

ABSTRACT
Screening a library of 1,200 preselected kinase inhibitors for anti-human rhinovirus 2 (HRV-2) activity in HeLa cells identified a class of epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKI) as effective virus blockers. These were based on the 4-anilinoquinazoline-7-oxypiperidine scaffold, with the most potent representative AZ5385 inhibiting the virus with EC50 of 0.35 µM. Several structurally related analogs confirmed activity in the low µM range, while interestingly, other TKIs targeting EGFR lacked anti-HRV-2 activity. To further probe this lack of association between antiviral activity and EGFR inhibition, we stained infected cells with antibodies specific for activated EGFR (Y1068) and did not observe a dependency on EGFR-TK activity. Instead, consecutive passages of HRV-2 in HeLa cells in the presence of a compound and subsequent nucleotide sequence analysis of resistant viral variants identified the S181T and T210A alterations in the major capsid VP1 protein, with both residues located in the vicinity of a known hydrophobic pocket on the viral capsid. Further characterization of the antiviral effects of AZ5385 showed a modest virus-inactivating (virucidal) activity, while anti-HRV-2 activity was still evident when the inhibitor was added as late as 10 h post infection. The RNA copy/infectivity ratio of HRV-2 propagated in AZ5385 presence was substantially higher than that of control HRV indicating that the compound preferentially targeted HRV progeny virions during their maturation in infected cells. Besides HRV, the compound showed anti-respiratory syncytial virus activity, which warrants its further studies as a candidate compound against viral respiratory infections.
KEYWORDS: human rhinovirus, hydrophobic pocket, epidermal growth factor receptor, kinase inhibitor, escape mutant
INTRODUCTION
Human rhinovirus (HRV) is one of the leading causes of upper respiratory tract infections in humans. HRV infects predominantly the ciliated cells of nasal and posterior nasopharynx epithelium causing usually a mild and self-limiting disease. However, in some cases, the pathogen can spread to the lower airways, causing bronchiolitis and pneumonia (1), which may result in the deterioration of pre-existing respiratory diseases such as asthma or COPD [reviewed in reference (2, 3)]. The HRV particle comprises a single-stranded positive sense RNA genome within an icosahedral capsid. The capsid is assembled from 60 capsomers each composed of viral proteins VP1, VP2, VP3, and VP4. Genetic relatedness analysis (4) and antigenic studies reveal that HRV currently exists in over 170 different types which are traditionally classified as HRV-A, HRV-B, or HRV-C species. The majority of HRV-A and all HRV-B types use the intracellular cell adhesion molecule 1 (ICAM-1) as the cellular receptor (5–7). The ICAM-1 binding site is provided by specific amino acid residues in the VP1 protein, which form a deep cleft or canyon at the capsid surface around the fivefold axes (8, 9). Another common structural feature of the HRV-A and HRV-B species is a surface-accessible hydrophobic pocket that is located beneath the floor of the canyon (10). This cavity is frequently occupied by a hydrophobic molecule referred to as the “pocket factor,” as exemplified by a C12 fatty acid in HRV-A type 16 (HRV-16) (11) and HRV-A type 2 (HRV-2) (12). The pocket factor is believed to stabilize the virus particles; however, its exact role in the HRV life cycle is unclear (13). Nonetheless, this hydrophobic pocket is also a site of activity for several classes of anti-HRV compounds, including pleconaril with observed efficacy in clinical studies but where progress has been slow due to safety concerns (10, 14–16). Besides ICAM-1, other receptors can mediate HRV binding. Members of the minor group of HRV-A, exemplified by HRV-2, use different proteins from the low-density lipoprotein receptor (LDLR) family (17). These engage with a site of the capsid located at the star-shaped dome on the fivefold axis (18, 19) outside the ICAM-1 binding site in the HRV major group. In addition, the HRV-C types are known to use the cadherin-related family member 3 as cellular receptor (20).
Viruses as strict intracellular parasites are critically dependent on specific cellular components and metabolic pathways. These among others include cellular kinases that play a regulatory role in a cell. Several kinases exemplified by phosphatidylinositol 4 kinase IIIB (21), phosphatidylinositol 3 kinase (22), and protein kinase D (23) are known to be exploited by HRV to promote specific aspects of the virus life cycle. Some kinases, such as the epidermal growth factor receptor-tyrosine kinase (EGFR-TK), are activated by many different viruses and are therefore regarded as potentially druggable targets for the development of pan-antiviral compounds [reviewed in reference (24, 25)]. Building on this knowledge, here, we screened a collection of 1,200 preselected kinase inhibitors for anti-HRV-2 activity in HeLa cells. Several EGFR-TK inhibitors (EGFR-TKI), each comprising the 4-anilinoquinazoline-7-oxypiperidine scaffold, were identified as hits. Additional studies on the hit exhibiting the most pronounced anti-HRV-2 activity showed that it targets the hydrophobic pocket of the viral capsid.
MATERIALS AND METHODS
Cells and viruses
Human epithelial adenocarcinoma cells of uterine cervix (HeLa; ATCC CCL-2) were grown in Eagle’s minimum essential medium (EMEM) supplemented with 5% fetal calf serum and 1% pest stock. The H1-HeLa cell subline (ATCC CRL-1958) was grown in Dulbecco modified Eagle’s medium (DMEM) supplemented with 10% fetal and 1% pest stock. The HRV-A type 2 (HRV-2; ATCC VR-482) was propagated at 34°C in nearly confluent (60%–90%) HeLa cells maintained in EMEM supplemented with 2% fetal calf serum, 1% pest stock, 1% L-glutamine stock, 30 mM MgCl2, and 20 mM HEPES (pH 7.1) (EMEM-M). The HRV-A type 16 (HRV-16; ATCC VR-283) and HRV-B type 14 (HRV-14; ATCC VR-284) were propagated in H1-HeLa cells in the same manner as HRV-2 except that DMEM was used instead of EMEM. When the virus-induced cytopathic effect (CPE) was 80%–90% advanced, the cells were harvested and subjected to three freeze/thaw cycles at −80°C ethanol/37°C water bath. Following centrifugation at 1,000 × g for 5 min, the infectious supernatant was aliquoted and stored at −80°C. In some experiments, human respiratory syncytial virus (RSV) strain A2 (ATCC VR-1540), SARS-2 coronavirus clinical isolate DE-P3 (Section for Clinical Virology, Gothenburg, 2020), herpes simplex virus type 2 (HSV-2) strain 333 (26), and poliovirus Sabin type 1 (obtained from National Institute for Infectious Diseases, Stockholm, Sweden) were used.
Anti-HRV-2 screening of kinase inhibitor library
As part of the Open Innovation program instituted by AstraZeneca, a library of 1,200 well-annotated public and proprietary kinase inhibitors in 96-well V-type plate format was made available for performing antiviral screening. Compounds were provided as 150 nL aliquots of 1 and 10 mM solutions, dissolved in DMSO and stored at −20°C. The screening assay was performed using ~70%–90% confluent monolayers of HeLa cells which were seeded (2 × 104 cells/100 µL of medium) in 96-well F-type plates the day prior to the experiment. The library plates were placed at 37°C for 5 min and then centrifuged shortly, and 30 µL of EMEM-M was added. After shaking the plates (750 rotation cycles for 30 sec), 20-µL volumes of each inhibitor were transferred to HeLa cells maintained in 60 µL of EMEM-M. Following shaking, the cells were incubated with inhibitors for 3 h at 34°C in humidified atmosphere comprising 5% CO2 (the CO2 incubator), and then, 20 µL of EMEM-M comprising 100 tissue culture infectious doses (TCID50) of HRV-2 was added. After shaking, the plates were placed in the CO2 incubator for 3 days. The cells were then stained with 1% crystal violet solution, and the plates were inspected under the microscope for the development of the HRV-induced CPE.
Anti-HRV-2 activity of screening hits
The 50% effective concentration (EC50) was determined as follows. The hit compounds at serial threefold dilutions (0.03–60 mM) in 150 nL of DMSO in the V-bottom 96-well plates were supplemented with 30 µL of EMEM-M, and after shaking, 20 µL of hit dilutions was transferred to HeLa cells maintained in 60 µL of EMEM-M. Following shaking, the plates were left for 3 h in the CO2 incubator. The cells were then inoculated with HRV-2 at 100 TCID50/20 µL of EMEM-M and placed in the CO2 incubator for 3 days. Finally, the cells were stained with 1% solution of crystal violet and the cell protection against HRV-induced CPE was recorded based on microscopic observation. The EC50 was calculated by the Reed and Muench formula.
In some experiments, the EC50 of test compounds was also determined by the tetrazolium (MTS)-based colorimetric assay (27). Briefly, 20 µL of serial twofold dilution of the test compound in EMEM-M was added to HeLa cell monolayer cultures maintained in 60 µL of the same medium in a 96-well plate. After incubation for 3 h in the CO2 incubator, 20 µL of EMEM-M comprising 100 TCID50 of HRV-2 was added and incubated in the CO2 incubator for 3 days. Then, 20 µL of the MTS reagent (Promega, Madison, USA) was added, and following further incubation for 1 h in the CO2 incubator, an absorbance at 490 nm was recorded. The percentage of cell protection was calculated from the following formula: (absorbance of compound-treated cells – absorbance of virus control) × 100/(absorbance of cell control – absorbance of virus control). The hit AZ5385 was selected for extended studies. The compound purity, assessed by mass spectrometry, was 100%.
The virus yield reduction assay was performed as follows. HeLa cells, seeded in a 24-well plate (9 × 104 cells/well) the day prior to experiment, were rinsed with 200 µL of EMEM-M, and 412 µL of the same medium was added. Then, the cells received 50 µL of EMEM-M comprising the hit compound at a final concentration of 3.5 µM or the corresponding volume of DMSO. After 3 h of incubation in a CO2 incubator, HRV in 38 µL of EMEM-M was added at multiplicity of infection (MOI) 1 or 0.01 and incubated with cells for 2 h in the CO2 incubator. The virus inoculum was then aspirated, the cells were rinsed twice with EMEM-M, and 500 µL of the fresh medium comprising the same concentrations of the hit compound or DMSO was added. After incubation of infected cells for 24 h in a CO2 incubator, the infectious culture fluid and infected cells were harvested and subjected to the titer determination. To this end, HeLa cells growing in 96-well plates were inoculated in triplicates with 50 µL of serial 10-fold dilutions of collected samples. After incubation for 3 days in the CO2 incubator, the cells were stained with crystal violet and viral titer (TCID50) calculated according to Reed and Muench formula. In some experiments, the virus harvested from infectious culture fluid and virus released from infected cells by three freeze-thaw cycles were combined and centrifuged at 250 × g for 5 min. The supernatant fluid was clarified by centrifugation at 5,000 × g for 10 min and then centrifuged at for 2 h at 300,000 × g (Ti 70.1 rotor, Beckman). The pelleted material was rinsed twice with PBS and left overnight covered with 200 µL of PBS. Then, the pellet was resuspended and tested for infectivity and the number of RNA copies by qPCR. Plasmid LV1 (GenScript) comprising specific HRV sequence was used for calculation of the viral RNA copy number.
Anti-HRV-14 and anti-HRV-16 activity was determined in H1-HeLa cells by the tetrazolium (MTS)-based colorimetric assay (27) as described in the first paragraph of this section. Anti-RSV activity was tested by the viral plaque assays as described previously (28). Anti-poliovirus, anti-HSV-2, and anti-SARS-2 coronavirus activities of the hit compound were determined in GMK AH1 and Vero cells, respectively, by assessing protection of cells against the virus-induced CPE.
Cell toxicity assays
HeLa cells (1.5 × 104 cells/well) were seeded in a 96-well plate and incubated overnight in the CO2 incubator at 37°C. The cells were rinsed with 100 µL of EMEM-M, and 50 µL of fresh medium was added. Subsequently, 50 µL of EMEM-M comprising twofold dilutions of the hit compounds (final concentration ranging from 0.16 to 20 µM) was added and incubated with cells for 72 h in the 37°C CO2 incubator. Then, in the cell viability assay, 20 µL of the MTS reagent was added and incubated for further 1 h at 37°C in a CO2 incubator. Finally, an absorbance was recorded at 490 nm with a multiple microplate reader (Thermo Fisher Scientific). The absorbance detected with the test compounds was related to that recorded in the mock-treated cells, and the 50% effective cytotoxic concentration (CC50) was interpolated from the dose response curves. The cell proliferation assay was performed in a similar manner, except that after incubation of compound with cells for 72 h in the 37°C CO2 incubator, the cells were rinsed twice with PBS (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, and 1.5 mM KH2PO4), then dispersed with 0.54 mM EDTA in PBS, and counted.
Time-of-addition assay
The assay was performed as described previously for RSV (28). Briefly, HeLa cells (7 × 104 cells/well) were seeded in 24-well plates the day prior to the experiment. The cells were rinsed with 200 µL of EMEM-M, and 300 µL of fresh medium was added. The cells were then inoculated with HRV-2 at a MOI of 1 (the time point 0 h). Following incubation of cells with the virus in the CO2 incubator for 2 h (the time point 2 h), the inoculum was removed, the cells were rinsed twice with 200 µL of EMEM-M, and 270 µL of fresh medium was added. The test compound (3.5 µM), diluted in 30µL of EMEM-M, was added to cells at the time points −3, 0, 2, 3, 4, 5, 6, 7, 8, 10, and 12 h relative to inoculation with the virus. After incubation of cells for 24 h in the CO2 incubator, an infectious extracellular medium and the infected cells were collected to determine the infectious titers as described in section ”Anti-HRV activity of screening hits”.
The virus binding and entry assays
The suspension of HeLa cells prepared by the treatment of cell monolayers with 0.54 mM EDTA in PBS was rinsed with EMEM-M and stored at 4°C. HRV-2 (105 TCID50) and AZ5385 (3.5 µM) were mixed in 450 µL of cold EMEM-M, and following 1 h of preincubation, 50 µL of HeLa cell suspension (106 cells) in cold EMEM-M was added. The mixture was gently agitated and incubated for further 1 h at 4°C. Finally, the mixture was centrifuged at 250 g for 5 min, and the cells were triple rinsed by repeating centrifugation steps and then subjected to quantification of viral RNA by qPCR. The virus entry assay was performed in a similar manner as the virus binding assay; however, following centrifugation of the virus, compound and cell, mixture, and triple rinsing, the cells were resuspended in warm EMEM-M and incubated for 2 h at 37°C water bath. Finally, the cells were tested for infectivity in monolayers of HeLa cells.
Isolation of HRV variants resistant to a hit compound
All the details of this assays were described for RSV in a previous publication (28). Briefly, 500 µL of EMEM-M comprising the test compound at 3.5 µM or a corresponding volume of DMSO was added to HeLa cells and incubated for 2 h in the CO2 incubator. Subsequently 1.2 × 105 TCID50 of HRV-2 was added, and the cells were left in the CO2 incubator until the development of complete CPE or in case of poorly progressing CPE for a period of ≤5 days. The cells and the supernatant fluid were then harvested and used for subsequent passage. The selection for the resistant variants was monitored by assessing the compound sensitivity of passaged virus. After 11 consecutive passages in the presence of test compound or DMSO, the virus variants were purified by limiting dilutions. RNA was extracted using the QIAamp viral RNA purification kit (Qiagen). The corresponding cDNA was synthesized using a reverse transcriptase kit (Thermo Scientific). Sequencing was performed using the Big Dye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems) according to the manufacturer’s protocol. The list of primers used for amplification and sequencing is shown in Table S1. The Sequencher software version 5.4 (Gene Codes Corporation, Michigan, USA) was used for primer design, alignment, and analysis of sequences.
The virus-inactivating (virucidal) activity assay
The test compound at a concentration of 8.7 µM or a corresponding volume of DMSO control in 500 µL of EMEM-M was mixed with 105 TCID50 of HRV-2 and incubated at 37°C water bath for 15 and 60 min. Subsequently, the residual infectivities of the virus-compound or the virus-DMSO mixtures were immediately titrated in HeLa cells.
Immunofluorescence assay
One-day-old cultures of HeLa cells at ~60%–90% confluency in 24-well plates (7 × 104 cells/well) were rinsed with EMEM-M, and 500 µL of the same medium comprising the test compound at 5 and 10 µM or corresponding volumes of DMSO control were added and incubated in the 34°C CO2 incubator for 3 h. The cells were then inoculated with 5 × 103 TCID50/well of HRV-2, and placed in the CO2 incubator for further 20 h. Then, the cells were rinsed twice with 500 µL of PBS and fixed with 0.5 mL of 4% paraformaldehyde in PBS for 10 min at room temperature. After triple rinsing with 500 µL of PBS, the cells were permeabilized by incubation with 0.5 mL of 0.2% Triton-X-100 for 10 min at room temperature. Following triple rinsing, the cells were blocked with 500 µL of 3% BSA in PBS for 2 h at room temperature. The cells were rinsed once and received 250 µL of 0.1% BSA in PBS comprising the rabbit monoclonal anti-pEGFR (Abcam, ab40815, 1:250) and mouse monoclonal anti-VP3 of HRV (Thermo Fisher, MA5-18249, 1:50) antibodies. After overnight incubation at 4°C, the cells were rinsed thrice with PBS and supplemented with 250 µL of 0.1% BSA in PBS containing goat-anti-rabbit Alexa fluor 555 (1:250) and goat anti-mouse FITC (1:250) antibodies. After incubation for 2 h at 4°C, the cells were rinsed thrice with PBS and analyzed using the Evos FL microscope. In some experiments, the cells were treated for 15 min and 120 min at 37°C with 100 ng/mL of recombinant human epidermal growth factor (EGF; Sigma-Aldrich E-9644) in the presence of 5 µM AZ5385. In parallel experiments, the cells were inoculated for 15 min and 120 min at 34°C with HRV (MOI 50) in the presence of 5 µM AZ5385. The cells were then stained with anti-pEGFR and anti-HRV VP3 antibodies as described in the preceding paragraph.
Statistical analysis
The graphs were prepared by using the GraphPad prism version 9.0.2 software, and the statistical significance was determined based on one-way ANOVA and multiple t-test analysis. P values ≤0.05 were considered significant.
RESULTS
Screening of kinase inhibitor library for anti-HRV-2 activity
Knowing that viruses may usurp cellular kinases to facilitate their life cycle in cells, a library of 1,200 kinase inhibitors was screened twice both at 10 µM and at 1 µM for their ability to protect HeLa cells against HRV-induced CPE. This resulted in the identification of 22 hits, 7 of which exhibited potent anti-HRV activity (EC50 < 1 µM). These inhibitors were specific for different protein and lipid kinases; however, 12 of them possessed the 4-anilinoquinazoline scaffold that is found in some inhibitors of EGFR-TK activity. The structures and anti-HRV-2 potencies (EC50) of these 12 inhibitors are shown in Table 1. All but one (AZ5719) were modified with N-substituted oxypiperidines at position C7 or C6 of the quinazoline. AZ5385, with the most pronounced anti-HRV-2 activity (EC50 0.5 µM), had 2-hydroxypropanone on the oxypiperidine at C7 in combination with a hydroxyl group at C6 of the quinazoline (Table 1). The analogs retained low micromolar antiviral activity with modifications at the oxypiperidine with 2-methoxypropanone (AZ4514, AZ4516), 3-hydroxypropanone (AZ6252), ethanone (AZ2538), 2-ethoxyethanone (AZ5756), 3-methoxypropanone (AZ5765), or oxolanylmethanone (AZ6610, AZ6636) in addition to possessing the methoxyl group at C6 of the quinazoline. Compounds AZ8146, AZ3426, and AZ5719 that were extensively modified at C6 of the quinazoline showed decreased anti-HRV-2 activity compared with AZ5385. Because these compounds inhibit the tyrosine kinase activity of EGFR and/or certain closely related receptors of the EbrB family, we also tested two structurally related and potent EGFR-TKI, gefitinib and AG1478, for their potential anti-HRV-2 activity. Both inhibitors showed no anti-HRV-2 activity at the highest tested non-cytotoxic concentration (Table 1), emphasizing a clear preference for the 4-anilinoquinazoline scaffold modified with oxypiperidine at C7 or C6 of quinazoline for achieving anti-HRV activity, while inhibitory potency on EGFR is not critical.
TABLE 1.
Anti HRV-2 activities of anilinoquinazoline-based kinase inhibitor hitsa
| Compound | Structure | EC50 (µM) |
|---|---|---|
| AZ5385 |
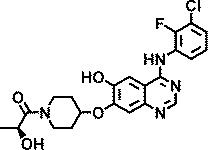
|
0.5 |
| AZ4514 |
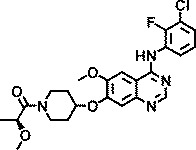
|
4.4 |
| AZ4516 |
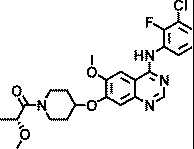
|
3 |
| AZ6252 |
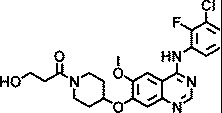
|
1.5 |
| AZ2538 |
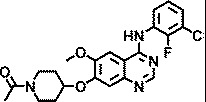
|
1.5 |
| AZ5756 |
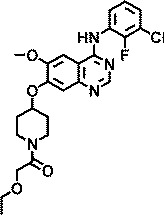
|
1.5 |
| AZ5765 |
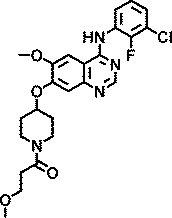
|
1.5 |
| AZ6610 |
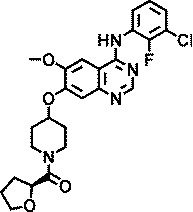
|
1.5 |
| AZ6636 |
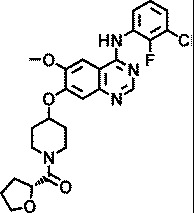
|
1.5 |
| AZ8146 |
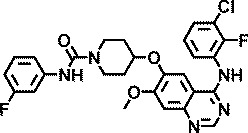
|
4.4 |
| AZ3426 |
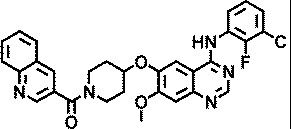
|
5.8 |
| AZ5719 |
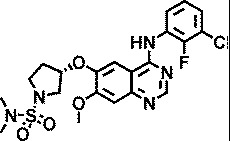
|
4.4 |
| Tyrphostin AG1478 |
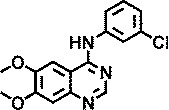
|
>4b |
| Gefitinib |
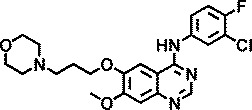
|
>4b |
Specific hits, identified by screening of the kinase inhibitor library against HRV-2, were tested for anti-HRV-2 activity in a concentration-dependent inhibition of viral cytopathic effect assay in HeLa cells. Three separate determinations were performed, and the EC50 values were calculated according to a Reed and Muench formula. Known EGFR-TKI AG1478 and gefitinib were also included for comparative purposes.
Lack of anti-HRV-2 activity at the highest non-cytotoxic concentration.
Antiviral potency of AZ5385
The hit AZ5385 that exhibited the most pronounced anti-HRV-2 activity (Table 1) was subjected to extended studies of antiviral potency. In the cell protection colorimetric assay, AZ5385 affected HRV-2 with an EC50 of 0.35 µM, while showing a CC50 for HeLa cells at 9 µM in both the MTS-based cell viability assay and the cell-count based cell proliferation assay (Selectivity index of 26) (Fig. 1A). Moreover, the compound reduced yields of HRV-2 progeny virus in infectious culture medium and in infected HeLa cell by ~1 log10 and ~2 log10 units upon infection at MOI 1 (Fig. 1B) and MOI 0.01 (Fig. 1C), respectively.
Fig 1.

Anti-HRV-2 activity of AZ5385 in HeLa cells. (A) The compound protects cells against HRV. The compound was incubated with HeLa cells for 3 h prior to the addition of 100 TCID50 of HRV-2 and then incubated at 34°C for further 72 h. After addition of MTS reagent, an absorbance at 490 nm was recorded. Two separate experiments, each in triplicate, were performed, and the % of cell protection was calculated by a formula described in Materials and Methods under Anti-HRV-2 activity of screening hits. In toxicity assays, the cells were incubated with compound for 72 h at 37°C prior to their dispersion and counting (cell proliferation assay) or addition of the MTS reagent (cell viability assay). Two separate experiments, each in triplicate, were performed, and each data point is expressed as % of absorbance (cell viability assay) or % of cell count (cell proliferation assay) detected with the compound as related to mock-treated control cells. (B and C) AZ5385 reduces HRV-2 yield in HeLa cells. The cells were inoculated with HRV-2 at MOI 1 (B) or 0.01 (C) in the presence of compound (3.5 µM) or DMSO. After 24 h, the virus released to culture medium (extracellular HRV) and the total virus (extracellular and cell-associated HRV) were harvested, and the viral TCID50 titer was quantified. The data are mean of four separate experiments. (D) Anti-HRV and cytotoxic activities of AZ5385 in H1-HeLa cells. The compound was tested for anti HRV-2, HRV-14, and HRV-16 activity. For explanations, see legend to Fig. 1A. Two separate experiments, each in triplicates, were performed. (E) Anti-RSV and cytotoxic activities of AZ5385 in HEp-2 cells. The test compound was incubated with cells for 3 h prior to the addition of ~100 plaque-forming units (PFU) of RSV A2. Following incubation at 37°C for 72 h, the cells were stained and the viral plaques were counted. Two separate experiments, each in duplicates, were performed, and the results are expressed as a % of the number of viral PFU or % of OD490 values found with compound-treated cells relative to mock-treated controls. (F) Expression of HRV-2 VP3 in HeLa cells treated with AZ5385. The cells infected with HRV-2 in the absence of AZ5385 (DMSO) or presence of 5 µM or 10 µM of AZ5385 were probed with anti-VP3 of HRV antibody (Thermo Fisher Sci., MA5-18249, 1:50). Two separate experiments each in duplicates were performed, and the number of HRV-2-infected cells were counted in 10 microscopic fields/well (Evos FL microscope 10×, NA 0.3) by the two independent investigators. Statistically significant differences at P < 0.05(*), P < 0.01(**), or P < 0.001 (***).
Besides HRV-2, we found that AZ5385 was able to inhibit infection of cells by another HRV-A, i.e., HRV-16 (Table 2; Fig. 1D), and by RSV strain A2 (Table 2; Fig. 1E) but not by HRV-B type 14 (HRV-14), poliovirus type 1, SARS-2 coronavirus isolate DE-P3, and HSV-2 strain 333 (Table 2). Besides reducing the HRV-2 infectivity (Fig. 1A), AZ5385 significantly decreased the number of virus-infected cells as revealed by staining of cells with anti-VP3 antibody (Fig. 1F).
TABLE 2.
Antiviral spectrum of AZ5385a
| Virus | Cells | EC50 (µM) | CC50 (µM) | SI (CC50/EC50) |
|---|---|---|---|---|
| HRV-A type 16 | H1-HeLa | 3.6 | >20 | >5.5 |
| HRV-B type 14 | H1-HeLa | >20 | >20 | |
| Poliovirus type 1 | GMK AH1 | >40b | 23 | |
| RSV strain A2 | HEp-2 | 0.4 | >50 | >125 |
| SARS-2 coronavirus isolate DE-P3 | Vero | ≥40c | >50 | |
| HSV-2 strain 333 | GMK AH1 | >40b | 23 |
The compound was tested for antiviral activity by the MTS-based colorimetric assay (HRV-16 and HRV-14), the viral plaque assay (RSV), and by the cytopathic effect inhibition assay (poliovirus, HSV-2, and SARS-2 coronavirus). The cytotoxicity of the compound for different cell lines was tested by using the MTS reagent, and the CC50 was interpolated from the dose-response curve.
Lack of antiviral activity at the highest non-cytotoxic concentration tested.
Partial inhibition (~50%) of the virus-induced CPE at 40 µM.
Mode of anti-HRV-2 activity of AZ5385
A time-of-addition experiment was performed to identify the stage of the viral life cycle in cells that is affected by AZ5385. In particular, the cells were inoculated with HRV-2 (MOI 1) at time point 0 h and the virus was left on cells at 34°C until time point 2 h, while the compound was added to cells at different time points counting from the virus inoculation at 0 h. After 24 h, the virus yield in the infectious culture supernatant was quantified. Compared with DMSO-treated controls, the compound reduced the virus yield by ~1 log10 even when added to cells as late as 10 h after the virus inoculation (Fig. 2), while addition of the compound at 12 h post inoculation resulted in lesser but still a statistically significant decrease in virus yield. This suggests that AZ5385 affects the late stage of viral life cycle. Furthermore, to assess involvement of possible cellular targets and early events of the virus-cell interaction, the compound was added to cells at 3 h prior to inoculation with HRV-2, and the amount of progeny virus in infectious culture medium (extracellular virus) and infected cells (cell-associated virus) was determined at 24 h post inoculation (Fig. 2B). Addition of AZ5385 at 3 h prior to HRV inoculation (−3 h) resulted in approx. 1 log10 decrease in infectious yield (Fig. 2B), a difference maintained in the following times of AZ5385 addition (Fig. 2B) until 10 h (Fig. 2A). Again, these data suggest that the event of the HRV-2 life cycle occurring between 10 and 12 h post inoculation is targeted by AZ5385.
Fig 2.

AZ5385 affects the late stage of HRV-2 life cycle. The compound at a concentration of 3.5 µM was added to HeLa cells at indicated time points relative to the beginning of virus inoculation (MOI 1) at 0 h. Two separate experiments were performed, and the data are presented as the amounts of infectious virus present in infectious medium (A) or the virus present in infectious medium (extracellular) and in infected cells (cell associated) (B) relative to the control DMSO-treated cells (24 h). Statistically significant differences ***P < 0.001.
Since AZ5385 is a known inhibitor of EGFR-TK activity, we sought to investigate whether the decrease of HRV-2 yield (Fig. 2) was due to cellular kinase suppression or alternatively the targeting of viral components. This seemed especially pertinent given that these observations were not consistent across all tested EGFR-TKIs, despite structural resemblance. To investigate involvement of viral components, the HRV-2 was subjected to 11 consecutive passages in HeLa cells in the presence of 3.5 µM of compound. The HRV-2 was also passaged in the absence of compound to serve as controls. The AZ5385 sensitivities (EC50) of these viruses were 0.46 µM for original non-passaged virus, 0.55 µM for control passage 11 virus, and >5 µM for AZ5385 passage 11 virus (Fig. 3A) indicating that the latter virus was relatively more resistant to compound manifested as >10-fold increase in EC50. Three viral variants of each original, control-passaged, and AZ5385-passaged virus were prepared by the limiting dilution method and then subjected to nucleotide sequencing. The GeneBank accession numbers for these sequences are shown in the Data Availability section.
Fig 3.

The drug resistance of AZ5385-passaged HRV-2. (A) The virus was subjected 11 consecutive passages in the presence of 3.5 µM AZ5385, and the sensitivity of original, control passage 11 (p.11), AZ5385 p.11, and AZ5385 p.3, 6, and 7 viruses was examined as described in Fig. 1A. (B) Schematic representation of emergence of specific mutations in AZ5385-passaged HRV-2.
All nucleotide sequences of the control-passaged and AZ5385-passaged virus variants V1, V2, and V3 were related to the respective variant of the original virus, and only mutations that occurred in all three variants of AZ5385-passaged but not in any of the control-passaged variants were regarded as possible cause of resistance (Table 3). These include two mutations in the genome fragment coding for the viral capsid protein VP1, i.e., the t2852a nucleotide change resulting in the S181T amino acid substitution and the a2939g alteration with a predicted T210A amino acid change, as well as a single silent mutation t6610c in the genome fragment coding for the viral polymerase (Table 3). To investigate whether both S181T and T210A are required for AZ5385 resistance, we traced the emergence of these mutations in passages 1 through 11. Surprisingly, a new mutation, the c2931a resulting in T207K amino acid substitution, occurred in passage 3 and dominated throughout passages 4, 5, and 6, i.e., until the advent of S181T and T210A mutations. These two alterations began to emerge concurrently in passage 6 and increasingly became dominant in the following passages (Fig. 3B). The emergence of both the T207K alteration (exemplified by virus passages 3 and 6 in Fig. 3A) and the S181T and T210A mutations (exemplified by virus passage 7 and passage 11 in Fig. 3A) was accompanied by a substantial decrease in their sensitivity to AZ5385 as compared with original virus and the control passage 11 virus (Fig. 3A).
TABLE 3.
Mutations in HRV-2 variants selected by passaging in the presence of AZ5385
| Protein | Alterationa in | |||||
|---|---|---|---|---|---|---|
| Control virus passage 11 | AZ5385 virus passage 11 | |||||
| V1b | V2 | V3 | V1 | V2 | V3 | |
| VP1 | t2852a S181T |
t2852a S181T |
t2852a S181T |
|||
| VP1 | a2939g T210A |
a2939gc T210A |
a2939g T210A |
|||
| Polymerase | t6610c silent | t6610c silent | t6610c silent | |||
Mutations found in HRV-2 passaged in the absence (control virus passage) or the presence of inhibitor as related to original HRV-2. Nucleotide substitutions are denoted with lowercase letters and followed by predicted amino acid changes. Numbering of nucleotide is based on the GenBank HRV-A2 strain X02316. Numbering of amino acid residues is based on published cleavage sites of HRV-2 polyprotein (UniProt, P04936).
Viral variants (V1–V3) purified by limiting dilution method.
Substitution accompanied by a background of original base.
Besides the drug-selected escape (resistance) alterations, we detected other alterations (Table S2) that were due to (i) variability among V1, V2, and V3 variants of original virus (e.g., the t941c change in V1 of control and V1 of AZ5385-passaged virus was due to alteration of V1 of the original virus) (Table S2 in italics), (ii) HeLa cell passage-dependent selection (the V265I alteration in 2C ATP-ase) found in all variants of the control virus passage but not in the compound-passaged virus, and (iii) random mutations occurring in some variants of control- but not in the compound-passaged virus (e.g., the a1519g mutation in V2 of control virus passage).
HRV-2 resistance to AZ5385 was related to alterations at VP1 amino acids S181 and T210 (Table 3). The localization of these residues in the published crystal structure of HRV-2 (12) is shown in Fig. 4A. These amino acids are located close to a hydrophobic pocket in HRV-2, which accommodates a C12 fatty acid (“pocket factor”) and is a known site of interaction for antiviral compounds. In particular, T210 is found at the entry of the pocket, where the hydrophilic head group of the fatty acid binds the side chain of N211, while S181 is more deeply embedded in the pocket, where the aliphatic tail of the fatty acid interacts with hydrophobic residues of the pocket wall (12). The targeting of hydrophobic pocket suggests that early events of the HRV life cycle could be affected by AZ5385. While the results of the time-of-addition experiment (Fig. 2) did not support this assumption, we anyhow assayed the capability of AZ5385 to inactivate the virus infectivity. The compound showed modest virus-inactivating (virucidal) activity, which was significant after incubation of HRV with AZ5385 for 1 h at 37°C (Fig. 4C) but not after 15 min at 37°C (Fig. 4B). A similar experiment performed with the passage 11-resistant HRV showed that incubation of this virus with AZ5385 for 1 h at 37°C caused no inactivation of virus infectivity (Fig. 4D) thus confirming specificity of the HRV-inactivating (virucidal) activity of AZ5385. We also investigated the capability of AZ5385 to inactivate infectivity of RSV, another pathogen affected by the compound (Table 2). Under experimental conditions similar to these used in HRV-2 assay, the mean titer (n = 3) of control (DMSO)-treated virus was 2.65 × 105 PFU/500 µL while that of the AZ5385-treated one was 2.52 × 105 PFU/500 µL thus demonstrating a lack of virucidal activity of AZ5385 against RSV particles. Furthermore, we studied the effect of AZ5385 on HRV-2 attachment to cells. To this end, the virus was preincubated with AZ5385 (3.5 µM) followed by its adsorption to HeLa cell suspension for 1 h at 4°C and quantification of attached virus by qPCR (Fig. 4E). Under experimental conditions of this experiment, the compound had no effect on the virus attachment to cells. Likewise, HeLa cells with attached HRV, prepared as in the attachment assay, were incubated for 2 h at 37°C to trigger the virus entry. These cells were then tested for their infectivity in cultures of HeLa cells. Under these experimental conditions, AZ5385 showed no effect on HRV-2 entry into HeLa cells (Fig. 4F). Because our data indicate that AZ5385 selected for drug-resistant mutations in VP1 hydrophobic pocket and exhibited specific virucidal activity at a concentration of 8.7 µM but to our surprise had no effect on the virus attachment to and entry into the cells, we sought, in line with the drug time-of-addition data, to examine the virus progeny particles which are produced in late stages of viral life cycle. To this end, the virus was propagated in the presence of AZ5385 (3.5 µM) for 20 h and the viral particles present in culture medium and these released from infected cells by repeated freeze-thaw cycles were combined and pelleted by ultracentrifugation. Assessment of infectivity (TCID50) and the number of viral RNA copies revealed that the RNA copy/TCID50 ratio was significantly higher for the virus produced in the presence of AZ5385 than the control (DMSO) virus. In particular, upon inoculation of cells at MOI 1, the RNA copy/TCID50 ratio was 199 for control and 2,296 for AZ5385-treated virus (Fig. 4G). The corresponding data for MOI 0.01 were 98 for control and 8370 for AZ5385-treated virus (Fig. 4H). We also found that AZ5385 affected the progeny virus particles already in infected cells, since HRV-2 prepared by collecting the cells, rinsing, and releasing the virus by freezing and thawing exhibited a substantial increase in RNA/infectivity ratio (Fig. 4I). Since the pelleted viral particle represents a source of RNA, these data suggest that the substantial number of viral particles was produced in the presence of AZ5385; however, only 1 particle per 2,296 (MOI 1) or per 8,370 (MOI 0.01) was infectious. Interestingly, the corresponding data for AZ5385-resistant passage 11 virus yielded the RNA copy/TCID50 ratio of 75 for control and of 111 for the compound-treated virus, a difference that was not significant (Fig. 4J).
Fig 4.

Mode of anti-HRV-2 activity of AZ5385. (A) Structure of the VP1 hydrophobic pocket of HRV-2 showing localization of the S181 and T210 residues (in yellow), the T207 residue (in blue), and the C12 fatty acid “pocket factor” (in pink). Neighboring residues to S181 at the pocket entry gate and to T210 at the pocket wall are specified. Image was created based on crystal structure of HRV-2 (12; PDB ID: 1FPN) using UCSF ChimeraX. (B and C) AZ5385 exhibits modest HRV2-inactivating (virucidal) activity. (D) The drug-resistant passage 11 virus is not inactivated by AZ5385. The virus at 105 TCID50 was incubated for 15 min (B) or 60 min (C and D) at 37°C with 8.7 µM of AZ5385 or a corresponding volume of DMSO, and the mixture was immediately subjected to determination of virus titer. Three separate experiments were performed. (E) AZ5385 exhibited no effect on the virus attachment to cells. The compound-treated virus was attached to HeLa cells for 1 h at 4°C. The amount of bound virus was quantified by qPCR and expressed as a number of RNA copies. (F) AZ5385 did not block HRV-2 entry into cells. The HeLa cell-bound HRV-2 was incubated for 2 h at 34°C in the presence of AZ5385 or DMSO control, and the cells were quantified for infectivity and RNA copies. (G–J) The virus propagated in the presence of AZ5385 exhibits high RNAcopy/infectivity ratio. The progeny HRV-2 particles prepared from culture supernatant and infected cells after inoculation at MOI 1 (G) and 0.01 (H) or prepared from infected cells only (I) were concentrated by ultracentrifugation, and their infectivity and RNA copy number were determined. (J) AZ5385-resistant passage 11 virus showed no increased RNA copy/infectivity ratio. The virus was prepared and analyzed as in Fig. 4H. Statistically significant difference **P < 0.01; ns, non-significant.
Furthermore, because AZ5385 is also an inhibitor of EGFR-TK activity, we investigated by using an immunofluorescence (IF) assay whether HRV-2 could activate (phosphorylate) EGFR in HeLa cells. Using antibodies against phosphorylated Y1068 in the TK domain of EGFR (pEGFR) and against VP3 of HRV, we found that EGFR-TK was activated already in non-infected HeLa cells, as manifested by nuclear staining and as small elongated pEGFR clusters present at the cell periphery (Fig. 5A). The pEGFR clusters were less frequent, or even absent, in HeLa cells infected with HRV-2 (Fig. 5B). AZ5385 both at 5 µM (Fig. 5C) and at 10 µM (Fig. 5D) did not block the background pEGFR staining in HRV-2-infected or in non-infected HeLa cells. To clarify why AZ5385 had no effect on activation of EGFR (Fig. 5C and D), we studied the activation of EGFR by its native ligand EGF. To this end, HeLa cells were incubated for 15 min or 120 min at 37°C with EGF (100 ng/mL) and then immunostained with anti-pEGFR(Y1068) antibody. Again, in control, non-treated cells, we detected pEGFR at the same sites as shown in Fig. 5A, and treatment of control cells with AZ5385 (5 µM) had no effect on pEGFR expression (Fig. 5E). This basic level of pEGFR (Y1068) visible at the cell membrane most likely represents a fraction of constitutively recycling pEGFR (29). Treatment of HeLa cells for 15 min but not for 120 min with EGF greatly enhanced activation of EGFR manifested as a cloud of punctate pEGFR around the nucleus (Fig. 5F) while incubation of cells with EGF and AZ5385 ablated this activation (Fig. 5G). Therefore, we tested whether activation of EGFR by HRV could occur shortly after inoculation of cells. Incubation of cells with HRV-2 for 15 min (Fig. 5H) caused no activation of EGFR. These results indicate that HRV-2 did not activate EGFR in HeLa cells, so the capability of AZ5385 to inhibit EGF-induced activation of this receptor may not be relevant to its anti-HRV-2 activity.
Fig 5.

Expression of activated EGFR-TK (pEGFR) in HRV-2 infected HeLa cells. Native HeLa cells, the cells infected with HRV-2 (MOI = 0.05), and the cells infected with HRV-2 and treated with 5 µM or 10 µM of AZ5385 were probed with rabbit monoclonal anti-pEGFR (Abcam, ab40815, 1:250) and mouse monoclonal anti-VP3 of HRV (Thermo Fisher Sci., MA5-18249, 1:50) antibodies. Images were captured using the Evos FL microscope (20×, NA 0.4). (A) Native HeLa cells showing pEGFR expression in the nucleus and at small protrusions at the cell periphery. (B) HRV-2-infected cells showing no enhanced pEGFR expression (see insets) as related to uninfected cells. (C and D) AZ5385 did not inhibit the pEGFR expression in cells but reduced the number of HRV-2-infected cells. (E) AZ5385 did not inhibit pEGFR expression in native HeLa cells. (F) EGF at 100 ng/mL activates pEGFR after 15 min of incubation with cells at 37°C. Note presence of a cloud of punctate pEGFR around the nuclei. (G) AZ5385 inhibits activation of pEGFR by EGF. (H) HRV-2 did not activate pEGFR after incubation with cells for 15 min at 34°C.
DISCUSSION
The increasing number of diagnosed HRV infections and their contribution to exacerbation of asthma and COPD (2, 3) fuel continuous searches for novel effective and safe anti-HRV candidate drugs. In the present study, an anti-HRV-2 screening of a preselected kinase inhibitor library identified a group of structurally similar EGFR-TKIs, the most potent of them affected HRV-2 infectivity with an EC50 value of 0.35 µM, while showing apparent toxicity for HeLa cells at 9 µM. Most of these inhibitors possessed the 4-anilinoquinazoline-7-oxypiperidine scaffold and, interestingly, the first-generation EGFR-TKIs gefitinib and AG1478 while being structurally related to the identified active compounds; they do not carry an oxypiperidine at C7 or C6 of the quinazoline, resulting in a loss of anti-HRV-2 activity. This suggests that the observed antiviral activity is not related to anti-EGFR activity and that the observed effects are instead mediated by targeting a virus component or the host cell, such as other receptor kinases. To further rule out EGFR involvement, we employed an antibody specific for phosphorylated tyrosine 1068 in the TK domain of EGFR and found that (i) this kinase shows background activation in native HeLa cells (29) and EGF enhances activation of EGFR, (ii) HRV-2 infection of these cells caused no further activation of kinase, and (iii) the background-activated EGFR-TK in native HeLa cells but not the EGF-activated receptor was resistant to the hit at the tested concentration. Hence, no experimental evidence was found that the anti-HRV-2 activity of AZ5385 is related to its inhibition of EGFR, with reservation that this kinase can be activated at several other tyrosines besides Y1068 (30), which were not tested in the present study. In this context, it is important to note that the resistance of cancer cells to EGFR-TKI is not a rare event (31) and AZ5385 may well show dual anti-HRV and anti-EGFR-TK effects on sensitive cells. Moreover, it should be noted that AZ5385 shows some structural similarities to vandetanib, an anti-cancer drug, that apart from EGFR-TK also targets several other kinases including vascular endothelial growth factor receptor 2 (32).
Antiviral compounds that target host cellular components are less likely to select for drug-resistant viral variants, although there are some reports that the HRV passaging in the presence of a PI4 kinase IIIB inhibitor selected for an escape mutation in the viral non-structural protein 3A (21). Here, we identified the S181T and the T210A amino acid substitutions in VP1 and a silent mutation in the viral polymerase, after 11 passages of HRV-2 in the presence of the AZ5385 inhibitor. Interestingly, the T207K mutation emerged at passage 3 and conveyed resistance to AZ5385 until passage 6, i.e., at the advent of the S181T and T210A alterations. Mapping of these VP1 alterations on the available crystal structure of HRV-2 (12) points to a known antiviral binding site within a hydrophobic pocket of the viral capsid. The T210A and the T207K alterations occurred at the entry to this pocket, just next to the N211 residue that binds the polar head group of the C12 fatty acid in HRV-2 (12). The analogous position (N219) in HRV-14 was found to be involved in the polar interaction with the pocket-binding WIN compound (10). The second alteration S181T occurred more deeply in the pocket between two hydrophobic residues L182 and F180, which line the pocket wall. We postulate that the S181 and T210 residues are important for binding of AZ5385 on the VP1 protein of the viral capsid, in line with other antiviral compounds acting at this site. Besides HRV-2, we observed that also HRV-16 was affected by AZ5385. These two viruses are known to exhibit some structural similarity of hydrophobic pocket and to bind the same pocket factor (11, 12). Sensitivities of different HRV types to pocket-binding antivirals, including pleconaril, are dictated by the nature of the amino acids forming the drug binding site (16), and alterations of two of these residues the Y152T and V191L conferred HRV resistance to pleconaril (33).
In HRVs that exploit ICAM-1 as the cellular receptor, the pocket-targeting compounds are known to alter the canyon that constitutes the binding site, thus blocking receptor interaction and thereby attachment of virus particles to cells (34). Instead, for LDLR binding HRVs, these compounds are known to inhibit the uncoating of viral particles (35). Here, we found that AZ5385 did not inhibit the virus attachment to and entry into the cells; however, the compound exhibited virus-inactivating (virucidal) activity, i.e., an established functional feature of the HRV pocket-targeting compounds (15). Interestingly, AZ5385 was shown to require a relatively slow onset for neutralizing HRV-2 infectivity, as the virucidal activity only became significant after 60 min of co-incubation with the virus at 37°C. Building on this finding, the results of the time-of-addition assay revealed that the compound was able to interfere with late stages of the HRV-2 life cycle, as significant antiviral effects were still observed when added as late as 10 h post infection. This is in contrast to the expected behavior of compounds that interfere primarily with the early stage of infection or inactivate the infectivity of viral particles. Indeed, we found that the progeny HRV-2 particles were severely impaired as regards their RNA/infectivity ratio which was substantially greater than that of native virions. This defect was evident already in infected cells, and the AZ5385-resistant passage 11 virus showed no such impairment. We therefore hypothesize that the compound competes with the pocket factor for binding to the hydrophobic pocket during maturation of HRV particles, such that infectivity and cell-cell spread of produced virions are impaired. A greater reduction of HRV-2 yield by AZ5385 after inoculation of cells with a low, rather than high, multiplicity of infection seemed to support the possible impact of the compound on the cell-cell spreading activity of the virus.
Finally, given that finding escape mutations in the VP1 capsid protein constitutes the most plausible explanation for the observed antiviral effects, we have not invested in further studies of the potential importance of inhibition of other kinases besides EGFR. Hence, we cannot exclude potential dual modes of activity, especially since compounds combining specific antiviral activity with inhibition of cellular kinases that facilitate the virus life cycle can be of interest. In this context, it should be noted that besides activity toward HRV, the compound also showed activity against respiratory syncytial virus, a pathogen that is known to activate EGFR-TK activity (36).
ACKNOWLEDGMENTS
This work was supported by the Swedish Medical Research Council (grant numbers: 2018–03918 and 2021–06386) and the Swedish Fund for Research Without Animal Experiments (grant numbers: N2021-00012 and F2022-00009).
Contributor Information
Joanna S. Said, Email: joanna.said@microbio.gu.se.
Miguel Angel Martinez, IrsiCaixa Institut de Recerca de la Sida, Barcelona, Spain.
DATA AVAILABILITY
The GeneBank accession numbers for HRV nucleotide sequences are OR296610 for input, original HRV2 variant 1; OR296611 for input, original HRV2 variant 2; OR296612 for input, original HRV2 variant 3; OR296613 for control, mock-passaged HRV2 variant 1; OR296614 for control, mock-passaged HRV2 variant 2; OR296615 for control, mock-passaged HRV2 variant 3; OR296616 for AZ5385-passaged HRV2 variant 1; OR296617 for AZ5385-passaged HRV2 variant 2; and OR296618 for AZ5385-passaged HRV2 variant 3.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/aac.01064-23.
Table S1 (Primers) and Table S2 (Nucleotide and amino acid alterations).
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Pitkäranta A, Hayden FG. 1998. Rhinoviruses: important respiratory pathogens. Ann Med 30:529–537. doi: 10.3109/07853899709002600 [DOI] [PubMed] [Google Scholar]
- 2. Bizot E, Bousquet A, Charpié M, Coquelin F, Lefevre S, Le Lorier J, Patin M, Sée P, Sarfati E, Walle S, Visseaux B, Basmaci R. 2021. Rhinovirus: a narrative review on its genetic characteristics, pediatric clinical presentations, and pathogenesis. Front Pediatr 9:643219. doi: 10.3389/fped.2021.643219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wedzicha JA. 2004. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc 1:115–120. doi: 10.1513/pats.2306030 [DOI] [PubMed] [Google Scholar]
- 4. McIntyre CL, Knowles NJ, Simmonds P. 2013. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J Gen Virol 94:1791–1806. doi: 10.1099/vir.0.053686-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW, Kamarck ME, McClelland A. 1989. The major human rhinovirus receptor is ICAM-1. Cell 56:839–847. doi: 10.1016/0092-8674(89)90688-0 [DOI] [PubMed] [Google Scholar]
- 6. Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. 1989. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell 56:849–853. doi: 10.1016/0092-8674(89)90689-2 [DOI] [PubMed] [Google Scholar]
- 7. Tomassini JE, Graham D, DeWitt CM, Lineberger DW, Rodkey JA, Colonno RJ. 1989. cDNA cloning reveals that the major group rhinovirus receptor on HeLa cells is intercellular adhesion molecule 1. Proc Natl Acad Sci U S A 86:4907–4911. doi: 10.1073/pnas.86.13.4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colonno RJ, Condra JH, Mizutani S, Callahan PL, Davies ME, Murcko MA. 1988. Evidence for the direct involvement of the rhinovirus canyon in receptor binding. Proc Natl Acad Sci U S A 85:5449–5453. doi: 10.1073/pnas.85.15.5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olson NH, Kolatkar PR, Oliveira MA, Cheng RH, Greve JM, McClelland A, Baker TS, Rossmann MG. 1993. Structure of a human rhinovirus complexed with its receptor molecule. Proc Natl Acad Sci U S A 90:507–511. doi: 10.1073/pnas.90.2.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith TJ, Kremer MJ, Luo M, Vriend G, Arnold E, Kamer G, Rossmann MG, McKinlay MA, Diana GD, Otto MJ. 1986. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science 233:1286–1293. doi: 10.1126/science.3018924 [DOI] [PubMed] [Google Scholar]
- 11. Hadfield AT, Lee W m, Zhao R, Oliveira MA, Minor I, Rueckert RR, Rossmann MG. 1997. The refined structure of human rhinovirus 16 at 2.15 A resolution: implications for the viral life cycle. Structure 5:427–441. doi: 10.1016/s0969-2126(97)00199-8 [DOI] [PubMed] [Google Scholar]
- 12. Verdaguer N, Blaas D, Fita I. 2000. Structure of human rhinovirus serotype 2 (HRV2). J Mol Biol 300:1179–1194. doi: 10.1006/jmbi.2000.3943 [DOI] [PubMed] [Google Scholar]
- 13. Katpally U, Smith TJ. 2007. Pocket factors are unlikely to play a major role in the life cycle of human rhinovirus. J Virol 81:6307–6315. doi: 10.1128/JVI.00441-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andries K, Dewindt B, Snoeks J, Willebrords R, Stokbroekx R, Lewi PJ. 1991. A comparative test of fifteen compounds against all known human rhinovirus serotypes as a basis for a more rational screening program. Antiviral Res 16:213–225. doi: 10.1016/0166-3542(91)90001-8 [DOI] [PubMed] [Google Scholar]
- 15. Andries K, Dewindt B, Snoeks J, Willebrords R, van Eemeren K, Stokbroekx R, Janssen PA. 1992. In vitro activity of pirodavir (R 77975), a substituted phenoxy-pyridazinamine with broad-spectrum antipicornaviral activity. Antimicrob Agents Chemother 36:100–107. doi: 10.1128/AAC.36.1.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ledford RM, Patel NR, Demenczuk TM, Watanyar A, Herbertz T, Collett MS, Pevear DC. 2004. VP1 sequencing of all human rhinovirus serotypes: insights into genus phylogeny and susceptibility to antiviral capsid-binding compounds. J Virol 78:3663–3674. doi: 10.1128/jvi.78.7.3663-3674.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, Blaas D. 1994. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci U S A 91:1839–1842. doi: 10.1073/pnas.91.5.1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hewat EA, Neumann E, Conway JF, Moser R, Ronacher B, Marlovits TC, Blaas D. 2000. The cellular receptor to human rhinovirus 2 binds around the 5-fold axis and not in the canyon: a structural view. EMBO J 19:6317–6325. doi: 10.1093/emboj/19.23.6317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verdaguer N, Fita I, Reithmayer M, Moser R, Blaas D. 2004. X-ray structure of a minor group human rhinovirus bound to a fragment of its cellular receptor protein. Nat Struct Mol Biol 11:429–434. doi: 10.1038/nsmb753 [DOI] [PubMed] [Google Scholar]
- 20. Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, Palmenberg AC, Gern JE. 2015. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci U S A 112:5485–5490. doi: 10.1073/pnas.1421178112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spickler C, Lippens J, Laberge M-K, Desmeules S, Bellavance É, Garneau M, Guo T, Hucke O, Leyssen P, Neyts J, Vaillancourt FH, Décor A, O’Meara J, Franti M, Gauthier A. 2013. Phosphatidylinositol 4-kinase III beta is essential for replication of human rhinovirus and its inhibition causes a lethal phenotype in vivo. Antimicrob Agents Chemother 57:3358–3368. doi: 10.1128/AAC.00303-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bentley JK, Newcomb DC, Goldsmith AM, Jia Y, Sajjan US, Hershenson MB. 2007. Rhinovirus activates interleukin-8 expression via a Src/p110beta phosphatidylinositol 3-kinase/Akt pathway in human airway epithelial cells. J Virol 81:1186–1194. doi: 10.1128/JVI.02309-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guedán A, Swieboda D, Charles M, Toussaint M, Johnston SL, Asfor A, Panjwani A, Tuthill TJ, Danahay H, Raynham T, Mousnier A, Solari R. 2017. Investigation of the role of protein kinase D in human rhinovirus replication. J Virol 91:e00217-17. doi: 10.1128/JVI.00217-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. García-Cárceles J, Caballero E, Gil C, Martínez A. 2022. Kinase inhibitors as underexplored antiviral agents. J Med Chem 65:935–954. doi: 10.1021/acs.jmedchem.1c00302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schor S, Einav S. 2018. Repurposing of kinase inhibitors as broad-spectrum antiviral drugs. DNA Cell Biol 37:63–69. doi: 10.1089/dna.2017.4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duff R, Rapp F. 1971. Oncogenic transformation of hamster cells after exposure to herpes simplex virus type 2. Nat New Biol 233:48–50. doi: 10.1038/newbio233048a0 [DOI] [PubMed] [Google Scholar]
- 27. Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. 1988. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 20:309–321. doi: 10.1016/0166-0934(88)90134-6 [DOI] [PubMed] [Google Scholar]
- 28. Lundin A, Bergström T, Trybala E. 2013. Screening and evaluation of anti-respiratory syncytial virus compounds in cultured cells. Methods Mol Biol 1030:345–363. doi: 10.1007/978-1-62703-484-5_27 [DOI] [PubMed] [Google Scholar]
- 29. Gosney JA, Wilkey DW, Merchant ML, Ceresa BP. 2018. Proteomics reveals novel protein associations with early endosomes in an epidermal growth factor-dependent manner. J Biol Chem 293:5895–5908. doi: 10.1074/jbc.RA117.000632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Downward J, Parker P, Waterfield MD. 1984. Autophosphorylation sites on the epidermal growth factor receptor. Nature 311:483–485. doi: 10.1038/311483a0 [DOI] [PubMed] [Google Scholar]
- 31. Cooper AJ, Sequist LV, Lin JJ. 2022. Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat Rev Clin Oncol 19:499–514. doi: 10.1038/s41571-022-00639-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee D. 2005. Phase II data with ZD6474, a small-molecule kinase inhibitor of epidermal growth factor receptor and vascular endothelial growth factor receptor, in previously treated advanced non-small-cell lung cancer. Clin Lung Cancer 7:89–91. doi: 10.1016/S1525-7304(11)70394-1 [DOI] [PubMed] [Google Scholar]
- 33. Ledford RM, Collett MS, Pevear DC. 2005. Insights into the genetic basis for natural phenotypic resistance of human rhinoviruses to pleconaril. Antiviral Res 68:135–138. doi: 10.1016/j.antiviral.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 34. Pevear DC, Fancher MJ, Felock PJ, Rossmann MG, Miller MS, Diana G, Treasurywala AM, McKinlay MA, Dutko FJ. 1989. Conformational change in the floor of the human rhinovirus canyon blocks adsorption to HeLa cell receptors. J Virol 63:2002–2007. doi: 10.1128/JVI.63.5.2002-2007.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim KH, Willingmann P, Gong ZX, Kremer MJ, Chapman MS, Minor I, Oliveira MA, Rossmann MG, Andries K, Diana GD. 1993. A comparison of the anti-rhinoviral drug binding pocket in HRV14 and HRV1A. J Mol Biol 230:206–227. doi: 10.1006/jmbi.1993.1137 [DOI] [PubMed] [Google Scholar]
- 36. Kalinowski A, Galen BT, Ueki IF, Sun Y, Mulenos A, Osafo-Addo A, Clark B, Joerns J, Liu W, Nadel JA, Dela Cruz CS, Koff JL. 2018. Respiratory syncytial virus activates epidermal growth factor receptor to suppress interferon regulatory factor 1-dependent interferon-Lambda and antiviral defense in airway epithelium. Mucosal Immunol 11:958–967. doi: 10.1038/mi.2017.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Primers) and Table S2 (Nucleotide and amino acid alterations).
Data Availability Statement
The GeneBank accession numbers for HRV nucleotide sequences are OR296610 for input, original HRV2 variant 1; OR296611 for input, original HRV2 variant 2; OR296612 for input, original HRV2 variant 3; OR296613 for control, mock-passaged HRV2 variant 1; OR296614 for control, mock-passaged HRV2 variant 2; OR296615 for control, mock-passaged HRV2 variant 3; OR296616 for AZ5385-passaged HRV2 variant 1; OR296617 for AZ5385-passaged HRV2 variant 2; and OR296618 for AZ5385-passaged HRV2 variant 3.