

Abstract
Macrophages are a major source of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), which are expressed during conditions of inflammation, infection, or injury. We identified an activity secreted by a macrophage tumor cell line that negatively regulates bacterial lipopolysaccharide (LPS)-induced expression of TNF-α. This activity, termed TNF-α-inhibiting factor (TIF), suppressed the induction of TNF-α expression in macrophages, whereas induction of three other proinflammatory cytokines (interleukin-1β [IL-1β], IL-6, and monocyte chemoattractant protein 1) was accelerated or enhanced. A similar or identical inhibitory activity was secreted by IC-21 macrophages following LPS stimulation. Inhibition of TNF-α expression by macrophage conditioned medium was associated with selective induction of the NF-κB p50 subunit. Hyperinduction of p50 occurred with delayed kinetics in LPS-stimulated macrophages but not in fibroblasts. Overexpression of p50 blocked LPS-induced transcription from a TNF-α promoter reporter construct, showing that this transcription factor is an inhibitor of the TNF-α gene. Repression of the TNF-α promoter by TIF required a distal region that includes three NF-κB binding sites with preferential affinity for p50 homodimers. Thus, the selective repression of the TNF-α promoter by TIF may be explained by the specific binding of inhibitory p50 homodimers. We propose that TIF serves as a negative autocrine signal to attenuate TNF-α expression in activated macrophages. TIF is distinct from the known TNF-α-inhibiting factors IL-4, IL-10, and transforming growth factor β and may represent a novel cytokine.
Proinflammatory cytokines such as interleukin-1 (IL-1), IL-6, and tumor necrosis factor alpha (TNF-α) regulate systemic responses to microbial infection or tissue injury (2, 49). These signals stimulate immune functions and induce expression of acute phase reactants in the liver, among other effects. Activated macrophages are a major source of cytokines and produce these and other inflammatory mediators upon exposure to viruses or bacterial endotoxins (e.g., lipopolysaccharide [LPS]) and priming factors such as gamma interferon. Induction of cytokine gene expression by LPS occurs primarily at the level of transcription and involves the action of several transcription factors, including members of the NF-κB/rel, C/EBP, Ets, and AP-1 protein families (reviewed in reference 48).
Although induction of proinflammatory cytokine expression is critical for a rapid response to tissue trauma or infection, prolonged or deregulated production of these factors may have serious adverse consequences. TNF-α, for example, can be highly cytotoxic, and inappropriate expression of this cytokine has been linked to a variety of serious pathological conditions, including septic shock, acute inflammation, cachexia (49), autoimmune disease (42), and neuronal degeneration associated with Alzheimer’s syndrome (33). Indeed, sepsis is estimated to cause 175,000 deaths per year in the United States alone (47). In view of its potentially injurious effects, production of TNF-α must be stringently controlled by negative as well as positive mechanisms. One factor that inhibits TNF-α expression is IL-10, an anti-inflammatory cytokine produced by LPS-activated macrophages that suppresses LPS-induced expression of several proinflammatory cytokines (14, 18, 53). IL-4, transforming growth factor β (TGF-β), prostaglandin E2 (PGE2), and glucocorticoids also possess anti-inflammatory activities and inhibit production of TNF-α and other cytokines (5, 23, 38, 41, 46).
Kinetic studies of cytokine mRNA accumulation in cultured macrophages stimulated with LPS show that induction is often transitory, despite the continuous presence of LPS in the culture medium. Peak levels of TNF-α transcripts occur a few hours after stimulation, after which they rapidly decrease and return to near baseline by 8 to 12 h (Fig. 1). In principle, this strict attenuation of TNF-α expression could be controlled either by cell-autonomous mechanisms or by production of negative feedback signals such as IL-10. However, little is known about the specific regulatory pathways that down-regulate TNF-α gene transcription after its activation by LPS.
FIG. 1.

Identification of TNF-α-inhibitory activity in CM from P388D1(IL1) macrophages. (A) Analysis of TNF-α, IL-6, MCP-1, and IL-1β RNA expression in IC-21 macrophages. IC-21 cells were pretreated with P388D1(IL1) CM (concentrated by ultrafiltration) or unconditioned medium (UCM) for 16 h and induced with LPS (10 μg/ml), and RNA was harvested over an 8-h time course. One microgram of total RNA from each time point was blotted onto a nylon membrane (slot blot), and duplicate blots were hybridized with the indicated cytokine probes. Cytokine RNA expression was quantitated with a PhosphorImager. Cytokine inductions were normalized to actin mRNA and are expressed as percent maximal induction in control (UCM-treated) cells. (B) Effect of CM on TNF-α expression in murine bone marrow (BM) and peritoneal macrophages. Primary macrophages were cultured for 3 to 4 days and treated for 16 h with CM or UCM. The cells were then stimulated with LPS, and RNA was prepared at 0, 3, and 6 h as described for panel A. TNF-α expression was analyzed by slot blotting and quantitated (normalized to actin) with a radioanalytical scanner.
Suppression of TNF-α expression is also associated with the phenomenon of LPS tolerance. Macrophages may be tolerized, or desensitized, to the effects of LPS by prior exposure to suboptimal amounts of this agent (56). Cells treated in this way are unable to produce TNF-α in response to subsequent high doses of LPS. Similarly, mice can be protected against the lethal effects of LPS, which are mainly mediated by TNF-α, by prior injection of sublethal doses of endotoxin (56). While LPS tolerization is believed to occur at the level of the macrophage in vivo, the molecular basis for tolerance to LPS has not been established.
These observations suggest the existence of potent and specific mechanisms to attenuate the expression of proinflammatory cytokines. Here we identify an activity secreted by macrophages that inhibits LPS-induced expression of TNF-α. We propose that this activity functions as a negative feedback signal to attenuate the transcription of TNF-α and provide evidence that NF-κB p50 is a downstream effector of this inhibitory pathway.
MATERIALS AND METHODS
Cells and cell culture.
P388D1(IL1) (ATCC TIB 63) (13), IC-21 (ATCC TIB 186) (51), and ANA-1 (12) are murine macrophage cell lines. L cells (ATCC CCL 1.3) are a murine fibroblastic cell line. P388D1(IL1) and IC-21 cells were grown in RPMI 1640 (BioWhittaker) supplemented with 10% FetalClone I serum (FCS; HyClone). ANA-1 and L cells were grown in Dulbecco modified Eagle medium (DMEM; Difco/Life Technologies) supplemented with 10% fetal bovine serum (FBS; HyClone). Escherichia coli LPS (serotype O26:B6) was obtained from Sigma. Biologically active recombinant cytokines and growth factors were obtained from the NCI Preclinical Repository, Frederick, Md.
Primary macrophages were isolated from adult C57/BL6 mice. Peritoneal macrophages were prepared by abdominal lavage with growth medium (RPMI 1640). After plating, adherent cells were used for further manipulations. Bone marrow-derived macrophages were obtained by growing bone marrow cells for 48 h in DMEM–10% FBS containing 20% L-cell conditioned medium (CM), a source of macrophage colony-stimulating factor, transferring the nonadherent cells to fresh plates, and culturing them for 5 to 7 days in DMEM–10% FBS–20% L-cell CM. The resulting adherent cells were used for LPS stimulation experiments.
Preparation and fractionation of macrophage CM.
CM was collected from confluent P388D1(IL1) cells grown for 3 to 5 days in RPMI 1640 with 5% FCS. CM was concentrated either in an Amicon stirred cell concentrator using a 30,000-molecular-weight cutoff membrane and then filtered with a 0.45-μm-pore-size syringe filter (Nalgene) or by using a Centriprep 30 centrifugal concentrator (Grace). CM was concentrated 10-fold and added to cells at a 2× dose (e.g., 40 ml of CM was concentrated to 4 ml and added to a 15-cm-diameter plate of cells containing 20 ml of fresh medium). As a control in each experiment, unconditioned medium was concentrated 10-fold and added to cells at a 2× dose. LPS+ CM from IC-21 cells (Fig. 2) was prepared similarly, except that the medium was conditioned for 16 h in the absence (control) or presence of 20 μg of LPS per ml.
FIG. 2.

A TNF-α-inhibiting activity is secreted by LPS-stimulated IC-21 macrophages. (A) CM was prepared from control and LPS-treated (10 μg/ml, 16 h) IC-21 cells and concentrated by ultrafiltration. The CM preparations were then added to naive IC-21 cells together with 20 μg of LPS per ml, and RNA was harvested over an 8-h time course. Parallel inductions were performed on cells treated with LPS alone, pretreated with P388D1(IL1) CM for 16 h, or treated simultaneously with P388D1(IL1) CM and LPS (see key at right). One microgram of total RNA from each time point was analyzed by slot blot hybridization using the indicated cytokine probes. Hybridization signals were quantitated by scanning (normalized to actin) and are expressed as a percentage of the maximal induction observed in untreated cells (no CM pretreatment). TNF-α expression was determined in two independent experiments (top panels). (B) Cytokine expression in IC-21 macrophages treated with LPS− CM. The culture medium was removed from naive IC-21 macrophages and replaced with LPS− CM or control CM (see Materials and Methods). After 16 h, the cells were treated with LPS and cytokine mRNA levels were analyzed as described for Fig. 1A.
LPS− CM was prepared by addition of 1 μg of LPS per ml to confluent plates of IC-21 cells. After 20 min at 37°C, the medium was discarded and the cells were washed twice with RPMI 1640 prewarmed to 37°C. RPMI 1640 supplemented with 10% FCS was then added, and the cells were incubated for 60 min at 37°C. CM was collected and used immediately or stored at 4°C. Control CM was prepared similarly except that no LPS was added. LPS− CM was applied to recipient cells at a 100% dose, after removal of the culture medium. LPS− CM was used in the experiment represented in Fig. 2B and in all subsequent CM experiments.
Quantitation of cytokines in CM.
Cytokine levels in CM (Table 1) were determined by the Lymphokine Testing Laboratory, Clinical Services Program, SAIC Frederick, Frederick, Md., using enzyme-linked immunosorbent assay (ELISA) kits for mouse IL-4, IL-6, and IL-10 (Endogen), mouse TNF-α (Genzyme), and human TGF-β1 (R&D Systems).
TABLE 1.
Cytokine levels in conditioned and control media from macrophage cell linesa
| Medium | Concn (pg/ml)
|
||
|---|---|---|---|
| IL-6 | TNF-α | TGF-β1 | |
| UCM | 0 | 0 | <32 |
| P388D1(IL1) CM | 6,100 | 3,446 | 4,424 |
| IC-21 control CM | 404 | 386 | 616 |
| IC-21 LPS+ CM | >600,000 | 6,092 | 802 |
Cytokine levels were determined by ELISA in unconditioned medium (UCM), P388D1(IL1) CM, IC-21 CM (control CM), and CM from LPS-stimulated IC-21 cells (LPS+ CM), all of which had been concentrated and partially purified by ultrafiltration. The lower limits of detection for IL-4 and TGF-β1 were 5.8 and 32 pg/ml, respectively. In all media, the IL-4 level was below the limit of detection and IL-10 was not detected.
Nuclear extracts.
Nuclear extracts were prepared by a detergent lysis procedure. Cells were scraped, washed once with phosphate-buffered saline, resuspended in lysis buffer (buffer A; 20 mM HEPES [pH 7.9], 1 mM EDTA, 10 mM NaCl, 1 mM dithiothreitol, 0.1% [vol/vol] Nonidet P-40, 0.4 mM phenylmethylsulfonyl fluoride, 0.1 μg of leupeptin per ml, 5 μg of antipain per ml), and incubated on ice for 10 min. Nuclei were pelleted by centrifugation at 3,500 × g for 10 min. Proteins were extracted from nuclei by incubation with high-salt buffer (buffer C; 420 mM NaCl, 1 mM EDTA, 20 mM HEPES [pH 7.9], 25% glycerol, 1 mM dithiothreitol, 0.4 mM phenylmethylsulfonyl fluoride, 0.1 μg of leupeptin per ml, 5 μg of antipain per ml) at 4°C for 20 min with vigorous shaking. Nuclear debris was pelleted by centrifugation at 14,000 × g for 5 min, and the supernatant was collected and stored at −70°C.
EMSA.
The following double-stranded oligonucleotides were used as electrophoretic mobility shift assay (EMSA) probes:
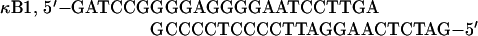 |
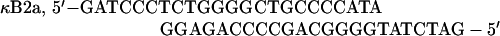 |
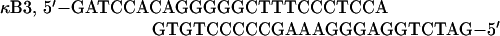 |
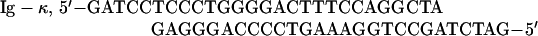 |
Ig-κ is the κB element from the immunoglobulin κ light-chain gene.
The probes were labeled with [32P]dCTP and Klenow polymerase. DNA binding reactions were performed for 20 min at room temperature in a 25-μl reaction mixture containing 100 mM NaCl, 10 mM HEPES (pH 7.5), 1 mM EDTA, 1 mM EGTA, 6% (vol/vol) glycerol, 0.06% bromophenol blue, 0.25 μg of bovine serum albumin, 1 μg of poly(dI-dC), 32P-labeled probe, and 4 μg of nuclear extract. NF-κB–DNA complexes were separated from free probe by electrophoresis through 6% polyacrylamide gels in 1× TBE (90 mM Tris base, 90 mM boric acid, 0.5 mM EDTA) at 160 V for 2 h (20, 21). Gels were dried and exposed to Kodak XAR film. For antibody supershift assays, 1 μl of rabbit antiserum was incubated with the protein extract on ice for 20 min prior to addition to the binding reaction mixture. Antibodies specific for NF-κB p50 and p65 were kindly provided by N. Rice (39, 40).
Western blotting.
Nuclear extracts were prepared by the detergent lysis method described above. Samples were mixed with sample buffer (30), heated to 100°C for 10 min, and loaded on precast sodium dodecyl sulfate–12% polyacrylamide gels (Novex). Proteins were transferred to Immobilon membranes (Millipore) and probed with antibodies specific for either p50 or p65 (39, 40). The blots were developed by using the Amersham enhanced chemiluminescence detection system.
RNA isolation and Northern or slot blot analysis.
Total RNA was isolated from cells as described by Kingston et al. (26). For Northern blot assays, 10 μg of RNA was analyzed. One microgram of RNA was used for slot blot assays, and separate filters were prepared for each probe. Slot blot signals were quantitated with an Ambis Radioanalytic Scanner or a Molecular Dynamics PhosphorImager and were normalized to actin expression. Hybridization probes were labeled by using a random priming kit (United States Biochemical). The IL-6, monocyte chemoattractant protein 1 (MCP-1), and IL-1β probes have been described elsewhere (8). The TNF-α probe was a 1.3-kb BamHI-PstI fragment from a murine cDNA clone (10). The β-actin probe was a 2-kb HindIII fragment excised from plasmid β2000 (11).
Plasmid constructs.
The TNF-Luc reporter plasmid was constructed by inserting a BamHI-HindIII fragment containing the murine TNF-α promoter (from plasmid pMAC 1260 [45]) into the luciferase vector pXP1 (34), which had been digested with BamHI and HindIII. TNF-Luc contains sequences extending to nucleotide (nt) −1260 of the TNF-α promoter. The BamHI-HindIII insert fragment was also ligated into a pBlueScript (Stratagene) vector digested with the same enzymes to generate the construct pBS-TNF-1. 5′ deletion mutants of the TNF-α promoter were created by PCR amplification using pBS-TNF-1 as the template, the BlueScript T3 sequencing primer as the 3′ amplimer, and the following oligonucleotides as 5′ primers: −646 (5′-GGTCAGGATCCCTCTGGGGCTGCCCCATA-3′), −527 (5′-GGTCAGGATCCACAGGGGGCTTTCCCTCC-3′), −527m (5′-GGTCAGGATCCACAacacaCTTTCCCTCC-3′), −514 (5′-GGTCAGGATCCTCCTCAATATCATGTCT-3′), and −210 (5′-GGTCAGGATCCTGCCTGGGTTCCCACTTT-3′). The PCR products were digested with BamHI and HindIII, gel purified, and ligated into pXP1. Candidate clones were sequenced to verify the 5′ endpoints.
Artificial NF-κB promoter constructs.
The double-stranded NF-κB oligonucleotides that were used for EMSA analysis were phosphorylated and self-ligated to generate oligomers. The oligomers were then digested with BamHI and BglII to select for molecules ligated in the head-to-tail orientation (direct repeats), and the products were separated by 10% polyacrylamide gel electrophoresis. The appropriate concatemers were eluted and ligated into the BamHI of TK-Luc, upstream of the thymidine kinase (TK) promoter. TK-Luc is based on the luciferase vector pXP2, into which a 5′ truncated TK promoter (−81) was inserted (34). Candidate clones were screened by restriction analysis and verified by sequencing.
Expression plasmids for p50 and p65 (Rc/CMV-p50 and Rc/CMV-p65), which are derived from Rc/CMV (Invitrogen), were kindly provided by N. Rice and A. Israël. pRSV-βgal (6) is a control vector expressing β-galactosidase. pGL2 promoter (Promega), referred to as pGL2-Luc in this study, is a control luciferase reporter driven by the simian virus 40 early promoter.
Transfection assays.
Nonadherent ANA-1 cells were transfected by using DEAE-dextran sulfate as follows. Cells were transfected in batch (2 × 106 cells/60-mm-diameter dish for each time point) and then divided for CM and LPS treatments. This procedure eliminates any differences in plate-to-plate transfection efficiency. Plasmid DNAs were prepared by a polyethylene glycol precipitation method or by using a commercial kit (Qiagen). The cells and DNA were incubated with 0.5 mg of DEAE-dextran per ml in DMEM–50 mM Tris (pH 8.0) for 75 min at 37°C on a rotator. Dimethyl sulfoxide was then added to a final concentration of 10%, and the cells were incubated at room temperature for 2 min. The cells were diluted 10-fold in serum-free DMEM, pelleted, washed twice in DMEM, and plated in RPMI 1640 with 10% FCS. Where appropriate, CM was added to the cells 16 h prior to LPS treatment. Forty hours after transfection, the cells were treated with LPS for the indicated time periods, lysed, and analyzed for luciferase activity by using an Enhanced Luciferase Assay kit (Analytical Luminescent Laboratory) or, where appropriate, for both luciferase and β-galactosidase activities by using the Luminescent β-Galactosidase Genetic Reporter System II (Clontech). In experiments lacking the β-galactosidase control, the protein concentration of each lysate was measured (Bio-Rad) and used to normalize luciferase activity.
RESULTS
An activity secreted by P388D1(IL1) macrophages inhibits LPS-induced expression of TNF-α mRNA.
We previously observed that the murine macrophage cell line P388D1(IL1) secretes a factor, termed AMF (autocrine macrophage factor), that alters the subnuclear localization and activity of the C/EBPβ transcription factor (3). Since C/EBPβ has been implicated in the regulation of proinflammatory cytokine gene expression in myeloid cells (1, 8), we examined the effects of P388D1(IL1) CM on LPS-induced expression of IL-1β, IL-6, MCP-1, and TNF-α in IC-21 macrophages (Fig. 1A). As expected, LPS alone elicited expression of each cytokine mRNA. However, CM pretreatment of the cells for 16 h prior to LPS stimulation suppressed the induction of TNF-α. In contrast, CM-treated cells expressed detectable levels of MCP-1 and IL-1β prior to LPS, and these levels were further increased by LPS treatment. Although IL-6 expression was not directly elicited by CM, LPS induction of IL-6 was augmented by pretreating the cells with CM. These findings show that an activity (or activities) secreted by P388D1(IL1) cells inhibits LPS-induced expression of TNF-α, increases MCP-1 basal expression, and accelerates LPS induction of IL-1β and IL-6. We provisionally refer to the TNF-α-inhibitory activity as TNF-α inhibiting factor (TIF). The relationship between TIF and AMF is presently unknown.
We also tested the ability of CM to inhibit TNF-α mRNA expression in primary murine macrophages. Figure 1B shows that TNF-α induction was suppressed by CM in peritoneal and bone marrow-derived macrophage preparations. CM therefore exerts similar effects on TNF-α expression in macrophage cell lines and primary macrophages.
A TNF-α-inhibitory activity is secreted by LPS-stimulated macrophages.
TNF-α expression is rapidly attenuated after its initial induction by LPS, whereas IL-1β and IL-6 mRNAs remain high 8 h after induction (Fig. 1A). This pattern of cytokine gene expression at 8 h is similar to that observed in CM-treated macrophages after brief (1-h) exposure to LPS (Fig. 1A); i.e., IL-1β, IL-6, and MCP-1 levels are enhanced and TNF-α is suppressed. These observations suggested that the TNF-α-inhibitory activity might be produced by LPS-stimulated macrophages and serve as an autocrine feedback signal to attenuate TNF-α transcription.
We tested this hypothesis first by comparing the TNF-α-inhibitory activity of CM from IC-21 cells exposed to LPS for 16 h (LPS+ CM) with that of CM from untreated cells (control CM). CM preparations were concentrated by ultrafiltration and administered to naive IC-21 cells, and the effect on LPS-induced cytokine expression was evaluated. Since LPS+ CM contained LPS, it was not possible to pretreat the cells with CM prior to LPS induction. Therefore, we first determined whether P388D1(IL1) CM could inhibit TNF-α transcription when cells were exposed to CM and LPS simultaneously. P388D1(IL1) CM suppressed the induction of TNF-α mRNA in this experiment (Fig. 2A), albeit less efficiently than when the cells were pretreated with CM. Since CM can partially inhibit TNF-α expression when applied together with LPS, we next compared the abilities of control CM and LPS+ CM to inhibit TNF-α transcription. Control CM did not affect TNF-α induction, indicating that unstimulated IC-21 cells do not secrete the inhibitory activity. However, LPS+ CM partially suppressed TNF-α expression, generating a profile nearly identical to that of cells treated with P388D1(IL1) CM and LPS simultaneously. Induction of IL-6 and MCP-1 mRNAs was not inhibited by P388D1(IL1) CM or LPS+ CM.
In a related experiment, IC-21 cells were briefly stimulated with LPS (20 min), washed extensively to remove the LPS, and then incubated in growth medium for 60 min. The resulting CM (LPS− CM) was compared to control CM (prepared from untreated cells) for its effects on cytokine expression in IC-21 cells (Fig. 2B). As observed for P388D1(IL1) CM, LPS− CM suppressed TNF-α but did not inhibit induction of IL-6, IL-1β, or MCP-1; instead, it stimulated or accelerated expression of these cytokine genes. A comparison of Fig. 1A and 2B shows that the effects of LPS− CM are nearly identical to those of P388D1(IL1) CM. Collectively, the results of Fig. 2 demonstrate that LPS stimulates the secretion of a TNF-α-inhibiting factor.
Analysis of known TNF-α-inhibitory factors.
P388D1(IL1) CM and LPS+ CM from IC-21 cells were analyzed by ELISA for the presence of factors that are known to inhibit production of TNF-α protein (Table 1). IL-4 and IL-10 were undetectable in CM preparations, eliminating these two cytokines as candidates for the secreted factor. TGF-β1 was present (4.4 ng/ml) in P388D1(IL1) CM and was detected at low levels in LPS+ CM. However, recombinant TGF-β inhibited IL-6 and MCP-1 induction but not TNF-α mRNA expression in IC-21 cells (data not shown), indicating that TIF is distinct from TGF-β.
Of the proinflammatory cytokines tested, TNF-α and IL-6 were present at 3.4 and 6.1 ng/ml, respectively, in P388D1(IL1) CM, while IL-1α and IL-1β were undetectable (data not shown). LPS+ CM contained IL-6 (>600 ng/ml) and TNF-α (6.1 ng/ml). Because IL-6 and TNF-α were highly expressed in P388D1(IL1) CM and these two cytokines were found to stimulate C/EBPβ protein expression in IC-21 cells (3), we examined whether they could recapitulate the effects on cytokine gene expression observed for CM (Fig. 2). In contrast to CM, neither cytokine was able to activate expression of IL-1β mRNA in the absence of LPS (Fig. 3, 0 h), nor was LPS-induced expression of TNF-α inhibited by these factors. The principal effect on cytokine transcription was enhanced expression of TNF-α mRNA in IL-6- and TNF-α-treated cells. These data demonstrate that CM contains a factor(s) that modulates cytokine production in macrophages and is distinct from IL-1, IL-6, and TNF-α (proinflammatory cytokines) and from IL-4, IL-10, and TGF-β (anti-inflammatory factors). At present, TIF activity has not been attributed to any known cytokine.
FIG. 3.

Effects of IL-6 and TNF-α pretreatment on cytokine mRNA expression in IC-21 cells. IC-21 cells were grown for 16 to 20 h in fresh medium (control) or fresh medium supplemented with recombinant IL-6 or TNF-α (10 ng/ml). The cells were then stimulated with LPS, and RNA was harvested over an 8-h time course. Northern blots were prepared (10 μg of RNA per lane) and hybridized with the indicated cDNA probes.
CM inhibits TNF-α promoter activity.
To further explore the mechanism by which CM inhibits TNF-α expression, we constructed a reporter gene (TNF-Luc) containing 1,260 bp of the murine TNF-α promoter fused to luciferase. This construct was transiently transfected into ANA-1 macrophages which, unlike IC-21 cells, can be transfected efficiently. The transfected cells were pretreated with LPS− CM or control CM for 16 h and stimulated with LPS, and luciferase activity was measured over a 12-h time course (Fig. 4). Luciferase expression in cells treated with control CM was induced by LPS approximately 16-fold over the basal level. CM decreased the magnitude of this induction by ∼50% but did not inhibit a control β-galactosidase reporter, pRSV-βgal. These results demonstrate that an inhibitory factor in LPS− CM specifically suppresses the TNF-α promoter, indicating that the repressive mechanism operates primarily at the transcriptional level.
FIG. 4.

CM suppresses LPS-induced transcription from the TNF-α promoter in transfected macrophages. (A) ANA-1 macrophages were transfected with the TNF-Luc reporter plasmid (1 μg/2 × 106 cells), treated with LPS− CM or control CM for 16 h, and then stimulated with LPS over a 12-h time course. Relative luciferase expression was calculated by normalizing to luciferase activity in control cells at 0 h. The data represent the average of three independent experiments. (B) The same experiment was performed with a control reporter plasmid, pRSV-βgal. Relative β-galactosidase (β-gal) expression was calculated as described above for luciferase activity.
CM preferentially induces NF-κB p50.
LPS-induced transcription of the murine TNF-α gene in macrophages is strongly dependent on NF-κB proteins (15, 45), and multiple NF-κB binding sites have been identified in the TNF-α promoter (reviewed in reference 37). To determine if inhibition of TNF-α transcription involves changes in the composition of NF-κB subunits, we analyzed nuclear levels of NF-κB p65 and p50 in IC-21 cells exposed to LPS− CM (Fig. 5A and B). p65 and p50 were induced by LPS− CM but not control CM within 1 h of treatment, and p65 levels in the LPS− CM-stimulated cells remained relatively constant thereafter. However, p50 expression continued to increase and reached high levels by 12 to 24 h. This pattern of NF-κB induction was distinct from that of cells treated with a combination of IL-6 and TNF-α, two factors in LPS− CM that are capable of activating NF-κB. IL-6 and TNF-α induced nuclear expression of p50 and p65 but did not cause prolonged induction of p50 (Fig. 5C). Thus, LPS− CM promotes the rapid activation of p50 and p65, probably due to the presence of IL-6 and/or TNF-α, but also contains an activity that elicits a sustained increase in nuclear p50 levels. We infer that this activity is distinct from (and cannot be induced by) TNF-α and IL-6.
FIG. 5.

CM preferentially activates nuclear NF-κB p50 expression. IC-21 cells were treated with control CM (A), LPS− CM (B), or a combination of IL-6 (10 ng/ml) and TNF-α (20 ng/ml) (C), and nuclear extracts were prepared over a 24-h time course. Four micrograms of each protein extract was assayed by Western blotting. The blots were probed simultaneously with polyclonal antibodies specific for p50 and p65.
If LPS stimulates the production of an autocrine factor that enhances p50 expression, p50 levels should eventually become elevated in nuclei of cells exposed to LPS. As expected, LPS activated nuclear expression of both p65 and p50 in IC-21 cells within 1 h (Fig. 6A). More importantly, p50 became hyperexpressed after 4 h of LPS treatment and continued to increase thereafter, while p65 levels declined slightly over time. These data further support the idea that an autocrine feedback system in LPS-activated macrophages stimulates nuclear NF-κB p50 expression. Interestingly, the p50 response was not observed in another cell line, L fibroblasts. LPS stimulation of L cells caused rapid induction of p65 and p50 in the nucleus, but levels of both proteins subsequently declined in a coordinate manner (Fig. 6B). Furthermore, LPS− CM from IC-21 cells failed to enhance p50 induction in L cells (data not shown). Thus, the ability to respond to TIF appears to be a cell-specific property.
FIG. 6.

Nuclear NF-κB p50 is selectively induced in LPS-stimulated macrophages. IC-21 macrophages (A) or L fibroblasts (B) were treated with LPS (1 μg/ml), and nuclear extracts were prepared over a 24-h time course. Samples were analyzed for p50 and p65 expression by Western blotting as described in the legend to Fig. 5.
p50 overexpression inhibits TNF-α promoter activity.
We next examined the functional relationship between p50 levels and inhibition of TNF-α-transcription by analyzing the effects of p50 or p65 overexpression on TNF-Luc activity. Figure 7 shows that LPS-induced transcription from the TNF-α promoter was strongly repressed by cotransfecting a p50 expression vector. In contrast, p65 enhanced both basal and LPS-induced luciferase expression. Cotransfection of equal amounts of the p50 and p65 vectors also resulted in nearly complete suppression of promoter activity. This observation indicates that the TNF-α promoter is highly sensitive to the repressive effects of p50, even when p65 levels are also elevated.
FIG. 7.

Overexpression of NF-κB p50 inhibits LPS induction of the TNF-α promoter. ANA-1 macrophages were transfected with 1 μg of TNF-Luc reporter DNA and 2 μg of either the Rc/CMV control vector, Rc/CMV-p50, Rc/CMV-p65, or Rc/CMV-p50 plus Rc/CMV-p65 (1 μg of each). Forty hours later, the cells were stimulated with LPS (1 μg/ml), and lysates were prepared at 0, 3, and 6 h. Relative luciferase expression was normalized to the luciferase activity of Rc/CMV-transfected cells prior to LPS treatment (0 h). Expression of a reporter gene driven by the simian virus 40 early promoter, pGL2-Luc, was compared as a control (lower panel). The data are the means ± standard errors of the means from three independent experiments.
κB sites in the TNF-α promoter mediate inhibition and preferentially bind p50 dimers.
The observation that TNF-α transcription is inhibited by overexpression of p50 or by LPS− CM, which activates endogenous p50, suggests that the repressive mechanism involves κB sites in the promoter. Four NF-κB sites (κB1, κB2, κB2a, and κB3) have been identified in the murine TNF-α promoter (Fig. 8A). To determine whether these sites are required for transcriptional repression, several 5′ deletion mutants were generated and tested for inhibition by LPS− CM. Deletions removing the first (i.e., most distal) κB site (−646) or the three most distal sites (−527) did not noticeably affect repression by CM (Fig. 8B). However, deletion mutants lacking all four NF-κB sites (−514 and −210) were not inhibited by CM (Fig. 8B, 3-h time points). In addition, a −527 deletion construct in which the κB3 site was inactivated by clustered point mutations (−527m) was also refractory to CM inhibition. Statistical analysis shows significant inhibition by CM at 3 h for promoter constructs that contain κB3. Although the mutants lacking κB elements were inducible by LPS, their induced expression levels were approximately one-half of that of the full-length promoter (data not shown). Thus, the proximal promoter sequences are sufficient for at least partial LPS inducibility, whereas one or more of the upstream κB sites are required for inhibition by CM.
FIG. 8.

Localization of TNF-α promoter sequences required for inhibition by CM. (A) Diagram of 5′ deletions and point mutations and a summary of their repression by LPS− CM. The locations of known κB elements are depicted. The name of each construct indicates the position of the deletion endpoint relative to the TNF-α transcription startsite. (B) LPS− CM inhibition assays. Each plasmid construct (1 μg/2 × 106 cells) was transfected into ANA-1 macrophages, and 24 h later the cells were treated with control CM (□) or LPS− CM (•) for 16 h. The cells were then stimulated with LPS, and lysates were prepared over a 6-h time course. Relative luciferase expression (fold induction) for each reporter construct was normalized to luciferase activity from cells treated with control CM before LPS stimulation (0 h). The data represent the average means ± standard errors of the means from three independent experiments. Significance was calculated by Student’s t test (∗, P ≤ 0.02; ∗∗, P ≤ 0.14 versus control CM).
Since the TNF-α promoter is highly sensitive to repression by p50, we analyzed the protein binding specificities of three TNF-α κB sites in assays using IC-21 nuclear extracts prepared at various times after LPS stimulation (Fig. 9A). For comparison, we tested the well-characterized Ig-κ element (44). Gel shift analysis showed that the most proximal TNF-α κB site (κB3) bound two major complexes in the 1-h extract (lane 2). At later time points, the amount of the faster-migrating complex increased and became the predominant binding species (lanes 3 to 7). Supershift analysis using specific antibodies identified the fast-migrating complex as a p50 homodimer and the slower complex as a p65-p50 heterodimer (Fig. 9B, lanes 2 and 3). Thus, the increase in p50 levels that occurs with delayed kinetics after LPS stimulation results in preferential binding of p50 homodimers to the TNF-κB3 site in vitro. In contrast, the Ig-κ probe exhibited significantly lower affinity for p50 homodimers. The upper (p65-p50) and lower (p50-p50) complexes occurred in nearly equimolar amounts, even at late times when p50 levels were elevated (Fig. 9A, lanes 8 to 14).
FIG. 9.

Three NF-κB sites in the TNF-α promoter preferentially bind NF-κB p50 homodimers. (A) EMSA analysis of nuclear extracts from LPS-stimulated IC-21 cells. The cells were induced with LPS, and nuclear extracts were prepared over a 24-h time course. Double-stranded oligonucleotide probes corresponding to the one of the distal NF-κB sites of the TNF-α promoter (TNF-κB3) or the Ig-κ light-chain NF-κB site (Ig-κ) were incubated with the nuclear extracts, and protein-DNA complexes were separated from free probe by electrophoresis. (B) Supershift analysis of complexes bound to κB sites from the TNF-α promoter or the Ig-κ element. Nuclear extract harvested 8 h after LPS treatment (A) was incubated in the presence or absence of p50- or p65-specific antibodies (Ab), and the indicated oligonucleotide probes were added prior to gel electrophoresis.
Probes corresponding to sites κB1 and κB2a were also examined for protein binding specificity. Although κB1 was a weaker site overall, the two probes were similar to κB3 in their preference for p50 homodimers (Fig. 9B, lanes 4 to 9). Thus, three κB elements in the distal TNF-α promoter region display higher affinity for p50 homodimers than for p65-p50 heterodimers. This binding specificity contrasts markedly with that of the Ig-κ element, which under the same binding conditions exhibits much greater affinity for p65-p50. The κB2 element was shown to be a relatively weak site for p50 homodimers, and its functional properties have been addressed elsewhere (28a). In addition, the κB2 site binds p65-p65 and p65–c-Rel more strongly than any of sites 1, 2a, and 3, and thus this site may mediate the response to p65 in the context of the entire promoter (28a, 29, 36).
The functional specificities of the three TNF α-κB elements were further examined by creating artificial promoters containing four copies of these sites upstream of the TK minimal promoter. For comparison, we generated an analogous construct with four copies of the Ig-κ element. We analyzed the response of each NF-κB-dependent promoter to p50 expression in transfected cells. An increasing ratio of p50 to p65 vectors was cotransfected with each reporter gene into ANA-1 cells, and luciferase activity was measured after LPS stimulation (Fig. 10). (Ig-κ)4-TK-Luc was strongly activated by p65 alone, and its expression initially increased when the p50 vector was included. Transcription peaked at a p50-p65 ratio of 0.33, after which reporter expression declined. In contrast, the three TNF-α NF-κB constructs were activated much less efficiently by p65, and increased ratios of p50 to p65 caused a continuous reduction in luciferase activity. The results of this experiment are thus consistent with the NF-κB subunit specificities observed in DNA binding experiments (Fig. 9). The Ig-κ element, which has high affinity for p65-p50 heterodimers, was activated most effectively by a combination of p65 and p50, whereas the p50-specific TNF-α κB sites were much less responsive to p65 and were inhibited by even low doses of p50.
FIG. 10.

The distal TNF-α κB sites are poorly activated by p65. Reporter plasmids (1 μg) containing four tandem copies of the indicated κB sites upstream of a minimal promoter reporter gene (TK-Luc) were cotransfected into ANA-1 cells with various ratios of p50 and p65 expression vectors (1 μg in total) and an internal standard, pRSV-βgal (0.5 μg). After 2 days, the cells were induced with LPS for 4 h and harvested, and the lysates were assayed for luciferase and β-galactosidase activities. Luciferase activity was normalized to β-galactosidase activity for each sample. The data are from a representative experiment; similar results were obtained in two independent experiments.
DISCUSSION
We describe an autocrine activity, TIF, that suppresses LPS-induced transcription of the TNF-α gene in macrophages. Our studies suggest that TIF functions as a negative feedback signal to attenuate transcription of the TNF-α gene, and perhaps other genes, in activated macrophages. TNF-α inhibition is associated with enhanced expression of NF-κB p50 in the nucleus, and several lines of evidence indicate that p50 causes repression of the TNF-α promoter. Since TIF is released from LPS-stimulated cells, we propose that this factor controls the decrease in TNF-α mRNA levels that begins approximately 4 h after LPS administration. Although TIF appears to be rapidly released from activated cells (at least within 90 min after LPS treatment), the increase in p50 levels in the nucleus becomes significant only several hours after exposure to the factor (Fig. 5 and 6). We suggest that this delayed response allows a burst of TNF-α transcription to occur before the inhibitory mechanism is fully activated.
The use of an autocrine mechanism to attenuate TNF-α production may have important implications for the inflammatory response in vivo. If activated monocytes/macrophages at sites of infection or injury release TIF, monocytic cells subsequently recruited to the affected region would encounter locally elevated levels of TIF, which would suppress their production of TNF-α. However, expression of chemotactic factors such as MCP-1 and less toxic cytokines like IL-1β would not be repressed and might even be enhanced in this environment (Fig. 1 and 2). In view of the deleterious effects of TNF-α overexpression, the TIF-mediated attenuation mechanism may be critical for a properly regulated response to microbial pathogens and other inflammatory stimuli. The identification of an activity that specifically suppresses TNF-α expression in macrophages, which are a major source of TNF-α in vivo (4), may have future therapeutic value for chronic and acute inflammatory diseases such as rheumatoid arthritis, asthma, Alzheimer’s disease, and bacterial sepsis.
It is notable that responsiveness to TIF was not observed in L fibroblasts, suggesting that this pathway is restricted to specific cell types. However, preliminary experiments indicate that L cells produce a TIF-like activity when stimulated with LPS (data not shown). Thus, L fibroblasts may produce TIF even though these cells are refractory to its effects. Further studies should reveal whether other cell types react to or express this factor. The fact that TIF accumulates in the medium of P388D1(IL1) macrophages in the absence of LPS stimulation may indicate that its expression is inappropriately activated in the transformed P388D1(IL1) cell line.
TIF may be a novel TNF-α-inhibitory factor.
Several factors have been shown to repress TNF-α synthesis in macrophages. IL-4 suppresses TNF-α production in LPS-stimulated human monocytes, yet unlike AMF, it also inhibits IL-1β expression (23); furthermore, macrophages do not normally express IL-4. IL-10 also inhibits synthesis of TNF-α, as well as IL-1, IL-6, and IL-8, in monocytes/macrophages and is secreted by these cells in response to LPS (5, 14, 18, 52, 53). These findings led to the proposal that IL-10 functions as an autocrine feedback signal that attenuates production of proinflammatory cytokines in activated macrophages (14). IL-10 was not detected in P388D1(IL1) CM or LPS− CM, however (Table 1). TGF-β is also known to suppress proinflammatory cytokine production in macrophages (16), but its inhibition of TNF-α expression occurs posttranscriptionally (5). A TGF-β-related cytokine, MIC-1, that inhibits macrophage TNF-α production was recently identified, although secretion of MIC-1 by macrophages was not induced by LPS (7). Finally, PGE2, a nonpeptide immune/inflammatory mediator, has been reported to inhibit TNF-α production in macrophages (38, 41, 46). However, PGE2 is a small molecule that should not be retained by 30-kDa-cutoff ultrafiltration. In summary, due to their biological properties and/or absence from CM, we infer that IL-4, IL-10, TGF-β, MIC-1, and PGE2 are distinct from TIF and that TIF is likely to be a novel TNF-α-inhibiting factor.
Studies of macrophages tolerized by chronic treatment with low levels of LPS have suggested the existence of a secreted factor that inhibits TNF-α induction upon subsequent exposure to high levels of LPS (19). This activity was not immunoreactive with an IL-10-specific antibody, ruling out IL-10 as a candidate. Tolerized macrophages also displayed elevated levels of NF-κB p50 (57). Similarly, Fahmi and Chaby (17) reported that LPS tolerance could be transferred to naive macrophages by exposure to a secreted activity from LPS-stimulated cells. This heat-labile factor suppressed the LPS-dependent release of TNF-α but not the production of IL-6 or IL-1. Although the inhibition of TNF-α expression was not analyzed at the mRNA level in the latter study, the parallels between the above findings and our observations are striking. It seems likely that these previously described inhibitory activities are identical or highly related to TIF. Indeed, the phenomenon of LPS tolerance may reflect chronic induction of the autocrine attenuation mechanism that normally operates in acutely activated macrophages.
Selective activation of NF-κB p50.
The preferential induction of p50 occurs with delayed kinetics in LPS− CM-treated macrophages as well as in LPS-stimulated cells. Watanabe et al. (54) recently demonstrated that the oncoprotein BCL-3, which was previously shown to interact with homodimers of p50 and p52, caused the specific induction of nuclear p50 homodimers when expressed in cells containing the p50 precursor, p105. This process involves the conversion of cytoplasmic p50-p105 heterodimers to nuclear p50 homodimers and may involve a subunit reassortment mechanism promoted by BCL-3. It is possible that the superinduction of p50 in macrophages exposed to TIF involves the ability of BCL-3 to mobilize p50 homodimers. TIF could also function by increasing the expression of p105; this possibility is supported by the finding that LPS-tolerized macrophages contain elevated levels of p105 mRNA (57).
Stimulatory and inhibitory elements in the TNF-α promoter.
Negative regulation by CM was eliminated in promoter mutants lacking the upstream κB elements. The loss of repression that occurred when sequences between nt −527 and −514 were deleted or when the κB3 site was mutated (−527m) shows that the κB3 site is critical for TIF-mediated inhibition. The roles of κB1 and κB2a in repression are unclear and will be the subject of further investigation. Other studies have shown that a proximal κB site (nt −99 to −89), together with an adjacent cyclic AMP response element, is critical for LPS inducibility of the human TNF-α promoter in monocytic cells (55). This κB element binds a heterodimeric p50-p65 complex in nuclear extracts from stimulated cells (50, 55). A corresponding element has not yet been identified in the murine TNF-α promoter. However, the presence of a proximal site that binds p50-p65 heterodimers would provide a plausible explanation for the LPS inducibility of deletion mutants lacking the upstream κB elements. Additional studies have shown that the entire promoter region, including distal sites 2 and 2a as well as the downstream κB enhancer element (29), are necessary for maximal activation of the human and murine TNF-α promoters by LPS (28a).
The distal κB elements preferentially bind p50 homodimers, which are activated in cells exposed to LPS− CM (TIF), and mediate repression by p50 when fused to a heterologous promoter. p50 lacks transcriptional activation domains and has been found to have neutral or inhibitory effects on other promoters (24, 25, 43). Inhibition by TIF via distal TNF-α promoter sequences appears to be dominant to the positive effects of the proximal region, suggesting an active repression mechanism. We do not know whether other proteins or binding sites are required for p50-mediated repression. However, a p50-specific corepressor protein, DSP-1 (Dorsal switch protein), has been cloned from a Drosophila cDNA library by using a functional screen in yeast (32). Dominant repression involving p50 and an adjacent DSP-1-like binding site was observed for the beta interferon promoter, using HeLa cells cotransfected with a Drosophila DSP-1 expression vector (32). These data indicate that an inhibitory mechanism involving a DSP-1-like protein and p50 may exist in mammalian cells. The possibility that a DSP-1-related factor is involved in p50-mediated repression of the TNF-α promoter will be explored in future studies.
Binding specificity of κB sites in the TNF-α promoter.
Three TNF-α κB sites show a marked preference for p50 homodimers compared to p50-p65, the other major NF-κB species activated in LPS-stimulated macrophages. Preferential binding of p50 to the TNF-α promoter was also observed by Brown et al. (9), who reported that the κB3 site binds p50 dimers in extracts from LPS-stimulated bone marrow macrophages. κB1 is a low-affinity binding site, while κB2a and κB3 bind NF-κB proteins more strongly.
There are now several examples in which κB sites exhibit preference for specific subsets of NF-κB complexes. For instance, the regulatory region of the IL-8 gene contains a κB element that binds p65, c-Rel, and p52 homodimers but not p50 homodimers or p50-p65 heterodimers (27). Similarly, induction of the ICAM-1 gene occurs in response to inflammatory signals, such as TNF-α, that act through an NF-κB site in the proximal promoter region. This site binds only p65 homodimers and p50-p65 heterodimers in vitro. p65 homodimers appear to be the transcriptionally active NF-κB complex for the ICAM-1 promoter, suggesting that p50-p65 may be excluded from binding in vivo (31). Furthermore, a κB-related sequence that is required for LPS inducibility in monocytic cells was identified in the tissue factor gene promoter. This motif bound p65 and c-Rel homodimers and a heterodimer of the two, but not p50-containing complexes (35). Thus, at least three types of κB elements have been described: (i) sites that bind p65–c-Rel complexes (and perhaps also p52 dimers); (ii) sites that exhibit specificity for p65-p50 heterodimers (exemplified by the Ig-κ element); and (iii) a class of p50-specific sites typified by the κB1, κB2a, and κB3 motifs in the TNF-α promoter. Other classes, such as sites that prefer p65 homodimers, may also exist.
Binding site selection experiments to determine the optimal specificities of p50, p65, and c-Rel homodimers revealed that certain sequences can bind to p50 but not p65, and vice versa (28). These homodimer sites tended not to bind p50-p65 heterodimers, indicating that both subunits of a dimer contribute to sequence specificity and that the p50 and p65 subunits are not interchangeable in terms of binding selectivity. These findings are consistent with the properties of the three classes of naturally occurring NF-κB sites described above. A comparison of p50-selected sites generated the consensus sequence 5′-GGGGATYCCC-3′. The three upstream TNF-α sites (κB1 [5′-GGGGAATCCT-3′], κB2a [5′-GGGGCTGCCC-3′], and κB3 [5′-GGGGCTTTCCC-3′]) conform well to this consensus element, thus accounting for their high affinity for p50 homodimers.
CM enhances IL-1β, MCP-1, and IL-6 expression.
In addition to inhibiting TNF-α transcription, macrophage CM activated expression of IL-1β and MCP-1 mRNAs in the absence of LPS and also enhanced the LPS-dependent expression of IL-6, IL-1β, and MCP-1. While these responses could be partly explained by the mobilization of p65-p50 heterodimers by the presence of IL-6 and TNF-α in CM, LPS-independent activation of IL-1β, and MCP-1 expression was not elicited by recombinant IL-6 or TNF-α. Moreover, an anti-TNF-α antibody did not alter the effects of CM on cytokine expression (data not shown), further supporting the idea that TNF-α is distinct from these stimulatory activities. Most of the known factors that enhance the expression of proinflammatory cytokines do not act autonomously but rather augment induction by LPS (22). Thus, the LPS-independent activation of IL-1β and MCP-1 expression by CM suggests the existence of a novel secreted factor or an unrecognized activity of a known factor produced by macrophages.
While CM pretreatment initially augments LPS-stimulated expression of IL-1β, MCP-1, and IL-6 mRNAs, at later time points these transcripts are suppressed relative to control cells (Fig. 1A and 2A). These results suggest that the enhancing activity is different from TIF. The TNF-α promoter may be especially sensitive to TIF repression because it contains three κB sites with selective affinity for p50 homodimers. Alternatively, it is possible that TIF activates other transcription factors in addition to mobilizing p50 and that the composition of the promoter determines the type (positive or negative) and duration of the transcriptional response for each cytokine gene. P388D1(IL1) CM contains an activity, AMF, that alters the subnuclear localization and transcriptional activity of C/EBPβ (3). We do not know if AMF and the cytokine-enhancing activity in CM are identical factors, nor is the relationship between TIF and AMF clear. The biochemical purification of these activities from CM will ultimately establish whether they correspond to the same or different proteins.
ACKNOWLEDGMENTS
We are indebted to Howard Young for cDNAs and advice, Nancy Rice for NF-κB expression vectors, antibodies, and helpful discussions, Dmitri Kuprash for advice and discussion, Craig Reynolds for recombinant cytokines, and Lori Sewell and Barbara Shankle for assistance in plasmid constructions. We also thank Carla Weinstock and Hilda Marusiodis for expert secretarial assistance.
This research was sponsored by the National Cancer Institute, DHHS, under contract with ABL and under contract NO1-CO-56000. R.C.S. is supported by ACS research grant DB-110.
REFERENCES
- 1.Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990;9:1897–1906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akira S, Taga T, Kishimoto T. Interleukin-6 in biology and medicine. Adv Immunol. 1993;54:1–78. doi: 10.1016/s0065-2776(08)60532-5. [DOI] [PubMed] [Google Scholar]
- 3.Baer, M., S. C. Williams, R. C. Schwartz, A. J. Dillner, and P. F. Johnson. Autocrine signals control C/EBPβ expression, localization, and activity in macrophages. Blood, in press. [PubMed]
- 4.Beyaert R, Fiers W. Molecular mechanisms of tumor necrosis factor-induced cytotoxicity. What we do understand and what we do not. FEBS Lett. 1994;340:9–16. doi: 10.1016/0014-5793(94)80163-0. [DOI] [PubMed] [Google Scholar]
- 5.Bogdan C, Paik J, Vodovotz Y, Nathan C. Contrasting mechanisms for suppression of macrophage cytokine release by transforming growth factor-β and interleukin-10. J Biol Chem. 1992;267:23301–23308. [PubMed] [Google Scholar]
- 6.Bonnerot C, Rocancourt D, Briand P, Grimber G, Nicolas J F. A β-galactosidase hybrid protein targeted to nuclei as a marker for developmental studies. Proc Natl Acad Sci USA. 1987;84:6795–6799. doi: 10.1073/pnas.84.19.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bootcov M R, Bauskin A R, Valenzuela S M, Moore A G, Bansal M, He X Y, Zhang H P, Donnellan M, Mahler S, Pryor K, Walsh B J, Nicholson R C, Fairlie W D, Por S B, Robbins J M, Breit S N. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-β superfamily. Proc Natl Acad Sci USA. 1997;94:11514–11519. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bretz J D, Williams S C, Baer M, Johnson P F, Schwartz R C. C/EBP-related protein 2 confers lipopolysaccharide-inducible expression of interleukin-6 and monocyte chemoattractant protein-1 to a lymphoblastic cell line. Proc Natl Acad Sci USA. 1994;91:7306–7310. doi: 10.1073/pnas.91.15.7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown M C, Tomaras G D, Vincenti M P, Taffet S M. Two forms of NF-κB1 (p105/p50) in murine macrophages: differential regulation by lipopolysaccharide, interleukin-2, and interferon-γ. J Interferon Cytokine Res. 1997;17:295–306. doi: 10.1089/jir.1997.17.295. [DOI] [PubMed] [Google Scholar]
- 10.Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci USA. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleveland D W, Lopata M A, MacDonald R J, Cowan M J, Rutter W J, Kirschner M W. Number and evolutionary conservation of α- and β-tubulin and cytoplasmic β- and γ-actin genes using specific cloned cDNA probes. Cell. 1980;20:95–105. doi: 10.1016/0092-8674(80)90238-x. [DOI] [PubMed] [Google Scholar]
- 12.Cox G W, Mathieson B J, Gandino L, Blasi E, Radzioch D, Varesio L. Heterogeneity of hematopoietic cells immortalized by v-myc/v-raf recombinant retrovirus infection of bone marrow or fetal liver. J Natl Cancer Inst. 1989;81:1492–1496. doi: 10.1093/jnci/81.19.1492. [DOI] [PubMed] [Google Scholar]
- 13.Dawe C J, Potter M. Morphologic and biologic progression of a lymphoid neoplasm of the mouse in vivo and in vitro. Am J Pathol. 1957;33:603. [Google Scholar]
- 14.de Waal Malefyt R, Abrams J, Bennett B, Figdor C G, de Vries J E. Interleukin-10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drouet C, Shakhov A N, Jongeneel C V. Enhancers and transcription factors controlling the inducibility of the tumor necrosis factor-α promoter in primary macrophages. J Immunol. 1991;147:1694–1700. [PubMed] [Google Scholar]
- 16.Espevik T, Figari I S, Shalaby M R, Lackides G A, Lewis G D, Shepard H M, Palladino M A., Jr Inhibition of cytokine production by cyclosporin A and transforming growth factor β. J Exp Med. 1987;166:571–576. doi: 10.1084/jem.166.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fahmi H, Chaby R. Selective refractoriness of macrophages to endotoxin-induced production of tumor necrosis factor, elicited by an autocrine mechanism. J Leukoc Biol. 1993;53:45–52. doi: 10.1002/jlb.53.1.45. [DOI] [PubMed] [Google Scholar]
- 18.Fiorentino D F, Zlotnik A, Mosmann T R, Howard M, O’Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 19.Frankenberger M, Pechumer H, Ziegler-Heitbrock H W. Interleukin-10 is upregulated in LPS tolerance. J Inflamm. 1995;45:56–63. [PubMed] [Google Scholar]
- 20.Fried M, Crothers D M. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 1981;9:6505–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garner M M, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981;9:3047–3060. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton J A. Colony stimulating factors, cytokines and monocyte-macrophages—some controversies. Immunol Today. 1993;14:18–24. doi: 10.1016/0167-5699(93)90319-G. [DOI] [PubMed] [Google Scholar]
- 23.Hart P H, Vitti G F, Burgess D R, Whitty G A, Piccoli D S, Hamilton J A. Potential antiinflammatory effects of interleukin 4: suppression of human monocyte tumor necrosis factor α, interleukin 1, and prostaglandin E2. Proc Natl Acad Sci USA. 1989;86:3803–3807. doi: 10.1073/pnas.86.10.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang S M, Tran A C, Grilli M, Lenardo M J. NF-κB subunit regulation in nontransformed CD4+ T lymphocytes. Science. 1992;256:1452–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 25.Kieran M, Blank V, Logeat F, Vandekerckhove J, Lottspeich F, Le Bail O, Urban M B, Kourilsky P, Baeuerle P A, Israel A. The DNA binding subunit of NF-κB is identical to factor KBF1 and homologous to the rel oncogene product. Cell. 1990;62:1007–1018. doi: 10.1016/0092-8674(90)90275-j. [DOI] [PubMed] [Google Scholar]
- 26.Kingston R E, Chomczynski P, Sacchi N. Guanidium methods for total RNA preparation. In: Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Greene Publishing and Wiley-Interscience; 1991. pp. 4.2.1–4.2.8. [Google Scholar]
- 27.Kunsch C, Rosen C A. NF-κB subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13:6137–6146. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunsch C, Ruben S M, Rosen C A. Selection of optimal κB/Rel DNA-binding motifs: interaction of both subunits of NF-κB with DNA is required for transcriptional activation. Mol Cell Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Kuprash, D. V., I. A. Udalova, and S. Nedospasov. Unpublished data.
- 29.Kuprash D V, Udalova I A, Turetskaya R L, Rice N R, Nedospasov S A. Conserved kappa B element located downstream of the tumor necrosis factor alpha gene: distinct NF-kappa B binding pattern and enhancer activity in LPS activated murine macrophages. Oncogene. 1995;11:97–106. [PubMed] [Google Scholar]
- 30.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Ledebur H C, Parks T P. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-κB site and p65 homodimers. J Biol Chem. 1995;270:933–943. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 32.Lehming N, Thanos D, Brickman J M, Ma J, Maniatis T, Ptashne M. An HMG-like protein that can switch a transcriptional activator to a repressor. Nature. 1994;371:175–179. doi: 10.1038/371175a0. [DOI] [PubMed] [Google Scholar]
- 33.Meda M, Cassatella M A, Szendrel G I, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rosel F. Activation of microglial cells by β-amyloid and interferon-γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 34.Nordeen S K. Luciferase reporter gene vectors for analysis of promoters and enhancers. BioTechniques. 1988;6:454–457. [PubMed] [Google Scholar]
- 35.Oeth P A, Parry G C, Kunsch C, Nantermet P, Rosen C A, Mackman N. Lipopolysaccharide induction of tissue factor gene expression in monocytic cells is mediated by binding of c-Rel/p65 heterodimers to a κB-like site. Mol Cell Biol. 1994;14:3772–3781. doi: 10.1128/mcb.14.6.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohmori Y, Tebo J, Nedospasov S, Hamilton T A. Kappa B binding activity in a murine macrophage-like cell line. Sequence-specific differences in kappa B binding and transcriptional activation functions. J Biol Chem. 1994;269:17684–17690. [PubMed] [Google Scholar]
- 37.Pauli U. Control of tumor necrosis factor gene expression. Crit Rev Eukaryotic Gene Expr. 1994;4:323–344. doi: 10.1615/critreveukargeneexpr.v4.i2-3.20. [DOI] [PubMed] [Google Scholar]
- 38.Renz H, Gong J H, Schmidt A, Nain M, Gemsa D. Release of tumor necrosis factor-alpha from macrophages. Enhancement and suppression are dose-dependently regulated by prostaglandin E2 and cyclic nucleotides. J Immunol. 1988;141:2388–2393. [PubMed] [Google Scholar]
- 39.Rice N R, Ernst M K. In vivo control of NF-κB activation by IκBα. EMBO J. 1993;12:4685–4695. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rice N R, MacKichan M L, Israel A. The precursor of NF-κB p50 has IκB-like functions. Cell. 1992;71:243–253. doi: 10.1016/0092-8674(92)90353-e. [DOI] [PubMed] [Google Scholar]
- 41.Scales W E, Chensue S W, Otterness I, Kunkel S L. Regulation of monokine gene expression: prostaglandin E2 suppresses tumor necrosis factor but not interleukin-1 alpha or beta-mRNA and cell-associated bioactivity. J Leukoc Biol. 1989;45:416–421. [PubMed] [Google Scholar]
- 42.Schattner A. Short analytical review. Lymphokines in autoimmunity—a critical review. Clin Immunol Immunopathol. 1994;70:177–189. doi: 10.1006/clin.1994.1027. [DOI] [PubMed] [Google Scholar]
- 43.Schmitz M L, Baeuerle P A. The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. [PubMed] [Google Scholar]
- 45.Shakhov A N, Collart M A, Vassalli P, Nedospasov S A, Jongeneel C V. κB-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor α gene in primary macrophages. J Exp Med. 1990;171:35–47. doi: 10.1084/jem.171.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spengler R N, Spengler M L, Lincoln P, Remick D G, Strieter R M, Kunkel S L. Dynamics of dibutyryl cyclic AMP- and prostaglandin E2-mediated suppression of lipopolysaccharide-induced tumor necrosis factor alpha gene expression. Infect Immun. 1989;57:2837–2841. doi: 10.1128/iai.57.9.2837-2841.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone R. Search for sepsis drugs goes on despite past failures. Science. 1994;264:365–367. doi: 10.1126/science.8153620. [DOI] [PubMed] [Google Scholar]
- 48.Sweet M J, Hume D A. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60:8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 49.Tracey K J, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med. 1994;45:491–503. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- 50.Trede N S, Tsytsykova A V, Chatila T, Goldfeld A E, Geha R S. Transcriptional activation of the human TNF-α promoter by superantigen in human monocytic cells: role of NF-κB. J Immunol. 1995;155:902–908. [PubMed] [Google Scholar]
- 51.Walker W S, Demus A. Antibody-dependent cytolysis of chicken erythrocytes by an in vitro-established line of mouse peritoneal macrophages. J Immunol. 1975;114:765–769. [PubMed] [Google Scholar]
- 52.Wang P, Wu P, Anthes J C, Siegel M I, Egan R W, Billah M M. Interleukin-10 inhibits interleukin-8 production in human neutrophils. Blood. 1994;83:2678–2683. [PubMed] [Google Scholar]
- 53.Wang P, Wu P, Siegel M I, Egan R W, Billah M M. IL-10 inhibits transcription of cytokine genes in human peripheral blood mononuclear cells. J Immunol. 1994;153:811–816. [PubMed] [Google Scholar]
- 54.Watanabe N, Iwamura T, Shinoda T, Fujita T. Regulation of NFKB1 proteins by the candidate oncoprotein BCL-3: generation of NF-κB homodimers from the cytoplasmic pool of p50-p105 and nuclear translocation. EMBO J. 1997;16:3609–3620. doi: 10.1093/emboj/16.12.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao J, Mackman N, Edgington T S, Fan S T. Lipopolysaccharide induction of the tumor necrosis factor-α promoter in human monocytic cells. Regulation by Egr-1, c-Jun, and NF-κB transcription factors. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 56.Ziegler-Heitbrock H W. Molecular mechanism in tolerance to lipopolysaccharide. J Inflamm. 1995;45:13–26. [PubMed] [Google Scholar]
- 57.Ziegler-Heitbrock H W, Wedel A, Schraut W, Strobel M, Wendelgass P, Sternsdorf T, Bauerle P A, Haas J G, Riethmuller G. Tolerance to lipopolysaccharide involves mobilization of nuclear factor κB with predominance of p50 homodimers. J Biol Chem. 1994;269:17001–17004. [PubMed] [Google Scholar]