprived germ-free mice of such enhanced wound healing [63]. These conflicting results suggest that the effect of skin commensals in wound healing is complicated and versatile, and recognition of the precise role of defined microorganisms is vital.
As the most abundant symbiotic bacteria, the contribution of S. epidermidis to enhanced wound healing has been appreciated (Figure 2a). In the innate arm of the immune system, S. epidermidis colonization induces the expression of many antimicrobials, such as AMPs [64, 65] and LTA, which attenuate cutaneous injury-induced TLR-3-mediated inflammation through TLR-2 signaling [1]. S. epidermidis activates the keratinocyte aryl hydrocarbon receptor and downstream IL-1α, IL-1β and β-defensin 3 expression [66]. In the adaptive arm, S. epidermidis-specific skin Tc17 cells expressed high levels of genes associated with wound healing, including angiogenesis (Csf2, Fgf2, Vegfa, Pdgfb), keratinocyte proliferation (Areg, Fgf2, Fgf16, Fgf18, Tgfb1, furin), chemotaxis (Pdgfb, Csf2, Tgfb1, furin), tissue remodeling (Mmp10, Mmp25) and ECM production (Fgf2, Pdgfb, Tgfb1, furin) [26]. In a mouse wound model, S. epidermidis-specific Tc17 cells rapidly accumulated at the wound edge postinjury and promoted faster wound closure compared to wounds associated with an S. epidermidis strain incapable of inducing such a cell subset [26]. When exposed to cytokines and alarmins previously shown to be associated with tissue damage, skin-resident S. epidermidis-elicited Tc17 cells demonstrate significant plasticity and rapidly release the type 2 cytokines IL-4 and IL-13, which are involved in tissue repair [2]. Furthermore, S. epidermidis strains expressing staphylococcal aromatic amino acid decarboxylase (SadA) are able to accelerate wound healing by converting aromatic amino acids into trace amines, which abrogate the cell motility inhibition effect of epinephrine produced by keratinocytes [67]. S. epidermidis also secretes a sphingomyelinase that facilitates host production of ceramides to maintain skin barrier integrity and prevent water loss in damaged skin [68].
Figure 2.

Microbe–host and microbe–microbe interactions in wound healing. (a) S. epidermidis promotes wound healing by converting AAA into TA and facilitating host ceramide production. S. epidermidis mediates neutrophil secretion of CXCL10 to induce pDC production of type 1 interferons, which stimulate growth factor production by fibroblasts and macrophages to promote tissue regeneration. S. epidermidis stimulates keratinocyte regeneration through the IL-1R/MyD88 pathway and induces IL-1α, IL-1β and β-defensin-3 production. S. epidermidis-specific CD17+ T cells express genes related to multiple facets of wound healing, including angiogenesis, chemotaxis, tissue remodeling and the release of type 2 cytokines. (b) S. aureus induces an inflammatory phenotype of keratinocytes with inflammatory cytokine production, AIM2 inflammasome activation and AMP (BD-3 and RNase 7) production and causes keratinocyte death. S. aureus inhibits fibroblast migration and induces apoptosis. S. aureus increases the level of MMP-2, which leads to collagen degradation. (c) P. aeruginosa impairs neutrophil motility and effector function. P. aeruginosa-derived elastase degrades thrombin and releases FYT21, which inhibits TLR dimerization. During the chronic infection phase, P. aeruginosa alters metabolic pathways to facilitate biofilm matrix formation. (d) S. epidermidis inhibits S. aureus growth in wounds via the protease Esp and SCFA and upregulates host expression of perforin-2. S. aureus promotes P. aeruginosa attachment, while P. aeruginosa increases the virulence and antibiotic resistance of S. aureus. Together, these bacteria reduce the level of growth factors related to regeneration. C. acnes either promotes S. aureus biofilm formation via coporphyrin III or inhibits its growth through fermentation. S. epidermidis Staphylococcus epidermidis, AAA aromatic amino acids, TA trace amine, IL-1 interleukin-1, CXCL10 CXC ligand 10, pDC plasmacytoid dendritic cell, IFN interferon, MyD88 myeloid differentiation primary response 88, S. aureus Staphylococcus aureus, MMP-2 matrix metalloproteinase-2, AIM2 absent in melanoma 2, BD-3 β-defensin-3, P. aeruginosa Pseudomonas aeruginosa, S100A8/9 S100 calcium binding protein A8–A9 complex, VEGF vascular endothelial growth factor, TLR toll-like receptor, acetyl-CoA acetylcholine A, SCFA short-chain fatty acid, C. acnes Cutibacterium acnes
Probiotics, defined as ‘live microorganisms which when administered in adequate amounts confer a health benefit on the host’ [69], are host-friendly microbes that are able to restore the body’s microbiome balance, such as the perturbed microbiome in the wound bed. In vitro studies have demonstrated the ability of Lactobacilus spp., mainly Lactobacillus plantarum, or their supernatant to promote keratinocyte proliferation and migration [70] and prevent biofilm formation [71–73]. Evidence from animal models also supports the role of probiotics, including Lactobacilus spp., Bifidobacteria, kefir and Saccharomyces cerevisiae, in reducing pathogen colonization and biofilm formation, promoting tissue repair, decreasing excessive scarring and lowering the mortality rate of infected wounds [74–81].
The microbiome in chronic wounds
Biofilm formation characterizes chronic wounds
Chronic wounds usually occur in elderly individuals with diabetes mellitus, vascular disease and obesity, as well as individuals with compromised immune and nutritional status [82, 83]; furthermore, care for nonhealing wounds leads to high healthcare expenditure [84]. The hallmark of impaired healing wounds is persistent inflammation, with microbial infection being the leading cause. Microbes colonizing the wound exist either as single, planktonic cells or as a complex structure called a biofilm, which consists of polysaccharides, lipids, proteins and nucleic acids and harbors a single species or various types of bacteria and fungi [85]. In contrast to acute wounds, a higher rate of biofilm formation was observed in chronic wounds (60 vs. 6%) by light and scanning electron microscopy techniques [86]. A systemic review and meta-analysis assessing 185 chronic nonhealing wounds reported an average biofilm prevalence of 78.2% [87]. A biofilm provides a habitable environment for microbial growth and is far more resistant to antimicrobial agents and host immune defense. It provides a continuous stimulus to the immune system, contributes to local hypoxic niche establishment and impedes the natural process of wound healing [86,88–91]. Even if a biofilm-infected wound is closed, the skin post-closure exhibits compromised epithelial tight-junction function [92] and reduced tensile strength [93]. As a result, the low-quality healed skin is likely to have higher risk of future complications such as wound recidivism.
Bacterial community and clinical relevance in chronic wounds
A large-scale retrospective clinical study of 2963 chronic wounds of various etiologies, including 916 venous leg ulcers, 767 decubitus ulcers and 370 nonhealing surgical wounds, revealed a distinct microbial profile compared to that of the surrounding healthy skin, but the composition of microbiota was similar among different types of wounds and was not affected by patient demographics [89]. Staphylococcus, Pseudomonas and Corynebacterium were identified as the most common genera [89], which is well supported by evidence from other studies [83,94–98]. S. aureus and S. epidermidis were the predominant species, and methicillin-resistant S. aureus (MRSA) was identified in 25% of chronic wounds. Pseudomonas is not only the most dominant genus in chronic wound polymicrobial biofilms but also the most common microbe seen in single-species biofilms [89]. Although chronic wounds are usually exposed to high levels of oxygenation, deep ulcers and those of a longer duration contain a large number of anaerobic bacteria, including Anaerococcus, Fingelodia, Prevotella, Preptonipihlus, Peptostreptococcus and Clostridium [83, 86, 89, 95, 97]. Importantly, facultative anaerobes, especially Enterobacter, were significantly associated with nonhealing wounds [99].
Among all types of chronic wounds, diabetic foot ulcers (DFUs) are the most studied. Using 16S rRNA gene sequencing technology, a reduced microbial diversity [94, 100] and a significantly increased relative abundance of Staphylococcus spp. [83, 96, 98, 100] was found in DFUs in contrast to healthy skin. Changes in the skin microbiome likely occur prior to ulcer development or skin infection. Surprisingly, unlike diabetic wounds, the plantar foot of diabetic men showed a higher bacterial diversity and a lower relative abundance of Staphylococcus spp. compared to nondiabetic men, but they possessed a higher quantity of more virulent forms of S. aureus [101]. Diabetic patients also had a lower relative abundance of Firmicutes and a higher abundance of Actinobacteria in their feet, but the composition and total bacterial counts were similar in arm samples from the two groups [101]. These observations have been linked to the risk of future DFU development.
When connected to clinical presentation and outcomes, Gardner et al. found that a high microbial diversity and an increase in the relative abundance of anaerobic bacteria and gram-negative Proteobacteria spp. were associated with deep DFUs and long disease duration, whereas an increase in the relative abundance of Staphylococcus spp., mainly S. aureus, was found in shallow ulcers and in those with a short disease duration [97]. A longitudinal study further illustrated that rapid and dynamic changes in the skin microbiota were associated with rapid and improved healing of DFUs [98]. This may seem counterintuitive, but microbiota community instability might reflect efficient clearance of wound-colonized bacteria that prevents dominance of the wound bed by a single aggressive bacterial strain. Moreover, systemic antibiotics that destabilize the chronic wound bed microbiome foster wound healing [98]. Using metagenomic shotgun sequencing, Kalan et al. discovered two isolates of S. aureus, SA10757 and SA7372, that were associated with nonhealing DFUs [83]. These strains harbored multiple antibiotic resistance genes and expressed staphylococcal enterotoxins. Sharp debridement, rather than antibiotics, significantly shifted the wound microbiota by depleting anaerobic bacteria and decreasing the bacterial diversity, which accelerated wound healing [83].
Fungi and viruses in chronic wounds
In contrast to bacteria, fewer studies have explored fungal communities in chronic wound healing. Since the foot is colonized by large amounts of fungi, fungal foot infections are common in diabetic patients [102]. Fungi were cultured from 27.2% of the lower-limb wounds of patients with type 2 diabetes [103]. Dowd et al. reported a positive rate of 23% fungal colonization in chronic wounds with different etiologies using deep molecular sequencing methods [104]. Of note, fungi were detected in 40.8% of DFUs. Unlike bacteria, fungi are extremely diverse in DFUs. The most abundant fungal members were Asomycota (Cladosporidium herbarum and C. albicans) and Basidomycota (Trichosporon asahii and Rodhosporidium diobovatum) [105]. Interestingly, fungal diversity increased upon the administration of systemic antibiotics and wound deterioration, and polymicrobial biofilms consisting of bacteria and fungi were associated with poor clinical outcomes [105].
Little attention has been given to the virome in wounds due to the difficulty of achieving sufficient sequencing depths, and only the phageome has been investigated. A recent metagenomic study evaluated 20 chronic wound patients with different etiologies, including diabetic, venous, arterial and pressure ulcers, and discovered a virome of greater diversity and containing more pathogen-targeting phages instead of commensal-targeting phages compared to that of healthy skin [106]. In addition, wounds that healed in 6 months were associated with two phage species targeting Staphylococcus strains, while unhealed ones were associated with phages targeting Streptococcus and putative Enterobacter phage spp. [106]. Clearly, further studies are desperately needed to elucidate the make-up of the virome in wounds.
Pathogens hinder wound healing
Mechanistic studies further confirmed the pathogenicity of defined microbes in chronic wounds, as was seen in correlative observations. As a prevalent pathogen persisting in nonhealing wounds, the role of S. aureus in wound healing has been extensively investigated (Figure 2b). Using coculture methods, Kirker et al. showed that S. aureus-induced biofilms promoted keratinocyte secretion of proinflammatory cytokines, led to keratinocyte death and impaired scratch closure [107]. It also hampered dermal fibroblast migration and resulted in fibroblast apoptosis [108]. In a porcine wound model, S. aureus-induced biofilm repressed microRNA (miR)-143, causing upregulation of the expression of the downstream target gene MMP-2. MMP-2 degrades collagen type 1 in granulation tissue and compromises wound healing [93]. Furthermore, reduction in collagen type 1 in the wound bed decreased the tensile strength of the healed skin, predisposing to wound recidivism [93]. S. aureus impaired DNA repair mechanisms via induction of miR-15b-5p and led to a sustained inflammatory response and delayed wound closure in DFUs [109]. Intracellular accumulation of S. aureus in the epidermis also triggered absent in melanoma 2 (AIM2) inflammasome activation and pyroptosis, which correlated with nonhealing DFUs [110].
The persistence of S. aureus in chronic wounds implies its ability to escape host defense, and this has recently been linked to the suppression of perforin-2 expression [110, 111]. Perforin-2, an AMP, is an antibacterial protein expressed by γδ T cells, keratinocytes, endothelial cells and fibroblasts in human skin [110, 111]. It is released from endosomal vesicles and fuses with bacteria-containing phagosomes to eliminate intracellular bacteria. A keratinocyte cell line overexpressing perforin-2 cleared intracellular S. aureus infection more efficiently than control cells [111]. However, perforin-2 expression was inhibited in both hematopoietic cells and skin tissue cells in S. aureus-infected human wounds [110, 111]. Using perforin-deficient mice, Tomic-Canic et al. showed that MRSA epicutaneously inoculated only translocates to internal organs and leads to death in the absence of perforin-2 expression [112], confirming the importance of perforin-2 in preventing S. aureus invasion.
Pseudomonas aeruginosa (P. aeruginosa) is an opportunistic gram-negative bacterium implicated in inducing biofilm formation in chronic wounds (Figure 2c). In contrast to S. aureus, which usually resides in the superficial layers of wounds, P. aeruginosa is localized in deeper tissues of the wound bed [113]. Wound infection with P. aeruginosa initiates an immune response characterized by early recruitment of neutrophils to sites of injury and later activation of systemic adaptive immunity [114]. P. aeruginosa-induced biofilms impair neutrophil effector function both in vitro [115] and in vivo in murine wound models [116] and delay wound closure by reducing VEGF and AMP (S100A8/9) levels [116, 117]. In addition, P. aeruginosa-derived elastase inhibits host inflammatory responses by digesting thrombin and releasing the C-terminal thrombin-derived peptide FYT21, which prevents TLR dimerization [118]. Compromised host defense against P. aeruginosa then leads to the chronicity of P. aeruginosa wound infection. Surprisingly, P. aeruginosa biofilms were not more virulent than planktonic P. aeruginosa in a murine chronic wound model [114].
Metabolome analysis of the log-phase P. aeruginosa (PAO1) strain, from colonizing to acute infecting and then to biofilm-infecting full-thickness excision wounds, has unveiled a dynamic regulation of gene expression related to metabolism, especially carbon-utilization pathways, and cell replication [119]. Pathway of ‘branched-chain amino acids’ was increasingly upregulated along with progression to chronic infection. From earlier to later biofilm infection, pathway associated with ‘fatty acid degradation’ were upregulated. Both metabolic pathways produce the extracellular polysaccharide acetyl-CoA, a component of the biofilm matrix [119].
Microbial crosstalk in chronic wounds
Since most chronic wounds are inhabited by more than one bacterial or fungal species, understanding the dialog between these microorganisms is essential (Figure 2d). As previously mentioned, perforin-2 plays an essential role in combating intracellular S. aureus infection and is downregulated in S. aureus-infected wounds [110, 111]. S. epidermidis aids in host defense against S. aureus infection by upregulating perforin-2 expression in γδ T cells, keratinocytes and fibroblasts [65]. S. epidermidis inhibits the formation of S. aureus-induced biofilm, destroys S. aureus-induced biofilms through the serine protease Esp and eliminates S. aureus nasal colonization [120]. Some strains of S. epidermidis also limit S. aureus-induced neutrophil recruitment and cytokine production [121]. Interestingly, commensal S. aureus can suppress pathogenic MRSA growth by several mechanisms; S. aureus ferments carbohydrates into short-chain fatty acids, which diffuse into the bacterial cell to reduce the intracellular pH and finally kill MRSA USA300. Antibodies induced upon exposure to commensal S. aureus offer protection against pathogenic S. aureus infection [122].
Pastar et al. evaluated the interaction between the two most common bacteria, S. aureus and P. aeruginosa, in chronic wounds using a porcine wound model [43]. P. aeruginosa diminished MRSA USA300 growth both in vitro and in vivo, but not vice versa. However, the presence of P. aeruginosa promoted MRSA expression of the virulence factors Panton–Valentine leukocidin and α-hemolysin, and coinfection with these bacteria significantly delayed re-epithelialization through suppression of keratinocyte growth factor 1 expression [43]. In vitro coculture experiments further demonstrated that S. aureus mediated an increase in the attachment of P. aeruginosa to human keratinocytes, while P. aeruginosa promoted an invasive phenotype in S. aureus [123]. P. aeruginosa also decreased the sensitivity of S. aureus biofilms to vancomycin [124]. Collectively, these results confirm both competitive and reciprocally beneficial interactions between these two bacteria in biofilm formation.
In other cases, Cutibacterium spp. can serve as an accomplice to S. aureus pathogenicity as coproporphyrin III secreted by Cutibacterium spp. induces the formation of S. aureus biofilms [125]. However, C. acnes can also suppress the growth and wound colonization of MRSA USA300 through glycerol fermentation [126]. Coculture of S. aureus with another common skin commensal, Corynebacterium striatum, induces a broad shift in S. aureus gene expression, including the downregulation of quorum sensing genes, which shifts the bacteria toward a commensal phenotype [127]. Enterococcus faecalis and P. aeruginosa have a synergistic effect on biofilm matrix production [128]. Most current studies regarding microbial crosstalk have been limited to dual-species models, but given that wound biofilms are frequently colonized by multiple microbial species, the development of novel multispecies biofilm models is urgently needed.
The microbiome in scar formation
Excessive ECM deposition by fibroblasts and imbalanced ECM degradation by recruited leukocytes in wound healing leads to scarring/fibrosis, an unintended result of aberrant tissue repair. Persistent pathogen-induced chronic inflammation characterized by massive alternatively activated macrophage (M2) infiltration and a T helper 2 cell (Th2) cytokine response is believed to be the key driver of unrestrained fibrotic responses in the lungs, liver, kidney, spleen, intestines and brain [129]. This is evidenced by the existence of pathogens in fibrotic patients, the successful induction of organ fibrosis by infectious agents using animal models, the exacerbation of fibrosis by infectious agents and the promising effects of antimicrobial treatment on fibrotic diseases [130]. Classic examples of pathogen-induced fibrosis include pulmonary tuberculosis and chronic viral hepatitis. Unfortunately, no specific pathogen has been linked to hypertrophic scars and keloids, although they share a similar dysregulated immune signature (Th2 activation and M2 infiltration) with other fibrotic diseases [131, 132]. Indeed, little is presently known about the role of the microbiome in scar formation, as the main focus of skin microbiome research is skewed toward nonhealing wounds instead of overly healed ones (Figure 3).
Figure 3.

Schematic diagram representing the interplay between the microbiota and different types of skin wounds. Skin commensals interact with host immunity to promote timely healing with normal scarring in acute wounds. Various pathogens form biofilms in chronic wounds and cause a strong inflammatory response, preventing healing from progressing to the next phase. Microbe-induced dermal inflammation might be responsible for inciting an uncontrolled fibrotic response that gives rise to hypertrophic scars and keloids. C. acnes Cutibacterium acnes, S. epidermidis Staphylococcus epidermidis, S. aureus Staphylococcus aureus, spp. species, P. aeruginosa Pseudomonas aeruginosa
The assumption that microbial infection plays a role in scar formation has some theoretical support. First, inflammatory skin diseases explicitly associated with bacteria such as acne and folliculitis and virus vaccine inoculation are common risk factors for keloids. Although the broad spectrum of pathogens capable of inducing keloids indicates that there might not be a single culprit, microbial-induced chronic local inflammation may still be responsible. Therefore, infection-based theories of keloidogenesis are constantly proposed [133, 134]. Second, scarless tissue regeneration is commonly seen in fetuses who live in the sterile environment of the amniotic fluid [135]. The introduction of bacteria into rabbit fetal wounds induces neovascularization and fibroplasia, which are classic features of scar formation in adults [15]. The oral mucosa represents another place where scar-free healing usually occurs. Similar to amniotic fluid, saliva provides a wet and bacteria-inhibiting milieu for wound healing [135].
Supporting this hypothesis, recent studies have shed some light on this area. As previously mentioned, depletion of skin commensals in mice leads to scarless wound healing [63]. Similarly, a hydrogel loaded with growth factors and antimicrobial components to create a sterile, moist microenvironment has been developed to treat skin wounds, and it indeed results in accelerated healing in the absence of scarring [136]. A study group compared the bacteriology of scalp folliculitis with different severities. S. aureus infection was detected in acne keloidalis nuchae, while less severe folliculitis was associated with C. acnes [137]. In a porcine burn model, infection of burn wounds with S. aureus led to delayed re-epithelialization and increased dermal injury and deep scarring [138]. Moreover, TLRs associated with classical viral infection are highly expressed in keloids [139], highlighting the potential involvement of viruses in skin fibrosis. This is intriguing because fibroblasts isolated from other fibrotic tissues showed high levels of TLR activation and subsequent release of profibrotic chemokines [129]. In another skin fibrotic disease, parvovirus B19 persists in systemic sclerosis fibroblasts and is able to stimulate human dermal fibroblast migration and the production of fibrotic cytokine metalloproteases [140]. Whether a similar mechanism exists in hypertrophic scars remains an interesting research question.
Modulating the microbiota following skin injury
Acute wound management
There is no doubt that timely empiric use of antibiotics is essential for reducing bacterial load and preventing infection in circumstances such as severe burn injury and trauma. However, currently, topical antibiotics are routinely used for even minor skin injuries by some patients and even physicians. In addition to the development of pharmacologic resistance due to the abuse of antibiotics, new evidence has suggested a counterproductive effect of topical antibiotic use in wound treatment. Contrary to our previous understanding, resident commensal bacteria may promote wound healing, and disruption of the skin microbiota delays tissue regeneration [3, 4]. Therefore, topical prophylactic antibiotic use for acute wounds with no signs of infection should be discarded in future clinical practice.
Chronic wound management
Management of chronic wounds requires comprehensive wound assessment and appropriate care. Basic strategies include wound cleansing, debridement, moisture control, nutritional support and microbial regulation, yet the effect is suboptimal. Because chronic wounds are characterized by an altered microbiome with an overgrowth of certain bacteria [83, 98], elimination or destabilization of the existing microbiota is of vital importance. Anti-fungal treatment is also necessary when there is concurrent fungal infection [58]. However, established biofilms are seldom removed via antibiotics alone, which conversely may lead to the selection of more resistant bacterial species. Alternatives or adjuvants to conventional antibiotics in inhibiting bacterial growth are needed (Figure 4).
Figure 4.
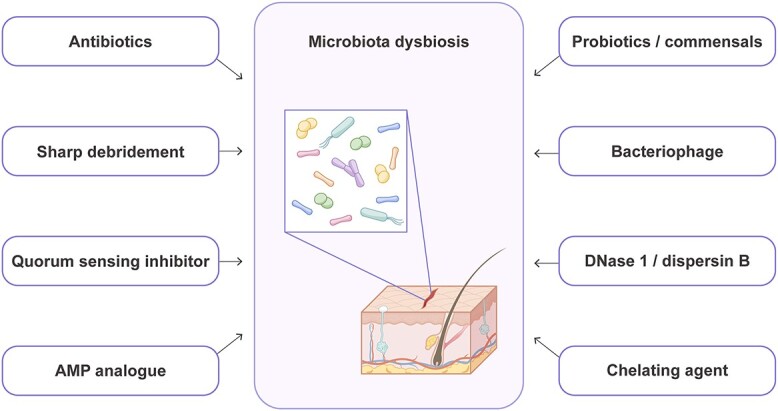
Biocontrol strategies in wound management. AMP antimicrobial peptides
Sharp debridement is recommended in all diabetic wounds to remove debris and necrotic and infected tissues. This procedure aims to induce an acute wound by creating sterile tissue damage and ‘reactivating’ healing pathways [141]. Sharp debridement is effective in reducing wound microbial diversity and disrupting anaerobic networks and has been linked to more favorable DFU clinical outcomes [83]. Several other approaches have also been developed to induce an acute wound healing response. Topical application of pathogen-associated molecular patterns, such as lipopolysaccharide and exopolysaccharide, rescues the wound from stalled status to amplify inflammation and subsequent tissue regeneration [142, 143].
One unique way in which bacterial communities communicate with each other in terms of population density is by quorum sensing, which regulates both biofilm formation and bacterial toxin production. Using murine wound infection models, several studies demonstrated that exposure to quorum sensing inhibitors prevented Staphylococcus spp.-induced biofilm formation, decreased bacterial bioburden and restored normal wound healing kinetics [88, 144]. Similarly, a synthetic quorum sensing inhibitor inhibits P. aeruginosa virulence factor pyocyanin production and biofilm formation [145]. Quorum sensing inhibitors also increase the susceptibility of bacterial biofilms to antibiotics, resulting in an enhanced therapeutic effect [146].
Engineered synthetic peptides derived from naturally produced AMPs represent another promising approach in inhibiting bacterial biofilms [147]. In addition to their antimicrobial function, AMPs improve the host’s immune response by recruiting antigen-presenting cells and enhancing their differentiation, facilitating neutrophil extracellular trap activation, enhancing phagocytosis, inducing proinflammatory cytokines and promoting T-cell polarization [147]. AMPs also accelerate wound healing by modulating the proliferation, migration and activation of keratinocytes and fibroblasts, facilitating re-epithelialization and angiogenesis [148, 149]. Approaches with these molecules are currently under development, and several peptides have entered clinical trials for treating infected burn wounds, DFUs and venous leg ulcers [147].
Correcting dysbiosis via probiotics has demonstrated satisfactory clinical efficacy in treating many intestinal diseases, such as Clostridium difficile infection and irritable bowel diseases; however, the same strategy has not been well developed in skin disorders, including wounds. In vitro studies have demonstrated the ability of probiotics, mainly L. plantarum, or their supernatants to promote keratinocyte proliferation and migration [70] and prevent biofilm formation [71–73]. Evidence from animal models also supports the role of probiotics in reducing pathogen colonization, promoting tissue repair, decreasing excessive scarring and lowering the mortality rate of infected wounds [74–77]. To date, only a few small-scale clinical studies involving burn wounds, chronic venous ulcers and oral wounds have evaluated the impact of the topical use of probiotics on wound healing, yet they have demonstrated promising therapeutic effects [150–153]. Moreover, probiotics can also serve as a drug-delivery system. CXCL12-delivering Lactobacillus promoted murine wound healing by inducing the proliferation of TGF-β-producing macrophages [154].
Harnessing the competitive relationship between commensals and pathogens by counteracting pathogens with commensals and their products is a viable option for bacterial control with unexploited potential in infected wound treatment. Accordingly, active colonization/transplantation of commensals with probiotic properties, such as S. epidermidis or C. acnes, or application of bacterial products such as commensal-derived short-chain fatty acids or proteases might be helpful in combating pathogens in wound infection and thus accelerating healing [3, 155]. This field awaits further investigation and more mechanistic studies evaluating microbe–microbe crosstalk could provide novel insights into microbial control.
Phages are natural enemies of bacteria. They target specific bacterial strains, and once they enter the bacterial host, they inject phage DNA to induce bacterial lysis and interfere with bacterial metabolism [156]. Therefore, they are potentially useful with minimal side-effects in treating wounds of a known pathogen infection. Cocktail therapy containing various phages has been considered a plausible biocontrol strategy for phytobacteria [157]. In a phase 1 trial, injection with phages targeting P. aeruginosa, S. aureus and Escherichia coli was proven to be safe and efficient in treating chronic venous leg ulcers [158].
Apart from eradicating bacteria themselves, disrupting the biofilm matrix that they attach to is another approach for microbial control. Deoxyribonuclease I and dispersin B degrade extracellular DNA (eDNA) and poly-β 1,6-N-acetylglucosamine, respectively, which are involved in microbial surface attachment [159, 160]; these agents have been found to be highly effective in inhibiting pathogens and promoting wound healing [161]. Ethylenediaminetetraacetic acid (EDTA) chelates Mg2+ and Ca2+ from the outer cell wall of gram-negative bacteria and destabilizes the negative charge of lipopolysaccharide [162, 163]. The application of a hydrogel containing EDTA and bacteriocin significantly reduced the bacterial load in a S. aureus Xen-31-infected murine model [164].
Reducing scar formation
Except for chronic nonhealing wounds, scar-related disfigurement and disability remain major challenges for medicine and cosmetics. Both normal and hypertrophic scars are difficult to treat and even less likely to be prevented. A variety of strategies have been applied in clinical practice, but their effects are still limited. Surgery, usually in combination with postoperative radiation, remains the major approach for reducing scarring; however, this strategy causes secondary trauma to the skin and the cost is high. A less invasive treatment is topical use and intralesional injections of corticosteroids, but it comes with many adverse effects, such as hypopigmentation, dermal atrophy, telangiectasia, widening of the scar and delayed wound healing [165]. Mechanical offloading is another attempt to limit fibrosis after skin injury, as mechanical tension activates many mechanoresponsive signaling pathways to induce inflammation and promote ECM deposition [166], but mechanical offloading is seldom used as a single therapy.
Due to the lack of active infection in most hypertrophic scars, antimicrobial treatment is not routinely used. Whereas antibiotics together with other therapies are effective in treating acne, their efficacy in preventing acne-associated keloid formation is largely unknown. Therefore, with its elusive pathogenesis, we are currently in no position to manipulate microbes in scar prevention. Further research characterizing the composition and function of the skin microbiome in hypertrophic scars and keloids would be informative in achieving the ultimate goal of scarless wound healing.
Current status and outlook for future research
Reviewing the past research literature shows that many skin microbes play a dual role in wound healing, depending on the bacterial load, strain-level heterogeneity (strain-level differences in the gene content determine functional differences of the same species) and interaction with other microbes. This is consistent with our new understanding about a microbe’s pathogenicity being contextual [167], and understanding factors that foster the transition from commensalism to pathogenicity may be the key to more effective and specific therapeutic approaches. It must be kept in mind that wound healing is an extremely complex process with a series of variables being potential confounding factors. Hence, similar to many other skin disorders, an overly simplified one-to-one mapping of microbes to poor wound healing is inappropriate.
The methodologies used in microbiome research should be prudently evaluated and utilized with caution given their huge impact on results. For example, skin specimens are typically low in bioburden and extremely susceptible to reagent and environmental contamination [168]. While skin swabbing is the most commonly used approach for microbial sampling because it is noninvasive, tissue biopsies are the gold standard to capture the whole microbial population. Therefore, standardized sampling and analysis are required to ensure reproducibility across studies. In addition, differences exist in the healing process of rodents and humans, and most animal wound models illustrate the process of acute wound healing instead of chronic wound healing. Therefore, care must be taken when interpreting the results obtained from descriptive human studies as well as experimental animal studies.
Most wound microbial studies have focused on chronic DFUs, and the bacterial communities in acute wounds, other types of chronic wounds and hypertrophic scars/keloids are only partially understood. With technological progress in profiling fungi, future studies should also encompass these microorganisms considering their potency in inducing cutaneous inflammation and interacting with bacteria via unknown mechanisms. More large-scale longitudinal in vivo studies are also desperately needed to elucidate the temporal dynamics of the microbiome in wounds. Follow-up meta-transcriptomics studies that identify real-time gene expression profiles may delineate the host–microbiome interaction in wounds more thoroughly. Additionally, to date, our understanding of host–microbe interplay mainly stems from pathogen-induced inflammation and infection. However, a symbiotic relationship uncoupled from inflammation is more common and yet largely underestimated in both healthy and certain disease conditions, including noninfected well-healed wounds. How the immune system coordinates with commensals to establish skin homeostasis and re-establish the balance in wound repair remains an important and fruitful question for future study.
Conclusions
The progress made in identifying skin microbiota using culture-independent methods in the past decade has been remarkable, leading to a more complex and thorough understanding of the association of wound healing with previously unappreciated microbial composition and diversity. Current evidence from germ-free and gnotobiotic animal studies as well as human wound studies indicates that efficient clearance of microbes with high inflammatory potential is detrimental to avoiding a stalled inflammation state and prolonged healing duration, while colonization of moderate nonaggressive commensals may aid rapid healing. Persistent dermal inflammation caused by unknown microbes may underscore the pathogenesis of exaggerated fibroproliferative responses in hypertrophic scars and keloids. These findings have revealed potential biomarkers that are associated with the quality of wound healing and provide the rationale and targets for manipulating the skin microbiome, paving the way for possible satisfactory wound healing induction.
Funding
This work was supported by National High Level Hospital Clinical Research Funding, grant Nos. 2022-PUMCH-B-041, 2022-PUMCH-A-210 and 2022-PUMCH-C-025, and National Natural Science Foundation of China, grant No. 81971846.
Authors’ contributions
Conceptualization: YY, JH, NY and XW; writing–original draft: YY; writing–review and editing: YY, JH, AZ, NY and XW; visualization: YY and NY; funding acquisition: JH, XL and XW; and supervision: XW
Conflict of interest
None declared.
Contributor Information
Yuyan Yang, Department of Plastic and Reconstructive Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuaifuyuan, Dongcheng District, Beijing, 100005, China.
Jiuzuo Huang, Department of Plastic and Reconstructive Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuaifuyuan, Dongcheng District, Beijing, 100005, China.
Ang Zeng, Department of Plastic and Reconstructive Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuaifuyuan, Dongcheng District, Beijing, 100005, China.
Xiao Long, Department of Plastic and Reconstructive Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuaifuyuan, Dongcheng District, Beijing, 100005, China.
Nanze Yu, Department of Plastic and Reconstructive Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuaifuyuan, Dongcheng District, Beijing, 100005, China.
Xiaojun Wang, Department of Plastic and Reconstructive Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuaifuyuan, Dongcheng District, Beijing, 100005, China.
References
- 1. Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, et al. Commensal bacteria regulate toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harrison OJ, Linehan JL, Shih HY, Bouladoux N, Han SJ, Smelkinson M, et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science. 2019;363:eaat6280. 10.1126/science.aat6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Di Domizio J, Belkhodja C, Chenuet P, Fries A, Murray T, Mondéjar PM, et al. The commensal skin microbiota triggers type I IFN-dependent innate repair responses in injured skin. Nat Immunol. 2020;21:1034–45. [DOI] [PubMed] [Google Scholar]
- 4. Wang G, Sweren E, Liu H, Wier E, Alphonse MP, Chen R, et al. Bacteria induce skin regeneration via IL-1β signaling. Cell Host Microbe. 2021;29:777–791.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Foulongne V, Sauvage V, Hebert C, Dereure O, Cheval J, Gouilh MA, et al. Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS One. 2012;7:e38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wylie KM, Mihindukulasuriya KA, Zhou Y, Sodergren E, Storch GA, Weinstock GM. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 2014;12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hannigan GD, Meisel JS, Tyldsley AS, Zheng Q, Hodkinson BP, SanMiguel AJ, et al. The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. MBio. 2015;6:e01578–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Graham EH, Clarke JL, Fernando SC, Herr JR, Adamowicz MS. The application of the skin virome for human identification. Forensic Sci Int Genet. 2022;57:102662. [DOI] [PubMed] [Google Scholar]
- 12. Oh J, Byrd AL, Park M, Kong HH, Segre JA. Temporal stability of the human skin microbiome. Cell. 2016;165:854–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mueller NT, Bakacs E, Combellick J, Grigoryan Z, Dominguez-Bello MG. The infant microbiome development: mom matters. Trends Mol Med. 2015;21:109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marples RR, Downing DT, Kligman AM. Control of free fatty acids in human surface lipids by Corynebacterium acnes. J Invest Dermatol. 1971;56:127–31. [DOI] [PubMed] [Google Scholar]
- 15. Gribbon EM, Cunliffe WJ, Holland KT. Interaction of Propionibacterium acnes with skin lipids in vitro. J Gen Microbiol. 1993;139:1745–51. [DOI] [PubMed] [Google Scholar]
- 16. Aly R, Shirley C, Cunico B, Maibach HI. Effect of prolonged occlusion on the microbial flora, pH, carbon dioxide and transepidermal water loss on human skin. J Invest Dermatol. 1978;71:378–81. [DOI] [PubMed] [Google Scholar]
- 17. Ying S, Zeng DN, Chi L, Tan Y, Galzote C, Cardona C, et al. The influence of age and gender on skin-associated microbial communities in urban and rural human populations. PLoS One. 2015;10:e0141842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bay L, Barnes CJ, Fritz BG, Thorsen J, Restrup MEM, Rasmussen L, et al. Universal dermal microbiome in human skin. MBio. 2020;11:e02945–19. 10.1128/mBio.02945-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prast-Nielsen S, Tobin AM, Adamzik K, Powles A, Hugerth LW, Sweeney C, et al. Investigation of the skin microbiome: swabs vs. biopsies. Br J Dermatol. 2019;181:572–9. [DOI] [PubMed] [Google Scholar]
- 21. Harris-Tryon TA, Grice EA. Microbiota and maintenance of skin barrier function. Science. 2022;376:940–5. [DOI] [PubMed] [Google Scholar]
- 22. Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. 2017;9:eaah4680. 10.1126/scitranslmed.aah4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laborel-Préneron E, Bianchi P, Boralevi F, Lehours P, Fraysse F, Morice-Picard F, et al. Effects of the Staphylococcus aureus and Staphylococcus epidermidis Secretomes isolated from the skin microbiota of atopic children on CD4+ T cell activation. PLoS One. 2015;10:e0141067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell. 2018;172:784–796.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nicoll MP, Proença JT, Efstathiou S. The molecular basis of herpes simplex virus latency. FEMS Microbiol Rev. 2012;36:684–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vella V, Galgani I, Polito L, Arora AK, Creech CB, David MZ, et al. Staphylococcus aureus skin and soft tissue infection recurrence rates in outpatients: a retrospective database study at 3 US medical Centers. Clin Infect Dis. 2021;73:e1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Muñoz-Planillo R, Hasegawa M, et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang HW, Yan D, Singh R, Liu J, Lu X, Ucmak D, et al. Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome. 2018;6:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bantz SK, Zhu Z, Zheng T. The atopic march: progression from atopic dermatitis to allergic rhinitis and asthma. J Clin Cell Immunol. 2014;05:202. 10.4172/2155-9899.1000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ubags ND, Trompette A, Pernot J, Nibbering B, Wong NC, Pattaroni C, et al. Microbiome-induced antigen-presenting cell recruitment coordinates skin and lung allergic inflammation. J Allergy Clin Immunol. 2021;147:1049–1062.e7. [DOI] [PubMed] [Google Scholar]
- 33. Yu J, Oh MH, Park JU, Myers AC, Dong C, Zhu Z, et al. Epicutaneous exposure to staphylococcal superantigen enterotoxin B enhances allergic lung inflammation via an IL-17A dependent mechanism. PLoS One. 2012;7:e39032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patrick GJ, Liu H, Alphonse MP, Dikeman DA, Youn C, Otterson JC, et al. Epicutaneous Staphylococcus aureus induces IL-36 to enhance IgE production and ensuing allergic disease. J Clin Invest. 2021;131:e143334. 10.1172/jci143334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y, Gao H, Loyd CM, Fu W, Diaconu D, Liu S, et al. Chronic skin-specific inflammation promotes vascular inflammation and thrombosis. J Invest Dermatol. 2012;132:2067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Evensen K, Slevolden E, Skagen K, Rønning OM, Brunborg C, Krogstad AL, et al. Increased subclinical atherosclerosis in patients with chronic plaque psoriasis. Atherosclerosis. 2014;237:499–503. [DOI] [PubMed] [Google Scholar]
- 37. Golden JB, Wang Y, Fritz Y, Diaconu D, Zhang X, Debanne SM, et al. Chronic, not acute, skin-specific inflammation promotes thrombosis in psoriasis murine models. J Transl Med. 2015;13:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hurabielle C, Link VM, Bouladoux N, Han SJ, Merrill ED, Lightfoot YL, et al. Immunity to commensal skin fungi promotes psoriasiform skin inflammation. Proc Natl Acad Sci U S A. 2020;117:16465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–21. [DOI] [PubMed] [Google Scholar]
- 40. Clark RAF. Wound repair: overview and general considerations. The molecular biology of wound repair. The Moleclar and Cellular Biology of Wound Repair. New York and London: Plenum Press, 1996, 3–50. [Google Scholar]
- 41. Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127:514–25. [DOI] [PubMed] [Google Scholar]
- 42. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pastar I, Nusbaum AG, Gil J, Patel SB, Chen J, Valdes J, et al. Interactions of methicillin resistant Staphylococcus aureus USA300 and Pseudomonas aeruginosa in polymicrobial wound infection. PLoS One. 2013;8:e56846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16:585–601. [DOI] [PubMed] [Google Scholar]
- 45. Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012;49:35–43. [DOI] [PubMed] [Google Scholar]
- 46. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63. [DOI] [PubMed] [Google Scholar]
- 47. Sadtler K, Estrellas K, Allen BW, Wolf MT, Fan H, Tam AJ, et al. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science. 2016;352:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rani M, Schwacha MG. The composition of T-cell subsets are altered in the burn wound early after injury. PLoS One. 2017;12:e0179015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Desmoulière A, Badid C, Bochaton-Piallat ML, Gabbiani G. Apoptosis during wound healing, fibrocontractive diseases and vascular wall injury. Int J Biochem Cell Biol. 1997;29:19–30. [DOI] [PubMed] [Google Scholar]
- 50. Greenhalgh DG. The role of apoptosis in wound healing. Int J Biochem Cell Biol. 1998;30:1019–30. [DOI] [PubMed] [Google Scholar]
- 51. Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol. 2008;40:1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Plichta JK, Gao X, Lin H, Dong Q, Toh E, Nelson DE, et al. Cutaneous burn injury promotes shifts in the bacterial microbiome in autologous donor skin: implications for skin grafting outcomes. Shock. 2017;48:441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lima KM, Davis RR, Liu SY, Greenhalgh DG, Tran NK. Longitudinal profiling of the burn patient cutaneous and gastrointestinal microbiota: a pilot study. Sci Rep. 2021;11:10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu SH, Huang YC, Chen LY, Yu SC, Yu HY, Chuang SS. The skin microbiome of wound scars and unaffected skin in patients with moderate to severe burns in the subacute phase. Wound Repair Regen. 2018;26:182–91. [DOI] [PubMed] [Google Scholar]
- 55. Bartow-McKenney C, Hannigan GD, Horwinski J, Hesketh P, Horan AD, Mehta S, et al. The microbiota of traumatic, open fracture wounds is associated with mechanism of injury. Wound Repair Regen. 2018;26:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hannigan GD, Hodkinson BP, McGinnis K, Tyldsley AS, Anari JB, Horan AD, et al. Culture-independent pilot study of microbiota colonizing open fractures and association with severity, mechanism, location, and complication from presentation to early outpatient follow-up. J Orthop Res. 2014;32:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ashrafi M, Xu Y, Muhamadali H, White I, Wilkinson M, Hollywood K, et al. A microbiome and metabolomic signature of phases of cutaneous healing identified by profiling sequential acute wounds of human skin: an exploratory study. PLoS One. 2020;15:e0229545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chellan G, Neethu K, Varma AK, Mangalanandan TS, Shashikala S, Dinesh KR, et al. Targeted treatment of invasive fungal infections accelerates healing of foot wounds in patients with type 2 diabetes. Diabet Med. 2012;29:e255–62. [DOI] [PubMed] [Google Scholar]
- 59. Warkentien TE, Shaikh F, Weintrob AC, Rodriguez CJ, Murray CK, Lloyd BA, et al. Impact of Mucorales and other invasive Molds on clinical outcomes of Polymicrobial traumatic wound infections. J Clin Microbiol. 2015;53:2262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lewandowski LR, Weintrob AC, Tribble DR, Rodriguez CJ, Petfield J, Lloyd BA, et al. Early complications and outcomes in combat injury-related invasive fungal wound infections: a case-control analysis. J Orthop Trauma. 2016;30:e93–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wanke I, Steffen H, Christ C, Krismer B, Götz F, Peschel A, et al. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J Invest Dermatol. 2011;131:382–90. [DOI] [PubMed] [Google Scholar]
- 62. Zhang M, Jiang Z, Li D, Jiang D, Wu Y, Ren H, et al. Oral antibiotic treatment induces skin microbiota dysbiosis and influences wound healing. Microb Ecol. 2015;69:415–21. [DOI] [PubMed] [Google Scholar]
- 63. Canesso MC, Vieira AT, Castro TB, Schirmer BG, Cisalpino D, Martins FS, et al. Skin wound healing is accelerated and scarless in the absence of commensal microbiota. J Immunol. 2014;193:5171–80. [DOI] [PubMed] [Google Scholar]
- 64. Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol. 2010;130:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pastar I, O'Neill K, Padula L, Head CR, Burgess JL, Chen V, et al. Staphylococcus epidermidis boosts innate immune response by activation of Gamma Delta T cells and induction of Perforin-2 in human skin. Front Immunol. 2020;11:550946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rademacher F, Simanski M, Hesse B, Dombrowsky G, Vent N, Gläser R, et al. Staphylococcus epidermidis activates aryl hydrocarbon receptor Signaling in human keratinocytes: implications for cutaneous Defense. J Innate Immun. 2019;11:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Luqman A, Muttaqin MZ, Yulaipi S, Ebner P, Matsuo M, Zabel S, et al. Trace amines produced by skin bacteria accelerate wound healing in mice. Commun Biol. 2020;3:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zheng Y, Hunt RL, Villaruz AE, Fisher EL, Liu R, Liu Q, et al. Commensal Staphylococcus epidermidis contributes to skin barrier homeostasis by generating protective ceramides. Cell Host Microbe. 2022;30:301–313.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, et al. Expert consensus document. The international scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506–14. [DOI] [PubMed] [Google Scholar]
- 70. Mohammedsaeed W, Cruickshank S, McBain AJ, O'Neill CA. Lactobacillus rhamnosus GG lysate increases re-epithelialization of keratinocyte scratch assays by promoting migration. Sci Rep. 2015;5:16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lopes EG, Moreira DA, Gullón P, Gullón B, Cardelle-Cobas A, Tavaria FK. Topical application of probiotics in skin: adhesion, antimicrobial and antibiofilm in vitro assays. J Appl Microbiol. 2017;122:450–61. [DOI] [PubMed] [Google Scholar]
- 72. Chan AP, Choi Y, Brinkac LM, Krishnakumar R, DePew J, Kim M, et al. Multidrug resistant pathogens respond differently to the presence of co-pathogen, commensal, probiotic and host cells. Sci Rep. 2018;8:8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Onbas T, Osmanagaoglu O, Kiran F. Potential properties of lactobacillus plantarum F-10 as a bio-control strategy for wound infections. Probiotics Antimicrob Proteins. 2019;11:1110–23. [DOI] [PubMed] [Google Scholar]
- 74. Valdéz JC, Peral MC, Rachid M, Santana M, Perdigón G. Interference of lactobacillus plantarum with Pseudomonas aeruginosa in vitro and in infected burns: the potential use of probiotics in wound treatment. Clin Microbiol Infect. 2005;11:472–9. [DOI] [PubMed] [Google Scholar]
- 75. Satish L, Gallo PH, Johnson S, Yates CC, Kathju S. Local probiotic therapy with lactobacillus plantarum mitigates scar formation in rabbits after burn injury and infection. Surg Infect. 2017;18:119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Olofsson TC, Butler É, Lindholm C, Nilson B, Michanek P, Vásquez A. Fighting off wound pathogens in horses with honeybee lactic acid bacteria. Curr Microbiol. 2016;73:463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Argenta A, Satish L, Gallo P, Liu F, Kathju S. Local application of probiotic bacteria prophylaxes against sepsis and death resulting from burn wound infection. PLoS One. 2016;11:e0165294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Oryan A, Alemzadeh E, Eskandari MH. Kefir accelerates burn wound healing through inducing fibroblast cell migration in vitro and modulating the expression of IL-1ß, TGF-ß1, and bFGF genes in vivo. Probiotics Antimicrob Proteins. 2019;11:874–86. [DOI] [PubMed] [Google Scholar]
- 79. Han N, Jia L, Su Y, Du J, Guo L, Luo Z, et al. Lactobacillus reuteri extracts promoted wound healing via PI3K/AKT/β-catenin/TGFβ1 pathway. Stem Cell Res Ther. 2019;10:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li M, Xiao H, Su Y, Cheng D, Jia Y, Li Y, et al. Synergistic inhibitory effect of honey and lactobacillus plantarum on pathogenic bacteria and their promotion of healing in infected wounds. Pathogens. 2023;12:501. 10.3390/pathogens12030501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rodrigues KL, Caputo LR, Carvalho JC, Evangelista J, Schneedorf JM. Antimicrobial and healing activity of kefir and kefiran extract. Int J Antimicrob Agents. 2005;25:404–8. [DOI] [PubMed] [Google Scholar]
- 82. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6:265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kalan LR, Meisel JS, Loesche MA, Horwinski J, Soaita I, Chen X, et al. Strain- and species-level variation in the microbiome of diabetic wounds is associated with clinical outcomes and therapeutic efficacy. Cell Host Microbe. 2019;25:641–655.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nussbaum SR, Carter MJ, Fife CE, DaVanzo J, Haught R, Nusgart M, et al. An economic evaluation of the impact, cost, and Medicare policy implications of chronic nonhealing wounds. Value Health. 2018;21:27–32. [DOI] [PubMed] [Google Scholar]
- 85. Versey Z, da CruzNWS, Russell E, Zigic S, DeZeeuw KG, Marek JE, et al. Biofilm-innate immune Interface: contribution to chronic wound formation. Front Immunol. 2021;12:648554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. James GA, Swogger E, Wolcott R, Pulcini E, Secor P, Sestrich J, et al. Biofilms in chronic wounds. Wound Repair Regen. 2008;16:37–44. [DOI] [PubMed] [Google Scholar]
- 87. Malone M, Bjarnsholt T, McBain AJ, James GA, Stoodley P, Leaper D, et al. The prevalence of biofilms in chronic wounds: a systematic review and meta-analysis of published data. J Wound Care. 2017;26:20–5. [DOI] [PubMed] [Google Scholar]
- 88. Schierle CF, De la Garza M, Mustoe TA, Galiano RD. Staphylococcal biofilms impair wound healing by delaying reepithelialization in a murine cutaneous wound model. Wound Repair Regen. 2009;17:354–9. [DOI] [PubMed] [Google Scholar]
- 89. Wolcott RD, Hanson JD, Rees EJ, Koenig LD, Phillips CD, Wolcott RA, et al. Analysis of the chronic wound microbiota of 2,963 patients by 16S rDNA pyrosequencing. Wound Repair Regen. 2016;24:163–74. [DOI] [PubMed] [Google Scholar]
- 90. James GA, Ge Zhao A, Usui M, Underwood RA, Nguyen H, Beyenal H, et al. Microsensor and transcriptomic signatures of oxygen depletion in biofilms associated with chronic wounds. Wound Repair Regen. 2016;24:373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nakagami G, Schultz G, Kitamura A, Minematsu T, Akamata K, Suga H, et al. Rapid detection of biofilm by wound blotting following sharp debridement of chronic pressure ulcers predicts wound healing: a preliminary study. Int Wound J. 2020;17:191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Roy S, Elgharably H, Sinha M, Ganesh K, Chaney S, Mann E, et al. Mixed-species biofilm compromises wound healing by disrupting epidermal barrier function. J Pathol. 2014;233:331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Roy S, Santra S, Das A, Dixith S, Sinha M, Ghatak S, et al. Staphylococcus aureus biofilm infection compromises wound healing by causing deficiencies in granulation tissue collagen. Ann Surg. 2020;271:1174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, et al. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008;8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Misic AM, Gardner SE, Grice EA. The wound microbiome: modern approaches to examining the role of microorganisms in impaired chronic wound healing. Adv Wound Care (New Rochelle). 2014;3:502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rhoads DD, Wolcott RD, Sun Y, Dowd SE. Comparison of culture and molecular identification of bacteria in chronic wounds. Int J Mol Sci. 2012;13:2535–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gardner SE, Hillis SL, Heilmann K, Segre JA, Grice EA. The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. Diabetes. 2013;62:923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Loesche M, Gardner SE, Kalan L, Horwinski J, Zheng Q, Hodkinson BP, et al. Temporal stability in chronic wound microbiota is associated with poor healing. J Invest Dermatol. 2017;137:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Verbanic S, Shen Y, Lee J, Deacon JM, Chen IA. Microbial predictors of healing and short-term effect of debridement on the microbiome of chronic wounds. NPJ Biofilms Microbiomes. 2020;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gardiner M, Vicaretti M, Sparks J, Bansal S, Bush S, Liu M, et al. A longitudinal study of the diabetic skin and wound microbiome. PeerJ. 2017;5:e3543. 10.7717/peerj.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Redel H, Gao Z, Li H, Alekseyenko AV, Zhou Y, Perez-Perez GI, et al. Quantitation and composition of cutaneous microbiota in diabetic and nondiabetic men. J Infect Dis. 2013;207:1105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Eckhard M, Lengler A, Liersch J, Bretzel R G, and Mayser P. Fungal foot infections in patients with diabetes mellitus--results of two independent investigations. Mycoses 2007;50 :14–9. doi: [DOI] [PubMed] [Google Scholar]
- 103. Chellan G, Shivaprakash S, Karimassery Ramaiyar S, Varma AK, Varma N, Thekkeparambil Sukumaran M, et al. Spectrum and prevalence of fungi infecting deep tissues of lower-limb wounds in patients with type 2 diabetes. J Clin Microbiol. 2010;48:2097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dowd SE, Delton Hanson J, Rees E, Wolcott RD, Zischau AM, Sun Y, et al. Survey of fungi and yeast in polymicrobial infections in chronic wounds. J Wound Care. 2011;20:40–7. [DOI] [PubMed] [Google Scholar]
- 105. Kalan L, Loesche M, Hodkinson BP, Heilmann K, Ruthel G, Gardner SE, et al. Redefining the chronic-wound microbiome: fungal communities are prevalent. Dynamic, and Associated with Delayed Healing mBio. 2016;7:e01058–16. 10.1128/mBio.01058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Verbanic S, Deacon JM, Chen IA. The chronic wound Phageome: phage diversity and associations with wounds and healing outcomes. Microbiol Spectr. 2022;10:e0277721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kirker KR, Secor PR, James GA, Fleckman P, Olerud JE, Stewart PS. Loss of viability and induction of apoptosis in human keratinocytes exposed to Staphylococcus aureus biofilms in vitro. Wound Repair Regen. 2009;17:690–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kirker KR, James GA, Fleckman P, Olerud JE, Stewart PS. Differential effects of planktonic and biofilm MRSA on human fibroblasts. Wound Repair Regen. 2012;20:253–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ramirez HA, Pastar I, Jozic I, Stojadinovic O, Stone RC, Ojeh N, et al. Staphylococcus aureus triggers induction of miR-15B-5P to diminish DNA repair and deregulate inflammatory response in diabetic foot ulcers. J Invest Dermatol. 2018;138:1187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pastar I, Sawaya AP, Marjanovic J, Burgess JL, Strbo N, Rivas KE, et al. Intracellular Staphylococcus aureus triggers pyroptosis and contributes to inhibition of healing due to perforin-2 suppression. J Clin Invest. 2021;131:e133727. 10.1172/jci133727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Strbo N, Pastar I, Romero L, Chen V, Vujanac M, Sawaya AP, et al. Single cell analyses reveal specific distribution of anti-bacterial molecule Perforin-2 in human skin and its modulation by wounding and Staphylococcus aureus infection. Exp Dermatol. 2019;28:225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tomic-Canic M, Burgess JL, O'Neill KE, Strbo N, Pastar I. Skin microbiota and its interplay with wound healing. Am J Clin Dermatol. 2020;21:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fazli M, Bjarnsholt T, Kirketerp-Møller K, Jørgensen B, Andersen AS, Krogfelt KA, et al. Nonrandom distribution of Pseudomonas aeruginosa and Staphylococcus aureus in chronic wounds. J Clin Microbiol. 2009;47:4084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sweere JM, Ishak H, Sunkari V, Bach MS, Manasherob R, Yadava K, et al. The immune response to chronic Pseudomonas aeruginosa wound infection in immunocompetent mice. Adv Wound Care (New Rochelle). 2020;9:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jesaitis AJ, Franklin MJ, Berglund D, Sasaki M, Lord CI, Bleazard JB, et al. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol. 2003;171:4329–39. [DOI] [PubMed] [Google Scholar]
- 116. Trøstrup H, Lerche CJ, Christophersen LJ, Thomsen K, Jensen P, Hougen HP, et al. Chronic Pseudomonas aeruginosa biofilm infection impairs murine S100A8/A9 and neutrophil effector cytokines-implications for delayed wound closure? Pathog Dis. 2017;75:1–10. 10.1093/femspd/ftx068. [DOI] [PubMed] [Google Scholar]
- 117. Trøstrup H, Lerche CJ, Christophersen LJ, Thomsen K, Jensen P, Hougen HP, et al. Pseudomonas aeruginosa biofilm hampers murine central wound healing by suppression of vascular epithelial growth factor. Int Wound J. 2018;15:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. van der PlasMJ, Bhongir RK, Kjellström S, Siller H, Kasetty G, Mörgelin M, et al. Pseudomonas aeruginosa elastase cleaves a C-terminal peptide from human thrombin that inhibits host inflammatory responses. Nat Commun. 2016;7:11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. D'Arpa P, Karna SLR, Chen T, Leung KP. Pseudomonas aeruginosa transcriptome adaptations from colonization to biofilm infection of skin wounds. Sci Rep. 2021;11:20632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, et al. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010;465:346–9. [DOI] [PubMed] [Google Scholar]
- 121. Bitschar K, Staudenmaier L, Klink L, Focken J, Sauer B, Fehrenbacher B, et al. Staphylococcus aureus skin colonization is enhanced by the interaction of neutrophil extracellular traps with keratinocytes. J Invest Dermatol. 2020;140:1054–1065.e4. [DOI] [PubMed] [Google Scholar]
- 122. Yang JJ, Chang TW, Jiang Y, Kao HJ, Chiou BH, Kao MS, et al. Commensal Staphylococcus aureus provokes immunity to protect against skin infection of methicillin-resistant Staphylococcus aureus. Int J Mol Sci. 2018;19:1290. 10.3390/ijms19051290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Alves PM, Al-Badi E, Withycombe C, Jones PM, Purdy KJ, Maddocks SE. Interaction between Staphylococcus aureus and Pseudomonas aeruginosa is beneficial for colonisation and pathogenicity in a mixed biofilm. Pathog Dis. 2018;76:1–10. 10.1093/femspd/fty003. [DOI] [PubMed] [Google Scholar]
- 124. Orazi G, O'Toole GA. Pseudomonas aeruginosa alters Staphylococcus aureus sensitivity to vancomycin in a biofilm model of cystic fibrosis infection. MBio. 2017;8:e00873–17. 10.1128/mBio.00873-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wollenberg MS, Claesen J, Escapa IF, Aldridge KL, Fischbach MA, Lemon KP. Propionibacterium-produced coproporphyrin III induces Staphylococcus aureus aggregation and biofilm formation. MBio. 2014;5:e01286–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shu M, Wang Y, Yu J, Kuo S, Coda A, Jiang Y, et al. Fermentation of Propionibacterium acnes, a commensal bacterium in the human skin microbiome, as skin probiotics against methicillin-resistant Staphylococcus aureus. PLoS One. 2013;8:e55380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ramsey MM, Freire MO, Gabrilska RA, Rumbaugh KP, Lemon KP. Staphylococcus aureus shifts toward commensalism in response to Corynebacterium species. Front Microbiol. 2016;7:1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mottola C, Mendes JJ, Cristino JM, Cavaco-Silva P, Tavares L, Oliveira M. Polymicrobial biofilms by diabetic foot clinical isolates. Folia Microbiol (Praha). 2016;61:35–43. [DOI] [PubMed] [Google Scholar]
- 129. Meneghin A, Hogaboam CM. Infectious disease, the innate immune response, and fibrosis. J Clin Invest. 2007;117:530–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chioma OS, Drake WP. Role of microbial agents in pulmonary fibrosis. Yale J Biol Med. 2017;90:219–27. [PMC free article] [PubMed] [Google Scholar]
- 131. Wu J, Del Duca E, Espino M, Gontzes A, Cueto I, Zhang N, et al. RNA sequencing keloid transcriptome associates keloids with Th2, Th1, Th17/Th22, and JAK3-skewing. Front Immunol. 2020;11:597741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Feng C, Shan M, Xia Y, Zheng Z, He K, Wei Y, et al. Single-cell RNA sequencing reveals distinct immunology profiles in human keloid. Front Immunol. 2022;13:940645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Agbenorkul PT, Kus H, Myczkowski T. The keloid triad hypothesis (KTH): a basis for keloid etiopathogenesis and clues for prevention. Eur J Plast Surg. 1995;18:301–4. [Google Scholar]
- 134. Alonso PE, Rioja LF, Pera C. Keloids: a viral hypothesis. Med Hypotheses. 2008;70:156–66. [DOI] [PubMed] [Google Scholar]
- 135. Pratsinis H, Mavrogonatou E, Kletsas D. Scarless wound healing: from development to senescence. Adv Drug Deliv Rev. 2019;146:325–43. [DOI] [PubMed] [Google Scholar]
- 136. Kong X, Fu J, Shao K, Wang L, Lan X, Shi J. Biomimetic hydrogel for rapid and scar-free healing of skin wounds inspired by the healing process of oral mucosa. Acta Biomater. 2019;100:255–69. [DOI] [PubMed] [Google Scholar]
- 137. Frenard C, Dagnelie MA, Khammari A, Saint-Jean M, Boisrobert A, Corvec S, et al. Do Cutibacterium acnes and Staphylococcus aureus define two different types of folliculitis?: bacteriological study of scalp folliculitis. J Eur Acad Dermatol Venereol. 2018;32:e266–8. [DOI] [PubMed] [Google Scholar]
- 138. Singer AJ, McClain SA. Persistent wound infection delays epidermal maturation and increases scarring in thermal burns. Wound Repair Regen. 2002;10:372–7. [DOI] [PubMed] [Google Scholar]
- 139. Bagabir RA, Syed F, Rautemaa R, McGrouther DA, Paus R, Bayat A. Upregulation of toll-like receptors (TLRs) 6, 7, and 8 in keloid scars. J Invest Dermatol. 2011;131:2128–30. [DOI] [PubMed] [Google Scholar]
- 140. Arvia R, Margheri F, Stincarelli MA, Laurenzana A, Fibbi G, Gallinella G, et al. Parvovirus B19 activates in vitro normal human dermal fibroblasts: a possible implication in skin fibrosis and systemic sclerosis. Rheumatology (Oxford). 2020;59:3526–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ashrafi M, Sebastian A, Shih B, Greaves N, Alonso-Rasgado T, Baguneid M, et al. Whole genome microarray data of chronic wound debridement prior to application of dermal skin substitutes. Wound Repair Regen. 2016;24:870–5. [DOI] [PubMed] [Google Scholar]
- 142. Kostarnoy AV, Gancheva PG, Logunov DY, Verkhovskaya LV, Bobrov MA, Scheblyakov DV, et al. Topical bacterial lipopolysaccharide application affects inflammatory response and promotes wound healing. J Interf Cytokine Res. 2013;33:514–22. [DOI] [PubMed] [Google Scholar]
- 143. Sahana TG, Rekha PD. A novel exopolysaccharide from marine bacterium Pantoea sp. YU16-S3 accelerates cutaneous wound healing through Wnt/β-catenin pathway. Carbohydr Polym. 2020;238:116191. [DOI] [PubMed] [Google Scholar]
- 144. Simonetti O, Cirioni O, Cacciatore I, Baldassarre L, Orlando F, Pierpaoli E, et al. Efficacy of the quorum sensing inhibitor FS10 alone and in combination with Tigecycline in an animal model of staphylococcal infected wound. PLoS One. 2016;11:e0151956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. O'Loughlin CT, Miller LC, Siryaporn A, Drescher K, Semmelhack MF, Bassler BL. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc Natl Acad Sci U S A. 2013;110:17981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Brackman G, Cos P, Maes L, Nelis HJ, Coenye T. Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrob Agents Chemother. 2011;55:2655–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ. Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov. 2020;19:311–32. [DOI] [PubMed] [Google Scholar]
- 148. Mangoni ML, McDermott AM, Zasloff M. Antimicrobial peptides and wound healing: biological and therapeutic considerations. Exp Dermatol. 2016;25:167–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Takahashi M, Umehara Y, Yue H, Trujillo-Paez JV, Peng G, Nguyen HLT, et al. The antimicrobial peptide human β-Defensin-3 accelerates wound healing by promoting angiogenesis, cell migration, and proliferation through the FGFR/JAK2/STAT3 Signaling pathway. Front Immunol. 2021;12:712781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Peral MC, Martinez MA, Valdez JC. Bacteriotherapy with lactobacillus plantarum in burns. Int Wound J. 2009;6:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Peral MC, Rachid MM, Gobbato NM, Huaman Martinez MA, Valdez JC. Interleukin-8 production by polymorphonuclear leukocytes from patients with chronic infected leg ulcers treated with lactobacillus plantarum. Clin Microbiol Infect. 2010;16:281–6. [DOI] [PubMed] [Google Scholar]
- 152. Twetman S, Keller MK, Lee L, Yucel-Lindberg T, Pedersen AML. Effect of probiotic lozenges containing lactobacillus reuteri on oral wound healing: a pilot study. Benef Microbes. 2018;9:691–6. [DOI] [PubMed] [Google Scholar]
- 153. Wälivaara D, Sjögren I, Gerasimcik N, Yucel-Lindberg T, Twetman S, Abrahamsson P. Effects of lactobacillus reuteri-containing lozenges on healing after surgical removal of mandibular third molars: a randomised controlled trial. Benef Microbes. 2019;10:653–9. [DOI] [PubMed] [Google Scholar]
- 154. Vågesjö E, Öhnstedt E, Mortier A, Lofton H, Huss F, Proost P, et al. Accelerated wound healing in mice by on-site production and delivery of CXCL12 by transformed lactic acid bacteria. Proc Natl Acad Sci U S A. 2018;115:1895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Severn MM, Horswill AR. Staphylococcus epidermidis and its dual lifestyle in skin health and infection. Nat Rev Microbiol. 2022;21:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Matsuzaki S, Yasuda M, Nishikawa H, Kuroda M, Ujihara T, Shuin T, et al. Experimental protection of mice against lethal Staphylococcus aureus infection by novel bacteriophage phi MR11. J Infect Dis. 2003;187:613–24. [DOI] [PubMed] [Google Scholar]
- 157. Kering KK, Kibii BJ, Wei H. Biocontrol of phytobacteria with bacteriophage cocktails. Pest Manag Sci. 2019;75:1775–81. [DOI] [PubMed] [Google Scholar]
- 158. Rhoads DD, Wolcott RD, Kuskowski MA, Wolcott BM, Ward LS, Sulakvelidze A. Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J Wound Care. 2009;18:237-8–40-3. [DOI] [PubMed] [Google Scholar]
- 159. Ramasubbu N, Thomas LM, Ragunath C, Kaplan JB. Structural analysis of dispersin B, a biofilm-releasing glycoside hydrolase from the periodontopathogen Actinobacillus actinomycetemcomitans. J Mol Biol. 2005;349:475–86. [DOI] [PubMed] [Google Scholar]
- 160. Qin Z, Ou Y, Yang L, Zhu Y, Tolker-Nielsen T, Molin S, et al. Role of autolysin-mediated DNA release in biofilm formation of Staphylococcus epidermidis. Microbiology (Reading). 2007;153:2083–92. [DOI] [PubMed] [Google Scholar]
- 161. Patel KK, Surekha DB, Tripathi M, Anjum MM, Muthu MS, Tilak R, et al. Antibiofilm potential of silver sulfadiazine-loaded nanoparticle formulations: a study on the effect of DNase-I on microbial biofilm and wound healing activity. Mol Pharm. 2019;16:3916–25. [DOI] [PubMed] [Google Scholar]
- 162. Percival SL, Kite P, Eastwood K, Murga R, Carr J, Arduino MJ, et al. Tetrasodium EDTA as a novel central venous catheter lock solution against biofilm. Infect Control Hosp Epidemiol. 2005;26:515–9. [DOI] [PubMed] [Google Scholar]
- 163. Raad I, Chatzinikolaou I, Chaiban G, Hanna H, Hachem R, Dvorak T, et al. In vitro and ex vivo activities of minocycline and EDTA against microorganisms embedded in biofilm on catheter surfaces. Antimicrob Agents Chemother. 2003;47:3580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Thapa RK, Winther-Larsen HC, Ovchinnikov K, Carlsen H, Diep DB, Tønnesen HH. Hybrid hydrogels for bacteriocin delivery to infected wounds. Eur J Pharm Sci. 2021;166:105990. [DOI] [PubMed] [Google Scholar]
- 165. Reish RG, Eriksson E. Scar treatments: preclinical and clinical studies. J Am Coll Surg. 2008;206:719–30. [DOI] [PubMed] [Google Scholar]
- 166. Agha R, Ogawa R, Pietramaggiori G, Orgill DP. A review of the role of mechanical forces in cutaneous wound healing. J Surg Res. 2011;171:700–8. [DOI] [PubMed] [Google Scholar]
- 167. Chen YE, Fischbach MA, Belkaid Y. Skin microbiota-host interactions. Nature. 2018;553:427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Grogan MD, Bartow-McKenney C, Flowers L, Knight SAB, Uberoi A, Grice EA. Research techniques made simple: profiling the skin microbiota. J Invest Dermatol. 2019;139:747–752.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]