

Abstract
INTRODUCTION
Tau and neurodegeneration strongly correlate with cognitive impairment, as compared to amyloid. However, their contribution in explaining cognition and predicting cognitive decline in memory clinics remains unclarified.
METHODS
We included 94 participants with Mini‐Mental State Examination (MMSE), tau positron emission tomography (PET), amyloid PET, fluorodeoxyglucose (FDG) PET, and MRI scans from Geneva Memory Center. Linear regression and mediation analyses tested the independent and combined association between biomarkers, cognitive performance, and decline. Linear mixed‐effects and Cox proportional hazards models assessed biomarkers’ prognostic values.
RESULTS
Metabolism had the strongest association with cognition (r = 0.712; p < 0.001), followed by tau (r = ‐0.682; p < 0.001). Neocortical tau showed the strongest association with cognitive decline (r = ‐0.677; p < 0.001). Metabolism mediated the association between tau and cognition and marginally mediated the one with decline. Tau positivity represented the strongest risk factor for decline (hazard ratio = 32).
DISCUSSION
Tau and neurodegeneration synergistically contribute to global cognitive impairment while tau drives decline. The tau PET superior prognostic value supports its implementation in memory clinics.
Highlights
Hypometabolism has the strongest association with concurrent cognitive impairment.
Neocortical tau pathology is the main determinant of cognitive decline over time.
FDG‐PET has a superior value compared to MRI as a measure of neurodegeneration.
The prognostic value of tau‐PET exceeded all other neuroimaging modalities.
Keywords: Alzheimer's disease biomarkers, cognitive decline, neuroimaging, positron emission tomography
1. BACKGROUND
The A/T/N model is a research framework proposed to investigate Alzheimer's disease (AD) main biomarkers (i.e., amyloidosis, A; neurofibrillary tangles, T; and neurodegeneration, N) defining AD as a biological rather than a clinical construct. 1 A can be measured through amyloid positron emission tomography (PET) or cerebrospinal fluid (CSF) amyloid beta 42 (Aβ42); T through tau‐PET or CSF phosphorylated tau; and N through MRI, fluorodeoxyglucose (FDG)‐PET, or CSF total tau. The addition of cognitive status (C) results in the A/T/N/C model, 2 offering the opportunity to improve subject diagnosis/prognosis as well as to elucidate the cognitive profile associated with different combinations of A/T/N biomarkers.
According to the temporal sequence of biomarker progression, 1 A is linked to increased T and promotes T propagation from the medial temporal lobes to neocortical regions, where T is linked to local N which further drives cognitive dysfunction. 3 Thus, T deposition initially localized in the medial temporal lobes, subsequently spreads stereotypically into the temporal, parietal, and frontal cortices in patients with AD. Notably, different T topographies underlie different clinical phenotypes, involving brain regions that are critical for cognitive functions differently affected in distinct AD variants, conforming to established brain‐behavior relationships. 4 , 5 , 6 T distribution strongly coincides with hypometabolism and atrophy patterns, both explicative of cognitive symptoms, rather than A which starts earlier and is widely distributed throughout the association cortices at the symptomatic stage. 7 , 8 , 9 , 10 Although associations have been found at the preclinical stage, 11 , 12 either absent or weak associations exist between regional amyloid plaques and specific cognitive impairment and cognitive decline in AD. The temporal dynamics of AD biomarkers 1 might explain the lack of a close association between A and C, unlike to what occurs with T and N. 13 Since also T, like A, accumulates and spreads years before the development of clinical symptoms, 14 questions have been raised about its specific contribution to cognitive deficits, which can pass through a variety of mechanisms. Specifically, the association between T and C seems to be in part mediated by N, as grey matter atrophy, but not (or only slightly) by A burden, 6 leaving open the question of other tau‐mediated pathological processes. N has been demonstrated to be crucial for exacerbating cognitive decline, in the presence of A and T, and furthering its progression to clinical AD. 15 Among N measures, FDG‐PET brain hypometabolism reveals disease‐specific alterations since the very early stage allowing the identification of subjects who will develop AD or other form of dementia and those who will remain stable over time. 16 , 17
Previous studies focusing on the A, T, and N in the AD spectrum, supported tau‐PET as a superior diagnostic and prognostic marker compared with MRI and amyloid‐PET, 18 , 19 especially in the prodromal and preclinical stages of AD. 20 , 21 , 22 , 23 Notably, most of these studies included only MRI as an N measure, which reflects late atrophy, without considering FDG‐PET representing an early biomarker of neural dysfunction. Despite this evidence, the association between individual A/T/N biomarkers and their combinations in explaining C, and their potential in predicting rates of cognitive decline in a memory clinic cohort is still poorly understood.
The role of PET imaging biomarkers in clinical settings and for the inclusion in clinical trials for disease–modifying therapy is becoming more and more relevant. The objectives of this study were to (1) assess the specific relationships between regional A/T/N abnormalities and C; (2) investigate the dynamic interplay between A/T/N and C applying mediation models; (3) elucidate the prognostic value of A/T/N biomarkers for predicting future cognitive change in a memory clinic setting.
RESEARCH IN CONTEXT
Systematic review: According to the temporal sequence of biomarker progression, amyloid promotes tau propagation from the medial temporal lobe to neocortical regions, where tau is linked to local neurodegeneration which further drives cognitive dysfunction in Alzheimer's disease. The association between individual biomarkers and their combinations in explaining cognition and predicting cognitive decline in a memory clinic cohort is still poorly understood.
Interpretation: When used individually, all neuroimaging biomarkers were associated with cognitive impairment and decline, supporting each of them as a robust indicator of Alzheimer's pathology. Tau and neurodegeneration synergistically contribute to cognitive impairment; however, neurodegeneration drives cognitive impairment while neocortical tau drives decline. The prognostic value of tau‐PET exceeded all other neuroimaging modalities, supporting its implementation in memory clinics’ routine workup of patients.
Future directions: The role of tau‐PET for inclusion in clinical trials for disease‐modifying therapy needs to be defined.
2. METHODS
2.1. Participants
The study included 94 subjects who consulted the Memory Center of Geneva University Hospitals, ranging from cognitively unimpaired (CU) over mild cognitive impairment (MCI) to dementia. Each subject underwent the memory center's routine clinical workup, including clinical and neurological assessment, neuropsychological testing, and MRI. Additional procedures, such as amyloid‐PET, tau‐PET, and FDG‐PET have been performed if deemed clinically useful, or in the context of other research projects. 24
A total of 94 subjects were classified as CU (N = 17), MCI (N = 57), 25 and dementia (DEM) (N = 20). 26 Inclusion criteria were the availability of at least one Mini‐Mental State Examination (MMSE), a tau‐PET scan and a FDG‐PET scan performed within 1 year from tau‐PET. A subset of participants also underwent amyloid‐PET (number = 90) and/or MRI (number = 81) at baseline within 1 year from tau‐PET, and/or follow‐up MMSE at 2 ± 1 years (number = 64). The subsample with clinical follow‐up, including 10 CU, 42 MCI, and 12 DEM patients, did not differ in any demographic, clinical, and biomarker data from the baseline sample without follow‐up (p > 0.05).
The local Ethics Committee approved the imaging studies, which have been conducted under the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice. Each subject or their relatives provided voluntary written informed consent to participate in the studies.
2.2. Imaging acquisition
2.2.1. MRI
MRI was performed at Geneva University Hospitals’ division of radiology using a 3 Tesla scanner (Magnetom Skyra, Siemens Healthineers, Erlangen, Germany), equipped with a 20‐ or 64‐channel head coil. The following acquisition parameters were used: repetition time [TR] = 1810–1930 ms, echo time [TE] = 2.19–2.36 ms, field of view = 256 × 256 mm, flip angle = 8°, slice thickness = 0.9–1 mm, matrix size = 288 × 288 pixels, or 256 × 230 pixels.
2.2.2. PET
All PET scans using 18F‐labeled tracers were performed at the Nuclear Medicine and Molecular Imaging division at Geneva University Hospitals with Biograph128 mCT, Biograph128 Vision 600 Edge, Biograph40 mCT, or Biograph64 TruePoint PET scanners (Siemens Medical Solutions, Malvern, PA, USA). All scanners were thus from the same vendor and of the same generation, harmonized regarding their performance and reconstructions, and cross‐calibrated.
Amyloid‐PET: Amyloid‐PET images were acquired using [18F]florbetapir (FBP) or [18F]flutametamol (FMM) tracers. FBP late images were acquired 50 min after the intravenous administration of 210 ± 18 MBq (3 × 5 min image frames). FMM late images were acquired 90 min after the intravenous administration of 166 ± 16 MBq (4 × 5 min image frames). Images were then averaged into a single 15 or 20 min frame.
Tau‐PET: [18F]flortaucipir (18F‐AV1451), synthesized at the Center for Radiopharmaceutical Sciences in Villigen, Switzerland, under license from the intellectual property (IP) owner (Avid subsidiary of Lilly, Philadelphia, PA, USA), was used for the tau‐PET scans. Subjects received 180 MBq of 18F‐AV1451, with image acquisition performed 75 min after injection (acquisition time 30 min). 27 Each emission frame was reconstructed in 6 × 5 min frames.
FDG‐PET: FDG‐PET was performed according to the European Association of Nuclear Medicine (EANM) guidelines. 28 , 29 Briefly, subjects fasted for at least 4 h. Before radiopharmaceutical injection, blood glucose was checked and was 7 mmol/L (at maximum) for all subjects. Subjects were injected with 203 ± 15 MBq of [18F]FDG via a venous cannula, eyes open in a dimly lit room. The required minimum time interval between injection and scan start was 30 min. Scanning time was 20 min.
For all tracers, data were acquired in list mode and were reconstructed using 3D OSEM, corrected for randoms, dead time, normalization, scatter, attenuation, and sensitivity, and averaged after motion correction. A 2 mm Gaussian filter at full width at half maximum (FWHM) was applied resulting in images with 400 × 400 matrix with 1.01 mm isotropic voxels.
2.3. Imaging processing
PET processing was performed using Statistical Parametric Mapping (SPM12, Wellcome Trust Centre for Neuroimaging, London, UK), running in MATLAB R2018b Version 9.5 (MathWorks Inc., Sherborn, MA, USA). MRI 3D T1 images were aligned to a reference plane passing through the anterior commissure, segmented into gray matter, white matter, and cerebrospinal fluid tissue compartments, and normalized to the Montreal Neurologic Institute (MNI) space using tissue probability maps. All PET images were aligned to the subject's respective T1 MRI scan and normalized to the MNI space using the transformation matrix that was generated during the registration of the MRI images to the standard space. Standardized uptake values for each PET modality were extracted in AD‐ or cognitive‐related regions of interest (ROIs) obtained using the automated anatomic labeling atlas 3, 30 as described in the following paragraphs.
2.4. A/T/N/C measures
MMSE score at baseline was used as the cognitive global assessment (C). MMSE annual rate of change (C‐long) was calculated by subtracting the MMSE score at baseline from the last follow‐up MMSE score and then dividing the result by the number of years of follow‐up, thus expressing the average number of MMSE points lost per year.
A/T/N measures were used both as dichotomous and continuous variables depending on the analyses. Both visual‐based or semi‐quantitative measures for A/T/N were tested in the different models for longitudinal analyses.
A: All amyloid–PET late images were visually assessed by two expert nuclear medicine physicians (V.G., D.P.) applying the standard operating procedures approved by the European Medicines Agency (https://www.ema.europa.eu/documents/product‐information/vizamyl‐epar‐product information_en.pdf; https://www.ema.europa.eu/documents/product‐information/amyvid‐epar‐product information_en.pdf) and classified into “A+” or “A‐”. As a semi‐quantitative measure, standardized uptake value ratios (SUVr) were calculated using the whole cerebellum as the reference region. SUVr was extracted from the Centiloid volume‐of interest (VOI) and converted into Centiloid units as recommended by Klunk. 31 A Centiloid value of 19 was used as the cut‐off point to define A+ versus A‐. 32
T: Tau distribution was visually assessed by two expert nuclear medicine physicians (V.G., D.P.), according to published recommendations, 33 describing regions of increased [18F]flortaucipir uptake. As semi‐quantitative measures, the SUVr was calculated using the cerebellar crus as a reference region. We considered the global SUVr calculated from the entorhinal cortex, lateral occipital cortex, inferior temporal cortex, and amygdala, 34 and, separately, from a lateral temporal ROI. Intensity‐normalized PET images were saved for further voxel‐wise analyses. An internally validated global SUVr cut‐off point of 1.24 was used to define T+ and T‐. 35
NFDG : The visual assessment of FDG‐PET images was performed following a validated SPM single‐subject procedure, 36 according to which each image was tested for relative hypometabolism by means of a two‐sample t‐test in comparison with images of 112 healthy controls on a voxel‐by‐voxel basis, including age as a covariate. The statistical threshold for the resulting hypometabolic SPM maps was set at p = 0.05, considering significant clusters containing more than 100 voxels. The resulting single‐subject SPM hypometabolic maps were visually inspected by nuclear medicine physicians (DP, VG) and classified into hypometabolism patterns suggestive of AD (NFDG+), or other neurodegenerative conditions, or absence of neurodegeneration (NFDG‐). 37 , 38 As semi‐quantitative measures, the SUVr was calculated from a lateral temporal ROI 39 and AD metaROI (including left and right angular gyrus, left and right temporal, and bilateral posterior cingulate ROIs), 40 using the pons and cerebellar vermis as the reference region. Intensity‐normalized PET images were saved for further voxel‐wise analyses. A metaROI SUVr cut‐off point of 1.41 was used to define NFDG+ and NFDG‐. 32
NMRI : T1 MRI images were segmented and cortical thickness and volumes extracted using Freesurfer (v.7.0; surfer.nmr.mgh.harvard.edu/). 41 Hippocampal volume (HPV) was extracted and adjusted for intracranial volume. An AD cortical signature (weighted average cortical thickness in the entorhinal, inferior temporal, middle temporal, and fusiform ROIs) was created, 42 setting 2.57 as the cut‐off point for discriminating between NMRI+ and NMRI‐. 32 To use gray matter (GM) MRI images for further voxel‐wise analyses, the images were normalized and modulated using the DARTEL toolbox 43 in SPM12 and smoothed with an 8‐mm FWHM Gaussian kernel.
2.5. Statistical analyses
Baseline demographics, clinical, cognitive, and biomarkers differences among syndromic diagnoses were assessed using a Kruskal‐Wallis rank sum test for continuous variables and a proportion test for categorical variables. All analyses were performed using R, version 4.0.2 (https://www.r‐project.org/). A p‐value of 0.05 was considered the significance threshold for all analyses.
First, to assess the correlation between A/T/N and C/C‐long in the whole sample, general linear models were performed. MMSE scores at baseline (C) and MMSE annual rate of change (C‐long) were used as dependent variables, and continuous semiquantitative A, T, NFDG, or NMRI measures as predictors. Namely, A was measured through centiloid values, T through the global SUVr and the lateral temporal SUVr, NFDG through the SUVr in the AD MetaROI and the lateral temporal ROI, and NMRI through HPV and AD cortical signature. Age, sex, education, and clinical status (CU, MCI, DEM) were included as nuisance variables in the models.
Second, to examine whether associations between T and C, and T and C‐long were mediated by NFDG and NMRI, we performed causal mediation analyses controlling for age, sex, education, and clinical status. Additional causal mediation analyses were run to examine whether associations between A and C, and A and C‐long were mediated by T and N.
Third, to investigate the effect of the A/T/N biomarkers on cognitive changes over time we applied linear mixed‐effects models with random intercepts and slopes using longitudinal MMSE as a dependent variable, adjusting for age, sex, education, and clinical status. For the longitudinal model, we included either A, T, NFDG, or NMRI binary status (defined by visual rating or semi‐quantification (see Methods Section 2.4) in different models) as predictors, both separately and together. Then, applying the longitudinal model, we examined differences in cognitive trajectories between groups stratified according to different T/N profiles, using T‐/N‐ as the reference group.
Last, Kaplan‐Meier analysis was performed to estimate the times to decline in the different T/N profiles and survival curves were compared by means of log‐rank. Hazard ratios (HR) for the variables of interest, namely A, T, N, C, and T/N profiles were estimated via Cox proportional hazard models adjusting for age, sex, education, and clinical status. We classified subjects into decliners or stable based on the change from MMSE at baseline and MMSE at the last follow‐up, considering the follow‐up duration and the participant's age. 44 The proportional hazard assumptions were tested, and the threshold was set at p < 0.05, where the lower limit of 95% HR confidence interval > 1 for risk factors, and the upper limit < 1 for protective factors.
2.5.1. Voxel‐wise analyses
To evaluate the correlation between A/T/N biomarkers and C or C‐long, we performed voxel‐wise regressions, controlling for age, sex, education, and clinical status. The statistical threshold was set at p < 0.005, FWE‐corrected at the cluster level. Voxel‐wise analyses were run in SPM12 and results were visualized using BrainNet Viewer software. 45 Based on the voxel‐wise results, we performed further mediation analyses (data‐driven) to explore the intermediary role of NFDG in the relationship between T and C/C‐long in the brain regions where T correlated with C/C‐long. For these analyses, SUVr of T and NFDG were extracted for each participant from the overlapping clusters (both T and NFDG correlated with C/C‐long), and the discrepancy clusters (only T or NFDG correlated with C/C‐long) obtained from the voxel‐wise regression analyses. The same analyses were performed using MRI as a measure of N.
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
3. RESULTS
Participants’ characteristics of our cohort at baseline and at follow‐up are displayed in Table 1 and 2, respectively. Out of 94 participants, 47 (50%) were female, the mean age was 70 ± 7, the majority was A+ (67%) and 50% were T+. Sixty‐one percent were MCI, 21% demented, and 18% cognitively unimpaired. A typical AD temporoparietal hypometabolism (N+) was expressed by 39.4% of subjects, whereas 14% presented a limbic‐predominant pattern, 3% a temporoparietal and occipital pattern, 7% a frontal pattern, the other patterns were negative or not suggestive of neurodegeneration.
TABLE 1.
Characteristics of the sample at baseline.
| Whole sample (N = 94) | Cognitively unimpaired (N = 17) | Mild cognitive impairment (N = 57) | Dementia (N = 20) | p‐Value | |
|---|---|---|---|---|---|
| Sex, female N (%) | 47 (50%) | 9 (52.9%) | 26 (45.6%) | 12 (60%) | = 0.522 |
| Age, y (mean ± SD) | 70 ± 7 | 69 ± 7 | 72 ± 5 | 67 ± 8 | = 0.008 |
| Education, y | 13 ± 3 | 15 ± 3 | 13 ± 3 | 11 ± 3 | = 0.034 |
| MMSE score | 24 ± 4 | 27 ± 2 | 26 ± 2 | 18 ± 6 | < 0.001 |
| A status, positivity (+) | 63 (67%) | 3 (17.6%) | 42 (73.6%) | 16 (80%) | < 0.001 |
| T status (+) | 47 (50%) | 0 (0%) | 32 (56%) | 15 (75%) | < 0.001 |
Note: Continuous variables are reported as mean and standard deviation, categorical variables as number and percentage. All p‐values are obtained by Kruskal‐Wallis test for continuous variables and proportion test for frequencies. Reported A, T statuses are based on visual assessment of amyloid‐PET, and tau‐PET scans, respectively.
Abbreviations: A, amyloid; MMSE, mini‐mental state examination; N, number; SD, standard deviation; y, year; T, tau.
TABLE 2.
Characteristics of the sample at follow‐up.
| Whole sample (N = 64) | Cognitively unimpaired (N = 10) | Mild cognitive impairment (N = 42) | Dementia (N = 12) | p‐Value | |
|---|---|---|---|---|---|
| Sex, female N (%) | 32 (50%) | 5 (10%) | 23 (54.7%) | 4 (33%) | = 0.424 |
| Age, y (mean ± SD) | 71 ± 6 | 68 ± 5 | 72 ± 6 | 68 ± 8 | = 0.041 |
| Education, y | 14 ± 3 | 15 ± 3 | 14 ± 3 | 11 ± 3 | = 0.011 |
| MMSE score at BL | 25 ± 5 | 27 ± 2 | 26 ± 2 | 18 ± 8 | < 0.001 |
| MMSE score at FU | 22 ± 7.2 | 26 ± 2 | 24 ± 4 | 13 ± 10 | < 0.001 |
| A status, positivity (+) | 45 (70%) | 2 (20%) | 33 (78.5%) | 10 (83%) | < 0.001 |
| T status (+) | 34 (53%) | 0 (0%) | 25 (59.5%) | 9 (75%) | < 0.001 |
Note: Continuous variables are reported as mean and standard deviation, categorical variables as number and percentage. All p‐values are obtained by Kruskal‐Wallis test for continuous variables and proportion test for frequencies. Reported A and T statuses are based on visual assessment of amyloid‐PET and tau‐PET scans, respectively.
Abbreviations: A, amyloid; BL, baseline; FU, follow‐up; MMSE, mini‐mental state examination; N, number; SD, standard deviation; y, year; T, tau.
3.1. Correlations between A/T/N biomarkers and MMSE
Baseline: All A/T/N biomarkers showed significant correlations with C (p < 0.001, Table 3). The NFDG SUVr in the AD metaROI showed the highest correlation (r = 0.712, p < 0.001), followed by NFDG SUVr in the lateral temporal ROI (r = 0.709, p < 0.001) and the T SUVr in the lateral temporal ROI (r = −0.682, p < 0.001). The voxel‐wise regressions between C and NFDG showed a widespread significant relationship involving the precuneus, posterior cingulate, temporoparietal cortices, and, to a lesser extent, the orbitofrontal cortex (Figure 1A). Similar voxel‐wise results were obtained using NMRI instead of NFDG (Figure S1A). T voxel‐wise correlation results extended to the same regions and, in addition, the dorsolateral prefrontal cortices (Figure 1B).
TABLE 3.
Correlations between Alzheimer's disease biomarkers and MMSE at baseline and MMSE rate of change controlling for age, sex, education, and clinical status.
| Biomarkers | Correlation coefficient | p‐Value |
|---|---|---|
| Correlations with MMSE at baseline | ||
| N: FDG AD meta‐ROI | 0.712 | <0.001 |
| N: FDG Lateral temporal ROI | 0.709 | <0.001 |
| T: Tau lateral temporal ROI | −0.682 | <0.001 |
| T: Tau meta‐ROI | −0.628 | <0.001 |
| N: MRI AD cortical signature | 0.673 | <0.001 |
| N: MRI HPV | 0.667 | <0.001 |
| A: Amyloid centiloid | −0.644 | <0.001 |
| Correlations with MMSE annual rate of change | ||
| T: Tau lateral temporal ROI | −0.677 | <0.001 |
| T: Tau meta‐ROI | −0.669 | <0.001 |
| A: Amyloid centiloid | −0.564 | 0.002 |
| N: FDG lateral temporal ROI | 0.498 | 0.015 |
| N: FDG AD meta‐ROI | 0.491 | 0.018 |
| N: MRI AD cortical signature | 0.447 | NS |
| N: MRI HPV | 0.438 | NS |
Abbreviations: A, amyloid; HPV, hippocampal volume; MMSE, mini‐mental state examination; N, neurodegeneration; NS, not significant; AD, Alzheimer's disease; T, tau.
FIGURE 1.
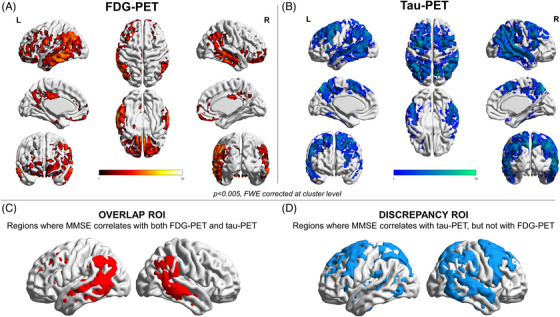
Voxel‐wise correlation between baseline MMSE and PET neuroimaging. Panels A and B show the voxel‐wise correlation results with MMSE for brain metabolism and tau uptake, respectively. Thresholded SPM images (p < 0.005 FWE‐corrected at the cluster level, minimum cluster extent k = 100 voxels) are over‐imposed on a standard ICBM152 surface with BrainNet Viewer. Panels C and D show the binary masks including the voxel correlating with MMSE for both brain metabolism and tau uptake (overlap ROI, C), and the ones correlating only with tau uptake (discrepancy ROI, D).
MMSE annual rate of change: The correlation between AD biomarkers and C‐long did not follow the same hierarchy as with C (Table 3). T SUVr in the lateral temporal ROI showed the highest correlation with C‐long (r = −0.677, p < 0.001), followed by the T SUVr in the meta‐ROI (r = −0.669, p < 0.001) and A Centiloid (r = −0.564, p = 0.002). The NMRI was the only biomarker not correlating with C‐long (p > 0.05). The voxel‐wise regressions between C‐long and NFDG showed results restricted to the posterior cingulate and orbitofrontal cortex (Figure 2A). When we used NMRI instead of NFDG, no voxels survived at p < 0.005, with FWE corrections at cluster level. T voxel‐wise correlation showed a widespread relationship, involving the same FDG regions but also the temporoparietal cortices (Figure 2B).
FIGURE 2.
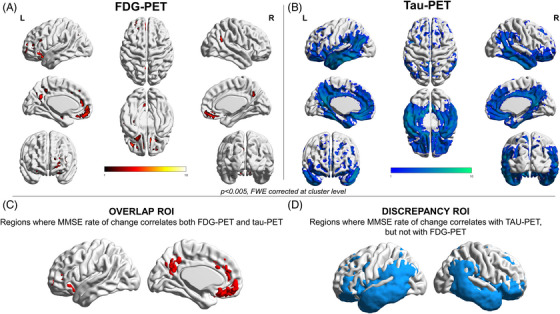
Voxel‐wise correlation between the MMSE annual rate of change and PET neuroimaging. Panels A and B show the voxel‐wise correlation results with MMSE annual rate of change for brain metabolism and tau uptake, respectively. Thresholded SPM images (p < 0.005 FWE‐corrected at the cluster level, minimum cluster extent k = 100 voxels) are over‐imposed on a standard ICBM152 surface with BrainNet Viewer. Panels C and D show the binary masks including the voxel correlating with MMSE and both brain metabolism and tau uptake (overlap ROI, C), and the ones correlating only with tau uptake (discrepancy ROI, D).
3.2. Mediation results between A/T/N biomarkers and MMSE
Baseline: Figure 3A shows path diagrams assessing NFDG SUVr in the overlap ROI as a potential mediator of associations between T SUVr (in the overlap (Figure 1C) and then, the discrepancy (Figure 1D) ROIs), and C. Significant mediation effects in the total group for both T SUVr in the overlap and discrepancy ROIs (39% and 30% of the total effects, respectively; p < 0.05) were observed. Mediation effects were lower (25%–26% of the total effects, p < 0.05) considering NMRI volume in the overlap ROI as the mediator between T SUVr (overlap and discrepancy ROIs) and C (Figure S1E).
FIGURE 3.
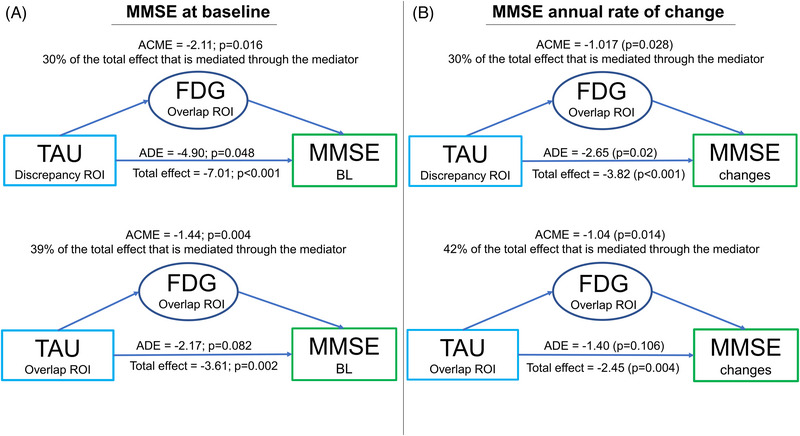
Mediation path diagrams. Path diagrams indicate whether brain metabolism mediates the associations between tau uptake and MMSE at baseline (A) and its annual rate of change (B). Brain metabolism and tau uptake have been extracted from the regions obtained in the voxel‐wise correlation analyses (overlap ROI, both tau and metabolism correlate with MMSE, and discrepancy ROI, only tau correlates with MMSE). The average direct effect (ADE) reflects the extent to which MMSE changes when baseline tau positron emission tomography (PET) increases by 1 unit while baseline metabolism remains unaltered. The indirect average causal mediation effect (ACME) reflects the extent to which MMSE changes when baseline tau PET is held constant and baseline metabolism changes by the amount it would have changed had baseline tau PET increased by 1 unit. The total effect represents the sum of the direct and indirect effects.
When we assessed the potential mediation of NFDG SUVr in the lateral temporal ROI or AD metaROI on the association between T SUVr in the lateral temporal ROI or metaROI and C, we found similar mediation effects ranging from 30% to 46% (p < 0.05) (Table 4). Instead, NMRI AD cortical signature did not mediate the total effects of T in the lateral temporal ROI and metaROI on C (Table S1). Both T and NFDG strongly mediated the association between A and C ranging from 61%–92% (p < 0.05) (Table 4 and Table S2).
TABLE 4.
Summary of all significant results of mediation analyses.
| Model | Total effect | Average direct effect‐ADE | Average causal mediation effect‐ACME | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Independent variable | Tested N mediator | Dependent variable | Estimate | p‐Value | Estimate | p‐Value | Estimate | p‐Value | Proportion mediated |
| Tau | N | MMSE BL | |||||||
| Lateral temporal ROI | FDG lateral temporal ROI | −3.72 | <0.001 | −2.47 | 0.06 | −1.24 | 0.02 | 33% | |
| MetaROI | FDG lateral temporal ROI | −3.57 | 0.008 | −1.91 | 0.23 | −1.65 | 0.008 | 46% | |
| Lateral temporal ROI | FDG AD metaROI | −3.72 | <0.001 | −2.60 | 0.05 | −1.12 | 0.006 | 30% | |
| MetaROI | FDG AD metaROI | −3.57 | 0.02 | −2.15 | 0.16 | −1.42 | 0.006 | 40% | |
| Amyloid | Tau or N | MMSE BL | |||||||
| Centiloid | Tau lateral temporal ROI | −0.017 | 0.13 | −0.001 | 0.91 | −0.016 | 0.02 | 92% | |
| Centiloid | FDG AD metaROI | −0.011 | 0.27 | −0.004 | 0.69 | −0.007 | 0.04 | 61% | |
| Centiloid | FDG lateral temporal ROI | −0.011 | 0.27 | −0.002 | 0.79 | −0.009 | 0.008 | 81% | |
| Amyloid | Tau or N | MMSE rate of change | |||||||
| Centiloid | Tau lateral temporal ROI | −0.018 | 0.01 | −0.005 | 0.430 | −0.012 | 0.004 | 70% | |
| Centiloid | Tau metaROI | −0.018 | 0.01 | −0.005 | 0.412 | −0.012 | 0.002 | 70% | |
Abbreviation: AD, Alzheimer's disease; BL, baseline; MMSE, mini‐mental state examination; N, neurodegeneration; ROI, region of interest.
MMSE annual rate of change: Figure 3B shows path diagrams assessing NFDG SUVr in the overlap ROI as a potential mediator of associations between T SUVr (in the overlap [Figure 2C] and then, the discrepancy [Figure 2D] ROIs) and C‐long. There were significant mediation effects in the total group for both T SUVr in the overlap and discrepancy ROIs (42% and 30% of the total effects, respectively; p < 0.05). When we assessed the potential mediation of NFDG SUVr in the lateral temporal ROI or AD metaROI on the association between T SUVr in the lateral temporal ROI or metaROI and C‐long, we did not find any significant mediation effect (Table 4 and Table S1). NMRI AD cortical signature did not significantly mediate the total effects of T in the lateral temporal ROI or metaROI on the C‐long (Table 4 and Table S1). T strongly mediated the association between A and C‐long (70% of the total effects, p < 0.004), whereas N (both NFDG and NMRI) did not (Table 4 and Table S2).
3.3. Longitudinal cognitive trajectories
Linear mixed effect models indicated that the T+ (standardized β [stb] of interaction with time in years = −2.73, p < 0.001), NFDG+ (stb = −2.61, p < 0.001), and A+ (stb = −2.02, p = 0.008) groups showed a faster decline over time on MMSE compared to the T‐, NFDG‐, and A‐ (reference) groups, respectively. When we considered N according to MRI, we did not find any significant differences between groups. Comparing different T/NFDG profiles, we found that T+/NFDG+ (stb = −2.7, p < 0.001) and T+/NFDG‐ (stb = −2.75, p = 0.004) groups showed a faster cognitive decline over time compared to the T‐/NFDG‐ (reference) groups (Figure 4A). Considering NMRI instead of NFDG, the previous results were still confirmed (T+/NMRI+: stb = −3.14, p < 0.001, T+/NMRI‐: stb = −2.68, p < 0.001 vs. T‐/NMRI‐).
FIGURE 4.
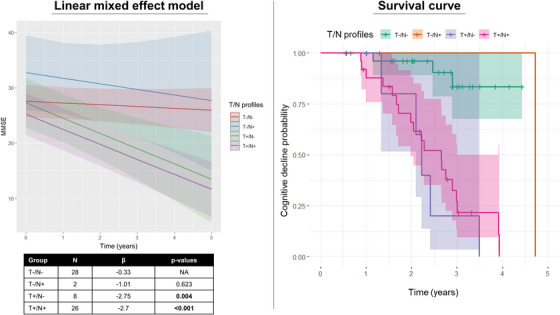
Longitudinal results. Panel A shows different cognitive trajectories of MMSE scores over time in the different T/N categories, where N is defined based on FDG‐PET and T on tau‐PET. Panel B shows survival curves in relation to declining or not in MMSE in the different T/N categories, where N is defined based on FDG‐PET and T on tau‐PET.
After defining 36 individuals as stable (CU = 10, MCI = 24, DEM = 2) and 28 as decliners (MCI = 18, DEM = 10) based on the change from MMSE at baseline and MMSE at follow‐up (considering the follow‐up duration and the participant's age), Cox proportional hazard models demonstrated an increased risk for cognitive decline in the T+ (HR = 15.2 [4.01–57.8], p < 0.001), NFDG+ (HR = 2.64 [0.97–7.16], p = 0.05), A+ (HR = 4.92 [1.07–22.7], p = 0.041) groups compared to the T‐, NFDG‐, and A‐ (reference) groups, respectively. When we considered N according to MRI instead of FDG‐PET, we did not find any significant differences. After including all biomarkers and the baseline MMSE (C) in the same model, T+ resulted as the highest significant risk factor (HR = 31.9 [5.89–173], p < 0.001) (Table S3). Accordingly, we found an increased risk for cognitive decline in the T+/NFDG+ (HR = 10.5 [2.63–41.5], p < 0.001), T+/NFDG‐ (HR = 24.7 [2.24–144], p < 0.001) compared to the T‐/NFDG‐ (reference) group. Figure 4B shows the survival curves for the four T/N profiles, revealing that patients T+/N+ and T+/N‐ have a shorter survival time than those with T‐/N‐ (p < 0.001). In this context, survival pertains to remaining free from cognitive decline. A similar increased risk for cognitive decline was found in T+/NMRI+ (HR = 27.4 [2.59–209], p = 0.006), T+/NMRI‐ (HR = 45.4 [4.27–483], p = 0.002) compared to the T‐/NMRI‐ (reference) group. The small size of T‐/N+ subgroups limited any inference on their clinical trajectories and risk for cognitive decline.
Comparable results were obtained considering the A, T, and NFDG binary status according to semi‐quantification, instead of visual rating (Tables S3 and S4). Moreover, since the clinical status was a significant predictor of cognitive decline over time, all the longitudinal analyses have been performed excluding DEM patients, confirming the results obtained in the whole sample. The longitudinal results regarding the subsample without DEM patients are fully reported in the Supplementary Materials (S1).
4. DISCUSSION
AD is a polygenic and multifactorial disease that is subtended by several partly independent pathological processes, each under the influence of both independent and common risk factors, and able to interact with each other. 46 , 47 Many pathological mechanisms, such as intracellular oligomers, neurofibrillary tangles, neuronal mitochondria dysfunction, neurotoxic effect of neuroinflammation, calcium channel changes, besides the extra‐cellular fibrillar amyloid deposition, contribute to neural and synaptic dysfunction with a direct impact on symptoms and progression in AD. 48 PET imaging provides the opportunity to assess in vivo the role of different pathological mechanisms to cognitive impairment and decline. FDG‐PET, as a proxy of neural and synaptic dysfunction, is the most established biomarker in clinical practice supporting the diagnosis of many dementing disorders. 49 The most recent addition for the assessment of proteinopathy is tau‐PET which holds great potential as a diagnostic and prognostic marker in AD. 50 In line with the promising evidence, this study explored the value of T in comparison with other AD biomarkers in explaining cognitive impairment and predicting the rate of cognitive decline in a memory clinic cohort. Focusing on the interplay between T and N, assessed as brain metabolism, we found that N drove concurrent C while cognitive longitudinal changes were mostly driven by neocortical T. T and N synergistically contributed to cognitive impairment with N mediating the relationship between T and C. A key finding of this study was the added value of tau‐PET in predicting cognitive worsening in a head‐to‐head comparison with other neuroimaging biomarkers.
In our sample, mainly composed of MCI subjects (61%), biomarkers from the A/T/N framework have different importance in explaining the concurrent cognitive dysfunction and predicting decline, with NFDG revealing the strongest association with concomitant cognitive impairment and T the best performance in predicting cognitive worsening (Table 3). The stronger relationship between hypometabolism and cognitive deficit is supported by neuropathological data showing that synaptic density is the strongest predictor of antemortem cognition. 51 Moreover, this result is in line with the temporal ordering of biomarkers preceding clinical symptomatology of AD, 52 and the evidence of cognitive impairment as initially triggered by A and T but further exacerbated by hypometabolism. 15 Our results showed a correlation between A and C, strongly mediated by T, consistent with the evidence that found A burden to be more relevant for cognition and decline in cognitively normal subjects. 11 , 12 , 53 A strong correlation was found between T and concurrent cognition according to the fact that tau pathology in the neocortex closely parallels the symptomatic stage of the disease. Given the relevance of the topographical distribution of T in defining clinical phenotypes, 4 , 5 , 6 , 54 tau pathology should require a regional approach able to capture distinctive regional patterns, as brain metabolism does. 55 , 56 Our voxel‐wise results showed that decreased performance in global cognitive assessment was related to increased tau deposition and decreased metabolism (and brain volumes) in similar expected brain areas based on the well‐known brain‐behavior relationship (Figure 1 and Figure S1). The associations between cognition and hypometabolism/atrophy or tau deposits reflected similar though not entirely overlapping patterns, consistent with previously published works. 5 , 6 , 54 , 57 T showed more spatially extensive relationships with cognitive impairment than N, according to T as an earlier event than neural dysfunction. Notably, only T correlated with cognitive performance in the dorsolateral prefrontal cortex, known to become hypometabolic in the later stages of AD. 58
Brain metabolism was found to be a significant mediator of the relationship between T and C (Table 4 and Figure 3), converging with previous works using another tau‐PET tracer ([18F]THK5317) 57 or considering GM atrophy 6 in AD. These mediating effects were observed in several hypometabolic AD‐related regions and were maintained in the cognition‐related atrophic regions (Figure S1). These results were in line with the regional causal sequence where the local accumulation of T triggers neuronal dysfunction, which in turn leads to cognitive impairment. The partial mediation, not exceeding 46%, did not rule out the existence of other tau‐mediated mechanisms on cognition, in need to be investigated, as well as a direct effect of T accumulation on cognitive performance. 21 , 59 Our results enrich previous atrophy‐based findings by showing an intermediate role for NFDG by applying a voxel‐wise approach. We found evidence of a mediating role for metabolism in the relationship between T and C, not only in colocalized regions but also in discrepant regions (Figures 1 and 3). This is consistent with the hypothesis that tau spreading precedes future neurodegeneration processes, 60 however, in need of further longitudinal proof. Instead, the mediation effect of brain metabolism on the relationship between T and cognitive changes over time emerged in the precuneus and orbitofrontal cortex (Figure 3) where both hypometabolism and tau correlated with longitudinal cognitive changes (Figure 2). While hypometabolism correlating voxel‐wise with cognitive decline was limitedly spread, widespread neocortical T strongly correlated with longitudinal cognitive changes (Figure 2). No atrophic regions correlated with MMSE changes over time by applying voxel‐wise analyses, showing NMRI as a less sensitive N measure for longitudinal changes as compared to NFDG. 61 Because tau pathology is thought to occur before the onset of neurodegeneration, T may be more sensitive to early pathological changes. The modest (or null in some regions) (Figure 3 and Table S1) mediation of N on the relationship between T and C‐long found here is in line with previous atrophy‐based results showing a disease stage‐specific mediation effect only in AD dementia and A+ MCI. 62 Thus, our results argue in favor of a strong independent effect (i.e., neither atrophy‐ nor metabolism‐mediated) of tau pathology on subsequent cognitive decline.
The longitudinal results found tau‐PET to be superior to all other investigated neuroimaging biomarkers in predicting cognitive decline. When considered separately, also increased A and decreased N levels predicted cognitive decline, but these relationships were likely predominantly driven by T (i.e., when all biomarkers were included in the model, T remained most significant among imaging biomarkers) (Table S4). When we considered different profiles, we found that T+/N+ and T+/N‐ groups exhibited a faster decline in global cognition, suggesting that T is sufficient to predict cognitive decline (Figure 4). Thus, the present longitudinal results supported the added value of tau‐PET in detecting individuals progressing to cognitive decline, since the T+/N‐ group likely included patients who will develop neurodegeneration later. Cox hazard regression results further confirmed that T+/N+ and T+/N‐ individuals were at the highest risk of deterioration based on their biomarker profile (Figure 4). Given the clinical stability of T‐/N+ group, it could be representative of limbic‐predominant non‐AD pathologies that are frequently seen in amnestic MCI and commonly associated with proteinopathy TAR DNA‐binding protein 43 (TDP‐43). 63 Since their clinical manifestation mimics AD and protein‐specific biomarkers are still lacking, tau‐PET ability to predict their stability is promising, particularly for the selection of candidates in clinical trials to avoid misclassifications.
Given the small sample size of T‐/N+ profile, any inference on clinical trajectories should be taken with caution. We cannot exclude that other biomarkers may have similar or even better predictive effects in larger samples. For instance, blood‐based biomarkers that we could not include are emerging as having a valuable role in diagnostic and prognostic procedures. 64 However, a recent head‐to‐head study 65 demonstrated that tau‐PET outperformed plasma (and CSF) phospho‐tau in the prediction of cognitive decline in patients with amnestic MCI or dementia. Blood‐based biomarkers might likely be more useful during the earliest stages, where phospho‐tau can predict future increases in tau‐PET 66 , 67 and can play a key role in prescreening subjects for further neuroimaging examinations. 68 In this context, the use of plasma as a gateway to traditional biomarkers could avoid additional investigations in relevant proportions of cases, making the diagnostic workup more cost‐effective and improving patient care. 68
There are some limitations and strengths to this study. First, we used MMSE as a measure of cognitive decline, although we are aware that MMSE is a global measure characterized by a ceiling effect, less sensitive than other tests or test combinations. 69 , 70 Second, we evaluated our subjects with a relatively short follow‐up period, and further studies with longer follow‐up and longitudinal neuroimaging measures are needed. We showed a similar ability of two approaches (visual‐based and semiquantitative methods for biomarkers status) in discriminating cognitively decliners and stable individuals. However, we are aware that the cut‐off points applied here are not internally validated (apart from for tau‐PET) and that only for volumetric MRI and amyloid‐PET standardized measures translatable from other studies exist. For FDG‐PET, we acknowledge that the imaging processing pipeline may influence the SUVR and we aligned our procedure with the original paper's one 32 to minimize this risk.
We consider the heterogeneity of our sample from a memory clinic as a strength of the study for the applicability of our results to clinical settings. Although our memory clinic sample was enriched for AD and our results are likely driven by AD subjects, we acknowledge that non‐AD pathologies could account for a few cases. We deliberately included a heterogenous sample without selecting based on the presence of amyloid pathology since our focus was on tau and neurodegeneration and their ability to predict cognitive decline in the perspective of clinical use.
5. CONCLUSION
In our cohort, all A/T/N biomarkers, when used individually, were associated with cognitive impairment and decline, supporting each of them as a robust indicator of AD pathology. Our results support T as the necessary determinant for cognitive decline, as previously suggested in preclinical and very early asymptomatic AD cohorts. 21 Complementing the evidence of tau‐PET outperformance over MRI and amyloid‐PET, 62 our study also found that tau‐PET is a better prognostic tool than FDG‐PET. Although our results (both baseline and longitudinal) support a superior value of FDG‐PET compared to MRI as a measure of neurodegeneration in its correlation with cognitive performance and decline, the prognostic value of tau‐PET exceeded both modalities, supporting its implementation in memory clinics’ routine workup of patients.
AUTHOR CONTRIBUTIONS
Cecilia Boccalini, Federica Ribaldi, Daniela Perani, and Valentina Garibotto were responsible for the study concept and design. Cecilia Boccalini was responsible for drafting the original report which was reviewed and revised by all coauthors (Federica Ribaldi, Ines Hristovska, Annachiara Arnone, Débora Elisa Peretti, Linjing Mu, Max Scheffler, Daniela Perani, Giovanni B. Frisoni, and Valentina Garibotto). Valentina Garibotto was responsible for the acquisition of the PET scan. Max Scheffler was responsible for the acquisition of the MRI scan. Cecilia Boccalini, Ines Hristovska, Annachiara Arnone, and Débora Elisa Peretti were responsible for all neuroimaging analyses and statistical analyses. Federica Ribaldi and Giovanni B. Frisoni were responsible for the acquisition of the clinical data. Cecilia Boccalini, Ines Hristovska, Annachiara Arnone, Débora Elisa Peretti, and Valentina Garibotto were responsible for the interpretation of data for the work.
CONFLICT OF INTEREST STATEMENT
V.G. received research support and speaker fees through her institution from GE Healthcare, Siemens Healthineers and Novo Nordisk. G.B.F. has received support, payment, consulting fees or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from: Biogen, Roche, Diadem, Novo Nordisk, GE HealthCare, OM Pharma, and Eisai (all through his institution). The other authors have no conflicts of interest pertinent to this manuscript. Author disclosures are available in the Supporting Information
CONSENT STATEMENT
The local Ethics Committee approved the imaging studies, which have been conducted under the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice. Each subject or their relatives provided voluntary written informed consent to participate in the studies.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
The Clinical Research Centre, at Geneva University Hospital and Faculty of Medicine provides valuable support for regulatory submissions and data management, and the Biobank at Geneva University Hospital for biofluid processing and storage. The authors thank the Centre for Radiopharmaceutical Sciences of ETH and USZ for providing the PET tracer for tau imaging. Avid radiopharmaceuticals provided access to the [18F]flortaucipir radiotracer but were not involved in data analysis or interpretation. The Centre de la mémoire is funded by the following private donors under the supervision of the Private Foundation of Geneva University Hospitals: A.P.R.A.: Association Suisse pour la Recherche sur la Maladie d'Alzheimer, Genève; Fondation Segré, Genève; Race Against Dementia Foundation, London, UK; Fondation Child Care, Genève; Fondation Edmond J. Safra, Genève; Fondation Minkoff, Genève; Fondazione Agusta, Lugano; McCall Macbain Foundation, Canada; Nicole et René Keller, Genève; Fondation AETAS, Genève.
Competitive research projects have been funded by: H2020 (projects n. 667375), Innovative Medicines Initiative (IMI contract n. 115736 and 115952), IMI2, Swiss National Science Foundation (projects n.320030_182772 and n. 320030_169876), VELUX Foundation.
Open access funding provided by Universite de Geneve.
Boccalini C, Ribaldi F, Hristovska I, et al. The impact of tau deposition and hypometabolism on cognitive impairment and longitudinal cognitive decline. Alzheimer's Dement. 2024;20:221–233. 10.1002/alz.13355
REFERENCES
- 1. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14(4):535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR, Hampel HJ, Universities S, Cu M, Petersen RC. A new classification system for AD, independent of cognition A /T / N : an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;0(July):1‐10. [Google Scholar]
- 3. Bilgel M, Wong DF, Moghekar AR, Ferrucci L, Resnick SM. Causal links among amyloid, tau, and neurodegeneration. Brain Commun. 2022;4(4):1‐13. doi: 10.1093/braincomms/fcac193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016;139(5):1551‐1567. doi: 10.1093/brain/aww027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanner JA, Iaccarino L, Edwards L, et al. Amyloid, tau and metabolic PET correlates of cognition in early and late‐onset Alzheimer's disease. Brain. 2022:awac229. doi: 10.1093/brain/awac229. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140(12):3286‐3300. doi: 10.1093/brain/awx243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whitwell JL, Graff‐Radford J, Tosakulwong N, et al. Imaging correlations of tau, amyloid, metabolism, and atrophy in typical and atypical Alzheimer's disease. Alzheimer's Dement. 2018;14(8):1005‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ossenkoppele R, Zwan MD, Tolboom N, et al. Amyloid burden and metabolic function in early‐onset Alzheimer's disease: parietal lobe involvement. Brain. 2012;135(7):2115‐2125. doi: 10.1093/brain/aws113 [DOI] [PubMed] [Google Scholar]
- 9. Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early‐onset Alzheimer's disease is not related to amyloid burden. Brain. 2010;133(2):512‐528. doi: 10.1093/brain/awp326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iaccarino L, Edwards L, et al. Spatial relationships between molecular pathology and neurodegeneration in the Alzheimer's disease continuum. Cereb Cortex. 2021;31(1):1‐14. doi: 10.1093/cercor/bhaa184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donohue MC, Sperling RA, Petersen R, Sun CK, Weiner M, Aisen PS. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA ‐ J Am Med Assoc. 2017;317(22):2305‐2316. doi: 10.1001/jama.2017.6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sperling RA, Mormino EC, Schultz AP, et al. The impact of amyloid‐beta and tau on prospective cognitive decline in older individuals. Ann Neurol. 2019;85(2):181‐193. doi: 10.1002/ana.25395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo T, Korman D, Baker SL, Landau SM, Jagust WJ. Longitudinal cognitive and biomarker measurements support a unidirectional pathway in Alzheimer's disease pathophysiology. Biol Psychiatry. 2021;89(8):786‐794. doi: 10.1016/j.biopsych.2020.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain. 2015;138(10):2814‐2833. doi: 10.1093/brain/awv236 [DOI] [PubMed] [Google Scholar]
- 15. Hammond TC, Xing X, Wang C, et al. β‐amyloid and tau drive early Alzheimer's disease decline while glucose hypometabolism drives late decline. Commun Biol. 2020;3(1):1‐13. doi: 10.1038/s42003-020-1079-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perani D, Caminiti SP, Carli G, Tondo G. PET Neuroimaging in dementia conditions. PET SPECT Neurol. 2020:211‐282. Published online. [Google Scholar]
- 17. Chételat G, Arbizu J, Barthel H, et al. Amyloid‐PET and 18F‐FDG‐PET in the diagnostic investigation of Alzheimer's disease and other dementias. Lancet Neurol. 2020;19(11):951‐962. doi: 10.1016/S1474-4422(20)30314-8 [DOI] [PubMed] [Google Scholar]
- 18. Ossenkoppele R, Smith R, Mattsson‐Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal alzheimer disease: a head‐to‐head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78(8):961‐971. doi: 10.1001/jamaneurol.2021.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tosun D, Demir Z, Veitch DP, et al. Contribution of Alzheimer's biomarkers and risk factors to cognitive impairment and decline across the Alzheimer's disease continuum. Alzheimer's Dement. 2021:1‐13. doi: 10.1002/alz.12480. August. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76(8):915‐924. doi: 10.1001/jamaneurol.2019.1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aschenbrenner AJ, Gordon BA, Benzinger TLS, Morris JC, Hassenstab JJ. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology. 2018;91(9):e859‐e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ossenkoppele R, Pichet Binette A, Groot C, et al. Amyloid and tau PET‐positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat Med. 2022:1‐7. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strikwerda‐Brown C, Hobbs D, Gonneaud J, et al. Association of elevated amyloid and tau positron emission tomography signal with near‐term development of Alzheimer disease symptoms in older adults without cognitive impairment. JAMA Neurol. 2023;79(10):975‐985. doi: 10.1001/jamaneurol.2022.2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ribaldi F, Chicherio C, Altomare D, et al. Brain connectivity and metacognition in persons with subjective cognitive decline (COSCODE): rationale and study design. Alzheimer's Res Ther. 2021;13(1):1‐8. doi: 10.1186/s13195-021-00846-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 2011;7(3):270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jefferies E, Thompson H, Cornelissen P, Smallwood J. The neurocognitive basis of knowledge about object identity and events: dissociations reflect opposing effects of semantic coherence and control. Philos Trans R Soc B Biol Sci. 2020;375(1791):20190300. doi: 10.1098/rstb.2019.0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shcherbinin S, Schwarz AJ, Joshi A, et al. Kinetics of the tau PET tracer 18F‐AV‐1451 (T807) in subjects with normal cognitive function, mild cognitive impairment, and Alzheimer disease. J Nucl Med. 2016;57(10):1535‐1542. doi: 10.2967/jnumed.115.170027. Published online 2016. [DOI] [PubMed] [Google Scholar]
- 28. Guedj E, Varrone A, Boellaard R, et al. EANM procedure guidelines for brain PET imaging using [18F]FDG, version 3. Eur J Nucl Med Mol Imaging. 2021(0123456789). doi: 10.1007/s00259-021-05603-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boellaard R, Delgado‐Bolton R, Oyen WJG, et al. FDG PET/CT: eANM procedure guidelines for tumour imaging: version 2.0. Eur J Nucl Med Mol Imaging. 2015;42(2):328‐354. doi: 10.1007/s00259-014-2961-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rolls ET, Huang CC, Lin CP, Feng J, Joliot M. Automated anatomical labelling atlas 3. Neuroimage. 2020;206:116189. doi: 10.1016/j.neuroimage.2019.116189. August 2019. [DOI] [PubMed] [Google Scholar]
- 31. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimer's Dement. 2015;11(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimer's Dement. 2017;13(3):205‐216. doi: 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fleisher AS, Pontecorvo MJ, Devous MD, et al. Positron Emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. doi: 10.1001/jamaneurol.2020.0528. Published online 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mishra S, Gordon BA, Su Y, et al. AV‐1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. Neuroimage. 2017;161(July):171‐178. doi: 10.1016/j.neuroimage.2017.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dodich A, Mendes A, Assal F, et al. The A/T/N model applied through imaging biomarkers in a memory clinic. Eur J Nucl Med Mol Imaging. 2020;47(2):247‐255. doi: 10.1007/s00259-019-04536-9 [DOI] [PubMed] [Google Scholar]
- 36. Perani D, Della Rosa PA, Cerami C, et al. Validation of an optimized SPM procedure for FDG‐PET in dementia diagnosis in a clinical setting. NeuroImage Clin. 2014;6:445‐454. doi: 10.1016/j.nicl.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cerami C, Della Rosa PA, Magnani G, et al. Brain metabolic maps in Mild Cognitive Impairment predict heterogeneity of progression to dementia. NeuroImage Clin. 2015;7:187‐194. doi: 10.1016/j.nicl.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Teune LK, Bartels AL, De Jong BM, et al. Typical cerebral metabolic patterns in neurodegenerative brain diseases. Mov Disord. 2010;25(14):2395‐2404. doi: 10.1002/mds.23291 [DOI] [PubMed] [Google Scholar]
- 39. Morbelli S, Bauckneht M, Arnaldi D, et al. 18F–FDG PET diagnostic and prognostic patterns do not overlap in Alzheimer's disease (AD) patients at the mild cognitive impairment (MCI) stage. Eur J Nucl Med Mol Imaging. 2017;44(12):2073‐2083. doi: 10.1007/s00259-017-3790-5 [DOI] [PubMed] [Google Scholar]
- 40. Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG‐PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32(7):1207‐1218. doi: 10.1016/j.neurobiolaging.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. FreeSurfe Fischl. Neuroimage. 2012;62(2):774‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mattsson‐Carlgren N, Leuzy A, Janelidze S, et al. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology. 2020;94(21):e2233‐e2244. doi: 10.1212/WNL.0000000000009485 Published online 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38(1):95‐113. [DOI] [PubMed] [Google Scholar]
- 44. Schneider LS, Kennedy RE, Wang G, Cutter GR. Differences in Alzheimer disease clinical trial outcomes based on age of the participants. Neurology. 2015;84(11):1121‐1127. doi: 10.1212/WNL.0000000000001376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xia M, Wang J, He Y. BrainNet Viewer: a network visualization tool for human brain connectomics. PLoS One. 2013;8(7):e68910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chételat G. Alzheimer disease: aβ‐independent processes‐rethinking preclinical AD. Nat Rev Neurol. 2013;9(3):123‐124. doi: 10.1038/nrneurol.2013.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frisoni GB, Altomare D, Thal DR, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci. 2022;23(1):53‐66. doi: 10.1038/s41583-021-00533-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hampel H, Cummings J, Blennow K, Gao P, Jack CR, Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol. 2021;17(9):580‐589. doi: 10.1038/s41582-021-00520-w [DOI] [PubMed] [Google Scholar]
- 49. Nobili F, Arbizu J, Bouwman F, et al. European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain 18F‐fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: delphi consensus. Eur J Neurol. 2018;25(10):1201‐1217. [DOI] [PubMed] [Google Scholar]
- 50. Groot C, Villeneuve S, Smith R, Hansson O, Ossenkoppele R. Tau PET imaging in neurodegenerative disorders. J Nucl Med. 2022;63(6):20S‐26S. doi: 10.2967/jnumed.121.263196 [DOI] [PubMed] [Google Scholar]
- 51. Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol Off J Am Neurol Assoc Child Neurol Soc. 1991;30(4):572‐580. [DOI] [PubMed] [Google Scholar]
- 52. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207‐216. doi: 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72(4):578‐586. doi: 10.1002/ana.23650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strom A, Iaccarino L, Edwards L, et al. Cortical hypometabolism reflects local atrophy and tau pathology in symptomatic Alzheimer's disease. Brain. 2022;145(2):713‐728. doi: 10.1093/brain/awab294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Villemagne VL, Lopresti BJ, Doré V, et al. What is T+? A gordian knot of tracers, thresholds, and topographies. J Nucl Med. 2021;62(5):614‐619. doi: 10.2967/jnumed.120.245423 [DOI] [PubMed] [Google Scholar]
- 56. Sala A, Caprioglio C, Santangelo R, et al. Brain metabolic signatures across the Alzheimer's disease spectrum. Eur J Nucl Med Mol Imaging. 2019. [DOI] [PubMed] [Google Scholar]
- 57. Saint‐Aubert L, Almkvist O, Chiotis K, Almeida R, Wall A, Nordberg A. Regional tau deposition measured by [ 18 F] THK5317 positron emission tomography is associated to cognition via glucose metabolism in Alzheimer ’ s disease. Alzheimers Res Ther. 2016:1‐9. doi: 10.1186/s13195-016-0204-z. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ossenkoppele R, Prins ND, Pijnenburg YAL, et al. Impact of molecular imaging on the diagnostic process in a memory clinic. Alzheimer's Dement. 2013;9(4):414‐421. doi: 10.1016/j.jalz.2012.07.003 [DOI] [PubMed] [Google Scholar]
- 59. Ossenkoppele R, Smith R, Ohlsson T, et al. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology. 2019;92(6):e601‐e612. doi: 10.1212/WNL.0000000000006875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ossenkoppele R, Iaccarino L, Schonhaut DR, et al. Tau covariance patterns in Alzheimer ’ s disease patients match intrinsic connectivity networks in the healthy brain. NeuroImage Clin. 2019;23:101848. doi: 10.1016/j.nicl.2019.101848. September 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chételat G, Desgranges B, Landeau B, et al. Direct voxel‐based comparison between grey matter hypometabolism and atrophy in Alzheimer's disease. Brain. 2008;131(1):60‐71. doi: 10.1093/brain/awm288 [DOI] [PubMed] [Google Scholar]
- 62. Ossenkoppele R, Smith R, Mattsson‐Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal alzheimer disease: a head‐to‐head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78(6):961‐971. doi: 10.1001/jamaneurol.2021.1858. Published online 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood‐based biomarkers for Alzheimer's disease: towards clinical implementation. Lancet Neurol. 2022;21(1):66‐77. doi: 10.1016/S1474-4422(21)00361-6 [DOI] [PubMed] [Google Scholar]
- 65. Smith R, Cullen NC, Pichet Binette A, et al. Tau‐PET is superior to phospho‐tau when predicting cognitive decline in symptomatic AD patients. Alzheimer's Dement. 2022:1‐11. doi: 10.1002/alz.12875. October. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leuzy A, Smith R, Cullen NC, et al. Biomarker‐based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2022;79(2):149‐158. doi: 10.1001/jamaneurol.2021.4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Groot C, Smith R, Stomrud E, et al. Phospho‐tau with subthreshold tau‐PET predicts increased tau accumulation rates in amyloid‐positive individuals. Brain. 2022;146(4):1580‐1591. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Altomare D, Stampacchia S, Ribaldi F, et al. Plasma biomarkers for Alzheimer's disease: a field‐test in a memory clinic. Submitted. 2022:1‐8. doi: 10.1136/jnnp-2022-330619. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Roalf DR, Moberg PJ, Xie SX, Wolk DA, Moelter ST, Arnold SE. Comparative accuracies of two common screening instruments for classification of Alzheimer's disease, mild cognitive impairment, and healthy aging. Alzheimer's Dement. 2013;9(5):529‐537. doi: 10.1016/j.jalz.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mormino EC, Papp KV, Rentz DM, et al. Early and late change on the preclinical Alzheimer's cognitive composite in clinically normal older individuals with elevated amyloid β. Alzheimer's Dement. 2017;13(9):1004‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information