

Abstract
Fibrosis is a major, but incompletely understood, component of many diseases. The most common vision-disrupting complication of cataract surgery involves differentiation of residual lens cells into myofibroblasts. In serum-free primary cultures of lens epithelial cells (DCDMLs), inhibitors of either ERK or of ErbB signaling prevent TGFβ from upregulating both early (fibronectin) and late (αSMA) markers of myofibroblast differentiation. TGFβ stimulates ERK in DCDMLs within 1.5 h. Kinase inhibitors of ErbBs, but not of several other growth factor receptors in lens cells, reduce phospho ERK to below basal levels in the absence or presence of TGFβ. This effect is attributable to constitutive ErbB activity playing a major role in regulating the basal levels pERK. Additional studies support a model in which TGFβ-generated reactive oxygen species serve to indirectly amplify ERK signaling downstream of tonically active ErbBs to mediate myofibroblast differentiation. ERK activity is in turn essential for expression of ErbB1 and ErbB2, major inducers of ERK signaling. By mechanistically linking TGFβ, ErbB, and ERK signaling to myofibroblast differentiation, our data elucidate a new role for ErbBs in fibrosis and reveal a novel mode by which TGFβ directs lens cell fate.
It is not understood how TGFβ contributes to secondary cataract (PCO), the most common vision-disrupting complication of cataract surgery.
Using our serum-free, primary lens epithelial cell culture system, we show that TGFβ leverages basal ERK activity downstream of tonic ErbB signaling to induce differentiation of lens epithelial cells into myofibroblasts.
By mechanistically linking TGFβ, oxidative stress, ErbB, and ERK signaling, our data elucidate a new role for ErbBs in fibrosis and support the use of ErbB inhibitors as therapeutics against PCO.
INTRODUCTION
Our vision is critically dependent on the proper development of lens cells. Epithelial cells on the anterior face of the organ differentiate into highly elongated, crystallin-rich lens fibers cells at what is referred to as the equator of the lens (Cvekl and Ashery-Padan, 2014). Defects in this process can result in lens opacity, although more common causes of cataract include older age, injury, and excessive UV exposure (Liu et al., 2017). The only treatment for the vast majority of cataracts regardless of etiology is surgery, in which the cells of the cloudy lens are removed from their encasing lens capsule and replaced with an artificial lens. Lens epithelial cells that survive this operation can undergo two types of abnormal differentiation (Apple et al., 2000; Awasthi et al., 2009; Wormstone et al., 2009; Findl et al., 2010; Fișuș and Findl, 2020). Whereas some of these residual lens cells attempt to become lens fiber cells, others appear to transform into contractile, extracellular matrix-producing myofibroblasts. If the latter accumulate at the rear of the lens capsule, they can interfere with transmission of light to the retina and lead to fibrotic posterior capsule opacification (PCO). PCO is the most common cause of long-term visual disruption after cataract surgery (Konopińska et al., 2021). Fibrosis of lens epithelial cells that postoperatively remain at the anterior of the capsule can result in anterior capsule opacification (ACO), which also adversely affects eye health (Trivedi et al., 2002). In addition, epithelial cells in the intact lens can also become fibrotic due to injury or disease, forming vision-impairing anterior subcapsular cataracts (Novotny and Pau, 1984; Shu et al., 2017).
It has been shown in in vitro and in vivo mammalian (Srinivasan et al., 1998; Lovicu et al., 2004; Robertson et al. 2007; West-Mays et al., 2010; Wormstone and Eldred, 2016; Tiwari et al., 2016) and avian (Boswell et al., 2017) systems that TGFβ induces lens epithelial cells to undergo an epithelial to mesenchymal transition (EMT) to myofibroblastic cells (EMyT; epithelial to myofibroblast transition; Masszi et al., 2010), in keeping with the major role of this growth factor in fibrosis in other organs (Eldred et al., 2011; Meng et al., 2016; Di Gregorio et al., 2020; Frangogiannis, 2020). TGFβ signaling in lens cells is known to be elevated after cataract surgery (Meacock et al., 2000; Saika et al., 2002; Wormstone et al., 2002; de Iongh et al., 2005), and is thought to first rise as part of a wounding response and then be maintained by chronic production and activation of endogenous TGFβ (Wormstone et al., 2006; Mamuya and Duncan, 2012; VanSlyke et al., 2018). In rat (Wojciechowski et al., 2017), human (Tiwari et al., 2016), and chick (Boswell et al., 2017) primary lens cell systems, TGFβ upregulation of EMyT is blocked by inhibitors of ERK activation. Conversely, conditional knock-out in the lens of two genes (sprouty 1and 2) that encode proteins that inhibit ERK signaling induce the formation of myofibroblasts (Shin et al., 2012).
More recently, it has been demonstrated that the ability of TGFβ to upregulate EMyT in primary rat (Shu et al., 2019) and chick (VanSlyke et al., 2023) lens cells is blocked by small molecule inhibitors of the ErbB family of receptor tyrosine kinases. ErbBs are widely expressed and regulate several essential processes including differentiation, proliferation, growth, survival, and migration in a cell type-specific manner (Wieduwilt and Moasser, 2008; Chen et al., 2016). We recently reported (VanSlyke et al., 2023) that serum-free, nonpassaged primary cultures of embryonic chick lens epithelial cells (DCDMLs) express on the cell surface all three functional tyrosine kinases in the ErbB family (namely, ErbB1/EGFR1, ErbB2/EGFR2/HER2, and ErbB4/EGFR4) and release ErbB-activating ligands into the medium. Our findings further indicated that most ErbB signaling under basal conditions is mediated by ErbB1/B2 heterodimers, and that upregulation of ErbB1 in response to TGFβ is likely to be important for myofibroblast differentiation. In contrast, TGFβ greatly reduces the total and cell surface levels of ErbB4, and there is no evidence that this species plays a causative role in TGFβ-induced EMyT (VanSlyke et al., 2023). ErbB inhibitors have previously been reported to have anti-PCO activity in human lens capsular bags (Maidment et al., 2004; Wertheimer et al., 2015, 2018), and siRNA knockdown of ErbB1 has been reported to reduce PCO in an in vivo rat model (Huang et al., 2011). In DCDMLs, TGFβ induces not only the formation of myofibroblasts, but also stimulates other lens cells in the same cultures to become lens fiber cells by an as yet incompletely characterized mechanism that is pharmacologically distinct from that involved in EMyT (Boswell et al., 2017). Notably, ErbB inhibitors do not prevent lens fiber cell differentiation downstream of TGFβ (VanSlyke et al., 2023).
Despite its relevance for PCO, we do not understand why ErbB activity is essential for TGFβ-induced EMyT. Here, we show that ongoing (i.e., tonic; basal) ErbB signaling is the major source of the ERK activity required for TGFβ to promote EMyT in primary cultures of lens epithelial cells. Our data support a model in which TGFβ indirectly increases the level of phospho ERK by altering a redox-dependent process downstream of ErbB autophosphorylation, but before activation of Raf-1. ERK activity is reciprocally required for expression of ErbB1 and ErbB2 protein. Together, these results elucidate how TGFβ, ErbB, and ERK signaling interact to direct the formation of myofibroblasts, a cell type central to the etiology of PCO and ACO. Given the widespread role of TGFβ, oxidative stress, ERK, and myofibroblasts in fibrosis throughout the body, our findings are also relevant to nonlenticular organs including lung, kidney, and liver.
RESULTS
Role of ERK and ErbB signaling in TGFβ-induced myofibroblast formation in DCDMLs
After 6 d of treatment with TGFβ (beginning on the day after plating, with the day of plating designated as d 0 of culture), some cells in DCDML primary lens epithelial cells undergo EMyT, including expression of the canonical myofibroblast marker αSMA in stress fibers and fibronectin in extracellular fibrils (Boswell et al., 2010, 2017). The highly selective ErbB inhibitor lapatinib blocks TGFβ-induced expression of αSMA and FN to near the level obtained when ERK is inactivated by the MEK inhibitor UO126, or if cells are cocultured with the TGFβR inhibitor SB-431542 (Figure 1A). In contrast, lapatinib does not inhibit the ability of TGFβ to promote lens fiber cell differentiation (Figure 1B; VanSlyke et al., 2023). Similar results were obtained with other mechanistically and structurally distinct ErbB inhibitors tested including erlotinib, and the irreversible blocker afatinib (Supplemental Figure S1). As expected (Liu et al., 2007), ErbB inhibitors do not prevent TGFβ from activating the downstream regulator Smad3, as assessed by pSer423/425 Smad3 immunoreactivity and expression of a Smad3 transcriptional reporter (VanSlyke et al., 2023). Taken together, these results support a specific, non-Smad3 role for ErbB signaling in TGFβ-induced EMyT.
FIGURE 1:

Effect of inhibition of ERK or ErbB on TGFβ stimulation of myofibroblast differentiation in DCDMLs. DCDML primary lens epithelial cells were treated starting on d 1 of culture with or without 4 ng/ml TGFβ1, in the presence or absence of the following kinase inhibitors: lapatinib (lap), UO126 (UO), or SB-431542 (SB4). Cells were lysed on d 7 and whole cell lysates analyzed for the EMyT markers FN and αSMA (A), or for FN, αSMA, and the lens fiber cell markers δ-crystallin, CP115, and CP49 (B) by Western blot. (C) Quantitation of EMyT marker data, expressed as the percent inhibition induced by lapatinib, UO126, or SB-431542 relative to cells treated with TGFβ and vehicle (0.1% DMSO) only. All data are normalized to tubulin in the same sample. For all, n ≥ 6; p = 0.000.
In DCDMLs, αSMA levels do not start to rise until ∼ 96 h after addition of TGFβ (Boswell et al., 2010, 2017). In contrast, expression of FN protein is stimulated within 48 h, consistent with initiation of protomyofibroblast differentiation (Tomasek et al., 2002; Eyden, 2008). Lapatinib and UO126 both prevented TGFβ from upregulating FN expression at this time (Figure 2). The finding that the activities of both ErbB and ERK are critical during the early stage of TGFβ-induced myofibroblast differentiation raises the possibility of a causal relationship between the two kinases in this process.
FIGURE 2:

Both ERK and ErbB activity are required for TGFβ to induce the early stage of EMyT. DCDMLs were cultured from d 1–3 in the presence or absence of TGFβ with or without lapatinib, UO126, or SB-431542 as indicated. Cells were lysed on d 3 and whole cell lysates analyzed for expression of FN. For all, p ≤ 0.004.
ErbBs are the major source of basal ERK activity in lens cells
To determine the extent to which growth factors produced by lens epithelial cells contribute to basal ERK activity in DCDMLs, we interrogated the ability of inhibitors of these pathways to reduce the level of phospho ERK. Addition of lapatinib to otherwise untreated DCDMLs elicited a striking decrease in the level of pERK within 90 min without affecting the amount of total ERK (Figure 3A) or the basal phosphorylation of p38 MAP kinase (Figure 3B) or the basal phosphorylation of AKT (Figure 3C). Avian cells express ERK2, but not ERK1, accounting for the single phospho ERK immunoreactive band (Lefloch et al., 2008). Notably, the reduction in pERK attained with lapatinib or erlotinib was greater than that observed after inhibition of FGFRs (Figure 3, A and D), a previously identified positive effector of ERK activity in lens cells in vivo (Govindarajan and Overbeek, 2001) and in culture (Wang et al., 2010; Boswell et al., 2017). Combining maximally effective concentrations of lapatinib with the FGFR inhibitor PD173074 resulted in an additive decrease in pERK to very near the level obtained with U0126 treatment, without affecting total ERK levels (Figure 3, A and D). Blockers of receptors for PDGF (crenolanib) or TGFβ (SB-431542) did not reduce pERK when tested under the same conditions (Figure 3D). These findings indicate that ErbB and FGFR signaling are together responsible for virtually all basal ERK activity in DCDMLs, with ErbB playing the major role. The finding that ErbBs are critical for the basal activity of ERK, but not of p38 or AKT, is indicative of a highly selective role for ErbBs in the regulation of lens cell signaling.
FIGURE 3:
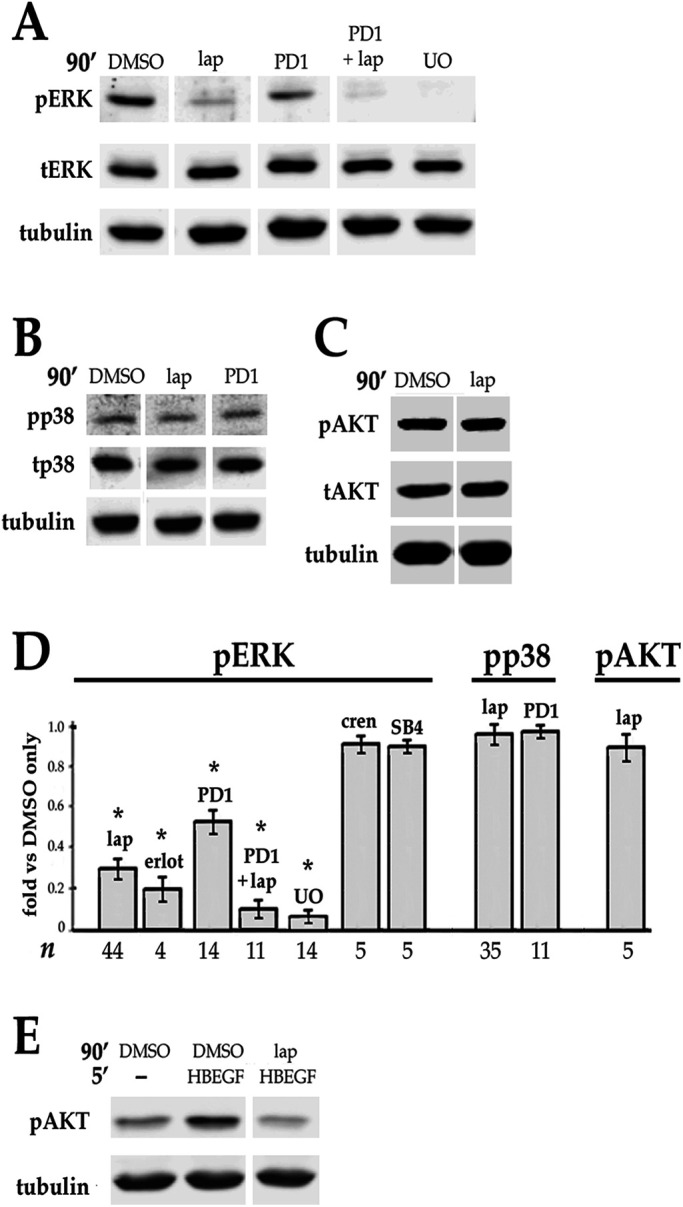
Effect of blocking ErbB and/or other growth factor receptors on the basal level of phospho ERK, p38, and AKT in DCDMLs. (A–C) DCDMLs were incubated for 90 min with DMSO, lapatinib, the selective FGFR inhibitor PD173074, lapatinib plus PD173074, or UO126 before Western blot analysis of phospho and total forms of ERK, p38, AKT, or of tubulin as indicated. (D) Results graphed as fold controls treated with DMSO only. Also included are data demonstrating that the ErbB inhibitor erlotinib (erlot), but not inhibitors of the receptors for TGFβ (SB-431542) or PDGF (crenolanib), reduces basal levels of pERK. Asterisks indicate p ≤ 0.001; for all other conditions, p ≥ 0.337. (E) DCDMLs were incubated with DMSO or lapatinib and lysed after a 5 min treatment with 10 ng/ml HB-EGF or medium only to demonstrate that the anti-pAKT antibody recognizes activated (pS475) AKT in DCDMLs.
ErbB signaling is specifically required for TGFβ-induced activation of ERK
We show here that, as expected, activation of ERK in response to the ErbB1/4 ligand HB-EGF is abolished by preincubation with UO126 or lapatinib, but not by the FGFR inhibitor PD173074 (Figure 4A). Notably, pERK levels drop to basal (e.g., no added HB-EGF) levels within 90 min of HB-EGF addition (Figure 4A), indicating that in DCDMLs, as in many other cell types (Traverse et al., 1992; Wang et al., 2010; Lemmon et al., 2016), activation of ERK by ErbBs is rapidly attenuated.
FIGURE 4:

ErbB and TGFβ signaling in lens cells. DCDMLs were pretreated for 1 h with DMSO or the indicated inhibitors before incubation for 5 or 90 min with either 10 ng/ml HB-EGF, 4 ng/ml TGFβ, 1 or 10 ng/ml FGF, or an equal volume of M199 medium (-). Whole cell lysates were analyzed for phospho and total forms of ERK, Smad3, and p38, or for tubulin by Western blot. (A) ErbB ligand induces a rapid and transient activation of ERK compared with TGFβ. Results graphed as fold pERK relative to DMSO only control. p < 0.001, except NS (p >0.325). Activation by phosphorylation decreases the apparent mobility of total (t) ERK(2) on SDS–PAGE, resulting in a doublet under conditions in which ERK is partially activated. (B) Inhibitor sensitivity of activation of ERK by TGFβ. Results graphed as fold pERK relative to DMSO only (no TGFβ) control. p < 0.001, except NS (p = 0.22). Compared to cells treated with TGFβ plus DMSO, the level of pERK in cells treated with TGFβ plus PD173074 is significantly lower (p = 0.000), whereas crenolanib (0.5 μM) and noggin (1 μg/ml) have no effect on activation of pERK by TGFβ (p > 0.628). Also graphed are data demonstrating that the ErbB inhibitor erlotinib and the TGFβR inhibitor SB-431542 reduce upregulation of pERK by TGFβ. (C) Lapatinib does not block activation of Smad3 by TGFβ. (D) Lapatinib does not block activation of p38 by TGFβ. (E) Quantitation of the data as compared with treatment with TGFβ and DMSO. In all cases, p ≥ 0.133. (F) Lapatinib does not block activation of pERK by FGF, as quantitated as percent inhibition of pERK in cells treated with FGF + lapatinib compared with cells treated with FGF + DMSO.
TGFβ also increases activation of ERK, but this response is slower than that elicited by ErbB ligand (Figure 4A). As previously reported (Boswell et al., 2017), UO126 pretreatment abolishes TGFβ-mediated elevation of pERK (Figure 4B). We show here that TGFβ upregulation of pERK in DCDMLs is also prevented by lapatinib or erlotinib (Figure 4B). Notably, the level of pERK in cells pretreated with either the MEK or ErbB inhibitor before the addition of TGFβ was below that of untreated controls. In contrast, blockers of two other growth factor receptors active in lens cells, namely PDGFR (crenolanib) and BMPRs (noggin), were ineffective in preventing TGFβ from increasing pERK (Figure 4B). Blocking FGF signaling partially (by ∼ 30%) reduced, but did not eliminate, ERK activation in response to TGFβ (Figure 4B). Notably, ErbB inhibition did not decrease the expression of total ERK (Figure 4, C and E), or diminish the ability of TGFβ to activate Smad3 or p38 (Figure 4, D and E) under the same treatment conditions. ErbB inhibitors also failed to reduce activation of ERK in response to physiologically relevant levels of FGF (Figure 4F; p ≥ 0.211; n = 10). Taken together, these results indicate that ErbB inhibitors specifically block activation of ERK downstream of TGFβ, and not in response to FGF.
In the intact lens and shortly after cataract surgery, central lens epithelial cells have access to the aqueous humor. FGF at the concentration thought to be present in aqueous humor (∼ 1 ng/ml) increases the level of pERK in mammalian (Iyengar et al., 2009) and avian (Le and Musil, 2001) primary lens cells more than fivefold within 30 min. This is considerably greater than the ∼2X increase in pERK observed after addition of TGFβ in either system (Boswell et al. 2017; Wojciechowski et al., 2017; Figure 4A). Given this more potent (and ErbB-independent; Figure 4F) source of ERK activity, how could stimulation of ERK by TGFβ be important for initiation of EMyT of lens epithelial cells under physiologically relevant conditions? The elevation of pERK induced by 1 ng/ml FGF in lens cells is transient, subsiding to basal levels within 6–12 h in cultured primary lens cells (Le and Musil, 2001; Iyengar et al., 2009). Such kinetics are consistent with the very low levels of active ERK detected in central lens epithelial cells in vivo (Zhao et al., 2008; Li et al., 2014) despite their expression of ligand-responsive FGFRs. Importantly, DCDMLs chronically exposed to 1 ng/ml FGF remain capable of increasing pERK in response to TGFβ in a process blocked by lapatinib (Figure 5A). After cataract surgery, this would allow wounding-induced TGFβ to upregulate pERK and initiate myofibroblast differentiation in an ErbB-dependent manner.
FIGURE 5:

Effect of long-term exposure to FGF or TGFβ on ERK. (A) TGFβ continues to stimulate ERK in DCDMLs chronically exposed to physiological levels of FGF. DCDMLs were cultured for 6 d in either the absence or presence of 1 ng/ml FGF2. Cells were then incubated for 60 min with DMSO or lapatinib, followed by a 90 min treatment with no additions (–), 1 ng/ml FGF2, or 4 ng/ml TGFβ. Whole cell lysates were assessed for phospho ERK, total ERK, or tubulin by Western blot. All results shown are from the same blot of a single experiment. Levels of pERK obtained under the indicated conditions are graphed relative to controls treated with DMSO without inhibitor or growth factor. NS, p = 0.593; for all other conditions, p < 0.012. (B) Prolonged exposure to TGFβ increases both phospho and total levels of ERK. DCDMLs were cultured for 6 d in either the absence or presence of TGFβ. Whole cell lysates were assessed for pERK, tERK, or tubulin by Western blot. pERK and tERK values were normalized to tubulin in the same sample.
A related physiologically important question is the effect on ERK of long-term exposure to TGFβ. We found that DCDMLs cultured for 6 d with TGFβ show a twofold elevation of pERK relative to untreated DCDMLs (Figure 5B). Unlike after a 1.5 h treatment with TGFβ (Figure 4C), this is accompanied by a similar increase in the amount of total ERK when normalized to tubulin (Figure 5B), demonstrating that prolonged exposure to TGFβ promotes the levels of both phospho and total ERK.
Lack of evidence for transactivation of ErbB signaling by TGFβ
An obvious mechanism by which TGFβ could stimulate ERK in a ErbB-dependent manner would be if TGFβ transactivated ErbBs, as has previously been implicated in EMT in some cell types (Uchiyama-Tanaka et al., 2002; Samarakoon et al., 2013a). In one mode of transactivation, TGFβ stimulates the posttranslational processing of transmembrane ErbB ligand proforms into soluble, active species, thereby allowing them to bind and stimulate ErbB1 and then pERK within 1–2 h of exposure to TGFβ (Wang et al., 2008; Ebi et al., 2010). To investigate if enhanced ligand shedding could be responsible for the observed elevation of pERK in DCDMLs in response to TGFβ, we transiently transfected DCDMLs with a plasmid encoding the full-length (i.e., transmembrane) proform of the ErbB 1/4 ligand HB-EGF (Figure 6). Transcripts for HB-EGF have been reported in primary rat lens epithelial cells (Shu et al., 2019). Cells were cultured for 2 d with or without TGFβ, after which the conditioned medium was collected and added to HEK293 cells at 4°C, a temperature that allows ligand-induced tyrosine autophosphorylation of endogenously expressed ErbBs but no other signaling events (Longva et al., 2002; Tong et al., 2014). Active ErbBs were detected by Western blot using the 4G10 antiphosphotyrosine antibody as previously reported (Samarakoon et al., 2013a; VanSlyke et al., 2023). This assay measures the ability of mature, soluble ligands in the DCDML conditioned medium to stimulate the autophosphorylation of HEK cell ErbB 1, 2, and 4, which comigrate under our SDS–PAGE conditions. Control experiments demonstrated that incubation of HEK cells with exogenous, recombinant soluble HB-EGF resulted in a dose-dependent increase in a ∼170 kD, phosphotyrosine-positive ErbB band (Figure 6, lanes 1–4). Conditioned medium from proHB-EGF-transfected DCDMLs cultured with TGFβ failed to activate ErbBs in HEK cells to a greater extent than conditioned medium from no TGFβ transfectants (compare lanes 6 and 7). These data indicate that TGFβ does not increase the generation of soluble HB-EGF ligand in DCDMLs and are in keeping with the reported constitutive expression in lens cells of ADAM-17 (Hodgkinson et al., 2010; Shu et al., 2019), the enzyme primarily responsible for maturation of HB-EGF and a subset of other ErbB ligands (Sahin et al., 2004).
FIGURE 6:

TGFβ does not stimulate the release of HB-EGF from DCDMLs. (A) DCDMLs were transiently transfected with plasmids encoding either a full-length, transmembrane proform of HB-EGF or an irrelevant control transmembrane protein (E208K Cx32; VanSlyke et al., 2023; Ctl) and cultured for 48 h in either the absence or presence of TGFβ. Untransfected HEK293 cells were then incubated at 4°C for 15 min with either 10–0 ng/ml mature, soluble recombinant HB-EGF (lanes 1–4), or the indicated DCDML transfectant conditioned medium (lanes 5–9). HEK whole cell lysates were analyzed by Western blot using antibodies against phosphotyrosine. To confirm the identity of the bracketed 170 kD pY band as autophosphorylated ErbB, recipient HEK cells were incubated at 37°C for 1 h with lapatinib before addition of conditioned medium (lanes 8 and 9); asterisk indicates a nonspecific band. Uniform expression of ErbB1, a major endogenous ErbB in HEKs, is also shown. All data shown are from the same blot of a single experiment. Phospho-Y values (in arbitrary units, normalized to β actin in the same samples) for the HB-EGF concentration curve are provided. (B) ErbB-associated 4G10 signal from three independent experiments was normalized to β actin and showed that conditioned medium from proHB-EGF transfectants cultured with TGFβ induced 0.893 ± 0.115 times the ErbB-associated 4G10 signal as was elicited by conditioned medium from cells cultured in the absence of TGFβ. Individual data points are plotted as black squares.
Next, we directly examined the activation state of ErbBs by subjecting DCDMLs to cell surface biotinylation, after which all active ErbBs on the plasma membrane were detected by Western blotting using the 4G10 antiphosphotyrosine antibody. In untreated DCDMLs, a ∼170 kD, lapatinib-sensitive band was detected that represents cell surface ErbBs basally activated by endogenous ligand (VanSlyke et al., 2023) (Figure 7A, lane 3). Pretreatment with TGFβ at 37°C for 5, 45 min, or 1.5 h did not significantly increase the amount of tyrosine autophosphorylated ErbB recovered (Figure 7A, lanes 5–10).
FIGURE 7:

Short-term treatment with TGFβ does not enhance activation of ErbBs in DCDMLs. (A) The level of tyrosine phosphorylated ErbBs on the cell surface is unaffected by TGFβ. DCDMLs were incubated at 37°C for 1 h with DMSO or lapatinib, or for 5, 45, or 90 min with or without TGFβ as indicated. In lanes 1 and 2, cells were then incubated at 4°C for 15 min with 100 ng/ml HB-EGF. All cultures were then subjected to cell surface biotinylation at 4°C, and plasma membrane pools of autoactivated ErbB analyzed by Western blotting using the antiphosphotyrosine 4G10 antibody. To demonstrate equal loading, biotinylated samples were reprobed with ErbB1 antibody and whole cell lysates were analyzed for β actin. (B) TGFβ does not increase autophosphorylation of ErbB1. DCDMLs were incubated at 37°C for 1 h with DMSO or lapatinib, or for 5, 45, or 90 min with or without TGFβ as indicated. In lanes 13-16, cells were then treated at 37°C for 5 min with 0.1, 1, or 10 ng/ml HB-EGF. All cultures were then lysed. Western blotting of whole cell lysates with a rabbit antibody specific to pY1068ErbB1 demonstrated that treatment with HB-EGF, but not with TGFβ, increased the activation of ErbB1. Blots were reprobed with a rat antibody against total ErbB1. (C). Results from (A) and (B) were graphed as fold value from cells incubated with TGFβ versus without TGFβ for the indicated time. In all cases, p > 0.2. Individual data points are plotted from experiments in which n = 3.
ErbB1 is a potent activator of ERK (Albeck et al., 2013; Pinilla-Macua et al., 2017). To specifically examine the activation state of this receptor, we used a rabbit polyclonal antibody specific for the tyrosine 1068 phosphorylated form of ErbB1 (Figure 7B). Autophosphorylation of this residue plays an essential role in downstream activation of many signaling pathways including ERK and AKT (Sebastian et al., 2006; Roskoski, 2014). Unlike the mouse reagent used in Figure 11A, this antibody readily detects endogenous active ErbB1 in whole cell lysates as demonstrated by its recognition of a Mr = 170 kD species (Figure 7B, lane 12) that was blocked by incubation with lapatinib (lane 11) and that is dose-dependently enhanced by exogenously added HB-EGF (lanes 13–15). The level of pY1068R1 immunoreactivity recovered from DCDMLs was unaffected by short-term treatment with pERK-inducing levels of TGFβ (lanes 17–22).
FIGURE 11:

ERK is required for expression of ErbB1 and ErbB2 protein. (A) DCDMLs were cultured for 6 d with DMSO, UO126, TGFβ, or both (DMSO or UO126 added 1 h before TGFβ). The cells were then incubated at 4°C for 15 min in either the absence or presence of 60 ng/ml TGFα as indicated. Whole cell lysates were then analyzed by Western blot with mouse anti-pY1068ErbB1 antibodies, followed by reprobing for total ErbB1. Data are graphed as fold obtained in cultures treated with DMSO instead of UO126 in the same experiment. For all conditions, p = 0.000. Note that the anti-pY1068ErbB1 antibody (#2236) employed in these experiments is not as sensitive as the rabbit reagent used in Figure 7B. (B) DCDMLs were cultured for 6 d with DMSO or the p38 inhibitor SB203580 before analysis of whole cell lysates for total ErbB1. (C) DCDMLs cultured for 6 d with DMSO or UO126 were lysed and analyzed for ErbB2 or ErbB4. (D) Effect of SB203580 on the level of ErbB1 (p = 0.012), or UO126 on the levels of ErbB2 (p = 0.000) and ErbB4 (p = 0.123) detected by Western blot expressed as fold DMSO only cultures in the same experiment.
A new model for TGFβ-induced activation of ERK
If transactivation of ErbBs is not the mechanism by which TGFβ stimulates ERK, then why do ErbB inhibitors prevent TGFβ from upregulating pERK levels? In addition to ErbBs and ERK itself, the core canonical ErbB-to-ERK pathway requires the sequential activity of two intermediate kinases, namely Raf-1 and MEK (Roskoski, 2014). We used specific inhibitors of each kinase in the core pathway to test their effect on pERK levels after stimulation of DCDMLs with either TGFβ or with sodium orthovanadate (NaV). The latter is a general inhibitor of tyrosine phosphatases (Swarup et al., 1982) that can directly block the activity of the phosphatases that inactive ERK (Caunt and Keyse, 2013). We found that sodium orthovanadate was capable of increasing the level of pERK in DCDMLs in the presence of the Raf inhibitor sorafinib, the MEK inhibitor UO126, or the ErbB inhibitor lapatinib (Figure 8A). In contrast, each of these compounds abolished the ability of TGFβ to activate ERK without affecting stimulation of Smad3 (Figure 8B). We conclude that upregulation of pERK by TGFβ requires the canonical ErbB-Raf-MEK-ERK cascade.
FIGURE 8:

Stimulation of ERK by TGFβ requires the canonical ErbB-to-ERK pathway and is not phenocopied by a tyrosine phosphatase inhibitor. DCDMLs were preincubated for 1 h with either DMSO, 10 uM sorafenib, UO126, or lapatinib before exposure to either the general tyrosine phosphatase inhibitor sodium orthovanadate (NaV; 0.2 mM) for 30 min (A), or to TGFβ for 1.5 h (B). The levels of pERK, tERK, pSmad3, and tubulin were assessed by Western blot. (C) The level of pERK in cells treated with either sodium orthovanadate or TGFβ in the presence of each kinase inhibitor was graphed relative to pERK in cells exposed to kinase inhibitor only. n = 5.
In DCDMLs, as in other cell types, activation of ERK by exogenously added ErbB ligand is short-lived (Figure 4A). This cannot be attributed to ErbB degradation, given that even very high concentrations of HB-EGF (10 nM; 100 ng/ml) cause a ∼25% decrease in ErbB1, ErbB2, and ErbB4 levels after a 4-h incubation (VanSlyke et al., 2023). The duration of ERK activation in other cell types is known to be limited by several negative feedback mechanisms (Lake et al., 2016; Muta et al., 2019). Some of these processes are mediated by effectors that are regulated by physiologically attainable levels of reactive oxygen species (ROS) (Son et al., 2013), and oxidative stress can prolong and/or enhance ERK signaling downstream of TGFβ (Rhyu et al., 2005; Liu et al., 2010). Given that ErbBs are the major source of basal ERK activity in lens cells (Figure 3), and that TGFβ is known to rapidly induce ROS in many cell types (Ohba et al., 1994; Junn et al., 2000; Rao et al., 2004; Rhyu et al., 2005: Yao et al., 2007; Joo et al., 2008; Liu et al. 2010; Michaeloudes et al., 2011; Samarakoon et al., 2013a; Raghavan and Nagaraj, 2016; Shin et al., 2017), we hypothesized that TGFβ may be inactivating or otherwise negating one or more negative feedback pathways in a ROS-dependent manner. This could lead to the relatively slow, ErbB-dependent increase in pERK we have observed. If such a model were correct, then TGFβ upregulation of pERK in DCDMLs should be blocked by an antioxidant. We, therefore, preincubated DCDMLs for 1 h with the commonly used cysteine source N-acetyl cysteine (NAC; Zhitkovich, 2019). Control experiments demonstrated that, as expected, this treatment prevented activation of pERK in response to the classical oxidant stressor hydrogen peroxide (H2O2; Figure 9A). We found that NAC pretreatment also prevented TGFβ from increasing the level of pERK (Figure 9A). In contrast, NAC had no effect on the ability of TGFβ to activate Smad3 under the same conditions. A 1-h preincubation with NAC also did not block activation of pERK in response to either FGF (Figure 9A) or HB-EGF (Figure 9B), further demonstrating a specific effect on stimulation of ERK by TGFβ. Similar results were obtained when NAC was replaced with the mechanistically distinct (Ezerina et al., 2018) antioxidant glutathione monoethyl ester (GEE; Supplemental Figure S2A).
FIGURE 9:

TGFβ upregulation of pERK is specifically inhibited by the antioxidant NAC. DCDMLs were preincubated for 1 h with or without 20 mM NAC before treatment with: (A) 4 ng/ml TGFβ, 1 ng/ml FGF, or 250 μM H2O2 for 90 min, or (B) 10 ng/ml HB-EGF for 5 min. Whole cell lysates were then analyzed for pERK, tERK, pSmad3, and tubulin. (C) Results were graphed as percent inhibition by NAC of upregulation of pERK or pSmad3 by the indicated treatment. Similar results were obtained with 10 mM NAC.
If EMyT of lens epithelial cells requires activation of ERK downstream of TGFβ-generated ROS, then NAC would be expected to prevent TGFβ from inducing myofibroblast differentiation. We found this to be that case as assessed by Western blot analysis (Figure 10A) and immunofluorescence microscopy (Figure 10B). The antioxidant GEE had a similar effect on EMyT marker expression (Supplemental Figure S2B). NAC did not inhibit the expression of the fiber differentiation marker δ-crystallin, demonstrating the specificity of the effect (Figure 10A). As after short-term treatments (Figure 9A), coculture with NAC for 6 d prevented TGFβ from increasing the levels of pERK (Figure 10A). Reduced glutathione was previously shown to block EMT in mammalian lens cells (Chamberlain et al., 2009).
FIGURE 10:

Oxidative stress is required for upregulation of EMyT by TGFβ and is enhanced by TGFβ. (A) EMyT in response to TGFβ is blocked by the antioxidant NAC. DCDMLs were cultured for 6 d with no additions (-), 20 mM NAC, TGFβ, or both (NAC added 1 h before TGFβ). Whole cell lysates were then analyzed by Western blot for FN, αSMA, δ-crystallin, phospho and total ERK, and tubulin. Results graphed as fold values obtained in cells cultured with NAC plus TGFβ relative to values from cells cultured with TGFβ only. (B) DCDMLs cultured as in (A) were fixed and immunostained to demonstrate the lack of αSMA-containing stress fibers in NAC treated cells. NAC also prevented TGFβ from inducing the expression of the fibrotic marker collagen I as assessed by an antibody specific to procollagen I. (C) Multi-day exposure to TGFβ increases pERK levels in response to oxidative stress. DCDMLs cultured for 6 d in the presence or absence of TGFβ were preincubated with or without 20 mM NAC for 1 h before a 1.5 h incubation in the absence or presence of 250 μM H2O2. Cell lysates were analyzed for phospho and total ERK, or tubulin. NAC inhibited the activation of ERK by H2O2 by 100% ± 0 in cells cultured in the absence of TGFβ, and by 84% ±10.8 in cells cultured with TGFβ (n = 9; p = 0.00).
In many cell types, TGFβ decreases the expression of multiple antioxidant enzymes (Liu and Desai, 2015). This also appears to be the case in lens epithelial cells. DCDMLs cultured with TGFβ for 2–6 d had ∼8.5-fold (8.56 ± 3.06; n = 8; p = 0.000) higher levels of pERK after exposure to H2O2 than no TGFβ control cells, an increase that was blocked by preincubation with NAC (Figure 10C). This effect was not observed if cells were exposed to TGFβ for only 90 min (unpublished data).
ErbB1 and ErbB2 are targets of ERK signaling
We have previously shown that TGFβ increases the expression of total as well as cell surface, ligand-activatable pools of ErbB1, a process that appears to be involved in TGFβ upregulation of EMyT (VanSlyke et al., 2023). Given the importance of ERK and ErbBs for TGFβ-induced EMyT, we considered the possibility that ERK activity could be required for TGFβ to enhance ErbB1 levels. Indeed, we found that inactivating ERK in DCDMLs by addition of UO126 prevented TGFβ from increasing ErbB1 levels (Figure 11A). Decreased expression of ErbB1 was associated with reduced function as assessed by autoactivation of ErbB1 in response to exposure to TGFα ligand (Figure 11A). Notably, UO126 also reduced basal levels of ErbB1 protein and function in the absence of TGFβ (Figure 11A). Culturing DCDMLs with an inhibitor of another class of MAP kinases, namely p38, increased instead of reduced ErbB1 levels (Figure 11B), demonstrating the specificity of this effect for ERK. UO126 also decreased the level of ErbB2, the favored dimerization partner of ErbB1 in the activation of ERK (Graus-Porta et al.,1995; Pinkas-Kramarski et al., 1996; Olayioye et al., 2000; Figure 11C). In contrast, blocking ERK had no significant effect on the level of ErbB4, the other functional ErbB kinase expressed in lens epithelial cells (Figure 11C) that does not appear to play a causative role in in TGFβ-induced EMyT (VanSlyke et al., 2023).
DISCUSSION
DCDMLs are maintained under serum-free conditions to mimic the avascular environment of the lens, allowing us to carry out studies on ERK and its growth factor signaling effectors without the confounding effects of exogenous serum or serum withdrawal. Multiple ERK-stimulating growth factors including PDGF (Potts et al., 1994; Reneker and Overbeek, 1996; Wang et al., 2010), FGF (Le and Musil, 2001; Lovicu and McAvoy, 2001; Robinson, 2006), and ErbB ligands (VanSlyke et al., 2023) are active in lens epithelial cells, likely a prerequisite for their ability to survive and differentiate in the avascular environment of the lens. The absence of ERK- or AKT- activating ErbB ligands in the aqueous humor (Iyengar et al., 2009) implies that ErbB signaling in lens central epithelial cells in vivo is controlled by their endogenously produced ligands. The level of ErbB-dependent pERK we observe in DCDMLs is therefore likely to reflect physiologically relevant conditions. Basal ERK activity downstream of tonic ErbB signaling has previously been shown to be required for TGFβ to upregulate the production of prostaglandin E2 in human bronchial cells (Liu et al., 2007). Our studies extend such a role to a new cell type (lens epithelial cells) and downstream process (EMyT).
Mechanism of ERK activation by TGFβ
In chick (Boswell et al., 2017), rodent (Wojciechowski et al., 2017), and human (Tiwari et al., 2016) primary lens epithelial cells, TGFβ increases the level of pERK within 1–2 h. In the same systems, blocking ERK activity prevents TGFβ from inducing EMyT. A causal relationship between TGFβ upregulation of ERK activity and of EMT has been established in some nonlenticular cell types (Xie et al., 2004; Iea-Flores et al., 2019). Derynck and colleagues (Lee et al., 2007; Muthusamy et al., 2015) have described a pathway by which ligand-stimulated TGFβRs phosphorylate the adaptor protein ShcA, which (via a ShcA/Grb2/SOS complex) directly activates the canonical Ras/Raf/MEK/ERK cascade. This is not the mechanism by which TGFβ increases pERK in lens cells, because: (1) induction of pERK mediated by ShcA is detectable within 5–10 min of TGFβ addition, a much shorter period than is required in lens cells, and (2) the aforementioned ShcA pathway would not be blocked by ErbB inhibitors. A major means by which TGFβ can less directly stimulate ERK is via transactivation of receptor tyrosine kinases upstream of the canonical Ras-to-ERK pathway. A simple explanation for the ability of ErbB inhibitors to block TGFβ-induced ERK activation in lens cells would be if TGFβ transactivated ErbBs (Uchiyama-Tanaka et al., 2002; Wang et al., 2008; Ebi et al., 2010; Samarakoon et al., 2013a).
However, we failed to obtain any evidence that TGFβ increases the level of autophosphorylated, activated ErbBs before or at the time at which it upregulates pERK levels (Figure 7). Addition of exogenous ErbB ligands to DCDMLs stimulates the phosphorylation of AKT within 5 min (Figure 3C). If TGFβ transactivated ErbBs, then we would expect to observe an increase in pAKT levels within 1.5 h of TGFβ addition. Such rapid, concomitant activation of AKT and ERK due to transactivation of ErbB has been reported in other cell types (Eguchi et al., 1999; Chiu et al., 2005). Instead, upregulation of pAKT by TGFβ in DCDMLs is only detectable after a 24-h treatment (Boswell et al., 2017). Our data also indicate that TGFβ does not stimulate ERK by transactivating either FGFRs or PDGFRs, given that TGFβ can still increase the level of pERK in the presence of inhibitors of these receptors (Figure 4). If TGFβ induced the transactivation of some other major class of receptor tyrosine kinase, it might be expected to lead to an increase in a [gt] 80 kD phosphotyrosine-positive species on the plasma membrane. No such band was detectable by cell surface biotinylation (Figure 7A). TGFβ-induced transactivation of a non-ErbB receptor also cannot explain why TGFβ stimulation of ERK is abolished by ErbB inhibitors.
The canonical core ErbB-to-ERK pathway involves multiple kinase-dependent steps (Figure 12). First, ligand activation of ErbBs leads to their lapatinib-inhibitable autophosphorylation on multiple tyrosine residues. These phosphotyrosines form binding sites for various molecules that initiate the assembly of a pY ErbB scaffold/effector complex that ultimately acts to increase the activation of the serine/threonine Raf-1 kinase. Raf-1 then activates MEK. The latter dual-specific kinase in turn phosphorylates ERK on threonine and tyrosine residues, thereby activating its kinase activity. In principle, one mechanism by which TGFβ could enhance pERK levels would be by inhibiting tyrosine phosphatases that directly or indirectly reduce the activity of one or more of these kinases. Several enzymes with such dephosphorylating activity have been reported to be inhibited by ROS (e.g., MKP1/2/3; Cdc25; Xia et al., 1999; Liu et al., 2010; Ostman et al., 2011; Tonks, 2013; Okoh et al., 2015). If this were the case, then TGFβ should phenocopy the ability of the general tyrosine phosphatase inhibitor sodium orthovanadate to increase pERK levels even if an upstream kinase is blocked. We found instead that inhibitors of ErbB (lapatinib), Raf-1 (sorafinib), or MEK (U0126) each completely abolished upregulation of pERK levels in response to TGFβ (Figure 8). Moreover, sodium orthovanadate did not prolong or increase activation of pERK when added immediately after a 5-min treatment with HB-EGF (unpublished data), implying that protein tyrosine phosphatases are not major negative determinants of ErbB-to-ERK signaling strength in DCDMLs. This contention is supported by evidence that lens epithelial cells have low levels of endogenous tyrosine phosphatase activity (Bozulic et al., 2004).
FIGURE 12:

Kinases in the core canonical ErbB-to-ERK pathway, and proposed step at which TGFβ/ROS acts to stimulate ERK in lens epithelial cells in an ErbB-dependent manner.
Another potential means by which TGFβ could increase pERK levels in lens cells would be by positively upregulating the activity of either Raf-1, MEK, or ERK in a ROS-dependent manner. Such a mechanism is, however, also inconsistent with our finding that blocking an upstream kinase prevents TGFβ from enhancing pERK levels (Figure 8). A stimulatory effect of TGFβ on the Raf/MEK/ERK signaling module would also not explain why upregulation of pERK by TGFβ is more dependent on ErbB than on the other lens growth factor receptors (e.g., FGFR; PDGFR) that use the same pathway to activate ERK. We therefore, favor the possibility that TGFβ acts at the level of the pY ErbB scaffold/effector complex. An attractive mechanism would be if TGFβ oxidatively activates Src, which then activates a positive effector (e.g., Gab1) that acts upstream of Raf-1 in the ErbB-to-ERK pathway (Furcht et al., 2015). However, Src is not detectably activated (i.e., phosphorylated on Y416) in DCDMLs in response to a 1.5-h treatment with TGFβ, nor does the Src inhibitor dasatinib reduce the level of pERK in the absence or presence of TGFβ (unpublished data). More than a dozen proteins have been identified to bind directly to tyrosine autophosphorylated ErbB1, which then recruit multiple other molecules to the complex in a cell type-specific manner. Consequently, over a hundred proteins have been reported to become associated with activated ErbB1 based on biochemical evidence, with many more proposed as putative ErbB-interacting proteins (Morandell et al., 2008; Tong et al., 2014). Determining the precise mechanism by which TGFβ upregulates pERK levels downstream of ErbB autoactivation is therefore likely to be challenging, especially in a limited through-put primary cell system.
Regardless of the underlying signaling pathways, our results support a model in which oxidative stress plays a central role in the upregulation of ERK and EMyT by TGFβ in lens epithelial cells (Figures 9, 10; Supplemental Figure S2). Although we favor TGFβ as a key source of such stress (discussed below), even low-level activation of ErbBs is well known to transiently generate ROS (Rao et al., 2004; Koseska and Bastiaens, 2020; Joshi et al., 2023). It has been proposed that isolated lens epithelial cells are constitutively in a state of relative oxidative stress, in part due to the absence of antioxidants provided by lens fiber cells in the intact organ (discussed by Wei et al., 2017). Our data suggest that in DCDMLs, as in many other cell types (Liu and Desai, 2015), TGFβ reduces the expression of antioxidants (Figure 10C). After cataract surgery, the loss of lens fiber cells and the TGFβ-induced decrease in antioxidants in lens epithelial cells could therefore combine with the generation of ROS downstream of TGFβ and ongoing basal ErbB signaling to create an environment particularly conducive to TGFβ-induced, ErbB-dependent activation of ERK and subsequent induction of EMyT.
Comparison with previous results
As previously reviewed (Musil, 2012), growth factor signaling is conserved between DCDMLs and primary mammalian lens epithelial cell systems. Blocking ErbB or ERK activation at the time of initial addition of TGFβ prevents TGFβ from inducing EMyT in both rat central lens epithelial explants (Wojciechowski et al., 2017) and in chick DCDMLs (Boswell et al., 2017). Lovicu and coworkers (Shu et al., 2019) have reported that an 18-h treatment of rat lens central epithelial explants with TGFβ increased the intensity of an anti-pY1068 ErbB1 immunoreactive species. The significance of this finding, which we have failed to reproduce in DCDMLs, is difficult to assess and could be a consequence of the time-dependent increase in ErbB1 transcripts they observed 24 h after addition of TGFβ in this system. We have recently reported (VanSlyke et al., 2023) that a 6-d treatment with TGFβ also does not increase anti-pY1068 ErbB1 immunoreactivity in otherwise untreated DCDMLs. More relevant to our current study is the observation of Shu et al. (2019) that a 15-min or 2-h treatment of rat lens explants with TGFβ failed to increase pY1068 ErbB1 levels to a statistically significant extent. Thus in rat, as in chick, lens epithelial cells (Figure 7B), there is no evidence that TGFβ stimulates autophosphorylation of ErbB1 on a major site of receptor activation at or before the time at which TGFβ increases pERK levels. Our finding that a 5–90 min exposure to TGFβ also fails to increase ErbB immunoreactivity detected with a pan-phosphotyrosine antibody (Figure 7A) rules out the possibility that Y1068 of ErbB1 is somehow insensitive to TGFβ-induced modification. Instead, our results indicate that TGFβ has no short-term effect on the overall activation state of ErbBs in lens cells.
The ability of TGFβ to induce oxidative stress has previously been examined in rodent lens epithelial explants using the ROS probe dihydroethidium (DHE). It was reported that a minimum of 6 h of exposure to TGFβ was required for a detectable increase in DHE signal (Das et al., 2016). Such a delay is unlikely to be attributable to a lack of enzymes capable of generating ROS in untreated cells, given the basal expression of NADPH oxidase subunits in lens epithelium (Rao et al., 2004; Wang and Lou, 2009), including Nox4 protein (Das et al., 2020).
As is true for many assays used to measure ROS, DHE has limitations, including a tendency to autoxidize, rapidly photobleach, and because of the nonstoichiometric yield of ethidium. It has, therefore, been considered to provide a qualitative instead of quantitative readout of superoxide generation (Ortega-Villasante et al., 2018). It may be that under the conditions used by Das et al., (2016), the DHE assay is only sufficiently sensitive to detect relatively large increases in ROS secondary to TGFβ enhancement of Nox4 levels. This possibility is supported by studies using other probes that demonstrate that induction of ROS by TGFβ is detectable within 1–2 h in many other systems (Ohba et al., 1994; Junn et al., 2000; Rhyu et al., 2005; Yao et al., 2007; Joo et al., 2008; Liu et al., 2010; Michaeloudes et al., 2011; Samarakoon et al., 2013a; Shin et al., 2017). This includes human lens epithelial cell lines in which TGFβ has been reported to increase ROS within 2 min (Rao et al., 2004) or 2 h (Raghavan and Nagaraj, 2016) as assessed using 2′,7′-dichlorofluorescein diacetate (DCFDA). Our attempts to use DCFDA in DCDMLs were unsuccessful due to limited retention of the deacetylated carboxydichlorofluorescein (CDCF) indicator dye within the cell, a known limitation of this assay in some systems (Ng and Ooi, 2021). Of note, primary lens epithelial cells have been reported to express functional MRP5 (multidrug resistance-associated protein 5), a major cellular exporter of CDCF (Umapathy et al. 2015; Li et al., 2020). Because of these limitations, we attempted to address the role of Nox activity in TGFβ upregulation of ERK activity using a Nox inhibitor (VAS2870) previously reported to reduce both ROS and EMyT in rodent primary lens epithelial cells (Das et al., 2016). These studies were uninformative because the drug increased pERK levels in the absence of TGFβ within 90 min; genetic knockout of NOX4 has recently been reported to have a similar effect in rodent lens (Das et al., 2020). As previously proposed in human lung fibroblasts (Junn et al., 2000), it is possible that generation of fibrosis-supporting levels of ROS is initiated by TGFβ stimulation of basally expressed ROS-generating enzymes, after which TGFβ upregulation of NOX4 transcription may play an important (but not in all cases essential; Das et al., 2020) role in EMyT progression.
ErbBs as major determinants of basal ERK signaling
Given the plethora of effectors that can stimulate downstream activation of ERK, why should ErbB signaling play such a major role in determining the level of ERK in lens epithelial cells? Partially occupied ErbB can signal and activate unliganded receptors (Koseska and Bastiaens, 2020). Moreover, ErbBs has been proposed to be subject to less negative regulation than FGFRs (Schlessinger, 2004; Raina et al., 2022). These properties make ErbBs particularly powerful activators of ERK, the functional levels of which are then restrained by multiple negative feedback pathways that prevent kinase overstimulation. It has been demonstrated in multiple cell types that terminating ErbB kinase activity with specific ErbB inhibitors results in a very rapid drop in pERK levels (Kleiman et al., 2011; Sparta et al., 2015). This underscores the importance of continuous ErbB activity for the maintenance of ERK function, and explains why exploiting ErbB signaling would be an effective means for TGFβ to stimulate ERK and promote ERK-dependent processes such as EMyT. Notably, we have reported evidence for the predominance of ErbB1/ErbB2 heterodimers in DCDMLs under basal conditions (VanSlyke et al., 2023). This species has been reported to be a more potent inducer of ERK activation than ErbB1 homodimers (Graus-Porta et al., 1995; Pinkas-Kramarski et al., 1996). Our finding (Figure 11) that ERK activity is required for basal levels of both ErbB1 and ErbB2 (but not of ErbB4) creates a reciprocal relationship to maintain ERK activity in which ErbB1 and ErbB2 are required for basal levels of pERK, and ERK activity is in turn required for expression of these receptors. Our results therefore establish a previously unrecognized TGFβ/ErbB/ERK axis in lens cells.
Therapeutic implications
As in other primary lens cell culture systems, the MEK inhibitor UO126 is an effective means to block TGFβ-induced EMyT in DCDMLs (Figure 1). Given the plethora of important roles of ERK in a wide variety of cell types, using such a global inhibitor of ERK signaling in the eye in vivo could, however, have unintended consequences in other ocular tissues (Méndez-Martínez et al., 2019). In this paper, we have shown that ErbBs are the main source of ERK activity in isolated lens epithelial cells. This finding raises the possibility that ErbB inhibitors would be as effective as UO126 in blocking the initiation of fibrotic PCO after cataract surgery without deleteriously reducing ERK activity in other ocular tissues in which ERK levels are driven by non-ErbB pathways. Notably, no ocular toxicity has been reported in humans after systemic administration of lapatinib despite its likely ability to enter the eye based on its permeability to the blood-brain barrier (Huillard et al., 2014).
Outside of the lens, fibrotic disorders are a leading cause of human death (Wynn, 2004). In at least some non-lenticular cell types, ERK, oxidative stress, TGFβ, and/or ErbB1 have been implicated in the development or progression of fibrosis (Leask, 2012; Samarakoon et al., 2013b; Vallath et al., 2014). Interestingly, these same factors have also been linked to cancer, consistent with the concept that tumors are “wounds that do not heal” (Rybinski et al., 2014).
Cooperation between TGFβ and ErbB1 in promoting fibrosis has been established in both kidney (Zhuang and Lui, 2011; Chen et al., 2012) and lung (Zhou et al., 2012; Cheng et al., 2022). Moreover, it has been reported that blocking ERK reduces lung fibrosis induced by overactivation of ErbB1 in both in culture and in vivo (Madala et al., 2012). It is therefore important to investigate whether the TGFβ/ErbB/ERK axis we have discovered in lens cells is also operative in other tissues.
MATERIALS AND METHODS
Materials
Recombinant human TGFβ1, TGFα, HB-EGF, noggin, and bovine FGF2 were from R & D Systems (Minneapolis, MN). The following antibodies were purchased from Cell Signaling Technology, Danvers, MA: antiphospho-p44/42 MAP kinase (#9106), antitotal p44/42 MAPK (#9102), antiphospho-p38 (#9211), antitotal AKT (#9272), and antiphospho (Ser473) AKT (#9275). The following antibodies against the cytoplasmic tail domains were used to detect ErbBs in DCDMLs: for ErbB1, rabbit SC-03 from Santa Cruz Biotechnology (Santa Cruz, CA) and the rat monoclonal 20.3.6 (Hayman et al., 1986; a kind gift of Dr Michael Hayman, Stonybrook University); for ErbB4, C18 (Santa Cruz Biotechnology), and for ErbB2, A0485 from Agilent (Santa Clara, CA). Two antibodies specific for the tyrosine1068 autophosphorylated, activated form of ErbB1 were used, a rabbit polyclonal (#2234) and a mouse monoclonal (#2236), both from Cell Signaling Technology. Other commercial antibodies used in this study: for phosphoSmad3, ab51451 from Abcam (Cambridge, MA); for total-Smad3, ab84177 from Abcam; for total p38, sc-535 from Santa Cruz Biotechnology; for α-tubulin, T5168 from Sigma-Aldrich (St Louis, MO); for αSMA, clone 1A4 from Agilent; for β actin, clone C4 (MilliporeSigma; Burlington, MA); for phosphotyrosine, clone 4G10 (MilliporeSigma); for chick fibronectin, B3/D6 (from D. Fambrough, Johns Hopkins University; Developmental Studies Hybridoma Bank, University of Iowa), and for procollagen I, SP1.D8 (from H. Furthmayr, Stanford University; Developmental Studies Hybridoma Bank). Rabbit antimouse CP49 polyclonal serum (#899 or 900) was a generous gift of Paul FitzGerald, University of California, Davis, as was the rabbit anti-CP115 antiserum (#76). Sheep anti–δ-crystallin antibody was produced in the laboratory of Joram Piatigorsky (National Institutes of Health) and was a gracious gift of Steve Bassnett (Washington University School of Medicine). SB-431542, glutathione monoethyl ester (GEE), and N-acetyl-l- cysteine (NAC) were from Sigma-Aldrich. UO126, PD173074, and SB-203580 were from Millipore Sigma. Lapatinib, erlotinib, and sorafinib were purchased from LC Labs (Woburn, MA), sodium orthovanadate from New England BioLabs (Ipswich, MA), and afatinib and crenolanib from Selleck (Houston, TX).
DCDML cell culture and treatments
DCDML cultures were prepared from E10 chick lenses as previously described (Le and Musil, 1998). During this process, cells exterior to the lens capsule are removed and mature lens fiber cells die, leaving a preparation of purified lens epithelial cells. Cell were plated at subconfluent density (0.9 × 105 cells/well) onto laminin-coated 96-well tissue culture plates, and cultured at 37°C in a 5% CO2 incubator in the absence of serum in 100–200 μl M199 medium supplemented with penicillin G, streptomycin, and BOTS (2.5 μg/ml bovine serum albumin, 25 μg/ml ovotransferrin, 300 nM selenium). Cells were fed every 2 d with fresh medium. Inhibitors were used at the following final concentrations: 4 μM lapatinib, 5 μM erlotinib, 3 μM SB-431542, 15 μM UO126, 0.1 μM PD173074, 20 μM SB-203580, 0.5 μM afatinib, 0.5 μM crenolanib, 1 ug/ml noggin, and 10 μM sorafenib.
Cell line
Human kidney HEK 293 cells were from the American Type Culture Collection (ATCC; Manassas, VA, USA).
Immunofluorescence microscopy
DCDMLs grown on laminin-coated glass coverslips were fixed in 2% paraformaldehyde in phosphate-buffered saline and processed as previously described (Le and Musil, 1998; 2001). Images were captured using a Leica DM LD photomicrography system and Scion Image 1.60 software.
Cell surface biotinylation
DCDMLs were biotinylated on ice using freshly prepared 0.25 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Thermoscientific #21331) as previously described (VanSlyke et al., 2023).
Plasmids and transient transfection
One day after plating, DCDML cultures were transfected in M199 medium without BOTS or antibiotics using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) following the manufacturer’s suggested protocol. Control experiments confirmed that the efficiency of transient transfection of DCDMLs is consistently ∼70% (Boswell et al., 2009). The pro HB-EGF WT plasmid (Nguyen et al., 2000) was a gift from Rosalyn Adam (Addgene plasmid # 11600).
Detection of HB-EGF bioactivity in conditioned medium
Medium was collected from transfected DCDML cells cultured for 2 d with or without TGFβ and immediately centrifuged at 3000 rpm in a microfuge (0.8 rcf) for 5 min to remove any cell debris before incubation with HEK cell recipients at 4°C for 15 min. Medium was snap-frozen and stored at -80°C before use.
Immunoblot analysis
Whole cell lysates were solubilized directly into SDS–PAGE sample buffer and boiled. Equal volumes of total cell lysate were transferred to polyvinylidene fluoride membranes, and the blots probed with primary antibodies. Immunoreactive proteins were detected using secondary antibodies conjugated to either IRDye800 conjugated to either IRDye800 (Rockland Immunochemicals, Pottstown, PA) or Alexa Fluor 680 (Invitrogen, Waltham, MA) and directly quantified using the LI-COR Biosciences Odyssey infrared imaging system (Lincoln, NE) and associated software. The level of each protein was normalized to the level of β actin or α tubulin in the same sample. Data are graphed as the means ± SD obtained in the number of experiments indicated in each figure. Data were analyzed for significance using the two-tailed paired Student’s t test.
Supplementary Material
Acknowledgments
Funded by R01 EY028558 from the National Eye Institute of the National Institutes of Health to L.S.M. We would like to thank John Allen for valuable technical assistance in the latter stages of this project.
Abbreviations used:
- αSMA
α−smooth muscle actin
- DCDML
dissociated cell-derived monolayer
- DHE
dihydroethidium
- EMT
epithelial–mesenchymal transition
- EMyT
epithelial–myofibroblast transition
- ERK
extracellular signal–regulated kinase
- FGF
fibroblast growth factor
- FN
fibronectin
- MEK
mitogen-activated protein kinase kinase
- NAC
N-acetyl cysteine
- NaV
sodium orthovandate
- PCO
posterior capsule opacification
- PDGF
platelet-derived growth factor
- ROS
reactive oxygen species
- TGFβ
transforming growth factor β
- TGFβR
transforming growth factor β receptor.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E23-07-0294) on January 3, 2024.
REFERENCES
- Albeck JG, Mills GB, Brugge JS (2013). Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell 49, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apple DJ, Ram J, Foster A, Peng Q (2000). Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol 45(Suppl 1), S1–S196. [PubMed] [Google Scholar]
- Awasthi N, Guo S, Wagner BJ (2009). Posterior capsular opacification: a problem reduced but not yet eradicated. Arch Ophthalmol 127, 555–562. [DOI] [PubMed] [Google Scholar]
- Boswell BA, Korol A, West-Mays JA, Musil LS (2017). Dual function of TGFβ in lens epithelial cell fate: implications for secondary cataract. Mol Biol Cell 28, 907–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell BA, Le AC, Musil LS (2009). Upregulation and maintenance of gap junctional communication in lens cells. Exp Eye Res 88, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell BA, VanSlyke JK, Musil LS (2010). Regulation of lens gap junctions by Transforming Growth Factor beta. Mol Biol Cell 21, 1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozulic LD, Dean WL, Delamere NA (2004). The influence of protein tyrosine phosphatase-1B on Na,K-ATPase activity in lens. J Cell Physiol 200, 370–376. [DOI] [PubMed] [Google Scholar]
- Caunt CJ, Keyse SM (2013). Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J 280, 489–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain CG, Mansfield KJ, Cerra A (2009). Glutathione and catalase suppress TGFbeta-induced cataract-related changes in cultured rat lenses and lens epithelial explants. Mol Vis 15, 895–905. [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC (2012). EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol 23, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang MZ, Harris RC (2016). Expression and Function of the Epidermal Growth Factor Receptor in Physiology and Disease. Physiol Rev 96, 1025–1069. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Kao SY, Chen CL, Yuliani FS, Lin LY, Lin CH, Chen BC (2022). Amphiregulin induces CCN2 and fibronectin expression by TGF-β through EGFR-dependent pathway in lung epithelial cells. Respir Res 23, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu T, Santiskulvong C, Rozengurt E (2005). EGF receptor transactivation mediates ANG II-stimulated mitogenesis in intestinal epithelial cells through the PI3-kinase/Akt/mTOR/p70S6K1 signaling pathway. Am J Physiol Gastrointest Liver Physiol 288, G182–G194. [DOI] [PubMed] [Google Scholar]
- Cvekl A, Ashery-Padan R (2014). The cellular and molecular mechanisms of vertebrate lens development. Development 141, 4432–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SJ, Lovicu FJ, Collinson EJ (2016). Nox4 Plays a Role in TGF-β-Dependent Lens Epithelial to Mesenchymal Transition. Invest Ophthalmol Vis Sci 57, 3665–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SJ, Wishart TFL, Jandeleit-Dahm K, Lovicu FJ (2020). Nox4-mediated ROS production is involved, but not essential for TGFβ-induced lens EMT leading to cataract. Exp Eye Res 192, 107918. [DOI] [PubMed] [Google Scholar]
- de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW (2005). Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs 179, 43–55. [DOI] [PubMed] [Google Scholar]
- Di Gregorio J, Robuffo I, Spalletta S, Giambuzzi G, De Iuliis V, Toniato E, Martinotti S, Pio Conti P, Flati V (2020). The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front Cell Dev Biol 8, 607483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebi M, Kataoka H, Shimura T, Kubota E, Hirata Y, Mizushima T, Mizoshita T, Tanaka M, Mabuchi M, Tsukamoto H, et al (2010). TGFβ induces proHB-EGF shedding and EGFR transactivation through ADAM activation in gastric cancer cells. Biochem Biophys Res Commun 402, 449–454. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Iwasaki H, Ueno H, Frank GD, Motley ED, Eguchi K, Marumo F, Hirata Y, Inagamiet T (1999). Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J Biol Chem 274, 36843–36851. [DOI] [PubMed] [Google Scholar]
- Eldred JA, Dawes LJ, Wormstone IM (2011). The lens as a model for fibrotic disease. Philos Trans R Soc Lond B Biol Sci 366, 1301–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyden BP. (2008). The myofibroblast in health and disease. Revista Española de Patología 41, 3–10. [Google Scholar]
- Ezeriņa D, Takano Y, Hanaoka K, Urano Y, Dick TP (2018). N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem Biol 25, 447–459.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findl O, Buehl W, Bauer P, Sycha T (2010). Interventions for preventing posterior capsule opacification. Cochrane Database Syst Rev CD003738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fișuș AD, Findl O (2020). Capsular fibrosis: a review of prevention methods and management. Eye 34, 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis N (2020). Transforming growth factor-β in tissue fibrosis. J Exp Med 217, e20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furcht CM, Buonato JM, Lazzara MJ (2015). EGFR-activated Src family kinases maintain GAB1-SHP2 complexes distal from EGFR. Sci Signal 8, ra46. [DOI] [PubMed] [Google Scholar]
- Govindarajan V, Overbeek PA (2001). Secreted FGFR3, but not FGFR1, inhibits lens fiber differentiation. Development 128, 1617–1627 [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Hynes NE (1995). Single-chain antibody-mediated intracellular retention of ErbB-2 impairs Neu differentiation factor and epidermal growth factor signaling. Mol Cell Biol 15, 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman MJ, Kitchener G, Knight J, McMahon J, Watson R, Beug H (1986). Analysis of the autophosphorylation activity of transformation defective mutants of avian erythroblastosis virus. Virology 150, 270–275. [DOI] [PubMed] [Google Scholar]
- Hodgkinson LM, Wang L, Duncan G, Edwards DR, Wormstone IM (2010). ADAM and ADAMTS gene expression in native and wound healing human lens epithelial cells. Mol Vis 16, 2765–2776 [PMC free article] [PubMed] [Google Scholar]
- Huang WR, Fan XX, Tang X (2011). SiRNA targeting EGFR effectively prevents posterior capsular opacification after cataract surgery. Mol Vis 17, 2349–2355. [PMC free article] [PubMed] [Google Scholar]
- Huillard O, Bakalian S, Levy C, Desjardins L, Lumbroso-Le Rouic L, Pop S, Sablin MP, Le Tourneau C (2014). Ocular adverse events of molecularly targeted agents approved in solid tumours: a systematic review. Eur J Cancer 50, 638–648. [DOI] [PubMed] [Google Scholar]
- Iyengar L, Patkunanathan B, McAvoy JW, Lovicu FJ (2009). Growth factors involved in aqueous humour-induced lens cell proliferation. Growth Factors 27, 50–62. [DOI] [PubMed] [Google Scholar]
- Joo CK, Kim HS, Park JY, Seomun Y, Son MJ, Kim JT (2008). Ligand release-independent transactivation of epidermal growth factor receptor by transforming growth factor-beta involves multiple signaling pathways. Oncogene 27, 614–628. [DOI] [PubMed] [Google Scholar]
- Joshi MS, Stanoev A, Huebinger J, Soetje B, Zorina V, Roßmannek L, Michel K, Müller SA, Bastiaens PI (2023). The EGFR phosphatase RPTPγ is a redox-regulated suppressor of promigratory signaling. EMBO J 42, e111806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E, Lee KN, Ju HR, Han SH, Im J, Kang HS, Lee TH, Bae Y, Ha K, Lee Z, et al. (2000). Requirement of hydrogen peroxide generation in TGF-beta 1 signal transduction in human lung fibroblast cells: involvement of hydrogen peroxide and Ca2+ in TGF-beta 1-induced IL-6 expression. J Immunol 165, 2190–2197. [DOI] [PubMed] [Google Scholar]
- Kleiman LB, Maiwald T, Conzelmann H, Lauffenburger DA, Sorger PK (2011). Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol Cell 43, 723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopińska J, Młynarczyk M, Dmuchowska DA, Obuchowska I (2021). Posterior Capsule Opacification: A Review of Experimental Studies. J Clin Med 10, 2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseska A, Bastiaens PIH (2020). Processing Temporal Growth Factor Patterns by an Epidermal Growth Factor Receptor Network Dynamically Established in Space. Annu Rev Cell Dev Biol 36, 359–383. [DOI] [PubMed] [Google Scholar]
- Lake D, Corrêa SA, Müller J (2016). Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci 73, 4397–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AC, Musil LS (1998). Normal differentiation of cultured lens cells after inhibition of gap junction-mediated intercellular communication. Dev Biol 204, 80–96. [DOI] [PubMed] [Google Scholar]
- Le AC, Musil LS (2001). A novel role for FGF and extracellular signal-regulated kinase in gap junction-mediated intercellular communication in the lens. J Cell Biol 154, 197–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A (2012). MEK/ERK inhibitors: proof-of-concept studies in lung fibrosis. J Cell Commun Signal 6, 59–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck D (2007). TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J 26, 3957–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefloch R, Pouysségur J, Lenormand P (2008). Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol 28, 511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Freed DM, Schlessinger J, Kiyatkin A (2016). The Dark Side of Cell Signaling: Positive Roles for Negative Regulators. Cell 164, 1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Kim JY, Martis RM, Donaldson PJ, Lim JC (2020). Characterisation of Glutathione Export from Human Donor Lenses. Transl Vis Sci Technol 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tao C, Cai Z, Hertzler-Schaefer K, Collins TN, Wang F, Feng GS, Gotoh N, Zhang X (2014). Frs2α and Shp2 signal independently of Gab to mediate FGF signaling in lens development. J Cell Sci 127(Pt 3), 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Yang SC, Sharma S, Luo J, Cui X, Peebles KA, Huang M, Sato M, Ramirez RD, Shay JW, et al. (2007). EGFR signaling is required for TGF-beta 1 mediated COX-2 induction in human bronchial epithelial cells. Am J Respir Cell Mol Biol 37, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RM, Choi J, Wu JH, Gaston Pravia KA, Lewis KM, Brand JD, Reyes Mochel NS, Krzywanski DM, Lambeth JD, Hagood JS, et al. (2010). Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J Biol Chem 285, 16239–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RM, Desai LP (2015). Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol 6, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Wilkins M, Kim T, Malyugin B, Mehta JS (2017). Cataracts. Lancet 390, 600–612. [DOI] [PubMed] [Google Scholar]
- Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH (2002). Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 156, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovicu FJ, Ang S, Chorazyczewska M, McAvoy JW (2004). Deregulation of lens epithelial cell proliferation and differentiation during the development of TGFbeta-induced anterior subcapsular cataract. Dev Neurosci 26, 446–455. [DOI] [PubMed] [Google Scholar]
- Lovicu FJ, McAvoy JW (2001). FGF-induced lens cell proliferation and differentiation is dependent on MAPK (ERK1/2) signalling. Development 128, 5075–5084. [DOI] [PubMed] [Google Scholar]
- Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD (2012). MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol 46, 380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maidment JM, Duncan G, Tamiya S, Collison DJ, Wang L, Wormstone IM (2004). Regional differences in tyrosine kinase receptor signaling components determine differential growth patterns in the human lens. Invest Ophthalmol Vis Sci 45, 1427–1435. [DOI] [PubMed] [Google Scholar]
- Mamuya FA, Duncan MK (2012). aV integrins and TGF-β-induced EMT: a circle of regulation. J Cell Mol Med 16, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masszi A, Speight P, Charbonney E, Lodyga M, Nakano H, Szászi K, Kapus A (2010). Fate-determining mechanisms in epithelial-myofibroblast transition: major inhibitory role for Smad3. J Cell Biol 188, 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacock WR, Spalton DJ, Stanford MR (2000). Role of cytokines in the pathogenesis of posterior capsule opacification. Br J Ophthalmol 84, 332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Martínez S, Calvo P, Ruiz-Moreno O, Barón NP, Bueno JL, Del Rocío G, Ruiz M, Pablo L (2019). Ocular adverse events associated with MEK inhibitors. Retina 39, 1435–1450. [DOI] [PubMed] [Google Scholar]
- Meng X, Nikolic-Paterson D, Lan H (2016). TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 12, 325–338. [DOI] [PubMed] [Google Scholar]
- Michaeloudes C, Sukkar MB, Khorasani NM, Bhavsar PK, Chung KF (2011). TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 300, L295–L304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandell S, Stasyk T, Skvortsov S, Ascher S, Huber LA (2008). Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signaling network. Proteomics 8, 4383–4401. [DOI] [PubMed] [Google Scholar]
- Musil LS (2012). Primary cultures of embryonic chick lens cells as a model system to study lens gap junctions and fiber cell differentiation. J Membr Biol 245, 357–368. [DOI] [PubMed] [Google Scholar]
- Muta Y, Matsuda M, Imajo M (2019). Divergent Dynamics and Functions of ERK MAP Kinase Signaling in Development, Homeostasis and Cancer: Lessons from Fluorescent Bioimaging. Cancers (Basel) 11, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthusamy BP, Budi EH, Katsuno Y, Lee MK, Smith SM, Mirza AM, Akhurst RJ, Derynck R (2015). ShcA Protects against Epithelial-Mesenchymal Transition through Compartmentalized Inhibition of TGF-β-Induced Smad Activation. PLoS Biol 13, e1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng NS, Ooi L. (2021). A Simple Microplate Assay for Reactive Oxygen Species Generation and Rapid Cellular Protein Normalization. Bio Protoc 11, e3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HT, Bride SH, Badawy AB, Adam RA, Lin J, Orsola A, Guthrie PD, Freeman MR, Peters CA (2000). Heparin-binding EGF-like growth factor is up-regulated in the obstructed kidney in a cell- and region-specific manner and acts to inhibit apoptosis. Am J Pathol 156, 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny GE, Pau H (1984). Myofibroblast-like cells in human anterior capsular cataract. Virchows Arch A Pathol Anat Histopathol 404, 393–401. [DOI] [PubMed] [Google Scholar]
- Ohba M, Shibanuma M, Kuroki T, Nose K (1994). Production of hydrogen peroxide by transforming growth factor-beta 1 and its involvement in induction of egr-1 in mouse osteoblastic cells. J Cell Biol 126, 1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoh VO, Garba NA, Penney RB, Das J, Deoraj A, Singh KP, Sarkar S, Felty Q, Yoo C, Jackson RM, Roy D (2015). Redox signalling to nuclear regulatory proteins by reactive oxygen species contributes to oestrogen-induced growth of breast cancer cells. Br J Cancer 112, 1687–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE (2000). The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19, 3159–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olea-Flores M, Zuniga-Eulogio MD, Mendoza-Catalan MA, Rodríguez-Ruiz HA, Castañeda-Saucedo E, Ortuño-Pineda C, Padilla-Benavides T, Navarro-Tito N (2019). Extracellular-signal regulated kinase: A central molecule driving epithelial-mesenchymal transition in cancer. Int J Mol Sci 20, 2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Villasante C, Burén S, Blázquez-Castro A, Barón-Sola Á, Hernández LE (2018). Fluorescent in vivo imaging of reactive oxygen species and redox potential in plants. Free Radic Biol Med 122, 202–220. [DOI] [PubMed] [Google Scholar]
- Ostman A, Frijhoff J, Sandin A, Böhmer FD (2011). Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem 150, 345–356. [DOI] [PubMed] [Google Scholar]
- Pinilla-Macua I, Grassart A, Duvvuri U, Watkins SC, Sorkin A (2017). EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. Elife 6, e31993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, Klapper L, Lavi S, Seger R, Ratzkin BJ, Sela M, et al. (1996). Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J 15, 2452–2467. [PMC free article] [PubMed] [Google Scholar]
- Potts JD, Bassnett S, Kornacker S, Beebe DC (1994). Expression of platelet-derived growth factor receptors in the developing chicken lens. Invest Ophthalmol Vis Sci 35, 3413–3421. [PubMed] [Google Scholar]
- Raghavan CT, Nagaraj RH (2016). AGE-RAGE interaction in the TGFβ2-mediated epithelial to mesenchymal transition of human lens epithelial cells [published correction appears in Glycoconj J. 2016 Oct;33(5):837-8]. Glycoconj J 33, 631–643. [DOI] [PubMed] [Google Scholar]
- Raina D, Fabris F, Morelli LG, Schröter C (2022). Intermittent ERK oscillations downstream of FGF in mouse embryonic stem cells. Development 149, dev199710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PV, Maddala R, John F, Zigler JS Jr. (2004). Expression of nonphagocytic NADPH oxidase system in the ocular lens. Mol Vis 10, 112–121. [PubMed] [Google Scholar]
- Reneker LW, Overbeek PA. (1996). Lens-specific expression of PDGF-A alters lens growth and development. Dev Biol 180, 554–565. [DOI] [PubMed] [Google Scholar]
- Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh S, Lee HB (2005). Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol 16, 667–675. [DOI] [PubMed] [Google Scholar]
- Robertson JV, Nathu Z, Najjar A, Dwivedi D, Gauldie J, West-Mays JA (2007). Adenoviral gene transfer of bioactive TGFbeta1 to the rodent eye as a novel model for anterior subcapsular cataract. Mol Vis 13, 457–469. [PMC free article] [PubMed] [Google Scholar]
- Robinson ML (2006). An essential role for FGF receptor signaling in lens development. Semin Cell Dev Biol 17, 726–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R Jr. (2014). The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 79, 34–74. [DOI] [PubMed] [Google Scholar]
- Rybinski B, Franco-Barraza J, Cukierman E (2014). The wound healing, chronic fibrosis, and cancer progression triad. Physiol Genomics 46, 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP (2004). Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164, 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saika S, Miyamoto T, Ishida I, Shirai K, Ohnishi Y, Ooshima A, McAvoy JM (2002). TGFbeta-Smad signalling in postoperative human lens epithelial cells. Br J Ophthalmol 86, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarakoon R, Dobberfuhl AD, Cooley C, Overstreet JM, Patel S, Goldschmeding R, Meldrum KK, Higgins PJ (2013a). Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal 25, 2198–2209. [DOI] [PubMed] [Google Scholar]
- Samarakoon R, Overstreet JM, Higgins PJ (2013b). TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal 25, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J (2004). Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 306, 1506–1507. [DOI] [PubMed] [Google Scholar]
- Sebastian S, Settleman J, Reshkin SJ, Azzariti A, Bellizzi A, Paradiso A (2006). The complexity of targeting EGFR signalling in cancer: from expression to turnover. Biochim Biophys Acta 1766, 120–139. [DOI] [PubMed] [Google Scholar]
- Shin HS, Ko J, Kim DA, Ryu ES, Ryu HM, Park SH, Kim YL, Oh ES, Kang DH (2012). Sprouty Is a Negative Regulator of Transforming Growth Factor β-Induced Epithelial-to-Mesenchymal Transition and Cataract. Mol Med 18, 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin EHH, Basson MA, Robinson ML, McAvoy JW, Lovicu FJ (2017). Metformin ameliorates the Phenotype Transition of Peritoneal Mesothelial Cells and Peritoneal Fibrosis via a modulation of Oxidative Stress. Sci Rep 7, 5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu DY, Ong K, Lovicu FJ (2017). Histopathology of Subcapsular Cataract in a Patient with Atopic Dermatitis. Optom Vis Sci 94, 270–276. [DOI] [PubMed] [Google Scholar]
- Shu DY, Wojciechowski M, Lovicu FJ (2019). ERK1/2-mediated EGFR-signaling is required for TGFβ-induced lens epithelial-mesenchymal transition. Exp Eye Res 178, 108–121. [DOI] [PubMed] [Google Scholar]
- Son Y, Kim S, Chung HT, HO P. (2013). Reactive oxygen species in the activation of MAP kinases. Methods Enzymol 528, 27–48. [DOI] [PubMed] [Google Scholar]
- Sparta B, Pargett M, Minguet M, Distor K, Bell G, Albeck JG (2015). Receptor Level Mechanisms Are Required for Epidermal Growth Factor (EGF)-stimulated Extracellular Signal-regulated Kinase (ERK) Activity Pulses. J Biol Chem 290, 24784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan Y, Lovicu FJ, Overbeek PA (1998). Lens-specific expression of transforming growth factor beta1 in transgenic mice causes anterior subcapsular cataracts. J Clin Invest 101, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup G, Cohen S, Garbers DL (1982). Inhibition of membrane phosphotyrosyl-protein phosphatase activity by vanadate. Biochem Biophys Res Commun 107, 1104–1109. [DOI] [PubMed] [Google Scholar]
- Tiwari A, Kumar R, Ram J, Sharma M, Luthra-Guptasarma M (2016). Control of fibrotic changes through the synergistic effects of anti-fibronectin antibody and an RGDS-tagged form of the same antibody. Sci Rep 6, 30872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3, 349–363. [DOI] [PubMed] [Google Scholar]
- Tong J, Taylor P, Moran MF (2014). Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol Cell Proteomics 13, 1644–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK (2013). Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J 280, 346–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse S, Gomez N, Paterson H, Marshall C, Cohen P (1992). Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J 288, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi RH, Werner L, Apple DJ, Pandey SK, Izak AM (2002). Post cataract-intraocular lens (IOL) surgery opacification. Eye (Lond) 16, 217–241 [DOI] [PubMed] [Google Scholar]
- Uchiyama-Tanaka Y, Matsubara H, Mori Y, Kosaki A, Kishimoto N, Amano K, Higashiyama S, Iwasaka T (2002). Involvement of HB-EGF and EGF receptor transactivation in TGF-beta-mediated fibronectin expression in mesangial cells. Kidney Int 62, 799–808. [DOI] [PubMed] [Google Scholar]
- Umapathy A, Li B, Donaldson PJ, Lim JC (2015). Identification and Functional Characterization of a GSH Conjugate Efflux Pathway in the Rat Lens. Invest Ophthalmol Vis Sci 56, 5256–5268. [DOI] [PubMed] [Google Scholar]
- Vallath S, Hynds RE, Succony L, Janes SM, Giangreco A (2014). Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur Respir J 44, 513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSlyke JK, Boswell BA, Musil LS (2018). Fibronectin regulates growth factor signaling and cell differentiation in primary lens cells. J Cell Sci 131, 217240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSlyke JK, Boswell BA, Musil LS (2023). ErbBs in Lens Cell Fibrosis and Secondary Cataract. Invest Ophthalmol Vis Sci 64, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lou MF (2009). The regulation of NADPH oxidase and its association with cell proliferation in human lens epithelial cells. Invest Ophthalmol Vis Sci 50, 2291–2300. [DOI] [PubMed] [Google Scholar]
- Wang Q, McAvoy JW, Lovicu FJ. (2010). Growth factor signaling in vitreous humor-induced lens fiber differentiation. Invest Ophthalmol Vis Sci 51, 3599–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Xiang B, Guix M, Olivares MG, Parker J, Chung CH, Pandiella A, Arteaga CL (2008). Transforming growth factor beta engages TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Mol Cell Biol 28, 5605–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Caty J, Whitson J, Zhang AD, Srinivasagan R, Kavanagh TJ, Yan H, Fan X (2017). Reduced Glutathione Level Promotes Epithelial-Mesenchymal Transition in Lens Epithelial Cells via a Wnt/β-Catenin-Mediated Pathway: Relevance for Cataract Therapy. Am J Pathol 187, 2399–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheimer C, Kueres A, Siedlecki J, Braun C, Kassumeh S, Wolf A, Mayer W, Priglinger C, Priglinger S, Eibl-Lindner K (2018). The intraocular lens as a drug delivery device for an epidermal growth factor-Receptor inhibitor for prophylaxis of posterior capsule opacification. Acta Ophthalmol 96, e874–e882. [DOI] [PubMed] [Google Scholar]
- Wertheimer C, Siedlecki J, Kook D, Mayer WJ, Wolf A, Klingenstein A, Kampik A, Eibl-Lindner K (2015). EGFR inhibitor Gefitinib attenuates posterior capsule opacification in vitro and in the ex vivo human capsular bag model. Graefes Arch Clin Exp Ophthalmol 253, 409–417. [DOI] [PubMed] [Google Scholar]
- West-Mays JA, Pino G, Lovicu FJ (2010). Development and use of the lens epithelial explant system to study lens differentiation and cataractogenesis. Prog Retin Eye Res 29, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieduwilt MJ, Moasser MM (2008). The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci 65, 1566–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowski MC, Mahmutovic L, Shu DY, Lovicu FJ (2017). ERK1/2 signaling is required for the initiation but not progression of TGFβ-induced lens epithelial to mesenchymal transition (EMT). Exp Eye Res 159, 98–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormstone IM, Anderson IK, Eldred JA, Dawes LJ, Duncan G (2006). Short-term exposure to transforming growth factor beta induces long-term fibrotic responses. Exp Eye Res 83, 1238–1245. [DOI] [PubMed] [Google Scholar]
- Wormstone IM, Eldred JA (2016). Experimental models for posterior capsule opacification research. Exp Eye Res 142, 2–12. [DOI] [PubMed] [Google Scholar]
- Wormstone IM, Tamiya S, Anderson I, Duncan G. (2002). TGF-beta2-induced matrix modification and cell transdifferentiation in the human lens capsular bag. Invest Ophthalmol Vis Sci 43, 2301–2308. [PubMed] [Google Scholar]
- Wormstone IM, Wang L, Liu CS (2009). Posterior capsule opacification. Exp Eye Res 88, 257–269. [DOI] [PubMed] [Google Scholar]
- Wynn TA (2004). Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K, Lee RS, Narsimhan RP, Mukhopadhyay NK, Neel BG, Roberts TM (1999). Tyrosine phosphorylation of the proto-oncoprotein Raf-1 is regulated by Raf-1 itself and the phosphatase Cdc25A. Mol Cell Biol 19, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL (2004). Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 6, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K, Tan J, Gu WZ, Ye PP, Wang KJ (2007). Reactive oxygen species mediates the apoptosis induced by transforming growth factor beta(2) in human lens epithelial cells. Biochem Biophys Res Commun 354, 278–283. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yang T, Madakashira BP, Thiels CA, Bechtle CA, Garcia CM, Zhang H, Yu K, Ornitz DM, Beebe DC, Robinson ML (2008). Fibroblast growth factor receptor signaling is essential for lens fiber cell differentiation. Dev Biol 318, 276–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A. (2019). N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem Res Toxicol 32, 1318–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lee JY, Lee CM, Cho WK, Kang MJ, Koff JL, Yoon PO, Chae J, Park HO, Elias JA, Lee CG (2012). Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. J Biol Chem 287, 41991–42000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang S, Liu N (2011). EGFR signaling in renal fibrosis. Kidney Int Suppl (2011) 4, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.