

Abstract
Attention deficit hyperactivity disorder (ADHD) is a prevalent neurodevelopmental disorder with a major genetic component. Here we present a genome-wide association study meta-analysis of ADHD comprising 38,691 individuals with ADHD and 186,843 controls. We identified 27 genome-wide significant loci, highlighting 76 potential risk genes enriched among genes expressed particularly in early brain development. Overall, ADHD genetic risk was associated with several brain-specific neuronal sub-types and midbrain dopaminergic neurons. In exome-sequencing data from 17,896 individuals, we identified an increased load of rare protein-truncating variants in ADHD for a set of risk genes enriched with likely causal common variants, potentially implicating SORCS3 in ADHD by both common and rare variants. Bivariate Gaussian mixture modeling estimated that 84–98% of ADHD-influencing variants are shared with other psychiatric disorders. Additionally, common variant ADHD risk was associated with impaired complex cognition such as verbal reasoning and a range of executive functions, including attention.
Attention deficit hyperactivity disorder (ADHD) is a prevalent neurodevelopmental disorder, affecting around 5% of children, and persists into adulthood in two-thirds of cases1,2. It is characterized by extensive hyperactive, impulsive and/or inattentive behaviors that impair daily functioning. The disorder is associated with multiple adverse outcomes, such as injuries3, accidents4, depression5, substance use disorders6, aggression7, premature death8 and high rate of unemployment9, and has large societal costs10–12.
ADHD has a major genetic component, with an estimated twin heritability of 0.7413. Despite this, ADHD’s complex polygenic architecture makes it difficult to unravel its underlying biological causes. Previously, we discovered the first 12 genome-wide significant loci for ADHD14 in a genome-wide association study (GWAS) of 20,183 cases and 35,191 controls (here referred to as ADHD2019) that combined the first wave of data from the Danish iPSYCH15 cohort (iPSYCH1) with 11 ADHD cohorts collected by the Psychiatric Genomics Consortium (PGC). We established the role of common variants in ADHD, explaining around 22% of the variance in the phenotype. The results implicated brain-expressed genes and demonstrated considerable genetic overlap of ADHD with a range of phenotypes, e.g., within psychiatric, cognitive, and metabolic domains. Additionally, a recent cross-disorder GWAS of ADHD and autism16 has identified shared and differentiating loci and showed that individuals with both ADHD and autism have distinctive patterns of genetic association with other traits compared to those with only a single diagnosis. This highlights that further mapping of the shared genetic risk component with other psychiatric disorders is important for understanding the complexity of the genetics underlying ADHD. Analyses of whole-exome sequencing data have shown that rare variants also contribute to the risk for ADHD17, especially in mutationally constrained genes.
To better understand the biological mechanisms underlying ADHD, it is fundamental to conduct large genetic studies, as has been demonstrated in other psychiatric disorders18–20. Here we present results from an updated GWAS meta-analysis of ADHD, combining data from the newly extended Danish iPSYCH cohort, the Icelandic deCODE cohort and the PGC, almost doubling the number of cases compared with ADHD2019. We fine-map identified risk loci and integrate with functional genomics data to pinpoint potential causal genes and evaluate the burden of rare deleterious variants in top-associated genes. We characterize the polygenic architecture of ADHD and its overlap with other phenotypes by bivariate mixture modeling and perform polygenic score (PGS) analyses to test for association of ADHD-PGS with neurocognitive measures in the Philadelphia Neurodevelopmental Cohort (PNC).
RESULTS
Identification of new ADHD risk loci by GWAS meta-analysis.
We conducted a GWAS meta-analysis based on expanded data from iPSYCH (25,895 cases; 37,148 controls), deCODE genetics (8,281 cases; 137,993 controls) and published data from 10 ADHD cohorts with European ancestry collected by the PGC (4,515 cases; 11,702 controls), resulting in a total sample size of 38,691 individuals with ADHD and 186,843 controls (effective sample size (neff_half) = 51,568; cohorts listed in Supplementary Table 1).
The GWAS meta-analysis identified 32 independent lead variants (i.e., with a squared correlation (r2) < 0.1 between variants) located in 27 genome-wide significant loci (Fig. 1, Table 1, locus plots in Supplementary Data 1, and forest plots in Supplementary Data 2), including 21 novel loci. No statistically significant heterogeneity was observed between cohorts (Supplementary Fig. 1). The three most strongly associated loci (P < 5 × 10−14) were located on chromosome 1 (in and around PTPRF), chromosome 5 (downstream of MEF2C) and chromosome 11 (downstream of METTL15); the latter is a novel ADHD risk locus. Four loci on chromosomes 1, 5, 11, and 20 had secondary genome-wide significant lead variants (r2 < 0.1 between the index variant and the secondary lead variant within a region of 0.5 Mb), but none remained genome-wide significant in analyses conditioning on the index variant using COJO21 (Supplementary Table 2).
Figure 1 |. Results from GWAS meta-analysis of iPSYCH, deCODE and PGC cohorts including 38,899 cases and 186,843 controls in total.
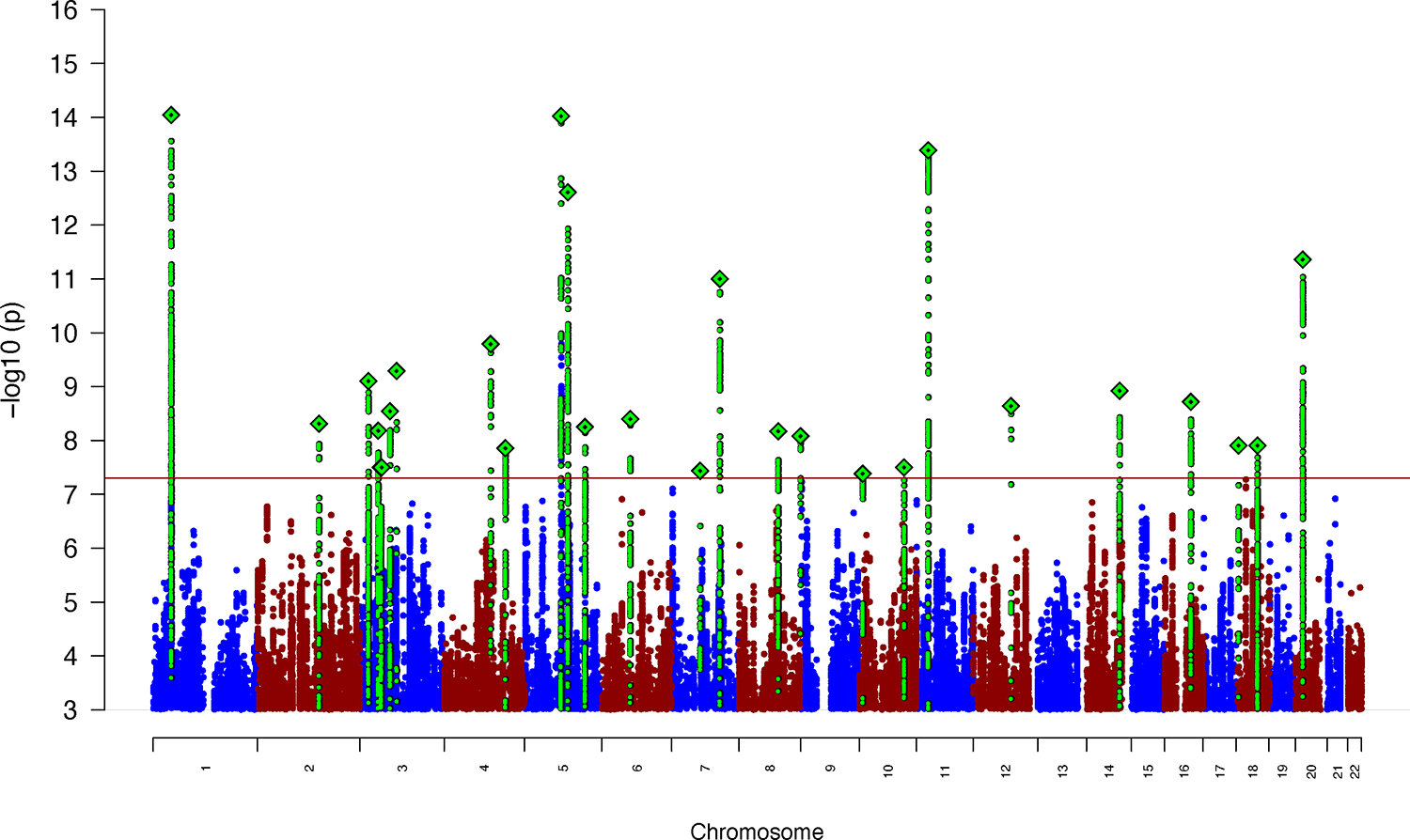
The y-axis represents −log10(two-sided P-values) from meta-analysis using an inverse-variance weighted fixed effects model. Index variants in each of the genome-wide significant loci are marked as a green diamond (note that two loci on chromosome 3, index variants rs7613360 and rs2311059, are located in close proximity and therefore appear as one diamond in the plot). The red horizontal line represents the threshold for genome-wide significant association (P = 5 × 10−8).
Table 1 |. Results for the 27 genome-wide significant index variants identified in the GWAS meta-analysis of 38,691 individuals with ADHD and 186,843 controls.
The location (chromosome (chr)) base position (bp) in hg19), alleles (A1 and A2), frequency (Freq.) of A1 in cases and controls, odds ratio (OR) of the effect with respect to A1, standard error (s.e.) and association P-values (two-sided) from inverse-variance weighted fixed effects model of the index variants are given. “Novel” indicates if the locus is a new ADHD risk locus i.e., not identified in ADHD2019 (ref. 14). Nearby genes located within 50 kb from index variants are listed (for a list of mapped genes based on other criteria see Supplementary Table 8).
| Genomic locus | chr | bp | rs ID | A1 | A2 | Nearby genes | Freq. cases | Freq. controls | OR | s.e. | P-value | Novel |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| 1 | 1 | 44076469 | rs549845 | G | A | PTPRF, KDM4A | 0.321 | 0.326 | 1.082 | 0.01 | 9.03E-15 | no |
| 2 | 2 | 145714354 | rs1438898 | A | C | 0.762 | 0.769 | 1.065 | 0.01 | 4.88E-09 | yes | |
| 3 | 3 | 20724204 | rs2886697 | G | A | 0.634 | 0.643 | 1.061 | 0.01 | 7.90E-10 | no | |
| 4 | 3 | 43691501 | rs9877066 | G | A | SNRK, ANO10, ABHD5 | 0.944 | 0.951 | 0.888 | 0.02 | 6.60E-09 | yes |
| 5 | 3 | 49916710 | rs7613360 | C | T | TRAIP, CAMKV, MST1R, CTD-2330K9.3, MON1A | 0.598 | 0.614 | 0.948 | 0.01 | 3.18E-08 | yes |
| 6 | 3 | 51884072 | rs2311059 | G | A | IQCF3, IQCF2, IQCF5, IQCF1 | 0.314 | 0.308 | 0.944 | 0.01 | 3.16E-08 | yes |
| 7 | 3 | 71499401 | rs17718444 | C | T | FOXP1 | 0.695 | 0.660 | 1.063 | 0.01 | 2.87E-09 | yes |
| 8 | 3 | 87015142 | rs114142727 | C | G | VGLL3 | 0.988 | 0.988 | 1.285 | 0.04 | 5.13E-10 | yes |
| 9 | 4 | 112217523 | rs17576773 | C | T | 0.888 | 0.880 | 1.101 | 0.02 | 1.63E-10 | yes | |
| 10 | 4 | 147099654 | rs6537401 | G | A | LSM6, RP11-6L6.2, SLC10A7 | 0.660 | 0.655 | 0.945 | 0.01 | 1.40E-08 | yes |
| 11 | 5 | 87854395 | rs4916723 | A | C | 0.553 | 0.573 | 0.918 | 0.01 | 9.48E-15 | no | |
| 12 | 5 | 103964585 | rs77960 | G | A | 0.665 | 0.682 | 0.929 | 0.01 | 2.46E-13 | yes | |
| 13 | 5 | 144474779 | rs10875612 | C | T | 0.483 | 0.470 | 0.947 | 0.01 | 5.62E-09 | yes | |
| 14 | 6 | 70858701 | rs2025286 | A | C | COL19A1 | 0.553 | 0.550 | 0.947 | 0.01 | 4.00E-09 | yes |
| 15 | 7 | 67685754 | rs73145587 | A | T | 0.910 | 0.901 | 1.107 | 0.02 | 3.67E-08 | yes | |
| 16 | 7 | 114158954 | rs9969232 | G | A | FOXP2 | 0.344 | 0.382 | 0.934 | 0.01 | 9.98E-12 | no |
| 17 | 8 | 93277087 | rs7844069 | T | G | 0.428 | 0.399 | 1.057 | 0.01 | 6.74E-09 | yes | |
| 18 | 8 | 145802447 | rs4925811 | T | G | C8orf82, ARHGAP39 | 0.515 | 0.531 | 0.944 | 0.01 | 8.30E-09 | yes |
| 19 | 10 | 8784773 | rs11255890 | C | A | 0.389 | 0.401 | 1.054 | 0.01 | 4.14E-08 | yes | |
| 20 | 10 | 106453832 | rs11596214 | G | A | SORCS3 | 0.597 | 0.569 | 1.054 | 0.01 | 3.17E-08 | no |
| 21 | 11 | 28602173 | rs2582895 | C | A | METTL15 | 0.634 | 0.618 | 1.075 | 0.01 | 4.09E-14 | yes |
| 22 | 12 | 89771903 | rs704061 | T | C | DUSP6, POC1B | 0.554 | 0.560 | 0.946 | 0.01 | 2.30E-09 | no |
| 23 | 14 | 98690923 | rs76284431 | T | A | 0.847 | 0.842 | 0.922 | 0.01 | 1.19E-09 | yes | |
| 24 | 16 | 61966703 | rs1162202 | C | T | CDH8 | 0.630 | 0.606 | 1.063 | 0.01 | 1.92E-09 | yes |
| 25 | 18 | 5871800 | rs76857496 | C | A | TMEM200C | 0.870 | 0.859 | 1.083 | 0.01 | 1.24E-08 | yes |
| 26 | 18 | 50625779 | rs7506904 | G | A | DCC | 0.343 | 0.372 | 0.946 | 0.01 | 1.24E-08 | yes |
| 27 | 20 | 21250843 | rs6082363 | T | C | XRN2, NKX2-4 | 0.296 | 0.291 | 1.073 | 0.01 | 4.38E-12 | yes |
Six of the previously identified 12 loci in the ADHD2019 study14 were significant in the present study (Table 1), and the remaining six loci demonstrated P-values < 8 × 10−4 (Supplementary Table 3). Overall, the direction of association of the top loci (726 loci with P < 1 × 10−4) was consistent with the direction of association in ADHD2019 for all loci but one (Supplementary Table 4).
Genetic correlations among cohorts and SNP-heritability.
Genetic correlation analyses supported a high consistency in the phenotype across cohorts (rg ranging from 0.82 to 0.93, Supplementary Table 5), and between iPSYCH1 and iPSYCH2 (rg = 0.97; s.e. = 0.06). None of the genetic correlations were significantly different from 1. LD score regression analysis found an intercept of 1.04 (s.e. = 0.009) and ratio of 0.092 (s.e. = 0.02), the latter indicating that around 90% of the deviation from null, in the distribution of the test statistics, reflects polygenicity (QQ-plot shown in Supplementary Fig. 2). The SNP heritability (h2SNP) was estimated to 0.14 (s.e. = 0.01), which is lower than the previously reported h2SNP of 0.2214. The h2SNP for iPSYCH (h2SNP = 0.23; s.e. = 0.01) was in line with the previous finding, but lower h2SNP was observed for PGC (h2SNP = 0.12; s.e. = 0.03) and deCODE (h2SNP = 0.08; s.e. = 0.014). The difference in h2SNP was not caused by different sex distributions across cohorts as there were no significant differences in h2SNP between males and females in the iPSYCH and deCODE cohorts (Supplementary Table 5). Between-cohort heterogeneity in h2SNP is not unusual and has been observed in other diagnoses such as major depressive disorder22.
Mapping risk variants to genes and enrichment analyses.
To link identified risk variants to genes, we first identified sets of Bayesian credible variants for each risk locus, with each set most likely (probability > 95%) including a causal variant (Supplementary Table 6). Credible variants were subsequently linked to genes based on genomic position, information about expression quantitative trait loci (eQTLs) and chromatin interaction mapping in human brain tissue as implemented in FUMA23 (datasets selected are listed in the Supplementary Note). We identified 76 plausible ADHD risk genes (Supplementary Table 7); four of the 76 were mapped by position alone. We found that this set of genes is significantly enriched among genes upregulated during early embryonic brain development (19th post-conceptual week; Pone_sided = 0.0008; Supplementary Fig. 3) and highly enriched for genes identified in GWASs of cognition-related phenotypes and reproduction (Supplementary Fig. 4). The role of the genes in synapses was evaluated using SynGO data24; nine genes mapped to SynGO annotations, and genes encoding integral components of the postsynaptic density membrane were borderline significantly enriched (P = 5.43 × 10−3; q-value 0.022; genes PTPRF, SORCS3, and DCC; Supplementary Fig. 5 and Supplementary Table 8). One SynGO-mapped gene was also a part of the upregulated genes during early embryonic brain development (ARHGAP39). Additionally, enrichment of the 76 genes in biological pathways was tested using data from 26 databases implemented in Enrichr25,26. No pathways showed significant enrichment after Bonferroni correction (database significant findings can be found in Supplementary Table 9). Finally, MAGMA27 gene-set analysis using gene-based P-values derived from the full GWAS summary statistics (i.e., no preselection of specific genes) did not reveal any significant findings (top gene sets can be found in Supplementary Table 10).
Transcriptome-wide association analysis.
To identify and prioritize ADHD risk genes, we also performed a transcriptome-wide association study (TWAS) of the genetically regulated gene expression using EpiXcan28 and expression data from the PsychENCODE Consortium29 on genes as well as isoforms detected in 924 samples from the dorsolateral prefrontal cortex (DLPFC). The TWAS identified 15 genes (Supplementary Table 11) and 18 isoforms (Supplementary Table 12), which together identified 23 distinct genes (Supplementary Fig. 6) with significantly different predicted gene expression levels in ADHD cases compared to controls (after Bonferroni correction for all the 34,646 genes and isoforms tested; Supplementary Fig. 6). Eight of the genes were among the 76 genes mapped by credible variants in FUMA. When using a less stringent correction (false discovery rate < 5%), we identified 237 genes with different predicted expression among cases and controls, of which 19 were also among the 76 prioritized risk genes. The B4GALT2-205 isoform located in the genome-wide significant locus on chromosome 1 showed the strongest association (P = 7 × 10−11), with lower predicted expression in ADHD compared to controls (Supplementary Fig. 7a). The expression model for B4GALT2-205 implicated four genome-wide significant variants. The second top gene was PPP1R16A (P = 1.4 × 10−8), which showed a predicted under-expression in cases compared to controls. The expression model for this gene implicated one genome-wide significant variant (Supplementary Fig. 7b).
Tissue- and cell type-specific expression of ADHD risk genes.
Gene-based association analysis using MAGMA27 identified 45 exome-wide significant genes (P < 2.72 × 10−6 (0.05/18,381 genes)) associated with ADHD (Supplementary Table 13). Gene association results across the entire genome were tested for a relationship with tissue-specific gene expression. This showed that brain-expressed genes, and in particular genes expressed in the cortex, are associated with ADHD (Supplementary Fig. 8). This result was supported by LDSC-SEG30 analysis, showing a significant enrichment in the heritability by variants located in genes specifically expressed in the frontal cortex (Supplementary Table 14).
Next, we examined neuronal cell type-specific gene expression in ADHD using two approaches. First, we tested for enrichment of variants located in cell-specific epigenomic peaks by intersecting our genetic associations with data from two recent catalogs of the human epigenome that profile major human body cell types31 as well as brain-specific cell types32. Here we found enrichment for genes expressed in major brain neuronal cell types, including both excitatory and inhibitory neurons (Supplementary Fig. 9). Second, we performed cell type-specific analyses in FUMA33 based on single cell RNA-sequencing data. This revealed a significant association (P = 0.005) between ADHD-associated genes and genes expressed in dopaminergic midbrain neurons (Linnarsson midbrain data34; Supplementary Fig. 10 and Supplementary Table 15).
Convergence of common and rare variant risk.
To test for convergence of risk conferred by common variants and rare protein-truncating variants (rPTVs), we analyzed whole-exome sequencing data from a subset of the iPSYCH cohort consisting of 8,895 ADHD cases and 9,001 controls. We tested three gene sets: (1) the 76 prioritized risk genes identified by positional and functional annotation, (2) the 45 significant genes in the MAGMA analysis, and (3) 18 genes with at least five credible variants located in the coding region (Supplementary Table 16). While there was no indication of increased burden of rPTVs in the first gene set (P = 0.39, OR = 1,30, s.e. = 0.16), the second gene set showed borderline nominal significant enrichment (P = 0.05, OR = 1.43, s.e. = 0.18), and the set of genes identified based on credible variants had a significantly increased burden of rPTVs in individuals with ADHD compared to controls (P = 0.015, OR = 2.19, s.e. = 0.32). For comparison, there was no enrichment in rare synonymous variants in the third gene set (P = 0.59). When evaluating the 18 genes from the “credible gene set” individually, SORCS3 was nominally significantly (P = 0.008; Supplementary Table 16) enriched in rPTVs in ADHD cases when compared to a combined group of iPSYCH controls and gnomAD individuals (non-psychiatric non-Finnish Europeans; n = 58,121); this suggests that SORCS3 might be implicated in ADHD both by common and rare deleterious variants.
Genetic overlap of ADHD with other phenotypes.
The genome-wide genetic correlation (rg) of ADHD with other phenotypes was estimated using published GWASs (258 phenotypes) and GWASs of UK Biobank data (514 phenotypes), available in LDhub35. ADHD showed significant genetic correlation (P < 2 × 10−4) with 56 phenotypes representing domains previously found to have significant genetic correlations with ADHD: cognition (e.g. educational attainment rg = −0.55, s.e. = 0.021), weight/obesity (e.g. body mass index rg = 0.27, s.e. = 0.03), smoking (e.g. smoking initiation rg = 0.48; s.e = 0.07), sleep (e.g. insomnia rg = 0.46, s.e. = 0.05), reproduction (e.g. age at first birth rg = −0.65, s.e. = 0.03) and longevity (e.g. mother’s age at death rg = −0.42, s.e. = 0.07). When considering other neurodevelopmental and psychiatric disorders, autism spectrum disorder (ASD) (rg = 0.42, s.e. = 0.05), schizophrenia (SCZ) (rg = 0.17, s.e. = 0.03), major depressive disorder (MDD) (rg = 0.31, s.e. = 0.07) and cannabis use disorder (CUD) (rg = 0.61, s.e. = 0.04) were significantly correlated with ADHD (Supplementary Table 17). In UK Biobank data, ADHD demonstrated the strongest genetic correlation with a low overall health rating (rg = 0.60, s.e. = 0.2; Supplementary Table 18).
Furthermore, we applied MiXeR36, which uses univariate and bivariate Gaussian mixture modeling to quantify the actual number of variants that: (1) explain 90% of the SNP heritability of ADHD and (2) overlap between ADHD and other phenotypes representing domains with high genetic correlation with ADHD (psychiatric disorders, smoking behavior, weight, reproduction, and sleep were evaluated). MiXeR considers all variants, i.e., variants with the same and opposite directions of effects. Approximately 7.3K (standard deviation (s.d.) = 324) common variants were found to influence ADHD, which is less than our estimates for SCZ (9.6K; s.d. = 199), MDD (11.7K; s.d. = 345) and ASD (10.3K; s.d. = 1,011), and less than previously reported for bipolar disorder (BD) (8.6K, s.d. = 200)18.
When considering the number of shared loci as a proportion of the total polygenicity of ADHD, the vast majority of variants influencing ADHD were also estimated to influence the other investigated psychiatric disorders (84%–98%; Fig. 2, Supplementary Fig. 11, and Supplementary Table 19). While the fraction of concordant variants (within the shared part) with ASD and MDD was at the high end (75–76%), it was lower for SCZ (59%). When considering other phenotypes, insomnia demonstrated the smallest overlap with ADHD in terms of actual number of variants (4.5K, s.d. = 1,281; 62% of ADHD variants shared), while almost all variants influencing ADHD also influence educational attainment, age at first birth and smoking (Fig. 2 and Supplementary Table 19). For insomnia and smoking, 83% and 79% of shared variants had concordant directions, respectively, while only 21% and 20% of ADHD risk variants were concordant with educational attainment and age at first birth associated variants, respectively (Supplementary Table 19).
Figure 2 |. Venn diagrams showing MiXeR results of the estimated number of variants shared between ADHD and psychiatric disorders (with significant genetic correlations with ADHD) and phenotypes representing other domains with high genetic correlation with ADHD.
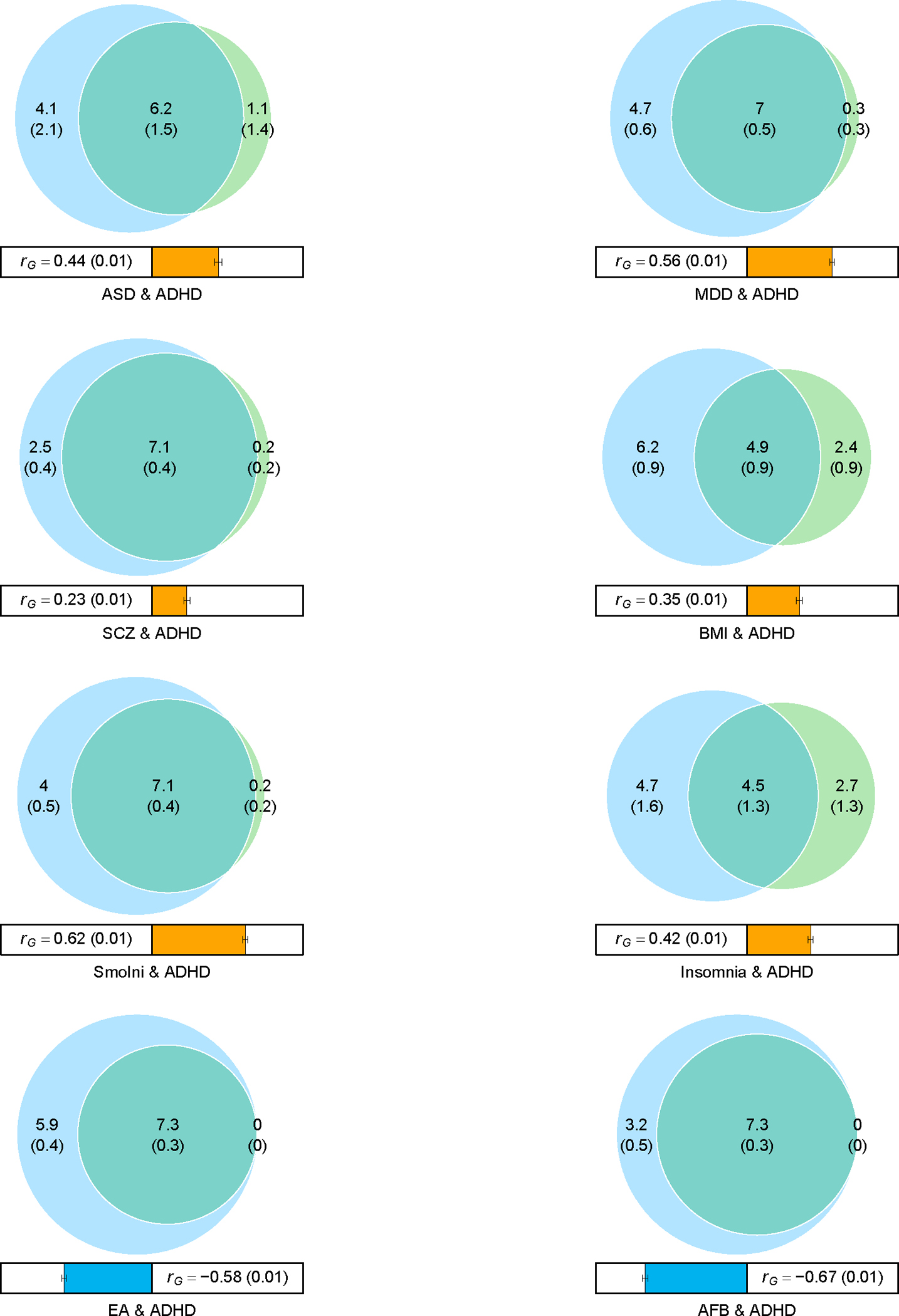
Circles represent shared variants (gray), unique to ADHD (light blue) and unique to the other phenotype of interest (orange). The number of shared variants (and standard deviations) is shown in thousands. The size of the circles reflects the polygenicity of each phenotype, with larger circles corresponding to greater polygenicity. The estimated genetic correlation (rg) between ADHD and each phenotype from LDSC is shown below the corresponding Venn diagram, with an accompanying scale (−1 to +1) with blue and red representing negative and positive genetic correlations, respectively. Bivariate results for ADHD, autism spectrum disorder (ASD), major depressive disorder (MDD), schizophrenia (SCZ), body mass index (BMI), smoking initiation (SmoIni), insomnia, educational attainment (EA) and age at first birth (AFB) are shown (see also Supplementary Table 17).
Impact of ADHD polygenic scores on cognitive domains.
Educational attainment is one of the phenotypes with the strongest negative genetic correlation with ADHD, as demonstrated above, and cognitive impairments in ADHD are well described37. To further explore how ADHD risk variants affect specific cognitive domains, we assessed the association of ADHD polygenic scores (PGS) with 15 cognitive measures in the Philadelphia Neurodevelopmental Cohort (PNC)38,39. This cohort is from the greater Philadelphia area and includes individuals, 8–21 years of age, who received medical care at the Children’s Hospital of Philadelphia Network. The subsample of the PNC cohort (v1 release) of 4,973 individuals with European descent was utilized in this study. The Computerized Neurocognitive Battery40 was used to assess cognitive performance in the study participants. The battery consists of 14 tests in five domains: executive control, episodic memory, complex cognitive processing, social cognition and sensorimotor speed. Additionally, the Wide Range Achievement Test (WRAT-4)41 was used as a proxy measure for overall IQ39.
ADHD-PGS was negatively associated with seven neurocognitive measures (Fig. 3), with the strongest association for the WRAT-4 test (beta = −0.09, P = 1.09 × 10−10). ADHD-PGS was associated with measures of executive control (attention: beta = −0.08, P = 3.94 × 10−8; working memory: beta = −0.05, P = 1.56 × 10−3), complex cognition (verbal reasoning: beta = −0.08, P = 1.31 × 10−10; non-verbal reasoning: beta = −0.05, P = 1.08 × 10−3; spatial reasoning: beta = −0.06, P = 5.15 × 10−5) and one measure of episodic memory (facial memory: beta = −0.05, P = 3.23 × 10−3) (Supplementary Table 20). The negative association of ADHD risk variants with executive functions, especially attention, is in line with the inattention problems often observed in individuals with ADHD.
Figure 3 |. Association of ADHD-PGS with measures of cognitive abilities in the PNC cohort (n = 4,973).
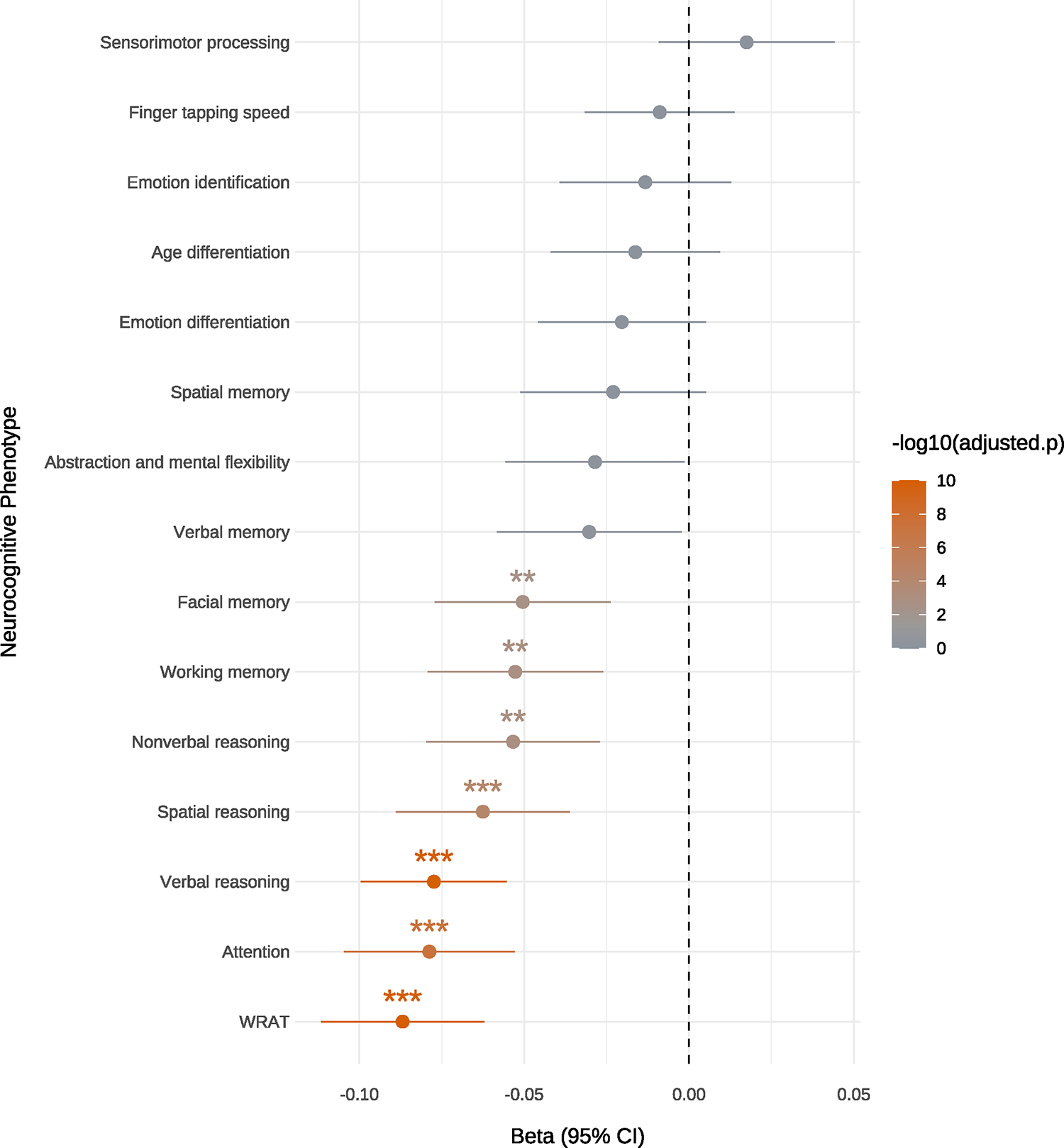
Beta values (represented as a dot and standard errors indicated as horizontal bars) from linear regression testing for the association of ADHD-PGS with the 15 neurocognitive measures listed on the y-axis (Wide Range Achievement Test-4 (WRAT). The color bar at the right indicates the −log10(Bonferroni adjusted two-sided P-value) and P-value thresholds are indicated by stars (*P = 0.05; **P = 0.01, ***P = 0.001).
DISCUSSION
The present study identified 27 genome-wide significant loci in the largest GWAS of ADHD to date. We analyzed around twice as many ADHD cases compared to the ADHD201914 study and more than doubled the number of associated loci, indicating that we have passed the inflection point for ADHD with respect to the rate of risk loci discovery.
Six of the 12 previously identified loci were also significant in this study. Even though some previously identified loci demonstrated weaker association here, their associations remained strong, and there was almost complete concordance in the direction of association between top-associated variants in this study and ADHD2019. In GWAS of complex disorders, it is not uncommon for some loci to fluctuate around the significance threshold with increasing sample sizes until they eventually achieve stable significance; this can often be attributed to the “winner’s curse” phenomenon, where effect size estimates close to the discovery threshold tend to be overestimated in initial GWAS42.
We report a lower h2SNP for ADHD (h2SNP = 0.14) than estimated previously (h2SNP = 0.22). This is driven by a lower h2SNP in the PGC and deCODE cohorts compared to iPSYCH. Different ascertainment and diagnostic strategies and designs among PGC cohorts could decrease the h2SNP, while a lower effective sample size43 in Iceland, and thus fewer recent variants, might bias h2SNP downwards in the deCODE cohort44.
We refined ADHD’s genetic architecture by estimating that around 7.3K (s.d. = 324) common variants can explain 90% of the h2SNP. This is a higher estimate than reported based on the 2019 ADHD GWAS (5.6K, s.d. = 400)45, but the current estimate is based on a better fit to the causal mixture model (AIC = 80 vs. AIC = 31 in Hindley et al.45). ADHD is often comorbid with other psychiatric disorders46, with 12–16% of cases also diagnosed with ASD16,47,48 and around 40% with depression49, which is also reflected in the genetic correlations reported here and previously14. Strikingly, when assessing both concordant and discordant allelic directions, over 90% of ADHD risk variants also seem to influence SCZ and MDD, and 84% influence ASD. This extensive sharing with SCZ, MDD, and ASD is at the same level as observed for SCZ and bipolar disorder36, which are among the most genetically correlated mental disorders50. Notably, for both MDD and ASD, around 75% of the variants shared with ADHD demonstrated concordant direction of association. The large sharing of variants influencing ADHD and other psychiatric disorders, when assessing both concordant and discordant allelic directions, suggests that the disorders are even more intermingled with respect to their common genetic architecture than previously thought based on their overall genetic correlations36,50. For common variants, the developmental trajectory towards ADHD might therefore be influenced by variants involved in several psychiatric disorders, but with disorder-specific allelic directions and effect sizes rather than actual ADHD-specific loci.
We also note that almost all variants that influence ADHD overlap with educational attainment51, and that the vast majority (79%) are associated with decreased educational attainment, consistent with the overall negative genetic correlation. For the models indicating a high number of shared variants (ADHD vs. MDD, SCZ, BMI, educational attainment, age at first birth and smoking), we found support (evaluated using the Akaike Information Criterion52) for the best fitting MiXeR models above the “minimal model”, which indicate that the data support the existence of a polygenic overlap, beyond the minimal level needed to explain the observed genetic correlations. For ADHD vs. ASD, the model had limited support, and the results should therefore be interpreted with caution.
Fine-mapping of the 27 loci identified credible variants, but only four variants had posterior probabilities greater than 0.5 in all three fine-mapping methods, and none were linked to specific genes based on our functional annotation analyses. Linking the credible variants to genes by integration with functional genomics data identified 76 prioritized risk genes, which were enriched among genes upregulated during early embryonic development and involved in cognitive abilities identified by GWAS of cognitive phenotypes. Among the 76 genes were PPP1R16A and B4GALT2 (mapped by psychENCODE eQTLs; Supplementary Fig. 12a,b), which were also the top-ranking genes in our TWAS of DLPFC expression, both showing a predicted decreased expression in cases compared to controls. These genes have not previously been linked to psychiatric disorders, but both have been linked to educational attainment51. The set of risk genes also included PTPRF, SORCS3 and DCC, which encode integral components of the postsynaptic density membrane. Involvement of postsynaptic components in the pathology of ADHD has been reported previously53 and also for SCZ54. We also highlight FOXP1 and FOXP2. The association signals were located within the transcribed regions of both genes and had credible variants being eQTLs (FOXP2, Supplementary Fig. 12c) or located in chromatin interacting regions (FOXP1, Supplementary Fig. 12d) in brain tissue. FOXP2 was identified in the ADHD2019 study14 and is also a risk gene for cannabis use disorder55, while FOXP1 is a new ADHD locus. Both FOXP1 and FOXP2 encode transcription factors that can heterodimerize to regulate transcription in brain tissues56,57 and have been implicated in speech disorders and intellectual disability58 by highly penetrant rare variants.
Overall, less than half of the TWAS Bonferroni significant genes overlapped with the 76 candidate risk genes (40% of “TWAS transcript genes”; and 47% of “TWAS genes”; Supplementary Fig. 13). This was not unexpected and could be due to noise in the data and/or that TWAS models are based on expression in adult brains whereas a large proportion of individuals in the GWAS are children. Additionally, eQTLs used to derive TWAS models might not overlap GWAS identified variants as the two types of methods are systematically biased toward identification of different types of variants59.
We report convergence of common and rare variants in a set of 18 genes defined by location of credible variants. Thirteen of the genes were hit by rPTVs, and eight had a higher load in cases compared to controls, and thus, the signal was not driven by a few genes but by several genes with an increased burden of rPTVs. Of particular note, SORCS3 seems to be implicated in ADHD by both common and rare variants. Common variants in SORCS3 show strong pleiotropic effects across several major psychiatric disorders50, but to our knowledge, rare variant analyses have not implicated SORCS3 in psychiatric disorders before. Our results add to the emerging picture of overlap between genes and pathways affected by common and rare variants in psychiatric disorders54,60–62.
We found that ADHD risk was associated with common variants located in genes significantly expressed in the brain, especially the frontal cortex. We also observed an enrichment of ADHD risk variants in genes expressed in major cell types of the brain, including both excitatory and inhibitory neurons and in midbrain dopaminergic neurons. The findings for frontal cortex and dopamine neurons fit well with the motor, reward and executive function deficits associated with ADHD; the frontal cortex is involved in executive functions including attention and working memory63, and midbrain dopaminergic neurons are essential for controlling key functions, such as voluntary movement64 and reward processing65. This interpretation is further supported by our ADHD-PGS analyses in PNC, which revealed that common ADHD risk variants impair several domains of cognitive abilities.
The PGS analyses in PNC identified strong association of polygenic ADHD risk with decreased overall IQ (approximated by the WRAT test scores), in line with the high negative genetic correlation of ADHD with educational attainment and the observation that 79% of all ADHD risk variants are associated with decreased educational attainment. Interestingly, we found that ADHD-PGS associates with decreased attention, which is a key ADHD symptom, and with impairments in measures of other cognitive traits such as working memory. Smaller studies have analyzed the impact of ADHD-PGS on executive functions with mixed results66–69. This study robustly identifies specific cognitive domains impacted by ADHD-PGS, and our results support ADHD-PGS being negatively associated with neurocognitive performance.
In summary, we identified new ADHD risk loci, highlighted candidate causal genes, and implicated genes expressed in frontal cortex and several brain specific neuronal subtypes in ADHD. Our analyses revealed ADHD to be highly polygenic, influenced by thousands of variants, of which the vast majority also influence other psychiatric disorders with concordant or discordant effects. Additionally, we demonstrated that common variant ADHD risk has an impairing impact on a range of executive functions. Overall, the results advance our understanding of the underlying biology of ADHD and reveal novel aspects of ADHD’s polygenic architecture, its relationship with other phenotypes, and its impact on cognitive domains.
METHODS
The study was approved by the local scientific ethics committees and IRBs. The iPSYCH study was approved by the Scientific Ethics Committee in the Central Denmark Region (Case No 1-10-72-287-12) and the Danish Data Protection Agency. In accordance with Danish legislation, the Danish Scientific Ethics Committee has, for this study, waived the need for specific informed consent in biomedical research based on existing biobanks. This deCODE study was approved by the National Bioethics Committee of Iceland (VSN 15-047) and all participants gave informed consent.
Samples, quality control and imputation.
iPSYCH.
The iPSYCH15,70 cohort consists of 129,950 genotyped individuals, among which 85,891 are cases diagnosed with at least one of six mental disorders (i.e., ADHD, SCZ, BD, MDD, ASD, post-partum disorder) and the remaining are population-based controls. Samples were selected from a baseline birth cohort comprising all singletons born in Denmark between May 1, 1981, and December 31, 2008, who were residents in Denmark on their first birthday and who have a known mother (n = 1,657,449). ADHD cases were diagnosed by psychiatrists according to the ICD10 criteria (F90.0, F90.1, F98.8 diagnosis codes) identified using the Danish Psychiatric Central Research Register71 and the Danish National Patient register72. Diagnoses were given in 2016 or earlier for individuals at least 1 year old. Controls were randomly selected from the same nationwide birth cohort and not diagnosed with ADHD.
Detailed information on genotyping, imputation and quality control can be found in the Supplementary Note. After QC, the iPSYCH1 ADHD sample included 38,899 individuals and iPSYCH2 included 24,144 individuals.
deCODE.
The deCODE cohort consisted of 8,281 individuals with ADHD. These were either individuals with a clinical diagnosis of ADHD (n = 5,583) according to the ICD10 criteria (ICD10-F90, F90.1, F98.8) or individuals that have been prescribed medication specific for ADHD symptoms (ATC-NA06BA, mostly methylphenidate) (n = 2,698). The control sample did not contain individuals with a diagnosis of SCZ, BD, ASD or self-reported ADHD symptoms or diagnosis. All participants who donated samples gave informed consent. Information about genotyping, QC and evaluation of potential genetic heterogeneity between individuals identified based on diagnosis codes and medication can be found in the Supplementary Note.
PGC cohorts.
We used summary statistics from the 10 PGC cohorts with European ancestry generated as a part of our previous GWAS meta-analysis of ADHD. Detailed information about cohort design, genotyping, QC, and imputation can be found in Demontis et al.14.
GWAS meta-analysis of ADHD.
GWASs were performed separately for iPSYCH1 (17,019 cases and 21,880 controls) and iPSYCH2 (8,876 cases and 15,268 controls) using dosages for imputed genotypes and additive logistic regression with the first 10 PCs (from the final PCAs) as covariates using PLINK v1.9.
GWAS of deCODE samples (8,281 ADHD cases; 137,993 controls) was done using dosage data and logistic regression with sex, year of birth, and county of origin as covariates. To account for inflation due to population stratification and cryptic relatedness, test statistics were divided by an inflation factor (lambda = 1.23) estimated from LD score regression as done previously55. Findings from analyses of the genetic structure of the Icelandic population by Price et al.73 support that lambda correction will ensure proper correction without false positives. Subsequently alleles were converted to match HRC alleles.
For the PGC cohorts, we used GWAS summary statistics for each of the 10 European PGC cohorts generated as a part of our previous GWAS meta-analysis14.
See Supplementary Note for sensitivity analyses related to the impact of using sex and age as covariates in the analyses. See Supplementary Figure 14 for the impact of including or excluding sex as covariate in GWAS of iPSYCH data.
Summary statistics from GWAS of the individual cohorts, containing variants with imputation quality (INFO score) > 0.8 and minor allele frequency > 0.01, were meta-analyzed with a fixed effects standard error weighted meta-analysis using METAL (version 2011-03-25)74. Only variants supported by an effective sample size greater than 60% were retained in the final summary statistics (6,774,224 variants).
Concordance in the direction of associations in the present GWAS with associations in the ADHD2019 data was evaluated by a sign-test at different P-value thresholds (see thresholds in Supplementary Table 4).
Conditional analysis.
We identified potentially independent genome-wide significant lead variants for four loci located on chromosome 1 (two secondary lead variants), 5, 11 and 20. To evaluate if these variants were independent from the lead variants, we performed association analyses of the secondary variants while conditioning on the index variant in the locus using COJO as implemented in GCTA21.
Identification of sets of credible variants.
To identify sets of causal variants, we fine-mapped each of the 27 genome-wide loci using three fine-mapping tools, FINEMAP v. 1.3.1 (ref. 75), PAINTOR v.3.0 (ref. 76) and CAVIARBF v.0.2.1 (ref. 77), using CAUSALdb-finemapping-pip downloaded from https://github.com/mulinlab/CAUSALdb-finemapping-pip78. Since no secondary lead variants remained genome-wide significant after conditional analyses, one causal variant was assumed per locus. Variants located in a region of 1 Mb around index variants were included in the analyses. We used a threshold of 95% for the total posterior probability of the variants included in the credible sets, and only variants claimed to be within the set by all three methods were included in the final credible set for each locus.
Genetic correlations among cohorts and SNP heritability.
SNP heritability (h2SNP) and pair-wise genetic correlation among the cohorts were calculated using LD score regression79 analysis of summary statistics from GWAS of deCODE samples, meta-analysis of iPSYCH1+iPSYCH2 and meta-analysis of the 10 PGC cohorts (applying the same approach as described for the meta-analysis of all cohorts). Conversion of h2SNP estimates from observed scale to the liability scale was done using a population prevalence of 5%. Test for significant differences in h2SNP between cohorts was done using a Z-test.
Mapping of risk genes, enrichment and pathway analyses.
To link identified risk variants to genes, we used the set of credible variants (identified as described above) for each locus and linked variants to genes based on genomic position and functional annotations in FUMA23. Protein coding genes were mapped if they were located with a distance of 10 kb upstream or downstream of the index variants or if a credible variant was annotated to the gene based on eQTL data or chromatin interaction data from human brain (datasets used are listed in the Supplementary Note). The mapping linked credible variants to 76 ADHD prioritized risk genes. These genes were used in gene-set enrichment analyses to evaluate if the candidate genes were enriched among (1) genes differentially expressed in specific brain tissues, (2) genes differentially expressed at specific brain developmental stages, (3) genes encoding proteins involved in synapses and (4) genes encoding proteins in specific biological pathways. We corrected for multiple testing separately for each of these hypotheses. The first two aims were addressed by performing enrichment analyses in the GENE2FUNC module in FUMA. Enrichment of ADHD risk genes among predefined sets of differentially expressed genes in GTEx (54 tissue types) and BrainSpan (29 different ages of samples and 11 general developmental stages) data using hypergeometric test, and protein-coding genes were chosen as background genes.
The third aim was addressed using SynGO24 (dataset version: 20210225) to test for enrichment among the 76 risk genes for genes involved in synaptic processes and locations. We analyzed for enrichment in two subsets: “biological process” (201 gene sets) and “cellular component” (92 gene sets). We controlled using a background set of “brain expressed” genes provided by the SynGo platform (defined as ‘expressed in any GTEx v7 brain tissues’) containing 18,035 unique genes, of which 1,225 overlap with SynGO annotated genes. For each ontology term, a one-sided Fisher exact test was performed to compare the list of ADHD risk genes and the selected background set. To find enriched terms within the entire SynGO ontology, the most specific term is selected where each ‘gene cluster’ (unique set of genes) is found and then multiple testing correction is applied using False Discovery Rate (FDR) on the subset of terms that contain these ‘gene clusters’. Only ontology terms with gene sets with a minimum of three genes were included in the enrichment analysis.
The fourth aim was addressed by testing if the 76 genes were enriched in pathways/gene sets using Enrichr25,26 and its implemented databases (26 databases). Only pathways enriched with more than two genes were considered. We took a conservative approach and only considered pathways to be significant if the within-database adjusted P-value was smaller than 0.002 (0.05/26 databases evaluated). After correction for the number of databases, no significantly enriched pathways were identified.
We also tested for enrichment among the 76 genes of genes reported from the GWAS catalog (2019) and UK Biobank GWASs (v1) and used https://appyters.maayanlab.cloud/Enrichr_Manhattan_Plot/ to visualize the results.
Finally, we conducted pathway enrichment analysis using results from the full GWAS meta-analysis (i.e., no preselection of genes) by performing MAGMA27 gene-set analysis in FUMA (see details in the Supplementary Note).
Transcriptomic imputation model construction and TWAS.
Transcriptomic imputation models were constructed as previously described28 for dorso-lateral prefrontal cortex (DLPFC) transcript levels80. The genetic dataset of the PsychENCODE cohort was uniformly processed for quality control (QC) steps before genotype imputation. The analysis was restricted to samples with European ancestry as previously described28. Genotypes were imputed using the University of Michigan server81 with the Haplotype Reference Consortium (HRC) reference panel82. Gene expression information (both at the level of gene and transcript) was derived from RNA-seq counts which were adjusted for known and hidden confounds, followed by quantile normalization80. For the construction of the transcriptomic imputation models, we used EpiXcan28, an elastic net-based method, which weighs SNPs based on available epigenetic annotation information83. We performed the transcript-trait association analysis for ADHD as previously described28. Briefly, we applied the S-PrediXcan method28 to integrate the ADHD GWAS meta-analysis summary statistics and the transcriptomic imputation models constructed above to obtain association results at both the level of genes and transcripts.
Gene-based association and tissue-specific gene expression.
We used MAGMA v1.08 implemented in FUMA v1.3.6a23 to perform gene-based association analysis using the full summary statistics from the GWAS meta-analysis. Genome-wide significance was assessed through Bonferroni correction for the number of genes tested (P = 0.05/18381 = 2.72 × 10−6).
The relationships between tissue-specific gene expression profiles and ADHD-gene associations were tested using MAGMA gene-property analysis of expression data from GTEx (54 tissue types) and BrainSpan (29 brain samples at different ages) available in FUMA (see Supplementary Note for datasets selected).
Enrichment in h2SNP of ADHD-associated variants located in or close to genes expressed in specific brain regions was estimated using LDSC-SEG30. Annotations indicating specific expression in 13 brain regions from the GTEx gene expression database were downloaded from https://alkesgroup.broadinstitute.org/LDSCORE/LDSC_SEG_ldscores/.
Cell type-specific expression of ADHD risk genes.
We tested for enrichment in the ADHD h2SNP of variants located in cell type-specific epigenetic peaks by examining the overlap of common genetic risk variants with open chromatin from a DHS (DNase I hypersensitive sites) study profiling major human cell types31 and an scATAC-seq (single-cell assay for transposase accessible chromatin)32 study using an LD-score partitioned heritability approach84. All regions of open chromatin were extended by 500 bp in either direction. The broad MHC region (hg19 chr6:25–35Mb) was excluded due to its extensive and complex LD structure, but otherwise default parameters were used for the algorithm. We applied Bonferroni correction (correcting for 23 cell types), and results below P = 0.0022 were considered significant.
Additionally, we performed cell type-specific analyses implemented in FUMA, using data from 13 single-cell RNA sequencing datasets from human brain. The method is described in detail in Watanabe et al.33. Datasets used and a short summary of the method can be found in the Supplementary Note).
Overlap of common ADHD risk variants with rare protein-truncating variants (rPTVs).
We analyzed the overlap of common variants with rPTVs in a subset of iPSYCH samples that have also been whole exome sequenced. A major part of the data (Pilot 1, Wave 1, Wave 2) was also included in the recent study by Satterstrom et al.17, and the same quality control procedure was applied in this study. Description of whole-exome sequencing procedure, QC and annotation can be found in the Supplementary Note. Variants were defined as PTVs if they were annotated as having large effects on gene function (nonsense variant, frameshift, splice site). We defined a variant as being rare if it had an allele count of five or less across the combination of the full iPSYCH exome-sequencing dataset (n = 28,448) and non-Finnish Europeans in the nonpsychiatric gnomAD exome database (n = 44,779).
We tested for increased burden of rPTVs in ADHD compared to controls in three gene sets: (1) the 76 genes linked to credible variants based on position and functional genomic data, (2) the 45 exome-wide significant genes identified in MAGMA analysis, and (3) genes with at least five credible variants within the coding regions. The requirement of five credible variants was chosen to prioritize the most likely causal genes. This threshold excluded eight genes located in the same locus covering a broad LD region on chromosome 3 (Supplementary Data 1; page 25). Additionally, two other genes with less than five credible variants were excluded located in two other loci on chromosome 3.
The burden of rPTVs and rare synonymous (rSYNs) in cases compared to controls was tested for the three gene sets with logistic regression corrected using the following covariates: birth year, sex, first ten principal components, number of rSYN, percentage of target with coverage > 20x, mean read depth at sites within the exome target passing VQSR, total number of variants, sequencing wave.
Only significant enrichment in the set of 18 genes identified based on credible variants was found. We therefore looked specifically into these genes to identify whether the signal was driven by specific genes. rPTVs were found in 13 of the genes, and eight of these genes had more rPTVs in cases than controls when looking at raw counts (Supplementary Table 16). We performed gene-based burden test using EPACTS (https://genome.sph.umich.edu/wiki/EPACTS) and a logistic Wald test (correcting using the covariates as described above). Additionally, in order to increase power to detect an increased burden of rPTVs at the gene level in ADHD cases, we combined iPSYCH controls with information about rPTVs in gnomAD (non-Finnish European individuals), done as described previously17. We performed these gene-based tests using Fisher’s exact test, and only the following genes were considered: (1) genes with a higher number rSYN in gnomAD controls compared to iPSYCH cases, and (2) genes with a higher rate of rPTVs in cases compared to controls in the iPSYCH data.
Genetic overlap with other phenotypes.
We estimated genetic correlations of ADHD with other phenotypes in LDhub35 (published GWASs: 255 phenotypes; UK Biobank GWASs: 514 phenotypes). Additionally, genetic correlations with three phenotypes not available in LDhub (cannabis use disorder55, smoking initiation85 and education attainment51) were estimated locally using LD score regression79.
We applied MiXeR36 to our ADHD GWAS summary statistics and GWAS from a selection of complex traits showing high genetic correlation with ADHD: ASD86, SCZ54, BMI87, educational attainmet88, age at first birth89, smoking initiation85, insomnia90 and a new GWAS meta-analysis of depression including 371,184 cases and 978,703 controls91 (Supplementary Table 19) to quantify (i) the number of variants influencing each trait and (ii) the genetic overlap between ADHD and each of the other traits. We used MiXeR with default settings (https://github.com/precimed/mixer) in a two-step process. First, we ran a univariate model for each trait to estimate the number of common variants having a non-zero genetic additive impact on the phenotype. The univariate model generates estimates of “polygenicity” (i.e., the proportion of non-null variants) and “discoverability” (i.e., the variance of effect sizes of non-null SNPs). Second, the variance estimates from the univariate step were used to run a bivariate model in a pairwise fashion (i.e. ADHD vs. each of the other traits), which produced estimates of SNPs with a specific effect on the first or on the second trait, and SNPs with a non-zero effect on both traits (for details on the method see also ref. 18 and Supplementary Note). The models were evaluated by the Akaike Information Criterion52 (AIC) and illustrated with modeled versus observed conditional quantile-quantile (Q-Q) plots (Supplementary Fig. 11). The AIC values can be found in Supplementary Table 19.
Polygenic score (PGS) analysis of cognitive measures in PNC.
PGS analysis was performed on 4,973 individuals of European ancestry from the Philadelphia Neurodevelopmental Cohort (PNC), ages 8–21. Information about imputation and QC of the PNC data can be found in the Supplementary Note.
The software PRS-CS92 was used to process ADHD GWAS summary statistics and assign per-allele posterior SNP effect sizes. A European LD reference panel generated from the 1000 Genomes Project data (https://github.com/getian107/PRScs) was utilized. The following default settings were used for PRS-CS: parameter a in the γ-γ prior = 1, parameter b in the γ-γ prior = 0.5, MCMC iterations = 1000, number of burn-in iterations = 500, and thinning of the Markov chain factor = 5. Additionally, the global shrinkage parameter phi was determined using a fully Bayesian method. Plink v2.093 was then used to calculate individual-level ADHD PGS. Linear regression was used to test the association between ADHD PGS and neurocognitive phenotypes measured in the PNC. Age (at time of neurocognitive testing), age2, genotyping batch, sex, and the first 10 MDS dimensions were used as covariates. The neurocognitive measures were obtained using the Computerized Neurocognitive Battery (CNB), which consists of 14 tests in 5 domains: executive control, episodic memory, complex cognitive processing, social cognition, and sensorimotor speed. The battery has been described in detail elsewhere40. Additionally, association of ADHD-PGS with results from the Wide Range Achievement Test (WRAT-4)41 were analyzed. See Supplementary Note regarding transformation of the CNB measures.
The total variance explained by ADHD-PGS and model covariates for each neurocognitive phenotype was reported using Adjusted R2. Additionally, the variance explained by ADHD-PGS was calculated in R using a variance partitioning tool (https://github.com/GabrielHoffman/misc_vp/blob/master/calcVarPart.R). Reported P-values were Bonferroni-adjusted to account for the number of independent tests performed.
Supplementary Material
ACKNOWLEDGEMENTS
We thank additional members of the ADHD working group of the Psychiatric Genomics Consortium, who are not named co-authors under the working group banner, for their contributions. We would like to thank the employees and research participants of 23andMe for making this work possible. D.D. was supported by the Novo Nordisk Foundation (NNF20OC0065561), the Lundbeck Foundation (R344-2020-1060) the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 965381(TIMESPAN). The iPSYCH team was supported by grants from the Lundbeck Foundation (R102-A9118, R155-2014-1724, and R248-2017-2003), NIH/NIMH (1U01MH109514-01 and 1R01MH124851-01 to A.D.B.) and the Universities and University Hospitals of Aarhus and Copenhagen. The Danish National Biobank resource was supported by the Novo Nordisk Foundation. High-performance computer capacity for handling and statistical analysis of iPSYCH data on the GenomeDK HPC facility was provided by the Center for Genomics and Personalized Medicine and the Centre for Integrative Sequencing, iSEQ, Aarhus University, Denmark (grant to A.D.B.).
Research reported in this publication was supported by the National Institute of Mental Health of the National Institutes of Health under Award Number R01MH124851. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The work was also supported by the European College of Neuropsychopharmacology (ECNP) Network “ADHD Across the Lifespan”. B.F. was also supported by funding from the European Community’s Horizon 2020 Programme (H2020/2014 – 2020) under grant agreements No. 728018 (Eat2beNICE) and No. 847879 (PRIME). B.F. also received relevant funding from the Netherlands Organization for Scientific Research (NWO) for the Dutch National Science Agenda NeurolabNL project (grant 400-17-602).
S.E.M. was funded by NHMRC grants APP1172917, APP1158125 and APP1103623.
This work was supported by the Instituto de Salud Carlos III (PI19/01224, PI20/0004), by the Pla Estratègic de Recerca i Innovació en Salut (PERIS), Generalitat de Catalunya (METAL-Cat; SLT006/17/287); by the Agència de Gestió d’Ajuts Universitaris i de Recerca AGAUR, Generalitat de Catalunya (2017SGR1461), Ministry of Science, Innovation and Universities (IJC2018-035346-I to M.S.A.); by the European Regional Development Fund (ERDF) and by “la Marató de TV3” (092330/31) and the ECNP Network ‘ADHD across the Lifespan’ (https://www.ecnp.eu/researchinnovation/ECNP-networks/List-ECNP-Networks/). T.Z. is funded by NIH, Grant No. R37MH107649-07S1 and by Research Council of Norway, NRC, Grant No. 288083.
This study was also supported by the National Institutes of Health (NIH), Bethesda, MD under award numbers T32MH087004 (to K.T.), K08MH122911 (to G.V.), R01MH125246 (to P.R.) and U01MH116442 (to P.R.).
CONSORTIUM AUTHORS
iPSYCH-Broad Consortium
Anders D. Børglum1,2,3, Benjamin M. Neale8,9, Daniel Howrigan8,9, David M. Hougaard2,17, Ditte Demontis1,2,3, Esben Agerbo2,20,21, F. Kyle Satterstrom8,9, Georgios Athanasiadis2,6,7, Jakob Grove1,2,16, Jinjie Duan1,2,3, Jonas Bybjerg-Grauholm2,17, Leila Farajzadeh1,2,3, Marie Bækved-Hansen2,17, Mark J. Daly8,9,38,39, Merete Nordentoft2,36, Ole Mors2,37, Preben Bo Mortensen2,20,21, Raymond Walters8,9, Tetyana Zayats8,9,34, Thomas D. Als1,2,3, Thomas Werge2,6, Trine Tollerup Nielsen1,2,3 and Veera M. Rajagopal1,2,3
ADHD Working Group of the Psychiatric Genomics Consortium
Alexandra Havdahl44,45, Alysa Doyle46, Andreas Reif47, Anita Thapar24, Barbara Franke31,32,43, Benjamin M. Neale8,9, Bru Cormand48,49,50,51, Calwing Liao52, Christie Burton53, Claiton H. D. Bau54,55, Diego Luiz Rovaris56, Ditte Demontis1,2,3, Edmund Sonuga-Barke57, Elizabeth Corfield44,45, Eugenio Horacio Grevet58,59, Georgios Athanasiadis2,6,7, Henrik Larsson60,61, Ian R. Gizer62, Irwin Waldman63, Isabell Brikell1,61, Jan Haavik64,65, Jennifer Crosbie53, Joanna Martin24, James McGough66, Joanna Kuntsi67, Joseph Glessner68,69, Kate Langley70, Klaus-Peter Lesch71,72,73, Luis Augusto Rohde74,75, Mara H. Hutz76, Maria Soler Artigas25,26,27,28, Marieke Klein31,32, Mark Bellgrove77, Marta Ribasés25,26,27,28, Martin Tesli44,78, Michael C. O’Donovan24, Nina Roth Mota31,32, Ole Andreas Andreassen79,80, Ole Mors2,38, Patrick W. L. Leung81, Pedro M. Pan82, Preben Bo Mortensen2,20,21, Raymond Walters8,9, Ridha Joober83, Russel Schachar53, Sandra Loo66, Sarah E. Medland33, Stephanie H. Witt84, Stephen V. Faraone41, Ted Reichborn-Kjennerud44,85, Tetyana Zayats8,9,34, Thomas Werge2,6, Tobias Banaschewski86 Veera M. Rajagopal1,2,3 and Ziarih Hawi77
44Department of Mental Disorders, Norwegian Institute of Public Health, Oslo, Norway.
45Nic Waals Institute, Lovisenberg Diaconal Hospital, Oslo, Norway.
46Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
47Department for Psychiatry, Psychosomatic Medicine and Psychotherapy, University Hospital Frankfurt, Frankfurt am Main, Germany.
48Department of Genetics, Microbiology and Statistics, University of Barcelona, Barcelona, Catalonia, Spain.
49Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Spain.
50Institut de Biomedicina de la Universitat de Barcelona (IBUB), Catalonia, Spain.
51Institut de Recerca Sant Joan de Déu (IR-SJD), Esplugues de Llobregat, Barcelona, Catalonia, Spain.
52Department of Human Genetics, McGill University, Montreal, QC, Canada.
53Neurosciences and Mental Health, Hospital for Sick Children, Toronto, ON, Canada.
54Department of Genetics, Instituto de Biociências, Universidade Federal do Rio Grande do Sul, Rio Grande do Sul, Brazil.
55ADHD and Developmental Psychiatry Programs, Hospital de Clínicas de Porto Alegre, Rio Grande do Sul, Brazil.
56Departamento de Fisiologia e Biofísica, Instituto de Ciencias Biomedicas, Universidade de São Paulo, São Paulo, Brazil.
57School of Psychiatry, Institute of Psychiatry, Psychology & Neuroscience, King’s College London, London, UK.
58Department of Psychiatry, Faculty of Medicine, Universidade Federal do Rio Grande do Sul, Rio Grande do Sul, Brazil.
59Adult ADHD Outpatient Program (ProDAH), Clinical Research Center, Hospital de Clínicas de Porto Alegre, Rio Grande do Sul, Brazil.
60School of medical sciences, Örebro University, Örebro, Sweden.
61Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden.
62Department of Psychological and Brain Sciences, University of Missouri, Columbia, MO, USA.
63Department of Psychology, Emory University, Atlanta, GA, USA.
64Department of Biomedicine, University of Bergen, Bergen, Norway.
65Division of Psychiatry, Haukeland University Hospital, Bergen, Norway.
66Semel Institute for Neuroscience and Human Behavior, UCLA David Geffen School of Medicine, Los Angeles, CA, USA.
67Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London, UK.
68Department of Pediatrics, Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
69Department of Genetics, Perelman School of Medicine, Philadelphia, PA, USA.
70School of Psychology, Cardiff University, Cardiff, UK.
71Division of Molecular Psychiatry, Center of Mental Health, University of Würzburg, Würzburg, Germany.
72Laboratory of Psychiatric Neurobiology, Institute of Molecular Medicine, I.M. Sechenov First Moscow State Medical University, Moscow, Russia.
73Department of Neuropsychology and Psychiatry, School for Mental Health and Neuroscience (MHeNS), Maastricht University, Maastricht, The Netherlands.
74ADHD Outpatient Program & Developmental Psychiatry Program, Hospital de Clinica de Porto Alegre, Federal University of Rio Grande do Sul, Rio Grande do Sul, Brazil.
75National Institute of Developmental Psychiatry, Brazil.
76Department of Genetics, Universidade Federal do Rio Grande do Sul Porto Alegre, Rio Grande do Sul, Brazil.
77Turner Institute for Brain and Mental Health, School of Psychological Sciences, Monash University, Melbourne, Australia.
78Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
79 NORMENT Centre, Division of Mental Health and Addiction, Oslo University Hospital & Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
80KG Jebsen Centre for Neurodevelopmental disorders, University of Oslo, Oslo, Norway.
81Department of Psychology, The Chinese University of Hong Kong, Shatin, Hong Kong, China.
82LiNC, Department of Psychiatry, Federal University of São Paulo, São Paulo, Brazil.
83Douglas Mental Health University Institute; Department of Psychiatry, McGill University, Montreal, QC, Canada.
84Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim / Heidelberg University, Mannheim, Germany.
85Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
86Department of Child & Adolescent Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim / Heidelberg University, Mannheim, Germany.
Footnotes
COMPETING INTERESTS
B.M.N. currently serves as a member of the scientific advisory board at Deep Genomics and Neumora (previously RBNC) and consultant for Camp4 Therapeutics, Takeda Pharmaceutical, and Biogen. All deCODE affiliated authors are employees of deCODE/Amgen. The remaining authors declare no competing interests.
Code availability
No previously unreported custom computer code or algorithms were used to generate results.
Data availability
Summary statistics from the ADHD GWAS meta-analysis are available for download at the PGC website (https://www.med.unc.edu/pgc/download-results/). All relevant iPSYCH data are available from the authors after approval by the iPSYCH Data Access Committee and can only be accessed on the secured Danish server (GenomeDK; https://genome.au.dk) as the data are protected by Danish legislation. For data access and correspondence, please contact Ditte Demontis (ditte@biomed.au.dk) or Anders D. Børglum (anders@biomed.au.dk).
REFERENCES
- 1.Faraone SV et al. Attention-deficit/hyperactivity disorder. Nature Reviews Disease Primers, 15020 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Franke B et al. The genetics of attention deficit/hyperactivity disorder in adults, a review. Mol Psychiatry 17, 960–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalsgaard S, Leckman JF, Mortensen PB, Nielsen HS & Simonsen M Effect of drugs on the risk of injuries in children with attention deficit hyperactivity disorder: a prospective cohort study. Lancet Psychiatry 2, 702–9 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Chang Z, Lichtenstein P, D’Onofrio BM, Sjolander A & Larsson H Serious transport accidents in adults with attention-deficit/hyperactivity disorder and the effect of medication: a population-based study. JAMA Psychiatry 71, 319–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babinski DE, Neely KA, Ba DM & Liu G Depression and Suicidal Behavior in Young Adult Men and Women With ADHD: Evidence From Claims Data. J Clin Psychiatry 81, 19m13130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capusan AJ, Bendtsen P, Marteinsdottir I & Larsson H Comorbidity of Adult ADHD and Its Subtypes With Substance Use Disorder in a Large Population-Based Epidemiological Study. J Atten Disord 23, 1416–1426 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Boomsma DI, van Beijsterveldt T, Odintsova VV, Neale MC & Dolan CV Genetically Informed Regression Analysis: Application to Aggression Prediction by Inattention and Hyperactivity in Children and Adults. Behav Genet 51, 250–263 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalsgaard S, Ostergaard SD, Leckman JF, Mortensen PB & Pedersen MG Mortality in children, adolescents, and adults with attention deficit hyperactivity disorder: a nationwide cohort study. Lancet 385, 2190–6 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Jangmo A et al. Attention-deficit/hyperactivity disorder and occupational outcomes: The role of educational attainment, comorbid developmental disorders, and intellectual disability. PLoS One 16, e0247724 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao X et al. Family Burden of Raising a Child with ADHD. J Abnorm Child Psychol 47, 1327–1338 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Le HH et al. Economic impact of childhood/adolescent ADHD in a European setting: the Netherlands as a reference case. Eur Child Adolesc Psychiatry 23, 587–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Libutzki B et al. Direct medical costs of ADHD and its comorbid conditions on basis of a claims data analysis. Eur Psychiatry 58, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Faraone SV & Larsson H Genetics of attention deficit hyperactivity disorder. Mol Psychiatry 24, 562–575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pedersen CB et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry 23, 6–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mattheisen M et al. Identification of shared and differentiating genetic architecture for autism spectrum disorder, attention-deficit hyperactivity disorder and case subgroups. Nat. Genet. 54, 1470–1478 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satterstrom FK et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat Neurosci 22, 1961–1965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mullins N et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet 53, 817–829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howard DM et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci 22, 343–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pardinas AF et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 50, 381–389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trzaskowski M et al. Quantifying between-cohort and between-sex genetic heterogeneity in major depressive disorder. Am J Med Genet B Neuropsychiatr Genet 180, 439–447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koopmans F et al. SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 103, 217–234 e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Z et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc 1, e90 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W et al. Integrative transcriptome imputation reveals tissue-specific and shared biological mechanisms mediating susceptibility to complex traits. Nat Commun 10, 3834 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meuleman W et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature 584, 244–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corces MR et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat Genet 52, 1158–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe K, Umicevic Mirkov M, de Leeuw CA, van den Heuvel MP & Posthuma D Genetic mapping of cell type specificity for complex traits. Nat Commun 10, 3222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.La Manno G et al. Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 167, 566–580.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng J et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frei O et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat Commun 10, 2417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franke B et al. Live fast, die young? A review on the developmental trajectories of ADHD across the lifespan. Eur Neuropsychopharmacol 28, 1059–1088 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satterthwaite TD et al. Neuroimaging of the Philadelphia neurodevelopmental cohort. Neuroimage 86, 544–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calkins ME et al. The Philadelphia Neurodevelopmental Cohort: constructing a deep phenotyping collaborative. J Child Psychol Psychiatry 56, 1356–1369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gur RC et al. Age group and sex differences in performance on a computerized neurocognitive battery in children age 8–21. Neuropsychology 26, 251–265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilkinson GS & Robertson GJ Wide range achievement test (WRAT4). Lutz, FL: Psychological Assessment Resources; (2006). [Google Scholar]
- 42.Uffelmann E et al. Genome-wide association studies. Nature Reviews Methods Primers 1, 59 (2021). [Google Scholar]
- 43.Bataillon T et al. The effective size of the Icelandic population and the prospects for LD mapping: inference from unphased microsatellite markers. Eur J Hum Genet 14, 1044–53 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Gazal S et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat Genet 49, 1421–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hindley G et al. The shared genetic basis of mood instability and psychiatric disorders: A cross-trait genome-wide association analysis. Am J Med Genet B Neuropsychiatr Genet 189, 207–218 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plana-Ripoll O et al. Exploring Comorbidity Within Mental Disorders Among a Danish National Population. JAMA Psychiatry 76, 259–270 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zablotsky B, Bramlett MD & Blumberg SJ The Co-Occurrence of Autism Spectrum Disorder in Children With ADHD. J Atten Disord 24, 94–103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen CM & Steinhausen HC Comorbid mental disorders in children and adolescents with attention-deficit/hyperactivity disorder in a large nationwide study. Atten Defic Hyperact Disord 7, 27–38 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Chen Q et al. Common psychiatric and metabolic comorbidity of adult attention-deficit/hyperactivity disorder: A population-based cross-sectional study. PLoS One 13, e0204516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 179, 1469–1482.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JJ et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 50, 1112–1121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akaike H A new look at the statistical model identification. IEE Transactions on Automatic Controls 19, 716–723 (1974). [Google Scholar]
- 53.Yao X et al. Integrative analysis of genome-wide association studies identifies novel loci associated with neuropsychiatric disorders. Transl Psychiatry 11, 69 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trubetskoy V et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson EC et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry 7, 1032–1045 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Araujo DJ et al. FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev 29, 2081–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fong WL, Kuo HY, Wu HL, Chen SY & Liu FC Differential and Overlapping Pattern of Foxp1 and Foxp2 Expression in the Striatum of Adult Mouse Brain. Neuroscience 388, 214–223 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Sollis E et al. Equivalent missense variant in the FOXP2 and FOXP1 transcription factors causes distinct neurodevelopmental disorders. Hum Mutat 38, 1542–1554 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Mostafavi H, Spence JP, Naqvi S & Pritchard JK Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. bioRxiv (2022). [Google Scholar]
- 60.Satterstrom FK et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh T et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 604, 509–516 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sazonovs A et al. Large-scale sequencing identifies multiple genes and rare variants associated with Crohn’s disease susceptibility. Nat. Genet. 54, 1275–1283 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bahmani Z et al. Prefrontal Contributions to Attention and Working Memory. Curr Top Behav Neurosci 41, 129–153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sonne J, Reddy V & Beato MR Neuroanatomy, Substantia Nigra. in StatPearls; (Treasure Island (FL), 2021). [PubMed] [Google Scholar]
- 65.Morales M & Margolis EB Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci 18, 73–85 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Chang S, Yang L, Wang Y & Faraone SV Shared polygenic risk for ADHD, executive dysfunction and other psychiatric disorders. Transl Psychiatry 10, 182 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nigg JT et al. Working Memory and Vigilance as Multivariate Endophenotypes Related to Common Genetic Risk for Attention-Deficit/Hyperactivity Disorder. J Am Acad Child Adolesc Psychiatry 57, 175–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aguilar-Lacasana S et al. Polygenic risk for ADHD and ASD and their relation with cognitive measures in school children. Psychol Med. 52, 1356–1364 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin J, Hamshere ML, Stergiakouli E, O’Donovan MC & Thapar A Neurocognitive abilities in the general population and composite genetic risk scores for attention-deficit hyperactivity disorder. J Child Psychol Psychiatry 56, 648–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 70.Bybjerg-Grauholm J et al. The iPSYCH2015 Case-Cohort sample: updated directions for unravelling genetic and environmental architectures of severe mental disorders. medRxiv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mors O, Perto GP & Mortensen PB The Danish Psychiatric Central Research Register. Scand J Public Health 39, 54–7 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Lynge E, Sandegaard JL & Rebolj M The Danish National Patient Register. Scand J Public Health 39, 30–3 (2011). [DOI] [PubMed] [Google Scholar]
- 73.Price AL et al. The impact of divergence time on the nature of population structure: an example from Iceland. PLoS Genet 5, e1000505 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benner C et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Greenbaum J & Deng HW A Statistical Approach to Fine Mapping for the Identification of Potential Causal Variants Related to Bone Mineral Density. J Bone Miner Res 32, 1651–1658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen W et al. Fine Mapping Causal Variants with an Approximate Bayesian Method Using Marginal Test Statistics. Genetics 200, 719–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang J et al. CAUSALdb: a database for disease/trait causal variants identified using summary statistics of genome-wide association studies. Nucleic Acids Res 48, D807–D816 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gandal MJ et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Das S et al. Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47, 1228–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu M et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 51, 237–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yengo L et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet 27, 3641–3649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Okbay A et al. Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat Genet 54, 437–449 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mills MC et al. Identification of 371 genetic variants for age at first sex and birth linked to externalising behaviour. Nat Hum Behav 5, 1717–1730 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watanabe K et al. Genome-wide meta-analysis of insomnia prioritizes genes associated with metabolic and psychiatric pathways. Nat. Genet. 54, 1125–1132 (2022). [DOI] [PubMed] [Google Scholar]
- 91.Als TD et al. Identification of 64 new risk loci for major depression, refinement of the genetic architecture and risk prediction of recurrence and comorbidities. medRxiv (2022). [Google Scholar]
- 92.Ge T, Chen CY, Ni Y, Feng YA & Smoller JW Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 10, 1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics from the ADHD GWAS meta-analysis are available for download at the PGC website (https://www.med.unc.edu/pgc/download-results/). All relevant iPSYCH data are available from the authors after approval by the iPSYCH Data Access Committee and can only be accessed on the secured Danish server (GenomeDK; https://genome.au.dk) as the data are protected by Danish legislation. For data access and correspondence, please contact Ditte Demontis (ditte@biomed.au.dk) or Anders D. Børglum (anders@biomed.au.dk).