

ABSTRACT
Bacillus anthracis is a Gram-positive Centers for Disease Control and Prevention category “A” biothreat pathogen. Without early treatment, inhalation of anthrax spores with progression to inhalational anthrax disease is associated with high fatality rates. Gepotidacin is a novel first-in-class triazaacenaphthylene antibiotic that inhibits bacterial DNA replication by a distinct mechanism of action and is being evaluated for use against biothreat and conventional pathogens. Gepotidacin selectively inhibits bacterial DNA replication via a unique binding mode and has in vitro activity against a collection of B. anthracis isolates including antibacterial-resistant strains, with the MIC90 ranging from 0.5 to 1 µg/mL. In vivo activity of gepotidacin was also evaluated in the New Zealand White rabbit model of inhalational anthrax. The primary endpoint was survival, with survival duration and bacterial clearance as secondary endpoints. The trigger for treatment was the presence of anthrax protective antigen in serum. New Zealand White rabbits were dosed intravenously for 5 days with saline or gepotidacin at 114 mg/kg/d to simulate a dosing regimen of 1,000 mg intravenous (i.v.) three times a day (TID) in humans. Gepotidacin provided a survival benefit compared to saline control, with 91% survival (P-value: 0.0001). All control animals succumbed to anthrax and were found to be blood- and organ culture-positive for B. anthracis. The novel mode of action, in vitro microbiology, preclinical safety, and animal model efficacy data, which were generated in line with Food and Drug Administration Animal Rule, support gepotidacin as a potential treatment for anthrax in an emergency biothreat situation.
KEYWORDS: Bacillus anthracis, biothreat, gepotidacin, FDA Animal Rule
INTRODUCTION
Bacillus anthracis, the etiologic agent of anthrax, is a Gram-positive spore-forming rod-shaped bacterium that is often found in the soil and commonly affects domestic and wild animals. Humans are incidentally infected through contact with infected animals or animal products. The major forms of anthrax are cutaneous, inhalational, gastrointestinal, and injectional. Cutaneous anthrax is the most frequent form of the disease and, although it can be fatal in 20% of cases, in the majority of the cases, it is self-limiting (1, 2). Inhalation anthrax is the most aggressive and lethal form of the disease, with a reported incubation period of 4–6 days from exposure to initial clinical symptoms and death; thus, treatment should be initiated as soon as possible (2). The bacterial spore is the infectious form leading to disease. The major virulence factors are the poly-γ-D-glutamic acid capsule, which is resistant to both phagocytosis and complement activation, and a tripartite toxin, which is composed of a receptor-binding subunit, protective antigen (PA), two enzymatic subunits, lethal factor (LF), a zinc metalloprotease and edema factor (EF), and a calmodulin-dependent adenylate cyclase. After binding to the receptor on the host’s cell surface, the PA is cleaved, resulting in the exposure of binding sites for which LF and EF compete. The PA forms a membrane-inserting heptamer, which translocates LF or EF into the cell. These subunits interact to form the active toxins: lethal toxin (PA +LF) and edema toxin (PA +EF). At the initial stages of infection, lethal toxin (LT) and edema toxin (ET) coordinately impair the host’s innate immune response, enabling the pathogen to establish infection. When elevated toxin concentrations are reached, LT and ET can directly cause host death by targeting both the cardiovascular system and the liver (3–5). Without aggressive prophylaxis or intervention, inhalational anthrax can result in mortality rates approaching 90% (6–8). Fatal anthrax is ultimately the result of acute toxemia, massive bacteremia, and the host’s response to the toxins and bacteria (1–3). The importance of anthrax toxins during the latter stages of disease becomes apparent as patients can still die from a systemic infection after bacteria are no longer detectable (3).
High-titer anthrax spores can be easily generated using basic microbiological techniques, and the ability of these spores to be rapidly disseminated by aerosolization has made anthrax a bio-weapon and military threat. An anthrax outbreak in 1979 in Sverdlosk, Russia, and the 2001 attacks in the United States (US) illustrate that inhalational anthrax can be rapidly fatal (6, 8, 9). Following the 2001 civilian attacks in the US, an emphasis on post-exposure therapeutics has become a research priority (10–12).
Gepotidacin is a novel first-in class bactericidal triazaacenaphthylene antibiotic. It was discovered by GSK, with further development supported by a public–private partnership between GSK, the Defense Threat Reduction Agency (DTRA, U.S. Department of Defense), and the Biomedical Advanced Research and Development Authority (U.S. Department of Health and Human Services), and it is being evaluated for use against both biothreat and conventional pathogens. Gepotidacin inhibits bacterial DNA replication by a distinct mechanism of action (13–15), demonstrating in vitro and in vivo activity against a range of Gram-positive and Gram-negative bacterial pathogens (16–22). Structural models of bacterial type II topoisomerase enzymes reveal the novel binding mode of this class of antibacterials and distinguish it from the binding mode of the fluoroquinolones (13, 15). As a consequence of this novel mode of action, in vitro activity is maintained against most target pathogens carrying resistance determinants to other antibacterials, including fluoroquinolones (16, 17). Gepotidacin is currently being evaluated for investigational oral treatment of uncomplicated urinary tract infection and uncomplicated gonorrhea, including those caused by pathogens resistant to currently used antimicrobials, (www.clinicaltrials.gov: NCT04020341, NCT04010539, and NCT04187144). In addition, gepotidacin has the potential to treat bacterial biothreat infections, including anthrax (23, 24).
It is neither practical nor ethical to conduct inhalational anthrax clinical trials in humans. Between 1900 and 1976, only 17 cases of inhalational anthrax were reported in the US, with an additional 11 cases reported in the 2001 anthrax attacks (25). Under the Food and Drug Administration’s (FDA’s) Animal Rule, establishing efficacy in well-characterized animal models is essential for the development of therapeutics directed against anthrax (26, 27). The New Zealand White (NZW) rabbit has been shown to be an appropriate model for inhalational anthrax, and the physiologic changes following aerosol challenge with B. anthracis in the rabbit have demonstrated that both the manifestations of the disease and the pathology were similar to those observed in humans, though the disease progresses more rapidly in rabbits. Furthermore, rabbits are predictive of the outcome of inhalational anthrax in nonhuman primate infection models. Refinements of the model have incorporated the use of biomarkers, such as a significant increase in body temperature (SIBT) and the presence of circulating PA, as triggers for therapeutic intervention (8, 12, 28, 29).
Gepotidacin has demonstrated in vitro activity against B. anthracis and efficacy in proof-of-concept rabbit inhalational challenge models (20). It was therefore of interest to evaluate gepotidacin in a study designed to provide pivotal efficacy data under the FDA Animal Rule and to predict effective clinical dosing regimens for gepotidacin in the treatment of inhalational anthrax.
RESULTS
Gepotidacin demonstrates in vitro activity against B. anthracis
A total of 160 isolates of B. anthracis from the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) (Study 1, n = 30; Study 2, n = 100) and Rutgers University (n = 30) were tested to evaluate the in vitro activity of gepotidacin (Table 1). The MIC90 values for gepotidacin ranged from 0.5 µg/mL to 1 µg/mL. Table 2 shows the activity of gepotidacin against the plasmid-cured B. anthracis Ames strain (ΔANR) and its isogenic mutants, S1-1 and S1-2. These single-step mutants have modifications in DNA gyrase at the following positions: S85L (S1-1) and E89K (S1-2). The MICs for the S1-1 and the S1-2 mutants increased 4- to 32-fold for levofloxacin and ciprofloxacin, respectively, compared to the parent strain. In contrast, there was only a modest 2-fold increase in the gepotidacin MIC for both mutants.
TABLE 1.
Gepotidacin MIC values for a collection of B. anthracis strains (N = 160)a
| Number of isolates | MIC range | MIC50 | MIC90 | |
|---|---|---|---|---|
| (μg/mL) | (μg/mL) | (μg/mL) | ||
| USAMRIID diversity set | 30 | 0.5–2 | 1 | 1 |
| USAMRIID collection | 100 | 0.12–1 | 0.5 | 0.5 |
| Rutgers | 30 | 0.25–1 | 0.5 | 1 |
MIC, minimum inhibitory concentration; USAMRIID, United States Army Medical Research Institute of Infectious Diseases.
TABLE 2.
Activity of gepotidacin, ciprofloxacin, and levofloxacin against attenuated B. anthracis single-step gyrase mutantsc
| Strain | Amino acid change | Frequency of isolationb | Gepotidacin | Ciprofloxacin | Levofloxacin |
|---|---|---|---|---|---|
| MIC | MIC | MIC | |||
| (μg/mL) | (μg/mL) | (μg/mL) | |||
| Parent ΔANRa | NA | NA | 1 | 0.03 | 0.06 |
| S1-1^ | S85L | 80% | 2 | 1 | 0.25 |
| S1-2^ | E89K | 20% | 2 | 1 | 0.25 |
Plasmid-cured B. anthracis Ames strain.
Laboratory-generated mutants; the frequency of isolation is from the original publication (30).
MIC: minimum inhibitory concentration; NA: not applicable.
Therapeutically administered gepotidacin enhances survival and reduces disease severity
NZW rabbits challenged with a targeted dose of 200 LD50 B. anthracis Ames spores received a mean inhalation exposure of 191 LD50 equivalents (range 106–258) for all study animals. All rabbits were negative for the PA electrochemiluminescence (PA-ECL) assay prior to challenge, and the median time for a positive PA-ECL post challenge was 28 h for both gepotidacin and saline groups. The time to trigger (positive PA-ECL) was comparable between groups (Table 3). All animals were bacteremic following trigger and prior to treatment, with an average bacterial burden of 3.0 Log10 colony forming unit (CFU)/mL in the blood. The median time to bacteremia was 25 h following challenge and was comparable between treatment groups: 24.7 h vs 25.5 h for gepotidacin and saline groups, respectively. No relationship between inhaled spore exposure and time to trigger was demonstrated. The median time to a significant increase in body temperature was comparable between the gepotidacin- and saline-treated groups: 29.3 h and 27.9 h, respectively (Table 3).
TABLE 3.
| Treatment | LD50 exposure average (range) | Time to triggera (hours post challenge) median (95%) | Time to onset of bacteremia (hours) median (95%) | % Bacteremic PTT |
Time to SIBT median (95%) |
|---|---|---|---|---|---|
| Gepotidacin | 180 (106–257) | 28.4 (24.5–35.4) | 24.7 (22.6–28.6) | 100 | 29.3 (25.2–30.6) |
| Saline | 212 (157–258) | 28.3 (23.3–38.3) | 25.5 (23.0–31.3) | 100 | 27.9 (23.9–33.4) |
Trigger = positive PA-ECL.
No statistical difference between the LD50 exposure, time from trigger to treatment, bacteremia onset, or challenge dose (two-tailed t-test).
LD50, lethal dose; PTT = prior to treatment; SIBT = significant increase in body temperature.
Gepotidacin was efficacious and provided a clear survival benefit compared to saline control (one-sided Boschloo’s test, P < 0.0001). As shown in Fig. 1, all six saline-treated animals succumbed 2 to 5 days post-challenge. Three animals died prior to euthanasia, and three were euthanized based on pre-defined euthanasia criteria. In contrast, 10 of the 11 (90.1%) gepotidacin-treated rabbits survived until the end of the study (day 28 post-challenge). The single animal that succumbed (euthanized) on day 3 post-challenge had catheter complications that impacted infusion start times and gepotidacin exposures (e.g., AUC, Cmax, and Tmax); in particular, the first dose exposure was approximately 10% of the AUC values noted for the other treated animals.
Fig 1.
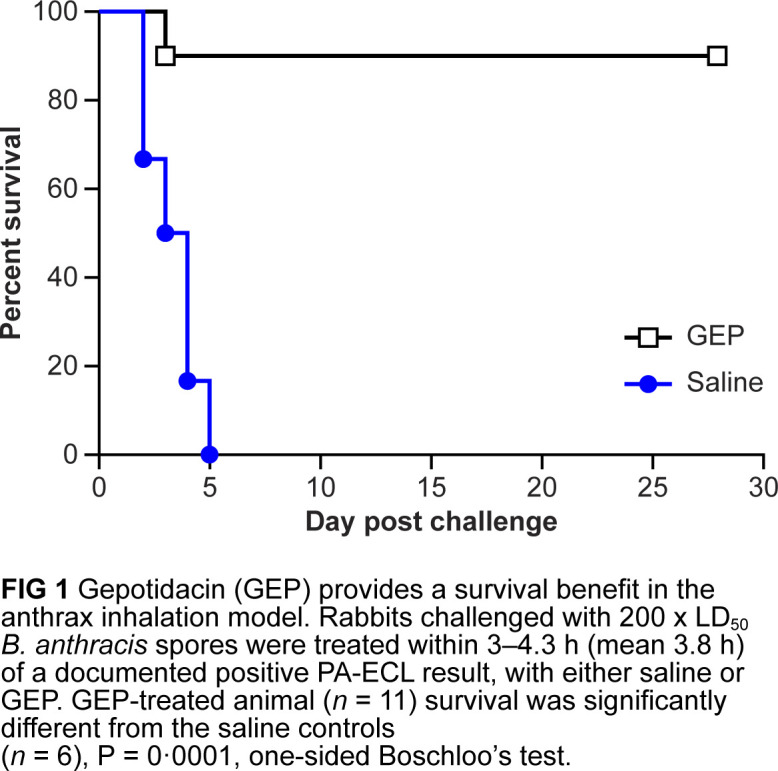
Gepotidacin (GEP) provided a survival benefit in the anthrax inhalation model. Rabbits challenged with 200 x LD50 B. anthracis spores were treated within 3 to 4.3 h (mean 3.8 h) of a documented positive protective antigen electrochemiluminescence result, with either saline or GEP. The survival of GEP-treated animals (n = 11) was significantly different from that of the saline control (n = 6), P = 0.0001, one-sided Boschloo’s test.
Gepotidacin treatment resolves bacteremia
Terminal blood cultures for all six saline–control animals were positive with high numbers of bacteria, (geometric mean 8.07 log10 CFU/mL), and all control animals demonstrated positive organ cultures at necropsy. In contrast, blood and tissues from all surviving animals and animals that succumbed during gepotidacin treatment were cultured for the presence of B. anthracis, and all survivors were blood culture-negative by day 7 post challenge. Blood cultures remained negative until the end of the study, i.e., day 28 post challenge. In addition, all organs from surviving gepotidacin-treated animals were culture-negative, with the exception of one animal that was positive for lung and one for spleen with colony-forming units below the limit that could be quantified by the methods used, likely representing latent spores. (Tables 4 and 5). The terminal blood culture from the single gepotidacin-treated animal that had catheter complications with reduced gepotidacin exposure and succumbed was positive; however, the numbers of bacteria were lower relative to those in controls (3.18 log10 CFU/mL compared to 8.07 log10 CFU/mL). Additionally, organ culture bacterial counts tended to be lower in this animal relative to the saline controls, with the exception of the brain, which had bacterial counts comparable to those in controls.
TABLE 4.
| Gepotidacin | Saline | |||
|---|---|---|---|---|
| Time point | No. of culture-positive/total no. of animals | Geometric mean (range) | No. of culture positive/total no. of animals | Geometric mean (range) |
| 24 h PC | 6/11 | 2.63 (<2.40, 3.04) | 3/6 | 2.88 (<2.40, 3.52) |
| 30 h PC | 6/7 | 2.78 (<2.40, 3.06) | 4/4 | 2.62 (<2.40, 3.17) |
| 36 h PC | 3/3 | 2.66 (<2.40, 3.11) | 2/2 | 2.83 (2.67, 3.0) |
| 42 h PC | 1/1 | <2.40 (--) | 0/0 | --(--) |
| PTT | 9/9b | 3.04 (<2.40, 3.82) | 6/6 | 3.30 (<2.40, 5.12) |
| 24 PTI | 9/11 | 3.70 (<2.40, 4.64) | 5/5 | 4.56 (4.05, 6.13) |
| Day 7 PC | 0/10 | --(--) | 0/0 | --(--) |
| Day 14 PC | 0/10 | --(--) | 0/0 | --(--) |
| Day 28 PC | 0/10 | --(--) | 0/0 | --(--) |
| Terminal | 1/1 | 3.18 (--) | 6/6 | 8.07 (6.15, 8.78) |
Parameter was log10-transformed for the analysis.
Blood samples were not available for two animals that were positive at time points prior to PTT.
-- Samples were not collected for this group at this time point, or all results were below the LLOQ, or there was a single observation for this group at this time point.
LLOQ = 2.40 log10 CFU/mL; LLOQ = lower limit of quantification; PC = post-challenge; PTT = prior to treatment ; PT1 = post first treatment.
TABLE 5.
| Gepotidacin | Saline | |||
|---|---|---|---|---|
| Tissue | No. of culture-positive/total no. of animals |
Geometric mean (range) |
No. of culture-positive/total no. of animals |
Geometric mean (range) |
| Heart | 1/11 | <3.40 (--) | 6/6 | 7.54 (<3.40, 7.94) |
| Brain | 1/11 | 6.84 (--) | 6/6 | 6.64 (3.68, 8.27) |
| Lung | 2/11b | <3.40 (<3.40) | 6/6 | 8.74 (4.49, 9.03) |
| Kidney | 1/11 | 3.44 (--) | 6/6 | 6.34 (<3.40, 8.15) |
| Spleen | 2/10b | 3.89 (<3.40, 4.22) | 6/6 | 8.31 (6.92, 9.20) |
| Mediastinal lymph node | 1/11 | 3.44 (--) | 6/6 | 7.75 (7.02, 8.48) |
Parameter was log10-transformed for the analysis. LLOQ = 3.40 log10 CFU/mL (triple plating was used for counts and <25 colonies were seen on two of three plates).
Includes a gepotidacin-treated survivor that was culture-positive at the study end.
-- Range of counts could not be determined since there was only one positive observation for this group at this time point or results were the same.
CFU, colony-forming units; LLOQ, lower limit of quantification.
Gepotidacin MICs were determined for isolates recovered from animals treated with gepotidacin and compared to those of the parent challenge strain to evaluate any development of resistance on therapy. All MICs were the same or within 1 dilution of the parent challenge strain, suggesting no development of resistance in this study.
Gepotidacin treatment resolves fever
The average baseline body temperature in the rabbit was 38.0 ± 0.7°C. After challenge, the mean body temperature for both groups of animals increased 1 to 2°C compared to that at baseline and resolved by day 7 post-challenge in the gepotidacin-treated group. The mean body temperature did not return to the baseline level for the saline–control group (Fig. 2).
Fig 2.
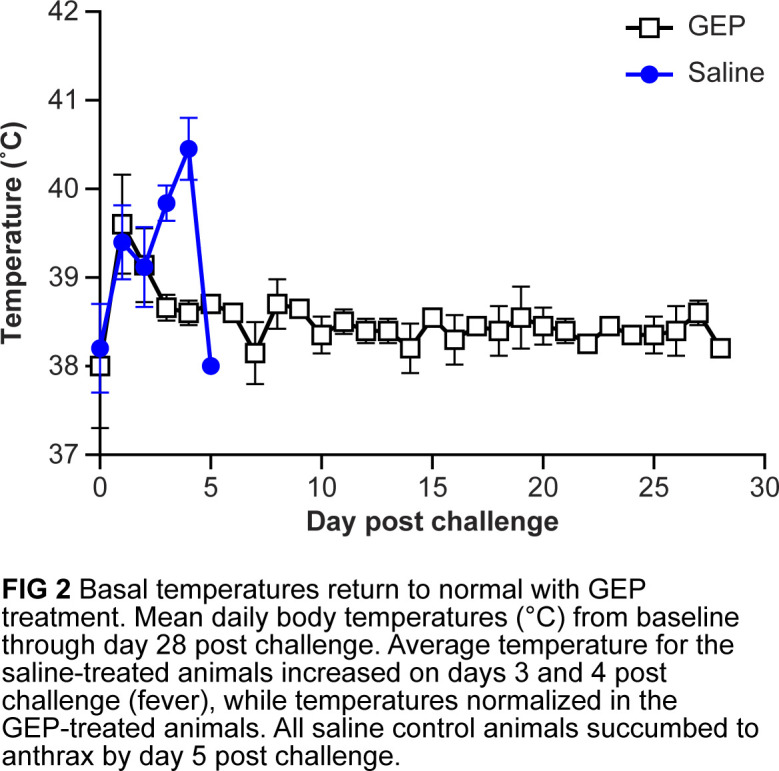
Basal temperatures returned to normal with gepotidacin (GEP) treatment. Mean daily body temperature (°C) from baseline through day 28 post-challenge. Average temperature for the saline-treated animals increased on days 3 and 4 post-challenge (fever), while temperatures normalized in the GEP-treated animals. All saline control animals succumbed to anthrax by day 5 post-challenge.
Gepotidacin-treated animals develop immune response to LT
Prior to challenge, no toxin neutralization assay (TNA) neutralizing titers were detected in any animals. Neutralization factor-50 (NF50) titers were detected only in gepotidacin-treated animals that survived beyond day 5. By day 10 post-challenge, all surviving animals had NF50 titers ranging from 0.23 to 9.5. At study termination (day 28 post-challenge), NF50 values increased and ranged from 1.55 to 17.5 (Fig. 3).
Fig 3.
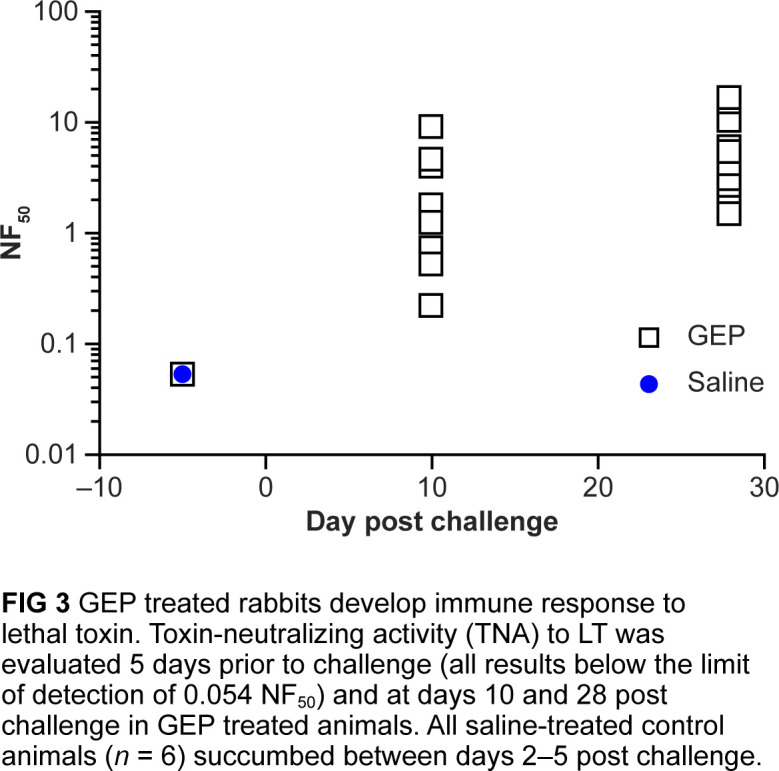
Gepotidacin (GEP)-treated rabbits developed an immune response to lethal toxin. Toxin-neutralizing activity to lethal toxin was evaluated 5 days prior to challenge (all results below the limit of detection of 0.054 NF50) and at days 10 and 28 post-challenge in GEP-treated animals. All saline-treated control animals (n = 6) succumbed between days 2 and 5 post-challenge.
Gepotidacin is efficacious at exposures below the predicted human exposure in rabbits
The administered gepotidacin dosing regimen of 114 (mg/kg/d) to rabbits provided free-drug Cmax (fCmax) and free-drug area under the concentration curve (fAUC) levels in plasma below exposures associated with the 1,000 mg i.v. TID human dose. Steady-state plasma pharmacokinetic profiles are shown in Fig. 4. Application of the previously determined rabbit plasma protein-binding value of 21.6% (20) and human plasma protein-binding value of 33.0% (31, 32) did not exceed fCmax in the rabbit compared to that in humans, fCmax (4.7 and 6.1 µg/mL, respectively), and the time to peak plasma concentration (Tmax) was 2 h in both rabbits and humans. The 24-h fAUC in the rabbit was also below the human fAUC (33 and 57 µg·h/mL, respectively).
Fig 4.
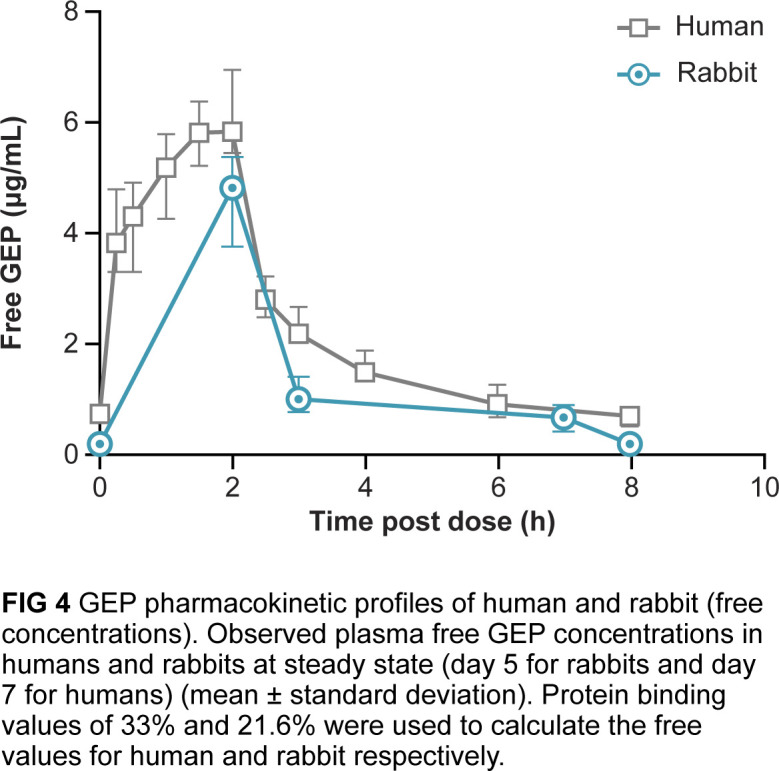
Gepotidacin (GEP) pharmacokinetic profiles of humans and rabbits (free concentrations). Observed plasma free gepotidacin concentrations in humans and rabbits at steady state (day 5 for rabbits and day 7 for humans) (mean ± standard deviation). Protein-binding values of 33% and 21.6% were used to calculate the free-drug values for human and rabbit respectively.
Gepotidacin treatment reduces the tissue bacterial burden and damage associated with B. anthracis infection
At necropsy, gross lesions were evident in three of six saline control animals that died or were euthanized due to anthrax. Gross lesions included enlarged mediastinal lymph nodes, with clear fluid in the abdominal, thoracic, and/or pericardial cavity, and red discoloration of the meninges. No gross lesions were seen in gepotidacin-treated rabbits, including the rabbit that succumbed to anthrax on day 3.
Microscopy findings of control animals and of the single gepotidacin-treated animal that succumbed were consistent with published reports of anthrax in rabbits (2). In these animals, there was fibrinous pneumonia with edema and intravascular and extravascular bacteria (Fig. 5). Intra- and/or extravascular bacteria were also identified in the meninges, brain, liver, spleen, and mediastinal lymph nodes, often associated with hemorrhage and necrosis.
Fig 5.
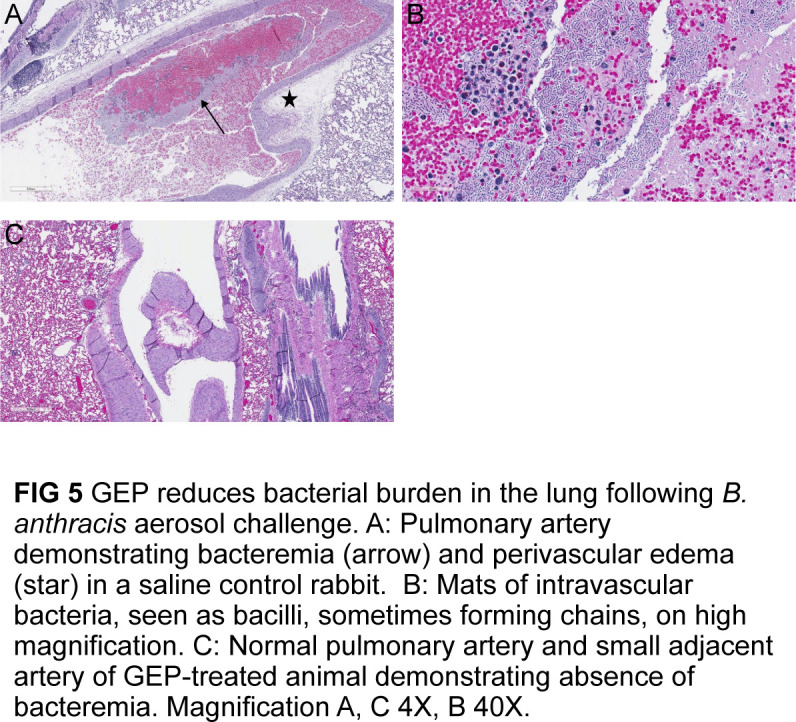
Gepotidacin reduced bacterial burden in the lungs following B. anthracis aerosol challenge. (A) Pulmonary artery demonstrating bacteremia (arrow) and perivascular edema (star) in the saline control rabbit. (B) Mats of intravascular bacteria, seen as bacilli, sometimes forming chains, on high magnification. (C) Normal pulmonary artery and small adjacent artery of gepotidacin-treated animals demonstrating absence of bacteremia. Magnification A and C, 4X; B, 40X.
DISCUSSION
Inhalational anthrax is a disease mediated by bacterial growth and toxemia, leading to inflammation, tissue damage, and, most often, acute death. In this study, gepotidacin, a novel, bactericidal, triazaacenaphthylene antibacterial rapidly cleared inhaled B. anthracis Ames strain from the blood and tissues of rabbits, reducing fever, inflammation, and subsequent tissue damage, resulting in enhanced survival. Animals that completed the gepotidacin dosing regimen survived until study end, whereas all saline controls succumbed to anthrax. Gepotidacin is widely distributed in tissues following i.v. infusion, with an alveolar macrophage:unbound plasma AUC ratio of 178:1(33). Once inside the lungs, anthrax spores are engulfed by alveolar macrophages and transported to mediastinal and peribronchial lymph nodes where the spores may germinate. Given the importance of macrophages in the progression of infection (1, 6), the high concentrations of gepotidacin in the macrophages could be beneficial in terms of cell killing once the spores have germinated, potentially limiting progression of the disease. Ionin et al. recently described a TNA NF50 threshold predictive of immunity to LT and survival of rabbits following anthrax vaccination with subsequent re-challenge (30). In those studies, a TNA NF50 titer of 0.56 corresponded to a 70% probability of survival in rabbits following re-challenge. In the current study, all gepotidacin-treated survivors assessed had an NF50 titer greater than 0.56 (1.5–17.5) on day 28 post-challenge. These data suggest that following treatment of an active infection with gepotidacin, surviving rabbits may mount an immune response that would enhance survival following exposure to a new aerosol challenge or potential germination of residual spores. In addition, gepotidacin demonstrated in vitro activity against diverse sets of B. anthracis isolates, suggesting gepotidacin would provide coverage against isolates that may be encountered in the real world.
One of the therapeutic challenges of anthrax infection is that it is the spore, rather than the vegetative cell, that is the infectious agent, and residual spores in the body may germinate after cessation of antibiotic therapy, producing an active infection. Following inhalational challenge, spores are phagocytosed by resident alveolar macrophages and dendritic cells and transported via the circulatory system to the mediastinal and peribronchial lymph nodes where they germinate. In this study, consistent with the hematologic dissemination from the lungs, intravascular bacteria were found in distal organs, such as the spleen, liver, and brain, of saline-treated controls. All animals had minimal to mild histiocytic cellular infiltrates within the lungs, indicative of an immune response to infection; however, there were no microscopic lesions consistent with anthrax in the gepotidacin-treated animals.
Steady-state plasma concentrations for rabbits given a dosing regimen designed to achieve plasma exposure profiles that simulate the human 1,000 mg i.v. TID regimen demonstrated Cmax and AUC values below those seen clinically in humans, demonstrating that the 1,000 mg i.v. TID regimen achieves gepotidacin exposures shown to be efficacious in these pivotal Animal Rule studies.
The FDA Animal Rule provides the basis to establish efficacy in support of human treatment, using a well-characterized animal efficacy model, in cases where it is not practical or ethical to conduct testing on humans (26, 27). The NZW rabbit model of inhalational anthrax utilized for this study has been shown to be an appropriate model for disease pathogenesis in humans. Furthermore, the similarities of the rabbit model to human disease make it suitable for evaluation of antibacterial therapies. The study presented here was blinded and randomized and designed to provide pivotal Animal Rule efficacy data for use of gepotidacin for anthrax treatment. In vitro studies against diverse collections of B. anthracis demonstrated no pre-existing gepotidacin resistance, supporting gepotidacin as a potential treatment option against anthrax strains resistant to standard antibiotic classes. The combination of the novel mode of action, in vitro activity, a favorable pharmacodynamic profile, and efficacy in the rabbit anthrax model supports gepotidacin as a potential treatment for anthrax, including infections caused by antibiotic-resistant strains.
MATERIALS AND METHODS
B. anthracis isolate collections and MIC testing
MICs were determined by the broth microdilution method on 130 isolates at USAMRIID, including at least 17 genotypes from a diverse geographic distribution (34). Thirty additional isolates were tested from the culture collection at Rutgers University. MICs were also determined against the attenuated (plasmid-cured ΔANR) B. anthracis Ames strain and two single-step DNA gyrase mutant isolates at USAMRIID (35, 36). All testing was conducted according to Clinical and Laboratory Standards Institute (CLSI) guidelines (37, 38). Gepotidacin MICs were determined for the in vivo B. anthracis Ames challenge strain and on positive cultures from terminal blood or tissue sample plates collected from gepotidacin-treated animals. Three to five colonies were assayed in triplicate from the same inoculum.
Circulating PA levels (PA-ECL)
The presence of B. anthracis PA suggests an active anthrax infection (28, 30, 39) and has been correlated with bacteremia. Prior to the challenge, and at 6-h intervals beginning 24 h post-challenge until 72 h post-challenge, 1 mL of blood was collected in serum separator tubes, and an aliquot of the serum was evaluated in the PA-ECL assay. A positive PA-ECL result was used as the trigger for treatment (29, 40).
Toxin neutralization assay
The TNA is designed to measure the functional ability of serum antibodies to neutralize B. anthracis lethal toxin activity. The assay was performed as previously described using an in vitro cytotoxicity assay from the serum sampled, as outlined in Fig. 3 (41).
Preparation of test article and dose formula analysis
Gepotidacin was dissolved in 0.9% sterile saline (pH 5.0 to 5.5) and filter-sterilized. All dosing solutions were prepared daily and maintained at ambient temperature for infusion. Aliquots from gepotidacin formulations were analyzed on the day of preparation to confirm the concentration of gepotidacin in the dose solution prior to use in the study.
In vivo NZW rabbit model of inhalational anthrax
All animal procedures were approved by the GSK’s and Battelle’s (a contract research laboratory with expertise in FDA Animal Rule studies) Institutional Animal Care and Use Committee and the U.S. Army Medical Research and Materiel Command’s Animal Care and Use Review Office and were conducted in an Association for Accreditation and Assessment Laboratory Animal Care-accredited facility in compliance with U.S. regulations governing the housing and use of animals. This study was conducted in coordination with the FDA and was completed under the agency’s Special Protocol Assessment process. All exposures and assays were performed in a Biosafety Level-3 laboratory registered and approved with the Centers for Disease Control and Prevention (CDC) and inspected by the U.S. Departments of Defense and Agriculture. The Battelle CDC principal investigator approved the use of B. anthracis on rabbits in this study. In addition, this study was conducted in compliance and reviewed by the Chair of Battelle’s Biological Safety Committee, which has oversight of all risk group 2 and 3 biological research at Battelle. All studies were conducted humanely and complied with national laws, guidelines, and company policies for the care, welfare, and treatment of animals.
Test system
Male and female NZW rabbits (Oryctolagus cuniculus) weighing at least 3 kg were obtained from Covance (Denver, PA), a U.S. Department of Agriculture-licensed facility. All rabbits were confirmed negative for prior exposure to B. anthracis using the TNA and for the presence of an active B. anthracis infection using the PA-ECL assay prior to challenge. Rabbits were housed individually, in stainless steel cages, on racks equipped with automatic watering systems. Rabbits were surgically implanted with dual cath-in-cath vascular access ports (VAPs) for antibacterial dosing and for blood collection. A tether was attached to the jacket and to a swivel in the top of the cage to allow for free movement of the animal without twisting the infusion line. Rabbits were observed for clinical signs either twice daily [BID (before challenge and from day 7 through day 28 post-challenge)] or four times daily [QID (from day 1 to day 7 post-challenge)]. Any animal judged to be moribund was humanely euthanized.
Surgical procedures
Prior to animal arrival, two VAPs were surgically implanted into rabbits by the vendor, one in the jugular vein and the other in the femoral vein. The port from the jugular vein catheter was utilized for blood sampling, and the port from the femoral vein catheter was used for dosing.
Body temperature
Rabbits were sedated with acepromazine (1 to 5 mg/kg, intramuscularly), and transponders (IPTT-300, BMDS, Seaford, DE) were injected subcutaneously into the rump of each rabbit. Mean baseline temperatures were established for each rabbit from day 5 until just prior to challenge. Following challenge, temperatures were recorded hourly for the first 3 days post-challenge and BID from day 4 post-challenge through study termination (day 28 post-challenge).
Inhalational challenge
Rabbits were exposed (muzzle only) to an aerosolized dose of B. anthracis spores, targeting 200 × LD50s (2.1 × 107 spores) by real-time plethysmography (42). Post-exposure aerosol concentrations of B. anthracis were quantified by determination of the CFU from the effluent streams of the animal exposure port by an in-line impinger (Model 7541, Ace Glass Incorporated, Vineland, NJ) (29). The gepotidacin MIC for the Ames in vivo challenge strain determined at Battelle was 2 µg/mL.
Randomization
Animals were randomized to the treatment group at the time of a positive PA-ECL result (trigger).
Test article administration
Intravenous infusions of gepotidacin or vehicle (0.9% saline) were administered based on a positive serum PA result in the ECL assay (trigger for treatment). The first treatment was initiated within 3 to 4.3 h (mean 3.8 h) of a documented positive ECL result. Gepotidacin dose or a corresponding volume of the saline vehicle was based on the individual body weights collected on the day of aerosol exposure. Gepotidacin was administered as a 2-h infusion (30 mg/kg), followed 1 h later by a 4-h infusion of 8 mg/kg. This dose regimen was repeated TID every 24 h, for a total daily dose of 114 mg/kg (six total infusions). Treatment was continued for 5 consecutive days. Doses were designed to simulate plasma exposures consistent with a human 1,000 mg i.v. TID dose in terms of the shape of the exposure profile and daily fAUC values.
Gepotidacin plasma analysis
On treatment days 1 and 5, blood samples were collected prior to treatment (PTT) and at 2 h, 3 h, 7 h, and 8 h after start of treatment. Blood samples collected from the VAP were chilled, centrifuged for plasma processing, filtered, tested for sterility, and maintained at ≤–70°C until assayed. Gepotidacin concentrations in plasma were analyzed using ultra-high performance liquid chromatography (UHPLC) with tandem mass spectrometry (MS/MS) detection. Tmax was observed at 2 h after the start of the first and 25th infusion for all animals. There was no marked (>2 fold) difference in systemic exposure (mean Cmax and AUC0-t values) between females and males after either the first or 25th infusions. Free gepotidacin pharmacokinetic analysis results were based on steady-state sampling on day 5 for rabbits and day 7 for humans.
Human pharmacokinetics
The pharmacokinetics of gepotidacin following repeat i.v. dosing were previously determined in healthy human volunteers in accordance with the International Conference on Harmonization-Good Clinical Practice guidelines and applicable subject privacy requirements and guidelines (43). Eight-hour dosing intervals were maintained for TID regimens.
Bacteriology assessments
At predetermined time points, whole blood was collected in sodium polyanethol sulfonate tubes and was assessed quantitatively for the presence of bacteria with colony morphology consistent with that of B. anthracis. Following necropsy and prior to fixation, ~1 cm3 samples of the heart, brain, lung, kidney, spleen, lymph nodes, and gross lesions from each animal were aseptically collected, weighed, homogenized, and cultured by spread plate enumeration and assessed quantitatively for colony morphology consistent with that of B. anthracis. The bacterial burden was reported as CFU/g tissue or CFU/mL of blood. Gepotidacin MICs were determined on any isolates recovered from blood and/or tissues of gepotidacin-treated animals by selecting three to five colonies and testing MICs in triplicate from the same inoculum.
Necropsy/histopathology
A complete necropsy was performed on all study animals found dead, euthanized in moribund condition, or at study termination. Gross necropsies included examinations of the external surfaces of the body; all orifices; and the cranial, thoracic, and abdominal cavities and their contents. Target tissues including those of the brain, lungs, liver, spleen, and mediastinal lymph nodes along with gross lesions were collected, preserved in 10% neutral-buffered formalin, stained with hematoxylin and eosin, and examined microscopically to confirm death or illness due to anthrax, by a study pathologist who was blinded to the treatment group.
Statistical analyses
Boschloo’s tests were conducted in R (version 3.3.1 64-bit). For a one-sided test, the 0.025 level was considered significant. Analyses were also conducted with SAS (version 9.4 64-bit). Other analyses were conducted using GraphPad Prism version 7.03, where a P-value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This work was largely funded by the DTRA under Agreement No. HDTRA1-07-9-0002. We would especially like to thank the DTRA project manager Amanda Horstman Smith for her technical expertise and insights into biothreat Animal Rule studies. We also acknowledge the laboratory staff at USAMRIID who performed the in vitro work presented in this manuscript, including Lynda Miller and Stephanie Halasohoris. In addition, we acknowledge the staff at Battelle, including Denise Pfefferle, Megan Posey, Joe Wickham, Michelle Sunderman, Jill Staugler, Andy Puttmann, Brent McCracken, Meredith Kirkbride, Mike Mladineo, and Dennis Pak. Further, we thank the entire GSK team, including Jennifer Hoover, Scott Sucoloski, Sean Maguire, Lorrie Day, Steve Lerman, and Kitaw Negash. In particular, we acknowledge Mick Gwynn, the discoverer of the novel bacterial topoisomerase inhibitors (NBTI) class of antibacterials, who passed away on December 16, 2013.
The funders had no role in study design, data collection, and interpretation or the decision to submit the work for publication. Editorial support, under the guidance of the authors, was provided by Pearl Okungbowa, BSc, of Ashfield MedComms, an Inizio company (Manchester, UK), and was funded by GSK. Trademarks are the property of their respective owner(s) Aerodynamic Particle Sizer Spectrometer (TSI Incorporated); WinNonlin (Certara).
This work was supported by GSK and largely funded by the DTRA under Agreement No. HDTRA1-07-9-0002. Editorial support for the manuscript was funded by GSK.
J.J.H., K.O.D., C.J., F.M., M.H., and L.H. designed the experiments. C.F. reviewed pathology data and generated images. M.H. and L.Q. performed the gepotidacin modeling studies. J.J.H., K.O.D., C.J., F.M., M.H., C.F., and J.N. analyzed the data. J.J.H. and C.J. co-wrote the manuscript. K.O.D., L.H., and C.F. edited the manuscript and provided critical review. S.D., C.S., and J.H. oversaw the execution of the M.I.C. studies at USAMRIID. R.R. oversaw the execution of the M.I.C. study at Rutgers University School of Medicine. All authors have seen and approved the final version of the manuscript.
Contributor Information
Karen O'Dwyer, Email: Karen.m.odwyer@gsk.com.
James E. Leggett, Providence Portland Med Ctr, Portland, Oregon, USA
DATA AVAILABILITY
Data associated with this study are presented in the paper. Requests for materials will be reasonably considered, and such requests should be addressed to the corresponding author.
REFERENCES
- 1. Cote CK, Welkos SL. 2015. Anthrax toxins in context of Bacillus anthracis spores and spore germination. Toxins (Basel) 7:3167–3178. doi: 10.3390/toxins7083167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Twenhafel NA. 2010. Pathology of inhalational anthrax animal models. Vet Pathol 47:819–830. doi: 10.1177/0300985810378112 [DOI] [PubMed] [Google Scholar]
- 3. Friebe S, van der Goot FG, Bürgi J. 2016. The ins and outs of anthrax toxin. Toxins (Basel) 8:69. doi: 10.3390/toxins8030069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moayeri M, Leppla SH. 2009. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med 30:439–455. doi: 10.1016/j.mam.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu S, Moayeri M, Leppla SH. 2014. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol 22:317–325. doi: 10.1016/j.tim.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kamal SM, Rashid A, Bakar MA, Ahad MA. 2011. Anthrax: an update. Asian Pac J Trop Biomed 1:496–501. doi: 10.1016/S2221-1691(11)60109-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. World Health Organization . 2008. Edited by Turnbull P. Anthrax in humans and animals. 4th ed. WHO Press, Geneva Switzerland. [Google Scholar]
- 8. Yee SB, Hatkin JM, Dyer DN, Orr SA, Pitt MLM. 2010. Aerosolized Bacillus anthracis infection in New Zealand white rabbits: natural history and intravenous levofloxacin treatment. Comp Med 60:461–468. [PMC free article] [PubMed] [Google Scholar]
- 9. Lovchik JA, Drysdale M, Koehler TM, Hutt JA, Lyons CR. 2012. Expression of either lethal toxin or edema toxin by Bacillus anthracis is sufficient for virulence in a rabbit model of inhalational anthrax. Infect Immun 80:2414–2425. doi: 10.1128/IAI.06340-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kammanadiminti S, Patnaikuni RK, Comer J, Meister G, Sinclair C, Kodihalli S. 2014. Combination therapy with antibiotics and anthrax immune globulin intravenous (AIGIV) is potentially more effective than antibiotics alone in rabbit model of inhalational anthrax. PLoS One 9:e106393. doi: 10.1371/journal.pone.0106393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Migone T-S, Subramanian GM, Zhong J, Healey LM, Corey A, Devalaraja M, Lo L, Ullrich S, Zimmerman J, Chen A, Lewis M, Meister G, Gillum K, Sanford D, Mott J, Bolmer SD. 2009. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med 361:135–144. doi: 10.1056/NEJMoa0810603 [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto BJ, Shadiack AM, Carpenter S, Sanford D, Henning LN, O’Connor E, Gonzales N, Mondick J, French J, Stark GV, Fisher AC, Casey LS, Serbina NV. 2016. Efficacy projection of obiltoxaximab for treatment of inhalational anthrax across a range of disease severity. Antimicrob Agents Chemother 60:5787–5795. doi: 10.1128/AAC.00972-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewis CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, Gwynn MN. 2010. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466:935–940. doi: 10.1038/nature09197 [DOI] [PubMed] [Google Scholar]
- 14. Gibson EG, Bax B, Chan PF, Osheroff N. 2019. Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect Dis 5:570–581. doi: 10.1021/acsinfecdis.8b00315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, Leydon VR, Miles TJ, Pearson ND, Perera RL, Shillings AJ, Gwynn MN, Bax BD. 2010. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol 17:1152–1153. doi: 10.1038/nsmb.1892 [DOI] [PubMed] [Google Scholar]
- 16. Biedenbach DJ, Bouchillon SK, Hackel M, Miller LA, Scangarella-Oman NE, Jakielaszek C, Sahm DF. 2016. In vitro activity of gepotidacin, a novel triazaacenaphthylene bacterial topoisomerase inhibitor, against a broad spectrum of bacterial pathogens. Antimicrob Agents Chemother 60:1918–1923. doi: 10.1128/AAC.02820-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flamm RK, Farrell DJ, Rhomberg PR, Scangarella-Oman NE, Sader HS. 2017. Gepotidacin (GSK2140944) in vitro activity against Gram-positive and Gram-negative bacteria. Antimicrob Agents Chemother 61:e00468-17. doi: 10.1128/AAC.00468-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobsson S, Golparian D, Scangarella-Oman N, Unemo M. 2018. In vitro activity of the novel triazaacenaphthylene gepotidacin (GSK2140944) against MDR Neisseria gonorrhoeae. J Antimicrob Chemother 73:2072–2077. doi: 10.1093/jac/dky162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mushtaq Shazad VA, Zahra S, Michelle C, Helen F, Do NV, Martin DElizabeth De PClaire J, Gauri G, Neil W. 2019. In-vitro activity of gepotidacin, a novel triazaacenaphthylene topoisomerase IV and DNA gyrase inhibitor, against multidrug-resistant gram-negative bacteria and Staphylococcus saprophyticus Abstr European Congress of Clinical Microbiology & Infectious Diseases (ECCMID), p 13–16, Amsterdam, The Netherlands [Google Scholar]
- 20. Gruber R, O’Dwyer K, Qian L, Henning L, Demons S, Jakielaszek C. 2016. Novel antibiotic gepotidacin is active in vitro and efficacious in a rabbit aerosol challenge treatment model of Bacillus anthracis Abstr ASM Biodefense and Emerging Diseases Research Meeting, p 8–10, Arlington, VA, USA [Google Scholar]
- 21. So W, Crandon JL, Nicolau DP. 2015. Pharmacodynamic profile of GSK2140944 against methicillin-resistant Staphylococcus aureus in a murine lung infection model. Antimicrob Agents Chemother 59:4956–4961. doi: 10.1128/AAC.00625-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. VanScoy BD, Scangarella-Oman NE, Fikes S, Min S, Huang J, Ingraham K, Bhavnani SM, Conde H, Ambrose PG. 2020. Relationship between gepotidacin exposure and prevention of on-therapy resistance amplification in a Neisseria gonorrhoeae hollow-fiber in vitro infection model. Antimicrob Agents Chemother 64:e00521-20. doi: 10.1128/AAC.00521-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jakielaszek C, Hilliard JJ, Mannino F, Hossain M, Qian L, Fishman C, Chou Y-L, Henning L, Novak J, Demons S, Hershfield J, O’Dwyer K. 2023. Efficacy of intravenously administered gepotidacin in cynomolgus macaques following a francisella tularensis inhalational challenge. Antimicrob Agents Chemother 67:e0138122. doi: 10.1128/aac.01381-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jakielaszek C, Hossain M, Qian L, Fishman C, Widdowson K, Hilliard JJ, Mannino F, Raychaudhuri A, Carniel E, Demons S, Heine HS, Hershfield J, Russo R, Mega WM, Revelli D, O’Dwyer K. 2022. Gepotidacin is efficacious in a nonhuman primate model of pneumonic plague. Sci Transl Med 14:eabg1787. doi: 10.1126/scitranslmed.abg1787 [DOI] [PubMed] [Google Scholar]
- 25. Hupert N, Bearman GML, Mushlin AI, Callahan MA. 2003. Accuracy of screening for inhalational anthrax after a bioterrorist attack. Ann Intern Med 139:337–345. doi: 10.7326/0003-4819-139-5_part_1-200309020-00009 [DOI] [PubMed] [Google Scholar]
- 26. U.S. Department of Health and Human Services Food and Drug Administration . 2018. Anthrax: developing drugs for prophylaxis of inhalational anthrax guidance for industry. Center for Drug Evaluation and Research (CDER), US Food and Drug Administration. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/anthrax-developing-drugs-prophylaxis-inhalational-anthrax-guidance-industry [Google Scholar]
- 27. U.S. Department of Health and Human Services Food and Drug Administration . 2015. Product development under the animal rule guidance for industry. Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), FDA. Available from: http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm [Google Scholar]
- 28. Nagy CF, Mondick J, Serbina N, Casey LS, Carpenter SE, French J, Guttendorf R. 2017. Animal-to-human dose translation of obiltoxaximab for treatment of inhalational anthrax under the US FDA animal rule. Clin Transl Sci 10:12–19. doi: 10.1111/cts.12433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Comer JE, Ray BD, Henning LN, Stark GV, Barnewall RE, Mott JM, Meister GT. 2012. Characterization of a therapeutic model of inhalational anthrax using an increase in body temperature in New Zealand white rabbits as a trigger for treatment. Clin Vaccine Immunol 19:1517–1525. doi: 10.1128/CVI.00292-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ionin B, Hopkins RJ, Pleune B, Sivko GS, Reid FM, Clement KH, Rudge TL, Stark GV, Innes A, Sari S, Guina T, Howard C, Smith J, Swoboda ML, Vert-Wong E, Johnson V, Nabors GS, Skiadopoulos MH. 2013. Evaluation of immunogenicity and efficacy of anthrax vaccine adsorbed for postexposure prophylaxis. Clin Vaccine Immunol 20:1016–1026. doi: 10.1128/CVI.00099-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoover JL, Singley CM, Elefante P, Rittenhouse S. 2019. Efficacy of human exposures of gepotidacin (GSK2140944) against Escherichia coli in a rat pyelonephritis model. Antimicrob Agents Chemother 63:e00086-19. doi: 10.1128/AAC.00086-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hossain M, Zhou M, Tiffany C, Dumont E, Darpo B. 2017. A phase I, randomized, double-blinded, placebo-and moxifloxacin-controlled, four-period crossover study to evaluate the effect of gepotidacin on cardiac conduction as assessed by 12-lead electrocardiogram in healthy volunteers. Antimicrob Agents Chemother 61:e02385-16. doi: 10.1128/AAC.02385-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barth A, Hossain M, Tiffany C, Perry CR, Dumont EF. 2021. “Distribution and exposure of Gepotidacin (Gsk2140944) in tissues and body fluids of healthy and infected participants” Abstr 31st European Congress of Clinical Microbiology & Infectious Diseases (ECCMID) Virtual Meeting [Google Scholar]
- 34. Keim P, Price LB, Klevytska AM, Smith KL, Schupp JM, Okinaka R, Jackson PJ, Hugh-Jones ME. 2000. Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J Bacteriol 182:2928–2936. doi: 10.1128/JB.182.10.2928-2936.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grohs P, Podglajen I, Gutmann L. 2004. Activities of different fluoroquinolones against Bacillus anthracis mutants selected in vitro and harboring topoisomerase mutations. Antimicrob Agents Chemother 48:3024–3027. doi: 10.1128/AAC.48.8.3024-3027.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Price LB, Vogler A, Pearson T, Busch JD, Schupp JM, Keim P. 2003. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob Agents Chemother 47:2362–2365. doi: 10.1128/AAC.47.7.2362-2365.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clinical and Laboratory Standards Institute . 2015. M45-A3: methods for antimicrobial dilution and disk susceptibility testing of infrequently isolated or fastidious bacteria. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania. [DOI] [PubMed] [Google Scholar]
- 38. Clinical and Laboratory Standards Institute . 2018. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania. [Google Scholar]
- 39. Corey A, Migone TS, Bolmer S, Fiscella M, Ward C, Chen C, Meister G. 2013. Bacillus anthracis protective antigen kinetics in inhalation spore-challenged untreated or levofloxacin/ raxibacumab-treated New Zealand white rabbits. Toxins (Basel) 5:120–138. doi: 10.3390/toxins5010120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Henning LN, Comer JE, Stark GV, Ray BD, Tordoff KP, Knostman KAB, Meister GT. 2012. Development of an inhalational Bacillus anthracis exposure therapeutic model in cynomolgus macaques. Clin Vaccine Immunol 19:1765–1775. doi: 10.1128/CVI.00288-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Henning LN, Carpenter S, Stark GV, Serbina NV. 2018. Development of protective immunity in New Zealand white rabbits challenged with Bacillus anthracis spores and treated with antibiotics and obiltoxaximab, a monoclonal antibody against protective antigen. Antimicrob Agents Chemother 62:e01590-17. doi: 10.1128/AAC.01590-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zaucha GM, Pitt LM, Estep J, Ivins BE, Friedlander AM. 1998. The pathology of experimental anthrax in rabbits exposed by inhalation and subcutaneous inoculation. Arch Pathol Lab Med 122:982–992. [PubMed] [Google Scholar]
- 43. Tiffany HM, McDonald M, Lerman S, Dumont EF. 2014. “Safety and pharmacokinetics of repeat escalating IV doses of Gsk2140944, a novel bacterial Topoisomerase inhibitor” Abstr 54th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy American Society for Microbiology, Washington, DC [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data associated with this study are presented in the paper. Requests for materials will be reasonably considered, and such requests should be addressed to the corresponding author.