

Abstract
Chronic obstructive pulmonary disease (COPD) remains a major public health challenge that contributes greatly to mortality and morbidity worldwide. Although it has long been recognized that the epithelium is altered in COPD, there has been little focus on targeting it to modify the disease course. Therefore, mechanisms that disrupt epithelial cell function in patients with COPD are poorly understood. In this study, we sought to determine whether epigenetic reprogramming of the cell-cell adhesion molecule E-cadherin, encoded by the CDH1 gene, disrupts epithelial integrity. By reducing these epigenetic marks, we can restore epithelial integrity and rescue alveolar airspace destruction. We used differentiated normal and COPD-derived primary human airway epithelial cells, genetically manipulated mouse tracheal epithelial cells, and mouse and human precision-cut lung slices to assess the effects of epigenetic reprogramming. We show that the loss of CDH1 in COPD is due to increased DNA methylation site at the CDH1 enhancer D through the downregulation of the ten-eleven translocase methylcytosine dioxygenase (TET) enzyme TET1. Increased DNA methylation at the enhancer D region decreases the enrichment of RNA polymerase II binding. Remarkably, treatment of human precision-cut slices derived from patients with COPD with the DNA demethylation agent 5-aza-2′-deoxycytidine decreased cell damage and reduced air space enlargement in the diseased tissue. Here, we present a novel mechanism that targets epigenetic modifications to reverse the tissue remodeling in human COPD lungs and serves as a proof of concept for developing a disease-modifying target.
Keywords: COPD, epigenetic, CDH1, E-cadherin, DNA methylation
Clinical Relevance
There is evidence of increased methylation of CDH1 (the gene for E-cadherin) in moderate-severe chronic obstructive pulmonary disease (COPD). We provide molecular evidence that targeted changes in the enhancer element decreases protein production, and targeting this improved lung epithelial function and tissue function. These findings suggest that reversing CDH1 DNA methylation could be a potential therapeutic strategy for COPD.
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease that currently has no disease-modifying therapies. It afflicts 13% of the world’s population and is on track to be the third leading cause of death worldwide. There is both structural and functional dysregulation of the epithelium, although the molecular basis mediating these changes is less well understood. However, the epithelium serves as the first line of defense of the lung and is in constant contact with the external environment. Therefore, it is highly susceptible to epigenetic modifications from these exposures (1, 2). As the molecular underpinnings of the pathogenesis of chronic lung diseases continue to be investigated, there is increasing evidence that epigenetic marks can drive the cellular phenotype in disease.
Epigenetic reprogramming caused by environmental exposure perturbs gene expression (3, 4) and can be transmitted across generations (5, 6). Notably, the heritable epigenetic modifications affect stem cell regeneration, differentiation, and repair (2, 7). DNA methylation, the addition of a methyl group to the five-carbon position of the cytosine of a cytosine–guanine dinucleotide (CpG), is a well-known epigenetic mark (8). A cluster of CpG dinucleotide repeats called CpG islands (CGIs) near the gene promoter is a primary site for DNA methylation for transcriptional regulation (9), and methylation of these sites blocks the binding of transcription factors and attracts methyl-binding proteins that initiate chromatin compaction (10, 11). Additional regions of DNA methylation outside CGIs, such as enhancer regions, can play a vital role by regulating the binding of transcription factors and RNA polymerase II (RNAPII) to activate gene transcription (12).
Both genetic and environmental factors affect DNA methylation, thus altering gene expression and impacting cellular phenotype and function, which can result in chronic diseases (13, 14). Changes in DNA methylation in response to environmental exposures, such as cigarette smoking (CS), are associated with the development of COPD (1, 15–17), although the mechanism mediating this association is not clear (15, 16, 18–20).
To address the gaps in knowledge, we sought to identify a mechanism resulting in reduced epithelial cadherin (E-cadherin) in COPD and CS-injured epithelia (21–23). E-cadherin, encoded by the CDH1 gene, is a transmembrane protein that initiates intercellular contacts through transpairing between cadherins on opposing cells and is critical in establishing epithelial integrity (24). We delineated the epigenetic machinery causing E-cadherin loss in COPD epithelia. We identified increased methylation in critical regions regulating CDH1 expression. Moreover, using the Food and Drug Administration (FDA)–approved epigenetic modulator, 5-aza-2′-deoxycytideine (decitabine; AZA), we found that DNA demethylation improves the integrity of epithelia in patients with COPD and drives pulmonary remodeling in mouse-derived precision-cut lung slices (PCLS). As proof of concept, we then extended our studies to human lungs using PCLS derived from patients with COPD to determine whether targeting methylation could serve as a therapeutic strategy. Our study identifies a novel epithelial target for disease-modifying agents in COPD.
Methods
Detailed methods are presented elsewhere (see the data supplement).
CDH1 Candidate Gene CpG Methylation Association Study
The proportion of methylation (β-methylation) at CpG loci within and adjacent to CDH1 in lower airway epithelial cells was evaluated in the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS) cohort (characteristics; see Table E3 in the data supplement).
Human Epithelial Cell Culture
Epithelial cells (passage 2, demographics; see Table E1) were cultured at air–liquid interface (ALI) for 4–6 weeks (22, 25). COPD cells were treated with 1 μM AZA (Sigma-Aldrich) or DMSO (vehicle) for 10 days. Cells from the cell line 16HBE14o- (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (Corning) supplemented with 2 mM L-glutamine, 10% fetal bovine serum (Sigma), and 1% penicillin–streptomycin (ThermoFisher Scientific).
PCLSs
Human lungs were obtained from the International Institute for the Advancement of Medicine. All animal procedures were performed according to the Johns Hopkins University Institutional Animal Care and Use Committee (MO22M343). Mouse (adult C57BL/6J female) and human lung PCLSs were prepared as described elsewhere (26, 27).
Acute CS Exposures
CS aerosol or humidified air exposures were conducted as described elsewhere (21–23, 25, 28) (six CS/air for PCLSs, two CS/air for ALI cultures).
Quantification of Mean Linear Intercept (MLI)
PCLS morphometric changes were measured using the MLI, as described elsewhere (29).
Lactate Dehydrogenase (LDH) Assay
Media from PCLS were collected, and cytotoxicity analysis was performed using an LDH assay (Abcam).
Knockdown of Ten-Eleven Translocase Methylcytosine Dioxygenase (TET) Enzyme TET1 in 16HBE Cells Using Lentivirus
The 16HBE cells were transduced with lentiviruses with shRNA TET1 (ACACAACTTGCTTCGATAATT) and scramble (CCTAAGGTTAAGTCGCCCTCG).
Mouse Tracheal Epithelial Cell (mTEC) Culture
The Cdh1fl/fl, Tet1+/−, and wild-type (WT) C57BL/J mice were killed, and the mTECs were cultured at ALI for 2 weeks (22).
In Vitro Knockdown of Cdh1 in mTECs
The mTECs of Cdh1fl/fl mice were transfected with adenovirus Ad-5CMVeGFP (GFP) or Ad5CMVCre-eGFP (Cre) at a titer of 2 × 107 plaque-forming units, as described in our previous study (22).
Immunofluorescence
The immunofluorescence analysis was performed as described elsewhere (22). The primary antibodies against E-cadherin, 5-methylcytosine (5mC), and 5-hydroxymethylcytosine (5hmC) were from Cell Signaling Technology. The images were taken using an LSM880-AiryscanFAST (30).
Quantification of Lung Epithelial Plasticity
The epithelial integrity was measured by transepithelial electrical resistance and FITC-dextran (21).
Gene Expression Measurement by Quantitative PCR (RT-qPCR)
Total RNA was reverse transcribed with the High-Capacity cDNA Reverse Transcription Kit (ABI) before we performed SYBR Green–based qPCR. We used the 2−ΔΔCt method (31) to calculate the relative gene expression ratio normalized by the housekeeping gene GAPDH. The qPCR primers are listed elsewhere (see Table E2).
5mC and 5hmC Measurement
5mC and 5hmC levels were measured using the MethylFlash Global DNA Methylation (5mC) ELISA Kit (EpiGentek) and MethylFlash Global DNA Hydroxymethylation (5hmC) ELISA (EpiGentek).
Bisulfite Sequencing
Genomic DNA was subjected to bisulfite conversion (EZ DNA Methylation Kit; Zymo) and PCR amplification and subcloned into the pCR2.1 vector (ThermoFisher Scientific) (32). The primer sequences are listed in Table E2.
Chromatin Immunoprecipitation (ChIP)-qPCR
We performed ChIP analysis using the ChIP-IT Express Kit (Active Motif) (32). Isolated chromatin was sheared after immunoprecipitation with antibodies against RNAPII (Active Motif), histone H3 monomethylated at lysine 4 (H3K4 me1) (Abcam), and IgG (a negative control). The primers are listed in Table E2.
Statistical Analysis
Results were expressed as the mean ± SEM. For the data analysis and figures, we used Prism 9.4 (GraphPad).
Results
DNA Demethylation Restores Remodeling in Both Mouse and Human Lung Tissues
Our group has demonstrated that altered epithelial integrity is critical to inducing the lung remodeling that occurs with CS-related injury and in COPD (22, 23, 25). In an epigenetic study from the SPIROMICS cohort (33), in moderate to severe COPD and control to mild COPD using a dataset of 73 COPD epithelial cells, we found that the DNA methylation was increased (chr16: 68789786–68789788, cg10313337) at the CDH1 gene body; effect size = 0.040; P value as genomic control (gc) corrected P = 0.0093 (Table 1). Baseline characteristics for patients in the epigenome-wide association study by COPD status are described elsewhere (Table E3). Next, we investigated whether global DNA demethylation with a DNA demethylation agent, AZA, can protect epithelial integrity in epithelial cells derived from patients with COPD. We utilized primary cells derived from patients with COPD, which were differentiated at ALI to study the effects of AZA. Treatment with 1 μM AZA, showing a significant induction in global DNA demethylation in human bronchial epithelial cells (34) without affecting cell viability (see Figure E1A), for 10 days significantly improved epithelial transepithelial electrical resistance (1.13-fold in AZA vs. vehicle; P = 0.0251) (Figure 1A) and reduced monolayer permeability as measured by FITC–dextran flux (0.76-fold in AZA vs. vehicle; P = 0089) (Figure 1B) in the differentiated cells derived from patients with COPD, which was not seen in normal cells. Notably, the addition of AZA significantly restored both CDH1 gene expression (1.44-fold in AZA vs. vehicle; P = 0.0063) (Figure 1C) and E-cadherin protein expression (Figure 1D), the loss of which we have demonstrated to be sufficient to induce the alveolar tissue destruction and airway tissue remodeling that occurs in COPD (22). Of note, other markers of COPD-induced epithelial plasticity, such as cell movement, percentage of ciliary cells, and ciliary beat frequency, which are more reflective of cytoskeletal rearrangement (25), were not changed with AZA treatment (see Figure E2). To determine whether the effect of AZA on the epithelial barrier occurred through its effects on CDH1, we examined mTECs derived from Cdh1fl/fl mice with Cre-induced Cdh1 knockdown (∼40% reduction in Cdh1 gene expression; data not shown) using Ad5CMVCre-eGFP (Cre) adenoviruses versus Ad5CMVeGFP (GFP) (∼50% transfection efficiency) (Figure 1E, left). For the differentiated GFP- or Cre-infected mTECs, Cdh1 knockdown decreased the AZA-induced improvement in barrier by 0.5-fold (P = 0.002; Figure 1E). Furthermore, the improvement in monolayer barrier properties recapitulates overexpression of E-cadherin in lung epithelial monolayer integrity (22, 23). These results indicate that the inhibition of DNA methylation mediates improved barrier function in COPD by CDH1 transcriptional activation.
Table 1.
Comparisons of Proportion DNA Methylation at CpG Sites within the CDH1 Genomic Region
| CpG Site ID | Chr | UCSC RefGene Name | UCSC RefGene Group | Stand hg38 | Effect.Size | SE | P Value | gc. P Value |
|---|---|---|---|---|---|---|---|---|
| cg10313337 | 16 | CDH1 | Body | + | 0.040 | 0.015 | 0.009 | 0.009 |
| cg05303053 | 16 | CDH1 | Body | − | 0.011 | 0.011 | 0.306 | 0.306 |
| cg20750889 | 16 | CDH1 | Body | − | −0.001 | 0.013 | 0.937 | 0.937 |
| cg00632260 | 16 | CDH1 | Body | − | −0.008 | 0.011 | 0.440 | 0.440 |
| cg25218831 | 16 | CDH1 | Body | − | −0.011 | 0.017 | 0.528 | 0.528 |
Definition of abbreviations: Chr = chromosome; COPD = chronic obstructive pulmonary disease; CpG = cytosine–guanine dinucleotide; gc = genomic control; SPIROMICS = Subpopulations and Intermediate Outcomes in COPD Study; UCSC = University of California, Santa Cruz.
β-methylation for five CpG sites within the CDH1 genomic region was compared between 36 SPIROMICS patients with moderate to severe COPD and 36 SPIROMICS patients without COPD or with mild COPD who had DNA isolated from lower airway epithelial cells from research bronchoscopic brushings. Regression-based models included age, sex, body mass index, smoking pack-years, current smoking status, and first five principal components of ancestry. DNA methylation at one CpG near the CDH1 enhancer D region (chr16: 68789786–68789788, cg10313337) was significantly increased in the DNA from epithelial cells from patients with moderate-to-severe COPD compared with cells from control patients and those with mild COPD. Bold value represents a region of significance. The “+” and “−” refer to the positive and negative DNA strands.
Figure 1.
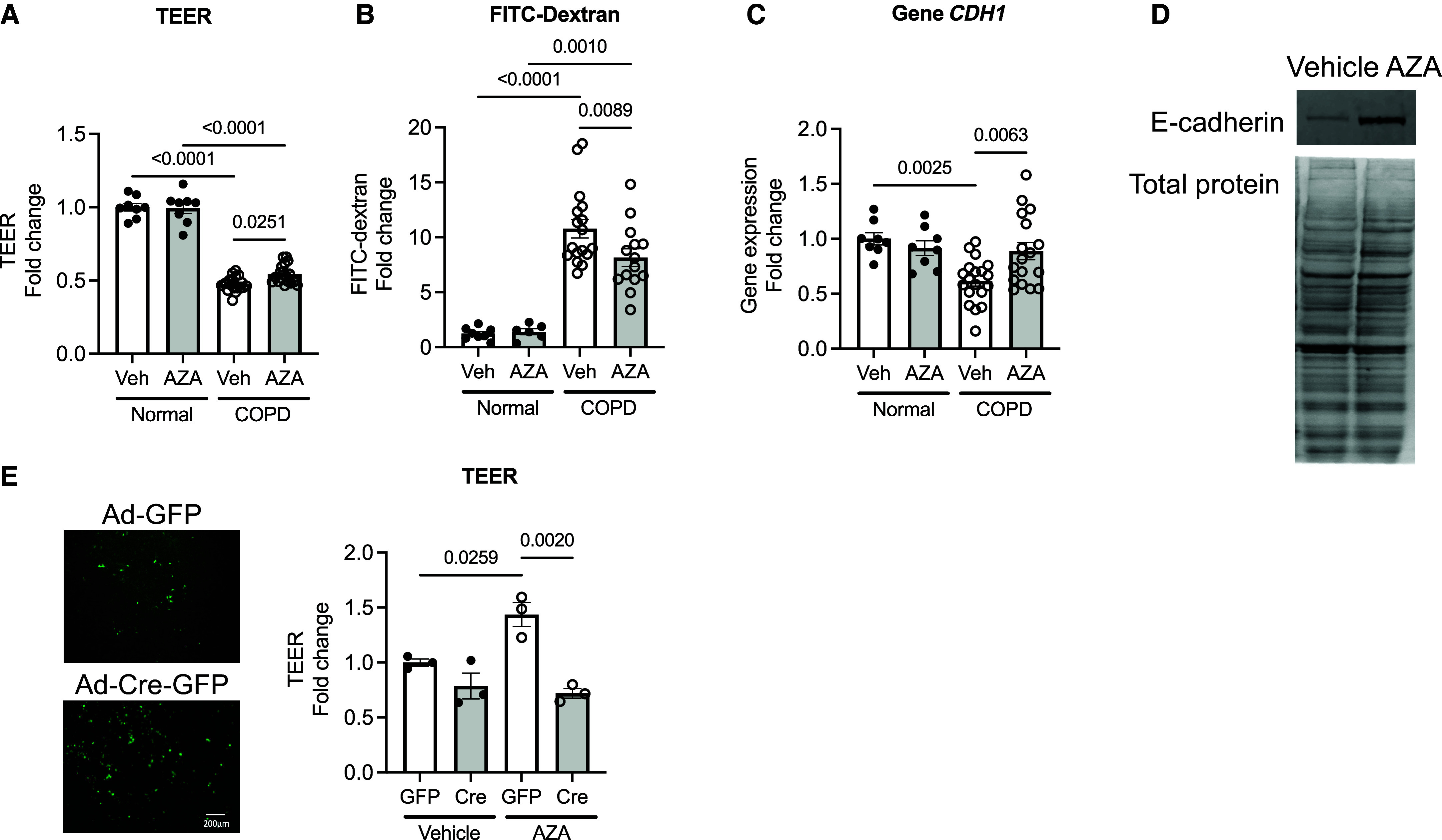
Global DNA demethylating agent 5-aza-2′-deoxcytidine (AZA) restores destructive epithelial integrity in chronic obstructive pulmonary disease (COPD)-derived epithelia. (A and B) Measurement of the barrier function of AZA-treated normal/COPD-derived epithelial cells by (A) transepithelial electrical resistance (TEER) and (B) FITC–dextran flux (n = 12–22). (C and D) Expression of both the cadherin 1 (CDH1) (C) gene and (D) protein. (E) Measurement of TEER in mouse tracheal epithelial cells (mTECs) derived from Cdh1fl/fl mice with Cre-induced Cdh1 knockdown at the air–liquid interface culture (n = 3). Scale bar, 200 μm. The error bars represent SEM. Statistics were determined by ordinary one-way ANOVA with Sidak’s multiple comparison test, with P < 0.05 considered statistically significant. Veh = vehicle.
Downregulation of CDH1 Transcript Is Associated with TET1
Given the evidence of increased methylation near CDH1 in patients (Table 1) and the improvement seen with AZA, we investigated whether the loss of CDH1 is due to modifiers of DNA methylation. Gene expressions of CDH1 and epigenetic modulators—TETs, DNA methyltransferases, histone acetyltransferase p300, and histone deacetylases—in human bronchial epithelial cells derived from healthy donors and those with COPD at ALI were detected using qRT-PCR and were assessed with Spearman correlations. Among all the modulators, we identified an association with CDH1 and TET1, in the five normal and COPD age- and sex-matched samples (r = 0.8, P = 0.1333) (Figure 2A). TET1 catalyzes the oxidation of 5mC for DNA demethylation pathways. Of note, we did not find a correlation between CDH1 and the histone modifications (see Table E4), supporting the primary role of DNA methylation in CDH1 regulation in COPD. In the acute CS-exposed normal human epithelial cells (two cigarettes per day), CDH1 expression was downregulated (0.54-fold in CS vs. air; P = 0.0513) (Figure 2B) with a corresponding reduction in TET1 (0.4-fold in CS vs. air; P = 0.035; Figure 2C), but not in TET2 (Figure 2D) or in TET3 (Figure 2E). In the human epithelial cell line 16HBE, knockdown of TET1 showed a trend toward downregulation of CDH1 gene expression (0.59-fold in shTET1 vs. shCTL; P = 0.0832) (Figure 2F), indicating that TET1 was, at least, one of the factors involved in the transcriptional regulation of CDH1. To delineate the role of TET1 on CDH1 transcriptional regulation, we acquired differentiated mTECs from WT and Tet1+/− mice. Tet1 expression was decreased after acute CS exposure (0.49-fold in WT-CS vs. WT-air; P = 0.0076) (Figure 3A). As expected, mTECs derived from Tet1+/− mice demonstrated low levels at baseline (0.45-fold in Tet1+/−-air vs. WT-CS; P = 0.0046) (Figure 3A). Tet2 gene expression was not significantly altered by CS exposure and was not altered by loss of Tet1 (Figure 3B). Tet3 gene expression in the mTECs was not detectable (data not shown). The reduced Tet1, as seen in both CS-exposed WT and Tet1+/−-derived mTECs, was associated with a significant decrease in Cdh1 expression (0.52-fold in WT-CS vs. WT-air; P = 0.0467; 0.51-fold in Tet1+/−-air vs. WT-air; P = 0.032), but Cdh1 expression was not further decreased in CS-exposed Tet1+/−-derived mTECs (Figure 3C). The downregulated of Cdh1 corresponded to the reduced 5hmC level (0.42-fold in WT-CS vs. WT-air; P = 0.0493; 0.42-fold in Tet1+/−-air vs. WT-air; P = 0.0493) (Figure 3D). The positive association of Tet1 and Cdh1 was also shown as Tet1 deficiency in the lung tissues derived from WT and Tet+/− mice (0.68-fold reduction in Tet1+/− lung vs. WT; P = 0.0281) (Figure 3E); Cdh1 gene expression was also downregulated (0.4-fold in Tet1+/− lung vs. WT; P = 0.0379) (Figure 3F). These results further suggest that Tet1-mediated DNA demethylation regulates Cdh1 expression in epithelia.
Figure 2.
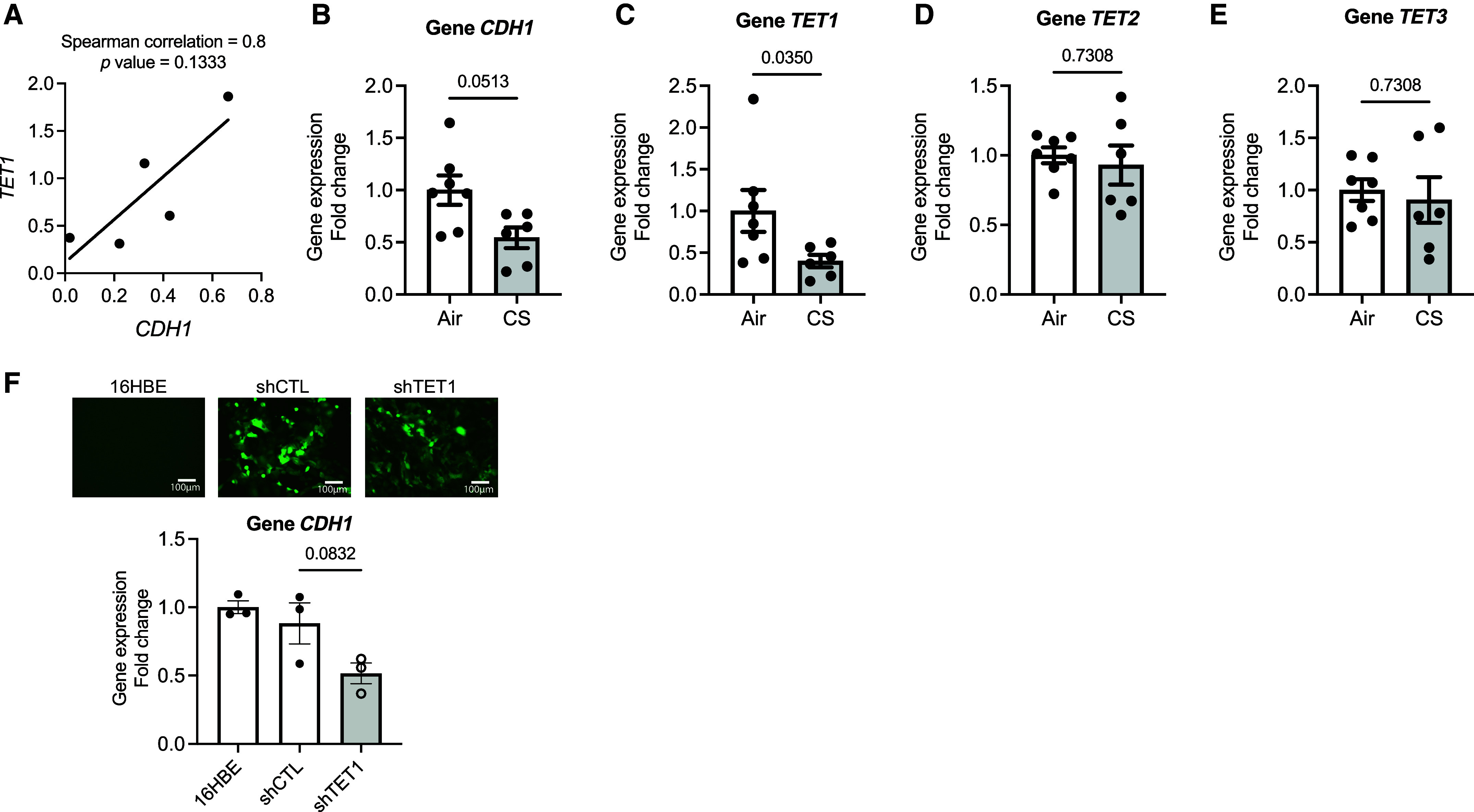
CDH1 gene expression correlates with the expression of ten-eleven translocase dioxygenase (TET1). (A) Correlation of CDH1 gene expression with TET1 in human epithelial cells by Spearman rank correlation analysis (n = 5). (B–E) Gene expressions of (B) CDH1, (C) TET1, (D) TET2, and (E) TET3 in the cigarette-smoking (CS)–exposed normal epithelial cells (n = 6–7). (F) Gene expression of CDH1 in the 16HBE cell line with TET1 knockdown (n = 3). Top: lentivirus transduction efficiency indicated as GFP-tagged fluorescence, 10×. Scale bars, 100 μm. Bottom: CDH1 expression decreases with knockdown of TET1. Error bars represent SEM. Statistics were determined by using Mann-Whitney test in (B–E) and an ordinary one-way ANOVA with Sidak’s multiple comparison test in (F), with P < 0.05 considered statistically significant. Scale bars, 100 μm.
Figure 3.
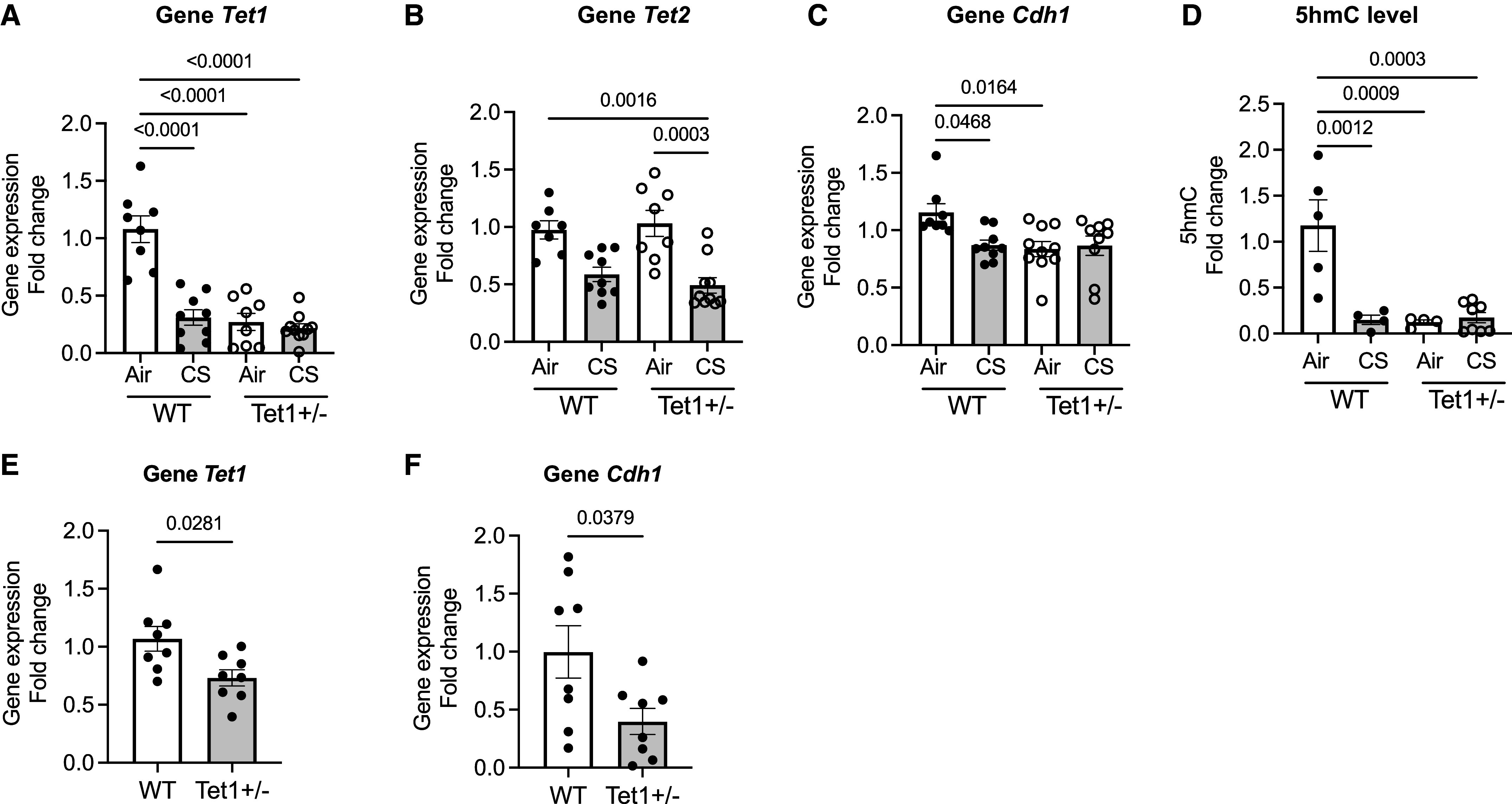
Tet1 mediates Cdh1 expression in mTECs. (A–D) Measurement of gene expressions of (A) Tet1, (B) Tet2, and (C) Cdh1, as well as (D) 5-hydroxymethylcytosine (5hmC) level in mTECs derived from wild-type (WT) C57BL/6J or Tet1+/− mice after exposure to acute CS (two cigarettes per day) versus humidified air. (E and F) Gene expressions of (E) Tet1 and (F) Cdh1 in WT C57BL/6J and Tet1+/− mouse lung tissues (n = 8). Error bars represent SEM (four to nine from two independent experiments). Statistics were determined by ordinary one-way ANOVA with Sidak’s multiple comparison tests, compared with WT-air, with P < 0.05 considered statistically significant.
DNA Hypermethylation in the CDH1 Enhancer D Region Blocks RNA Polymerase Binding
Because the DNA methylation changes are tissue and cell type specific, we sought to determine whether DNA methylation is involved in the loss of E-cadherin in the lung epithelium of patients with COPD. In an in silico analysis with MethPrimer (35), four CGIs (percentage of guanine and cytosine >60%) at 5′ CDH1 were found to encompass the transcription start site (TSS): CGI 1 (NC_000016.10: 68737081–68737235, −211 to −57 TSS), CGI 2 (NC_000016.10: 68737303–68737871, +11 to +279 TSS), CGI 3 (NC_000016.10: 68738057–68738255, +765 to +963 TSS), and CGI 4 (NC_000016.10: 68738317– 68738443, +1025 to +1151 TSS) (Figure 4A, blue). To determine whether the transcriptional decreases in E-cadherin are due to hypermethylation of these CGIs, we investigated the methylation status of 135 CpG sites at the 5′ promoter region of CDH1 using bisulfite sequencing with three sets of primers. We calculated the average methylation percentage of each CGI by taking the average methylation percentage of all CpG sites in each donor (unmethylated, 0%; methylated, 100%) (three donors per group) (Figure 4A). We found that COPD-derived epithelial cells had a trend toward increased DNA methylation at CGI 2 to CGI 4 but that methylation percentage was very low and no specific methylation pattern was observed, as shown in the graphs (6–10 clones per donor) in Figure 4B and the dot plots (see Figure E3). We did not observe any enrichment of RNAPII (Figure 4C) and H3K4 me1, an active enhancer mark (36) (Figure 4D) at the TSS of the CDH1 gene using ChIP assay. On the basis of the evidence of enhancer methylation of E-cadherin in other systems (37), we investigated whether DNA methylation at specific enhancers was involved in CDH1 transcriptional activation. Using bisulfite sequencing, we assessed the DNA methylation status at the enhancers A (chr16: 68732016–68732406, hg38), B (chr16: 68744856–68745117, hg38), and D (chr16: 68760873–68761125, hg38) of the CDH1 gene in epithelial cells from both normal donors and those with COPD. We found the average methylation percentage of the enhancers (A, B, and D) by assessing the CpG sites of each enhancer in every donor. There was a significant increase in enhancer methylation (2.76-fold in COPD vs. normal; P = 0.0015; Figure 5A). Although there was patient-to-patient variability, of all the enhancers, the average methylation percentage was significantly increased at the enhancer D region (2.12-fold in COPD vs. normal; P = 0.0469) (Figure 5B). Each CpG site is shown in the graphs (left) and the dot plots (right) of Figure 5C. Notably, we found that the increased methylation percentage of CDH1 enhancer D was located at CpG Site 1 at Position 9 (4.66-fold in COPD vs. normal; P = 0.039) (Figure 5D), not at CpG Site 2 at Position 74 (P = 0.1094) (Figure 5E). The methylation at the enhancer D region showed a decrease in RNAPII binding (0.42-fold in COPD vs. normal; P = 0.0079) (Figure 5F) with the reduction of an active enhancer mark, H3K4 me1 (0.73-fold in COPD vs. normal; P = 0.0317) (Figure 5G). AZA treatment in COPD-derived epithelial cells reduced the DNA hypermethylated at the enhancer D region (0.39-fold in AZA vs. vehicle; P = 0.0364) (Figure 5H), as shown in the dot plots (left) and the graph (right) with the enrichment in the RNAPII binding (1.18-fold in AZA vs. vehicle; P = 0.0286) in Figure 5I. Further supporting this notion, the DNA methylation at Site 1 of the enhancer D region was also correlated with the CDH1 expression among A549 and HEK293T cells (see Figure E4). These data suggested that DNA methylation at the enhancer D region reduces RNAPII binding for transcriptional activation.
Figure 4.
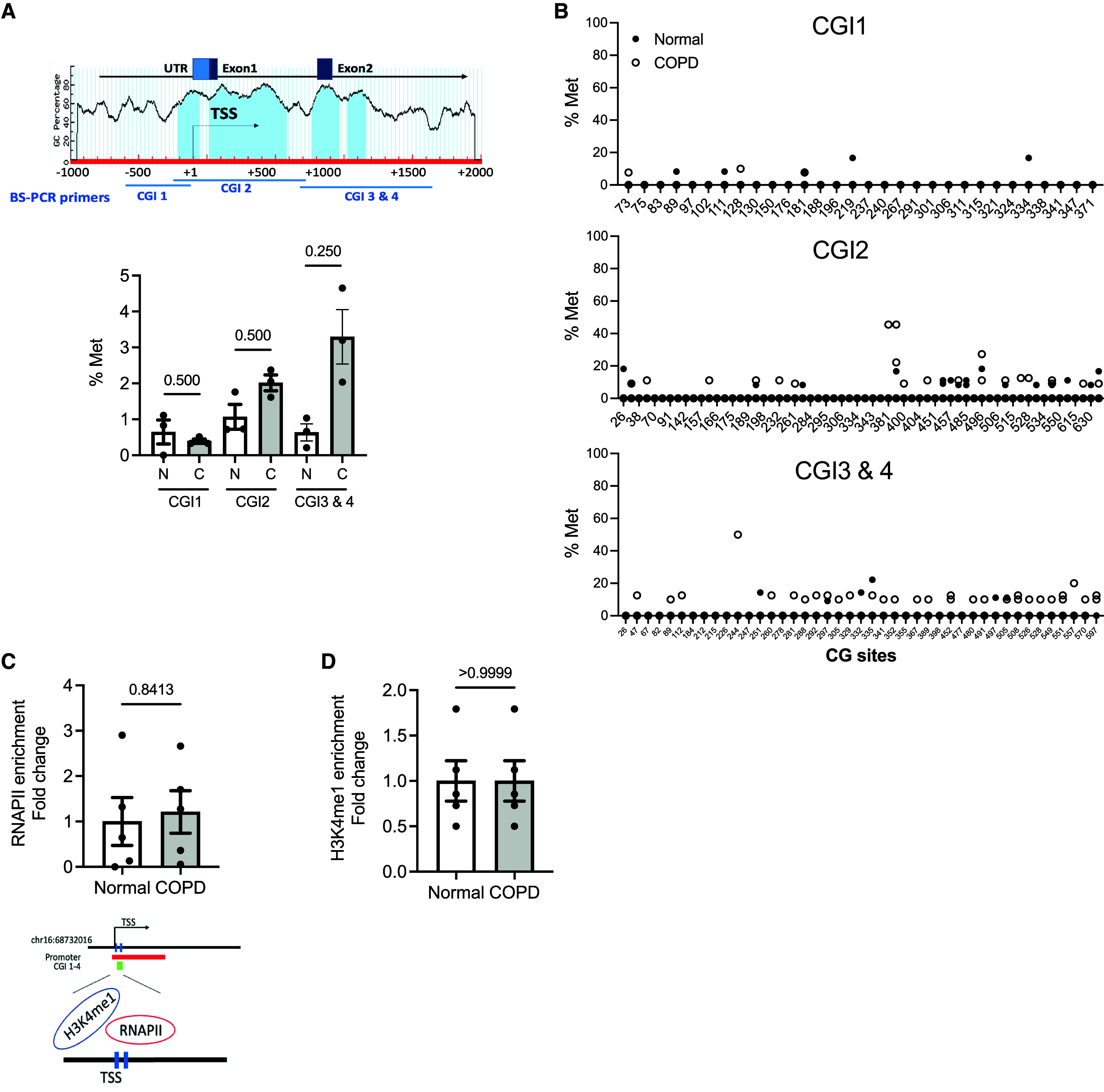
The promoter methylation level of CDH1 is not significantly changed in human epithelial cells derived from normal donors and those with COPD. (A) Top: The schematic diagram displays the percentage of cytosine–guanine dinucleotide (CpG) dinucleotide (CG) content in the 5′ promoter region of the CDH1. In an in silico analysis by MethPrimer, four CpG islands (CGIs) were identified (blue) (CG content >60%, an observed:expected ratio = 0.6). Bottom: The average methylation percentage (% Met) of the CDH1 promoter is shown among CGIs. % Met was calculated by taking an average of the methylation level of all CpG sites within CGI1, CGI2, and CGI3 and CGI4 of 3 normal donors (N) and 3 with COPD (C) by bisulfite sequencing. (B) The % Met of individual CpG sites at the CDH1 promoter (6–10 clones per donor). (C and D) Chromatin immunoprecipitation of (C) RNA polymerase II (RNAPII) and (D) histone H3 monomethylated at lysine 4 (H3K4 me1) at the TSS region (5 normal donors and 5 with COPD). Error bars represent SEM. Statistics were determined using (A) Wilcoxon matched-pairs signed rank tests and (C and D) the Mann–Whitney test, with P < 0.05 considered statistically significant. TSS = transcription start site; UTR = untranslated region.
Figure 5.
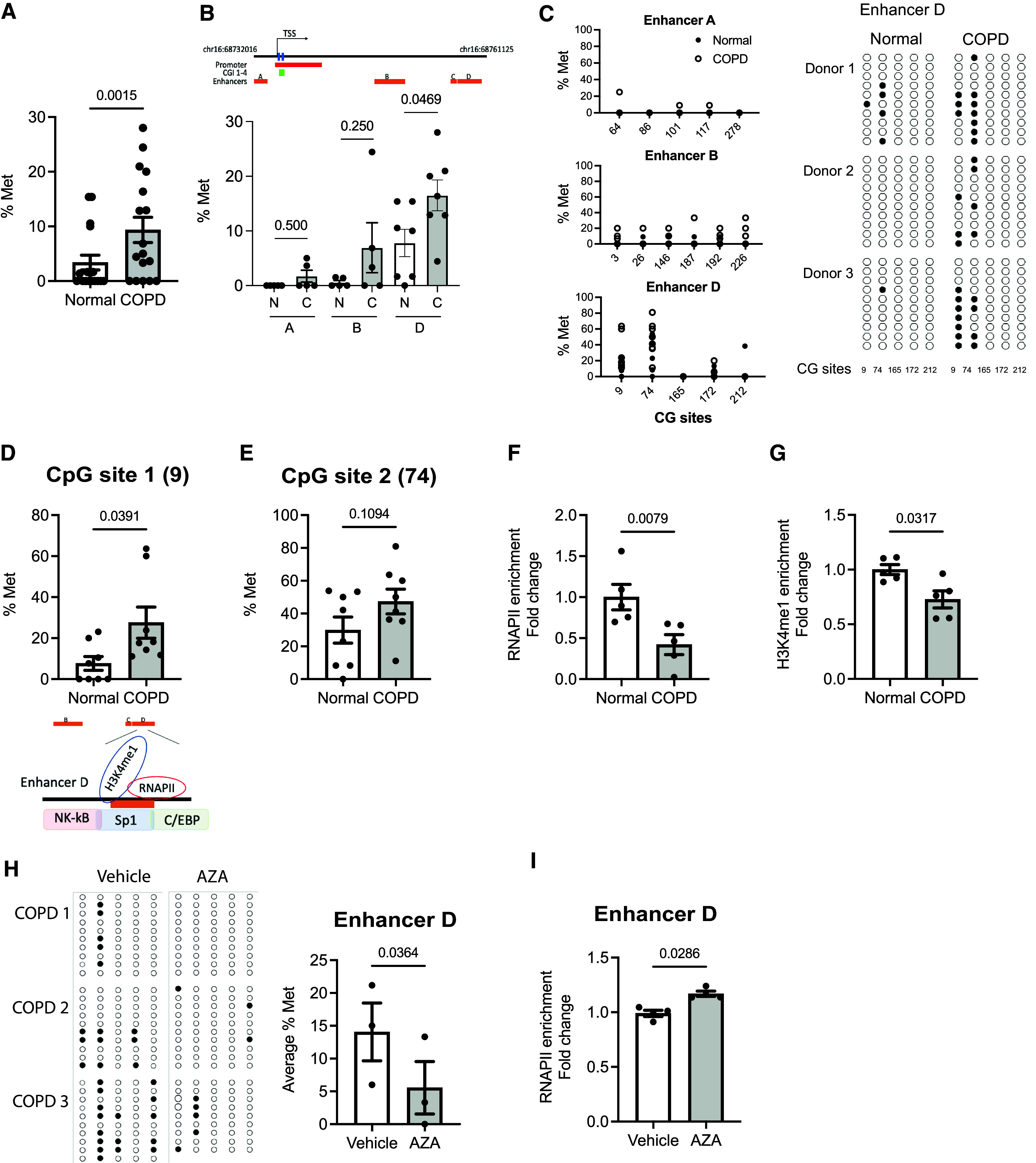
DNA hypermethylation in the CDH1 enhancer D region blocks RNAPII binding. (A) The average % Met of the CDH1 enhancers A, B, and D was calculated by taking an average of the methylation level of all CpG sites within enhancers A, B, and D of each donor (N: 5–6 normal donors; C: 5–6 donors with COPD) by bisulfite sequencing. (B) The average % Met of CDH1 enhancers A, B, and D. (C) % Met of each CG site of enhancers A, B, and C are shown in graphs (left) and in dot plots (right) (3 normal donors and 3 with COPD). Unmethylated (open circles) and methylated (closed circles) nucleotides are indicated. Each row of circles corresponds to an individual clone sequenced (9–10 clones per donor). (D and E) The % Met (D) Site 1 (CG Site 9) and (E) CpG Site 2 (CG Site 74) of enhancer D in donors with COPD versus normal donors (8 normal and 8 with COPD). (F and G) Enrichment of (F) RNAPII and (G) H3K4 me1 at the enhancer D region (5 normal and 5 with COPD). (H) The methylation at the CDH1 enhancer D in AZA-treated COPD cells is shown in dot plots (left) and in % Met (right) (9–10 clones per group; 3 donors). (I) Enrichment of RNAPII in AZA-treated COPD cells (n = 3). Error bars represent SEM. Statistics were determined by (A, B, D, E, and H) Wilcoxon matched-pairs signed rank tests and (F, G, and I) Mann–Whitney test, with P < 0.05 considered statistically significant.
We found that acute CS exposure increased global DNA methylation in mTECs derived from WT mice (1.92-fold in CS vs. air; P = 0.0012) (Figure 6A) as evidenced by 5mC localization in the alveolar tissue of the ex vivo human lung slices from a healthy donor after CS exposure to six cigarettes (see Figure E5A), indicating that the pulmonary remodeling caused by CS is linked to DNA methylation. Exposing ex vivo PCLSs derived from WT C57BL/6J mice to the acute CS exposure, we found that 1 μM AZA (noncytotoxic dose, as shown in Figure E1B) for 24 hours attenuated the CS-induced cytotoxicity as measured by the release of LDH (0.36-fold in CS-AZA vs. CS-vehicle; P = 0.002) (Figure 6B). We assessed for the localization of 5mC (indicated in red) on mouse lung slices and counterstained for nuclei with Hoechst 33342 (indicated in blue) by immunofluorescence (Figure 6C). The acute CS exposure increased 5mC expression in the alveolar epithelium, which was reversed by AZA. To determine whether this resulted in a decrease in TET1-mediated DNA demethylation, we assessed 5-hydroxymethylcytosine (5hmC) colocalization with E-cadherin in the lung sections, as 5hmC is a stable mark of DNA base modification from the conversion of 5mC by means of the TET enzyme (38). In the presence of AZA, the abundance of both E-cadherin (indicated in green) and 5hmC (indicated in red) in the air-/CS-exposed mouse PCLSs was increased in the airway (Figure 6D). Negative controls for rabbit or mouse IgG are shown in Figure 6D (bottom). In PCLSs derived from patients with COPD, we compared the ex vivo treatment of AZA (1 μM) with the vehicle control in adjacent slices (250 μm). AZA did not significantly increase 5mC fluorescence (Figure 6E), but it increased E-cadherin (twofold in AZA vs. vehicle; P = 0.0127) as well as 5hmC (7.54-fold in AZA vs. vehicle; P = 0.0003) in the airways (Figure 6F). Besides, we examined whether AZA affects cell viability and cell death in lung epithelium and tissues. We observed that AZA did not alter cell death, as measured using the alamarBlue cell viability assay, in either the COPD-derived PCLSs nor the COPD-derived epithelial cells (Figure E1), but its improvement was likely due to the increase in cell proliferation caused by AZA as measured using KI67 (Figure E5B).
Figure 6.
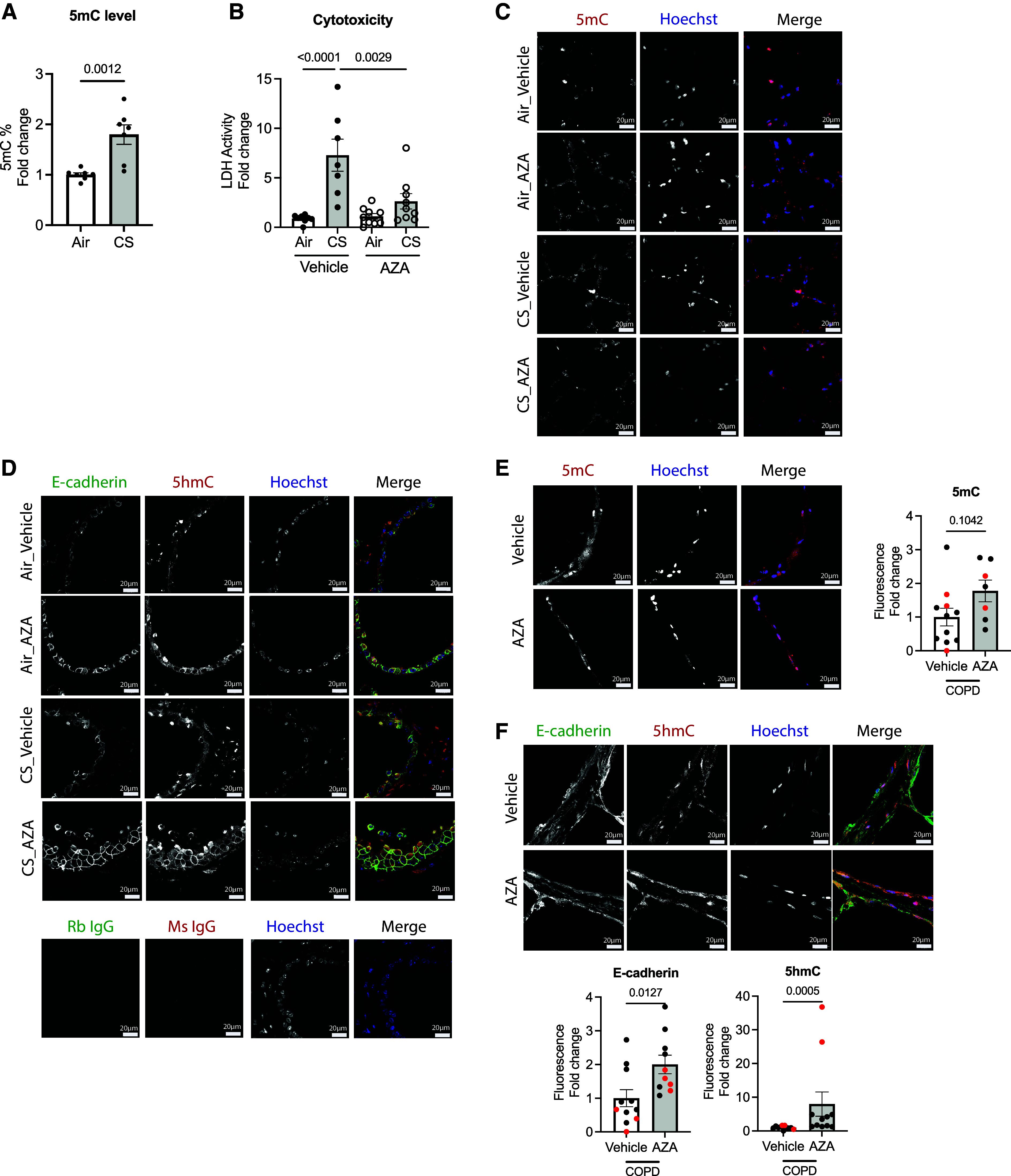
Global DNA demethylating agent AZA restores lung pulmonary remodeling in both mouse and human lungs. (A) Global DNA methylation in WT C57BL/6J-derived mTECs after acute exposure to CS (two cigarettes or humidified air in a day) (n = 7 from two independent experiments). (B) Cytotoxicity in mouse precision-cut lung slices (PCLSs) from WT C57BL/6J with acute exposure to CS (six cigarettes or humidified air in a day) by LDH level (seven to nine slices from three independent experiments). (C and D) Immunofluorescence of (C) 5-methylcytosine (5mC; red) and nucleus (blue) and of (D) E-cadherin (green), 5hmC (red), and nucleus (blue) of PCLSs derived from WT mice (n = 3 from three independent experiments). Negative controls against rabbit IgG (Rb IgG) and mouse IgG (Ms IgG) are indicated at the bottom. (E and F) Immunofluorescence of (E) 5mC (red) and nucleus (blue) and of (F) E-cadherin (green), 5hmC (red), and nucleus (blue) in human PCLSs derived from patients with COPD (n = 3 from 2 donors). Relative fluorescent intensity is indicated on the right in (E) and at the bottom in (F). The immunofluorescence images were taken at a 63× oil objective. Scale bars, 20 μm. Error bars represent SEM. Statistics were determined by using (A, E, and F) the Mann–Whitney test and (B) one-way ANOVA with Sidak’s multiple comparison tests, with P < 0.05 considered statistically significant. LDH = lactate dehydrogenase.
Utilizing the MLI, we assessed whether AZA protects or repairs lung injury in mice and patients with COPD. The presence of AZA significantly reduced the CS-induced alveolar destruction in the PCLS tissues (0.84-fold in CS-AZA vs. CS-vehicle; P = 0.0159; Figure 7A [representative images are shown on the left]). The alveolar lung destruction was also diminished by AZA (0.88-fold in AZA vs. vehicle; P = 0.0195) (Figure 7B). As ex vivo PCLSs provide a more physiologically relevant context, these data suggest that AZA is a potential therapy to improve lung tissue architecture.
Figure 7.
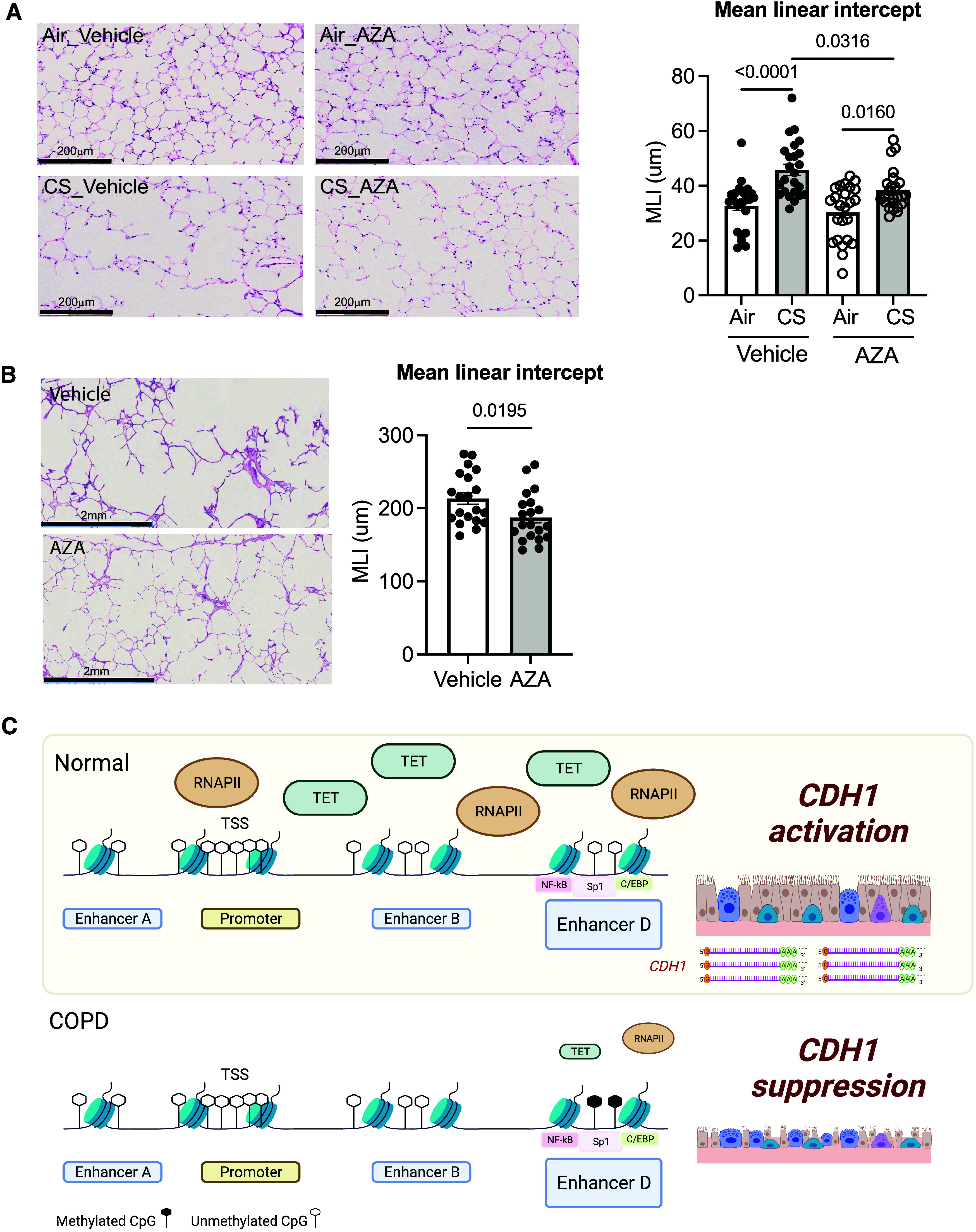
Global DNA demethylating agent AZA restores lung pulmonary remodeling in both mouse and human lungs. (A) In mouse PCLSs from WT C57BL/6J mice with acute exposure to CS (six cigarettes or humidified air in a day), AZA was found to attenuate CS-induced alveolar destruction by measuring the MLI and by hematoxylin and eosin staining (left) at 20× (3 slices from 3 mice). Scale bars, 200 μm. (B) In human PCLSs from patients with COPD, AZA was found to reduce alveolar destruction (10 images per group, 58 chords per image; total, 580 chords per group from two donors) by hematoxylin and eosin staining (left) at 2×. Scale bars, 2 mm. Error bars represent SEM. Statistics were determined by using (A) a one-way ANOVA with Sidak’s multiple comparison tests and (B) the Mann–Whitney test, with P < 0.05 considered statistically significant. (C) A schematic of the epigenetic regulations underlying CDH1 transcription in COPD. DNA hypermethylation at the CDH1 enhancer D region, not the CDH1 promoter, is accompanied by the reduction of RNAPII binding. The downregulation of TET1 in COPD contributed to the methylation pattern at the CDH1 enhancer, leading to barrier dysfunction (created from BioRender.com). MLI = mean linear intercept.
Discussion
It has been well recognized that environmental exposures increase DNA methylation, promoting the development of metabolic disorders and chronic diseases (39), and have even been associated with age-related chronic conditions (40–42). The airway epithelium, the first line of defense of the lung, is in constant contact with the external environment and, therefore, at high risk for epigenetic reprogramming. Although the epithelium has long been known to be dysregulated in COPD with chronic remodeling and cellular loss, the therapeutic focus has been on immune regulation and smooth muscle relaxation. Here, we have evidence that reversing the epigenetic reprogramming that occurs by DNA methylation can restore epithelial integrity and reverse the alveolar damage observed in COPD lungs, independent of CS exposure (Figure 7C).
Aberrant methylation is a fundamental epigenetic mechanism to silence a gene. Enhancers, which can work independently on the orientation, distance, and location of the corresponding target gene, activate the gene transcription by binding RNAPII (43). In silico analysis with LASAGNA-Search 2.0 (44) suggests that NF-κB, p50, Sp1, and CCAAT/enhancer-binding protein (C/EBP) α are putative binding transcription factors at the enhancer D region. Alteration in the methylation status at the enhancer D region may affect NF-κB– and Sp1-involved inflammatory response in COPD (45, 46). Notably, C/EBP activity shows a decrease in airway epithelium from patients with COPD and exerts severe COPD phenotypes in a rodent model (47). To our knowledge, this is the first demonstration of DNA methylation on the CDH1 enhancer element in epithelial cells derived from patients with COPD, with evidence that AZA can reverse these changes and improve tissue architecture. Future studies are needed to dissect the downstream binding of the transcription factors at the methylated enhancer D region.
TET enzymes catalyze the oxidation of 5mC to 5hmC, 5-formylcytosine, and 5-carboxycytosine as intermediates for the process of both active and passive DNA demethylation pathways in an α-ketoglutarate– and iron-dependent manner (previously reviewed by our group) (38). Loss of TETs has been shown in various chronic diseases such as childhood immunodeficiency with lymphoma (48), obesity (49), allergic rhinitis (50, 51), and childhood asthma (34). As our findings demonstrate, the expression of TET1 in lung epithelial cells correlates with CDH1 expression. Tet1 can cause dynamic changes in DNA methylation in response to exposures, as has been demonstrated in house dust mite–induced airway hyperresponsiveness (52, 53) and inflammatory responses in human lung epithelial cells (53, 54), as well as in lung cancer development (55). Tet-mediated DNA demethylation is thought to mainly occur at the distally located enhancers to fine-tune the transcription (56, 57) and activate transcription through the enhancer enrichment of 5hmC (58), which occurs as we have found in the COPD-derived epithelia.
E-cadherin plays a key role in maintaining the proliferation capabilities of alveolar type 2epithelial cells (22). It is plausible that maintenance of DNA hypomethylation in the CDH1 enhancer is critical for cell regeneration and that increased DNA methylation at this site abrogates the repair mechanisms necessary for alveolar regeneration in the injured epithelium of patients with COPD. We demonstrated that DNA demethylation increases CDH1 gene expression in epithelial cells derived from patients with COPD. Although we are currently limited by the technologies to demethylate specific nucleic acids in primary cells and human tissues, these strategies would significantly further both our molecular understanding of the disease and future therapies. There are several demethylation drugs in development for the treatment of specific cancers. We chose to study 5-AZA, as 5-Aza-CR and 5-Aza-CdR have already been approved by the FDA for the treatment of certain subtypes of myelodysplastic syndrome and chronic myelomonocytic leukemia. Whether future strategies are needed to increase the specificity of the drug or modalities for inhaled versions to target the lung epithelium, this study provides the proof of concept needed to show that the treatment of COPD lungs with AZA causes improvement in lung architecture. As a proof of concept, we studied the effects of AZA in the two different donors and found significant improvements in alveolar destruction. As we see increases in cell proliferation, these studies demonstrate evidence of repair and regeneration in the ex vivo lung slices. To our knowledge, this is the first demonstration of disease-modifying therapy in human lungs using a current FDA-approved demethylating agent. Moreover, this raises the possibility that these strategies are relevant in age-associated lung function decline.
There are several limitations to the study. Epigenetic regulation of E-cadherin is not the only mechanism by which E-cadherin is regulated in COPD, nor is it the only protein that is altered in COPD, which is substantiated by the fact that the effect of AZA is modest. Moreover, given the small number of human PCLS samples, our data serve as a proof of principle that these proteins, identified in vitro and in mouse models, have some relevance in human lungs, although a more comprehensive analysis of patient characteristics and responses is needed before these strategies can be translated clinically. There is likely to be patient-to-patient variation in gene expression and DNA methylation patterns, and future studies will likely have to consider this while considering therapeutic options. Finally, we recognize that the improvement of transepithelial electrical resistance with AZA is small, which is likely due to its minimal effect on other junctional proteins. Despite these limitations, our study presents, to our knowledge, the first evidence that the loss of E-cadherin is due to the DNA methylation at the CDH1 enhancer element as a result of loss of TET1 in COPD epithelium and that reversing DNA methylation improves alveolar destruction in COPD. The SPIROMICS cohort showed increased DNA methylation of CDH1 in moderate to severe COPD. These findings suggest that reversing CDH1 DNA methylation could be a potential therapeutic strategy for COPD.
Acknowledgments
Acknowledgment
The authors acknowledge the following current and former investigators of the SPIROMICS sites and reading centers: Neil E. Alexis, Wayne H. Anderson, Mehrdad Arjomandi, Igor Barjaktarevic, R. Graham Barr, Patricia Basta, Lori A. Bateman, Christina Bellinger, Surya P. Bhatt, Eugene R. Bleecker, Richard C. Boucher, Russell P. Bowler, Russell G. Buhr, Stephanie A. Christenson, Alejandro P. Comellas, Christopher B. Cooper, David J. Couper, Gerard J. Criner, Ronald G. Crystal, Jeffrey L. Curtis, Claire M. Doerschuk, Mark T. Dransfield, M. Bradley Drummond, Christine M. Freeman, Craig Galban, Katherine Gershner, MeiLan K. Han, Nadia N. Hansel, Annette T. Hastie, Eric A. Hoffman, Yvonne J. Huang, Robert J. Kaner, Richard E. Kanner, Mehmet Kesimer, Eric C. Kleerup, Jerry A. Krishnan, Wassim W. Labaki, Lisa M. LaVange, Stephen C. Lazarus, Fernando J. Martinez, Merry-Lynn McDonald, Deborah A. Meyers, Wendy C. Moore, John D. Newell Jr., Elizabeth C. Oelsner, Jill Ohar, Wanda K. O’Neal, Victor E. Ortega, Robert Paine III, Laura Paulin, Stephen P. Peters, Cheryl Pirozzi, Nirupama Putcha, Sanjeev Raman, Stephen I. Rennard, Donald P. Tashkin, J. Michael Wells, Robert A. Wise, and Prescott G. Woodruff. The project officers from the Lung Division of the National Heart, Lung, and Blood Institute were Lisa Postow and Lisa Viviano. The authors thank the Johns Hopkins University School of Medicine Microscope Facility for providing access to and training on the Zeiss LSM 880 Confocal with Airyscan FAST Module. The authors also thank Johns Hopkins University School of Medicine Reference Histology Laboratory for assisting in hematoxylin and eosin staining in precision-cut lung tissue slices.
Footnotes
Supported by the National Heart, Lung, and Blood Institute (NHLBI; grants R01HL124099 and HLR01HL151107 to V.K.S.); National Institute of Environmental Health Sciences (grant 5U01ES026721 to S.B.); a Ludwig Family Department of Medicine Physician-Scientist grant (to V.K.S.); and the Office of the Director of the National Institutes of Health, National Institute of General Medical Sciences, under award number S10OD023548 (to S.C. Kuo, Johns Hopkins University Microscope Facility). The Subpopulations and Intermediate Outcomes in COPD Study (or, SPIROMICS) was supported by contracts from the NHLBI (HHSN268200900013C, HHSN268200900014C, HHSN268200900015C, HHSN268200900016C, HHSN268200900017C, HHSN268200900018C, HHSN268200900019C, and HHSN268200900020C) and grants from the NHLBI (U01HL137880, U24HL141762, R01HL182622, and R01HL144718) and was supplemented by contributions made through the Foundation for the National Institutes of Health and the COPD Foundation from Amgen; AstraZeneca/MedImmune; Bayer; Bellerophon Therapeutics; Boehringer Ingelheim; Chiesi Farmaceutici; Forest Research Institute; Genentech; GlaxoSmithKline; Grifols; Ikaria; MGC Diagnostics; Novartis Pharmaceuticals Corporation; Nycomed GmbH; Polarean; ProterixBio; Regeneron Pharmaceuticals; Sanofi; Sunovion; Takeda Pharmaceutical Company; and Theravance Biopharma US and Mylan/Viatris.
Author Contributions: B.H.Y.-L., W-Y.T., and V.K.S. conceived and designed the experiments. B.H.Y.-L., A.W., S.J., E.L., R.Z., D.C., N.U., E.T., E.S., D.S., M.L., and B.G. performed experiments. B.H.Y.-L., A.W., D.C., S.H.L., C.S., K.W., A.K.S., S.C., S.A.C., Y.J.H., and V.E.O. analyzed data. B.H.Y.-L. and A.W. prepared the figures. W-Y.T., S.B., and V.K.S. contributed to the mice and lab supplies. B.H.Y.-L. and V.K.S. wrote the manuscript. All authors revised the manuscript.
This article has an online data supplement, which is accessible from this issue’s table of contents online at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2023-0147OC on November 17, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet . 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paksa A, Rajagopal J. The epigenetic basis of cellular plasticity. Curr Opin Cell Biol . 2017;49:116–122. doi: 10.1016/j.ceb.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet . 2012;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 4. Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol . 2010;126:453–465. doi: 10.1016/j.jaci.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell . 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 6. Morgan HD, Sutherland HGE, Martin DIK, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet . 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 7.Ti D, Li M, Fu X, Han W. Causes and consequences of epigenetic regulation in wound healing. Wound Repair Regen. 2014;22:305–312. doi: 10.1111/wrr.12160. [DOI] [PubMed] [Google Scholar]
- 8.2022. https://www.cdc.gov/genomics/update/current.htm
- 9. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev . 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science . 2017;356:eaaj2239. doi: 10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci . 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 12. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet . 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 13. Poursafa P, Kamali Z, Fraszczyk E, Boezen HM, Vaez A, Snieder H. DNA methylation: a potential mediator between air pollution and metabolic syndrome. Clin Epigenetics . 2022;14:82. doi: 10.1186/s13148-022-01301-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet . 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vucic EA, Chari R, Thu KL, Wilson IM, Cotton AM, Kennett JY, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol . 2014;50:912–922. doi: 10.1165/rcmb.2013-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Casas-Recasens S, Noell G, Mendoza N, Lopez-Giraldo A, Garcia T, Guirao A, et al. Lung DNA methylation in chronic obstructive pulmonary disease: relationship with smoking status and airflow limitation severity. Am J Respir Crit Care Med . 2021;203:129–134. doi: 10.1164/rccm.201912-2420LE. [DOI] [PubMed] [Google Scholar]
- 17. Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med . 2012;185:373–381. doi: 10.1164/rccm.201108-1382OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Vries M, van der Plaat DA, Vonk JM, Boezen HM. No association between DNA methylation and COPD in never and current smokers. BMJ Open Respir Res . 2018;5:e000282. doi: 10.1136/bmjresp-2018-000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huo X, Jin S, Wang Y, Ma L. DNA methylation in chronic obstructive pulmonary disease. Epigenomics . 2021;13:1145–1155. doi: 10.2217/epi-2021-0111. [DOI] [PubMed] [Google Scholar]
- 20. Chen X, Yan F, Lin X, Shi L, Wang X, Zeng Y. DNA methylation in chronic obstructive pulmonary disease. Adv Exp Med Biol . 2020;1255:83–98. doi: 10.1007/978-981-15-4494-1_7. [DOI] [PubMed] [Google Scholar]
- 21. Ghosh B, Reyes-Caballero H, Akgün-Ölmez SG, Nishida K, Chandrala L, Smirnova L, et al. Effect of sub-chronic exposure to cigarette smoke, electronic cigarette and waterpipe on human lung epithelial barrier function. BMC Pulm Med . 2020;20:216. doi: 10.1186/s12890-020-01255-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ghosh B, Loube J, Thapa S, Ryan H, Capodanno E, Chen D, et al. Loss of E-cadherin is causal to pathologic changes in chronic lung disease. Commun Biol . 2022;5:1149. doi: 10.1038/s42003-022-04150-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nishida K, Brune KA, Putcha N, Mandke P, O’Neal WK, Shade D, et al. Cigarette smoke disrupts monolayer integrity by altering epithelial cell-cell adhesion and cortical tension. Am J Physiol Lung Cell Mol Physiol . 2017;313:L581–L591. doi: 10.1152/ajplung.00074.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci . 2008;65:3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghosh B, Nishida K, Chandrala L, Mahmud S, Thapa S, Swaby C, et al. Epithelial plasticity in COPD results in cellular unjamming due to an increase in polymerized actin. J Cell Sci . 2022;135:jcs258513. doi: 10.1242/jcs.258513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lyons-Cohen MR, Thomas SY, Cook DN, Nakano H. Precision-cut mouse lung slices to visualize live pulmonary dendritic cells. J Vis Exp . 2017:55465. doi: 10.3791/55465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lagowala DA, Wally A, Wilmsen K, Kim B, Yeung-Luk B, Choi J, et al. Microphysiological models of lung epithelium-alveolar macrophage co-cultures to study chronic lung disease. Adv Biol (Weinh) . 2023:e2300165. doi: 10.1002/adbi.202300165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chandrala LD, Afshar-Mohajer N, Nishida K, Ronzhes Y, Sidhaye VK, Koehler K, et al. A device for measuring the in-situ response of human bronchial epithelial cells to airborne environmental agents. Sci Rep . 2019;9:7263. doi: 10.1038/s41598-019-43784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crowley G, Kwon S, Caraher EJ, Haider SH, Lam R, Batra P, et al. Quantitative lung morphology: semi-automated measurement of mean linear intercept. BMC Pulm Med . 2019;19:206. doi: 10.1186/s12890-019-0915-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shihan MH, Novo SG, Le Marchand SJ, Wang Y, Duncan MK. A simple method for quantitating confocal fluorescent images. Biochem Biophys Rep . 2021;25:100916. doi: 10.1016/j.bbrep.2021.100916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔ C(T)) method. Methods . 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32. Yeung BHY, Griffiths K, Berger L, Paudel O, Shin MK, Rui L, et al. Leptin induces epigenetic regulation of transient receptor potential melastatin 7 in rat adrenal pheochromocytoma cells. Am J Respir Cell Mol Biol . 2021;65:214–221. doi: 10.1165/rcmb.2020-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Couper D, LaVange LM, Han M, Barr RG, Bleecker E, Hoffman EA, et al. SPIROMICS Research Group Design of the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS) Thorax . 2014;69:491–494. doi: 10.1136/thoraxjnl-2013-203897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Somineni HK, Zhang X, Biagini Myers JM, Kovacic MB, Ulm A, Jurcak N, et al. Ten-eleven translocation 1 (TET1) methylation is associated with childhood asthma and traffic-related air pollution. J Allergy Clin Immunol . 2016;137:797–805.e5. doi: 10.1016/j.jaci.2015.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li L-C, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics . 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 36. Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet . 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 37. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature . 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 38.Pulczinski J, Yeung BHY, Wu Q, Cheng RYS, Tang W. In: ToxicoEpigenetics: core principles and applications. McCullough S, Dolinoy D, editors. San Diego, CA: Elsevier Science and Technology; 2018. DNA hydroxymethylation: implications for toxicology and epigenetic epidemiology; pp. 191–214. [Google Scholar]
- 39. Rider CF, Carlsten C. Air pollution and DNA methylation: effects of exposure in humans. Clin Epigenetics . 2019;11:131. doi: 10.1186/s13148-019-0713-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA methylation clocks in aging: categories, causes, and consequences. Mol Cell . 2018;71:882–895. doi: 10.1016/j.molcel.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang K, Liu H, Hu Q, Wang L, Liu J, Zheng Z, et al. Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct Target Ther . 2022;7:374. doi: 10.1038/s41392-022-01211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morgan AE, Davies TJ, McAuley MT. The role of DNA methylation in ageing and cancer. Proc Nutr Soc . 2018;77:412–422. doi: 10.1017/S0029665118000150. [DOI] [PubMed] [Google Scholar]
- 43. Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell . 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 44. Lee C, Huang CH. LASAGNA-Search: an integrated web tool for transcription factor binding site search and visualization. Biotechniques . 2013;54:141–153. doi: 10.2144/000113999. [DOI] [PubMed] [Google Scholar]
- 45. Schuliga M. NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules . 2015;5:1266–1283. doi: 10.3390/biom5031266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baraniuk JN, Casado B, Pannell LK, McGarvey PB, Boschetto P, Luisetti M, et al. Protein networks in induced sputum from smokers and COPD patients. Int J Chron Obstruct Pulmon Dis . 2015;10:1957–1975. doi: 10.2147/COPD.S75978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Didon L, Roos AB, Elmberger GP, Gonzalez FJ, Nord M. Lung-specific inactivation of CCAAT/enhancer binding protein α causes a pathological pattern characteristic of COPD. Eur Respir J . 2010;35:186–197. doi: 10.1183/09031936.00185008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stremenova Spegarova J, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood . 2020;136:1055–1066. doi: 10.1182/blood.2020005844. [DOI] [PubMed] [Google Scholar]
- 49. Yuan Y, Liu C, Chen X, Sun Y, Xiong M, Fan Y, et al. Vitamin C inhibits the metabolic changes induced by Tet1 insufficiency under high fat diet stress. Mol Nutr Food Res . 2021;65:e2100417. doi: 10.1002/mnfr.202100417. [DOI] [PubMed] [Google Scholar]
- 50. Li H, Lu T, Sun W, Ma R, Zhong H, Wei Y, et al. Ten-eleven translocation (TET) enzymes modulate the activation of dendritic cells in allergic rhinitis. Front Immunol . 2019;10:2271. doi: 10.3389/fimmu.2019.02271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tan L, Qiu T, Xiang R, Cao C, Deng Y, Tao Z, et al. Down-regulation of Tet2 is associated with Foxp3 TSDR hypermethylation in regulatory T cell of allergic rhinitis. Life Sci . 2020;241:117101. doi: 10.1016/j.lfs.2019.117101. [DOI] [PubMed] [Google Scholar]
- 52. Cheng RY, Shang Y, Limjunyawong N, Dao T, Das S, Rabold R, et al. Alterations of the lung methylome in allergic airway hyper-responsiveness. Environ Mol Mutagen . 2014;55:244–255. doi: 10.1002/em.21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burleson JD, Siniard D, Yadagiri VK, Chen X, Weirauch MT, Ruff BP, et al. TET1 contributes to allergic airway inflammation and regulates interferon and aryl hydrocarbon receptor signaling pathways in bronchial epithelial cells. Sci Rep . 2019;9:7361. doi: 10.1038/s41598-019-43767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaur G, Batra S. Regulation of DNA methylation signatures on NF-κB and STAT3 pathway genes and TET activity in cigarette smoke extract-challenged cells/COPD exacerbation model in vitro. Cell Biol Toxicol . 2020;36:459–480. doi: 10.1007/s10565-020-09522-8. [DOI] [PubMed] [Google Scholar]
- 55. Xu Q, Wang C, Zhou J-X, Xu Z-M, Gao J, Sui P, et al. Loss of TET reprograms Wnt signaling through impaired demethylation to promote lung cancer development. Proc Natl Acad Sci USA . 2022;119:e2107599119. doi: 10.1073/pnas.2107599119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu F, Liu Y, Jiang L, Yamaguchi S, Zhang Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev . 2014;28:2103–2119. doi: 10.1101/gad.248005.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell . 2014;56:286–297. doi: 10.1016/j.molcel.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Putiri EL, Tiedemann RL, Thompson JJ, Liu C, Ho T, Choi JH, et al. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biol . 2014;15:R81. doi: 10.1186/gb-2014-15-6-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]