

Abstract
INTRODUCTION
The timing of plasma biomarker changes is not well understood. The goal of this study was to evaluate the temporal co‐evolution of plasma and positron emission tomography (PET) Alzheimer's disease (AD) biomarkers.
METHODS
We included 1408 Mayo Clinic Study of Aging and Alzheimer's Disease Research Center participants. An accelerated failure time (AFT) model was fit with amyloid beta (Aβ) PET, tau PET, plasma p‐tau217, p‐tau181, and glial fibrillary acidic protein (GFAP) as endpoints.
RESULTS
Individual timing of plasma p‐tau progression was strongly associated with Aβ PET and GFAP progression. In the population, GFAP became abnormal first, then Aβ PET, plasma p‐tau, and tau PET temporal meta‐regions of interest when applying cut points based on young, cognitively unimpaired participants.
DISCUSSION
Plasma p‐tau is a stronger indicator of a temporally linked response to elevated brain Aβ than of tau pathology. While Aβ deposition and a rise in GFAP are upstream events associated with tau phosphorylation, the temporal link between p‐tau and Aβ PET was the strongest.
Highlights
Plasma p‐tau progression was more strongly associated with Aβ than tau PET.
Progression on plasma p‐tau was associated with Aβ PET and GFAP progression.
P‐tau181 and p‐tau217 become abnormal after Aβ PET and before tau PET.
GFAP became abnormal first, before plasma p‐tau and Aβ PET.
Keywords: Alzheimer's disease, amyloid beta PET, plasma p‐tau, tau PET, temporal modeling
1. BACKGROUND
Plasma Alzheimer's disease (AD) biomarkers have recently emerged as a more feasible and less invasive biomarker compared to cerebrospinal fluid (CSF) or positron emission tomography (PET) for evaluation of AD‐related changes, amyloid beta (Aβ) and tau aggregation. 1 , 2 , 3 As clinical trials and future clinical practice seek to incorporate plasma biomarkers into screening, diagnosis, and staging, it is important to understand the relationship between the timing of plasma AD biomarker change and previously established in vivo metrics of CSF and PET. Plasma phosphorylated tau (p‐tau) is of particular interest as it has been shown to perform well in differentiating AD clinical severity and correlates well with validated metrics of AD‐related changes, though with variable performance based on the isoform as well as measurement platform and assay. 4 , 5 For example, plasma p‐tau has been shown to increase with disease severity and predict cognitive decline in AD 6 , 7 , 8 , 9 , 10 , 11 as well as differentiate AD patients from those with cognitive impairment secondary to other tauopathies. 7 , 12 While plasma p‐tau181, 217, and 231 are associated with both Aβ and tau PET standardized uptake value ratio (SUVR) in cross‐sectional analyses, studies suggest that plasma p‐tau is more closely associated with Aβ than tau deposition and rises in response to Aβ deposition. 9 , 13 , 14 , 15
Relatively little data exist on longitudinal changes of plasma p‐tau and, in particular, the relative associations of the timing of plasma and PET biomarker progression. Plasma p‐tau217 has been shown to be associated with worsening cognition and brain atrophy in early AD stages. 16 , 17 Limited longitudinal modeling studies suggest that plasma p‐tau181 and p‐tau217 become abnormal around the same time or within several years after Aβ PET and precede changes in tau PET. 18 , 19 , 20 While these studies provide information on the timing of change in the population, they do not explain how plasma p‐tau and PET biomarker changes are related on the individual level. Both are of interest. Associations of the timing of biomarker progression on the individual level indicate how closely two biomarkers are temporally linked and may inform disease mechanism. The relative timing of progression on the population level will inform where in the disease process we may detect plasma p‐tau change to guide its appropriate implementation, potentially in combination with other biomarkers.
RESEARCH‐IN‐CONTEXT
Systematic review: The authors reviewed the literature using traditional (eg, PubMed) sources. While cross‐sectional relationships of plasma p‐tau with Aβ and tau PET have been described, the temporal co‐evolution of these biomarkers on the individual and population levels is not well understood. In particular, these associations have not been evaluated using methods like accelerated failure time modeling that are designed to elucidate the temporal ordering of events. Prior cross‐sectional and limited longitudinal studies of plasma p‐tau and Aβ/tau PET have been cited.
Interpretation: Our findings indicate that on the individual level, the timing of plasma p‐tau progression is more closely related to Aβ than tau PET. On the population level, the relative timing of p‐tau progression depends on the chosen cut point, plasma analyte, and nature of the population sampled.
Future directions: Future studies will include evaluation of these temporal relationships using alternative plasma p‐tau assays and a more demographically diverse sample.
One approach to modeling biomarker temporal evolution is that in which each biomarker is assumed to progress along the same common curve for all individuals. 21 , 22 Multiple methods have been developed to model these trajectories, 20 , 23 , 24 , 25 , 26 , 27 and the approach our group has employed is an accelerated failure time (AFT) or non‐linear mixed‐effects model. 28 The AFT model differs from other approaches in that the primary result is the relative timing of biomarker progression on the individual level, whereas others model population‐level change. Additionally, we estimate a unique temporal shift for each biomarker per individual, whereas other methods estimate a single time offset per individual that affects all biomarkers equally. 27 Left‐right temporal shifts (individual adjustments) of the common curve for each individual and biomarker indicate how much earlier or later the individual is estimated to progress on that biomarker relative to the population mean. The association of individual adjustments between biomarkers provides insight into how much variation in the timing of progression of one biomarker can be accounted for by another; a greater association implies a stronger temporal and mechanistic relationship. An additional advantage of the AFT approach is that cut points are not used in the primary model fits. As with other methods, the AFT approach does require the use of cut points to estimate the ordering that biomarkers become abnormal on the population level.
The goal of this study was to describe the temporal evolution of plasma p‐tau181 and p‐tau217 in relation to Aβ and tau PET on the individual and population levels using an AFT model. Plasma glial fibrillary acidic protein (GFAP), a marker of astrocytic activation, is also included in models, since, although it is not specific to AD, it is also thought to rise in response to amyloidosis. 29 , 30 , 31 , 32 Understanding the relationship between the relative timing of plasma and PET biomarker change on the individual and population levels will help inform disease mechanisms and appropriate integration of plasma biomarkers in research, clinical trials, and clinical practice.
2. METHODS
2.1. Participants
This study included participants in the Mayo Clinic Study of Aging (MCSA), a longitudinal population‐based study of individuals residing in Olmsted County, Minnesota, or in the Mayo Alzheimer's Disease Research Center (ADRC) study, a longitudinal study of patients enrolled through the clinical practice. All participants were required to have at least two visits with plasma measured on the same platform; have at least one visit with PET (Aβ or tau) performed in 2009 or after; and have sex, education, and apolipoprotein E (APOE) genotype available. MCSA participants were required to be 50 years of age or older at their earliest visit with PET imaging and have a diagnosis of cognitively unimpaired (CU), mild cognitive impairment (MCI), or Alzheimer clinical syndrome (AlzCS) dementia. ADRC study participants, all of whom were cognitively impaired, were required to be 70 years of age or older at their earliest visit with PET imaging and have a diagnosis of MCI or AlzCS dementia at their most recent visit with either PET or plasma. We recognize that the ADRC study referral cohort is not the same as a population‐based cohort. With the exclusion of the younger, potentially early‐onset AD cases from this study's ADRC sample, it became more representative of symptom onset in the general population. We also excluded participants with Lewy body disease—MCI, dementia with Lewy bodies (DLB), frontotemporal dementia (FTD), and REM sleep behavior disorder (RBD). Clinical diagnoses were determined by an expert panel based on established criteria. 33 , 34 , 35
2.2. Standard protocol approvals, registrations, and patient consents
The study was approved by the Mayo Clinic and Olmsted Medical Center institutional review boards and was performed in accordance with the ethical standards of the Declaration of Helsinki and its later amendments. All participants or a legally authorized representative provided informed written consent.
2.3. Plasma processing and metrics
Plasma samples were collected after an overnight fast and centrifuged, and 500 uL plasma was aliquoted into polypropylene tubes and stored at −80°C. Plasma ptau‐181 and GFAP were measured using the Simoa® Neurology 4‐Plex E Advantage kit and run on a Quanterix HD‐X analyzer (Quanterix, Lexington, MA, USA). Plasma p‐tau217 was measured using proprietary assays on the Meso Scale Diagnostics (MSD) platform (Lilly Research Laboratories, Indianapolis, IN, USA). See prior publications for additional details on plasma handling and assays. 14
The plasma metrics of greatest interest in this work were p‐tau181 and p‐tau217. GFAP, a non‐specific marker of astrocytic activation, was included given the interest in its proposed association with amyloidosis. Aβ42/40 and NfL were also measured with the 4‐Plex but not used in this study. This Aβ42/40 assay has been shown to have suboptimal performance for differentiation of Aβ PET positive versus negative, 36 and the high level of noise in the measurement does not make it amenable to longitudinal modeling. NfL is a non‐specific marker of neuronal injury that is highly associated with age and, therefore, also of marginal value in this time shift analysis.
2.4. Aβ and tau PET
Aβ PET was performed with Pittsburgh compound B 37 and tau PET with [18F]flortaucipir (Avid Radiopharmaceuticals) on GE scanners (models Discovery 690XT, Discovery RX, and Discovery MI) or Siemens scanners (Biograph Vision 600). Four 5‐min frames were acquired after a 40‐min uptake period for Aβ and 80‐min uptake period for tau PET. The frames were averaged and processed using in‐house pipelines 38 , 39 using corresponding T1‐weighted magnetic resonance imaging (MRI) slices that were also acquired in these cohorts. The Aβ PET meta‐region of interest (ROI) was derived via the voxel number weighted average of the median uptake in each of the prefrontal, orbitofrontal, parietal, temporal, anterior, and posterior cingulate and precuneus regions normalized to the cerebellar crus gray matter. The tau PET temporal meta‐ROI was derived via the voxel number weighted average of the median uptake in each of the amygdala, fusiform, middle/inferior temporal, entorhinal, and parahippocampal regions normalized by the cerebellar crus gray matter. Harmonization between the PET scanners was performed by adjusting blurring parameters during the recon, according to the method of Joshi et al. 40 PET data were not partial‐volume corrected.
2.5. Primary model and individual adjustments
We implemented an AFT model, 28 a type of joint non‐linear mixed‐effects model. In this approach, we assumed that a given biomarker progressed along a similar trajectory or common curve, which was shifted left or right for each individual. The assumption of a hypothetical common curve is an extension of prior work described by Jack et al. 21 , 22 and has been similarly implemented in other recently described temporal modeling approaches. 20 , 23 , 25 , 27
In this study, we extended prior AFT models to accommodate five biomarker outcomes: Aβ PET meta‐ROI, tau PET temporal meta‐ROI, plasma p‐tau217, plasma p‐tau181, and GFAP. Aβ and tau PET were natural log‐transformed to account for skewness. Sex, APOE ε4 carrier status, and education were included as covariates. Given the known earlier average age of symptom onset of participants in the ADRC study relative to the MCSA, an overall ADRC effect/shift was applied for all biomarkers. The residual effect of referral for enrollment in the ADRC study on the individual biomarkers was reported as a covariate, as was done in prior work. 28 We included both MCSA and ADRC participants in a single model to include the full disease spectrum, given the relatively low number of participants with dementia in the MCSA.
The model included a smooth progression function for each biomarker (simplest possible non‐linear function), regression coefficients for each covariate and biomarker pair, and per‐participant age adjustments for each biomarker (individual‐level adjustment). The model output also included a correlation matrix of the random effects or the correlation (R) of the individual‐level adjustment between biomarker pairs. See the Supplemental Methods and prior publications 28 , 41 for additional information on the model fits.
TABLE 1.
Participant characteristics at most recent visit with PET or plasma, unless otherwise specified.
| CU (N = 1009) | MCI (N = 293) | AlzCS dementia (N = 106) | Total (N = 1408) | |
|---|---|---|---|---|
| Age, years | ||||
| Median (Q1, Q3) | 76 (68, 83) | 83 (78, 88) | 79 (76, 85) | 78 (71, 84) |
| Range | 50 to 101 | 54 to 102 | 70 to 94 | 50 to 102 |
| Female Sex | 467 (46%) | 124 (42%) | 49 (46%) | 640 (45%) |
| APOE ε4 carrier | 262 (26%) | 118 (40%) | 68 (64%) | 448 (32%) |
| Education, years | ||||
| Median (Q1, Q3) | 15 (13, 16) | 13 (12, 16) | 16 (12, 16) | 14 (12, 16) |
| Range | 6 to 20 | 0 to 21 | 8 to 25 | 0 to 25 |
| Study | ||||
| ADRC | 0 (0%) | 18 (6%) | 77 (73%) | 95 (7%) |
| MCSA | 1009 (100%) | 275 (94%) | 29 (27%) | 1313 (93%) |
| Total No. Aβ PET | ||||
| 0 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| 1 | 201 (20%) | 100 (34%) | 22 (21%) | 323 (23%) |
| 2 | 347 (34%) | 74 (25%) | 32 (30%) | 453 (32%) |
| ≥3 | 461 (46%) | 119 (41%) | 52 (49%) | 632 (45%) |
| Total No. tau PET | ||||
| 0 | 334 (33%) | 162 (55%) | 46 (43%) | 542 (38%) |
| 1 | 308 (31%) | 39 (13%) | 16 (15%) | 363 (26%) |
| 2 | 254 (25%) | 38 (13%) | 19 (18%) | 311 (22%) |
| ≥3 | 113 (11%) | 54 (18%) | 25 (24%) | 192 (14%) |
| Total No. p‐tau217 | ||||
| 0 | 280 (28%) | 87 (30%) | 85 (80%) | 452 (32%) |
| 1 | 6 (1%) | 1 (0%) | 0 (0%) | 7 (0%) |
| 2 | 172 (17%) | 76 (26%) | 11 (10%) | 259 (18%) |
| 3 | 551 (55%) | 129 (44%) | 10 (9%) | 690 (49%) |
| Total No. p‐tau181 | ||||
| 0 | 1 (0%) | 1 (0%) | 0 (0%) | 2 (0%) |
| 1 | 94 (9%) | 21 (7%) | 2 (2%) | 117 (8%) |
| 2 | 397 (39%) | 103 (35%) | 42 (40%) | 542 (38%) |
| ≥3 | 517 (51%) | 168 (57%) | 62 (58%) | 747 (53%) |
| Total No. GFAP | ||||
| 0 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| 1 | 94 (9%) | 21 (7%) | 2 (2%) | 117 (8%) |
| 2 | 395 (39%) | 106 (36%) | 42 (40%) | 543 (39%) |
| ≥3 | 520 (52%) | 166 (57%) | 62 (58%) | 748 (53%) |
| Aβ PET, SUVR | ||||
| Median (Q1, Q3) | 1.4 (1.4, 1.7) | 2.0 (1.5, 2.5) | 2.6 (2.2, 2.8) | 1.5 (1.4, 2.0) |
| Range | 1.1 to 3.3 | 1.2 to 3.6 | 1.3 to 3.5 | 1.1 to 3.6 |
| Tau PET, SUVR | ||||
| Median (Q1, Q3) | 1.2 (1.1, 1.3) | 1.2 (1.2, 1.4) | 1.9 (1.6, 2.1) | 1.2 (1.2, 1.3) |
| Range | 0.9 to 2.2 | 1.0 to 2.2 | 1.1 to 3.6 | 0.9 to 3.6 |
| * P‐tau 217, pg/ml | ||||
| Median (Q1, Q3) | 0.15 (0.12, 0.22) | 0.31 (0.20, 0.50) | 0.30 (0.16, 0.33) | 0.17 (0.13, 0.26) |
| Range | 0.02 to 1.28 | 0.09 to 1.20 | 0.10 to 0.74 | 0.02 to 1.28 |
| P‐tau 181, pg/ml | ||||
| Median (Q1, Q3) | 1.94 (1.45, 2.84) | 2.92 (2.13, 4.25) | 3.56 (2.80, 4.87) | 2.23 (1.56, 3.37) |
| Range | 0.32 to 18.92 | 0.46 to 13.44 | 1.13 to 11.22 | 0.32 to 18.92 |
| GFAP, pg/ml | ||||
| Median (Q1, Q3) | 115 (76, 163) | 175 (122, 244) | 218 (151, 279) | 134 (87, 195) |
| Range | 24 to 848 | 41 to 661 | 56 to 666 | 24 to 848 |
Abbreviations: AlzCS, Alzheimer clinical syndrome; CU, cognitive unimpaired; MCI, mild cognitive impairment.
p‐tau217 available only in MCSA participants.
2.6. Cut‐point determination and population‐level biomarker progression
We evaluated the relative timing of biomarker progression at the population level by applying cut points to the model output to estimate the proportion of individuals with abnormal biomarker levels versus age. Plasma analyte cut points are not well established and will vary with assay and platform. Therefore, we generated cut points for all biomarkers as two (lenient) and three (conservative) standard deviations (SDs) above the mean in a group of 142 MCSA CU participants aged 30 to 59 years who had all five biomarkers. We chose this method of cut‐point determination because it is less sensitive to the composition of a sample compared to other methods, such as mixture models, and can be applied to all included biomarkers. Since there was an ADRC referral effect on the relative timing of biomarker progression, the population‐level analyses were performed using the MCSA participants only.
2.7. Secondary analyses: regional Aβ and tau PET
Since regional PET measures may affect the observed association of individual adjustments and timing of PET abnormality relative to plasma p‐tau, we ran two additional models, one with regional Aβ PET in place of the meta‐ROI and one with regional tau PET in place of the temporal meta‐ROI. Aβ PET topographic regions were defined as A1 to A4 in accordance with the recommendations of Collij et al. 42 , 43 : A1 (anterior, mid, and posterior cingulum, retrosplenial cortex), A2 (superior, mid, medial, and inferior orbital frontal, olfactory, paracentral lobule, precuneus, rectus), A3 (angular, cuneus, inferior frontal operculum, inferior frontal triangularis, mid and superior frontal, medial superior frontal, fusiform, insula, lingual, parahippocampal, inferior parietal, precentral, supplementary motor, supramarginal, inferior temporal), and A4 (entorhinal cortex, Heschl, inferior, mid, and superior occipital, superior parietal, postcentral, superior and mid temporal, superior and mid temporal pole). We used tau PET Braak stages for regional tau PET: 1‐2 (entorhinal cortex), 3‐4 (parahippocampal, fusiform, lingual, insula, inferior temporal, superior temporal pole, middle temporal pole, middle temporal, posterior cingulate, retrosplenial cortex, anterior cingulate, and mid cingulate), and 5‐6 (inferior parietal, angular, middle orbitofrontal, inferior orbitofrontal, superior orbitofrontal, medial orbitofrontal, rectus, olfactory, superior temporal, Heschl, inferior frontal operculum, inferior frontal triangularis, supramarginal, superior occipital, middle occipital, inferior occipital, precuneus, superior parietal, superior frontal, superior medial frontal, supplemental motor area, middle frontal, paracentral lobule, postcentral, precentral, calcarine, cuneus, and rolandic operculum). 44 Using the groupings described above, we created staging regions by taking a voxel number weighted average of the median uptake across the regions normalized to the median uptake in the cerebellar crus grey matter. For the population‐level analyses, we determined regional cut points using the same method as that described earlier.
2.8. Secondary analysis: effect of GFAP level on Aβ PET and plasma p‐tau association
It has been proposed that astrocyte activation, as indicated by increased plasma GFAP levels, plays a role in the association of Aβ deposition and the phosphorylation of tau, 45 so we performed a secondary analysis to evaluate the effects of early versus late Aβ PET and plasma GFAP progression on the timing of plasma p‐tau181, plasma p‐tau217, and tau PET progression. We compared the plasma p‐tau181, plasma p‐tau217, and tau PET individual adjustments among four groups: early Aβ PET and early plasma GFAP, early Aβ PET and late plasma GFAP, late Aβ PET and early plasma GFAP, and late Aβ PET and late plasma GFAP. Early was defined as individual adjustments > 0 and late as individual adjustments < 0.
3. RESULTS
3.1. Participants
The study included 1408 MCSA and ADRC study participants with a median (Q1, Q3) age of 78 (71, 84) years, 640 (45%) female, 1009 (72%) CU, 293 (21%) with MCI, and 106 (8%) with AlzCS (Table 1). Diagnoses are provided for context, but the diagnosis was not included as a covariate in the model. The number of serial Aβ PET scans ranged from 1 to 9; 32% of participants had two Aβ PET scans, and 45% had three or more. Of the 1408 participants, 542 (38%) did not have a tau PET, 363 (26%) had one, and 503 (36%) had two or more. Most participants (92%) had two or more plasma p‐tau181 and GFAP measurements. Plasma p‐tau217 was available in 956 (68%) of participants, all of whom were from the MCSA. Of those participants, nearly all had two or three plasma p‐tau217 measurements.
3.2. Model fits
The relationships of the biomarker values with age and adjusted age (the participant's estimated age with respect to the biomarker of interest based on both the covariate and random effects, that is, biological age + random effect) are shown in Figure 1 and Supplemental Figure 1. The estimated mean curves show that Aβ and tau PET have a relatively sharp elbow beyond which SUVR rapidly increased. Plasma p‐tau and GFAP showed a more gradual increase with age.
FIGURE 1.
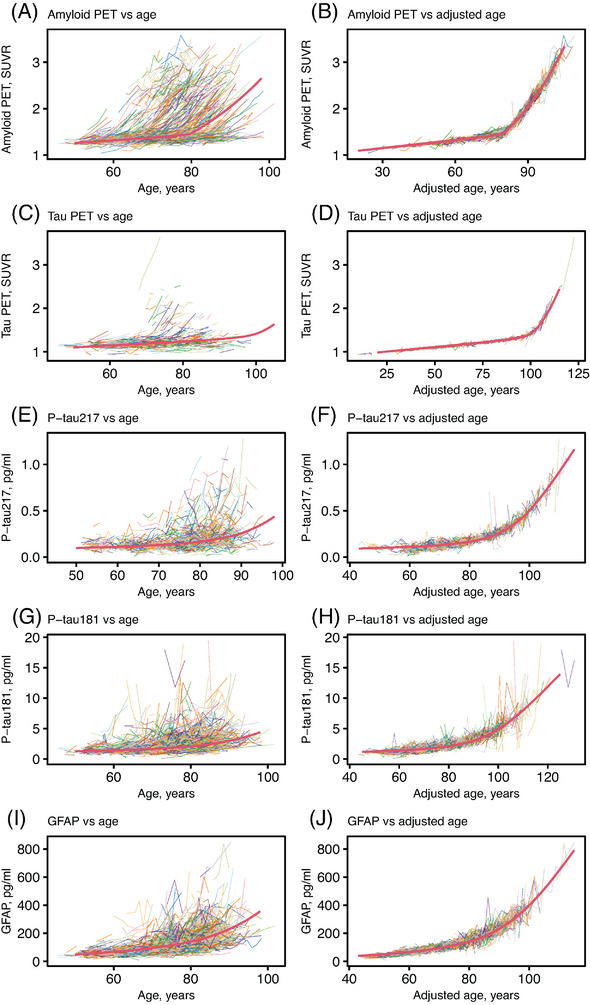
Relationships of PET and plasma biomarkers with age from an AFT model for a subset of participants. Individual trajectories of Aβ PET SUVR, tau PET SUVR, p‐tau217 (pg/ml), p‐tau181 (pg/ml), and GFAP (pg/ml) versus age (A, C, E, G, I) and adjusted age (B, D, F, H, J). The adjusted age is the participant's estimated age with respect to the biomarker of interest based on both the covariate and random effects. The red curves indicate a hypothetical common curve; we assumed all individuals followed this trajectory of biomarker progression with the curve shifted left or right based on the random effects and covariate effects.
3.3. Association of individual‐level adjustments
Associations of the measured biomarker values are shown in Supplemental Figure 2 and associations of the individual‐level adjustments for each biomarker in Figure 2 and Table 2. The individual‐level adjustments of p‐tau217 and Aβ PET were most strongly associated, R = 0.70 (0.65, 0.75); if particular individuals were early with respect to a rise in Aβ PET, they were also early with respect to a rise in p‐tau217. The individual adjustments of p‐tau217 and p‐tau181 were similarly strongly associated, R = 0.64 (0.58, 0.69). However, plasma p‐tau181 and Aβ PET individual level adjustments were less strongly associated, R = 0.36 (0.29, 0.42).
FIGURE 2.
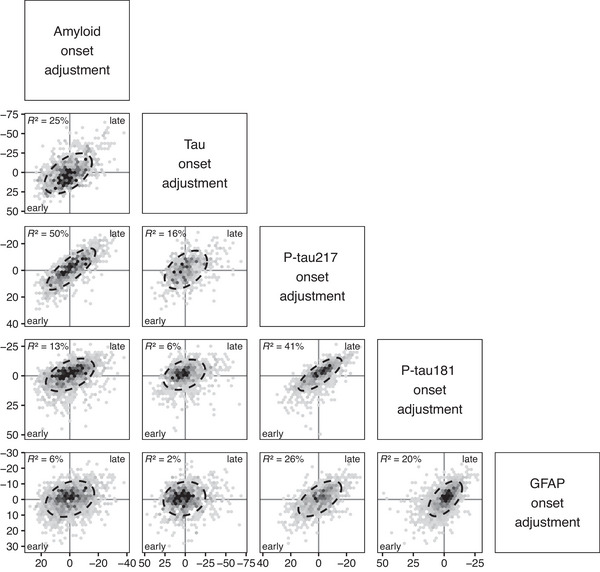
Relationships of individual adjustments between Aβ PET (meta‐ROI), tau PET (temporal meta‐ROI), p‐tau217 (MSD, Lilly), p‐tau181 (Quanterix, Simoa), and GFAP (Quanterix, Simoa). An 80% ellipse is included with a perfect circle indicating no relationship between adjustments. The percent variation explained (square of correlation×100) between individual‐level adjustments is given in the upper left. The x‐ and y‐axes are flipped for the individual adjustments. A higher positive value or earlier onset relative to the population mean is shown to the left of the x‐axis and bottom of the y‐axis. Each dot represents one participant, and the number of participants included in each comparison varies by data availability: Aβ versus tau PET, n = 866; Aβ versus p‐tau217, n = 955; Aβ versus p‐tau181, n = 1405; Aβ versus GFAP, n = 1408; tau PET versus p‐tau217, n = 548; tau PET versus p‐tau181, n = 863; p‐tau217 versus p‐tau181, n = 953; p‐tau217 versus GFAP, n = 955; p‐tau181 versus GFAP, n = 1405.
TABLE 2.
Correlation coefficient, R (95% credible interval), between individual‐level adjustments.
| Tau PET | P‐tau217 | P‐tau181 | GFAP | |
|---|---|---|---|---|
| Aβ PET | 0.50 (0.43, 0.55) | 0.70 (0.65, 0.75) | 0.36 (0.29, 0.42) | 0.25 (0.19, 0.31) |
| Tau PET | 0.39 (0.31, 0.48) | 0.25 (0.16, 0.33) | 0.13 (0.05, 0.21) | |
| P‐tau217 | 0.64 (0.58, 0.69) | 0.51 (0.45, 0.57) | ||
| P‐tau181 | 0.45 (0.39, 0.51) |
The individual‐level adjustments of plasma p‐tau with tau PET (temporal meta‐ROI) were modest and not as strong as the associations with Aβ; R = 0.39 (0.31, 0.48) for p‐tau217 and 0.25 (0.16, 0.33) for p‐tau181. As was seen in prior work, the Aβ and tau PET individual adjustments were strongly associated, R = 0.50 (0.43, 0.55).
The GFAP and p‐tau individual adjustments were strongly associated, R = 0.51 (0.45, 0.57) for p‐tau217 and 0.45 (0.39, 0.51) for p‐tau181. Timing of progression of GFAP showed modest to weak association with Aβ and tau PET, R = 0.25 (0.19, 0.31) and 0.13 (0.05, 0.21), respectively.
3.4. Covariate effects
Covariate effects are shown in Figure 3 and Supplemental Table 1. The greatest covariate effect across all biomarkers was enrollment in the ADRC, a known source of referral bias, which ranged from 8.1 (7.3, 9.0) years for GFAP to 17.4 (15.0, 20.0) years for tau PET. Positive values indicate earlier onset, so the ADRC effect is a much earlier/younger onset of these abnormalities compared with the overall sample average. After the referral effect, the covariate effects were greatest for APOE ε4 carriership with Aβ PET and p‐tau217, 8.5 (7.2, 9.7) and 4.4 (1.8, 7.1) years, respectively. The APOE ε4 carriership effect was weakest for GFAP, 2.2 (1.0, 3.3) years. There were opposing effects of female sex on p‐tau181, −3.1(−4.6, −1.5) years, and GFAP, 3.4 (2.3, 4.4) years. Notably, there was no effect of female sex on p‐tau217, −0.1 (−1.5, 1.3). Education had essentially no effect on the timing of biomarker progression.
FIGURE 3.
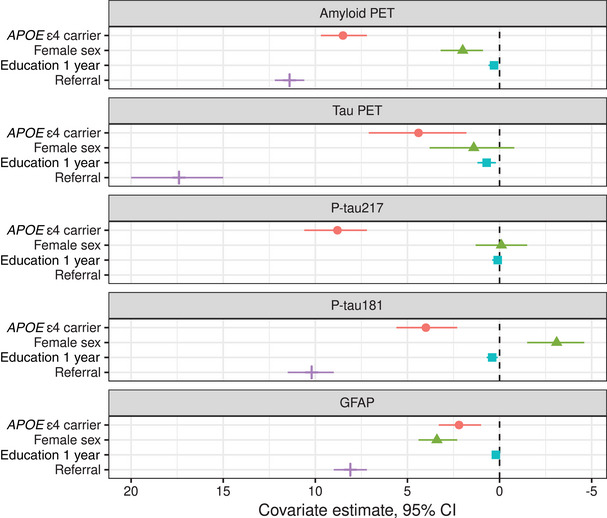
Estimated covariate effects with 95% credible interval. As in Figure 2, the x‐axis is flipped; a higher positive value or earlier onset relative to the population mean is shown to the left. For example, referral (to the ADRC) is associated with much earlier/younger onset of biomarker progression compared with the overall sample average.
3.5. Cut points and biomarker progression on population level
The determined cut points are shown in Table 3. The estimated relative timing of progression of biomarkers using each of the lenient and conservative cut points is shown in Figure 4. In this MCSA sample, GFAP became abnormal first, followed by Aβ PET, plasma p‐tau, and then tau PET. Using both sets of cut points, plasma p‐tau181 and p‐tau217 became abnormal after Aβ PET and before tau PET, though the estimated number of years between biomarker abnormality differed. Using the lenient cut points plasma p‐tau became abnormal approximately 5 to 7 years after Aβ PET and about 6 to 8 years before tau PET (Supplemental Table 2). Using the conservative cut points the time lag between Aβ PET, p‐tau, and tau abnormality were each prolonged by a few years (Figure 4 and Supplemental Table 3).
TABLE 3.
Biomarker cutpoints determined via mean of young (age 30‐59 years) cognitively unimpaired MCSA participants plus two (lenient) and three (conservative) standard deviations (SD).
| +2 SD | +3 SD | |
|---|---|---|
| Aβ PET (SUVR) | 1.45 | 1.53 |
| Tau PET (SUVR) | 1.29 | 1.37 |
| P‐tau217 (pg/ml) | 0.21 | 0.25 |
| P‐tau181 (pg/ml) | 2.76 | 3.42 |
| GFAP (pg/ml) | 93 | 114 |
FIGURE 4.
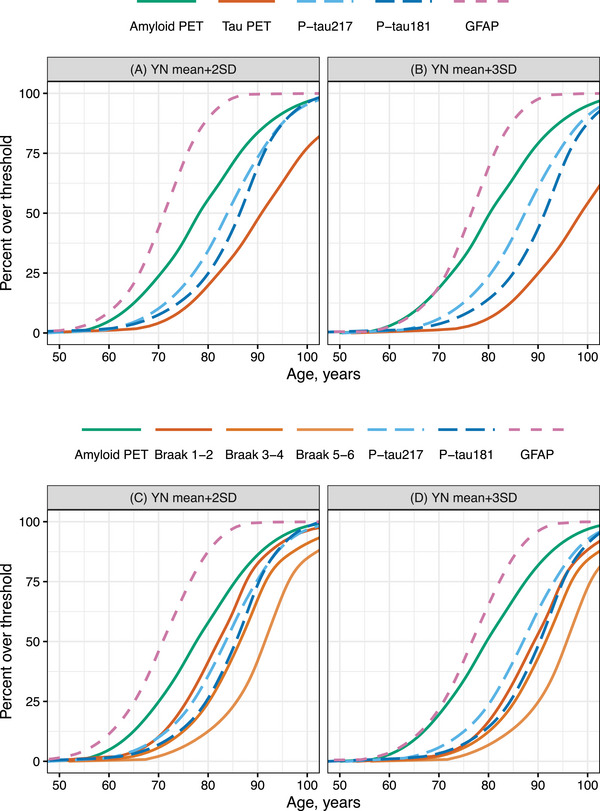
By year of age, predicted proportion of MCSA participants having abnormal biomarker levels using (A, C) 2SD and (B, D) 3SD above the mean of young normal (YN) participants, cognitively unimpaired participants aged 30 to 59 years, as cut points for the primary model (A, B) and model using the tau PET Braak regions in place of the temporal meta‐ROI (C, D).
Note these curves represent averages in the population. The time between biomarker abnormality may differ for certain subgroups, for example, by covariate effects. For example, APOE ε4 carriership differentially affects each of the biomarkers – shifts Aβ PET to the left on average 8.6 years and p‐tau181 4.4 years. Therefore, in APOE ε4 carriers, the average time between Aβ and p‐tau181 positivity will be about 4 years greater than the population average.
3.6. Regional Aβ and tau PET
The individual adjustments were highly correlated among the tau PET Braak regions (Supplemental Figure 3 and Supplemental Table 4). The associations of tau PET Braak region individual adjustments with Aβ PET and plasma p‐tau were strongest for Braak 1‐2, then 3‐4, and 5‐6: 0.47 (0.42, 0.52), 0.41 (0.35, 0.45), and 0.35 (0.30, 0.40) for Aβ PET, respectively. On the population level, Braak 1‐2 become abnormal before or near the same time as plasma p‐tau217 and p‐tau181 (Figure 4C‐D). Braak 3‐4 became abnormal near the same time or just after p‐tau181, and Braak 5‐6 trailed by, on average, about 5 years. Covariate effects showed similar trends to those in the primary model; the APOE ε4 carrier and referral effects were greatest on Braak 1‐2 (Supplemental Table 5).
There was essentially no difference in association of individual adjustments or timing of biomarker abnormality for the Aβ PET regions (Supplemental Figures 4 and 5, Supplemental Table 6). Covariate effects showed similar trends to those in the primary model, and there was minimal difference in the covariate effects on each of the Aβ PET regions (Supplemental Table 7). , ,
3.7. Effect of GFAP level on association of Aβ PET and plasma p‐tau
Figure 5 shows plasma p‐tau181, plasma p‐tau217, and tau PET individual adjustments grouped by early versus late progression on Aβ PET and GFAP. For plasma p‐tau181, individuals who were early on either Aβ PET or plasma GFAP were estimated to progress slightly earlier than those who were late on both. Those who progressed the earliest on plasma p‐tau181 were early on both Aβ PET and GFAP. For plasma p‐tau217, there was a stepwise acceleration of biomarker progression for those who were early on GFAP only, early on Aβ PET only, and early on both. For tau PET, the timing of progression depended on Aβ timing only; GFAP timing had no appreciable effect.
FIGURE 5.
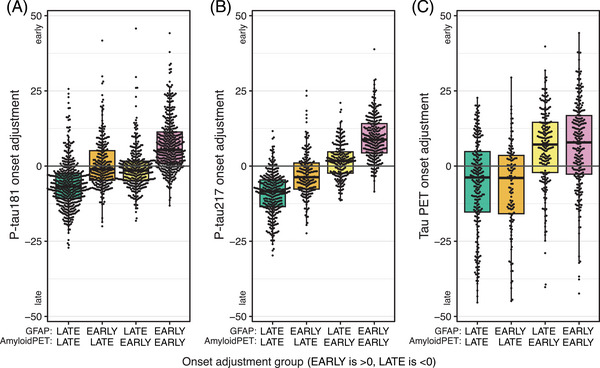
Effect of GFAP on temporal association of Aβ PET with plasma p‐tau and tau PET. Box plots of plasma p‐tau181 (A), plasma p‐tau217 (B), and tau PET (C) individual adjustments by combinations of early (individual adjustment > 0) versus late (individual adjustments < 0) progression of Aβ PET and plasma GFAP. These groups correspond to quadrants in plots of the associations of individual adjustments (Figure 2).
4. DISCUSSION
In this work we evaluated the temporal co‐evolution of plasma p‐tau217, p‐tau181, GFAP, Aβ PET, and tau PET on the individual and population levels. The primary findings were as follows. On the individual level, the timing of plasma p‐tau217 and p‐tau181 progression was more highly associated with Aβ than tau PET, and the associations with PET were stronger for p‐tau217 than p‐tau181. The timing of plasma p‐tau progression was also strongly associated with the timing of GFAP progression. On the population level, plasma p‐tau181 and p‐tau217 were estimated to progress after Aβ PET and before tau PET meta‐ROIs with the implemented cut points.
The stronger association of individual‐level adjustments of p‐tau217 and p‐tau181 with Aβ PET than tau PET meta‐ROIs supports the hypothesis that phosphorylation of tau is more closely linked temporally with Aβ than tau deposition. These findings provide further support to results of cross‐sectional studies demonstrating a higher association of plasma p‐tau217 and p‐tau181 with Aβ than tau PET. 13 , 14 , 15 , 46 An explanation for this phenomenon has been that tau phosphorylation at specific residues and secretions are neuronal responses to Aβ plaques. 47
The higher associations of p‐tau217 than p‐tau181 with Aβ and tau PET could reflect a closer mechanistic relationship of the p‐tau217 isoform with Aβ and tau deposition, less measurement noise in the p‐tau217 than p‐tau181 measurement, differences related to platform/antibody, or all of these. Prior cross‐sectional analyses have similarly shown that, regardless of assay, p‐tau217 is a better predictor of Aβ status than p‐tau181, and the Quanterix Simoa p‐tau181 was not one of the top performing p‐tau181 assays in a head‐to‐head comparison. 5 Although different p‐tau measurement platforms and antibodies may affect biomarker performance, similar trends were observed for the analytes used in this study. Therefore, we anticipate other p‐tau181 and 217 assays to show similar associations with PET as those described here.
In the population‐level analyses, we estimated the percentage of individuals with abnormal biomarkers versus age using cut points of 2 SD and 3 SD above the mean of young CU participants. The Aβ PET, tau PET, and p‐tau217 cut points closely corresponded to previously published cut points. 38 , 46 , 48 The p‐tau181 cut points spanned a larger range than the other biomarkers, though they cover a similar range to a prior study. 11 Using these cut points, Aβ PET was estimated to become abnormal on average 13 or 19 years before tau PET in the MCSA, which is similar to the Aβ to tau delay of 13 years in prior work. 28
Plasma p‐tau217 and p‐tau181 were estimated to become abnormal approximately 5 to 7 years after Aβ PET and 6 to 9 years before the tau PET temporal meta‐ROI using lenient/conservative cut points. The timing of p‐tau abnormality relative to PET corresponds to prior work that also showed that plasma p‐tau217 became abnormal between Aβ and tau PET in early AD 18 and that the time from Aβ PET to p‐tau abnormality was about 5 to 10 years. 19 The relative timing at which p‐tau181 and p‐tau217 became abnormal in this study corresponds to prior work in CSF, which showed p‐tau181 and 217 becoming abnormal around the same time 49 or, for p‐tau217 to become abnormal slightly before p‐tau181. 50 The temporal ordering of Aβ PET < plasma p‐tau < tau PET supports the potential utility of soluble p‐tau measures in disease staging. Rises in soluble p‐tau, indicative of pathologic tau processing, occur before tau aggregation in the brain can be detected in the tau PET temporal meta‐ROI.
The secondary analysis using the tau PET Braak stages supports their use in disease staging as well, as demonstrated in prior work. 51 , 52 , 53 , 54 Abnormal tau PET SUVR in the Braak 1‐2 region (entorhinal cortex) was detected prior to or at the same time as plasma p‐tau abnormality and Braak 3‐4 within 1 to 2 years after plasma p‐tau181 abnormality. The individual associations also support the idea that Braak 1‐2 tau progression is the region with the strongest temporal link with Aβ deposition and rise in plasma p‐tau. Regional Aβ PET did not provide any additional temporal information – all regions were closely associated on the individual level and on average became abnormal at the same time in the population.
GFAP was included in the models as a marker of astrocytic activation, which has been shown to be related to AD as well as other pathologies. When an individual progressed on GFAP was strongly associated with plasma p‐tau181 and 217 progression but only weakly associated with the timing of Aβ and tau PET progression. In the population‐level analyses, GFAP was estimated to be the first biomarker to become abnormal. Based on prior work showing that plasma GFAP was associated with Aβ deposition, 29 , 30 , 31 , 55 we had anticipated a stronger temporal association of these biomarkers on the individual level. The weak associations of GFAP and Aβ PET progression may be due to the presence of other pathologies 56 , 57 , 58 accounting for a component of the observed increases in GFAP in our cohort. As shown in prior work, 45 Figure 5 demonstrates that GFAP rise appears to play a role in the association of Aβ deposition and plasma p‐tau progression. However, the associations of Aβ and p‐tau217 show a more graded response in this work – individuals who progressed earlier on Aβ PET also progressed earlier on plasma p‐tau; this effect was accentuated by, but not entirely dependent on, early progression on GFAP. Additionally, individuals who progressed early on GFAP but not Aβ PET progressed on average earlier on p‐tau than those who were late on both, suggesting that GFAP and Aβ deposition may be associated with the phosphorylation of tau in part via independent mechanisms. In contrast, the timing of progression of tau deposition in the brain (tau PET) was only associated with the timing of Aβ progression; GFAP had no appreciable effect. Further work is needed to better understand the interactions of astrocyte activation and AD pathology in the brain.
The greatest covariate effect was referral bias attendant to being recruited to enroll in the ADRC. The ADRC sample represents a highly selected group with a substantial tertiary clinical referral bias. The ADRC patients progressed earlier on all biomarkers than the average MCSA participant, and this referral effect was greatest on tau PET, as seen in prior AFT models. 28 After the referral effect, the covariate effects were strongest for APOE ε4 genotype on Aβ PET and p‐tau217, which further supports mechanistic relationship between p‐tau217 and amyloidosis. The APOE ε4 carrier effect was lowest on GFAP, as anticipated, given its lack of specificity for Aβ deposition and AD‐related changes. A small sex effect was seen for some biomarkers: female sex was associated with earlier progression on Aβ PET and GFAP, later progression on p‐tau181, but had essentially no effect on p‐tau217. The variability in sex effects is of unclear etiology and should be further evaluated in future studies. Education serves as a control; we do not anticipate level of education to affect the mechanistic associations or underlying biology.
Longitudinal modeling of biomarker change has been performed via multiple approaches. 24 , 25 , 28 A primary advantage of the AFT model is that it addresses all biomarkers in a single unified fit. This allows estimation of the interrelation between biomarkers (information on individual‐level change). Because biomarkers are fit simultaneously, a participant with at least two measures (eg, one PET and one plasma) will provide some information regarding the relative timing of biomarker progression and could be included in the model. However, participants with serial data, and in particular those who progress, provide the most information toward model fits. The requirement for at least two plasma measurements was implemented in our cohort given the large number of participants with serial data.
As in other longitudinal modeling approaches, cut points must be applied to determine the order in which biomarkers become abnormal in the population, and the relative timing of biomarker change is dependent on those chosen cut points, regardless of the approach. The dependence of the relative timing of biomarker change on the chosen cut point was highlighted using two sets of cut points. The choice of cut points is of particular importance for plasma biomarkers, as they may vary not only by measurement technique but also with population given the effects of medical comorbidities. 46
There are limitations to this study. We assumed all participants progressed along the same curve for a given biomarker, which was supported by our data and similarly assumed in other modeling approaches. 23 If at some point sufficient longitudinal data were available to detect different trajectories of change within a biomarker, this would be of interest. The cohort was predominantly white; we plan to obtain data to study the temporal co‐evolution of these biomarkers in a more diverse sample in future work. However, the population‐based nature of the MCSA is a strength; people must be randomly selected from the local population to participate, which is much different from most studies that have a self‐referral bias. By definition, a population‐based sample must reflect the demographics of the geographic area from which the population is derived, which in this case is southeastern Minnesota. Another potential limitation relates to the sensitivity limits of biomarkers to mild levels of neuropathology. Aβ PET is insensitive to plaque burden that falls below moderate to severe. 59 The sensitivity of GFAP to astrocytic activation is unknown. It is possible that the earlier appearance of GFAP versus Aβ PET in these analyses could be due to effects of plaque burden on GFAP that fall below the detection threshold of Aβ PET.
In conclusion, we applied an AFT model to evaluate the relative timing of plasma and PET AD biomarker progression on the individual and population levels. On the individual level, the timing of progression of plasma p‐tau was more strongly associated with Aβ than tau PET. P‐tau and GFAP progression were also strongly associated. On the population level, the relative timing of when biomarkers become abnormal depends on the definition of abnormal, independent of the modeling approach. Using the chosen cut points, plasma p‐tau was estimated to become abnormal after Aβ PET and before tau PET in the temporal meta‐ROI. The individual‐ and population‐level findings support phosphorylation of tau as a downstream effect of both amyloidosis and astrocyte activation and plasma p‐tau as a stronger indicator of Aβ than tau pathology.
CONFLICTS OF INTEREST STATEMENT
P.M.C, E.S.L., T.M.T., H.J.W., and J.L.G. have no disclosures. J.G.R. serves on a data safety monitoring board for Strokenet and receives research support from the NIH. A.A.‐S. has participated in advisory boards for Roche Diagnostics, Fujirebio Diagnostics and Siemens Healthineers. C.G.S. receives research support from the NIH. M.L.S. holds stock in medical‐related companies, unrelated to the current work: Align Technology, Inc. He has also owned stock in these medical‐related companies within the past 3 years, unrelated to the current work: LHC Group, Inc. V.J.L. is a consultant for AVID Radiopharmaceuticals, Eisai Co. Inc., Bayer Schering Pharma, GE Healthcare, Piramal Life Sciences, and Merck Research and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, and NIH (NIA, NCI). M.M.M. has served on scientific advisory boards and/or has consulted for Biogen, LabCorp, Lilly, Merck, Siemens Healthineers, and Sunbird Bio and receives grant support from the NIH and Department of Defense. D.S.K. served on a data safety monitoring board for the DIAN study. He serves on a data safety monitoring board for a tau therapeutic for Biogen but receives no personal compensation. He was a site investigator in the Biogen aducanumab trials. He is an investigator in a clinical trial sponsored by Lilly Pharmaceuticals and the University of Southern California. He serves as a consultant for Samus Therapeutics, Third Rock, Roche, and Alzeca Biosciences but receives no personal compensation. He receives research support from the NIH. P.V. received speaker fees from Miller Medical Communications, Inc. and receives research support from the NIH. R.C.P. serves as a consultant for Roche Inc., Genetech, Inc., Nestle, Inc., Eli Llly and Co., and Eisai, Inc. He serves on the data safety monitoring board for Genentech, Inc. and receives royalties from Oxford University Press, UpToDate, and MedScape. He receives research support from the NIH and the Alzheimer's Association. C.R.J. receives no personal compensation from any commercial entity. He receives research support from the NIH and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Clinic. Author disclosures are available in the supporting information.
CODE AVAILABILITY
The code is available at github.com/Therneau/AFTmodel.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We would like to thank AVID Radiopharmaceuticals, Inc., for their support in supplying AV‐1451 precursor, chemistry production advice, and FDA regulatory cross‐filing permission and documentation needed for this work. This work was supported by the National Institutes of Health (U01 AG006786, P50 AG016574, R37 AG011378, RO1 AG041851, R01 NS097495, R01 AG056366, RF1 AG069052).
Cogswell PM, Lundt ES, Therneau TM, et al. Modeling the temporal evolution of plasma p‐tau in relation to amyloid beta and tau PET. Alzheimer's Dement. 2024;20:1225–1238. 10.1002/alz.13539
DATA AVAILABILITY STATEMENT
MRI, PET, and other data from the Mayo Clinic Study of Aging and the Alzheimer's Disease Research Center are available to qualified academic and industry researchers by request to the MCSA and ADRC Executive Committee (https://www.mayo.edu/research/centers‐programs/alzheimers‐disease‐research‐center/research‐activities/mayo‐clinic‐study‐aging/for‐researchers/data‐sharing‐resources).
REFERENCES
- 1. Angioni D, Delrieu J, Hansson O, et al. Blood biomarkers from research use to clinical practice: what must be done? A report from the EU/US CTAD task force. J Prev Alzheimers Dis. 2022;9:569‐579. doi: 10.14283/jpad.2022.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Bryant SE, Mielke MM, Rissman RA, et al. Blood‐based biomarkers in Alzheimer disease: current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 2017;13:45‐58. doi: 10.1016/j.jalz.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zetterberg H, Bendlin BB. Biomarkers for Alzheimer's disease—preparing for a new era of disease‐modifying therapies. Mol Psychiatry. 2021;26:296‐308. doi: 10.1038/s41380-020-0721-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bayoumy S, Verberk IMW, den Dulk B, et al. Clinical and analytical comparison of six Simoa assays for plasma P‐tau isoforms P‐tau181, P‐tau217, and P‐tau231. Alzheimers Res Ther. 2021;13:198. doi: 10.1186/s13195-021-00939-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Janelidze S, Bali D, Ashton NJ, et al. Head‐to‐head comparison of 10 plasma phospho‐tau assays in prodromal Alzheimer's disease. Brain. 2022;146(4):1592‐1601. doi: 10.1093/brain/awac333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barthélemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated‐tau isoforms track CNS change in Alzheimer's disease. J Exp Med. 2020;217(11):e20200861. doi: 10.1084/jem.20200861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26:379‐386. doi: 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 8. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurology. 2020;19:422‐433. doi: 10.1016/S1474-4422(20)30071-5 [DOI] [PubMed] [Google Scholar]
- 9. Meyer P‐F, Ashton NJ, Karikari TK, et al. Plasma p‐tau231, p‐tau181, PET biomarkers, and cognitive change in older adults. Ann Neurol. 2022;91:548‐560. doi: 10.1002/ana.26308 [DOI] [PubMed] [Google Scholar]
- 10. Mielke MM, Hagen CE, Xu J, et al. Plasma phospho‐tau181 increases with Alzheimer's disease clinical severity and is associated with tau‐PET and amyloid‐PET. Alzheimers Dement. 2018;14:989‐997. doi: 10.1016/j.jalz.2018.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tropea TF, Waligorska T, Xie SX, et al. Plasma phosphorylated tau181 predicts cognitive and functional decline. Ann Clin Transl Neurol. 2022;10(1):18‐31. doi: 10.1002/acn3.51695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387‐397. doi: 10.1038/s41591-020-0762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mattsson‐Carlgren N, Janelidze S, Bateman RJ, et al. Soluble P‐tau217 reflects amyloid and tau pathology and mediates the association of amyloid with tau. EMBO Mol Med. 2021;13:e14022. doi: 10.15252/emmm.202114022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mielke MM, Frank RD, Dage JL, et al. Comparison of plasma phosphorylated tau species with amyloid and tau positron emission tomography, neurodegeneration, vascular pathology, and cognitive outcomes. JAMA Neurol. 2021;78:1108‐1117. doi: 10.1001/jamaneurol.2021.2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Therriault J, Vermeiren M, Servaes S, et al. Association of phosphorylated tau biomarkers with amyloid positron emission tomography vs tau positron emission tomography. JAMA Neurol. 2022;80:188‐199. doi: 10.1001/jamaneurol.2022.4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mattsson‐Carlgren N, Janelidze S, Palmqvist S, et al. Longitudinal plasma p‐tau217 is increased in early stages of Alzheimer's disease. Brain. 2020;143:3234‐3241. doi: 10.1093/brain/awaa286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moscoso A, Grothe MJ, Ashton NJ, et al. Longitudinal associations of blood phosphorylated tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78:396‐406. doi: 10.1001/jamaneurol.2020.4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Janelidze S, Berron D, Smith R, et al. Associations of plasma phospho‐tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 2021;78:1‐8. doi: 10.1001/jamaneurol.2020.4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moscoso A, Grothe MJ, Ashton NJ, et al. Time course of phosphorylated‐tau181 in blood across the Alzheimer's disease spectrum. Brain. 2021;144:325‐339. doi: 10.1093/brain/awaa399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bilgel M, An Y, Walker KA, et al. Longitudinal changes in Alzheimer's‐related plasma biomarkers and brain amyloid. Alzheimers Dement. 2023;19(10):4335‐4345. doi: 10.1002/alz.13157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. doi: 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119‐128. doi: 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Betthauser TJ, Bilgel M, Koscik RL, et al. Multi‐method investigation of factors influencing amyloid onset and impairment in three cohorts. Brain. 2022;145(11):4065‐4079. doi: 10.1093/brain/awac213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Proust‐Lima C, Dartigues J‐F, Jacqmin‐Gadda H. Joint modeling of repeated multivariate cognitive measures and competing risks of dementia and death: a latent process and latent class approach. Stat Med. 2016;35:382‐398. doi: 10.1002/sim.6731 [DOI] [PubMed] [Google Scholar]
- 25. Koscik RL, Betthauser TJ, Jonaitis EM, et al. Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimers Dement (Amst). 2020;12:e12007. doi: 10.1002/dad2.12007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schindler S, Li Y, Buckles VD, et al. predicting symptom onset in sporadic Alzheimer disease with amyloid PET. Neurology. 2021;97(18):e1823‐e1834. doi: 10.1212/WNL.0000000000012775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jedynak BM, Lang A, Liu B, et al. A computational neurodegenerative disease progression score: method and results with the Alzheimer's disease neuroimaging initiative cohort. Neuroimage. 2012;63:1478‐1486. doi: 10.1016/j.neuroimage.2012.07.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Therneau TM, Knopman DS, Lowe VJ, et al. Relationships between β‐amyloid and tau in an elderly population: an accelerated failure time model. Neuroimage. 2021;242:118440. doi: 10.1016/j.neuroimage.2021.118440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benedet AL, Milà‐Alomà M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78:1471‐1483. doi: 10.1001/jamaneurol.2021.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐β but not tau pathology in Alzheimer's disease. Brain. 2021;144:3505‐3516. doi: 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Transl Psychiatry. 2021;11:1‐10. doi: 10.1038/s41398-020-01137-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chatterjee P, Pedrini S, Doecke JD, et al. Plasma Aβ42/40 ratio, p‐tau181, GFAP, and NfL across the Alzheimer's disease continuum: a cross‐sectional and longitudinal study in the AIBL cohort. Alzheimer's Dement. 2022;19(4):1117‐1134. doi: 10.1002/alz.12724 [DOI] [PubMed] [Google Scholar]
- 33. Blacker D, Albert MS, Bassett SS, Go RC, Harrell LE, Folstein MF. Reliability and validity of NINCDS‐ADRDA criteria for Alzheimer's disease. The National Institute of Mental Health Genetics Initiative. Arch Neurol. 1994;51:1198‐1204. doi: 10.1001/archneur.1994.00540240042014 [DOI] [PubMed] [Google Scholar]
- 34. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183‐194. doi: 10.1111/j.1365-2796.2004.01388.x [DOI] [PubMed] [Google Scholar]
- 35. Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, et al. The mayo clinic study of aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30:58‐69. doi: 10.1159/000115751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Janelidze S, Teunissen CE, Zetterberg H, et al. Head‐to‐head comparison of 8 plasma amyloid‐β 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78:1375‐1382. doi: 10.1001/jamaneurol.2021.3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol. 2004;55:306‐319. doi: 10.1002/ana.20009 [DOI] [PubMed] [Google Scholar]
- 38. Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13:205‐216. doi: 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwarz CG, Gunter JL, Lowe VJ, et al. A comparison of partial volume correction techniques for measuring change in serial amyloid PET SUVR. J Alzheimers Dis. 2019;67:181‐195. doi: 10.3233/JAD-180749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Joshi A, Koeppe RA, Fessler JA. Reducing between scanner differences in multi‐center PET studies. Neuroimage. 2009;46:154‐159. doi: 10.1016/j.neuroimage.2009.01.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cogswell PM, Lundt ES, Therneau TM, et al. Evidence against a temporal association between cerebrovascular disease and Alzheimer's disease imaging biomarkers. Nat Commun. 2023;14:3097. doi: 10.1038/s41467-023-38878-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Collij LE, Heeman F, Salvadó G, et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology. 2020;95:e1538‐e1553. doi: 10.1212/WNL.0000000000010256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jack CR Jr, Wiste HJ, Algeciras‐Schimnich A, et al. Predicting amyloid PET and tau PET stages with plasma biomarkers. Brain. 2023;146(5):2029‐2044. doi: 10.1093/brain/awad042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 45. Bellaver B, Povala G, Ferreira PCL, et al. Astrocyte reactivity influences amyloid‐β effects on tau pathology in preclinical Alzheimer's disease. Nat Med. 2023;29(7):1775‐1781. doi: 10.1038/s41591-023-02380-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mielke MM, Dage JL, Frank RD, et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nat Med. 2022;28:1398‐1405. doi: 10.1038/s41591-022-01822-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97:1284‐1298.e7. doi: 10.1016/j.neuron.2018.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lowe VJ, Lundt ES, Albertson SM, et al. Tau‐positron emission tomography correlates with neuropathology findings. Alzheimer's Dement. 2020;16:561‐571. doi: 10.1016/j.jalz.2019.09.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mattsson‐Carlgren N, Andersson E, Janelidze S, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer's disease. Sci Adv. 2020;6:eaaz2387. doi: 10.1126/sciadv.aaz2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barthélemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26:398‐407. doi: 10.1038/s41591-020-0781-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79:110‐119. doi: 10.1002/ana.24546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Therriault J, Pascoal TA, Lussier FZ, et al. Biomarker modeling of Alzheimer's disease using PET‐based Braak staging. Nat Aging. 2022;2:526‐535. doi: 10.1038/s43587-022-00204-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cho H, Choi JY, Hwang MS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol. 2016;80:247‐258. doi: 10.1002/ana.24711 [DOI] [PubMed] [Google Scholar]
- 54. Chen S‐D, Lu J‐Y, Li H‐Q, et al. Staging tau pathology with tau PET in Alzheimer's disease: a longitudinal study. Transl Psychiatry. 2021;11:1‐12. doi: 10.1038/s41398-021-01602-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Asken BM, Elahi FM, La Joie R, et al. Plasma glial fibrillary acidic protein levels differ along the spectra of amyloid burden and clinical disease stage. J Alzheimers Dis. 2020;78:265‐276. doi: 10.3233/JAD-200755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Axelsson M, Malmeström C, Nilsson S, Haghighi S, Rosengren L, Lycke J. Glial fibrillary acidic protein: a potential biomarker for progression in multiple sclerosis. J Neurol. 2011;258:882‐888. doi: 10.1007/s00415-010-5863-2 [DOI] [PubMed] [Google Scholar]
- 57. Heller C, Foiani MS, Moore K, et al. Plasma glial fibrillary acidic protein is raised in progranulin‐associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91:263‐270. doi: 10.1136/jnnp-2019-321954 [DOI] [PubMed] [Google Scholar]
- 58. Huebschmann NA, Luoto TM, Karr JE, et al. Comparing glial fibrillary acidic protein (GFAP) in serum and plasma following mild traumatic brain injury in older adults. Front Neurol. 2020;11:1054. doi: 10.3389/fneur.2020.01054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [11C]PIB‐PET Centiloid values and postmortem measures of Alzheimer's disease neuropathology. Alzheimers Dement. 2019;15:205‐216. doi: 10.1016/j.jalz.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
MRI, PET, and other data from the Mayo Clinic Study of Aging and the Alzheimer's Disease Research Center are available to qualified academic and industry researchers by request to the MCSA and ADRC Executive Committee (https://www.mayo.edu/research/centers‐programs/alzheimers‐disease‐research‐center/research‐activities/mayo‐clinic‐study‐aging/for‐researchers/data‐sharing‐resources).