

Abstract
Background
Okur‐Chung neurodevelopmental syndrome (OCNDS) is a rare autosomal dominant disorder caused by pathogenic variants in CSNK2A1. It is characterized by intellectual disability, developmental delay, and multisystemic abnormalities.
Methods
We performed the whole‐exome sequencing for a patient in a Chinese family. The co‐segregation study using the Sanger sequencing method was performed among family members. Reverse transcription and quantitative real‐time polymerase chain reaction were carried out using total RNA from blood samples of the proband and wild‐type control subjects. A review of patients with OCNDS harboring CSNK2A1 pathogenic variants was conducted through a comprehensive search of the PubMed database.
Results
We identified a novel CSNK2A1 frameshift variant p.Tyr323Leufs*16 in a Chinese family. The proband, a 31‐year‐old female, presented with abnormal eating habits, recurrent seizures, language impairment, and intellectual disability. Her mother exhibited postnatal hernias, splenomegaly, and a predisposition to infections, but showed no significant developmental impairments or intellectual disability. Genetic studies revealed the presence of this variant in CSNK2A1 in both the proband and her mother. Transcription analysis revealed this variant may lead to nonsense‐mediated mRNA decay, suggesting haploinsufficiency as a potential disease mechanism. We reviewed 47 previously reported OCNDS cases and discovered that individuals carrying CSNK2A1 null variants may exhibit a diminished frequency of symptoms linked to language deficits, dysmorphic facial features, or intellectual disability, consequently presenting an overall milder phenotype when compared to those with missense variants.
Conclusion
We report a novel frameshift variant, p.Tyr323Leufs*16, in an OCNDS family with a generally mild phenotype. This study may broaden the spectrum of clinical presentations associated with OCNDS and contribute novel insights into the genotype–phenotype correlation of this condition.
Keywords: CSNK2A1, nonsense‐mediated mRNA decay, OCNDS, Okur‐Chung neurodevelopmental syndrome, phenotype
Okur‐Chung neurodevelopmental syndrome is a rare autosomal dominant disorder caused by CSNK2A1 gene mutations. A novel frameshift variant in CSNK2A1 was identified in a Chinese family, with the proband showing intellectual disability, seizures, language impairment, and abnormal eating habits, while her mother had milder symptoms. Transcription analysis suggested haploinsufficiency as a potential disease mechanism, and a review of previously reported cases indicated that individuals with CSNK2A1 null mutations may exhibit milder symptoms compared to those with missense mutations.
1. INTRODUCTION
Okur‐Chung neurodevelopmental syndrome (OCNDS), initially described by Okur et al., is a rare genetic disorder resulting from pathogenic variants in CSNK2A1 (MIM: 115440) (Okur et al., 2016). Casein kinase II is a protein kinase involved in several cellular processes (Niefind et al., 2001). The alpha subunit of casein kinase II is encoded by the CSNK2A1 gene (Niefind et al., 2001; Wafik et al., 2023). CSNK2A1 predominantly consists of a single extensive functional domain (Figure 1a). OCNDS typically presents with developmental delays, mild‐to‐moderate intellectual impairment, hypotonia, feeding challenges, distinctive facial features, speech delay, and may include non‐specific clinical characteristics such as behavioral issues, disrupted sleep patterns, microcephaly, seizures, and short stature in some cases (Jafari Khamirani et al., 2022; Okur et al., 2016). Previously, OCNDS cases were diagnosed as simplex cases due to de novo heterozygous pathogenic variants in CSNK2A1 with unaffected parents (Chiu et al., 2018; Okur et al., 2016). However, more recent observations have revealed instances of familial transmission, indicating that affected individuals can be fertile (Belnap et al., 2023).
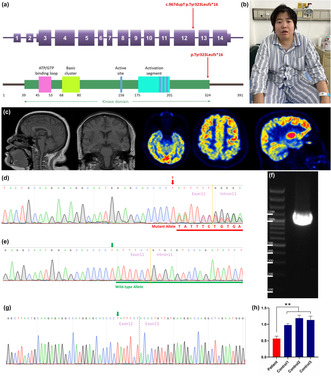
FIGURE 1.
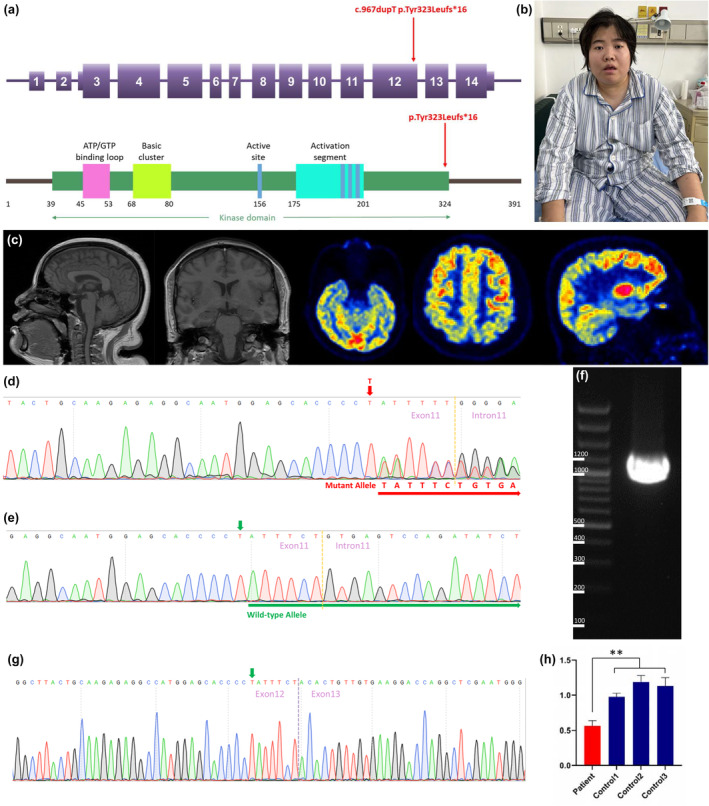
Clinical and genetic study. (a) Schematic representation of the 14 coding exons and UTR of CSNK2A1 (NM_177559.3). The c.967dupT variant identified in this study is indicated in red. (b) Schematic structure of the human CSNK2A1 protein. CSNK2A1 has a large kinase domain with functional segments including an ATP/GTP‐binding loop, basic cluster, active site, and activation segment. The truncating variant identified in this study is shown by a red arrow above the protein. (c) Neuroimaging of the proband: left: T1 weighted imaging; right: 18F‐FDG‐PET imaging. (d) Sequence analysis revealed a c.967dupT, p.Tyr323Leufs*16 frameshift variant in exon 12 of CSNK2A1 in the proband and her mother. The red arrow indicates the c.967 nucleotide. (e) Sequence analysis revealed no variant in exon 12 of CSNK2A1 in the proband's father. The green arrow indicates the c.967 nucleotide. (f) Transcription analysis of CSNK2A1 in peripheral leukocytes of the patient. Agarose gel electrophoresis of the long‐range RT‐PCR products revealed only the cDNA fragment of 1262 bp corresponding to the predicted canonical transcripts in the patient. (g) Sanger sequencing of long‐range RT‐PCR products revealed only the canonical transcript in the proband. The transcript with the frameshift variant was not detected. The green arrow indicates the c.967 nucleotide. (h) CSNK2A1 gene expression was analyzed by qRT‐PCR using total RNA extracted from peripheral leukocytes of the patient with primer sets corresponding to sequences within exons 8–10. A ∼50% decrease in the CSNK2A1 product (normalized to β‐actin level) was observed in the patient compared with the same product in the three wild‐type control subjects. Data are expressed as mean ± SD of three independent experiments. **p < 0.01 (Mann–Whitney U test).
To date, 47 patients with OCNDS have been described in detail in the literature. However, a clear genotype–phenotype correlation has not been established. Most variants in OCNDS are missense variants primarily clustered within the functional regions of the CSNK2A1 kinase domain, while less commonly, frameshift variants have been reported as well (Jafari Khamirani et al., 2022; Okur et al., 2016). The molecular mechanisms underlying CSNK2A1 variants remain incompletely elucidated (Wafik et al., 2023). The frameshift, splice site variants identified in OCNDS patients suggest that loss‐of‐function could be the plausible disease mechanism for the CSNK2A1 variants, but there may be other possible molecular mechanisms for missense variants within the protein kinase domain.
In the present study, we report two individuals within a Chinese family who presented with a unique, non‐conventional, and relatively mild OCNDS phenotype. We conducted mRNA expression analysis to preliminarily investigate the molecular mechanisms associated with this frameshift variant. Moreover, by systematically reviewing the literature and comparing the clinical features of all identified patients with OCNDS, we elucidated the phenotypic ranges linked to individual CSNK2A1 variants, thus offering insights into the genotype–phenotype correlations within OCNDS.
2. METHODS
2.1. Ethical compliance
The study was approved by the Ethics Committees of the Xuanwu Hospital of Capital Medical University, China and was conducted in accordance with the principles stated in the Declaration of Helsinki. Written informed consent for genetic testing and publication was obtained from each patient or their guardian.
2.2. Patients
Two patients in a family with OCNDS were investigated in this study. Detailed clinical information was obtained from the Department of Neurology of Xuanwu Hospital.
2.3. Whole‐exome sequencing (WES) study
The genomic DNA was isolated from peripheral blood leukocytes (QIAamp DNA Blood Kits, QIAGEN). Exome capture was performed with a SureSelect Human All Exon V6+UTR (89 Mb) Kit (Agilent Technologies, Santa Clara, CA, USA). Paired‐end sequencing was carried out on a HiSeq2500 (Illumina, San Diego, CA, USA) using a HiSeq SBS Kit V4 (Illumina), which generated 100‐bp reads. The average and minimum sequencing depths were 125× and 20×, respectively. The reference databases utilized included hg38 (GRCh38) (http://genome.ucsc.edu), HGMD (https://portal.biobase‐international.com), gnomAD (http://gnomad.broadinstitute.org), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and dbSNP (https://www.ncbi.nlm.nih.gov/SNP). We summarized intellectual disability, neurodevelopmental disorders, and dementia‐related genes using the Online Mendelian Inheritance in Man (OMIM) and PubMed databases (Table S1). Whole‐exome sequencing (WES) data were analyzed for single‐nucleotide variants and insertions/deletions in these genes. Minor allele frequency, conservation, predicted pathogenicity, and disease association were analyzed, and the results were confirmed by Sanger sequencing.
Finally, WES was performed using a triple diagnostic approach (proband, mother, and father) to identify potential de novo variants responsible for the phenotype in the proband. The disease‐causing variant frequency was set at less than 0.01%, a dominant inheritance mode with full penetrance in both males and females being assumed. Moreover, targeted analysis was carried out to determine the number of ATN1 CAG repeats in the proband. Testing was performed by polymerase chain reaction (PCR) amplification of the ATN1 trinucleotide repeat region followed by gel electrophoresis.
2.4. RNA isolation, reverse transcription, and quantitative real‐time PCR (qRT‐PCR)
Reverse transcription, Sanger sequencing, and quantitative real‐time (qRT)‐PCR were performed using total RNA from blood samples of the proband and three wild‐type control subjects. Total RNA was extracted from leukocytes using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Reverse transcription was performed using Bio‐Rad's iScript™ gDNA Clear cDNA Synthesis Kit (Bio‐Rad Laboratories, Hercules, CA). Complementary DNA (cDNA) was then amplified by long‐range PCR using a primer set designed to amplify from exon 3 to exon 14 of CSNK2A1 (NM_177559.3). qRT‐PCR was performed with SsoAdvanced™ Universal SYBR® Green Supermix (Bio‐Rad). Normalization and relative quantification of the expression levels of the CSNK2A1 gene were performed using the ΔCT method, with constitutively expressed gene β‐actin as an internal control. The primer sequences used in this study are listed in Table S2.
2.5. Literature data extraction and analysis
The PubMed database was searched on September 2023 for primary research articles and case studies reporting clinically diagnosed OCNDS patients with CSNK2A1 pathogenic variants using the search terms: ‘Okur‐Chung syndrome’, ‘Okur‐Chung neurodevelopmental syndrome’, ‘OCNDS’, or ‘CSNK2A1’.
We only included studies with available full texts and selected those that involved patients with OCNDS and CSNK2A1 germline variants with full details of phenotypes and genotypes. For clinically diagnosed OCNDS patients with CSNK2A1 pathogenic variants, the following demographic and clinical variables were extracted from the retrieved articles: gender, age at diagnosis (years), developmental delay, language deficits, dysmorphic facial features, intellectual disability, behavioral issues, eating disorders, musculoskeletal disorders, microcephaly, abnormal brain MRI, hypotonia, seizures, herniation, and inheritance pattern.
2.6. Statistical analysis
Statistical analyses were performed using SPSS version 22.0 (IBM, Armonk, NY, USA). Continuous data are presented as the mean value and analyzed using the Mann–Whitney U test. Dichotomous data are shown as percentages and were compared using the χ 2 test or Fisher test. Two‐tailed p‐values <0.05 were considered statistically significant when comparing the two groups.
3. RESULTS
3.1. Clinical study
A 31‐year‐old female presenting recurrent generalized tonic–clonic seizures and language impairment was referred to our center. The proband is the product of an uneventful pregnancy and a normal vaginal delivery, born to non‐consanguineous parents. There were no available records for her birth. Her father recalls she sat unsupported at 8 months (+1.8) and walked independently at around 18 months (+2.7). The speech was achieved on time. Her growth parameters were all within the normal range. She showed some facial features such as bilateral epicanthic folds, a depressed nasal bridge, a thin upper lip, mild retrognathia, and micrognathia with a continuous open‐mouth posture (Figure 1b). However, these characteristics do not qualify as facial dysmorphisms. She attended a mainstream primary school, with her academic performance consistently in the lower‐middle range, and later graduated from a technical secondary school. Subsequently, she worked as a cashier in a small grocery store. The patient exhibits abnormal eating habits, displaying a predilection for consuming sweets and sugary foods. She habitually consumes only sugary beverages and adamantly avoids drinking pure water. The patient was primarily raised by the grandmother in the absence of adequate care, making it likely that some potential early cognitive and behavioral symptoms remain unknown to us.
She experienced her first seizure at the age of 29 and subsequently had recurrent seizure episodes over the next two years. Her seizures were predominantly generalized tonic–clonic, lasting several minutes. Additional manifestations followed at age 30, including involuntary facial twitching, chewing coordination issues, alternating limb weakness with involuntary movements, and transient head deviation. Several months ago, the patient presented with memory decline, language impairment, anosognosia, and visual and auditory hallucinations. She experienced daily episodes of limb rigidity and spasms, lasting several minutes, without loss of consciousness or urinary/fecal incontinence. On examination, it was observed that her weight is 85 kg (+1.7), her height is 160 cm (−0.2), and her head circumference measures 56 cm (+2.0). She is capable of vocalization, but she exhibits complete motor aphasia. Limb strength assessments revealed asymmetrical weakness, with right upper limb strength graded at Medical Research Scale level 3/5, and left upper and both lower limbs graded at level 4/5. Involuntary movements were observed bilaterally, while muscle tone remained normal. The spinal cord and brain magnetic resonance imaging (MRI), along with cerebrospinal fluid examination, did not yield any notable abnormalities. A comprehensive chemistry panel, complete blood count with differential, blood lactate and pyruvate, ammonia, creatine kinase, hemoglobin A1C, plasma amino acid analysis, carnitine and acylcarnitine profile, lipoprotein profile, hormone screening studies, and urine studies, including urinalysis, urine organic acid analysis, and urine amino acid analysis, did not reveal any notable abnormalities. No elevations in blood lactic acid and pyruvate were found. Positron emission tomography computed tomography reveals a severe reduction in metabolic activity in the brainstem and a mild regional metabolic decrease in the cortical areas of both parietal and temporal lobes (Figure 1c). Cardiac ultrasound revealed enlargement of the left atrium and reduced left ventricular diastolic function. Video electroencephalograph findings reveal the presence of a substantial amount of low‐amplitude fast wave activity, predominantly localized in the anterior frontal region. Treatment with corticosteroid pulse therapy and antiviral agents showed limited effectiveness. Upon discharge, she continued to receive levetiracetam for effective seizure control, but no improvement was observed in other symptoms.
The patient's mother was abandoned by her biological parents after birth due to noticeable abdominal distention, as they believed her care would be challenging. After being adopted by foster parents, it was discovered that she had splenomegaly, umbilical hernia, and inguinal hernia. There were no available records for her birth and developmental history. Her umbilical hernia and inguinal hernia resolved and her splenomegaly subsequently improved. Her growth parameters were all within the normal range. Her weight is 58 kg (−0.1), her height is 160 cm (−0.2), and her head circumference is 57 cm (+2.6). No distinctive facial dysmorphisms were noted on examination. Her neurological examination was normal. Her academic performance has consistently been at a lower‐middle range, with no evident signs of learning difficulties. She graduated from a vocational high school and subsequently engaged in roles as a ticket seller and a retail assistant in a grocery store. She possesses a susceptibility to infections and has experienced recurrent urinary tract infections, which subsequently progressed into pyelonephritis at age 24. At the age of 57, a follow‐up abdominal ultrasound still indicates splenomegaly.
3.2. Genetic study
We carried out WES of genomic DNA from the proband. We examined gene pathogenic variants known to be involved in intellectual disability, neurodevelopmental disorders, and dementia‐related neurodegenerative diseases. Through this analysis, we identified a heterozygous frameshift variant (c.967dupT, p.Tyr323Leufs*16 [NM_177559.3]) in exon 12 of the CSNK2A1 gene in the proband and did not find any additional causal genetic variants in other genes. We then examined exon 12 of the CSNK2A1 gene in the proband, the proband's father, and mother by Sanger sequencing. On Sanger sequencing, we found the c.967dupT, p.Tyr323Leufs*16 variant in CSNK2A1 in a heterozygous state in the proband and her mother (Figure 1d). This variant was not detected in the proband's father without symptoms (Figure 1e), indicating the variant co‐segregated with the phenotype in this family. This variant was not present in HGMD (Last Access Date:2023‐9‐15), ClinVar (Last Access Date:2022‐9‐15), or GnomAD (Last Access Date:2022‐9‐15), thus we considered it a novel molecular variant causing OCNDS. Bioinformatic analyses using Mutation Taster (http://www.mutationtaster.org) predicted that this variant was disease‐causing: truncated protein might cause nonsense‐mediated mRNA decay (NMD). This variant is designated as pathogenic following the criteria of the American College of Medical Genetics and Genomics (ACMG) (PVS1 + PS3 + PM2 + PP1 + PP3).
Meanwhile, WES was conducted using a triple diagnostic approach involving the proband, mother, and father. Through this analysis, we identified 14 variants present in heterozygous states in the proband but absent in both her father and mother. However, upon further examination of the 14 variants, we determined that they were unrelated to neurological diseases or neuronal dysfunctions. Moreover, targeted analysis for an ATN1 CAG expansion revealed that the number of ATN1 CAG repeats in the proband was within the normal range for both alleles. Finally, the DNA from the buccal swab of the proband's mother was also subjected to variant testing using PCR. The analyses revealed the presence of the p.Tyr323Leufs*16 variant in CSNK2A1 in a heterozygous state in the buccal tissue (ectodermal origin) from the proband's mother, indicating a lower likelihood of somatic mosaicism for this variant in the mother.
3.3. Transcription analysis
To analyze the mRNA expression affected by the variant c.967dupT in CSNK2A1, we performed reverse transcription polymerase chain reaction (RT‐PCR) using total RNA from leukocytes of the proband. Agarose gel electrophoresis of the long‐range RT‐PCR products revealed only the 1, 262 bp band expected in the patient, but no aberrant bands (Figure 1f). Sanger sequencing from both directions of the RT‐PCR products identified only canonical transcripts in the patient (Figure 1g). Therefore, no additional CSNK2A1 isoforms were found, making alternative splicing less likely. Moreover, the truncated transcript was not detected in the patient. We then examined mRNA levels in the proband's leukocytes by quantitative RT‐PCR. The results showed that the expression of CSNK2A1 transcripts was decreased by approximately 0.5‐fold in the patient compared to the three wild‐type controls when normalized to the β‐actin levels (Figure 1h). Thus, the truncated transcript of CSNK2A1 may be significantly reduced due to NMD.
3.4. Review of the genotype–phenotype relationship in identified CSNK2A1 variants
Sixteen articles reporting 47 OCNDS patients with germline CSNK2A1 pathogenic variants were included in the study (Akahira‐Azuma et al., 2018; Belnap et al., 2023; Chiu et al., 2018; Colavito et al., 2018; Duan et al., 2019; Jafari Khamirani et al., 2022; Martinez‐Monseny et al., 2020; Murakami et al., 2022; Nakashima et al., 2019; Okur et al., 2016; Owen et al., 2018; Trinh et al., 2017; Wafik et al., 2023; Wu et al., 2020, 2021; Xu et al., 2020). Together with the two patients reported in our study, 49 patients were included in the final analysis. The clinical and auxiliary characteristics of these patients were reviewed in Table 1, in which the most common features observed in a large enough fraction of the cases were listed. We found that individuals with CSNK2A1 variants had various congenital anomalies and neurodevelopmental disorders with different ranges of phenotypic spectra. The most commonly reported phenotypes of congenital anomalies were developmental delay (48/48, 100%), language deficits (35/46, 76.1%), intellectual disability (34/47, 72.3%), and dysmorphic facial features (34/49, 69.4%).
TABLE 1.
A concise review of the phenotypes of the reported OCNDS patients.
| ID | Variants | Sex | Age at diagnosis (years) | Developmental delay | Language deficits | Dysmorphic facial features | Intellectual disability | Behavioral issues | Eating disorders | Musculoskeletal disorders | Microcephaly | Abnormal brain MRI | Hypotonia | Seizures | Herniation | Inheritance pattern | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | p.R47Q | F | 6 | Yes | Yes | Yes | Yes | Yes | No | No | Yes | Yes | Yes | No | Yes | de novo | Okur et al. |
| 2 | p.K198R | F | 4.5 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No | de novo | Okur et al. |
| 3 | p.D175G | F | 4 | Yes | Yes | Yes | Yes | Yes | No | No | Yes | Yes | No | Yes | No | de novo | Okur et al. |
| 4 | c.824 + 2 T > C | F | 13 | Yes | No | No | Yes | Yes | Yes | Yes | No | No | Yes | No | Yes | de novo | Okur et al. |
| 5 | p.Y50S | F | 2 | Yes | No | Yes | NA | No | Yes | Yes | Yes | No | Yes | Yes | No | de novo | Okur et al. |
| 6 | p.K198R | M | 5 | Yes | Yes | Yes | No | No | No | No | No | NA | No | No | No | de novo | Chiu et al. |
| 7 | p.E27K | M | 14 | Yes | No | No | Yes | Yes | Yes | Yes | No | No | Yes | No | Yes | de novo | Chiu et al. |
| 8 | p.M1V | F | 2.5 | Yes | Yes | Yes | No | Yes | No | Yes | No | No | No | No | No | de novo | Chiu et al. |
| 9 | p.P231R | M | 4 | Yes | Yes | Yes | No | Yes | Yes | No | Yes | NA | Yes | No | No | de novo | Chiu et al. |
| 10 | p.V73E | M | 8 | Yes | Yes | No | No | No | Yes | No | Yes | No | No | No | No | de novo | Chiu et al. |
| 11 | p.R47Q | F | 8 | Yes | Yes | Yes | No | No | Yes | Yes | Yes | Yes | Yes | Yes | No | de novo | Chiu et al. |
| 12 | p.R312Q | M | 14 | Yes | Yes | Yes | No | No | No | No | No | NA | No | No | No | de novo | Chiu et al. |
| 13 | p.S51R | M | 4 | Yes | Yes | Yes | No | Yes | Yes | Yes | No | NA | Yes | Yes | No | de novo | Chiu et al. |
| 14 | p.R47Q | M | 6.1 | Yes | Yes | Yes | Yes | No | Yes | Yes | No | No | No | No | Yes | de novo | Owen et al. |
| 15 | p.S51N | F | 11.1 | Yes | Yes | Yes | Yes | No | No | Yes | No | NA | No | No | No | de novo | Owen et al. |
| 16 | p.R80H | M | 10.7 | Yes | Yes | Yes | Yes | Yes | No | Yes | No | No | Yes | No | No | de novo | Owen et al. |
| 17 | p.I174M | M | 10.9 | Yes | Yes | No | Yes | Yes | Yes | No | No | NA | Yes | No | No | de novo | Owen et al. |
| 18 | p.R191Q | F | 10 | Yes | Yes | No | Yes | Yes | Yes | No | Yes | NA | Yes | No | No | de novo | Owen et al. |
| 19 | p.F197I | F | 8 | Yes | Yes | Yes | Yes | No | No | No | No | No | No | No | No | de novo | Owen et al. |
| 20 | p.K198R | F | 7.3 | Yes | Yes | Yes | Yes | Yes | No | No | No | No | Yes | Yes | No | de novo | Owen et al. |
| 21 | p.R312W | M | 10.9 | Yes | Yes | No | Yes | No | No | No | Yes | No | No | No | No | de novo | Owen et al. |
| 22 | p.K198R | M | 18.3 | Yes | NA | Yes | Yes | No | No | No | No | NA | Yes | Yes | No | de novo | Owen et al. |
| 23 | p.K198R | F | 18.7 | Yes | Yes | No | Yes | No | No | No | Yes | Yes | Yes | No | No | de novo | Owen et al. |
| 24 | p.R312W | F | 6 | Yes | No | Yes | Yes | Yes | No | Yes | No | NA | Yes | No | No | de novo | Owen et al. |
| 25 | p.K198R | F | 15 | Yes | Yes | Yes | Yes | No | Yes | Yes | No | Yes | Yes | Yes | No | de novo | Nakashima et al. |
| 26 | p.R191* | M | 1.7 | Yes | No | No | NA | NA | NA | No | Yes | No | Yes | Yes | No | de novo | Nakashima et al. |
| 27 | p.D156H | M | 7 | Yes | Yes | Yes | Yes | Yes | No | No | Yes | Yes | No | No | No | de novo | Trinh et al. |
| 28 | c.1061‐1G > C | M | 1 | Yes | No | No | No | Yes | No | No | No | Yes | Yes | No | No | de novo | Colavito et al. |
| 29 | p.K198R | M | 8 | Yes | Yes | Yes | Yes | Yes | No | No | No | Yes | Yes | No | No | de novo | Akahira‐Azuma et al. |
| 30 | p.Y50C | F | 5.8 | Yes | Yes | Yes | No | Yes | Yes | No | No | Yes | No | No | No | de novo | Martinez‐Monseny et al. |
| 31 | p.K198R | M | 6 | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | No | No | No | AD | Xu et al. |
| 32 | p.D175G | M | 1 | Yes | No | Yes | No | No | Yes | Yes | No | No | Yes | No | No | de novo | Duan et al. |
| 33 | p.Y50C | M | 8 | Yes | NA | Yes | Yes | NA | Yes | No | No | Yes | No | Yes | No | de novo | Wu et al. |
| 34 | p.R21Q | F | 8 | Yes | Yes | Yes | Yes | Yes | No | Yes | No | No | Yes | Yes | No | de novo | Khamirani et al. |
| 35 | p.R80C | F | 2 | Yes | Yes | Yes | Yes | NA | No | Yes | Yes | Yes | No | No | No | de novo | Wu et al. |
| 36 | p.H160R | F | 3 | Yes | No | Yes | Yes | No | No | Yes | No | Yes | No | Yes | No | de novo | Wu et al. |
| 37 | p.R107* | M | 5 | Yes | No | Yes | No | Yes | No | No | No | No | No | No | No | de novo | Murakami et al. |
| 38 | p.K198R | F | NA | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | NA | Yes | No | NA | AD | Belnap et al. |
| 39 | p.K198R | F | NA | Yes | Yes | Yes | Yes | Yes | No | No | No | NA | Yes | No | NA | AD | Belnap et al. |
| 40 | p.K198R | F | NA | Yes | Yes | No | Yes | No | No | Yes | NA | NA | NA | Yes | NA | AD | Belnap et al. |
| 41 | p.K198R | F | NA | Yes | Yes | Yes | Yes | No | Yes | Yes | No | NA | No | No | NA | AD | Belnap et al. |
| 42 | p.K198R | M | NA | Yes | Yes | Yes | Yes | No | No | No | No | NA | No | No | NA | AD | Belnap et al. |
| 43 | p.E27K | M | NA | Yes | Yes | No | Yes | Yes | Yes | Yes | NA | NA | No | No | NA | AD | Belnap et al. |
| 44 | p.E27K | M | NA | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | NA | Yes | No | NA | AD | Belnap et al. |
| 45 | p.E27K | F | NA | Yes | NA | No | Yes | Yes | NA | Yes | NA | NA | NA | No | NA | AD | Belnap et al. |
| 46 | p.G48S | M | 7 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No | No | No | de novo | Wafik et al. |
| 47 | p.H29Cfs*9 | F | 9 | Yes | No | No | No | No | No | No | No | No | No | Yes | No | de novo | Wafik et al. |
| 48 | p.Y323Lfs*16 | F | 31 | NA | Yes | Yes | Yes | Yes | Yes | Yes | No | No | No | Yes | No | AD | Present study |
| 49 | p.Y323Lfs*16 | F | 57 | NA | No | No | No | No | No | No | No | NA | No | No | Yes | AD | Present study |
Note: CSNK2A1 GenBank version number: NM_177559.3.
Abbreviations: AD: autosomal dominant; F, female; M, male; NA, not available.
Our investigation revealed only seven null variants (p.M1V, c.824+2T>C, p.R191*, c.1061−1G>C, p.R107*, p.H29Cfs*9, p.Y323Lfs*16) among eight patients (8/49, 16.3%), while the remaining patients (41/49, 83.7%) exhibited missense variants. Among the missense variants, the recurring p.Lys198Arg is the most frequently reported pathogenic variant of OCNDS (13/41, 31.7%), followed by p.E27K (4/41, 9.8%) and p.R47Q (3/41, 7.3%). The clinical and ancillary characteristics of the eight OCNDS patients with null variants were compared to 41 OCNDS patients with missense variants. We observed that individuals with CSNK2A1 null variants are less inclined to exhibit significant developmental and neurological symptoms when compared to those harboring missense variants. They display a significantly reduced frequency of symptoms associated with language deficits, dysmorphic facial features, or intellectual disability (Figure 2), which are substantial contributors to the debilitating and malforming characteristics of OCNDS. As a result, they may present an overall milder phenotype.
FIGURE 2.
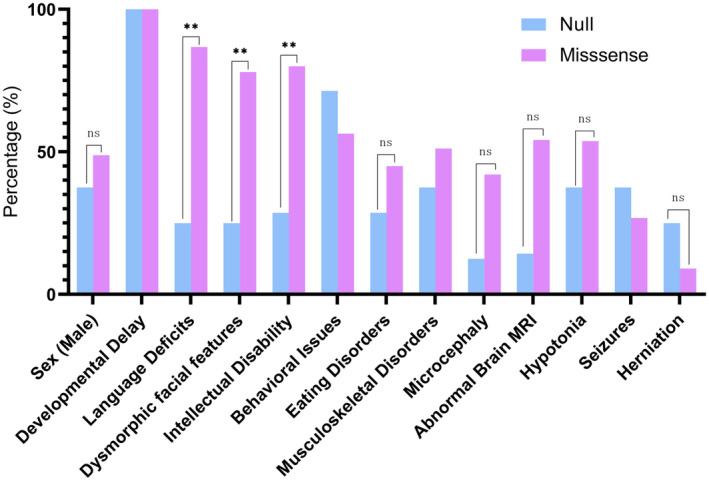
The comparison of clinical and auxiliary features between OCNDS patients with CSNK2A1 missense variants and OCNDS patients with CSNK2A1 null variants. *p < 0.05, **p < 0.01, ***p < 0.001, ns: not significant.
4. DISCUSSION
In this study, we present a novel frameshift variant, p.Tyr323Leufs*16, in an OCNDS family. The proband exhibited seizures, language impairment, and limb weakness. Her mother displayed postnatal hernias, splenomegaly, and susceptibility to infections, without significant developmental or intellectual impairments. In our review of previously reported OCNDS cases, individuals with CSNK2A1 null variants exhibited a milder phenotype compared to those with missense variants, showing reduced frequency of symptoms like language deficits, dysmorphic facial features, or intellectual disability.
The proband and her mother both exhibit a mild phenotype. Brain imaging revealed no structural abnormalities, and their neurodevelopment is within the normal range. This mirrors the phenotype of a patient with the p.H29Cfs*9 pathogenic variant in CSNK2A1, who displayed a well‐controlled generalized seizure disorder with no neurodevelopmental concerns, brain MRI abnormalities, intellectual disability, or facial dysmorphisms (Wafik et al., 2023). The p.M1V variant, affecting the start codon and predicted to cause a loss of protein from Position 1 to 137, where the next in‐frame start codon is located, was classified as a null variant in this study (Chiu et al., 2018). Apart from this patient, individuals with CSNK2A1 null variants showed a clinical profile similar to our patient's presentations with a generally mild phenotype. This observation was also acknowledged by Dr. Volkan Okur (Wafik et al., 2023).
No clear genotype–phenotype correlation has been previously established for the possible association between the nature of the pathogenic variants and the severity of OCNDS. The null variants reported to date include two splicing variants, two frameshift variants, two nonsense variants, and a start‐loss variant. Our transcription analysis suggests that mutant transcripts carrying these variants are likely subjected to NMD, indicating a haploinsufficiency mechanism. However, it is not yet known whether there exists a disparity in the pathogenic mechanisms between missense variants and frameshift variants. Recently, reduced CK2α kinase activity has been demonstrated in missense variants associated with OCNDS (Dominguez et al., 2021). Given missense variants' potential for more severe symptoms, their underlying pathogenic mechanism might be dominant negative or gain‐of‐function rather than haploinsufficiency (Dominguez et al., 2021). This parallels differences observed between missense and truncating variants in Rett syndrome caused by mutations in the MECP2 gene, with patients harboring missense variants being more likely to develop scoliosis than patients with truncating mutations (Amir et al., 2000). Further large‐scale studies are required to deepen our understanding of the clinical and genetic spectrum in OCNDS.
AUTHOR CONTRIBUTIONS
Haitian Nan and Liyong Wu designed and conceptualized the study. Min Chu, Jing Zhang, Deming Jiang, and Yihao Wang provided the patients of the study. Haitian Nan performed the genetic study. Haitian Nan and Min Chu performed a literature review. Haitian Nan and Liyong Wu drafted and revised the manuscript. The authors have read and approved the final manuscript.
FUNDING INFORMATION
This work was supported by grants from the National Natural Science Foundation of China [no. 82201573].
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
ETHICS STATEMENT
Written informed consent for publication was obtained from the guardian of the patient.
Supporting information
Table S1.
Table S2.
ACKNOWLEDGMENTS
The authors appreciate the patients for their participation in this study.
Nan, H. , Chu, M. , Zhang, J. , Jiang, D. , Wang, Y. , & Wu, L. (2024). Okur‐Chung neurodevelopmental syndrome: Implications for phenotype and genotype expansion. Molecular Genetics & Genomic Medicine, 12, e2398. 10.1002/mgg3.2398
DATA AVAILABILITY STATEMENT
All data relevant to the study are included in the article or uploaded as supplementary information.
REFERENCES
- Akahira‐Azuma, M. , Tsurusaki, Y. , Enomoto, Y. , Mitsui, J. , & Kurosawa, K. (2018). Refining the clinical phenotype of Okur‐Chung neurodevelopmental syndrome. Human Genome Variation, 5, 18011. 10.1038/hgv.2018.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir, R. E. , Van den Veyver, I. B. , Schultz, R. , Malicki, D. M. , Tran, C. Q. , Dahle, E. J. , Philippi, A. , Timar, L. , Percy, A. K. , Motil, K. J. , Lichtarge, O. , Smith, E. O. , Glaze, D. G. , & Zoghbi, H. Y. (2000, May). Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Annals of Neurology, 47(5), 670–679. [PubMed] [Google Scholar]
- Belnap, N. , Price‐Smith, A. , Ramsey, K. , Leka, K. , Abraham, A. , Lieberman, E. , Hassett, K. , Potu, S. , Rudy, N. , Smith, K. , Mikhail, F. M. , Monaghan, K. G. , Hendershot, A. , Mourmans, J. , Descartes, M. , Huentelman, M. J. , Sills, J. , Rangasamy, S. , & Narayanan, V. (2023, July 25). Inherited CSNK2A1 variants in families with Okur‐Chung neurodevelopmental syndrome. Clinical Genetics, 104, 607–609. 10.1111/cge.14408 [DOI] [PubMed] [Google Scholar]
- Chiu, A. T. G. , Pei, S. L. C. , Mak, C. C. Y. , Leung, G. K. C. , Yu, M. H. C. , Lee, S. L. , Vreeburg, M. , Pfundt, R. , van der Burgt, I. , Kleefstra, T. , Frederic, T. M. T. , Nambot, S. , Faivre, L. , Bruel, A. L. , Rossi, M. , Isidor, B. , Küry, S. , Cogne, B. , Besnard, T. , … Chung, B. H. Y. (2018, April). Okur‐Chung neurodevelopmental syndrome: Eight additional cases with implications on phenotype and genotype expansion. Clinical Genetics, 93(4), 880–890. 10.1111/cge.13196 [DOI] [PubMed] [Google Scholar]
- Colavito, D. , Del Giudice, E. , Ceccato, C. , Dalle Carbonare, M. , Leon, A. , & Suppiej, A. (2018, June). Are CSNK2A1 gene mutations associated with retinal dystrophy? Report of a patient carrier of a novel de novo splice site mutation. Journal of Human Genetics, 63(6), 779–781. 10.1038/s10038-018-0434-y [DOI] [PubMed] [Google Scholar]
- Dominguez, I. , Cruz‐Gamero, J. M. , Corasolla, V. , Dacher, N. , Rangasamy, S. , Urbani, A. , Narayanan, V. , & Rebholz, H. (2021, July). Okur‐Chung neurodevelopmental syndrome‐linked CK2alpha variants have reduced kinase activity. Human Genetics, 140(7), 1077–1096. 10.1007/s00439-021-02280-5 [DOI] [PubMed] [Google Scholar]
- Duan, H. L. , Peng, J. , Pang, N. , Chen, S. M. , Xiong, J. , Guang, S. Q. , & Yin, F. [(2019, May 2). A case of Okur‐Chung syndrome caused by CSNK2A1 gene variation and review of literature]. Zhonghua Er Ke Za Zhi, 57(5), 368–372. 10.3760/cma.j.issn.0578-1310.2019.05.010 [DOI] [PubMed] [Google Scholar]
- Jafari Khamirani, H. , Zoghi, S. , Motealleh, A. , Dianatpour, M. , Tabei, S. M. B. , Mohammadi, S. , & Dastgheib, S. A. (2022, December). Clinical features of Okur‐Chung neurodevelopmental syndrome: Case report and literature review. Molecular Syndromology, 13(5), 381–388. 10.1159/000522353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Monseny, A. F. , Casas‐Alba, D. , Arjona, C. , Bolasell, M. , Casano, P. , Muchart, J. , Ramos, F. , Martorell, L. , Palau, F. , García‐Alix, A. , & Serrano, M. (2020, January). Okur‐Chung neurodevelopmental syndrome in a patient from Spain. American Journal of Medical Genetics. Part A, 182(1), 20–24. 10.1002/ajmg.a.61405 [DOI] [PubMed] [Google Scholar]
- Murakami, H. , Uehara, T. , Enomoto, Y. , Nishimura, N. , Kumaki, T. , Kuroda, Y. , Asano, M. , Aida, N. , Kosaki, K. , & Kurosawa, K. (2022, February). Persistent hyperplastic primary vitreous with microphthalmia and coloboma in a patient with Okur‐Chung neurodevelopmental syndrome. Molecular Syndromology, 13(1), 75–79. 10.1159/000517977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima, M. , Tohyama, J. , Nakagawa, E. , Watanabe, Y. , Siew, C.’. G. , Kwong, C. S. , Yamoto, K. , Hiraide, T. , Fukuda, T. , Kaname, T. , Nakabayashi, K. , Hata, K. , Ogata, T. , Saitsu, H. , & Matsumoto, N. (2019, April). Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. Journal of Human Genetics, 64(4), 313–322. 10.1038/s10038-018-0559-z [DOI] [PubMed] [Google Scholar]
- Niefind, K. , Guerra, B. , Ermakowa, I. , & Issinger, O. G. (2001, October 1). Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. The EMBO Journal, 20(19), 5320–5331. 10.1093/emboj/20.19.5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okur, V. , Cho, M. T. , Henderson, L. , Retterer, K. , Schneider, M. , Sattler, S. , Niyazov, D. , Azage, M. , Smith, S. , Picker, J. , Lincoln, S. , Tarnopolsky, M. , Brady, L. , Bjornsson, H. T. , Applegate, C. , Dameron, A. , Willaert, R. , Baskin, B. , Juusola, J. , & Chung, W. K. (2016, July). De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Human Genetics, 135(7), 699–705. 10.1007/s00439-016-1661-y [DOI] [PubMed] [Google Scholar]
- Owen, C. I. , Bowden, R. , Parker, M. J. , Patterson, J. , Patterson, J. , Price, S. , Sarkar, A. , Castle, B. , Deshpande, C. , Splitt, M. , Ghali, N. , Dean, J. , Green, A. J. , Crosby, C. , Deciphering Developmental Disorders Study , & Tatton‐Brown, K. (2018, May). Extending the phenotype associated with the CSNK2A1‐related Okur‐Chung syndrome‐A clinical study of 11 individuals. American Journal of Medical Genetics. Part A, 176(5), 1108–1114. 10.1002/ajmg.a.38610 [DOI] [PubMed] [Google Scholar]
- Trinh, J. , Huning, I. , Budler, N. , Hingst, V. , Lohmann, K. , & Gillessen‐Kaesbach, G. (2017, November). A novel de novo mutation in CSNK2A1: Reinforcing the link to neurodevelopmental abnormalities and dysmorphic features. Journal of Human Genetics, 62(11), 1005–1006. 10.1038/jhg.2017.73 [DOI] [PubMed] [Google Scholar]
- Wafik, M. , Kuoppamaa, H. , Hirani, P. , Hignett, J. , Lillis, S. , Lascelles, K. , Sardesai, S. , Gomez, K. , & Holder‐Espinasse, M. (2023, July 1). Two novel CSNK2A1 variants associated with mild Okur‐Chung neurodevelopmental syndrome phenotype. Clinical Dysmorphology, 32(3), 116–123. 10.1097/MCD.0000000000000456 [DOI] [PubMed] [Google Scholar]
- Wu, R. , Tang, W. , Liang, L. , Li, X. , Ouyang, N. , & Meng, Z. [(2020, June 10). Identification of a novel de novo variant of CSNK2A1 gene in a boy with Okur‐Chung neurodevelopmental syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 37(6), 641–644. 10.3760/cma.j.issn.1003-9406.2020.06.011 [DOI] [PubMed] [Google Scholar]
- Wu, R. H. , Tang, W. T. , Qiu, K. Y. , Li, X. J. , Tang, D. X. , Meng, Z. , & He, Z. W. (2021, May). Identification of novel CSNK2A1 variants and the genotype‐phenotype relationship in patients with Okur‐Chung neurodevelopmental syndrome: A case report and systematic literature review. The Journal of International Medical Research, 49(5), 3000605211017063. 10.1177/03000605211017063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S. , Lian, Q. , Wu, J. , Li, L. , & Song, J. (2020, August 3). Dual molecular diagnosis of tricho‐rhino‐phalangeal syndrome type I and Okur‐Chung neurodevelopmental syndrome in one Chinese patient: A case report. BMC Medical Genetics, 21(1), 158. 10.1186/s12881-020-01096-w [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Table S2.
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.