

Abstract
INTRODUCTION
Biomarkers of TDP‐43 pathology are needed to distinguish frontotemporal lobar degeneration with TDP‐43 pathology (FTLD‐TDP) from phenotypically related disorders. While normal physiological TDP‐43 is not a promising biomarker, low‐resolution techniques have suggested truncated forms of TDP‐43 may be specific to TDP‐43 pathology. To advance biomarker efforts for FTLD‐TDP, we employed a high‐resolution structural technique to characterize TDP‐43 post‐translational modifications in FTLD‐TDP.
METHODS
High‐resolution mass spectrometry was used to characterize TDP‐43 proteoforms in brain tissue from FTLD‐TDP, non‐TDP‐43 dementias and neuropathologically unaffected cases. Findings were then verified in a larger cohort of FTLD‐TDP and non‐TDP‐43 dementias via targeted quantitative mass spectrometry.
RESULTS
In the discovery phase, truncated TDP‐43 identified FTLD‐TDP with 85% sensitivity and 100% specificity. The verification phase revealed similar findings, with 83% sensitivity and 89% specificity.
DISCUSSION
The concentration of truncated TDP‐43 proteoforms—in particular, in vivo generated C‐terminal fragments—have high diagnostic accuracy for FTLD‐TDP.
Highlights
Discovery: Truncated TDP‐43 differentiates FTLD‐TDP from related dementias.
Verification: Truncated TDP‐43 concentration has high accuracy for FTLD‐TDP.
TDP‐43 proteoforms <28 kDa have highest discriminatory power for TDP‐43 pathology.
Keywords: biomarkers, frontotemporal dementia, frontotemporal lobar degeneration, mass spectrometry, post‐translational protein processing, proteoforms, proteomics, TDP‐43
1. BACKGROUND
Frontotemporal dementia (FTD) presents with prominent changes in understanding social cues, behavior, and personality or as a language disorder (i.e., aphasia), and serves as an umbrella term for three clinical syndromes: behavioral variant FTD, and semantic or non‐fluent primary progressive aphasia. 1 The pathology in FTD, that is, frontotemporal lobar degeneration (FTLD), is categorized by specific pathological protein aggregates of: transactive response DNA binding protein (TDP‐43, FTLD‐TDP) in 50% of cases, tau (FTLD‐tau) in 45% of cases, and fused in sarcoma, e‐wing protein, and TATA‐binding proteins (FTLD‐FET) in ∼5% of cases. 2 This heterogeneity in both clinical presentation and pathology of FTD, with little correlation between the two, presents many diagnostic challenges. Moreover, it can be difficult to even differentiate FTD from phenotypically related non‐FTD dementias. 3 , 4 As such, a biomarker that can detect the most common FTD pathology, FTLD‐TDP, and distinguish it from disorders with overlapping clinical phenotypes is highly desired.
Toward that end, TDP‐43 has been investigated as a potential biomarker; however, this protein is neither specific to the central nervous system nor to FTLD‐TDP pathology. 5 Attempts to measure natively structured TDP‐43 in biofluids yielded inconsistent results and insufficient discriminatory power, even when comparing to healthy controls. 6 Other biomarkers, such as plasma phosphorylated tau, neurofilament light chain, glial fibrillary protein, and chitinase‐3‐like protein have been investigated and proven unsuccessful in discriminating FTLD‐TDP from other neurodegenerative diseases including FTLD‐tau. 7 , 8 , 9 , 10 , 11 , 12
RESEARCH IN CONTEXT
Systematic review: Via predominantly low‐resolution techniques like Western blotting, fragments of TDP‐43 in brain tissue, and in particular C‐terminal fragments, have been associated with TDP‐43 pathology. Exploration of these proteoforms as potential biomarkers has only been undertaken in a very small number of samples, with minimal control materials. To improve characterization of disease‐specific proteoforms to advance biomarker efforts, higher resolution techniques, such as mass spectrometry, and a greater number and diversity of cases should be explored.
Interpretation: In the largest cohort report to date, including disease mimics, biomarker discovery efforts identified disease‐specific TDP‐43 proteoforms. Via biomarker verification experiments, quantitative analysis demonstrated high diagnostic accuracy of these biomarkers for the identification of TDP‐43 pathology.
Future directions: This work provides strong evidence for the utility of proteolytically cleaved TDP‐43 proteoforms as a diagnostic biomarker target, and thus provides candidate disease‐specific biomarkers for exploration in biofluids.
Based on the success of disease‐specific biomarkers for other neurodegenerative diseases, like phosphorylated‐tau and amyloid‐β 1‐42 in Alzheimer's disease (AD), post‐translationally modified forms of TDP‐43 may also hold promise in TDP‐43 pathologies. Ubiquitinated, sumolyated, phosphorylated, cleaved, and aggregated proteoforms have in fact been linked to ALS and FTLD‐TDP pathology. 13 , 14 , 15 , 16 , 17 , 18 Previous efforts to characterize these proteoforms have largely focused on indirect and non‐specific detection methods, such as Western blot analysis where truncated TDP‐43 proteoforms are identified via immunoreactive bands below the expected molecular weight of TDP‐43 (i.e., <43 kDa). The observation of banding at ∼25 and ∼35 kDa in TDP‐43‐pathology‐affected brain tissue, gave rise to the terminology “TDP‐35” and “TDP‐25”. 14 , 15 , 17 , 18 Given that the poor specificity of commercial anti‐TDP‐43 antibodies has been documented, 5 , 19 , 20 caution should be exercised in the interpretation of anti‐TDP‐43 Western blots. 21 It is not surprising then that the actual structures of the purported “25 kDa” and “35 kDa” TDP‐43 proteoforms have yet to be resolved through higher resolution techniques.
In addition to data on TDP‐43 proteoforms from Western blots, a small number of mass spectrometric studies have been reported. These studies used small sample sizes, in some cases without controls, and yielded inconsistent and unverified findings. To overcome the challenges in TDP‐43 proteoform characterization, we applied an untargeted proteomics approach to brain tissue to identify TDP‐43 proteoforms in the largest cohort studied to date which included relevant neuropathologic controls. We then verified these findings using our targeted multiple reaction monitoring (MRM) method for TDP‐43 proteoform quantitation. Herein we describe the identification of candidate biomarkers—in vivo TDP‐43 proteolytic fragments—which differentiate FTLD‐TDP cases from both related dementias and unaffected controls with high diagnostic accuracy.
2. METHODS
2.1. Biospecimens and case selection
This study was undertaken with Providence Health Care Research Institute and University of British Columbia research ethics board approval. Frontal lobe brain tissue was collected at autopsy, fresh‐frozen, and stored at −70°C until analysis. For every sample used in this study, immunohistochemical (IHC) analysis was performed for α‐synuclein (Invitrogen 180215), amyloid‐β (Dako M08720), phosphorylated‐tau (Thermo Fisher MN1020), TDP‐43 (ProteinTech 10782‐2‐AP), and ubiquitin (Dako Z0458) (Table S1). Additional materials used can be found in the Supplemental Materials.
For biomarker discovery experiments, cases with pure pathologies were selected, that is, cases with characteristic primary pathologies without IHC evidence of secondary protein misfolding pathologies. All FTLD‐TDP cases demonstrated characteristic TDP‐43 aggregation in the frontal and temporal lobes (Table S1). Control tissues of related dementias were IHC positive for the characteristic protein aggregate and negative for TDP‐43 pathology except for one FTLD‐tau case with mild hippocampal‐restricted TDP‐43 staining (Table S1, Figure 1A). Unaffected controls were negative for pathological protein aggregates. For biomarker verification experiments, cases with both mixed and pure pathologies were selected, and were classified as TDP‐43 positive or negative based on the primary pathology.
FIGURE 1.
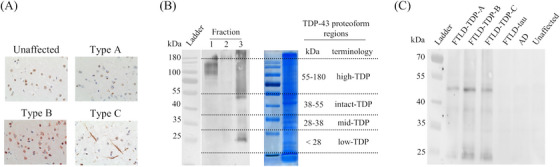
Biochemical characterization of pathological TDP‐43 proteoforms. (A) Representative characteristic immunohistochemical TDP‐43 staining of FTLD‐TDP and control tissues. (B) Tissue fractionation led to enrichment of pathological TDP‐43 in the insoluble fraction (i.e., fraction 3) and TDP‐43 proteoforms were excised from the sodium dodecyl sulfate‐polyacrylamide gel electrophoresis for high‐resolution mass spectrometry analysis. (C) Representative Western blot analysis of the pooled samples subjected to mass spectrometric analysis
Brain tissue was homogenized, and chemically fractionated prior to further analysis (see Supplemental Materials). For electrophoresis, duplicate gels were run with one used for Western blot confirmation of the enrichment of pathological TDP‐43 and to define regions for excision from the paired gel for mass spectrometry analysis (see Supplemental Materials). Four molecular weight regions were excised: 55–180 (high molecular weight [high‐TDP]), 38–55 (intact [intact‐TDP]), 28–38 (mid‐molecular weight [mid‐TDP]), and <28 kDa (low molecular weight [low‐TDP]) (Figure 1B).
2.2. Discovery phase: Unbiased discovery proteomics
Peptide extracts, re‐constituted in 0.1% FA containing 0.5% ACN, were analyzed by nanoflow LC‐QTOF mass spectrometry as previously described. 22 Mass spectrometry data analysis was performed using Byonic 3.4.0 and label‐free quantification was performed via MaxQuant 1.5.1.0 software using a 1% false discovery rate (FDR) against the UniProt Human entries database.
Identification of in vivo TDP‐43 proteolytic fragments was made based on the identification of non‐tryptic or non‐chymotryptic peptide termini, as described in Supplemental Materials. These peptides are denoted as TDP |xxx ‐xxx or TDPxxx‐ xxx| , where “|” and the bold type is used to denote the in vivo cleavage and, thus, either the N‐ or C‐in vivo terminus of the TDP‐43 proteoform. These identifications were further denoted as either high or low confidence identifications based on supporting data.
2.3. Verification phase: Quantitative targeted mass spectrometry
For the verification study, 51 cases (24 cases of TDP‐43 pathology and 27 cases without primary TDP‐43 pathology [Table S1]) were blinded and analyzed using our quantitative targeted mass spectrometry assay. Samples were processed in the same manner as the discovery cohort, except pestle homogenization was done with a motorized pestle, and at the SPE stage, extraction was performed using cation‐exchange mixed‐mode plates for higher throughput. Dried pellets were resuspended in 50 µL of 0.1% FA, 10 µL were used for each injection.
Quantification of TDP‐43 peptides was performed using our previously developed targeted high performance liquid chromatography‐mass spectrometry (LC‐MS/MS) method, 21 , 23 adapted for μLC‐MS/MS for increased analytical sensitivity and decreased sample volume requirements (see Supplemental Materials and Figure S1). In the MRM method, one quantifier and one qualifier ion are monitored per peptide, and quantification is performed via a single‐point internal calibrator via the addition of isotopically labeled internal standards (IS) of TDP‐43 peptides to each sample (Tables S2 and S3).
2.4. Statistical analysis
Sample groups were compared using a Kruskal–Wallis test with Bonferroni correction for multiple comparisons. Graphs were generated using GraphPad Prism or Microsoft Excel. Logistic regression was performed using R. Statistics were calculated using GraphPad Prism. p‐values less than 0.05 were considered significant.
3. RESULTS
3.1. Discovery: Identification of FTLD‐TDP‐specific TDP‐43 proteoforms
Phosphorylated TDP‐43 Western blot analysis confirmed enrichment of pathological TDP‐43 proteoforms in brain tissue homogenate. For FTLD‐TDP cases only (sarkosyl‐insoluble fraction), we observed smearing in the high molecular weight regions, bands around 55 and 20 kDa in FTLD‐TDP cases—all consistent with enrichment of pathological TDP‐43 proteoforms (Figure 1C).
Via high‐resolution MS (HRMS) analysis of pooled sample, the high‐TDP, intact‐TDP, and mid‐TDP regions were found to have similar TDP‐43 sequence coverage across all case types (Figure 2). Examining the low‐TDP fraction across FTLD‐TDP and the control groups, C‐terminal TDP‐43 peptides were more frequently detected in FTLD‐TDP cases. Comparing the four molecular weight fractions in the FTLD‐TDP cases, the low‐TDP fraction had reduced N‐terminal sequence coverage (Figure 2). As no substantial differences in proteolytic cleavages or sequence coverage were observed in the high‐TDP fraction, this fraction was not analyzed further.
FIGURE 2.

Sequence coverage of TDP‐43 in pooled brain tissue subjected to molecular weight fractionation by gel electrophoresis followed by high‐resolution mass spectrometric analysis. Gray bar represents the sequence of full‐length TDP‐43 (residues 1‐414) with proteolytic peptides detected using trypsin (dark blue) and chymotrypsin (light blue)
3.2. Discovery: Quantification of TDP‐43 proteoforms
Intact‐TDP concentration based on normalized relative peak area was similar across the 26 cases studied (p = 0.108; Figure 3A). Mid‐TDP and low‐TDP concentrations were increased in FTLD‐TDP types A‐C compared to related dementia and unaffected controls (p = 0.003 and 0.0003, respectively; Figure 3A). Notably, there was no apparent trend in TDP‐43 proteoform concentration when comparing the FTLD‐TDP types A‐C in the overall FTLD‐TDP group, nor when comparing FTLD‐tau with AD in the disease mimic group (Figure S2). The relative concentrations of mid‐TDP:intact‐TDP and low:intact‐TDP were not correlated (Pearson's correlation = 0.06 and −0.14, respectively; Figure S3), whereas mid:low‐TDP had a strong positive correlation (Pearson's correlation = 0.79; Figure S3). The area under the curve (AUC) of the receiver operating characteristic curve (ROC) for low‐ and mid‐TDP were 0.92 and 0.89, respectively, and 0.91 for total truncated TDP‐43 proteoforms (i.e., low‐ and mid‐TDP) (Figure 3B). Low‐TDP concentrations, identified FTLD‐TDP with 89% sensitivity and 100% specificity and total truncated TDP‐43 proteoform concentration identified FTLD‐TDP cases with 100% sensitivity and 84% specificity (Figure 3B).
FIGURE 3.
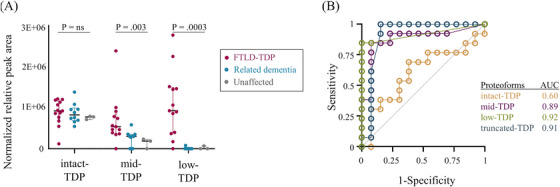
In the biomarker discovery phase, low‐TDP and mid‐TDP proteoforms distinguished FTLD‐TDP from controls as demonstrated by (A) normalized relative peak area and (B) receiver operating characteristics curve analysis
3.3. Discovery: TDP‐43 in vivo proteolytic cleavage sites
Eight in vivo proteolytic cleavage sites were identified from HRMS experiments in both pooled and individual samples (Figure 4). After examination of additional supporting evidence (i.e., in source fragmentation, cleavage specificity of known human proteases, and previous reports either in vitro or in vivo), this list was reduced to one high‐confidence target consisting of a C‐terminal fragment with N‐termini beginning at residue TDP| 280 , where the “|” denotes the in vivo cleavage site (Table 1). The TDP| 280 site was identified in the low‐TDP fraction of FTLD‐TDP type A, B, and C pooled samples and none of the controls through the peptide TDP| 280 ‐293 (Table 1). For individual cases analyzed, this truncation site was again found in FTLD‐TDP cases, across all subtypes, (n = 4), and not identified in control cases (n = 13).
FIGURE 4.

Identification of in vivo proteolytic fragments of TDP‐43 in FTLD‐TDP cases. TDP‐43 sequence (gray) with C‐ and N‐terminal in vivo truncation sites noted (magenta and cyan, respectively), with confidence level noted based on multiple supporting data points
TABLE 1.
Determination of in vivo TDP‐43 cleavage sites in FTLD‐TDP via the identification of semi‐tryptic/semi‐chymotryptic peptides
| Human protease cleavage site b | HRMS data | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TDP‐43 residues | Confidence level | C or N term a | TDP‐43 specific peptide | Predicted | Empirical | ISF ruled out c | MW gel fraction (kDa) | Maximal predicted MW (kDa) d | Appropriate MW (kDa) e | IDs f | Disease type |
| 28–42 | Low | C | ✓ | ✓ | х | nd | <28 | 41–163 | х | 2 | TDP‐A/C |
| 30–42 | Low | C | ✓ | х | х | nd | 38–55 | 41–163 | ✓ | 1 | Unaffected |
| 31–42 | Low | C | ✓ | ✓ | х | nd | 38–55 | 41–163 | ✓ | 1 | TDP‐C |
| 254–263 | Low | C | ✓ | ✓ | х | х | <28 | 16–18 | na | na | TDP‐B |
| 266–276 | Low | C | ✓ | х | х | nd | 38–55 | 15–17 | х | 5 | TDP‐A/B/C, tau, AD |
| 280–293 | High | C | ✓ | ✓ | ✓ 23 | ✓ | <28 | 13–15 | ✓ | 7 | TDP‐A/B/C |
| 294–316 | Low | N | ✓ | ✓ | х | nd | 38–55 | 35–157 | ✓ | 2 | TDP‐B |
| 304–313 | Low | C | ✓ | ✓ | х | ✓ | 38–55 | 11–12 | х | 1 | TDP‐C |
| 341–360 | Low | C | ✓ | ✓ | х | nd | 38–55 | 10–11 | х | 1 | TDP‐C |
In vivo cleavage site (i.e., non‐tryptic/chymotryptic site) on the N‐ or C‐terminus of the peptide.
Predicted or empirical evidence of human enzymatic cleavage site based on Expasy data and previously published experiments, respectively.
In source fragmentation (ISF) ruled out (✓) or in (x) using retention time differences and relative alterations in hydrophobicity.
Molecular weight of the protein sequence based on the in vivo truncation site is consistent with the gel electrophoresis band region from which the peptide was found, including consideration of other additive potential post‐translational modifications (e.g., phosphorylation).
Peptide maximal molecular weight (without—with maximal predicted PTMs) and including empirical evidence.
Number of times the peptide was identified in both pooled and individual experiments.
We then further examined our data for semi‐specific TDP‐43 peptides in previously published proteomic studies of brain tissue. We considered only studies that utilized appropriate mass spectrometric data search parameters, semi‐specific enzymes, and two or fewer missed cleavage sites (Table S4). Of the nine peptides previously reported in the literature, we identified two peptides: TDP |266 ‐276 and TDP |254 ‐263. TDP |266 ‐276 was deemed a low confidence identification as we could not rule out in source fragmentation, and no human enzyme is predicted to cleave TDP‐43 at this site (Table 1). We determined TDP |254 ‐263 to be an experimental artifact arising from in source fragmentation as it had a similar retention time to its fully‐tryptic counterpart, with retention times of 25.64 and 25.67 minutes, respectively.
3.4. Verification: truncated TDP‐43 proteoforms as a biomarker of FTLD‐TDP pathology
Using our quantitative targeted MRM method for TDP‐43 proteoforms, we verified the diagnostic performance of mid‐TDP, low‐TDP, and total truncated TDP‐43 proteoforms as biomarkers of FTLD‐TDP. From the mid‐ and low‐TDP fractions, TDP‐43 peptide concentrations were higher in FTLD‐TDP compared to related dementias (Figure 5A,B). As observed in the discovery phase, low‐TDP concentration had the largest effect size, with the median TDP‐43 concentration over 1000‐times higher in FTLD‐TDP cases (Figure 5A). Notably, low‐TDP was below the limit of the measuring interval in only 17% of FTLD‐TDP cases but 89% of controls. Similar to the discovery phase, the concentration of mid‐ and low‐TDP did not differ within the subgroups of either the FTLD‐TDP or non‐TDP control groups (Figure S2). Additionally, the concentration of the low‐ and mid‐TDP proteoforms were positively correlated (Figure S3). Mid‐TDP had insufficient diagnostic power (AUC 0.60), whereas low‐TDP identified FTLD‐TDP cases from related non‐primary TDP‐43 dementias and unaffected controls with 83% sensitivity and 89% specificity and an AUC of 0.85 (Figure 5C). Low‐TDP combined with mid‐TDP identified FTLD‐TDP cases with 79% sensitivity and 100% specificity and an AUC of 0.90 (Figure 5C).
FIGURE 5.
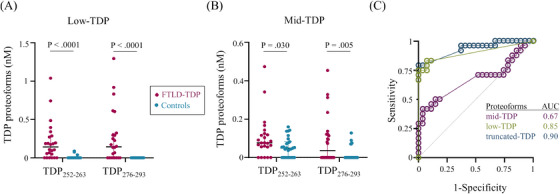
Targeted MRM analysis of truncated TDP‐43 proteoforms discriminated cases with (magenta) and without (cyan) TDP‐43 pathology as demonstrated by: (A,B) TDP‐43 concentration in the (A) low‐TDP and (B) mid‐TDP fractions, and (C) ROC curve analysis
4. DISCUSSION
Herein, we report the first combined biomarker discovery and verification study for an FTLD‐TDP specific biomarker. Brain tissue was selected as the preferred biospecimen for biomarker discovery efforts, as it enabled the use of specimens with confirmed presence/absence of the target pathology and due to the high relative concentration of the target pathology. The latter is particularly important in enabling investigations of TDP‐43 post‐translation modifications, which are anticipated to be in relatively low abundance compared to the natively structured intact TDP‐43.
4.1. Discovery: characterization of TDP‐43 proteoforms
Discovery analysis of TDP‐43 proteoforms, encapsuling intact‐TDP (38–55 kDa), mid‐TDP (28–38 kDa), and low‐TDP (<28 kDa), revealed that the concentration of truncated TDP‐43 differentiated FTLD‐TDP from related dementias and unaffected controls. Both mid‐ and low‐TDP proteoforms were significantly increased in FTLD‐TDP cases compared to related dementia and unaffected cases, however, low‐TDP had the better diagnostic accuracy owing to the larger relative increase in concentration. Using total truncated TDP‐43 proteoforms (low and mid) concentration provided no additional discriminatory power, likely owing to their positive correlation. Focusing on the best performing biomarker, low‐TDP, this biomarker resulted in no false positives and two false negatives of the 26 cases studied, with the latter comprised of two FTLD‐TDP type C cases that had low‐TDP concentrations below the lower limit of the measuring interval.
As anticipated, intact‐TDP proteoforms did not differentiate FTLD‐TDP from related dementias or unaffected controls, and intact‐TDP proteoform concentration did not correlate with either mid‐ or low‐TDP proteoform concentration. As we previously demonstrated in cell models that a ratio of the concentrations of C‐to‐N‐terminal TDP‐peptides can be used to detect TDP‐43 truncations, 21 we applied this analysis to this current cohort using the most extreme N‐ and C‐terminal peptides to create a C:N terminal total TDP‐43 profile (Figure S4); however, in human brain tissues, these internal peptide ratios did not provide any discrimination between FTLD‐TDP and non‐TDP pathological cases.
Consistent with other experimental data points from orthogonal techniques, including Western blot analyses and cryo‐electron microscopy, we identified a highly confident in vivo cleavage site of TDP‐43, corresponding to a C‐terminal fragment. This cleaved proteoform, TDP |280‐ 293, was detected across all FTLD‐TDP subtypes and not detected in controls. Notably, the N‐terminal cleavage site is consistent with the TDP‐43 fibril core (i.e., TDP282‐360) recently discovered in ALS‐FTD. 24 The TDP282‐360 fibril core does not preclude the sequence from extending further to the N‐terminus, as part of structure determination requires ragged ends to be trimmed in vitro by proteases to facilitate crystallization. In vivo, asparagine endopeptidase (cleavages N‐terminal to Asn) could create the fragment we observed, as residue 280 is an asparagine. Additional supporting evidence includes detection of TDP |280 ‐293 in a HEK293 cell model overexpressing TDP‐43 through mass spectrometric detection of the TDP |280 ‐293 peptide. 14 The pathophysiological implications of the cleavage of TDP‐43 at residue 280 requires further investigation but as this truncation would result in the loss of the nuclear import and export domains, as well as both RNA binding domains, normal nuclear‐cytoplasmic shuttling and RNA binding capacity would be lost.
We investigated an additional 14 TDP‐43 cleavage sites; however, all other cleavage sites were deemed low confidence because of a paucity of supporting evidence (Table 1 and Table S4). This finding highlights the need for proteomic data such as these to be carefully analyzed following best‐practices 25 and to incorporate, where possible, other supporting evidence to avoid over‐interpreting the data (i.e., claiming identification of more PTMs than the data supports). These 14 cleavage sites included 7 discovered herein as well as seven additional cleavage sites from four studies which reported discovery of new in vivo proteolytic fragments of TDP‐43. 13 , 14 , 15 , 18 These inconsistencies in claimed peptide identifications between our studies and those previously reported could be attributed to several factors, including that previous studies: (1) used much smaller sample sizes and in some cases no controls, (2) omitted rigorous data analysis procedures (e.g., consideration of in‐source fragmentation, and/or use of defined false‐discovery rates), (3) omitted verification of discovery findings through an alternative method, and (4) omitted the incorporation of supporting evidence from the literature (e.g., cleavage specificity of human proteases and evidence from in vitro studies). Given the number of confounding factors in the identification of confident in vivo proteolytic cleavage sites from mass spectrometric data, stringent data quality assessments, as followed herein, are a necessity.
4.2. Biomarker verification: truncated TDP‐43 proteoforms
After discovery, we pursued the important next step of biomarker verification. Notably, less than 10% of biomarker studies are advanced to the verification phase; thus, biomarker discovery efforts struggle to impact biomarker development. 26 , 27 Absolute quantification revealed that both mid‐ and low‐TDP in vivo proteolytic fragments were increased in FTLD‐TDP cases compared to related dementia cases, and as in the discovery cohort, low‐TDP had the highest diagnostic accuracy (AUC 0.92). Using total truncated TDP‐43 proteoforms concentration (i.e., low and mid) to discriminate between FTLD‐TDP and control cases provided an increase in specificity (89% to 100%) with slightly decreased sensitivity (83% to 79%). In assessing the relationship of low‐TDP concentration with age at death, sex, and genetic status in FTLD‐TDP cases, we found no strong correlation with or significant differences among these factors and low‐TDP concentration (Figure S5).
Examining the specific cases in more detail, using low‐TDP to discriminate between FTLD‐TDP cases and controls resulted in only two false positives and four false negatives. False positives included three AD cases, all with relatively low concentrations of the TDP252‐263 peptide and no detection of TDP276‐293; thus, establishing a concentration cut‐off of TDP252‐263 could boost specificity. False negatives included three FTLD‐TDP type C cases and one case of mild FTLD‐TDP type B pathology. In three of four of the false negatives, low‐TDP was detected but peptide transitions were not within predefined quality control limits (i.e., <15% of the expected peak area ratio), suggesting greater analytical sensitivity may be helpful in discriminating these cases. The neuropathological reports for the false negative type C cases were consistent with the common pathological finding in type C of milder pathology in the frontal cortex as compared to types A and B 28 , 29 , 30 ; this lower pathological burden likely contributed to the low‐TDP signal from these cases not meeting the pre‐specified analytical performance metrics. Using a biomarker combination of low‐TDP and mid‐TDP proteoforms eliminated false positives; however, false negatives rose to five cases. These five cases included the four mentioned in the low‐TDP analysis, and one FTLD‐TDP type A case. Analysis of low‐ and mid‐TDP concentration by subtype revealed no trends, thus, we cannot attribute these false negatives to subtype differences. TDP‐43 peptides were not quantifiable in the low‐TDP fraction in secondary TDP‐43 pathology restricted to the hippocampus or in FTLD‐TDP with mild pathology.
4.3. Strengths and limitations
To date, this is the largest proteomics study on immunohistochemically confirmed cases of FTLD‐TDP, which were compared to a range of relevant controls (i.e., phenotypically related dementias). Herein, we provide evidence of a biomarker specific for FTLD‐TDP pathology, a condition which currently has no disease‐specific biomarkers. We applied stringent data quality assessment criteria in the discovery and verification phases. Further, this is the first verification study of a TDP‐43‐based biomarker. Limitations of this study include that the cases used were at the end stage of disease; thus, it is unknown if they are also reflective of earlier disease stages, and general limitations in a bottom‐up mass spectrometric approach including limitations in observable peptides (i.e., sequence coverage) based on enzymes used in preparation for analysis. For the latter, we employed two different in vitro proteolytic enzymes in order to increase sequence coverage.
While non‐specific biomarkers of neurodegeneration have shown promise in detecting neurodegeneration in the broad group of molecular pathologies that comprise FTD, there remains a strong need for diagnostic biomarkers of FTD subgroups, including the largest subgroup, FTLD‐TDP. We found that truncated TDP‐43 proteoforms differentiate FTLD‐TDP from both related‐dementia and unaffected control cases with high diagnostic accuracy. We identified low molecular weight, C‐terminal fragments of TDP‐43 as the most promising proteoform. The knowledge of specific TDP‐43 proteoforms of pathological significance is of particularly high value in the translation of biomarker findings from post‐mortem investigations to biofluid diagnostics. For instance, knowledge of the structural and physiochemical properties of a target analyte can be leveraged to design enrichment strategies and optimize analytical procedures for large gains in analytical sensitivity. Analytical sensitivity along with molecular selectivity, based on our experience in developing TDP‐43 proteoform assays, 21 , 23 will be critical to these translational efforts. Thus, these new findings provide additional molecular‐level evidence needed to advance a much sought after antemortem, pathology‐specific biomarker for FTD with TDP‐43 pathology.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Written informed consent was obtained from all human subjects or, if applicable, their legally authorized representative (i.e., substitute decision‐maker).
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
Lauren M. Forgrave is supported by a fellowship from the Canadian Institutes for Health Research [201911FB‐D43495‐2295330, 2019]. Jordan Hamden was supported by Michael Smith Health Research Trainee Award [RT‐2022‐2717, 2022]. Leonard J. Foster receives operational support from Genome Canada and Genome BC [264PRO, 2019]. Mari L. DeMarco is the recipient of the Michael Smith Health Research Scholar Award [SCH‐2016‐1976, 2017]. Mass spectrometry infrastructure used herein (in the labs of Leonard J. Foster and Mari L. DeMarco) was supported by the Canada Foundation for Innovation and the British Columbia Knowledge Development Fund.
Forgrave LM, Moon K‐M, Hamden JE, et al. Truncated TDP‐43 proteoforms diagnostic of frontotemporal dementia with TDP‐43 pathology. Alzheimer's Dement. 2024;20:103–111. 10.1002/alz.13368
REFERENCES
- 1. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546‐1554. [DOI] [PubMed] [Google Scholar]
- 2. Radford RA, Morsch M, Rayner SL, Cole NJ, Pountney DL, Chung RS. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front Cell Neurosci. 2015;9(414). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seo SW, Thibodeau MP, Perry DC, et al. Early vs late age at onset frontotemporal dementia and frontotemporal lobar degeneration. Neurology. 2018;90(12):e1047‐e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329‐3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feneberg E, Steinacker P, Lehnert S, et al. Limited role of free TDP‐43 as a diagnostic tool in neurodegenerative diseases. Amyotroph Lateral Scler. 2014;15(5‐6):351‐356. [DOI] [PubMed] [Google Scholar]
- 6. Majumder V, Gregory JM, Barria MA, Green A, Pal S. TDP‐43 as a potential biomarker for amyotrophic lateral sclerosis: a systematic review and meta‐analysis. BMC Neurology. 2018;18(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Del Campo M, Galimberti D, Elias N, et al. Novel CSF biomarkers to discriminate FTLD and its pathological subtypes. Ann Clin Transl Neurol. 2018;5(10):1163‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baldacci F, Lista S, Cavedo E, Bonuccelli U, Hampel H. Diagnostic function of the neuroinflammatory biomarker YKL‐40 in Alzheimer's disease and other neurodegenerative diseases. Expert Rev Proteomics. 2017;14(4):285‐299. [DOI] [PubMed] [Google Scholar]
- 9. Ishiki A, Kamada M, Kawamura Y, et al. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer's disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J Neurochem. 2016;136(2):258‐261. [DOI] [PubMed] [Google Scholar]
- 10. Heller C, Foiani MS, Moore K, et al. Plasma glial fibrillary acidic protein is raised in progranulin‐associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91(3):263‐270. [DOI] [PubMed] [Google Scholar]
- 11. Forgrave LM, Ma M, Best JR, DeMarco ML. The diagnostic performance of neurofilament light chain in CSF and blood for Alzheimer's disease, frontotemporal dementia, and amyotrophic lateral sclerosis: a systematic review and meta‐analysis. Alzheimers Dement (Amst). 2019;11:730‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. 2019;25(2):277‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feneberg E, Charles PD, Finelli MJ, et al. Detection and quantification of novel C‐terminal TDP‐43 fragments in ALS‐TDP. Brain Pathol. 2021;31(4):e12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herskowitz JH, Gozal YM, Duong DM, et al. Asparaginyl endopeptidase cleaves TDP‐43 in brain. Proteomics. 2012;12(15‐16):2455‐2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Igaz LM, Kwong LK, Chen‐Plotkin A, et al. Expression of TDP‐43 C‐terminal fragments in vitro recapitulates pathological features of TDP‐43 proteinopathies. J Biol Chem. 2009;284(13):8516‐8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laferriere F, Maniecka Z, Perez‐Berlanga M, et al. TDP‐43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci. 2019;22(1):65‐77. [DOI] [PubMed] [Google Scholar]
- 17. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006:314. [DOI] [PubMed] [Google Scholar]
- 18. Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP‐43. Hum Mol Genet. 2009;18(18):3353‐3364. [DOI] [PubMed] [Google Scholar]
- 19. Kasai T, Tokuda T, Ishigami N, et al. Increased TDP‐43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathologica. 2009;117(1):55‐62. [DOI] [PubMed] [Google Scholar]
- 20. Steinacker P, Hendrich C, Sperfeld AD, et al. TDP‐43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Archives of Neurology. 2008;65(11):1481‐1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pobran TD, Forgrave LM, Zheng YZ, Lim JGK, Mackenzie IRA, DeMarco ML. Detection and characterization of TDP‐43 in human cells and tissues by multiple reaction monitoring mass spectrometry. Clin Mass Spectrom. 2019;14:66‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gibbs MR, Moon KM, Chen M, Balakrishnan R, Foster LJ, Fredrick K. Conserved GTPase LepA (elongation factor 4) functions in biogenesis of the 30S subunit of the 70S ribosome. Proc Natl Acad Sci U S A. 2017;114(5):980‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pobran TD, Yang D, Mackenzie IRA, DeMarco ML. Aptamer‐based enrichment of TDP‐43 from human cells and tissues with quantification by HPLC‐MS/MS. J Neurosci Methods. 2021;363:109344. [DOI] [PubMed] [Google Scholar]
- 24. Arseni D, Hasegawa M, Murzin AG, et al. Structure of pathological TDP‐43 filaments from ALS with FTLD. Nature. 2021;601:139‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakayasu ES, Gritsenko M, Piehowski PD, et al. Tutorial: best practices and considerations for mass‐spectrometry‐based protein biomarker discovery and validation. Nat Protoc. 2021;16(8):3737‐3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anderson NL. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin Chem. 2010;56(2):177‐185. [DOI] [PubMed] [Google Scholar]
- 27. Davis KD, Aghaeepour N, Ahn AH, et al. Discovery and validation of biomarkers to aid the development of safe and effective pain therapeutics: challenges and opportunities. Nat Rev Neurol. 2020;16(7):381‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bocchetta M, Espinosa MdM Iglesias, Lashley T, Warren JD, Rohrer JD. In vivo staging of frontotemporal lobar degeneration TDP‐43 type C pathology. Alzheimers Res Ther. 2020;12(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Borghesani V, Battistella G, Mandelli ML, et al. Regional and hemispheric susceptibility of the temporal lobe to FTLD‐TDP type C pathology. Neuroimage Clin. 2020;28:102369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mackenzie IRA, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. 2016;138(S1):54‐70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information